

Review on recent development of quinoline for anticancer activities
⁎Corresponding authors at: School of Advanced Sciences, Department of Chemistry, VIT University, Vellore - 632014, Tamil Nadu, India. aanapoleon@vit.ac.in (Ayyakannu Arumugam Napoleon) napoleonaa@gmail.com (Ayyakannu Arumugam Napoleon)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Quinoline is an efficient scaffold for anticancer drug development as its derivatives have shown potent results through several mechanisms like growth regulators through “apoptosis, disruption of cell migration, inhibition of angiogenesis, modulation of nuclear receptor responsiveness and cell cycle arrest, etc.,” The potential of quinoline derivatives has been proved in several cancer cell lines like breast cancer, colon cancer, lung cancer, colorectal cancer, renal cancer, etc. This review article aims to provide information about the synthesis, potential of the anticancer property, cytotoxicity, and level of clinical treatments, which could lay out the research to develop more effective quinoline-derived anticancer drugs.
Keywords
Cancer
Quinolines
Synthesis
Anticancer activity
Antiproliferative activity

1 Introduction
Cancer is a primary cause of death and a significant hindrance to extending life span across the world. In 2020, 19.3 million cancer cases were recorded, and around 10 million victims have died primarily due to cancer (Sung et al., 2021). It is the most dangerous disease characterized by the uncontrolled growth of cells spreading throughout the body. To date, various cancer causes have been proposed, including genetic factors (Giri et al., 2018) and environmental elements (Rudolph, Chang-Claude and Schmidt, 2016). It also occurs due to the habits of human beings like smoking tobacco (Didkowska et al., 2016), consuming alcohol (Choi, Myung and Lee, 2018), and eating red meat (Zhao et al., 2020), etc., A few kinds of cancer are also caused by Helicobacter pylori (Wroblewski, Peek and Wilson, 2010), HPV (Chan et al., 2019), and HBV infections (Mani and Andrisani, 2018). Anticancer drugs are used to prevent cancer cells from developing and multiplying; if this happens, the tumour cells usually die. for this reason a series of research have been employed to develop a more efficient anti-cancerous drug. Yet it is a continuous process for profound progress in developing a drug without any side effects (Tabassum, Ashfaq and Oku, 2020). Regardless, antitumor drugs like daunorubicin, trastuzumab (Vu and Claret, 2012), sunitinib (Motzer et al., 2017), etc have been authorized by the FDA. But those drugs contain some serious side effects like stasis of the lower bowel, mouth sores, diarrhea, full hair loss, neuropathy, bone marrow suppression, and life-threatening problems, that are difficult to identify. In the recent past, heterocyclic compounds have served as an effective pharmacophore in the treatment of a variety of illnesses (Afzal et al., 2015). Among the heterocyclic compounds, quino line is an efficient scaffold for the development of drugs.
Quinoline is an aromatic heterocyclic organic compound with the chemical formula C9H7N; whose structure is formed by a fusion of benzene ring with pyridine at two neighboring carbon atoms, giving a double-ring structure (Moor et al., 2021). Benzopyridine, benzo[b]pyridine, 1-benzazine, and benzazine are other name of quinoline. It's a hygroscopic, yellowish oily liquid that's soluble in alcohol, ether, and a variety of other solvents.(Jain et al., 2019) Quinoline plays a crucial role in medicinal chemistry due to the presence of nitrogen atom, which pulls electrons by resonance. This bicyclic aromatic compound is an electron-deficient system with weak tertiary base properties. It demonstrates both electrophilic and nucleophilic substitutions, and pyridine exhibits similar substitutions. The clinical observation proves that even when a human inhales a 0.001 ppm level of quinoline for 8 h was perfectly nontoxic and this case is quite opposite to benzene; even a smaller concentration of benzene leads to various biological disorders like cancer.
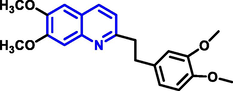
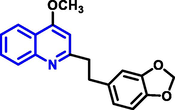
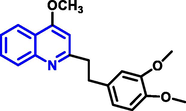
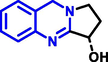
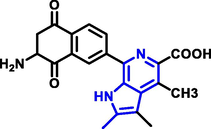
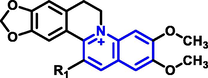
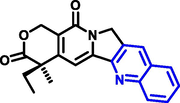
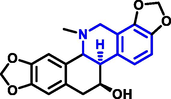
Quinoline can be derived from different natural sources. Naturally existing quinoline alkaloids like Cuspareine, Cusparine, Galipine (Angosturaalkaloide, 1911), Vasicine, Echinopsine, Fabrifugine (Mathada, 2022), Cinchona, Streptonigrin (Rao, Biemann and Woodward, 1963), Nitidine (Douglas and Hughes, 1991), Lavendamycin (Balitz et al., 1982), Chelerythrine, Berberine derivative (Guamán Ortiz et al., 2014), Dictamnine (Mariano et al., 2002), Camptothecin (Mao et al., 2020), and Chelidonine (Santos and Adkilen, 1932). These alkaloids have potent anticancer activity. Table 1 represents the various naturally existing significant quinoline compounds.
| S.No | Chemical Structures | Alkaloids/ Natural sources |
|---|---|---|
| 1 |
|
Cuspareine |
| 2 |
|
Cusparine |
| 3 |
|
Galipine |
| 4 |
|
Vasicine |
| 5 |
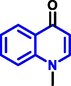
|
Echinopsine |
| 6 |
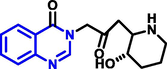
|
Fabrifugine |
| 7 |
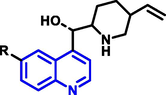
|
Cinchona |
| 8 |
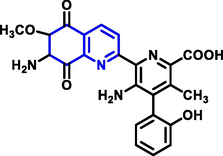
|
Streptonigrin |
| 9 |
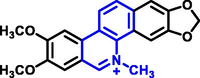
|
Nitidine |
| 10 |
|
Lavendamycin |
| 11 |

|
Chelerythrine |
| 12 |
|
Berberine derivative |
| 13 |
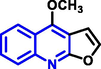
|
Dictamnine |
| 14 |
|
Camptothecin |
| 15 |
|
Chelidonine |
Table 2 represents the marketed anticancer compounds for various cancer. Quinoline is an essential system of several drugs used in therapeutic settings to treat a variety of disorders. Here we addressed the anticancer drug that is found in commercialized and clinical treatments. (Dittrich et al., 2003) (Singh et al., 2015) (Somagond et al., 2018) (Wang et al., 2016).
| S.No | Drug Names | Chemical Structures | Types of cancers |
|---|---|---|---|
| 1 | Neratinib |
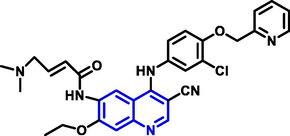
|
Breast cancer |
| 2 | Amsacrine |
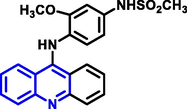
|
Acute leukemia |
| 3 | Quinoline analog |
|
Blood Cancer |
| 4 | Irinotecan |
|
Colorectal cancer |
| 5 | Cabozantinib |
|
Thyroid cancer |
| 6 | Acridine carboxamide |
|
Lunger cancer and brain and central nervous system tumors |
| 7 | Foretinib |
|
Breast cancer |
| 8 | AMG-208 |
|
Prostate cancer |
| 9 | Sitamaquine |
|
Prostate cancer |
| 10 | Talazoparib |
|
Breast cancer |
2 Traditional methods of making quinoline compounds
There are various traditional methods for quinoline synthesis. The majority of the naming reactions are usually used to develop quinolines from simple anilines. Aryl amine is used as one of the precursors in the following naming reactions.
-
Riehm synthesis: When aniline reacts with acetone in the presence of aluminium chloride to form quinoline compound
-
Gould-Jacobs reaction: When aniline reacts with ethyl ethoxy methylene malonate in the presence of Con.H2SO4 to form quinoline compound.
-
Doebner reaction: When aniline reacts with aldehyde and pyruvic acid to form quinoline compound.
-
Doebner-Miller reaction: When aniline reacts with
-
Conrad-Limpach synthesis: When aniline reacts with
-
Combes quinoline synthesis: When aniline reacts with
-
Skraup reaction: When aniline reacts with glycerol and nitrobenzene in the presence of sulfuric acid to form a quinoline compound.
-
Povarov reaction: When aniline reacts with benzaldehyde in the presence of activated alkene to form a quinoline compound. The synthetic approaches of these traditional methods are recapped in the Fig. 1
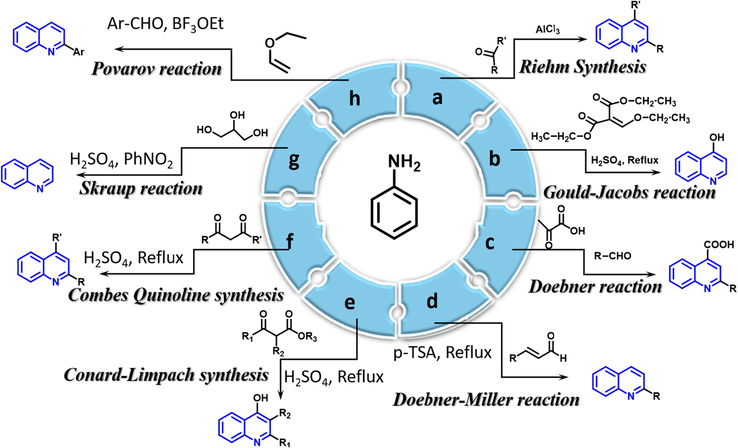
- Traditional methods for synthesis of quinoline compounds.
The remaining naming reactions are commonly used to develop quinoline compounds from substituted anilines or similar precursors in the following reactions.
-
Friedlander synthesis: When 2-amino benzaldehyde reacts with carbonyl compounds in the presence of trifluoroacetic acid to form a quinoline compound.
-
Knorr quinoline synthesis: When
-
Niementowski quinoline synthesis: When anthranilic acid reacts with carbonyl compound to form quinoline compound.
-
Pfitzinger reaction: When isatin reacts with carbonyl compound in the presence of potassium hydroxide as a base to form quinoline compound.
-
Camps quinoline synthesis: When o-acylaminoacetophenone reacts with the hydroxide to form a quinoline compound.
-
Meth-Cohn synthesis: When acylanilides react with dimethyl formamide in the presence of phosphorous oxychloride to form a quinoline compound. Fig. 2 presents an overview of the synthetic schemes of quinoline compounds.
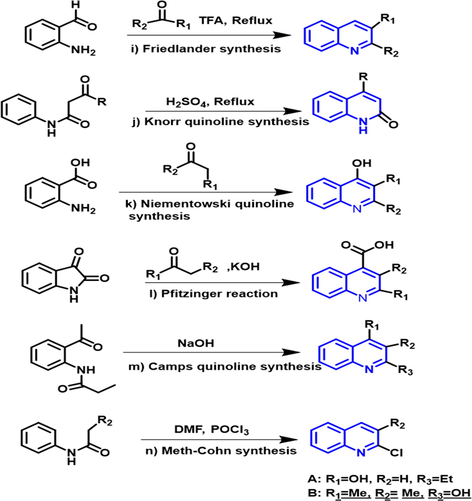
- Synthetic schemes of quinoline compounds.
The ability of a quinoline molecule to preferentially target cancer-specific signals or enzymatic routes will be essential for the development of efficient and safe anticancer treatments (Zhou et al., 2021). The wide application of biochemical and biological properties has been further aided by quinoline synthetic diversity permits the creation of various types of structurally diverse derivatives. Quinoline compounds have wide range of biological activities like anti-bacterial (Ranjbar-Karimi and Poorfreidoni, 2018), anti-fungal (Hamama et al., 2018), antimalarial (Kalaria, Karad and Raval, 2018), anti-leishmanial (Upadhyay et al., 2019), anti-cancer (Jain et al., 2019), etc.
Quinoline and its derivatives were demonstrated to inhibit tyrosine kinases (Karnik, Sarkate, Tiwari, Azad, Burra, et al., 2021), topoisomerase (Elbadawi et al., 2021), tubulin polymerization (Khelifi et al., 2017), and DHODH kinase (Parikh, Ghate and Vyas, 2015) in the latest research. Fig. 3 shows a schematic representation of different types of inhibitors for anticancer activity of quinoline compounds and Table 3 represents the overall outline of quinoline derivatives against various cancers. In this review article, we gathered the antitumor potential of quinoline-based compounds on a goal approach, which will aid biomedical scientists to progress in advanced antitumor medicine.
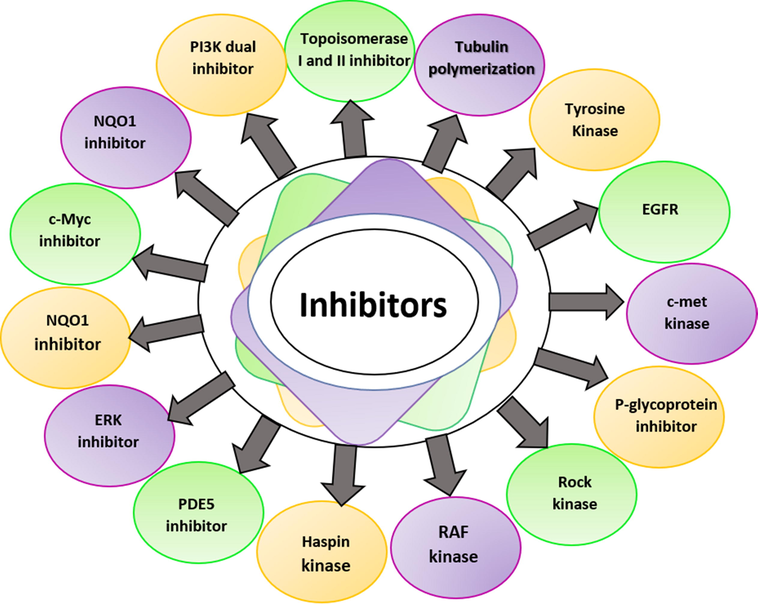
- Various types of inhibitors for anticancer activity.
| Type of Inhibitors/ Combinations | Quinolines | Type of cancers | IC50 values | Ref. |
|---|---|---|---|---|
| Topoisomerase-I [(2-(2-amino-4,5-dimethoxyphenyl)-2-oxoethan-1-ylium and halo substituted benzoyl chloride] | Q1 | Colon cancer, leukemic, and melanoma | 0.875, 0.904 and 0.926
|
(Elbadawi et al., 2021) |
| Topoisomerase-I [(2-(2-amino-4,5-dimethoxyphenyl)-2-oxoethan-1-ylium and halo substituted benzoyl chloride] | Q2 | Melanoma and renal caner | 1.40 and 1.66
|
(Elbadawi et al., 2021) |
| Topoisomerase-I [(2-(2-amino-4,5-dimethoxyphenyl)-2-oxoethan-1-ylium and halo substituted benzoyl chloride] | Q3 | Breast and melanoma | 1.13 and 1.19
|
(Elbadawi et al., 2021) |
| Topoisomerase-I [Bromoaniline and malonic acid] | Q4 | Epithelial carcinoma and breast cancer | 0.44 and 0.62
|
(Kardile et al., 2021) |
| Topoisomerase-I [Bromoaniline and malonic acid] | Q5 | Epithelial carcinoma and breast cancer | 0.69 and 0.54
|
(Kardile et al., 2021) |
| Topoisomerase-I [Diazene and thiocyanate] | Q6 | Breast cancer | 0.82
|
(Ribeiro et al., 2019) |
| Topoisomerase-I [Aromatic aldehyde and (2-aminophenyl)diphenylphosphine oxide ] | Q7 | Lung cancer | 0.25 ± 0.03
|
(Alonso et al., 2018) |
| Topoisomerase-I [Aromatic aldehyde and alkenyl substituted benzene] | Q8 | Lung cancer | 0.08 ± 0.01
|
(Alonso et al., 2018) |
| Topoisomerase-I [Aromatic aldehyde and (2-aminophenyl)diphenylphosphine sulfide] | Q9 | Lung cancer | 0.03 ± 0.04
|
(Alonso et al., 2018) |
| Topoisomerase-I [Aniline and benzaldehyde] | Q13 | Prostate cancer | 4.40
|
(Kouznetsov et al., 2017) |
| Topoisomerase-I [Aniline and benzaldehyde] | Q14 | Cervical cancer | 0.016
|
(Kouznetsov et al., 2017) |
| Topoisomerase-II [Phenyl acetamide and 2-bromothiophene] | Q15 | Breast cancer | 38.41
|
(Othman et al., 2019) |
| Topoisomerase-II [Phenyl acetamide and 2-bromothiophene] | Q16 | Breast cancer | 28.36
|
(Othman et al., 2019) |
| Tubulin polymerization [2-chloro-5-methoxyquinoline and m-nitroaniline] | Q17 | Colon cancer | 0.888
|
(El-Damasy et al., 2020) |
| Tubulin polymerization [2-chloro-5-methoxyquinoline and m-nitroaniline] | Q18 | Colon cancer | 0.229
|
(El-Damasy et al., 2020) |
| Tubulin polymerization [4-chloro-2-methylquinoline and N-tosylhydrazone | Q19 | Malignant gliomas, chronic myeloid leukemia, bone marrow, lung cancer, and colon caner |
|
(Khelifi et al., 2017) |
| Tubulin polymerization [4-chloro-2-methylquinoline and 4-methoxy-3-nitroaniline] | Q20 | Colon cancer |
|
(Zhou et al., 2017) |
| Tubulin polymerization [3,4,5-trimethoxybenzaldehyde, 2-oxopropanoic acid, and 3,4,5-trimethoxyaniline] | Q21 | Breast cancer, ovarian metastatic cancer | 16.28 and 11.44
|
(Shobeiri et al., 2016) |
| Tubulin polymerization [Halo substituted quinoline and alkyl substituted aniline] | Q25 | Human ovarian carcinoma, cisplatin-resistant human ovarian carcinoma, breast cancer, mitoxantrone-resistant human breast cancer, and normal Huvec cancer | 0.50 ± 0.07, 1.21 ± 0.22, 1.11 ± 0.26, 1.66 ± 0.27 and 9.94 ± 2.68
|
(Jafari et al., 2019) |
| Tubulin polymerization [Halo substituted quinoline and alkyl substituted aniline] | Q26 | Human ovarian carcinoma, cisplatin-resistant human ovarian carcinoma, breast cancer, mitoxantrone-resistant human breast cancer, and normal Huvec cancer | 1.44 ± 0.16, 1.13 ± 0.22, 1.79 ± 0.26, 1.67 ± 0.17 and 7.82 ± 2.44
|
(Jafari et al., 2019) |
| Tubulin polymerization [phenyl acetamide and 1-argiothiourea] | Q27 | Cervical cancer, lung, urinary bladder, and ovarian adenocarcinoma cancers | 4.4 to 8.7
|
(Fang et al., 2021) |
| Tubulin polymerization [Indole and methyl iodide] | Q28 | Hepatocellular carcinoma of the mouse | 0.008
|
(Li et al., 2019) |
| Tubulin polymerization [Indole and formaldehyde] | Q29 | Hepatocellular carcinoma of the mouse | 0.006
|
(Li et al., 2019) |
| Tyrosine kinase [mercaptoacetic acid and substituted quinoline derivative | Q30 | Colorectal cancer | 0.19
|
(Zhou et al., 2021) |
| Tyrosine kinase [2-chloro-5-methoxyquinoline and nitroamine] | Q31 | Human foreskin fibroblasts | 1.62
|
(El-Damasy et al., 2016) |
| EGFR-TK [4-substituted benzaldehyde and benzamide derivative] | Q32 | Breast cancer | 3.46
|
(Ibrahim et al., 2015) |
| EGFR-TK [ 2,2-dimethyl-1,3-dioxane-4,6-dione and trimethyl orthoformate] | Q33 | Lung and breast cancer | 1.03 ± 0.37 and 1.45 ± 0.19
|
(Li et al., 2022) |
| EGFR-TK [Aniline derivative and isoindoline] | Q34 | Lung cancer | 0.91
|
(Karnik, Sarkate, Tiwari, Azad and Wakte, 2021) |
| EGFR-TK [3-amino-2-bromobenzoic acid and diethyl 2-(ethoxymethylene)malonate] | Q35 | Breast and non-small lung cancer | 3.42 and 5.97
|
(Abdellatif et al., 2017) |
| EGFR-TK [4-(6,7-dichlorobenzo[d]thiazol-2-yl)aniline and 3-(2-bromoacetyl)-4-hydroxy-1-methylquinolin-2(1H)-one] | Q36 | Breast cancer and hepatic cancer | 0.398 and 02.25
|
(Bolakatti et al., 2021) |
| EGFR-TK [4-(6,7-dichlorobenzo[d]thiazol-2-yl)aniline and 3-(2-bromoacetyl)-4-hydroxy-1-phenylnaphthalen-2(1H)-one] | Q37 | Breast cancer and hepatic cancer | 2.97 and 2.48
|
(Bolakatti et al., 2021) |
| EGFR-TK [Indoline-2,3-dione and acetophenone] | Q38 | Lung, breast, and colon cancer | 6.1 ± 0.8, 7.5 ± 1.2 and 7.8 ± 1.4
|
(Mohassab et al., 2021) |
| EGFR-TK [Indoline-2,3-dione and acetophenone] | Q39 | Lung, breast, and colon cancer | 3.6 ± 0.05, 3.3 ± 0.08 and 3.5 ± 0.02
|
(Mohassab et al., 2021) |
| EGFR-TK [Indoline-2,3-dione and acetophenone] | Q40 | Lung, breast, and colon cancer | 6.9 ± 0.3, 6.8 ± 1.6 and 6.9 ± 1.1
|
(Mohassab et al., 2021) |
| EGFR-TK [Indoline-2,3-dione and acetophenone] | Q41 | Lung, breast, and colon cancer | 3.9 ± 0.6, 3.2 ± 0.08 and 3.8 ± 0.2
|
(Mohassab et al., 2021) |
| EGFR-TK [Indoline-2,3-dione and acetophenone] | Q42 | Lung, breast, and colon cancer | 4.7 ± 0.4, 4.9 ± 0.6 and 4.9 ± 0.2
|
(Mohassab et al., 2021) |
| EGFR-TK [8-nitro-2,3-dihydroquinolin-4(1H)-one, benzaldehyde and pyrrolidine] | Q43 | Breast and lung cancer | 14.26 and 15.41
|
(Arasakumar et al., 2017) |
| EGFR-TK [Amine substituted quinoline and Furaldehyde derivative] | Q44 | Breast cancer | 3.355
|
(Aly et al., 2017) |
| EGFR-TK [Amine substituted quinoline and Furaldehyde derivative] | Q45 | Breast cancer | 3.647
|
(Aly et al., 2017) |
| EGFR-TK [Amine substituted quinoline and morpholine] | Q46 | Breast cancer | 5.069
|
(Aly et al., 2017) |
| EGFR-TK [Amine substituted quinoline and allylisothiocyanate] | Q47 | Breast cancer | 3.617
|
(Aly et al., 2017) |
| EGFR-TK [Amine substituted quinoline and allylisothiocyanate] | Q48 | Breast cancer | 0.839
|
(Aly et al., 2017) |
| EGFR-TK [Amine substituted quinoline and benzyl chloride] | Q49 | Breast cancer | 10.85
|
(Aly et al., 2017) |
| EGFR-TK [6-methoxy-3,4-dihydronaphthalen-1(2H)-one and benzaldehyde] | Q50 | Breast cancer | 7.70 ± 0.39
|
(Abdelsalam et al., 2019) |
| EGFR-TK [(E)-2-(argiomethylene)-6-methoxy-3,4-dihydronaphthalen-1(2H)-one and ethyl 2-cyanoacetate] | Q51 | Breast cancer | 7.21 ± 0.43
|
(Abdelsalam et al., 2019) |
| EGFR-TK [6-bromo-8-iodo-4-oxo-1,4-dihydroquinoline-3-carbaldehyde and aromatic acetylene derivative] | Q53 | Breast cancer | 14.48 ± 0.48
|
(Mphahlele et al., 2020) |
| EGFR-TK [6-bromo-8-iodo-4-oxo-1,4-dihydroquinoline-3-carbaldehyde and aromatic acetylene derivative] | Q54 | Breast cancer | 11.33 ± 0.53
|
(Mphahlele et al., 2020) |
| EGFR-TK [Aniline and 2-chloronicotinic acid] | Q55 | Lung, hepatocellular carcinoma, and breast caner | 0.23, 0.42 and 0.21
|
(Zhou et al., 2020) |
| c-met kinase [mercaptoacetic acid and substituted quinoline derivative | Q56 | Colorectal cancer | 0.31
|
(Tang et al., 2018) |
| c-met kinase [2-chlorobenzoic acid and aniline] | Q57 | Lung cancer | 1.25
|
(Tang et al., 2016) |
| Rapidly accelerated fibrosarcoma kinase [2-amino-4,5-dimethoxybenzaldehyde and 1-(4-nitrophenoxy)propan-2-one] | Q60 | Leukemia, colon, melanoma, ovarian, renal, prostate, and breast cancer | 0.98, 1.16, 1.31, 1.09, 1.46, 1.08, 1.01, and 0.93
|
(El-Gamal et al., 2017) |
| Rapidly accelerated fibrosarcoma kinase [Bromo aniline and methyl vinyl ketone] | Q61 | Various types of breast caner | 0.80, 0.67,1.30, 5.0 and 1.30
|
(El-Gamal et al., 2017) |
| PI3K [1H-indazol-6-amine and benzaldehye] | Q62 | Colon cancer | 14
|
(He et al., 2021) |
| PDE5 [4-chloro-2-methylquinoline and (4-fluorophenyl)methanamine] | Q64 | Lung cancer | 0.96
|
(Ibrahim et al., 2020) |
| PDK1 [6-chloro-4-hydroxyquinolin-2(1H)-one and 2,3-dichloronaphthalene-1,4-dione] | Q65 | Melanoma caner | 3.7
|
(Vennila et al., 2018) |
| ERK [6-chloro-4-hydroxyquinolin-2(1H)-one and 7-hydroxy-5-phenylphenanthridine-6,9(5H,10H)-dione] | Q66 | Melanoma caner | 0.13
|
(Aly et al., 2019) |
| ERK [naphthalene-1,2-dione and 2,2-dimethyl-1,3-dioxane-4,6-dione] | Q67 | Breast cancer | 3.06 ± 0.38
|
(Aly et al., 2019) |
| NQOL [6,7-dihydroquinoline-5,8-dione and aniline] | Q68 | Cervical cancer | 0.44 ± 0.08
|
(Wu et al., 2020) |
| DHODH kinase [Aniline, 2-oxopropanoic acid, and benzaldehyde] | Q73 | Breast and Malignant melanoma | 4.14 ± 1.72, 24.03 ± 0.02
|
(Petrović et al., 2020) |
| DHODH kinase [Aniline, 2-oxopropanoic acid, and benzaldehyde] | Q74 | Breast and Malignant melanoma | 1.77 ± 0.57, 8.96 ± 0.05
|
(Petrović et al., 2020) |
| DHODH kinase [Aniline, 2-oxopropanoic acid, and benzaldehyde] | Q75 | Breast and Malignant melanoma | 3.47 ± 0.91, 10.75 ± 0.07
|
(Petrović et al., 2020) |
| DHODH kinase [Aniline, 2-oxopropanoic acid, and benzaldehyde] | Q76 | Breast and Malignant melanoma | 2.52 ± 0.72, 13.79 ± 0.05
|
(Petrović et al., 2020) |
| DHODH kinase [Aniline, 2-oxopropanoic acid, and benzaldehyde] | Q77 | Colon and breast cancer | 1.56
|
(Vyas et al., 2019) |
| DHODH kinase [Aniline, 2-oxopropanoic acid, and benzaldehyde] | Q78 | Colon and breast cancer | 1.22
|
(Vyas et al., 2019) |
| C-myc [Quinolin-4-ol and benzocaine] | Q79 | Liver cancer | 12.6 ± 0.1
|
(Zhao et al., 2013) |
| C-myc [Quinolin-4-ol and benzocaine] | Q80 | Liver cancer | 27.3 ± 1.7
|
(Zhao et al., 2013) |
2.1 Inhibitors for anticancer activity
2.1.1 Topoisomerase I and II inhibitor
DNA topoisomerase is classified into two kinds like topoisomerase-I (topos-I) and topoisomerase-II (topos-II). (Capranico, Marinello and Chillemi, 2017) A comparison of topoisomerase-I and topoisomerase-II is shown in Table 4.
| Topoisomerase-I | Topoisomerase-II | |
|---|---|---|
| Definition | Type-I topoisomerase is an enzyme that causes single-strand breaks and relegation to vary the degree of DNA supercoiling. | Type-II topoisomerase is an enzyme that causes double-strand breaks and relegation to vary the degree of DNA supercoiling |
| Structure | Monomer | Heterodimer |
| DNA breaking | It performs single-strand breaks | It performs double-strand breaks |
| ATP hydrolysis | Its actions aren’t dependent on hydrolysis | Its actions are dependent on hydrolysis |
| Found | It found in eukaryotic | It found in both eukaryotic and prokaryotic |
| Modify the linking number of circular deoxyribonucleic acid | It modifies the linking number of circular deoxyribonucleic acid by a unit of 1. | It modifies the linking number of circular deoxyribonucleic acid by units of + or − 2. |
| Potent anticancer compounds | Irinotecan drug used to cure colorectal cancer | Amacrine drug used to cure cute leukemia cancer |
The majority of these drugs target topoisomerase (types II) enzymes, which are currently used to treat human cancers. Topoisomerase inhibitors are known as “poisons” because they generate cleavable complexes with DNA, resulting from lasting DNA impairment that sets off a sequence of biological processes that progressively lead to programmed cell death or cell death in different ways. (Schmidt, Osheroff and Berger, 2012) They can relax positive or negatively supercoiled deoxyribonucleic acid, topos are common nuclear enzymes that regulate the topological form of DNA and perform a variety of other tasks such as replication and transcription (Wang, 2002) TopII enzymes are the target of most presently used anti-tumor medications in the therapy of cancer and many new medication sequences are now being developed for the treatment of human cancers, target topos enzymes.
A series of new methoxy-based quinoline compounds were found to have anti-cancer properties in vitro by Elbadawi and co-workers. (Elbadawi et al., 2021) taking 2-aminoacetophenone and benzoyl chloride as starting substrate in the presence of triethyl amine at 0 0C in overnight; further reacts with dioxane under nitrogen atmosphere to form a cyclized compound; it reacts with an alkyl halide from chloro substituted compound and then finally product reacts with the amine to form Q1-3 compounds (Scheme 1).
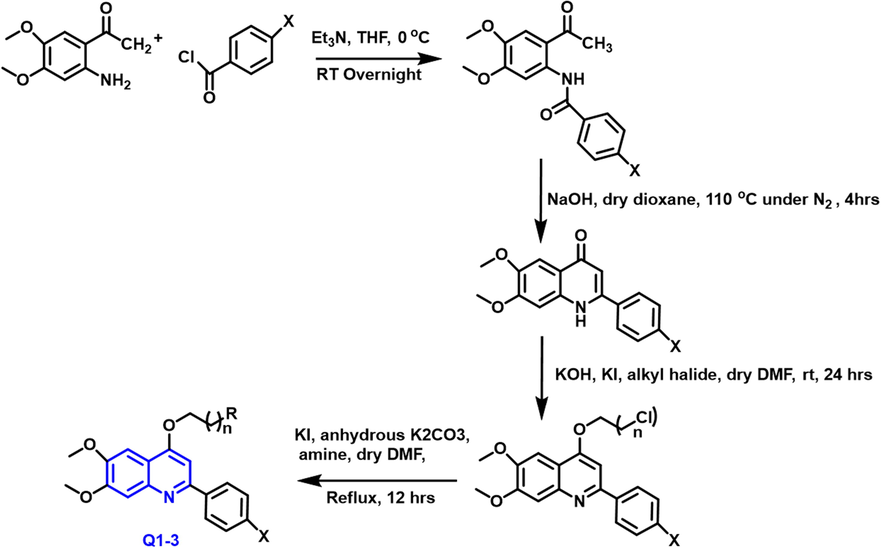
- Synthesis of 4-alkoxy-2-arylquinoline compounds.
Two novel sequences of quinoline derivatives were designed and synthesized to develop prospective antitumor drugs targeting Topoisomerase I (TOP1), depending on SAR of previously reported Topoisomerase I inhibitors and structural characteristics required for Topoisomerase I -deoxyribonucleic acid cleavage complex stabilization. The National Cancer Institute (NCI-60) tumor cell lines panel was used to investigate the antitumor properties of these two sequences of drugs in vitro at a single treatment stage. Most active compounds had a para-substituted phenyl at Carbon-2 and a propyl group link at Carbon-4 and were chosen for assay at five dose levels, where they showed potent anticancer activity at sub-micromolar levels vs various tumor cell lines. The NCI procedure was used to evaluate all substances for their anticancer potential. Q1 compound was found to be the most effective in causing rectal cancer, melanoma, and leukemia at submicromolar levels. As a result, Q2 and Q3 substances were discovered to be promising lead compounds for the creation of more active anticancer medicines with TOP1 inhibitory action, both substances have promising pharmacokinetics and drug-likeness properties.
Naphthyridine-based quinoline compounds (Scheme 2) were designed and developed by Ramakant. Q4 and Q5 compounds had high potent antiproliferative agents towards the epithelial carcinoma and breast cancer cells. (Kardile et al., 2021) Both the compound have a substantial binding site of the DNA topoisomerase I protein was confirmed by molecular docking. Assessing the Top-I inhibitory efficacy of Q4 (72.4 %) and Q5 (60.6 %) compounds vs camptothecin (61.4 %). From this result, both compounds have notable cytotoxicity by using MTT assay and inhibit the proliferation of cancer cells.
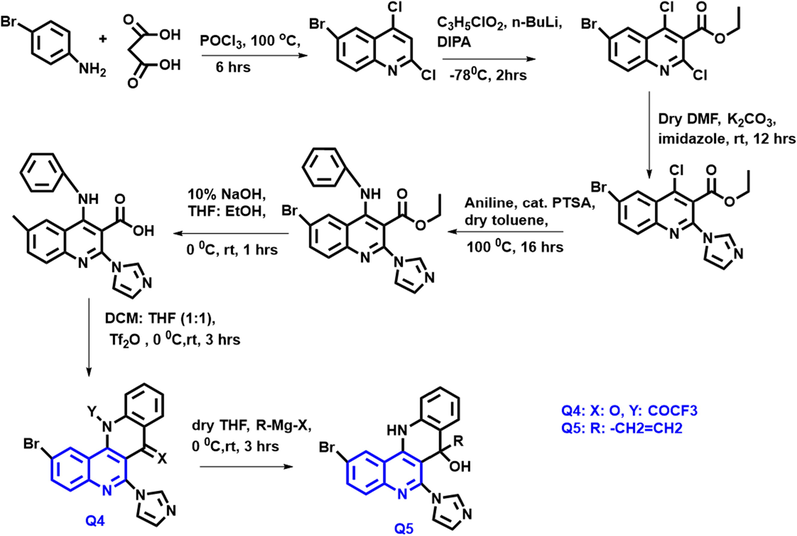
- Synthetic route of the quinoline derivatives.
Amelia Galdino Ribeiro et al.,(Ribeiro et al., 2019) reported a novel 4-quinoline-thiosemicarbazone (Q6) variant from hydrazine and substituted isothiocyanate at 25 0C; further condensation reaction between 4-quinolinal (Scheme 3). The thiosemicarbazone derivatives showed DNA and BSA binding ability. The N-(4-ethylphenyl)-2-(quinoline-4-ylmethylene) hydrazine carbothioamide molecule was emphasized because it had a higher Kb and Ksv value for DNA and BSA binding, indicating a possible method of DNA intercalation, which was confirmed by absorption methods, CD, and molecular docking. Q6 derivative also had a lower IC50 value for MCF7 lines (0.82
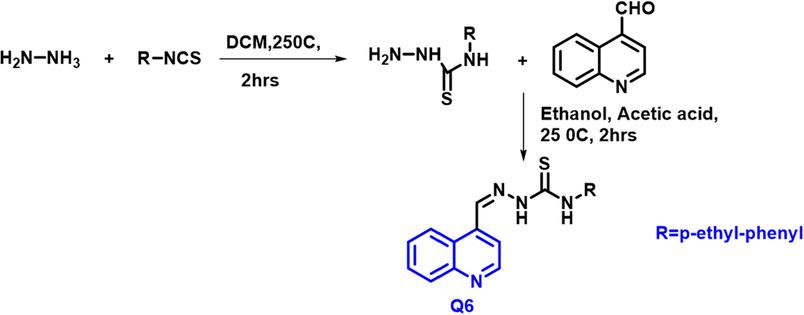
- Synthesis of the quinoline derivatives based on thiosemicarbazones.
Concepcion Alonso et al., (Alonso et al., 2018) have reported the synthesis of new hydroquinoline and quinoline having phosphorous linkers including phosphine oxide, phosphine, and phosphine sulphide. The “Povarov reaction” is used for synthesis (Scheme 4). A multicomponent reaction is used in the synthesis, which allows for the chosen creation of two stereocentres in a quick, effective and repeatable manner. Q-7–9 compounds were tested against kidney, lungs, and ovarian cancer cell lines for cytotoxicity; Q7 exhibited potent activity against the lung cancer cell line (0.25 ± 0.03
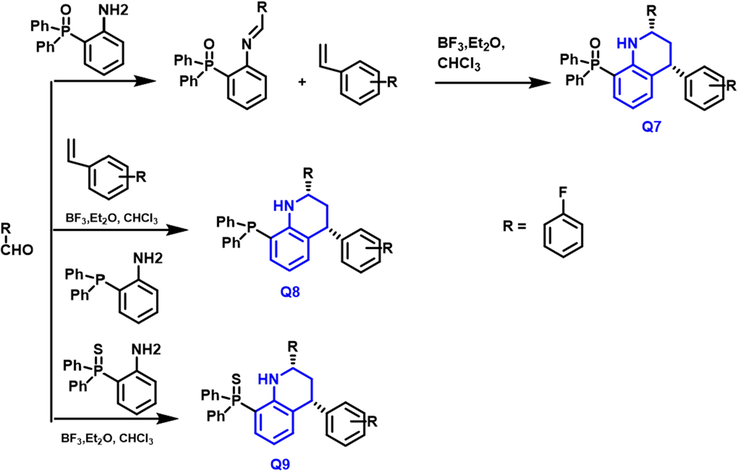
- Synthetic pathway of quinoline derivatives based on phosophorus.
The preparation of sequences of “2-amino, 5-amino, and 2,5-diamino substituted benzimidazo[1,2-a]quinolines” using microwave-assisted amination without catalysis was described by Natasa Perin et al., (Perin et al., 2016) (Scheme 5) 4-fluorobenzaldehyde reacts with benzimidazole derivative in the presetonce of piperidine and ethanol as a solvent to form intermediate; cyclized and then reacts with the amine to form product Q10 and Q11, similarly 4-fluoro benzoyl chloride taking as a substrate to form Q12 product. These compounds were tested against the colon, breast and lung cancer cell lines for anticancer properties due to the presence of 4-methyl and 3,5-dimethyl-1-piperazinyl groups. The compounds were designed and evaluated for 3D-QSAR (Quantitative structure–activity relationship) analysis, DNA binding abilities, and antiproliferative properties. 2-amino-substituted analogs had the highest antiproliferative activity while 5- amino and 2,5 diamino substituted compounds were significantly decreased.
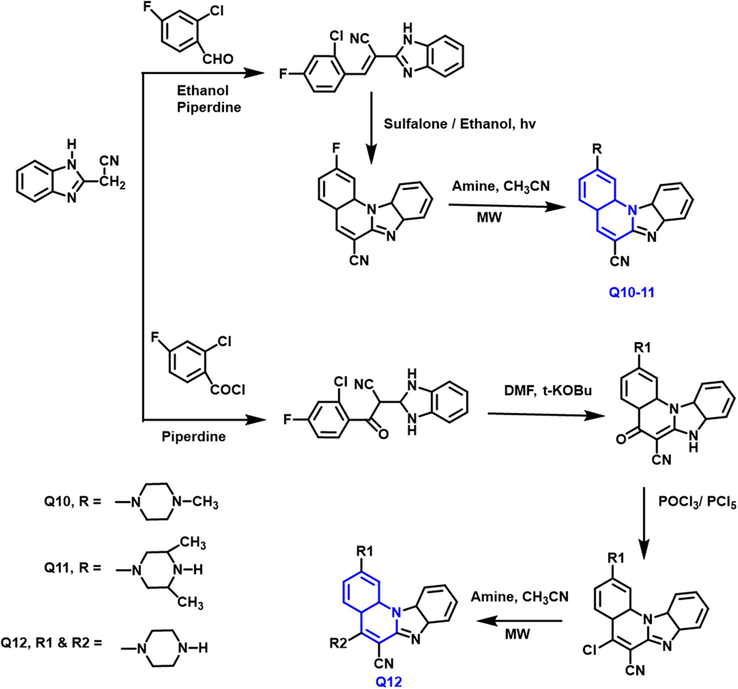
- Synthesis of 2-amino and 2,5 diamino substituted quinoline derivatives.
Vladimir V et al., (Kouznetsov et al., 2017) reported the synthesis of “4-methyl-2-(2-, 3- and 4-pyridinyl) quinolones” heterocycles from p-substituted anilines and pyridine carboxaldehyde in the presence of sodium sulfate under dichloromethane solvent. Scheme 6 Q13-14 compounds were synthesized using altered Kametani reaction and evaluated for biological activities in vitro. These compounds have significant anticancer activities, but they also had a small safe margin, as seen with doxorubicin. Human tumor cell lines like SKBR3, MCF7, PC3, and HeLa were evaluated in vitro for cytotoxicity, with human skin fibroblasts serving as non-cancer cells. As a promising method for anticancer medicines against prostate carcinoma, “4-methyl-2-(3-pyridinyl)quinoline (PQ)” is unique out for its low non-specific cytotoxicity and highly outstanding selectivity for PC3 cells (IC50 = 4.40
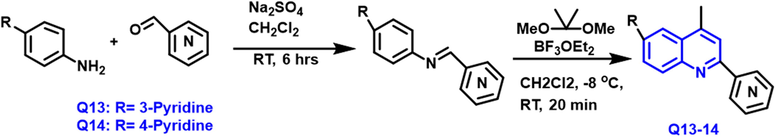
- Synthesis of quinoline compound based on pyridine moiety.
Dina I.A. Othman et al., (Othman et al., 2019) described the synthesis of thiophene-1-benzopyridine compounds with isooxazole,trizole, and phenyl molecules linked by the OH functional group using click chemistry methods (Scheme 7). In vitro testing of the produced molecules towards 4 human tumor cell lines such as HepG2, HeLa, HCT-116, and MCF7 was performed using the MTT assay. Q-15 and Q-16 compounds had very effective antitumor properties toward breast cancer (IC50 = 38.14
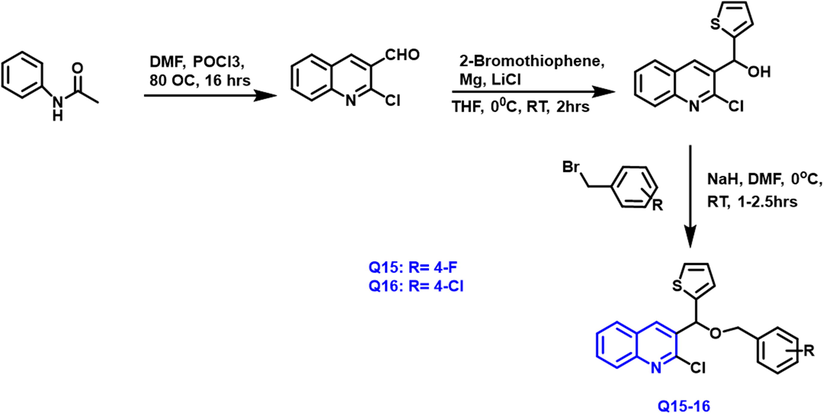
- Synthesis of quinoline compound based on thiophene group.
2.1.2 Tubulin polymerization
Microtubules are essential for all eukaryotic organisms. This structure is made up of α and β tubulin heterodimers organized in the form of a slender filamentous tube and tightly arranged both spatially and temporally. Cell cycle arrest in the G2-M phase and the development of aberrant mitotic spindles can both occur from microtubule disruption. This characteristic makes microtubules an appealing target for the development of anticancer drugs because it is crucial for mitosis and cell division.(Field, Kanakkanthara and Miller, 2015).
Ashraf K. El-Damasy and coworkers (El-Damasy et al., 2020) have designed and synthesized a unique sequence of n-phenylquinolin-2-amine-based acrylamide from 2-chloro-5-methoxy quinoline and meta or para nito aniline taking as starting material at 160 0C in two hours reaction (Scheme 8). These compounds were tested against various cancer such as colon, renal and ovarian. Q17 and Q18 compounds have a potent anticancer activity due to the presence of piperazine substituted phenyl ring; moreover, this compound is more active than imatinib drugs. At the National Cancer Institute, a set of potential compounds was tested for anticancer efficacy against a board of sixty tumor cell lines at a dose of 10
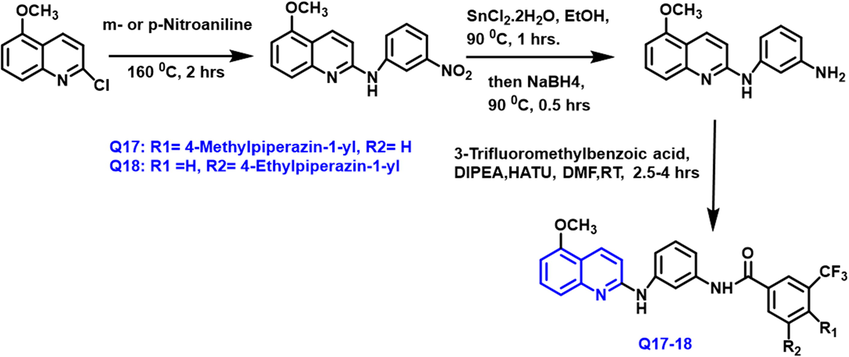
- Synthesis of quinoline compound based on arylamide group.
Ilhem Khelifi et al., (Khelifi et al., 2017) developed a method for the synthesis of Isocombreat quinoline derivative through the reaction of 4-chloroquinaldine and N-tosylhydrazones in the presence of palladium catalyst under a sealed tube reaction (Scheme 9). Q-19 compound binds to the colchicine binding site of tubulin by docking experiments. It showed significant cytotoxicity toward cancer cell lines like malignant gliomas, chronic myeloid leukemia, bone marrow, lung, and colon at 10 nM. This compound shows anticancer activity due to the presence of hydroxy and methoxy phenyl ring system.
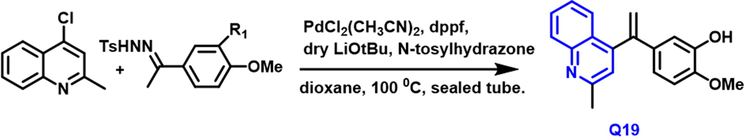
- Synthesis of quinoline compound as an anticancer agent.
Yuanyuan Zhou et al., (Zhou et al., 2017) developed a series of 4-anilinoquinoline compounds as potent anti-cancer properties (Scheme 10). Q-20 molecule had the high effective cytotoxic activity towards all tumor cell lines studied, with an IC50 = 1.5–3.9 nM, and in MDR (Multiple Drug Resistant ) tumor cells, demonstrated encouraging efficacy. This compound also demonstrated powerful anticancer activity without substantial weight loss in body weight by using in vivo effectiveness testing of the heterograft model. All the studies represented that the compound shows a potent anti-cancer treatment; especially for colon cancer.
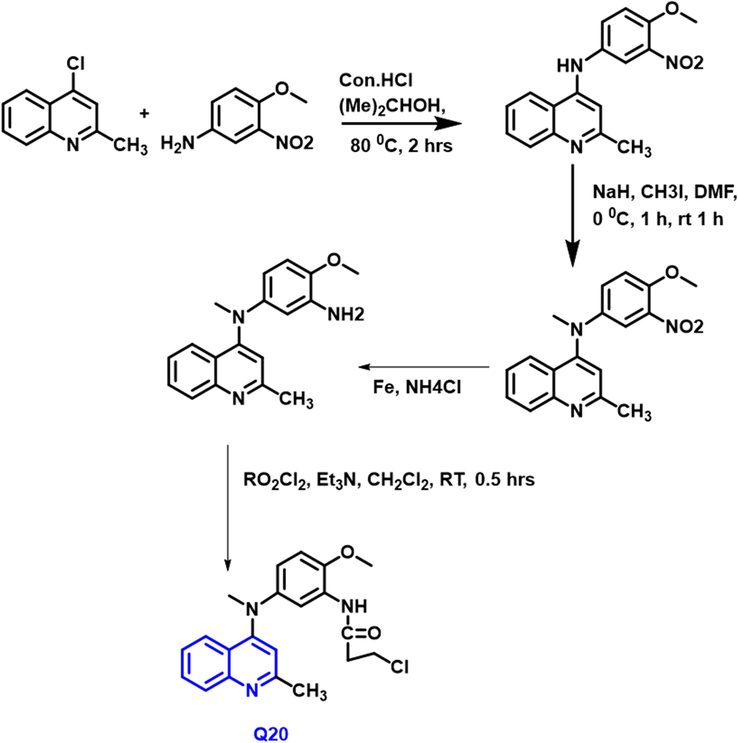
- Synthesis of quinoline compound as a novel potent tubulin depolymerization agent.
Nikta Shobeiri, and coworkers (Shobeiri et al., 2016) have reported that methoxylated flavones were utilized as lead compounds in the methods and synthesis of a new sequence of 2-aryl-trimethoxyquinoline analogs as tubulin inhibitors (Scheme 11). Their cytotoxic activity has been tested on 4 human beings tumor cell lines breast, resistant breast, ovarian, and human ovarian carcinoma cells. Q21 compound has a potent anticancer activity toward breast and ovarian cancer (IC50 = 16.28 and 11.44
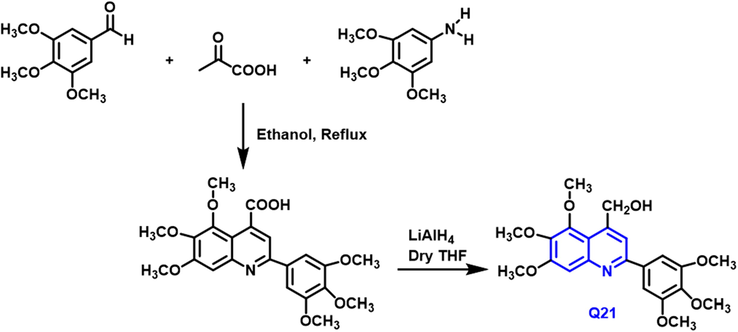
- Synthesis of quinoline compound based on flavones group.
It was discovered to be a highly active tubulin-inhibitor, causing cell growth arrest in the second growth phase process and apoptosis. Interaction between the Q-21 and colchicine-binding site of tubulin was studied by molecular docking.
Florian Schmitt et al., (Schmitt, Schobert and Biersack, 2019) developed a series of pyrano-based quinoline compounds from 8-hydroxyquinoline and aryl aldehyde in the presence of piperidine and ethanol as a solvent under reflux conditions for 30 min reaction (Scheme 12). m-nitro and m-halophenyl-based compounds were tested against the six human tumor cells for anticancer activity. The compounds were extremely active, having an IC50 in the nanomolar range. The Q-22 to Q-24 compounds showed promise in inhibiting tumor cell development while not affecting non-malignant fibroblasts. These compounds have a vascular-disrupting property that was confirmed in ovo studies, and also boosts the reactive oxygen species (ROS) in tumor cells.
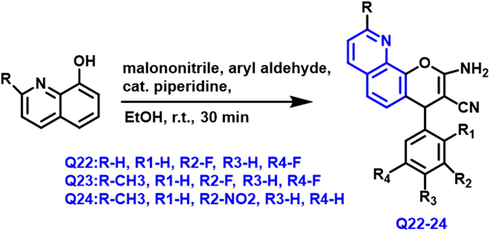
- Synthesis of quinoline compound based on pyrano group.
Salimeh Mirzaei et al., (Mirzaei et al., 2020) reported that anticancer drugs and tubulin polymerization inhibitors, a novel class of styrylquinolines were developed and produced (Scheme 13). 4 human tumor cells cultures were investigated in vitro with the synthesized quinolines. That cell lines are ovarian carcinoma, cisplatin-resistant human ovarian carcinoma, breast, and umbilical vein endothelical cells. In general, compounds containing N-trimethoxy phenyl demonstrated greater cytotoxic action towards the 4 tumor cell cultures, with Half-maximal inhibitory values ranging from 0.38 to 5.01
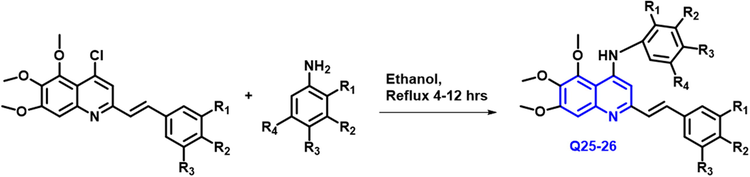
- Synthetic route of strylquinoline derivatives.
Yilin Fang et al., (Fang et al., 2021) reported that possible anticancer drugs, and sequences of new 2-quinolone compounds including 1,3-Thiazol-2-amine were developed and produced (Scheme 14). Using the MTT assay, the synthesized substances were tested for in vitro cytotoxicity towards the cervical, hypotriploid, urinary bladder, and ovarian tumor cell cultures. In these, the Q-27 compound displayed the most powerful effect on the tumor cell lines tested, with the half-maximal inhibitory concentration varying from 0.0044 to 0.0087 M. This compound inhibited the polymer of tubulin in vitro, according to the consequence of the polymer of tubulin assay. Meanwhile, molecular docking research demonstrated that the compound may bind to tubulin's colchicine site and create H-bonds with critical amino acids in the active center. This compound is a prospective candidate for developing effective microtubule inhibitors for tumor treatment.
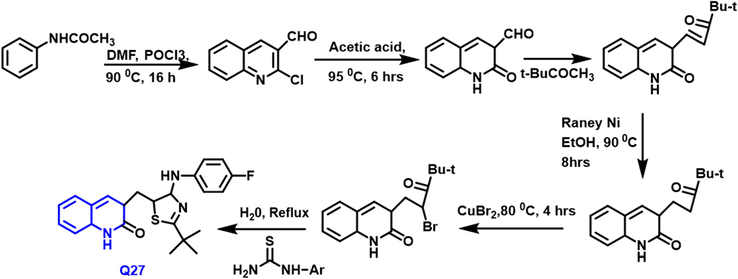
- Synthesis of quinoline derivative based on arylaminothiazole moiety.
Wenlong Li et al., (Li et al., 2019) reported that a new class of quinoline-indole compounds has been developed and produced (Scheme 15). Q-28 and Q29 compounds are the most effective for 5 tumor cell lines, with IC50 values ranging from 2 to 11 nM. Q-28 compound hindered the microtubule by attaching to the colcrys region. It had the strongest antitumor efficacy and anti-vascular property in vitro. Furthermore, those compounds dramatically reduced tumor development in liver xenograft mice design and absence of toxic effects, it indicating Q-28 and Q29 compounds are promising anticancer medicines for cancer treatment.
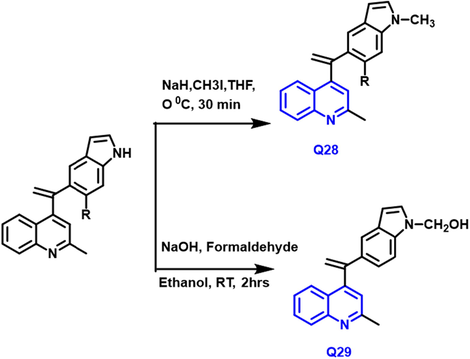
- Synthesis of quinoline derivative based on indole group.
2.1.3 Tyrosine kinase
PTKs are tyrosine kinase proteins that play a role in cell development, differentiation, and death, as well as a variety of physiological and biochemical activities (Wang and Cole, 2014) PTK expression abnormalities, may influence ‘carcinogenesis, tumor invasion and secondary tumor, tumor neovascularization, and tumor treatment resistance’. (Drake, Lee and Witte, 2014)(Knösel et al., 2014) As a result, it has emerged as a popular focus for anti-tumor medication development. (Jiao et al., 2018) Receptors and non-receptor are two types of tyrosine kinases (Gotink and Verheul, 2010).
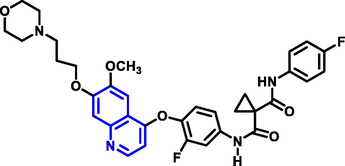
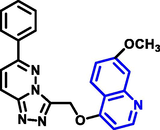
Yuting Zhou and coworkers (Zhou et al., 2021) reported the production of 1-benzopyridine derivatives based on thiazolidinone with biological activity (Scheme 16). Based on the findings of the biological assessment and docking investigation, a structure–activity connection analysis was carried out. A most effective carboxamide-based molecule has the potential to suppress the activity of recepteur d’origine natais, tyrosine-protein kinase met, and proto-oncogene tyrosine-protein kinase among other proteins. Q30 compound demonstrated good antitumor activity on human colorectal adenocarcinoma cells. Flow cytometry results showed that the compound may cause significant cell growth arrest in the growth 2 phase stage in human colorectal adenocarcinoma cells. Furthermore, there was neither antiproliferative nor cytotoxic effect. FHC was found in human normal colorectal mucosa epithelial cells(NCM460). The compound had toxicity to normal cells of 10.0
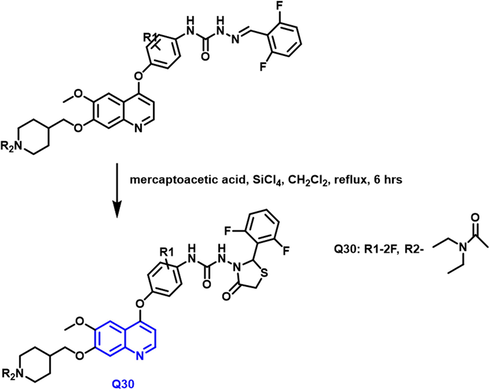
- Synthetic route of quinoline derivatives.
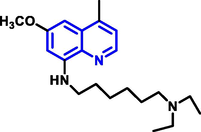
Ashraf Kareem El-Damasy et al (El-Damasy et al., 2016) have noted a sequence of novel ‘2-anilinoquinolines’ with a ‘3-(morpholino or 4-methylpiperazin-1-yl)propoxy’ group at the Carbon-5 of the 1-benzopyridine have been developed and prepared (Scheme 17).
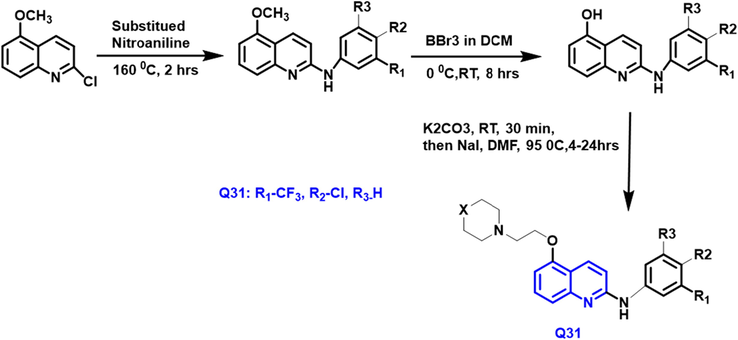
- Synthetic pathway of quinoline derivatives.
The majority of the substances examined had an antiproliferative activity that was both strong and wide in the spectrum. Q-31 was evaluated towards a board of 47 oncogenic kinases and shown to have a specific prohibitor impact (96 percent inhibition) on TrkA kinase. Furthermore, against the HFF-1 normal cell type, the most powerful chemicals had modest cytotoxic effects. At the NCI, their antitumor effects have been assessed towards sixty tumor cell lines. These compounds have more potential than gefitinib drugs during the several tested cell lines.
2.1.4 Epidermal growth factor receptor tyrosine kinase (EGFRTK)
The epidermal growth factor receptor is a member of the tyrosine kinase receptor family. Cell proliferative signaling system in cancer cells representing in Fig. 4.
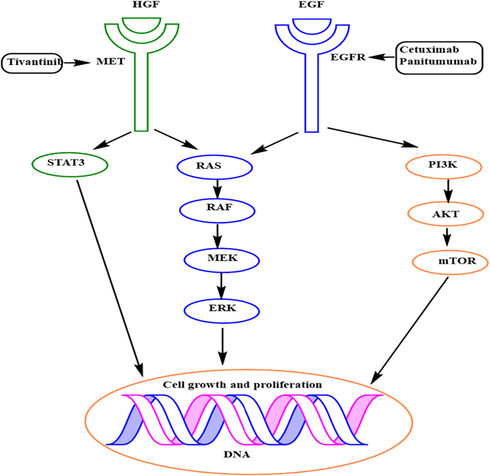
- Targeted EGFR mechanism against cancer.
When ligands like EGF connect to the epidermal growth factor receptor (EGFR), the receptor changes shape, releasing the dimerization domain further permitting homodimerization with a second EGFR or different dimerization with another group of the EGFR family. Now EGFR has activated the results of its autophosphorylation to the key tyrosine groups. These groups trigger important downstream signaling cascades, such as the PI3K/AKT pathways. These tracks act in a coordinated manner to promote cell survival. These kinds of receptors are broadly spread in cell membranes and they often influence a variety of functions like cell growth, cell death, and cell multiplication. This protein plays a significant role in the growth and development of many different forms of solid cancers, breast, lung, colorectal, neck, and head tumors types (Guardiola et al., 2019).
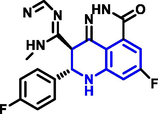
Diaa A. Ibrahim and colleagues (Ibrahim et al., 2015) have developed a new class of 4-anilinoquinoline-3-carboxamide derivatives from 6-acetyl quinoline-3-carboxamide reacts with 4-substituted benzaldehyde in the presence of sodium hydroxide under reflux condition at 90 0C. (Scheme 18). All the derivatives were tested against breast cancer but only the Q32 compound showed substantial activity toward breast cancer. The early results showed that carboxamide-based compound showed a powerful inhibitory effect in cancer development and highly powerful property on the EGFR-TK enzyme having sixty-seven percent growth inhibition comparison with ATP, which might be a promising antitumor drug.
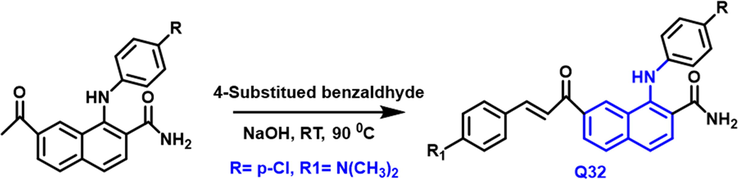
- Synthesis of quinoline derivatives as a anticancer agent.
The methoxy-based quinoline compound was the layout and developed by Xin-yang Li (Scheme 19). Q33 compound had potent antiproliferative activity toward lung and breast cancer. This drug can reduce breast tumor growth and angiogenesis by acting as a dual target inhibitor of EGFR and HER-2 (Li et al., 2022).
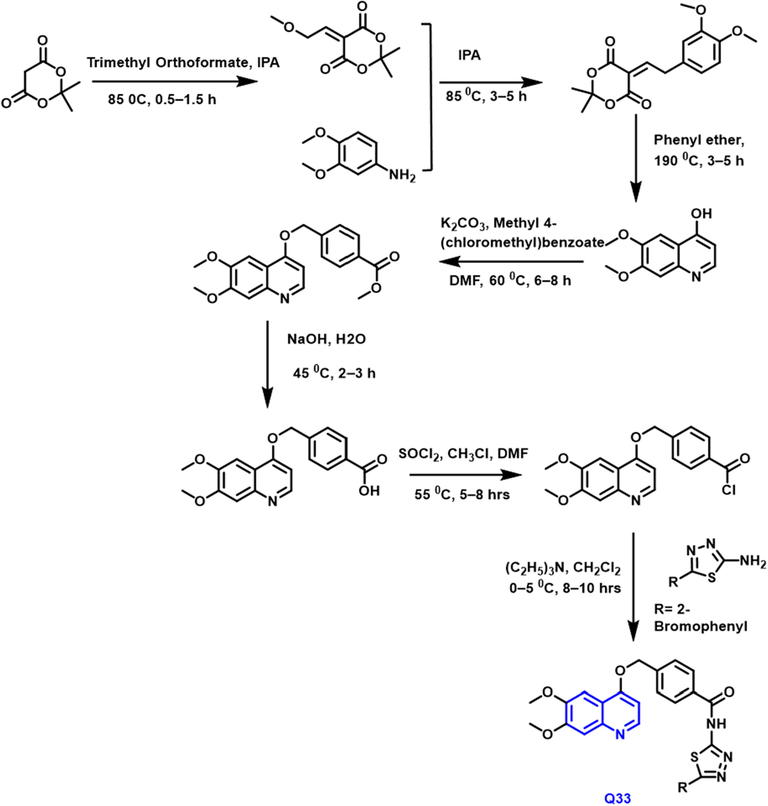
- Synthetic route of quinoline compound.
Karnik and coworkers designed and developed the quinoline-derived compounds from acetoacetanilide and polyphosphoric acid (Karnik, Sarkate, Tiwari, Azad and Wakte, 2021). (6-chloro-2-(isoindolin-2-yl)-4-methylquinoline) the compound had potent inhibitory activity towards the mutant EGFR kinase (IC50 = 0.91
Karnikand coworkers have developed a good antiproliferative activity towards lung cancer (Scheme 20) (Karnik, Sarkate, Tiwari, Azad and Wakte, 2021) When acetoacetanilide reacts with poly phosphoric acid to form hydroxy substituted quinoline compound; it was chlorinated using phosphorus oxychloride to form chloro substituted quinoline compound; it was further reacted with isoindoline in the presence of cuI to form quinoline derivative (Q34) as a potent anticancer property against lung cancer.
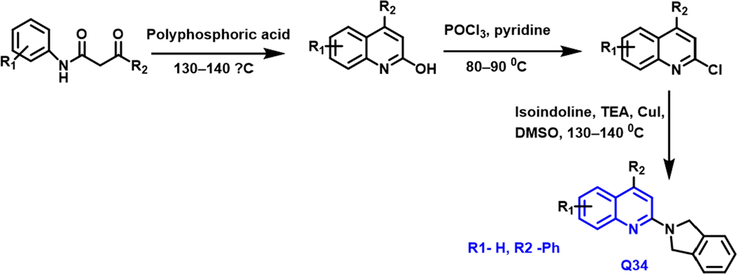
- Synthetic pathway of quinoline compound as anticancer agent.
The Gould–Jacobs reaction was used to make a novel class of ‘4-(4-substituted-anilino)quinoline’ derivatives from amine derivatives has been developed by Khaled R and collaborators (Scheme 21) (Abdellatif et al., 2017) The cytotoxic activity of all produced compounds was tested towards breast and lung cancer. The chemicals examined exhibited a wide variety of activity for MCF7 and A549. The Q-35 compound (IC50 = 3.42 and 5.97
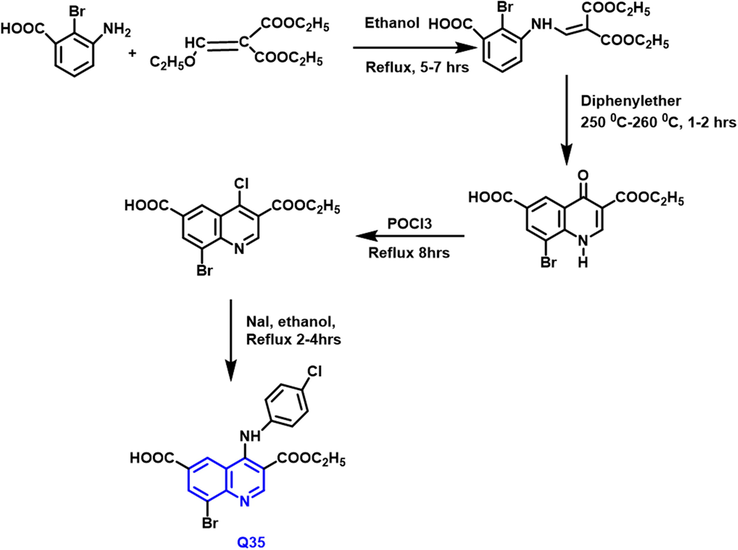
- Synthetic route of quinoline compound.
Girish Bolakatti and coworkers (Bolakatti et al., 2021) have developed a unique sequence of the quinoline-based compound (Scheme 22). All the derivatives were tested against the MCF7 and WRL-68 tumor cells for cytotoxicity studies; from these results, Q37 had a more potent antitumor action than other derivatives and the Q36 compound had a significant binding nature. Docking experiments were performed to better understand the binding mechanism of the title molecules at the target enzyme's active center. The new quinolone-based hybrid is a potential outcome that might be used to build potential anticancer/antimicrobial medicines.
![Synthesis of quinoline compound based on benzo[d]thiazoyl group.](/content/184/2022/15/11/img/10.1016_j.arabjc.2022.104168-fig26.png)
- Synthesis of quinoline compound based on benzo[d]thiazoyl group.
Aliaa et al.(Mohassab et al., 2021) had reported the synthesis of triazole-based quinoline derivatives from the istain and acetophenone taking as a starting material under reflux conditions (Scheme 23). The synthesized compounds have an average to better anticancer property on various cell lines. Q38-Q42 compounds exhibit a potent anticancer property towards lung, breast, and colon cancer. These compounds had a significant site center of EGFR and BRAF kinase was confirmed by docking studies; Q38, Q41, and Q42 compounds have a significant binding site; this pattern is similar to erlotinib drug.
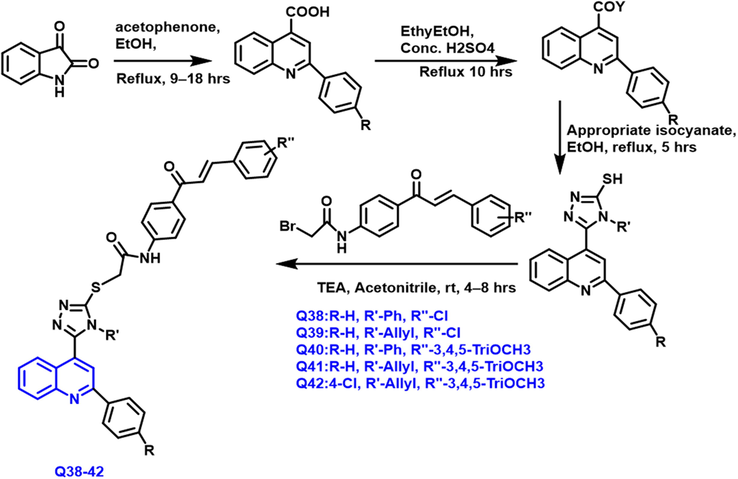
- Synthesis of quinoline compound based on 1,2,4-triazole group.
A novel family of ‘pyrazolo[4,3-c] and pyrano[3,2-c]quinoline’ derivatives have been produced by Thangaraj Arasakumar and coworkers.(Arasakumar et al., 2017) Microwave conditions were used to develop and synthesize derivatives in medium to outstanding yields (Scheme 24). A multicomponent one-pot process has been devised to increase the production of pyrano quinoline derivatives. Q43 compound was tested for cytotoxicity toward breast and lung tumor cell lines. Against these cell growth lines, the preponderance of the drugs had moderate to strong cytotoxic activity. In this experiment, the pyrano[3,2-c] quinoline derivative compound was shown to be very active and also caused apoptosis, which resulted in cell death. The molecular interactions of the EGFR inhibitor were determined using molecular docking experiments.
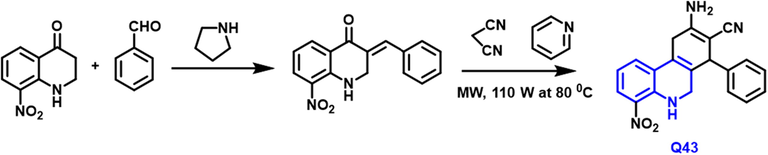
- Synthetic pathway of quinoline compound.
Rasha M. Aly et al (Aly et al., 2017) designed a 3,4,6-trisubstituted quinoline compound from 4-(N, N-dimethylamino)benzaldehyde with 5-(3- or 4-substituted phenyl) furan-2-carbaldehydes, malononitrile and benzyl chloride under reflux and stirring condition. Scheme 25 Q-44–49 have a good yield and showed a potent anticancer agent for breast cancer. Especially, Q-44 has a furan derivative showing IC50 2.61
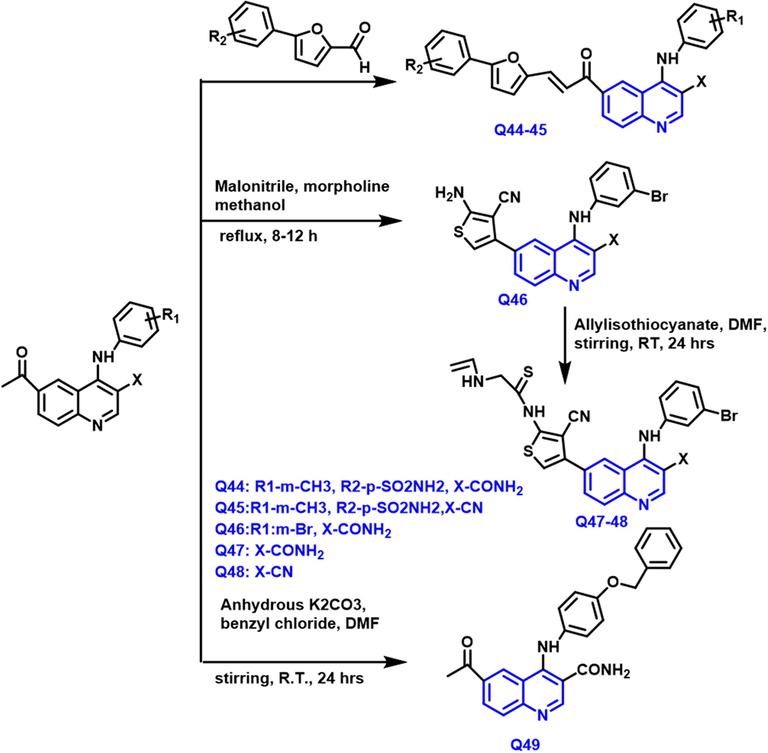
- Synthetic pathway of quinoline compound as an anticancer agent.
The preparation of benzo-based isoindazole, 1-benzopyridine, and 1,3-diazanapthalene derivatives (Scheme 26) has been described by Esraa A et al., (Abdelsalam et al., 2019) The antitumor properties of all the produced derivatives were tested towards tumor cell-growth lines like breast, HepG2, HCT116, and Caco-2. Q50 and Q51 compounds had potent antiproliferative properties against breast cancer (IC50 = 7.70 ± 0.39 and7.21 ± 0.43
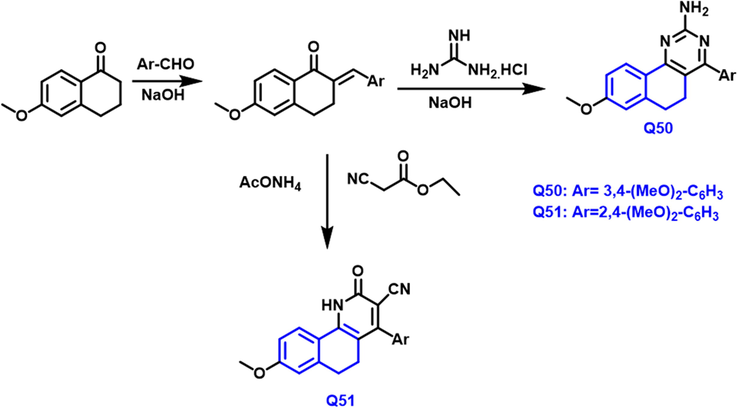
- Synthesis of novel quinoline compounds based on quinazoline and indazole groups.
Kshipra S and colleagues (Karnik, Sarkate, Tiwari, Azad, Burra, et al., 2021) have reported the synthesis of new substituted quinoline compounds from substituted chloride compounds that react with phenyl benzoate derivate in the presence of sodium methoxide and acetonitrile as a solvent under room temperature (Scheme 27); this reaction is Suzuki coupling reaction for C—C bond formation. Q52 compound had a significant antiproliferative property; and also acts as inhibitory property against the EGFR kinases.
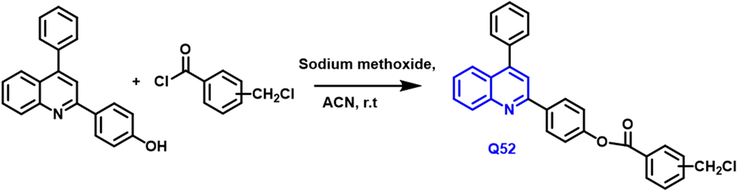
- Synthetic pathway of novel quinoline compound.
The novel sequence of pyrrolo- based quinoline compounds was described by Malose J. et al., (Mphahlele et al., 2020) (Scheme 28). Their structure was studied using IR, nuclear magnetic resonance (proton and carbon), and MS. In the solid-state, XRD single crystal was employed to identify their framework. The samples were screened in vitro for cytotoxicity toward breast cancer cell lines and human embryonic kidneys. These chemicals can cause cell death in the breast cell line, according to a preliminary apoptotic experiment utilizing a DNA laddering assay. Q-53 and Q-54 compounds had a significant inhibitory action on the VEGFR-2 and cyclooxygenase-2. The inhibitory nature of COX-2 controls the growth of breast cancer cells in both animals and human beings.
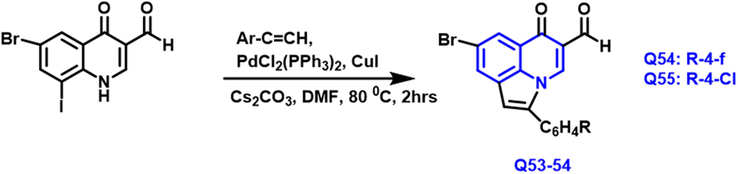
- Synthesis of novel quinoline compounds based on pyrrole moiety.
2.1.5 c-met kinase
The HGFR or (c-Met) was identified in the 1980 s. The tyrosine-protein kinase met belongs to the receptor tyrosine kinases.(Qi et al., 2013)(Liu et al., 2012) In the extracellular domain, the hepatocyte growth factor or c-MET kinase routes activate numerous complicated signaling pathways that play a crucial role.(Clavijo-Cornejo et al., 2013) This kind of kinase is used to detect liver, breast, pancreatic, ovarian, gastric, and prostate cancers; the c-Met signal provides for the growth, and cell division from its starting site and colonization. An appealing target for cancer treatment is c-Met. (Papa et al., 2017).
Yuting Zhou and colleagues (Zhou et al., 2020) discovered and produced a new family of quinoline analogs containing thiazolidinones was developed and their biological activities were assessed (Scheme 29). Supported by the findings of the biological assessment and docking investigation; a structure–activity relationship analysis was conducted. The powerful Q-55 molecule has been found as a multikinase inhibitor. The compound showed good antitumor, cyto-toxicity, and induction of HT-29 cell apoptosis. Furthermore, the Q-55 compound caused a minor cell cycle arrest in HT29 cells during the G2/M stage. As per the results of cancer progression and cell growth studies, other factors that promote cell death could be candidates for the molecule. Compound cell selectivity revealed that it was inactive against NCM356 cells at doses as low as 10.0
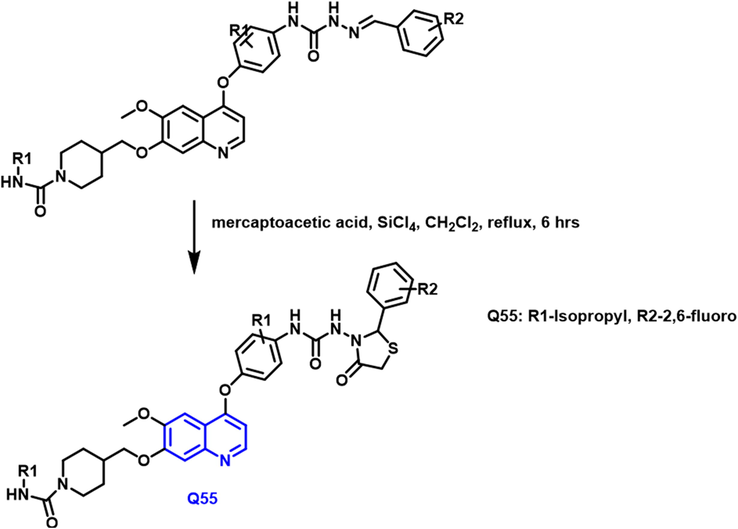
- Synthesis of quinoline derivative based on thiazolidinone group.
The biological activities of a series of ‘6,7-disubstituted-4-phenoxyquinoline’ compounds were developed, produced, and assessed by Qidong Tang et al., (Tang et al., 2018) (Scheme 30) Antitumor efficacy was evaluated in tumor cell-line like breast, lung, and liver. Quinoline-based derivatives were tested for c-Met kinase activity. A very effective molecule Q-56 (IC50 c-Met 14.36
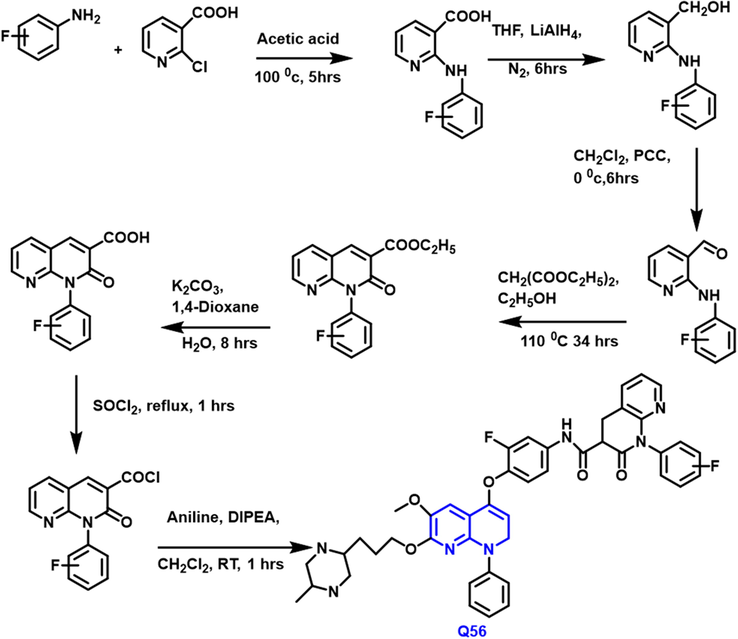
- Synthesis of novel quinoline compound.
Palak Parikh et al., (Parikh, Ghate and Vyas, 2015) reported that Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), a varied collection of seventy-four c-MET kinase inhibitors made up of phenoxy-based quinoline derivatives were employed (3D QSAR). These protein kinases are a well-known candidate for the treatment of cancer therapy. The activity of the compound is determined by the presence of H-bonding and hydrophobic groups; CoMSIA analysis gives this kind of information and on another side, CoMFA analysis provides information about the steric and electrostatic nature of the group. The CoMSIA design appeared to have a stronger performance than the CoMFA design.
The discovery of a sequence of ‘6,7-disubstituted-4-phenoxyquinoline’ derivatives with antitumor properties towards 5 tumor cell lines such as H460, HT-29, MKN-45, A549, and U87MG was reported by Qidong Tang and collaborators (Tang et al., 2016) (Scheme 31). Q57 phenoxy-based quinoline derivative demonstrated more antitumor properties against lung tumors due to the presence of amine and EWG at the 4-site of the phenyl ring was confirmed by the SAR study. Q57 was 6.1, 2.4, and 2.1 times more active than foretinib in its ability to kill lung, colon, and malignant cell lines, respectively.
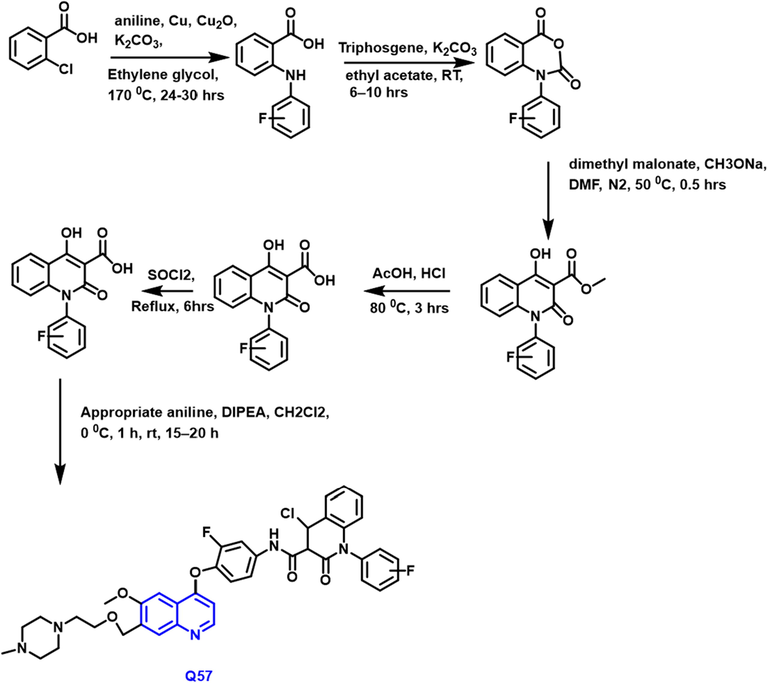
- Synthesis of novel quinoline compound as an anticancer agent.
Alanazi and colleagues (Alanazi et al., 2021) designed and prepared a novel sequence of bis([1,2,4]triazolo)[4,3-a:3′,4′-c] quinoxaline derivatives as a good anticancer agent for the human hepatoma and breast cancer cells. 4-methoxy benzene substituted quinoxaline compound have a more potent antiproliferative activity than other derivatives; IC50 is 3.2
Baohui Qi and coworkers (Qi et al., 2018) reported the biological activities of twenty-nine new molecules containing ‘N1 -(2-aryl-1, 3-thiazolidin-4-one)–N3 -aryl ureas’ were developed, produced, and assessed (Scheme 32). This set of chemicals SAR and binding processes were investigated simultaneously. Q-58 compound was discovered to be highly effective against a variety of TKIs. IncuCyte live-cell imaging showed this compound antiproliferative and cytotoxic activity in vitro against the A549 cancer cell line.
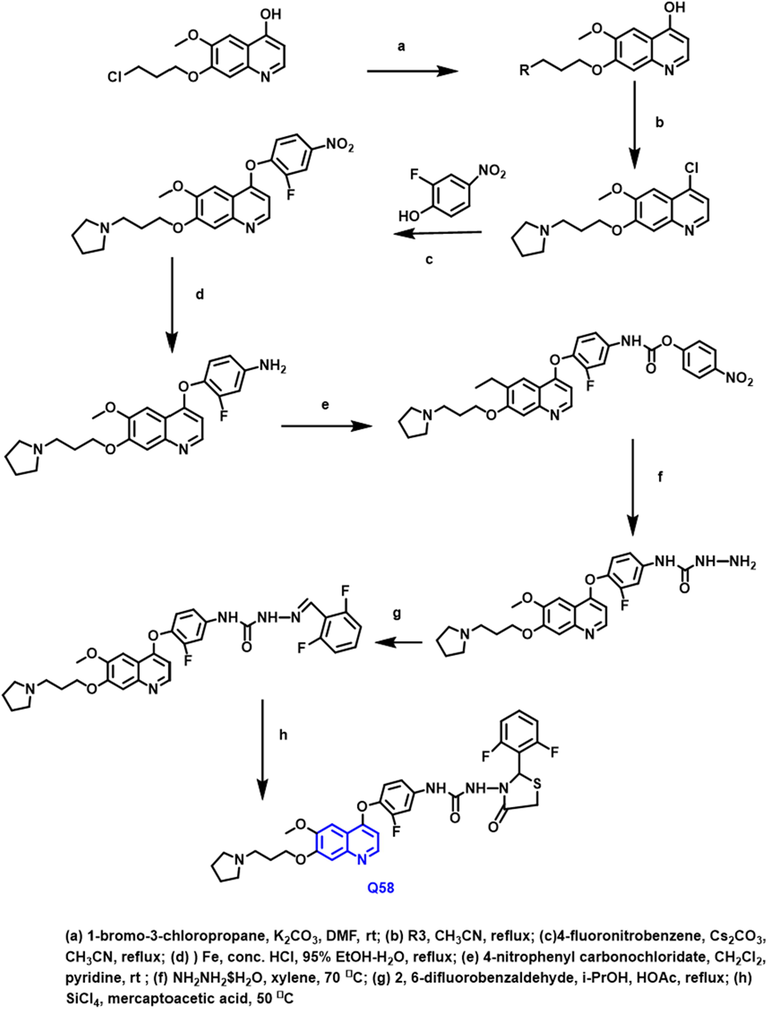
- Synthesis of novel quinoline compound as an anticancer agent.
2.1.6 P-glycoprotein inhibitors
Permeability glycoprotein is abbreviated as P-gp or Pgp. It is a 1280 protein encoded by the MDR1 gene. It's one of the ABC transporter families with the most research done on it. Pgp is play an important role in the body and distribution of xenobiotics and toxic compounds. It prevents the drug from absorbed, retained and permeabilized from the cells. Overexpression of P-gp, a critical protein controlling MDR in many types of tumor cells. As a result, decreasing P-gp activity and lowering anti-cancer medication efflux have become key strategies for reversing MDR. (Endicott and Ling, 1989)(Binkhathlan and Lavasanifar, 2013)(Nobili et al., 2012).
Mutta Kairuki et al., (Kairuki et al., 2019) developed and synthesized two sequences of isoquinoline component Pgp inhibitors based on tetrahydro and triazole (Scheme 33). MDR is a term that refers to a cancer cell's cross-resistance to one drug, as well as other medicines with distinct mechanisms and structures. It is one of the most significant challenges in clinical chemotherapy. P-glycoprotein (P-gp) overexpression has been investigated extensively as a cause of MDR. As a result, blocking P-gp has emerged as a key approach for reversing MDR. Q-59 compound showed impressive MDR reversal activity, and a preliminary mechanistic investigation was conducted. All of the findings suggested that the compound might be a viable P-gp-mediated MDR reversal option.
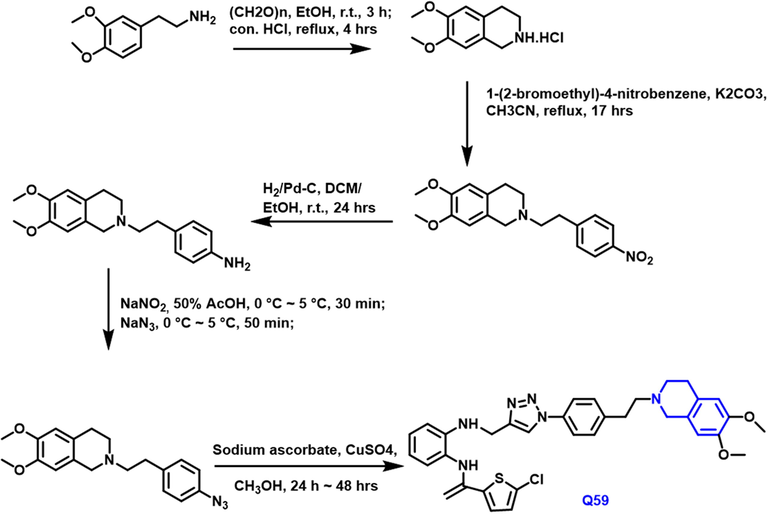
- Synthetic pathway of novel quinoline compound as an anticancer agent.
2.1.7 RAF kinase
Rapidly Accelerated Fibrosarcoma is known as RAF; it plays an important role in the development of organism, control of the cell cycle, the multiplication and differentiation of cells, lifespan of cell and mortality of cells as well as many other cellular and physiologicl processes.
Mohammed I. El-Gamal et al., (El-Gamal et al., 2017) have noted that a new family of diarylurea derivatives with quinoline nuclei has been developed, synthesized, and biological screening (Scheme 34).
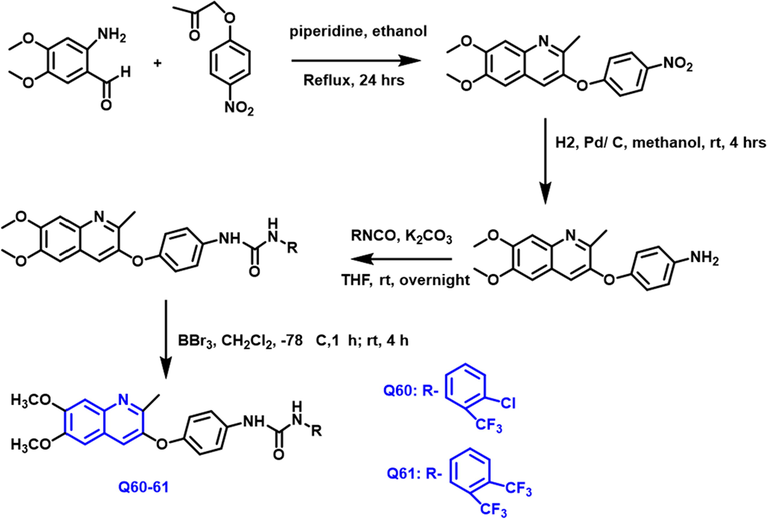
- Synthetic pathway of novel quinoline compound as an antiproliferative agent.
The National Cancer Institute chose nine target compounds for antiproliferative in vitro testing towards a board of fifty-eight tumor cell-lines from nine various tumors. The most promising counterparts were Q-60 and Q-61. Two substances were shown to have high efficacy and board anticancer action towards the various tumor examined.
2.1.8 PI3K/mTOR dual inhibitor
Ruoyu He et al., (He et al., 2021) reported that the amido-based 1-benzopyridine compounds were designed and synthesized as potent anticancer agents (Scheme 35).
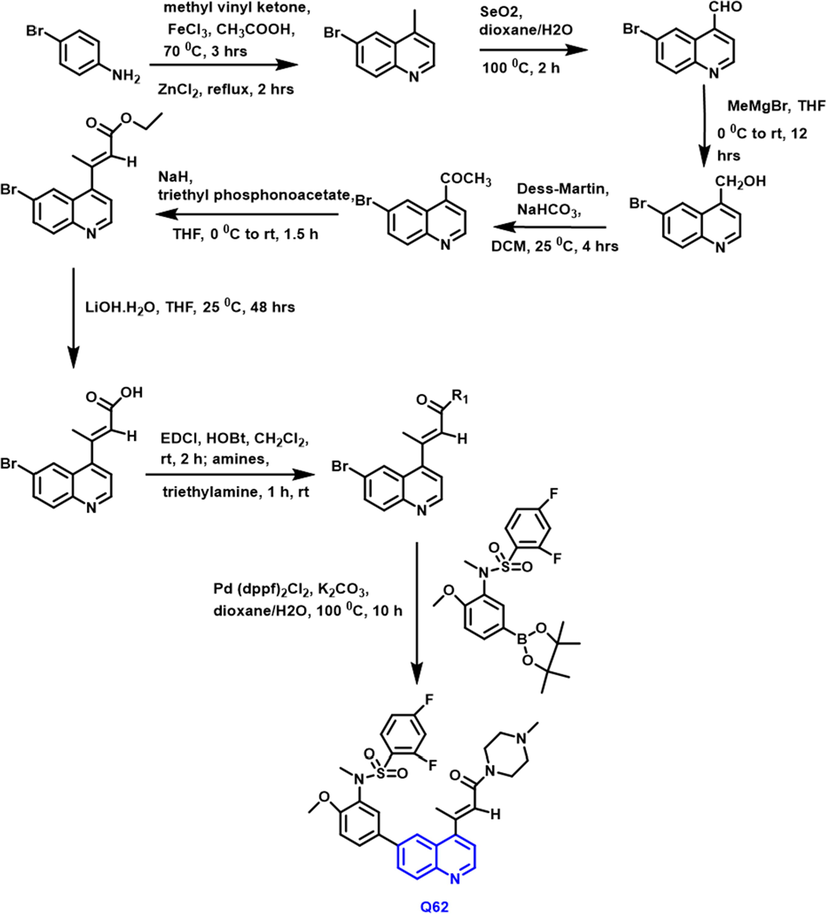
- Synthetic route of novel quinoline compound as an anticancer agents.
Q62 compound showed potent anti-proliferative property due to the presence of 4-acrylamido functional group; it create some steric hindrance to the molecules. As a consequence, the Q-62 compound demonstrated substantial anti-proliferative and enzymatic activity, as well as inhibiting PI3K/mTOR signaling in a Western Blot experiment in vitro at nanomole concentrations. In vivo, the compound was shown to have substantially better oral exposure than its beta-position unsubstituted analog. In an Uppsala 87 malignant glioma GBM xenograft design in vivo, this compound showed substantial therapeutic effectiveness.
2.1.9 Haspin kinase inhibitors
The serine/threonine kinase Haspin (haploid germ cell-specific protein kinase) is associated with histone phosphorylation, notably at Thr3 (H3T3) throughout mitosis (Dai et al., 2005) Haspin is expressed in a variety of cell types, and its activation (or) overexpression causes H3T3 phosphorylation.(Rossetto, Avvakumov and Côté, 2012) Depletion of has pin by RNA interference led to reduced H3T3 phosphorylation, resulting in chromosomal misalignment at metaphase, as reported by Dai et al. As a result, haspin is considered to have a function in chromosomal segregation and tumor development. Because haspin has a bright future as an anticancer target, there are only a few inhibitors known. (Dai et al., 2005).
The inhibitors of haspin kinase property can stop cancer cells from multiplying were reported by Clement Opoku-Temeng and coworkers (Opoku-Temeng et al., 2018). One pot synthesis of Doebner or Povarov reaction; Scheme 36 the synthesized compounds were tested against the HCT116 and HeLa and A375 cell lines. Q63 derivatives had a potent anticancer activity.
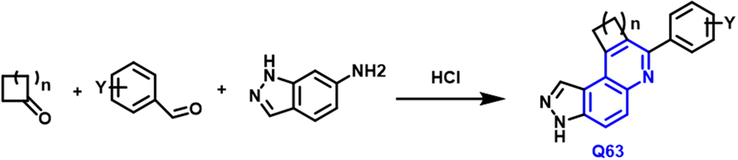
- Synthetic pathway of novel quinoline compound as an anticancer agents.
2.1.10 PDE5 inhibitor
Phosphodiesterase type 5 (PDE5) inhibition appears to be a viable strategy for restoring normal intracellular cyclic guanosine monophosphate (cGMP) signaling. As a result, several downstream compounds are activated, which limit cancer cell proliferation, motility, and invasion. PDE5 is linked to the aetiology and treatment of a wide range of cancers, according to several studies. (Barone et al., 2017) Because of its overexpression in several forms of human carcinomas; Phosphodiesterase type 5 provides a unique and effective approach for inducing cell death and inhibiting cancer cell proliferation.
The synthesis of 18 new innovative pyrazole-based 1-benzopyridine molecules Scheme 37 was reported by Tarek S. Ibrahim and colleagues. (Ibrahim et al., 2020) NCI characterizes them and tests them for cytotoxic properties towards sixty tumor cell lines. The majority of them exhibited effective PDE5 inhibition, especially the Q-64 compound, which was strengthened by a molecular docking analysis, making it a viable option for ED therapy. This compound shows strong PDE5 inhibition as a possible explanation for its robust anti-cancer activity. Generally, the anticancer and PDE5 inhibitory actions of new hybrids are strongly linked. Furthermore, Vivo and clinical studies on the compound are needed. This might pave the way for a deeper knowledge of these chemical substances and the development of new cancer treatments.
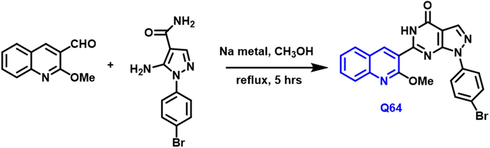
- Synthesis of novel quinoline compounds based on pyrazole moiety.
2.1.11 Pdk1:
Protein kinases provide a wealth of targets for the creation of chemical assay and therapies, as their abnormal regulations of protein phosphorylation contribute to the growth of a variety of malignancies. As a result, various 3-Phosphoinositide-dependent kinase 1 inhibition methods and pharmacological attempts are published regularly in the hopes of developing a highly effective therapeutic medication. Tamgüney et al., (Tamgüney et al., 2008) found that inhibiting PDK1 has no effect on cell proliferation in standard culture conditions while sensitizing cells to apoptotic triggers. Targeting PDK1 alone or in conjunction with conventional chemotherapeutics has also been considered a possible cancer therapy.
K.N. Vennilaa et al., (Vennila et al., 2018) have reported the conventional grid of amino-based 1-benzopyridine is decided to carry out and 8 derivatives are produced Scheme 38. The necessary H- bonding contacts with amino-acid sequences in the orthosteric region of PDK1 are formed as a result of the lock-key docking of the amino-based quinoline framework (PDK1). The binding mechanism of four crystal structures with PDK1′s adenosine triphosphate (ATP) site has been investigated. Q65 compound had potent anticancer properties against lung cancer (IC50 value of 0.96
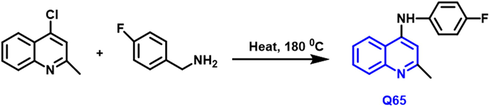
- Synthesis of novel 4-Substituted quinoline compound as an anticancer agent.
2.1.12 ERK inhibitors
The Renin-angiotensin system (RAS) / Rapidly Accelerated Fibrosarcoma (RAF) / mitogen-activated protein kinase kinase (MEK) / extracellular signal-regulated kinases (ERK) pathway has a high frequency of anomalous activation in human malignancies, making it a possible therapeutic target. (Blake et al., 2016)(Sullivan et al., 2018) ERK is a signaling nodes that connects two downstream kinases ERK1 and ERK2. (Morris et al., 2013) Various ERK inhibitors have been developed and have shown to be effective in the face of resistance. (Maik-Rachline and Seger, 2016).
The development and production of a new series of pyrano, trione, and tetraone-based 1-benzopyridine as possible Extracellular Signal-Regulated Kinase stimulators was disclosed by Ashraf A. Aly (Aly et al., 2019) (Scheme 39). The six novel prohibitions were evaluated in vitro towards the NCI-60 panel of tumor cell lines for their antitumor potential. Q-66 and Q-67 compounds were shown to be highly effective in the majority of human tumor cell lines examined. Furthermore, with IC50s of 3.7 and 0.13
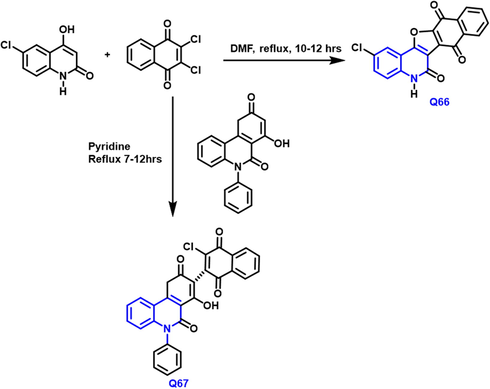
- Synthesis of novel substituted quinoline compound as an anticancer agent.
2.1.13 NQO1 inhibitor
NQO1 is a NAD(P)H quinone oxidoreductase-1 or DTD, and is one of the flavoproteins found in eukaryotic cells. The de electro reduction reaction, it contributes to the metabolism of different quinones as well as the biological activation of quinone medicines. (Danson et al., 2004).
Li-Qiang Wu et al., (Wu et al., 2020) have reported the NQO1-directed anticancer drugs, a new sequence of the quinoline-based compound was developed, prepared, and physiologically assessed (Scheme 40). Q-68 compound was shown to be excellent NQO1 substrates, with higher metabolic rates as compared to beta–lapachone. Furthermore, breast, liver, and lung tumor cells were all potently suppressed by compounds. The quinoline-based compound reduced tumor development in a HepG2 xenograft rat model at a dosage of 20 mg/kg without altering the animal weights. As a result, this compound might be a promising lead or medication candidate for further research.
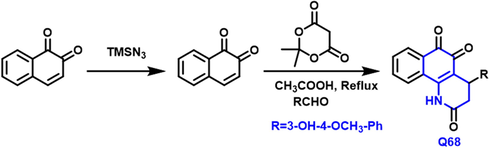
- Synthesis of novel quinoline compounds based on trione moitety.
Yong Ling et al., (Ling et al., 2018) have reported the new amino-based quinoline compounds from Quinoline-5,8-dione with amine or anilines in the presence of methanol under reflux conditions with a reaction time of 6 hrs (Scheme 41). Q-69 compound had a strong anticancer agent for cervical cancer; also exhibited potent cytotoxicity due to the presence of an amino group at 6 and 7 positions of quinoline compound.
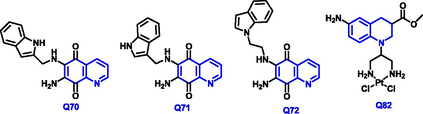
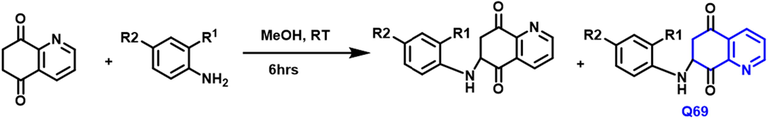
- Synthesis of novel substituted quinoline compound as an antiproliterative agent.
Maryam Gholampour et al., (Gholampour et al., 2021) have reported the synthesis of diaminoquinoline-5,8–dione derivatives using a molecular method based on silico design. Molecular docking was first utilized to investigate critical amino acid sequences and binding energies at the enzyme's active center. Depending on the outcomes, Q-70 to Q-72 molecules were chosen for a hundred molecular dynamics simulations because they have a high enzyme binding affinity and are oriented against the active center of this inhibitor. It has shown to be a viable treatment in antitumor drug development.
2.1.14 Dihydroorotate dehydrogenase inhibitor
Human dihydroorotate dehydrogenase (hDHODH) is a suitable target for chemotherapeutic medicines since it is one of the most significant enzymes in cancer cell growth. (Madak et al., 2019) Milena M. Petrovi et al., (Petrović et al., 2020) has reported sequences of new carboxylic acid-based quinoline compound were prepared by using the Doebner reaction (Scheme 42). Q-73 and Q-76 compounds were shown to be effective hDHODH inhibitors whereas other few compounds had strong cytotoxic activity and high selectivity towards breast and melanoma cells. Molecular docking was used to show the pharmacology of the molecule at the molecular level, revealing the structural distinctions that separate highly active hDHODH inhibitors from moderate and lesser potent prohibit.
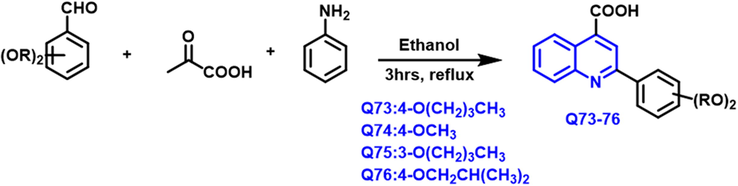
- Synthesis of novel quinoline-4-carboxylic acid derivates as an anticancer agents.
Vivek K et al., (Vyas et al., 2019) designed novel quinoline derivatives from benzaldehyde and substituted aniline in the presence of pyruvic acid and ethanol under reflux conditions to form 2-(4-substituted phenyl)-6,7-substituted-quinoline-4-Carboxylic acid by Doebner reaction. It further reacts with benzyl bromide in the presence of sodium hydride and dimethyl formamide as a solvent from the Q77 compound as a potent anticancer agent. The carboxylic derivative is reduced by lithium aluminum hydride to form alcoholic derivatives; it further reacts with benzyl bromide in the presence of sodium hydride and dimethyl formamide as a solvent to form Q78 compound as a potent anticancer agent (Scheme 43). Q77 and Q78 have an IC50 value of 1.56 µM and 1.22 µM towards hDHODH. Both the compounds have a notable antitumor agent against colon and breast cancer; non-toxicity in nature and potent hDHODH inhibitors.
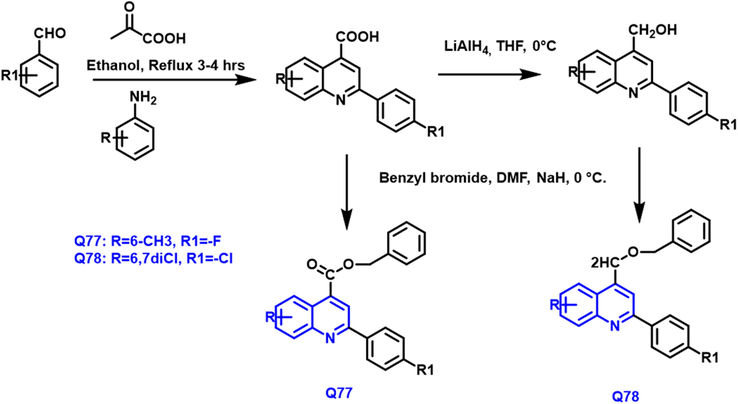
- Synthetic pathway of novel quinoline compound as an antiproliferative agent.
2.1.15 c-Myc inhibitor
The most prevalent kind of liver cancer, as well as the most frequent primary malignant tumor of the liver, is hepatocellular carcinoma. Hepatocellular carcinoma's morbidity and mortality have continued to rise in recent years, necessitating immediate action. As a result, it is critical to accelerating the development of hepatocellular cancer target medicines. C-Myc is a transcription factor that affects cell proliferation, cell growth promoter, development, cell death, and cell migration. c-Myc abnormal regulation is a frequent finding in several cancers (Dang, Le and Gao, 2009)(Eilers and Eisenman, 2008).
RNAi-mediated downregulation of c-Myc in HepG2 cells could be dramatically reduced HepG2 cell motility, invasion, and proliferation. As a result, c-Myc is being considered a possible therapeutic target for hepatocellular cancer. (Zhao et al., 2013) Baicun Lia and coworkers (Li et al., 2020) have reported that anticancer drugs were created by synthesizing novel N′-substituted methylene-4-(quinoline-4- amino) benzoylhydrazide derivatives (Scheme 44). Q79 and Q80 had strong antiproliferative activity toward lung cancer (IC50 = 12.6 ± 0.1 and 27.3 ± 1.7
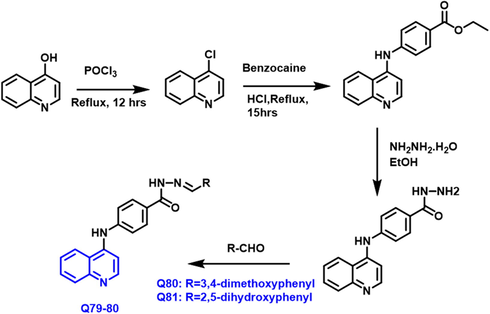
-
Synthesis of N'-substituted methylene-4- (quinoline-4-amino) benzoylhydrazides as an anticancer agent.
2.1.16 Metal ion chelator:
Yuxia Zhang and associates (Zhang et al., 2021) have reported the Q-81 compound from substituted quinoline reacts with (4-(bromomethyl) phenyl)boronic acid in the presence of potassium carbonate and acetone as a solvent under reflux conditions. Scheme 45 Q-81 has a strong inhibitor than 8-hydroxyquinoline was confirmed by in vivo studies; it inhibits the growth of pancreas tumors. Q-85 has a potent anticancer agent due to the presence of nitric oxide and 8-hydroxyquinoline conjugates. The cytotoxicity, metal-binding capacity, and (NO)-releasing effectiveness of these conjugates were all tested. Q-81 is a prodrug, that was shown to effectively limit the growth of several tumor cells. Several transition metal ions like copper, iron, and zinc metals are chelated by the 8-hydroxyquinoline molecule. Q-81 had potent cytotoxicity toward pancreatic cancer and was confirmed by MIT assays.
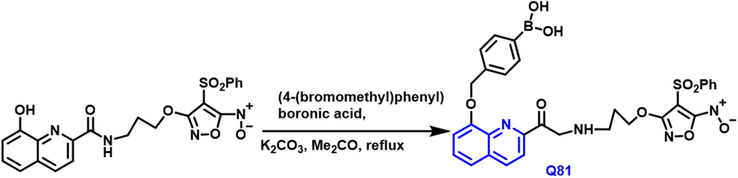
- Synthesis of novel quinohne compounds based on 8-hydroxy moiety.
2.1.17 Cis-platin-based quinoline derivatives
Cisplatin is still one of the most effective anticancer medicines on the market, and it is included in the World health organization of needed drugs. It’s used to treat malignancies of the testicles, bladder, ovary, cervix, head, neck, and esophagus.(Johnstone, Suntharalingam and Lippard, 2016) The capacity to construct deoxyribonucleic acid interconnection through nitrogen-7 purine bases and guanine bases is core to its antitumor property. However, the shortage of specificity among both tumor and non-cancer cells frequently causes side effects such as myelosuppression, immunosuppression, nephron-toxicity, hearing loss, and so on. (Jamieson and Lippard, 1999)(Florea and Büsselberg, 2011) (Yao et al., 2007).
Sateeshkumar Kumbhakonam et al.,(Kumbhakonam et al., 2020) have reported that a novel cisplatin-based quinoline derivative (Q82) was produced. When compared to mouse fibroblast NIH3T3 cells, they showed greater selectivity for cancer cells (A549), with cytotoxicity at the same level as cisplatin. ROS levels varied depending on the structure, and they may potentially kill cells, as indicated by the concentration of cells in the sub-G1 phase.
K. Pluta et al.,(Pluta et al., 2016) reported a unique preparation of thiazine-based quinoline derivatives. Certain molecules like the reference drug cisplatin showed antitumor efficacy in breast tumor cell lines. The anticancer activity of the quinobenzothiazine system is dependent on the nature of the substituents, as well as the position and kind of fusion.
2.2 Conclusion
This review, is a comprehensive overview of the quinoline-derived compounds, and their activity toward various cancer has been discussed in detail, in a nutshell, the synthesis involved in each compound is demonstrated. Plenty of quinoline-bearing compounds were developed and screened for anticancer activity. Currently, Bosutinib, Camptothecin, Lenvatinib, Cabozantinib, Topotecan, Irinotecan, and some other quinoline derivatives are being used as potential drugs for various cancer treatments. Though these drugs have greater potential for cancerous treatment, some of the newly reported quinoline-derived compounds show still higher anti-cancerous activity. Among these compounds, the Q4 compound shows greater potential and low cytotoxicity level for cancer therapy and there is a great possibility for this compound to be a successful clinical trial. In addition to these, most of the quinoline-based compounds are in laboratory trial and we hope these compounds will move on to clinical trial after required investigation. Despite several quinoline-derived compounds having potent progress reached, some of the quinoline derivatives mentioned in this review certain obstacles to overcome to be a better anticancer drug. The major challenge among many is drug resistance, low efficiency, and damage to normal cells. From our point of view, the researchers have to address these obstacles to make quinoline compounds commerically viable. As a result, we anticipate that the current study will be beneficial to medicinal chemists and the drug discovery community; since it will aid in the synthesis of novel anticancer drugs that are targeted at various human cancers.
CRediT authorship contribution statement
Mohan Ilakiyalakshmi: Writing – review & editing. Ayyakannu Arumugam Napoleon: Supervision, Conceptualization.
Acknowledgement
We have to thank VIT University, Tamil Nadu, India for their technical and financial support without which the current investigation would be incomplete.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Design, synthesis and biological evaluation of new 4-(4-substituted-anilino)quinoline derivatives as anticancer agents. Med. Chem. Res.. 2017;26(5):929-939.
- [CrossRef] [Google Scholar]
- Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg. Chem.. 2019;89
- [CrossRef] [Google Scholar]
- A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem.. 2015;97(1):871-910.
- [CrossRef] [Google Scholar]
- New bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem.. January 2021;112:104949
- [CrossRef] [Google Scholar]
- ‘Novel topoisomerase I inhibitors. syntheses and biological evaluation of phosphorus substituted quinoline derivates with antiproliferative activity’. Eur. J. Med. Chem.. 2018;149:225-237.
- [CrossRef] [Google Scholar]
- Novel quinoline-3-carboxamides (Part 2): Design, optimization and synthesis of quinoline based scaffold as EGFR inhibitors with potent anticancer activity. Bioorg. Chem.. 2017;75:368-392.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of fused naphthofuro[3,2-c] quinoline-6,7,12-triones and pyrano[3,2-c]quinoline-6,7,8,13-tetraones derivatives as ERK inhibitors with efficacy in BRAF-mutant melanoma. Bioorg. Chem.. 2019;82:290-305.
- [CrossRef] [Google Scholar]
- Angosturaalkaloide, E. Der (1911) ‘Erforschung der Angosturaalkaloide.’.
- Biologically active perspective synthesis of heteroannulated 8-nitroquinolines with green chemistry approach. Bioorg. Med. Chem. Lett.. 2017;27(7):1538-1546.
- [CrossRef] [Google Scholar]
- Isolation of lavendamycin a new antibiotic from streptomyces lavendulae. J. Antibiot.. 1982;35(3):259-265.
- [CrossRef] [Google Scholar]
- Barone, I. et al. (2017) Phosphodiesterase type 5 and cancers: progress and challenges. Available at: www.impactjournals.com/oncotarget
- Binkhathlan, Z. and Lavasanifar, A. (2013) Send Orders of Reprints at reprints@benthamscience.net P-glycoprotein Inhibition as a Therapeutic Approach for Overcoming Multidrug Resistance in Cancer: Current Status and Future Perspectives, Current Cancer Drug Targets.
- Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J. Med. Chem.. 2016;59(12):5650-5660.
- [CrossRef] [Google Scholar]
- Novel series of benzo[d]thiazolyl substituted-2-quinolone hybrids: design, synthesis, biological evaluation and in-silico insights. J. Mol. Struct.. 2021;1227
- [CrossRef] [Google Scholar]
- Human papillomavirus infection and cervical cancer: epidemiology, screening, and vaccination - review of current perspectives. J. Oncol.. 2019;2019
- [CrossRef] [Google Scholar]
- Light alcohol drinking and risk of cancer: a meta-analysis of cohort studies. Cancer Res. Treat.. 2018;50(2):474-487.
- [CrossRef] [Google Scholar]
- Biphasic regulation of the NADPH oxidase by HGF/c-Met signaling pathway in primary mouse hepatocytes. Biochimie. 2013;95(6):1177-1184.
- [CrossRef] [Google Scholar]
- The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev.. 2005;19(4):472-488.
- [CrossRef] [Google Scholar]
- MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res.. 2009;15(21):6479-6483.
- [CrossRef] [Google Scholar]
- DT-diaphorase: a target for new anticancer drugs. Cancer Treat. Rev.. 2004;30(5):437-449.
- [CrossRef] [Google Scholar]
- Lung cancer epidemiology: contemporary and future challenges worldwide. Ann. Translat. Med.. 2016;4(8):1-11.
- [CrossRef] [Google Scholar]
- Phase II study of XR 5000 (DACA), an inhibitor of topoisomerase I and II, administered as a 120-h infusion in patients with non-small cell lung cancer. Eur. J. Cancer. 2003;39(3):330-334.
- [CrossRef] [Google Scholar]
- Douglas, A. and Hughes, H. (1991) ‘Evaluation of Natural Products As Inhibitors of Human Immunodeficiency Virus Type 1’, (1)
- Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol. Cell. Biol.. 2014;34(10):1722-1732.
- [CrossRef] [Google Scholar]
- Discovery of 4-alkoxy-2-aryl-6,7-dimethoxyquinolines as a new class of topoisomerase I inhibitors endowed with potent in vitro anticancer activity. Eur. J. Med. Chem.. 2021;215
- [CrossRef] [Google Scholar]
- Novel 5-substituted-2-anilinoquinolines with 3-(morpholino or 4-methylpiperazin-1-yl)propoxy moiety as broad spectrum antiproliferative agents: synthesis, cell based assays and kinase screening. Bioorg. Med. Chem. Lett.. 2016;26(14):3307-3312.
- [CrossRef] [Google Scholar]
- 2-Anilinoquinoline based arylamides as broad spectrum anticancer agents with B-RAFV600E/C-RAF kinase inhibitory effects: design, synthesis, in vitro cell-based and oncogenic kinase assessments. Eur. J. Med. Chem.. 2020;208
- [CrossRef] [Google Scholar]
- Design and synthesis of new RAF kinase-inhibiting antiproliferative quinoline derivatives. part 2: diarylurea derivatives. Eur. J. Med. Chem.. 2017;127:413-423.
- [CrossRef] [Google Scholar]
- Endicott, J. A. and Ling, V. (1989) THE BIOCHEMISTRY OF P-GLYCOPROTEIN-MEDIATED MULTIDRUG RESISTANCE ! Available at: www.annualreviews.org
- Design, synthesis, and evaluation of new 2-oxoquinoline arylaminothiazole derivatives as potential anticancer agents. Bioorg. Chem.. 2021;106
- [CrossRef] [Google Scholar]
- Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem.. 2015;22(18):5050-5059.
- [CrossRef] [Google Scholar]
- Florea, A. M. and Büsselberg, D. (2011) ‘Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects’, Cancers, pp. 1351–1371. doi: 10.3390/cancers3011351.
- In silico design of novel diamino-quinoline-5,8-dione derivatives as putative inhibitors of NAD(P)H: quinone oxidoreductase 1 based on docking studies and molecular dynamics simulations. J. Mol. Struct.. 2021;1230
- [CrossRef] [Google Scholar]
- Role of genetic testing for inherited prostate cancer risk: philadelphia prostate cancer consensus conference 2017. J. Clin. Oncol.. 2018;36(4):414-424.
- [CrossRef] [Google Scholar]
- Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis 2010:1-14.
- [CrossRef] [Google Scholar]
- Multiple effects of berberine derivatives on colon cancer cells. Biomed. Res. Int.. 2014;2014
- [CrossRef] [Google Scholar]
- A third shot at EGFR: new opportunities in cancer therapy. Trends Pharmacol. Sci.. 2019;40(12):941-955.
- [CrossRef] [Google Scholar]
- Efficient synthesis, antimicrobial, antioxidant assessments and geometric optimization calculations of azoles- incorporating quinoline moiety. J. Heterocycl. Chem.. 2018;55(11):2623-2634.
- [CrossRef] [Google Scholar]
- Structural optimization towards promising β-methyl-4-acrylamido quinoline derivatives as PI3K/mTOR dual inhibitors for anti-cancer therapy: the in vitro and in vivo biological evaluation. Eur. J. Med. Chem.. 2021;214
- [CrossRef] [Google Scholar]
- Molecular design and synthesis of certain new quinoline derivatives having potential anticancer activity. Eur. J. Med. Chem.. 2015;102:115-131.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel pyrazolo[3,4-d]pyrimidin-4-one bearing quinoline scaffold as potent dual PDE5 inhibitors and apoptotic inducers for cancer therapy. Bioorg. Chem.. 2020;105
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of novel benzo- and tetrahydrobenzo-[h]quinoline derivatives as potential DNA-intercalating antitumor agents. Eur. J. Med. Chem.. 2019;164:292-303.
- [CrossRef] [Google Scholar]
- Comprehensive review on current developments of quinoline-based anticancer agents. Arabian J. Chem.. Elsevier B.V. 2019:4920-4946.
- [CrossRef] [Google Scholar]
- Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev.. 1999;99(9):2467-2498.
- [CrossRef] [Google Scholar]
- Jiao, Q. et al. (2018) ‘Advances in studies of tyrosine kinase inhibitors and their acquired resistance’, Molecular Cancer. BioMed Central Ltd. doi: 10.1186/s12943-018-0801-5.
- The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev.. Am. Chem. Soc. 2016:3436-3486.
- [CrossRef] [Google Scholar]
- Designed P-glycoprotein inhibitors with triazol-tetrahydroisoquinoline-core increase doxorubicin-induced mortality in multidrug resistant K562/A02 cells. Bioorg. Med. Chem.. 2019;27(15):3347-3357.
- [CrossRef] [Google Scholar]
- A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem.. 2018;158:917-936.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel conformationally constrained 7,12-dihydrodibenzo[b, h][1,6] naphthyridine and 7H-Chromeno[3,2-c] quinoline derivatives as topoisomerase I inhibitors: In vitro screening, molecular docking and ADME predictions. Bioorg. Chem.. 2021;115(July):105174
- [CrossRef] [Google Scholar]
- Computational and synthetic approach with biological evaluation of substituted quinoline derivatives as small molecule L858R/T790M/C797S triple mutant EGFR inhibitors targeting resistance in non-small cell lung cancer (NSCLC) Bioorg. Chem.. 2021;107
- [CrossRef] [Google Scholar]
- Free energy perturbation guided synthesis with biological evaluation of substituted quinoline derivatives as small molecule L858R/T790M/C797S mutant EGFR inhibitors targeting resistance in non-small cell lung cancer (NSCLC) Bioorg. Chem.. 2021;115(July):105226
- [CrossRef] [Google Scholar]
- Design, synthesis and anticancer properties of IsoCombretaQuinolines as potent tubulin assembly inhibitors. Eur. J. Med. Chem.. 2017;127:1025-1034.
- [CrossRef] [Google Scholar]
- Diverse C-6 substituted 4-methyl-2-(2-, 3- and 4-pyridinyl)quinolines: synthesis, in vitro anticancer evaluation and in silico studies. Med. Chem. Res.. 2017;26(3):551-561.
- [CrossRef] [Google Scholar]
- Reactive Pt(II) center as part of redox-active quinoline-based heterocyclic scaffolds toward new anticancer leads. Bioorg. Med. Chem. Lett.. 2020;30(22)
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of quinoline-indole derivatives as anti-tubulin agents targeting the colchicine binding site. Eur. J. Med. Chem.. 2019;163:428-442.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluations of N′-substituted methylene-4-(quinoline-4-amino) benzoylhydrazides as potential anti-hepatoma agents. Bioorg. Chem.. 2020;96
- [CrossRef] [Google Scholar]
- Li, X. yang et al. (2022) ‘Discovery of N-(1,3,4-thiadiazol-2-yl)benzamide derivatives containing a 6,7-methoxyquinoline structure as novel EGFR/HER-2 dual-target inhibitors against cancer growth and angiogenesis’, Bioorganic Chemistry, 119(36), p. 105469. doi: 10.1016/j.bioorg.2021.105469.
- Development of novel amino-quinoline-5,8-dione derivatives as NAD(P)H:quinone oxidoreductase 1 (NQO1) inhibitors with potent antiproliferative activities. Eur. J. Med. Chem.. 2018;154:199-209.
- [CrossRef] [Google Scholar]
- Structure-based design of novel class II c-Met inhibitors: 2. SAR and kinase selectivity profiles of the pyrazolone series. J. Med. Chem.. 2012;55(5):1868-1897.
- [CrossRef] [Google Scholar]
- Madak, J. T. et al. (2019) ‘Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer’, Pharmacology and Therapeutics. Elsevier Inc., pp. 111–131. doi: 10.1016/j.pharmthera.2018.10.012.
- Maik-Rachline, G. and Seger, R. (2016) ‘The ERK cascade inhibitors: Towards overcoming resistance’, Drug Resistance Updates. Churchill Livingstone, pp. 1–12. doi: 10.1016/j.drup.2015.12.001
- Hepatitis b virus-associated hepatocellular carcinoma and hepatic cancer stem cells. Genes. 2018;9(3):1-15.
- [CrossRef] [Google Scholar]
- An overview of privileged scaffold: quinolines and isoquinolines in medicinal chemistry as anticancer agents. Curr. Top. Med. Chem.. 2020;20(28):2599-2633.
- [CrossRef] [Google Scholar]
- Mariano, T. M. et al. (2002) ‘Contents of Volume 63 and bacterial pathogens of the skin’, 63(January), pp. 2208–2222
- The versatile quinoline and its derivatives as anti-cancer agents: an overview. Polycyclic Aromat. Compd. 2022:1-13.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of novel 5,6,7-trimethoxy-N-aryl-2-styrylquinolin-4-amines as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem.. 2020;98
- [CrossRef] [Google Scholar]
- Mohassab, A. M. et al. (2021) ‘Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases’, Bioorganic Chemistry, 106(October 2020), p. 104510. doi: 10.1016/j.bioorg.2020.104510
- Quinoline: an attractive scaffold in drug design. Mini-Rev. Med. Chem.. 2021;21(16):2209-2226.
- [CrossRef] [Google Scholar]
- Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discovery. 2013;3(7):742-750.
- [CrossRef] [Google Scholar]
- Sunitinib: ten years of successful clinical use and study in advanced renal cell carcinoma. Oncologist. 2017;22(1):41-52.
- [CrossRef] [Google Scholar]
- Synthesis, crystal structure, cytotoxicity and evaluation of the 6-oxo-6H-pyrrolo[3,2,1-ij]quinoline-5-carbaldehydes for inhibitory effect against protein kinases (VEGFR-2 and EGFR) and cyclooxygenase-2 (COX-2) activities. J. Mol. Struct.. 2020;1222
- [CrossRef] [Google Scholar]
- Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med. Res. Rev.. 2012;32(6):1220-1262.
- [CrossRef] [Google Scholar]
- 3H-pyrazolo[4,3-f]quinoline haspin kinase inhibitors and anticancer properties. Bioorg. Chem.. 2018;78:418-426.
- [CrossRef] [Google Scholar]
- Design, synthesis and anticancer evaluation of new substituted thiophene-quinoline derivatives. Bioorg. Med. Chem.. 2019;27(19)
- [CrossRef] [Google Scholar]
- Negative control of the HGF/c-MET pathway by TGF-β: a new look at the regulation of stemness in glioblastoma article. Cell Death Dis.. 2017;8(12)
- [CrossRef] [Google Scholar]
- CoMFA and CoMSIA studies on 6,7-disubstituted-4-phenoxyquinoline derivatives as c-Met kinase inhibitors and anticancer agents. Med. Chem. Res.. 2015;24(12):4078-4092.
- [CrossRef] [Google Scholar]
- Amino substituted benzimidazo[1,2-a]quinolines: antiproliferative potency, 3D QSAR study and DNA binding properties. Eur. J. Med. Chem.. 2016;122:530-545.
- [CrossRef] [Google Scholar]
- Potent human dihydroorotate dehydrogenase inhibitory activity of new quinoline-4-carboxylic acids derived from phenolic aldehydes: synthesis, cytotoxicity, lipophilicity and molecular docking studies. Bioorg. Chem.. 2020;105
- [CrossRef] [Google Scholar]
- Synthesis, spectroscopic structure identification, X-ray study and anticancer activities of new angularly fused quinobenzothiazines. J. Mol. Struct.. 2016;1122:62-71.
- [CrossRef] [Google Scholar]
- Discovery and optimization of novel 4-phenoxy-6,7-disubstituted quinolines possessing semicarbazones as c-Met kinase inhibitors. Bioorg. Med. Chem.. 2013;21(17):5246-5260.
- [CrossRef] [Google Scholar]
- Identification of novel N1-(2-aryl-1, 3-thiazolidin-4-one)-N3-aryl ureas showing potent multi-tyrosine kinase inhibitory activities. Eur. J. Med. Chem.. 2018;146:368-380.
- [CrossRef] [Google Scholar]
- Incorporation of fluorinated pyridine in the side chain of 4-aminoquinolines: synthesis, characterization and antibacterial activity. Drug Res.. 2018;68(1):17-22.
- [CrossRef] [Google Scholar]
- The structure of streptonigrin. J. Am. Chem. Soc.. 1963;85(16):2532-2533.
- [CrossRef] [Google Scholar]
- Novel 4-quinoline-thiosemicarbazone derivatives: synthesis, antiproliferative activity, in vitro and in silico biomacromolecule interaction studies and topoisomerase inhibition. Eur. J. Med. Chem.. 2019;182
- [CrossRef] [Google Scholar]
- Histone phosphorylation: A chromatin modification involved in diverse nuclear events. In: Epigenetics. Taylor and Francis Inc.; 2012. p. :1098-1108.
- [CrossRef] [Google Scholar]
- Gene-environment interaction and risk of breast cancer. Br. J. Cancer. 2016;114(2):125-133.
- [CrossRef] [Google Scholar]
- The alkaloids of argemone mexicana. J. Am. Chem. Soc.. 1932;54(7):2923-2924.
- [CrossRef] [Google Scholar]
- Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol.. 2012;19(11):1147-1154.
- [CrossRef] [Google Scholar]
- New pyranoquinoline derivatives as vascular-disrupting anticancer agents. Med. Chem. Res.. 2019;28(10):1694-1703.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of quinoline analogues of flavones as potential anticancer agents and tubulin polymerization inhibitors. Eur. J. Med. Chem.. 2016;114:14-23.
- [CrossRef] [Google Scholar]
- Population pharmacokinetics modeling and analysis of foretinib in adult patients with advanced solid tumors. J. Clin. Pharmacol.. 2015;55(10):1184-1192.
- [CrossRef] [Google Scholar]
- Design, docking, and synthesis of quinoline-2H-1,2,4-triazol-3(4H)-ones as potent anticancer and antitubercular agents. ChemistrySelect. 2018;3(7):2004-2016.
- [CrossRef] [Google Scholar]
- First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer Discovery. 2018;8(2):184-195.
- [CrossRef] [Google Scholar]
- Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.. 2021;71(3):209-249.
- [CrossRef] [Google Scholar]
- Current pharmaceutical aspects of synthetic quinoline derivatives. Mini-Rev. Medi. Chem.. 2020;21(10):1152-1172.
- [CrossRef] [Google Scholar]
- Analysis of 3-phosphoinositide-dependent kinase-1 signaling and function in ES cells. Exp. Cell Res.. 2008;314(11–12):2299-2312.
- [CrossRef] [Google Scholar]
- Synthesis and antiproliferative activity of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing the 2-oxo-4-chloro-1,2-dihydroquinoline-3-carboxamide moiety. Bioorg. Med. Chem. Lett.. 2016;26(7):1794-1798.
- [CrossRef] [Google Scholar]
- Synthesis and antiproliferative activity of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing the 1,8-naphthyridin-2-one moiety. Eur. J. Med. Chem.. 2018;158:201-213.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation, structure-activity relationship, and mechanism of action studies of quinoline-metronidazole derivatives against experimental visceral leishmaniasis. J. Med. Chem.. 2019;62(11):5655-5671.
- [CrossRef] [Google Scholar]
- Design, synthesis, crystal structures and anticancer activity of 4-substituted quinolines to target PDK1. Bioorg. Chem.. 2018;81:184-190.
- [CrossRef] [Google Scholar]
- ‘Trastuzumab: Updated mechanisms of action and resistance in breast cancer’, Frontiers. Oncology 2 JUN(June) 2012:1-6.
- [CrossRef] [Google Scholar]
- Synthesis of 2-,4,-6-, and/or 7-substituted quinoline derivatives as human dihydroorotate dehydrogenase (hDHODH) inhibitors and anticancer agents: 3D QSAR-assisted design. Bioorg. Med. Chem. Lett.. 2019;29(7):917-922.
- [CrossRef] [Google Scholar]
- Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002:430-440.
- [CrossRef] [Google Scholar]
- Discovery and characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) polymer. J. Med. Chem.. 2016;59(1):335-357.
- [CrossRef] [Google Scholar]
- Catalytic mechanisms and regulation of protein kinases. In: Methods in Enzymology. Academic Press Inc.; 2014. p. :1-21.
- [CrossRef] [Google Scholar]
- Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev.. 2010;23(4):713-739.
- [CrossRef] [Google Scholar]
- Design, synthesis, and biological evaluation of 4-substituted-3,4-dihydrobenzo[h]quinoline-2,5,6(1H)-triones as NQO1-directed antitumor agents. Eur. J. Med. Chem.. 2020;198
- [CrossRef] [Google Scholar]
- Nitric oxide-donating and reactive oxygen species-responsive prochelators based on 8-hydroxyquinoline as anticancer agents. Eur. J. Med. Chem.. 2021;212
- [CrossRef] [Google Scholar]
- Zhao, Y. et al. (2013) RNAi silencing of c-Myc inhibits cell migration, invasion, and proliferation in HepG 2 human hepatocellular carcinoma cell line: c-Myc silencing in hepatocellular carcinoma cell. Available at: http://www.cancerci.com/content/13/1/23
- Red and processed meat consumption and esophageal cancer risk: a systematic review and meta-analysis. Clin. Transl. Oncol.. 2020;22(4):532-545.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of 4-anilinoquinoline derivatives as novel potent tubulin depolymerization agents. Eur. J. Med. Chem.. 2017;138:1114-1125.
- [CrossRef] [Google Scholar]
- Identification of novel quinoline analogues bearing thiazolidinones as potent kinase inhibitors for the treatment of colorectal cancer. Eur. J. Med. Chem.. 2020;204
- [CrossRef] [Google Scholar]
- Discovery of 4-((4-(4-(3-(2-(2,6-difluorophenyl)-4-oxothiazolidin-3-yl)ureido)-2-fluorophenoxy)-6-methoxyquinolin-7-yl)oxy)-N, N-diethylpiperidine-1-carboxamide as kinase inhibitor for the treatment of colorectal cancer. Bioorg. Chem.. 2021;106
- [CrossRef] [Google Scholar]




