

Translate this page into:
Stereoselectivity of the captodative alkenes 1-acetylvinyl arenecarboxylates in Diels-Alder reactions with cyclic dienes and stereospecific rearrangement of their bicyclo[2.2.n] α-ketol adducts
⁎Corresponding author. jtamarizm@gmail.com (Joaquín Tamariz) jtamariz@woodward.encb.ipn.mx (Joaquín Tamariz)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Captodative alkene 1-acetylvinyl p-nitrobenzenecarboxylate 1a was evaluated for its reactivity and stereoselectivity with cyclohexadiene (10) in Diels-Alder reactions, showing exclusive endo preference. The two hydrolyzed products of the endo and the exo adducts obtained from the Diels-Alder cycloaddition between 1a and cyclopentadiene (7) were 8b and 9b. When treated with mCPBA, a ring expansion took place to stereospecifically yield the novel 3-oxatricyclo[3.3.1.02,4]nonanone acyloins 15 and 16, respectively. In the case of the α-ketol bicyclo[2.2.2]octanes 11b and 12b, the epoxidation/Baeyer-Villiger cascade process was preferred, resulting in the syn ketoepoxide 19b from each isomer. A synthetic application of this kind of transformation was carried out by reacting ketols 8b and 9b with an excess of mCPBA through a five-step cascade process to yield the bicyclic lactone 27 as a potential precursor of racemic β-carbaxylose.
Keywords
Captodative alkenes
1-Acetylvinyl p-nitrobenzoyloxy
α-Keto carbinols
α-Ketol rearrangement
Cascade process
mCPBA

1 Introduction
Diels-Alder reactions are one of the fundamental cornerstones in organic chemistry for the experimental and theoretical study of concerted reactions (Sauer and Sustmann, 1980; Herges et al., 1994; Damoun et al., 1997; Brocksom et al., 2001; Tantillo et al., 2001; Wang et al., 2009; Domingo et al., 2010) and for the regio- and stereoselective synthesis of six-membered rings (Carruthers, 1990; Fringuelli and Taticchi, 2002; Nicolaou et al., 2002; Reymond and Cossy, 2008; Ishihara and Sakakura, 2014). Although Diels-Alder additions are usually conducted through a concerted (synchronical and non-synchronical and diradicaloid) pericyclic transition state (Singleton, 2001; Jasiński, 2016a), there is evidence of non-polar stepwise diradical (Jasiński, 2016a) and polar stepwise (zwitterionic) (Jasiński, 2014, 2016b; Jasiński et al., 2014) mechanisms. The reactivity, selectivity and concertedness of this process are mainly subjected to the electronic and structural effects of the substituents in both cycloaddends: diene and dienophile (Sauer and Sustmann, 1980; Paquette et al., 1980; Pindur, et al., 1993; Kumar, 2001; Zhou et al., 2014).
Captodative alkenes have been subjected to experimental (De Boeck and Viehe, 1998; Rulev, 2002; Moody et al., 2002; Kozmin et al., 2002; Hatano et al., 2011; Kotha et al., 2014) and theoretical studies (Méndez et al., 1998; Geerlings et al., 2003; Domingo et al., 2008; Bouacha et al., 2013) due to the opposite electronic demand of their geminally substituted functional groups (Viehe, et al., 1985). Research has mainly focused on their reactivity and chemo-, stereo- and regioselectivity in [4 + 2] cycloaddition (Diels-Alder and 1,3-dipolar) reactions (Benavides et al., 2001; Herrera et al., 2005; Mendoza et al., 2006; Bǎdoiu and Kündig, 2012; Carmona et al., 2013; Zou et al., 2013) and on their application in organic synthesis (Thorat and Pansare, 2013; Wang et al., 2013; Sun et al., 2014).
In particular, captodative alkenes such as 1-acetylvinyl carboxylates 1 (Fig. 1) have proven to be highly reactive and selective in Diels-Alder (Reyes et al., 1990; Santoyo et al., 2007) and 1,3-dipolar cycloadditions (Nagarajan et al., 1996; Herrera et al., 2001a) as well as in Friedel-Crafts reactions (Zárate-Zárate et al., 2015). They undergo regioselective addition to acyclic nonsymmetrical dienes to give the corresponding adducts (Reyes et al., 1990), which have been employed as versatile synthons in natural product synthesis (Orduña et al., 1993; Dienes and Vogel, 1996). The captodative alkyl 2-aroyloxy acrylates 2a-2b have shown high reactivity and selectivity in Diels-Alder reactions as well (Herrera et al., 2001b).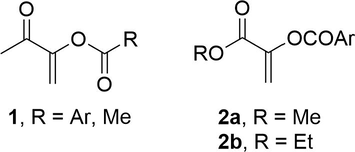
Captodative alkenes 1-acetylvinyl p-arenecarboxylates 1 and alkyl 2-aroyloxy acrylates 2a-2b.
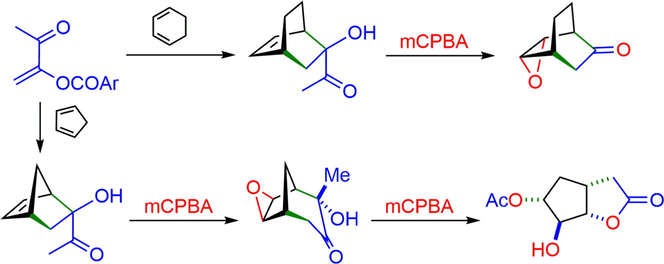
Moreover, a new synthetic approach has been developed for the preparation of γ-hydroxycyclohexenones 6 through an epoxidation/Baeyer-Villiger/hydrolysis cascade reaction of α-ketols 5 with m-chloroperbenzoic acid (mCPBA). In this process, alkene 1a behaved as a synthetic equivalent of ketenes in the Diels-Alder cycloaddition (Ochoa et al., 1999) (Scheme 1). The resulting bicyclo[2.2.n] adducts of the cycloaddition of a variety of ketene equivalents with cyclic dienes have sparked special interest in the study of the reactivity and selectivity of these systems (Ranganathan et al., 1977; Aggarwal et al., 1999; Moreno-Clavijo et al., 2013).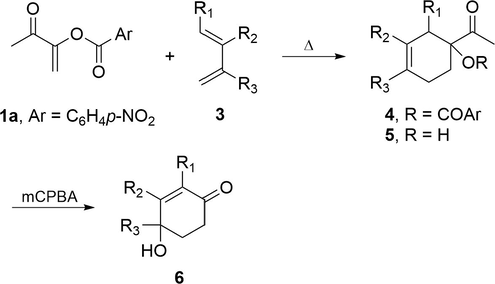
Regioselective Diels-Alder addition of captodative alkene 1a and conversion of adducts 5 into γ-hydroxycyclohexenones 6.
The stereoselectivity of captodative alkenes 1 in thermal Diels-Alder cycloadditions with cyclopentadiene (7) is reportedly low (Tamariz and Vogel, 1981). However, the use of Lewis acid catalysts dramatically increases either endo or the exo selectivity, depending on the choice of the Lewis acid (García de Alba et al., 1996). Thus, when 1a was added to 7 in the presence of TiCl4, the endo adduct 8a was obtained almost exclusively. With ZnI2 as the catalyst, on the other hand, the exo adduct 9a was yielded as a single product (Scheme 2).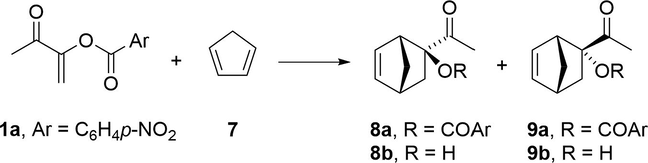
Preparation of adducts 8a-9a via Diels-Alder addition of captodative alkene 1a to cyclopentadiene (7), and structures of the α-ketol derivatives 8b and 9b.
As part of our ongoing interest in the stereoselective Diels-Alder reactions of olefins 1 with cyclic dienes, we undertook an extensive study of the cycloaddition between 1a and cyclohexadiene (10) under various different conditions. Moreover, to explore the reactivity of α-ketols and their application in organic synthesis, we investigated the reaction of the α-ketols arising from the Diels-Alder adducts with mCPBA. Finally, we examined the utilization of these compounds in a formal synthesis of the carbasugar β-carbaxylose (Marschner et al., 1995) via a cascade process.
2 Results and discussion
2.1 Diels-Alder reaction of olefin 1a with cyclohexadiene (10)
The thermal Diels-Alder cycloaddition of 1a to cyclohexadiene (10) was highly diastereoselective to give the endo isomer 11a as a single adduct (Table 1, entry 1). Contrarily, the thermal cycloaddition to cyclopentadiene (7) generated a mixture of 8a/9a (Scheme 2) in low diastereoisomeric endo/exo ratio (33:67) (Tamariz and Vogel, 1981). Also unlike the cycloaddition to 7, where the use of TiCl4 provided an endo/exo mixture of adducts 8a/9a (96:4) (García de Alba et al., 1996), the catalyzed cycloaddition with 10 yielded a nonselective mixture of endo/exo adducts, 11a/12a (44:56) (Table 1, entry 2). Similarly, the BF3·Et2O-catalyzed reaction was modestly diastereoselective, leading to an endo/exo mixture of 11a/12a (67:33) (Table 1, entry 3). However, a higher selectivity was found when the cycloadditions were carried out in the presence of AlCl3 or ZnI2 as the catalyst. Indeed, the endo isomer 11a was isolated as a single product (Table 1, entries 4 and 5).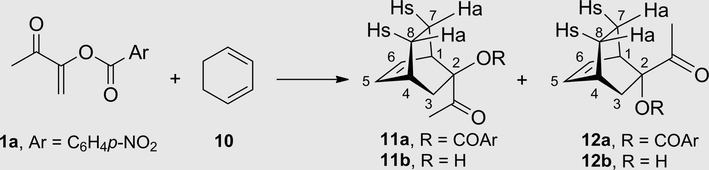
Entry
Solvent
Catalyst (mol equiv)
T (°C)
t (h)
11a/12a (ratio)b
Yield (%)c
1
C6H6
–
130
168
100:0
70
2
CH2Cl2
TiCl4 (8.6)
−50
3
44:56
92
3
CH2Cl2
BF3.Et2O (19)
−50
10
67:33
85
4
CH2Cl2
AlCl3 (2.4)
0
1
100:0
95
5
CH2Cl2
ZnI2 (24)
25
120
100:0
84
The amount of Lewis acids (shown in Table 1) was optimized to render the cycloadditions in the shortest reaction times. A large excess of Lewis acid was used in most cases, since the reaction time sharply increased with a lesser amount. It is likely that the multiple binding sites (oxygen atoms) can explain the need for such large catalyst loads. Regarding AlCl3, interestingly, only 2.4 mol equiv of the solid catalyst were required, and the ratio of the endo/exo adducts was not significantly modified by using greater amounts of this catalyst.
Besides identifying the most efficient trials (Table 1), several screening tests were carried out to determine the optimal reaction conditions, taking into account the catalyst, temperature and reaction time. As previously mentioned, the type of Lewis acid catalyst modifies reactivity and diastereoselectivity (Table 1). Although both temperature and reaction time affected the yields, the endo/exo diastereoselective ratio was not significantly modified by these factors. For example, when the TiCl4-catalyzed reaction took place at a temperature higher than −50 °C, there was a decomposition of the dienophile and the formation of a complex mixture of products. By reducing the temperature to −78 °C, the reaction time was extended 5–6 h with no considerable change of the endo/exo ratio. Likewise, by employing AlCl3 and ZnI2 as the catalysts at temperatures lower than those indicated in Table 1 (entries 4 and 5), either the cycloaddends did not react or the reaction time was longer, but the endo/exo ratios remained essentially the same. Similar behavior was observed for the thermal trials, in which a reaction temperature below or above 130 °C, augmented or diminished the reaction times, respectively. However, the yields and endo/exo ratios were comparable regardless of the temperature.
The structure of the major endo stereoisomer 11a was established by X-ray diffraction analysis (Fig. 2). Notably, the conformation of the acetyl group is such that the carbonyl group is oriented towards the C-3 methylene of the bicyclo[2.2.2]oct-5-ene (in conformation). An identical conformation is found in the endo adduct 8a (García de Alba et al., 1996). The minor exo adduct 12a was separated from the reaction mixture by column chromatography and was fully characterized by spectroscopy, elemental analysis and X-ray crystallography (Fig. 2). In the latter structure, the carbonyl group is also oriented towards the C-3 methylene of the bicyclo[2.2.2]oct-5-ene. This conformation seems to be adopted even in solution, as suggested by the 1H NMR high frequency chemical shift of the H-C3 proton syn to the carbonyl group in both adducts: 2.48 ppm for H-3n of 11a (1.59 ppm for H-3x) and 2.90 ppm for H-3x of 12a (1.33 ppm for H-3n). The proximity of the oxygen atom of the carbonyl group probably generates a deshielding effect on the syn protons.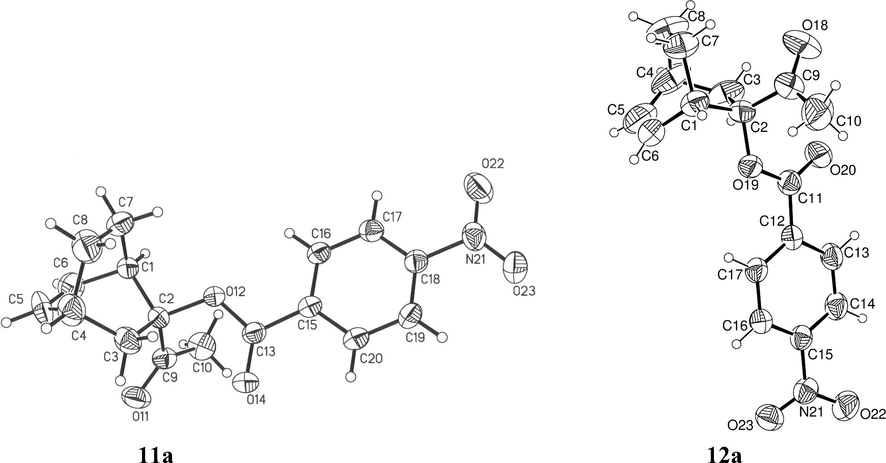
X-ray structures of the endo adduct 11a and exo adduct 12a (ellipsoids with 30% probability).
The observed endo diastereoselectivity for the thermal and some of the catalyzed trials could possibly be explained by steric interactions of the acetyl and the aroyloxy groups with the most hindered ethane bridge of the diene (Fig. 3). According to the X-ray diffraction and quantum calculations of 1a (Jiménez-Vázquez et al., 1997), the s-trans conformation is preferred by the conjugated enone system, with the p-nitrobenzoyl group out of the plane formed by the enone moiety. This preferred conformation probably is also adopted at the ground-state complexes of 1a with Lewis acids, including AlCl3 and ZnI2 (Loncharich et al., 1987; Denmark and Almstead, 1993; Ishihara et al., 1993), as well as at the transition states (Rebiere et al., 1990; Maruoka et al., 1994). Therefore, the exo approach would be prevented by non-bonding repulsive interactions of the methyl group and the bulky metal-coordinated carbonyl group with the ethane bridge. Additionally, at the endo transition state, the π-systems might be stabilized by secondary interactions, such as those promoted by orbitals (Arrieta et al., 2001; Caramella et al., 2002; Wannere et al., 2007) or by electrostatic forces (García et al., 2000; Sakata and Fujimoto, 2016).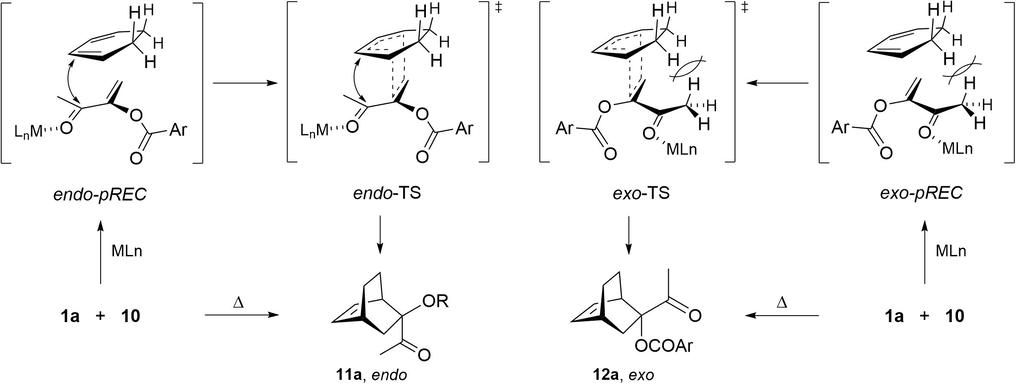
Possible interactions at the endo and exo pRECs and TSs between the MLn (n = 2, 3)-complexed and non-complexed dienophile 1a and cyclohexadiene (10).
In agreement with previous results (Suárez and Sordo, 1998), we have recently demonstrated the importance of the formation of pre-reactive electrostatic complexes (pREC) in the thermal Diels-Alder potential energy surfaces (PES). These complexes are a result of the π/π interaction between the reactants (Ramírez-Gualito et al., 2013), which is mainly controlled by dispersion forces (Cioslowski et al., 1993). These are to a great extent responsible for the endo preference (Ramírez-Gualito et al., 2013). It is likely that similar pRECs are formed in the presence of the Lewis acid catalyst, and that repulsive steric interactions are present at the exo-pREC. Such interactions would then destabilize not only this complex but its respective transition state (exo-TS) as well. Hence, the secondary interactions and dispersion forces stabilizing the endo-pREC may also stabilize the endo-TS, leading to the selective formation of the endo adduct 11a.
2.2 Stereospecific rearrangement of α-ketols 8b and 9b
α-Ketols 8b and 9b were prepared by saponification (K2CO3, MeOH, reflux, 10 h) of the corresponding adducts 8a and 9a in almost quantitative yields. Treatment of α-ketol 8b with 1.0 mol equiv of mCPBA in CH2Cl2 at room temperature led exclusively to the endo-hydroxy-exo-epoxy tricyclo[3.3.1.02,4]nonan-7-one (endo-hydroxy-α-ketol) 15, in 68% yield (Scheme 3). Although analogous bicyclo[2.2.1] α-ketols have given rise to the corresponding bicyclo[3.2.1] products through an α-ketol rearrangement (Paquette and Hofferberth, 2003), they do so by undergoing thermal or base-promoted processes under much more severe conditions (Stevens et al., 1972). In these cases, the stereoisomeric ratio of the bicyclo[3.2.1] products depends on the reaction conditions (Creary et al., 1985). The easy rearrangement of α-ketol 8b is probably associated with the strain on the ring systems brought about by epoxidation to give the epoxy intermediate 13. That is, the C—C bond migration might be accelerated to relieve the strain and furnish 15 (Brunner et al., 2001). This process may also be catalyzed by the acidic reaction medium generated both by mCPBA and the m-chlorobenzoic acid byproduct of the reaction.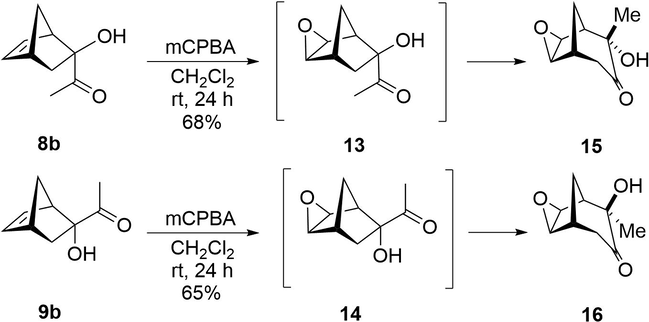
Epoxidation and stereospecific α-ketol rearrangement cascade reaction of α-ketols 8b and 9b.
These results suggest that the process is not stereoselective, but instead stereospecific. Indeed, when the exo-α-ketol 9b was submitted to a reaction under identical conditions, only exo-hydroxy-exo-epoxy tricyclo[3.3.1.02,4]nonan-7-one (exo-hydroxy- α-ketol) 16 was obtained, with a 65% yield (Scheme 3). The rearrangement may owe itself to the formation of the corresponding epoxy intermediate, in this case 14. The structure of exo-hydroxy- α-ketol 16 was unambiguously established by X-ray crystallography (Fig. 4), while the structure of endo-hydroxy- α-ketol 15 was determined by nOe and 2D NMR experiments. Interestingly, epoxidation takes place diastereoselectively on the exo face of the double bond, regardless of the stereochemistry of α-ketols 8b and 9b.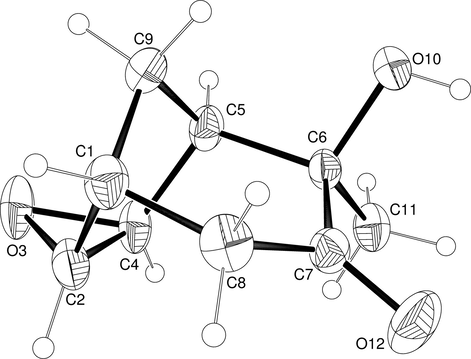
X-ray structure of exo-hydroxy-α-ketol 16 (ellipsoids with 30% probability).
2.3 Rearrangement of α-ketols 11b and 12b
Adducts 11a and 12a were hydrolyzed under basic conditions (K2CO3, MeOH/THF, reflux, 3–4 h) to afford α-ketols 11b and 12b, respectively, in good yields (81–94%). Reaction conditions analogous to those previously applied for 8b and 9b were used for the rearrangement of α-ketol 11b. Thus, upon reacting a solution of 11b in CH2Cl2 with 2.0 mol equiv of mCPBA, the syn-facial bicyclic ketoepoxide 19b was produced in 64% yield (Scheme 4). In contrast to the results when utilizing α-ketols 8b and 9b, only the syn-ketoepoxide 19b was formed with 11b, even in the presence of an excess of mCPBA. No syn-epoxyketol 20 was found. Interestingly, the anti-facial isomer 19a was not detected by 1H NMR analysis of the reaction crude. Under similar reaction conditions, α-ketol 12b was also able to exclusively provide 19b. Ketoepoxide 19b is presumably generated from a reaction sequence starting with the epoxidation of 11b to give 17, followed by the Baeyer-Villiger rearrangement and then the decomposition of the hemiketal 18. The structure of 19b was established by nOe experiments showing the relative configuration of the epoxy and keto groups within the bicyclo[2.2.2]octane framework. Therefore, the Baeyer-Villiger reaction was preferable to the possible rearrangement of the α-ketol 17, which would have induced the expansion of the bicyclo[2.2.2]octane to afford the bicyclo[3.2.2]nonane derivative 20.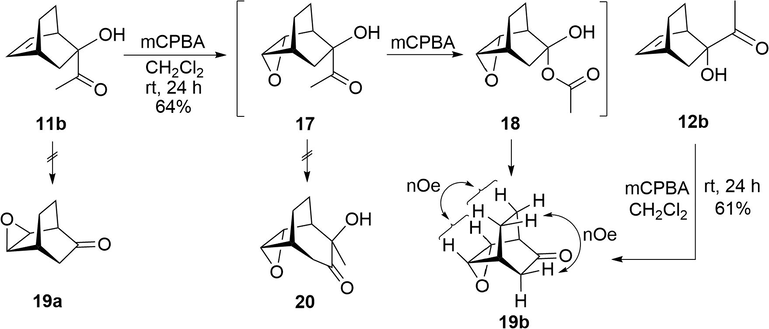
Epoxidation/Baeyer-Villiger cascade reaction of α-ketols 11b and 12b.
Hence, α-ketol 11b behaves mostly as a β,γ-unsaturated carbonyl compound, as we have found for unconjugated cyclohexenones (Ochoa et al., 1999). In the same sense, it is noteworthy that 19b did not undergo a subsequent Baeyer-Villiger reaction, even though there was an excess of the reagent.
Detailed studies by Ohwada and coworkers (Ohwada et al., 1996) on the stereofacial control of the epoxidation of monosubstituted bicyclo[2.2.2]oct-5-ene derivatives showed that epoxidation was favored on the syn face when employing exo electron-withdrawing groups, but that this preference decreased with electron-donating groups. The selectivity appears to be controlled by the perturbation of the σ bicyclo[2.2.2]oct-5-ene skeleton on the π double bond (Ohwada, 1999). In the case of the exo-isomer 12b, consequently, epoxidation seems to be controlled by the electron-demand of the acetyl group. Since using 11b in the reaction also leads to syn-epoxide 19b, it can be understood that the exo hydroxyl group and/or the endo acetyl group are probably acting as dominant σ electron-withdrawing groups, reinforcing the syn selectivity. Anchimeric assistance of the lone pairs of the acetyl or the hydroxyl groups, controlling the endo approach of the peroxyacid reagent, cannot be ruled out as another factor (Chamberlain et al., 1970; Hudlicky, 1990; Houk et al., 1997).
Vogel and coworkers (Fattori et al., 1993) also investigated kinetically controlled facial selectivity in reactions between substituted bicyclo[2.2.2]oct-5-en-2-ones and diverse electrophiles. They reported that the approach of the electrophile takes place with anti preference in relation to the oxo-substituted tether side, a behavior opposite to what was observed with isomers 11b and 12b. Thus epoxidation seems to occur before the Baeyer-Villiger/hydrolysis cascade reaction takes place to furnish the oxo group. Otherwise, the latter would direct the epoxidation towards the formation of the anti epoxide 19a.
These results reveal the synthetic potential of the captodative alkene 1a as an efficient ketene analogue in the formation of the bicyclo[2.2.1]heptan-2-one and bicyclo[2.2.2]octan-2-one frameworks, similar to other alkenes in Diels-Alder cycloadditions (Cantello et al., 1974; Trost and Tamaru, 1977; Viehe et al., 1985; Reymond and Vogel, 1990; Kernen and Vogel, 1995).
Additionally, we investigated the viable conditions that could lead to the acyloin rearrangement of 11b, finding that by maintaining the latter in a chromatography column over silica gel at room temperature for 120 h and eluting it with a mixture of hexane/EtOAc (9:1), the expansion product 22a was isolated as a single isomer (Scheme 5). The structure of the latter was determined by nOe experiments of the 4-nitrobenzoyl derivative 23, in which the signal size of the methylene protons H-8′ and H-9′ exhibited enhancements by irradiation of the aromatic H-2 protons.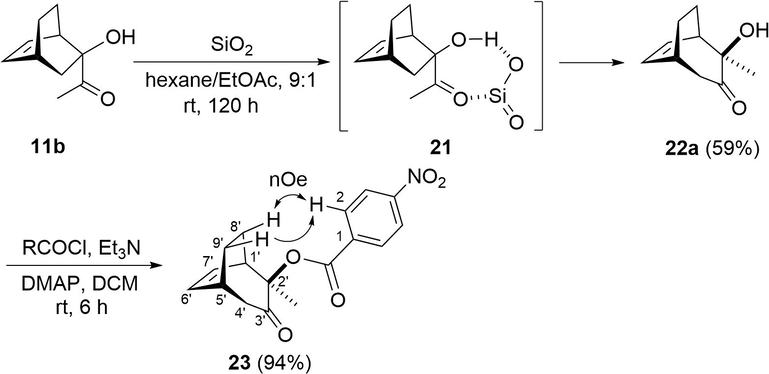
Possible stereospecific α-ketol rearrangement of 11b.
Notably, the stereoselectivity of this rearrangement was opposite to that found in the bicyclo[2.2.1]hept-5-ene of α-ketols 8b and 9b. In the case of the endo α-ketol 8b (with the acetyl group on the endo side) the expansion leads to endo-hydroxy-α-ketol product 15 (with the hydroxyl group on the endo side), whereas exo 9b rearranges to the exo-hydroxy-α-ketol 16. Contrarily, the endo α-ketol 11b provides the exo expansion product 22a. A coordination chelate like 21 could be involved in promoting such inverse stereoselectivity (Creary et al., 1985).
Unexpectedly, unchanged starting material was recovered when α-ketol 12b was treated under these conditions. By employing a more severe reaction temperature and longer reaction time, a mixture of starting material and several byproducts was isolated.
2.4 Cascade conversion of α-ketols 8b and 9b into the bicyclic γ-lactone 26 and a synthetic approach to racemic β-carbaxylose
The substituted bicyclo[2.2.1]-5-hepten-2-ones and bicyclo[2.2.1]-5-heptenes have proven to be versatile scaffolds in the synthesis of cyclic and acyclic natural and unnatural products (Corey et al., 1970; Greene et al., 1980; Grieco et al., 1982; Carreño, 1995; Mehta and Islam, 2002; Araki et al., 2003; Vincent et al., 2013). These bicyclic precursors have been obtained from stereoselective and/or regioselective Diels-Alder cycloadditions with substituted cyclopentadienes (Carruthers, 1990; Fringuelli and Taticchi, 2002), including the asymmetric version (Hudon et al., 2008; Reymond and Cossy, 2008; Mukherjee and Corey, 2010; Mehta and Shinde, 2012). Being inspired by these antecedents, we investigated the synthetic potential of α-ketols 8b and 9b and their stereospecific rearrangement in the presence of mCPBA.
As shown, the treatment of α-ketols 8b and 9b with 1.0 mol equiv of mCPBA in CH2Cl2 at room temperature resulted in the tricyclo[3.3.1.02,4] derivatives 15 and 16, respectively (Scheme 3). Furthermore, the reaction of 9b with 2.0 mol equiv of mCPBA generated the bicyclic γ-lactone 26 as a single diastereoisomeric product in 45% yield (Scheme 6). Similarly, when the reaction was carried out with a mixture of 8b/9b, compound 26 was isolated as the only product in 70% yield. This transformation likely takes place through a cascade process involving the isolated expanded tricyclo[3.3.1.02,4] epoxy intermediate 16, which undergoes a Baeyer-Villiger reaction to afford lactone 24. The regioselectivity of the latter rearrangement may be due to the higher migratory aptitude of the carbinol tertiary center in relation to the methylene group (Krow, 1993; Renz and Meunier, 1999). Lactone 24 is hydrolyzed within the acidic reaction medium, followed by the intramolecular proton-assisted opening of the epoxy group of 25, promoting stereoselective lactonization to yield the five-membered lactone 26.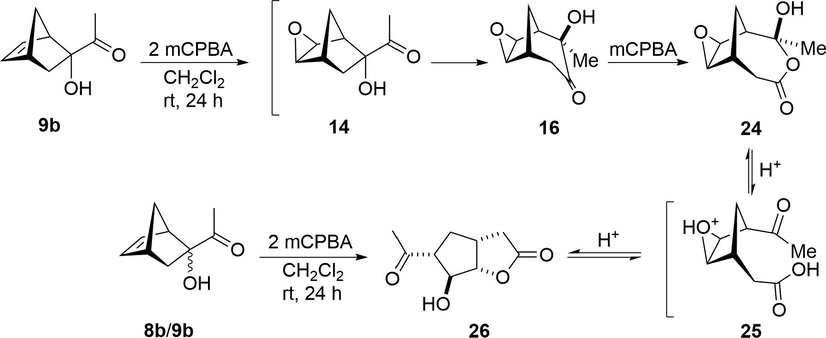
Conversion of α-ketols 8b and 9b into the bicyclic γ-lactone 26.
Carbasugars are hydrolysis-resistant analogues to carbohydrates with significant pharmacological activity (Arjona et al., 2007; Kurteva and Afonso, 2009). In particular, lactone 26 may be a potential precursor of racemic β-carbaxylose (Marschner et al., 1995). In order to propose a synthetic approach to the latter compound, we investigated the reactivity of 26 under diverse reaction conditions. Thus, the conversion of the acetyl group into the acetoxy group was carried out through a further Baeyer-Villiger reaction to give γ-lactone 27. For this purpose, attempts were made using various solvents (CH2Cl2, CHCl2CHCl2, MeOH, tert-BuOH, THF and C6H6), catalysts (TFA, TFA-H2O2 and p-TsOH), temperatures (25–70 °C), and other peracids (mCPBA, mono-o-perftalic acid and pertrifluroacetic acid) (Krow, 1993; Renz and Meunier, 1999). In each case, however, low yields of 27 were obtained. Unexpectedly, a better conversion occurred when the reaction was induced with mCPBA in a mixture of hexane/CH2Cl2 (8:2) employing a desiccant additive (Na2SO4) at room temperature (Scheme 7). The reduced reactivity of 26 may be due to the steric hindrance found at the acetyl group, which is located at the crowded endo face of the bicyclo[3.3.0]oxaoctane. Since the concerted Baeyer-Villiger rearrangement takes place through a bulky transition state (Mihovilovic et al., 2004), the abated reactivity of 26 is understandable.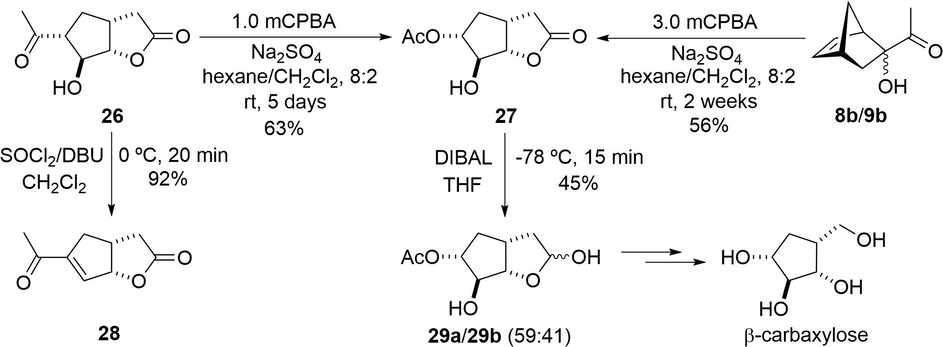
Conversion of α-ketols 8b/9b and bicyclic γ-lactone 26 into bicyclic γ-lactone 27, as a synthetic approach to racemic β-carbaxylose, and into bicycle 28.
These reaction conditions also efficiently furnished 27 from a mixture of α-ketols 8b/9b, although a long reaction time (2 weeks) was required. Despite the modest yield of γ-lactone 27, it is noteworthy that this conversion involved a least five-steps in a cascade process.
Further reactions carried out on γ-lactone 26 are summarized in Scheme 7. When 26 was reacted with thionyl chloride, enone 28 was obtained in high yield. Because lactone 27 is a potential precursor of β-carbaxylose (Marschner et al., 1995), the lactone group was reduced with DIBAL, generating a mixture of ketals 29a/29b (59:41).
2.5 Mechanism of the stereospecific acyloin rearrangement of α-Ketols 8b and 9b
Since we have established that the acyloin rearrangement of α-ketol 8b stereospecifically produces α-ketol 15 (Scheme 3), a plausible mechanism can be postulated for the C-1 migration pathway of the two possible acetyl group conformations, 8b-endo-in and 8b-endo-out (Scheme 8(a)). The NMR and X-ray analyses provided evidence of lower stability for the latter versus former conformation (García de Alba et al., 1996). From this ground state conformational bias, similar conformations are possibly maintained at the transition state with the most stable TS corresponding to 8b-endo-in, which results in the observed α-ketol 15.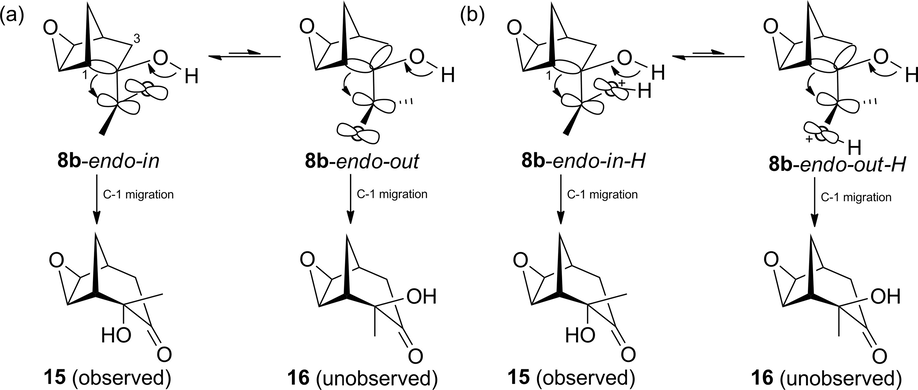
(a) Conformational equilibrium in the neutral species of conformers 8b-endo-in and 8b-endo-out, and possible C-1 migration products 15 and 16. (b) Conformational equilibrium in the protonated species of conformers 8b-endo-in-H and 8b-endo-out-H, and possible C-1 migration products 15 and 16.
Considering that the process is carried out under acidic conditions, a Brønsted acid-catalyzed acyloin rearrangement may also take place (Paquette and Hofferberth, 2003). In such case, both 8b-endo-in and 8b-endo-out conformations would be protonated at the carbonyl group (Scheme 8(b)), leading to the corresponding TSs with a relative stability similar to that of the neutral species. Therefore, the greater stability of the 8b-endo-in-H TS would afford α-ketol 15.
Regarding exo α-ketol 9b, the NMR analysis (García de Alba et al., 1996) also suggests that the ground state conformer 9b-exo-in would be more stable than 9b-exo-out. Presumably, the protonated species 9b-exo-in-H and 9b-exo-out-H and their corresponding transition states would display parallel stability (Scheme 9). The TS would be more stable for the former species than latter species, promoting the formation of the observed α-ketol 16. Some reports claim that the controlling factor for the TS is the proton transfer from the hydroxyl group to the oxygen carbonyl group via an intramolecular hydrogen bond between the former moiety and the oxygen of the latter, as in the A TS (Paquette and Hofferberth, 2003). Because the geometry of the 9b-exo-out-H conformer allows for the formation of such a hydrogen bond, the ring expansion step would be directed towards the unobserved α-ketol 15. The A TS is likely destabilized by the electron-deficiency of the protonated oxygen of the carbonyl group.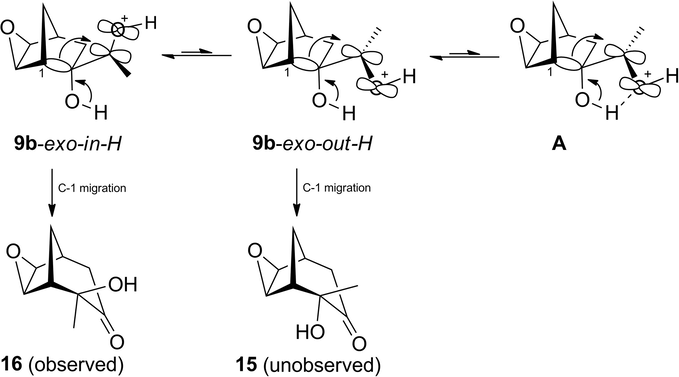
Conformational equilibrium in the protonated species of conformers 9b-exo-in-H and 9b-exo-out-H, and possible C-1 migration products 15 and 16.
3 Conclusions
The present results indicate that captodative alkene 1a undergoes highly stereoselective thermal and Lewis acid-catalyzed Diels-Alder reactions with cyclohexadiene (10) to furnish the corresponding endo adduct 11a as a single isomer in high yields. The reactivity of α-ketol bicyclo[2.2.1]heptanes 8b and 9b, the hydrolyzed products generated from the cyclopentadiene (7) adducts endo 8a and exo 9a, respectively, was evaluated under mCPBA-promoted acyloin rearrangement conditions. Such rearrangement was found to be efficient and stereospecific, rendering the ring expansion products endo- and exo-hydroxy-exo-epoxy tricyclo[3.3.1.02,4]nonan-7-ones 15 and 16, respectively. In the case of α-ketol bicyclo[2.2.2]octanes 11b and 12b, the epoxidation/Baeyer-Villiger cascade process was preferred, leading to the syn ketoepoxide 19b from each of the isomers. Only α-ketol bicyclo[2.2.2]octane 11b was able to undergo acyloin rearrangement by silica gel treatment to stereospecifically produce α-ketol bicyclo[3.2.2]nonane 22a. The structure of the latter reveals that the rearrangement took place through a pathway opposite to the one followed by α-ketol bicyclo[2.2.1]heptanes 8b and 9b. The rearrangement of these substrates occurred in a five-step cascade transformation to compound 27, a potential precursor in the synthesis of racemic β-carbaxylose. Quantum calculations may provide further insight into the controlling effects involved in the endo and exo transition states for the thermal and Lewis acid-catalyzed Diels-Alder reaction between 1a and cyclohexadiene (10), as well as into the effects promoting the stereospecific acyloin rearrangements of the bicyclic α-ketols 8b, 9b, 11b and 12b. Such calculations are in progress and will be reported in due course.
4 Experimental section
4.1 General methods
Melting points were determined with an Electrothermal capillary melting point apparatus. IR spectra were recorded on a Perkin-Elmer 2000 spectrophotometer. 1H (300 or 500 MHz) and 13C (75.4 or 125 MHz) NMR spectra were recorded on Varian Mercury-300 or Varian VNMR System instruments, with TMS as internal standard. Mass spectra (MS) and high resolution mass spectrometry were conducted in electron impact (70 eV) and fast atom bombardment (FAB) modes, on Hewlett-Packard 5971A and Thermo-Finnigan Polaris Q, and Jeol JSM-GCMat-eII and JMS-SX 102 spectrometers. X-Ray crystallographic measurements were collected on a Siemens P4 diffractometer with Mo Kα radiation (graphite crystal monochromator, λ = 0.7107 Å). Microanalyses were performed by a CE-440 Exeter elemental analyzer instrument. Analytical thin-layer chromatography was carried out with E. Merck silica gel 60 F254-coated 0.25 plates, visualized by a long- and short-wavelength UV lamp. For all air moisture sensitive reactions, a nitrogen atmosphere and oven-dried glassware were employed. THF was freshly distilled from sodium, and methylene chloride from calcium hydride prior to use. Triethylamine was freshly distilled from NaOH. All other reagents were used without further purification. Compounds 1a, 8a-b and 9a-b were prepared as previously described (Tamariz and Vogel, 1981; García de Alba et al., 1996).
4.2 (1R∗, 2S∗, 4R∗)-2-Acetylbicyclo[2.2.2]oct-5-en-2-yl 4-nitrobenzoate (11a)
4.2.1 (1R∗, 2R∗, 4R∗)-2-Acetylbicyclo[2.2.2]oct-5-en-2-yl 4-nitrobenzoate (12a)
Method A. Under N2 atmosphere and vigorous magnetic stirring, a mixture of 1a (0.050 g, 0.21 mmol), 10 (0.042 g, 0.53 mmol), and hydroquinone (3 mg) in dry C6H6 (3.0 mL) was placed at room temperature in a threaded ACE glass pressure tube with a sealed Teflon screw cap and kept in the dark. The mixture was heated to 130 °C for 168 h, then diluted with EtOAc (60 mL), and washed with H2O (3 × 15 mL). The organic layer was dried (Na2SO4) and the solvent was removed under vacuum. The residue was purified by column chromatography over silica gel (3 g, hexane/EtOAc, 9:1) to give 11a (0.047 g, 70%) as a pale yellow solid.
Method B. Under N2 atmosphere and vigorous magnetic stirring, to a solution of 1a (0.050 g, 0.21 mmol) in anhydrous CH2Cl2 (5.0 mL), BF3·Et2O (0.567 g, 3.99 mmol) and 10 (0.042 g, 0.53 mmol) were added dropwise at −50 °C. The mixture was stirred at the same temperature for 10 h, then diluted with EtOAc (60 mL) and successively washed with H2O (2 × 10 mL), a cold aqueous saturated solution of NaHCO3 (3 × 15 mL), and a cold saturated solution of NaCl until neutral. The organic layer was dried (Na2SO4) and the solvent was removed under vacuum. The residue was purified by column chromatography over silica gel (3 g, hexane/EtOAc, 9:1) to give a mixture of 11a/12a (67:33) (0.057 g, 85%) as a pale yellow solid.
Method C. Following Method B, with TiCl4 (0.345 g, 1.80 mmol) instead BF3·Et2O, and stirring the mixture for 3 h, a mixture of 11a/12a (44:56) (0.061 g, 92%) was provided as a pale yellow solid.
Method D. Following Method B, with AlCl3 (0.066 g, 0.50 mmol) instead BF3·Et2O, and stirring the mixture at 0 °C for 1 h, 11a (0.063 g, 95%) was furnished as a pale yellow solid.
Method E. Following Method B, with ZnI2 (1.595 g, 5.00 mmol) instead BF3·Et2O, and stirring the mixture at 25 °C for 120 h, 11a (0.056 g, 84%) was produced as a pale yellow solid.
Data of 11a. Rf 0.43 (hexane/EtOAc, 8:2); mp 149–150 °C. IR (film) 1722, 1522, 1348, 1279, 1105 cm−1. 1H NMR (500 MHz, CDCl3) δ 1.30 (dddd, J = 12.6, 12.2, 4.9, 2.8 Hz, 1H, H-7s′), 1.39 (tdd, J = 12.2, 6.3, 3.2 Hz, 1H, H-8s′), 1.59 (dd, J = 14.3, 2.4 Hz, 1H, H-3x′), 1.62–1.68 (m, 1H, H-8a′), 2.12–2.19 (m, 1H, H-7a′), 2.14 (s, 3H, CH3CO), 2.48 (dt, J = 14.3, 3.0 Hz, 1H, H-3n′), 2.77–2.80 (m, 1H, H-4′), 3.10–3.12 (m, 1H, H-1′), 6.14 (td, J = 6.9, 1.2 Hz, 1H, H-6′), 6.39 (d, J = 7.7, 6.9 Hz, 1H, H-5′), 8.22–8.25 (m, 2H, ArH-2), 8.32–8.34 (m, 2H, ArH-3). 13C NMR (125 MHz, CDCl3) δ 20.1 (C-7′), 23.7 (C-8′), 24.7 (CH3CO), 30.0 (C-4′), 36.2 (C-1′), 36.3 (C-3′), 89.2 (C-2′), 123.7 (Ar-3), 129.5 (C-6′), 130.9 (Ar-2), 135.0 (Ar-1), 135.9 (C-5′), 150.8 (ArC-4), 164.2 (ArCO2), 203.7 (CH3CO). MS (70 eV) m/z (%) 314 (M+−1, 2), 272 (36), 150 (100), 120 (4), 104 (12), 76 (5). Anal. Calcd for C17H17NO5: C, 64.75; H, 5.43; N, 4.44. Found: C, 64.96; H, 5.55; N, 4.31.
Data of 12a. Rf 0.64 (hexane/EtOAc, 8:2); mp 137–138 °C. IR (KBr) 1716, 1524, 1353, 1299, 1107, 722 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.17–1.28 (m, 2H, H-7s′, H-8′), 1.33 (dt, J = 14.1, 2.4 Hz, 1H, H-3n′), 1.38–1.47 (m, 1H, H-7a′), 1.55–1.62 (m, 1H, H-8a′), 2.16 (s, 3H, CH3CO), 2.73 (br s, 1H, H-4′), 2.90 (dd, J = 14.1, 2.4 Hz, 1H, H-3x′), 2.94 (br s, 1H, H-1′), 6.37–6.50 (m, 2H, H-5′, H-6′), 8.14–8.20 (m, 2H, ArH-2), 8.27–8.33 (m, 2H, ArH-3). 13C NMR (75.4 MHz, CDCl3) δ 19.8 (C-7′), 21.9 (C-8′), 23.9 (CH3CO), 30.1 (C-4′), 36.0 (C-1′), 37.2 (C-3′), 91.4 (C-2′), 123.6 (Ar-3), 130.0 (C-5′), 130.9 (Ar-2), 135.0 (Ar-1), 136.1 (C-6′), 150.7 (Ar-4), 163.9 (ArCO2), 204.3 (CH3CO). MS (70 eV) m/z (%) 150 (M+−164, 100), 134 (3), 120 (6), 104 (28), 92 (14), 76 (21). HRMS (EI) calcd for [M+1]+ C17H18NO5: 316.1185. Found: 316.1179.
4.3 1-((1R∗, 2S∗, 4R∗)-2-Hydroxybicyclo[2.2.2]oct-5-en-2-yl)ethanone (11b)
Under N2 atmosphere and vigorous magnetic stirring, to a solution of 11a (0.400 g, 1.27 mmol) in anhydrous THF (7.0 mL), K2CO3 (0.910 g, 6.60 mmol) in anhydrous MeOH (2.0 mL) was added dropwise at 25 °C. The mixture was stirred at the same temperature for 4 h, then diluted with EtOAc (60 mL) and successively washed with H2O until neutral. The organic layer was dried (Na2SO4), and the solvent was removed under vacuum. The residue was purified by column chromatography over silica gel (15 g, hexane/EtOAc, 8:2) to obtain 11b (0.198 g, 94%) as a pale yellow oil. Rf 0.35 (hexane/EtOAc, 8:2). IR (film) 3455, 2942, 1703, 1354, 1101 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.09 (tdd, J = 12.1, 5.4, 2.6 Hz, 1H, H-7s), 1.31 (tdd, J = 12.1, 6.5, 3.2 Hz, 1H, H-8s), 1.47 (dd, J = 13.6, 2.6 Hz, 1H, H-3x), 1.67–1.78 (m, 1H, H-8a), 1.92 (dt, J = 13.6, 2.9 Hz, 1H, H-3n), 2.19 (s, 3H, CH3CO), 2.19–2.30 (m, 1H, H-7a), 2.43–2.47 (m, 1H, H-1), 2.73–2.77 (m, 1H, H-4), 3.81 (br s, 1H, OH), 6.23–6.40 (m, 2H, H-5, H-6). 13C NMR (75.4 MHz, CDCl3) δ 20.6 (C-7), 23.4 (C-8), 25.6 (CH3CO), 30.4 (C-4), 38.9 (C-1), 39.0 (C-3), 80.5 (C-2), 132.7 (C-6), 134.6 (C-5), 211.3 (CH3CO). MS (70 eV) m/z (%) 166 (M+, 1), 123 (10), 105 (2), 95 (100), 94 (13), 80 (54), 79 (50), 77 (52), 67 (11). Anal. Calcd for C10H14O2: C, 72.26; H, 8.49. Found: C, 72.40; H, 8.47.
4.4 1-((1R∗, 2R∗, 4R∗)-2-Hydroxybicyclo[2.2.2]oct-5-en-2-yl)ethanone (12b)
According to the procedure used for the preparation of 11b, with 12a (0.134 g, 0.426 mmol) and K2CO3 (0.292 g, 2.12 mmol) in anhydrous MeOH (2.0 mL) and stirring for 4 h, 12b (0.057 g, 81%) was delivered as a pale yellow oil. Rf 0.34 (hexane/EtOAc, 8:2). IR (film) 3460, 2944, 1711, 1351, 1110, 722 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.04–1.25 (m, 3H, H-3n, H-7, H-8), 1.36–1.54 (m, 2H, H-7, H-8), 2.11 (br s, 1H, OH), 2.30 (s, 3H, CH3CO), 2.47 (dd, J = 13.8, 2.1 Hz, 1H, H-3x), 2.68–2.78 (m, 2H, H-1, H-4), 6.24 (ddd, J = 8.1, 6.6, 1.5 Hz, 1H, H-6), 6.54 (ddd, J = 7.8, 6.6, 1.2 Hz, 1H, H-5). 13C NMR (75.4 MHz, CDCl3) δ 19.9 (C-7), 23.0 (C-8), 25.0 (CH3CO), 30.4 (C-4), 39.2 (C-1), 39.7 (C-3), 82.9 (C-2), 129.8 (C-6), 138.1 (C-5), 210.3 (CH3CO). MS (70 eV) m/z (%) 167 (M++1, 100), 149 (54), 145 (34), 133 (40), 123 (50), 111 (46), 97 (56), 95 (82), 81 (54), 79 (38), 67 (31). Anal. Calcd for C10H14O2: C, 72.26; H, 8.49. Found: C, 72.24; H, 8.51.
4.5 (1R∗, 2R∗, 4S∗, 5R∗, 6S∗)-6-Hydroxy-6-methyl-3-oxatricyclo[3.3.1.02,4]nonan-7-one (15)
Under N2 atmosphere and vigorous magnetic stirring, to a solution of 8b (0.100 g, 0.66 mmol) in dry CH2Cl2 (3 mL), mCPBA (0.112 g, 0.65 mmol) and Na2SO4 (0.5 g) were added at room temperature. The mixture was stirred at the same temperature for 24 h and then diluted with CH2Cl2 (50 mL). The organic solution was successively washed with H2O (2 × 10 mL), an aqueous saturated solution of NaHCO3 (3 × 10 mL) and brine until neutral. The organic layer was dried (Na2SO4) and the solvent evaporated under vacuum. The residue was purified by column chromatography over silica gel (10 g, hexane/EtOAc, 7:3) to give 15 (0.075 g, 68%) as a white solid. Rf 0.54 (hexane/EtOAc, 7:3); mp 142–143 °C. IR (CH2Cl2) 3468, 2930, 1708, 1452, 1368, 1142, 1084, 1010, 984, 838 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.35 (s, 3H, CH3), 1.59 (br d, J = 12.9 Hz, 1H, H-9a), 1.67–1.77 (m, 1H, H-9s), 2.49 (ddd, J = 17.3, 3.3, 3.0 Hz, 1H, H-8 α), 2.59–2.67 (m, 3H, H-1, H-5, H-8β), 3.27 (br d, J = 3.0 Hz, 1H, H-2), 3.56 (s, 1H, HO), 3.61 (br d, J = 3.0 Hz, 1H, H-4). 13C NMR (75.4 MHz, CDCl3) δ 25.4 (C-9), 26.0 (CH3), 34.8 (C-1), 41.4 (C-8), 44.9 (C-5), 52.9 (C-4), 54.0 (C-2), 78.8 (C-6), 213.5 (CO); MS (70 eV) m/z (%) 168 (M+, 9), 140 (5), 111 (55), 97 (51), 82 (57), 69 (27), 55 (25), 43 (100). Anal. Calcd for C9H12O3: C, 64.27; H, 7.19. Found: C, 64.45; H, 7.30.
4.6 (1R∗, 2R∗, 4S∗, 5R∗, 6R∗)-6-Hydroxy-6-methyl-3-oxatricyclo[3.3.1.02,4]nonan-7-one (16)
According to the procedure used for the preparation of 15, with 9b (0.300 g, 1.97 mmol) and mCPBA (0.339 g, 1.97 mmol) in dry CH2Cl2 (7 mL) and Na2SO4 (0.5 g) 16 (0.215, 65%) was afforded as a white solid. Rf 0.56 (hexane/EtOAc, 7:3); mp 139–140 °C. IR (film) 3564, 2972, 1708, 1278, 1256 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.36 (s, 3H, CH3), 1.46–1.56 (m, 1H, H-9s), 1.88–1.96 (m, 1H, H-9a), 2.32 (dm, J = 18.0 Hz, 1H, H-8α), 2.53–2.60 (m, 2H, H-1, H-5), 2.75 (dm, J = 18.0 Hz, 1H, H-8β), 3.28 (dm, J = 3.9 Hz, 1H, H-4), 3.35 (dm, J = 3.9 Hz, 1H, H-2). 13C NMR (75.4 MHz, CDCl3) δ 22.3 (CH3), 22.8 (C-9), 33.5 (C-1), 40.9 (C-8), 44.9 (C-5), 51.9 (C-4), 55.1 (C-2), 75.8 (C-6), 209.9 (CO). MS (70 eV) m/z (%)168 (M+, 16), 140 (6), 111 (52), 97 (53), 82 (51), 81 (52), 69 (27), 43 (100). Anal. Calcd for C9H12O3: C, 64.27; H, 7.19. Found: C, 64.39; H, 6.95.
4.7 (1R∗, 2R∗, 4S∗, 5R∗)-3-Oxatricyclo[3.2.2.02,4]nonan-6-one (19b)
Method A: Under N2 atmosphere and vigorous magnetic stirring, a mixture of 11b (0.090 g, 0.54 mmol) and mCPBA (0.465 g, 2.70 mmol) in dry CH2Cl2 (5.0 mL) was stirred at 20 °C for 24 h. The mixture was diluted with CH2Cl2 (60 mL) and washed with H2O (2 × 10 mL), an aqueous saturated solution of NaHCO3 (3 × 10 mL) and with brine until neutral. The organic layer was dried (Na2SO4) and the solvent evaporated under vacuum. The residue was purified by column chromatography over silica gel (15 g, hexane/EtOAc, 7:3) to afford 19b (0.047 g, 64%) as a white solid.
Method B: According to Method A, with 12b (0.050 g, 0.30 mmol) and mCPBA (0.258 g, 1.50 mmol) in dry CH2Cl2 (3 mL), 19b (0.025 g, 61%) was furnished as a white solid. Rf 0.25 (hexane/EtOAc, 7:3); mp 147–148 °C. IR (film) 2927, 1727, 1467, 1377, 1275, 1126, 1073 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.62–1.82 (m, 3H, H-8, H-9), 1.84–1.92 (m, 1H, H-8), 1.96 (dd, J = 18.7, 2.7 Hz, 1H, H-7α), 2.29 (dm, J = 18.7 Hz, 1H, H-7β), 2.58–2.68 (m, 1H, H-1), 2.83–2.90 (m, 1H, H-5), 3.44–3.54 (m, 2H, H-2, H-4). 13C NMR (75.4 MHz, CDCl3) δ 19.6 (C-9), 21.4 (C-8), 28.3 (C-1), 40.5 (C-7), 44.8 (C-5), 49.2 (C-4), 51.9 (C-2), 209.7 (CO). Anal. Calcd for C8H10O2: C, 69.54; H, 7.30. Found: C, 69.53; H, 7.32.
4.8 (1R∗, 2R∗, 5R∗)-2-Hydroxy-2-methylbicyclo[3.2.2]non-6-en-3-one (22a)
A solution of 11b (0.400 g, 1.27 mmol) in a mixture of dry hexane/EtOAc (7:3) (2 mL) was poured into a chromatography column over silica gel (5 g, hexane/EtOAc, 7:3) and maintained at room temperature for 120 h. The column was eluted (hexane/EtOAc, 7:3), resulting in 22a (0.125 g, 59%) as a pale yellow oil. Rf 0.85 (hexane/EtOAc, 7:3). IR (film) 3431, 2950, 1639 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.26 (s, 3H, CH3), 1.43–1.73 (m, 3H, H-8, 2H-9), 1.80–1.95 (m, 1H, H-8), 2.50–2.74 (m, 4H, H-1, H-4, H-5), 4.32 (s, 1H, HO), 6.33 (dd, J = 9.1, 7.9 Hz, 1H, H-7), 6.42 (dd, J = 9.1, 6.3 Hz, 1H, H-6). 13C NMR (75.4 MHz, CDCl3) δ 20.2 (C-8), 23.6 (C-9), 26.3 (CH3), 29.3 (C-5), 41.2 (C-1), 45.6 (C-4), 81.4 (C-2), 132.7 (C-7), 135.6 (C-6), 215.1 (CO). MS (70 eV) m/z (%) 166 (M+, 2), 123 (21), 105 (5), 95 (100), 80 (90), 79 (60), 77 (41), 67 (13), 65 (11). HRMS (EI) m/z [M+] calcd for C10H4O2: 166.0994. Found: 166.0991.
4.9 (1R∗, 2R∗, 5R∗)-2-Methyl-3-oxobicyclo[3.2.2]non-6-en-2-yl 4-nitrobenzoate (23)
Under N2 atmosphere and vigorous magnetic stirring, to a solution of 22a (0.022 g, 0.13 mmol) in anhydrous CH2Cl2 (8 mL), triethylamine (0.013 g, 0.13 mmol), DMAP (0.016 g, 0.13 mmol) and p-nitrobenzoyl chloride (0.024 g, 0.13 mmol) were added dropwise at room temperature. The mixture was stirred at the same temperature for 6 h, then diluted with CH2Cl2 (50 mL) and washed with a cold 5% aqueous solution of HCl (2 × 20 mL) and brine until neutral. The organic layer was dried (Na2SO4) and the solvent was removed under vacuum. The residue was purified by column chromatography over silica gel (30 g, hexane/EtOAc, 95:15) to produce 23 (0.039 g, 94%) as a pale yellow solid. Rf 0.49 (hexane/EtOAc, 8:2); mp 99–101 °C. IR (KBr) 1719, 1524, 1350, 1289, 1103, 716 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.54–1.72 (m, 2H, H-8s′, H-9s′), 1.76 (s, 3H, CH3), 1.78–1.92 (m, 1H, H-9a′), 1.98–2.09 (m, 1H, H-8a′), 2.60–2.84 (m, 3H, H-4′, H-5′), 3.08–3.16 (m, 1H, H-1′), 6.30 (t, J = 8.4 Hz, 1H, H-7′), 6.50 (dd, J = 8.4, 6.3 Hz, 1H, H-6′), 8.17–8.26 (m, 2H, H-2), 8.26–8.35 (m, 2H, H-3). 13C NMR (75.4 MHz, CDCl3) δ 21.2 (C-8′), 22.8 (CH3), 24.3 (C-9′), 29.3 (C-5′), 40.2 (C-1′), 47.4 (C-4′), 90.5 (C-2′), 123.5 (C-3), 130.7 (C-7′), 130.8 (C-2), 136.3 (C-1), 137.1 (C-6′), 150.4 (C-4), 163.4 (ArCO2), 206.0 (CO). HRMS (EI) m/z [M+] calcd for C17H17NO5: 315.1107. Found: 315.1119.
4.10 (3aR∗, 5R∗, 6S∗, 6aS∗)-5-Acetyl-6-hydroxyhexahydro-2H-cyclopenta[b]furan-2-one (26)
Following the method for the preparation of 15, 9b (0.20 g, 1.3 mmol), mCPBA (0.448 g, 2.60 mmol) and Na2SO4 (0.5 g) were used in dry CH2Cl2 (3 mL). The crude of the reaction was purified by column chromatography over silica gel (6.0 g, hexane/EtOAc, 6:4), obtaining 26 (0.109 g, 45%) as a pale yellow oil. Following this method, with a mixture of 8a/9b (0.20 g, 1.3 mmol), mCPBA (0.448 g, 2.60 mmol) and Na2SO4 (0.5 g) in dry CH2Cl2 (3 mL), 26 (0.17 g, 70%) was delivered as a pale yellow oil. Rf 0.18 (hexane/EtOAc, 7:3). IR (CH2Cl2) 3417, 1771, 1705, 1362, 1170, 1034 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.68–1.80 (m, 1H, H-4β), 2.25 (s, 3H, CH3CO), 2.27–2.38 (m, 1H, H-4α), 2.39 (dd, J = 18.4, 2.9 Hz, 1H, H-3β), 2.77 (dd, J = 18.4, 9.9 Hz, 1H, H-3α), 2.95–3.11 (m, 2H, H-3a, H-5), 3.58 (br s, 1H, OH), 4.43 (dd, J = 8.0, 3.3 Hz, 1H, H-6), 4.76 (dd, J = 8.0, 3.3 Hz, 1H, H-6a). 13C NMR (75.4 MHz, CDCl3) δ 29.7 (CH3CO), 32.4 (C-4), 35.1 (C-3), 35.5 (C-3a), 58.2 (C-5), 79.0 (C-6), 80.3 (C-6a), 176.8 (C-2), 207.9 (CH3CO). MS (70 eV) m/z (%)166 (M+−18, 4), 151 (7), 123 (9), 121 (15), 107 (61), 79 (1 0 0), 77 (66). HRMS (EI) m/z [M+] calcd for C9H12O4: 184.0736. Found: 184.0742.
4.11 (3aR∗, 5R∗, 6S∗, 6aS∗)-6-Hydroxy-2-oxohexahydro-2H-cyclopenta[b]furan-5-yl acetate (27)
A mixture of 8b/9b (30:70) (0.40 g, 2.63 mmol), mCPBA (1.360 g, 7.88 mmol) and Na2SO4 (1.0 g) in a dry mixture of heptane/CH2Cl2 (8:2, 50 mL) was placed in a threaded ACE glass pressure tube with a sealed Teflon screw cap and kept in the dark. The mixture was stirred at room temperature for 2 weeks and the solvent was removed under vacuum. The crude of the reaction was diluted with CH2Cl2 (30 mL) and filtered, followed by removal of the solvent under vacuum. The crude was purified by column chromatography over silica gel (90 g, hexane/EtOAc, 7:3), leading to 27 (0.294 g, 56%) as a white solid. Rf 0.45 (EtOAc); mp 99–101 °C. IR (CH2Cl2) 3433, 1771, 1735, 1373, 1239, 1170, 1051 cm−1. 1H NMR (300 MHz, CDCl3) δ 1.81 (ddd, J = 14.2, 3.5, 3.3 Hz, 1H, H-4α), 2.05 (s, 3H, CH3CO2), 2.43 (dd, J = 18.3, 3.0 Hz, 1H, H-3α), 2.51 (ddd, J = 14.4, 8.7, 5.1 Hz, 1H, H-4β), 2.88 (dd, J = 18.3, 11.0 Hz, 1H, H-3β), 3.13–3.25 (m, 1H, H-3a), 3.40–3.72 (br, 1H, OH), 4.31 (br s, 1H, H-6), 4.82 (d, J = 7.5 Hz, 1H, H-6a), 4.95–5.02 (m, 1H, H-5). 13C NMR (75.4 MHz, CDCl3) δ 20.9 (CH3CO2), 35.1 (C-3a), 35.9 (C-3), 36.4 (C-4), 78.4 (C-6), 80.1 (C-5), 88.7 (C-6a), 171.1 (CH3CO2), 177.2 (C-2). MS (70 eV) m/z (%) 201 (M++1, 1), 182 (2), 140 (65), 112 (22), 97 (31), 84 (36), 43 (100). Anal. Calcd for C9H12O5: C, 53.99; H, 6.04. Found: C, 53.83; H, 5.87.
4.12 (3aR∗, 6aS∗)-5-Acetyl-3,3a,4,6a-tetrahydro-2H-cyclopenta[b]furan-2-one (28)
Under N2 atmosphere and vigorous magnetic stirring, to a solution of 26 (0.150 g, 0.85 mmol) in dry CH2Cl2 (1 mL), DBU (0.487 g, 3.20 mmol) and SOCl2 (0.19 g, 1.6 mmol) were added dropwise at 0 °C. At the same temperature, the mixture was stirred for 20 min and the solvent was removed under vacuum. The residue was purified by column chromatography over silica gel (10 g, hexane/EtOAc, 9:1), rendering 28 (0.103 g, 76%) as a pale yellow oil. Rf 0.85 (hexane/EtOAc, 7:3). IR (CH2Cl2) 1768, 1673, 1372, 1337, 1288, 1250, 1203, 1163, 1054, 1011, 904 cm−1. 1H NMR (300 MHz, CDCl3) δ 2.34 (dd, J = 18.5, 6.2 Hz, 1H, H-3), 2.40 (s, 3H, CH3CO), 2.54 (ddd, J = 17.5, 4.4, 2.3 Hz, 1H, H-4), 2.88 (dd, J = 18.5, 10.7 Hz, 1H, H-3), 2.86–2.97 (m, 1H, H-4), 3.21–3.33 (m, 1H, H-3a), 5.67 (dm, J = 7.7 Hz, 1H, H-6a), 6.60–6.64 (m, 1H, H-6). 13C NMR (75.4 MHz, CDCl3) δ 26.9 (CH3CO), 35.0 (C-3a), 35.4 (C-3 or C-4), 37.2 (C-4 or C-3), 88.7 (C-6a), 136.8 (C-6), 148.0 (C-5), 176.4 (C-2), 196.6 (CH3CO). MS (70 eV) m/z (%) 166 (M+, 37), 151 (31), 123 (18), 121 (34), 107 (91), 95 (30), 79 (88), 77 (65), 67 (31), 43 (100). HRMS (FAB) (mNBA) (MH)+ m/z calculated for C9H11O3: 167.0708; found: 167.0710.
4.13 (2R∗, 3aR∗, 5R∗, 6S∗, 6aS∗)-2,6-Dihydroxyhexahydro-2H-cyclopenta[b]furan-5-yl acetate (29a). (2S∗, 3aR∗, 5R∗, 6S∗, 6aS∗)-2,6-Dihydroxyhexahydro-2H-cyclopenta[b]furan-5-yl acetate (29b)
To a solution of 27 (0.050 g, 0.25 mmol) in anhydrous THF (1 mL), under N2 atmosphere and vigorous magnetic stirring, a 1.0 M of DIBAL in THF (0.5 mL, 0.5 mmol) was added at −78 °C. The mixture was stirred at the same temperature for 15 min, and a suspension of 5% of NH4Cl in MeOH (5 mL) was added. While the mixture was stirred for 2 h, the temperature rose to room temperature. The mixture was filtered and the solvent removed under vacuum. The crude was purified by column chromatography over silica gel (5 g, hexane/EtOAc, 7:3), generating a mixture of 29a/29b (59:41) (0.023, 45%) as a pale yellow oil. Rf 0.30 (EtOAc). IR (CH2Cl2) 3392, 1730, 1442, 1373, 1241, 1068, 1031 cm−1. 1H NMR (300 MHz, CDCl3) δ signals attributed to the major isomer: 1.52–1.65 (m, 1H, H-4), 1.84–2.05 (m, 1H, H-3), 2.07 (s, 3H, CH3CO2), 2.32–2.39 (m, 1H, H-4), 2.90–3.06 (m, 1H, H-3a), 3.64–3.82 (m, 1H, HO), 4.10 (dd, J = 5.1, 2.4 Hz, 1H, H-6), 4.52 (dd, J = 7.8, 2.4 Hz, 1H, H-6a), 4.79–4.87 (m, 1H, H-5), 5.62–5.66 (m, 1H, H-2); signals attributed to the minor isomer: 2.05–2.18 (m, 2H, H-3, H-4), 2.09 (s, 3H, CH3CO2), 2.39–2.46 (m, 1H, H-4), 2.69–2.82 (m, 1H, H-3a), 3.82–4.00 (m, 1H, HO), 4.34 (dd, J = 7.5, 4.2 Hz, 1H, H-6), 4.40 (dd, J = 8.7, 4.2 Hz, 1H, H-6a), 4.87–4.96 (m, 1H, H-5), 5.66–5.70 (m, 1H, H-2). 13C NMR (75.4 MHz, CDCl3) δ signals attributed to the major isomer: 21.1 (CH3CO2), 35.5 (C-4), 37.4 (C-3a), 40.7 (C-3), 80.6 (C-6), 81.7 (C-5), 88.0 (C-6a), 100.1 (C-2), 171.7 (CH3CO2); signals attributed to the minor isomer: 34.6 (C-4), 36.3 (C-3a), 39.2 (C-3), 80.0 (C-5), 82.6 (C-6), 89.4 (C-6a), 100.9 (C-2), 172.1 (CH3CO2); MS (70 eV) m/z (%) 185 (M+−17, 6), 184 (M+−18, 6), 142 (12), 124 (50), 96 (34), 95 (100), 82 (42), 81 (74), 67 (47). HRMS (FAB) (mNBA) m/z [MH]+ calculated for C9H15O5: 203.0919; found: 203.0918.
4.14 Single-crystal X-Ray crystallography
Compounds 11a and 12a were obtained as pale yellow crystals, and compound 16 as colorless crystals. These were mounted on glass fibers. Crystallographic measurements were carried out at room temperature in a Siemens P4 diffractometer equipped with a graphite monocromator, with Mo Kα radiation (λ = 0.7107 Å) for 11a and 16, and Cu Kα radiation (λ = 1.5418 Å) for 12a. In all cases, three standard reflections were monitored periodically and they showed no change during data collection. Unit cell parameters were obtained from least-squares refinement of 28 reflections in the range 4.63 < θ < 12.18° for 11a, 39 reflections in the range 11.87 < θ < 28.15° for 12a; and 41 reflections in the range 5.36 < θ < 15.58° for 16. Intensities were corrected for Lorentz and polarization effects. Although no absorption corrections were made to the diffraction data of 11a, empirical corrections (psi-scans) were applied to the data of 12a and 16. Anisotropic temperature factors were introduced for all non-hydrogen atoms. Hydrogen atoms were placed in idealized positions and their atomic coordinates refined by using unit weights. The structure was solved and refined with the SHELX-97 program package (Shedrick, 2008) running under the WinGX environment (Farrugia, 1999, 2012) and visualized and plotted with the Ortep-3 program (Farrugia, 1997, 2012). Data on 11a: (CCDC 1499434) Formula: C17H17NO5; molecular weight: 315.32; cryst. syst.: monoclinic; space group: P21/c; unit cell parameters: a, 8.3876(19), b, 17.584(3), c, 11.1855(15) (Å); α, 90, β, 108.93(2), γ, 90 (deg); V = 1560.5(5) Å3; temp. (K): 293(2); Z: 4; no. of reflections collected: 4365; no. of independent reflections: 3402; no. of reflections observed: 1662; data collection range: 2.25 < θ < 27.00°; R: 0.0697; wR: 0.1781; GOF: 1.010. Data on 12a: (CCDC 1499435) Formula: C17H17NO5; molecular weight: 315.32; cryst. syst.: monoclinic; space group: P21/n; unit cell parameters: a, 7.2332(12), b, 20.794(4), c, 10.7106(17) (Å); α, 90, β, 101.31(2), γ, 90 (deg); V = 1579.7(5) Å3; temp. (K): 293(2); Z: 4; no. of reflections collected: 2887; no. of independent reflections: 2124; no. of reflections observed: 1737; data collection range: 4.25 < θ < 56.93°; R: 0.0705; wR: 0.0836; GOF: 1.013. Data on 16: (CCDC 1499436) Formula: C9H12O2; molecular weight: 168.19; cryst. syst.: monoclinic; space group: P21/n; unit cell parameters: a, 6.8247(5), b, 9.7191(11), c, 12.2271(12) (Å); α, 90, β, 91.237(8), γ, 90 (deg); V = 810.83(14) Å3; temp. (K): 293(2); Z: 4; no. of reflections collected: 2818; no. of independent reflections: 2044; no. of reflections observed: 1591; data collection range: 2.68 < θ < 28.49°; R: 0.0486; wR: 0.1227; GOF: 1.043. The authors have deposited the atomic coordinates for these structures with the Cambridge Crystallographic Data Center (CCDC 1499434, 1499435, and 1499436). The coordinates can be obtained, on request, from the director of the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge, CB2 1EZ, UK.
Acknowledgments
We thank Fabiola Jiménez for her help in spectrometric measurements and Bruce A. Larsen for proofreading. J.T. would like to acknowledge the financial support provided by the Secretaría de Investigación y Posgrado (SIP)-IPN (Grants 20130686, 20140858, 20150917, 20160791, and 20170902) and CONACYT (Grants 83446 and 178319). M.A.V. thanks Dirección de Apoyo a la Investigación y al Posgrado (DAIP-UG) (Grant 811/2016). R.M.P. is grateful to CONACYT for a postdoctoral fellowship. R.A., B.M.S, and D.Z.-Z. are beholden to CONACYT for graduate fellowships, to the Programa Institucional de Formación de Investigadores (PIFI)-IPN program, and to the Ludwig K. Hellweg Foundation for scholarships awarded. J.T. and H.A.J.-V. are fellows of the Estímulos al Desempeño de los Investigadores (EDI)-IPN and Comisión de Operación y Fomento de Actividades Académicas (COFAA)-IPN programs.
References
- The development and use of ketene equivalents in [4+2] cycloadditions for organic synthesis. Tetrahedron. 1999;55:293-312.
- [Google Scholar]
- Synthesis and conformational and biological aspects of carbasugars. Chem. Rev.. 2007;107:1919-2036.
- [Google Scholar]
- Direct evaluation of secondary orbital interactions in the Diels-Alder reaction between cyclopentadiene and maleic anhydride. J. Org. Chem.. 2001;66:6178-6180.
- [Google Scholar]
- Electronic effects in 1,3-dipolar cycloaddition reactions of N-alkyl and N-benzyl nitrones with dipolarophiles. Org. Biomol. Chem.. 2012;10:114-121.
- [Google Scholar]
- Synthesis and cycloaddition reactions of new captodative olefins, N-substituted 5-alkylidene-1,3-oxazolidine-2,4-diones. Heterocycles. 2001;55:469-485.
- [Google Scholar]
- A theoretical study of the mechanism, stereoselectivity and Lewis acid catalyst on the Diels-Alder cycloaddition between furan and activated alkenes. Tetrahedron Lett.. 2013;54:4030-4033.
- [Google Scholar]
- Asymmetric catalysis. Part 137: Nickel catalysed enantioselective α-ketol rearrangement of 1-benzoylcycloalkanols. Tetrahedron: Asymmetry. 2001;12:497-499.
- [Google Scholar]
- Stereochemistry of the Diels-Alder reaction: effect of diene structure on endo-selectivity. J. Chem. Soc. Perkin. 1974;II:22-25.
- [Google Scholar]
- An unexpected bispericyclic transition structure leading to 4+2 and 2+4 cycloadducts in the endo dimerization of cyclopentadiene. J. Am. Chem. Soc.. 2002;124:1130-1131.
- [Google Scholar]
- Chiral octahedral phosphano-oxazoline iridium(III) complexes as catalysts in asymmetric cycloaddition reactions. Organometallics. 2013;32:1609-1619.
- [Google Scholar]
- Applications of sulfoxides to asymmetric synthesis of biologically active compounds. Chem. Rev.. 1995;95:1717-1760.
- [Google Scholar]
- Cycloaddition Reactions in Organic Synthesis. Oxford: Pergamon Press; 1990.
- Epoxidation of allylic alcohols with peroxy-acids. Attemps to define transition state geometry. J. Chem. Soc. B 1970:1374-1381.
- [Google Scholar]
- Ab initio quantum-mechanical and experimental mechanistic studies of Diels-Alder reactions between unsubstituted and phenyl-substituted acetylenes and 1,2,4,5-tetrazines. J. Am. Chem. Soc.. 1993;115:1353-1359.
- [Google Scholar]
- Total synthesis of prostaglandins F2α and E2 as the naturally occurring forms. J. Am. Chem. Soc.. 1970;92:397-398.
- [Google Scholar]
- Diels-Alder approach to bicyclic α-hydroxy ketones. Facile ketol rearrangements of strained α-hydroxy ketones. J. Org. Chem.. 1985;50:1932-1938.
- [Google Scholar]
- Local softness as a regioselectivity indicator in [4+2] cycloaddition reactions. J. Phys. Chem. A. 1997;101:886-893.
- [Google Scholar]
- The intermediate of “α-cyclization of tertiary amines” as a vinyl-1,3-dipole. Tetrahedron. 1998;54:513-520.
- [Google Scholar]
- Spectroscopic studies on the structure and conformation of Lewis acid-aldehyde complexes. J. Am. Chem. Soc.. 1993;115:3133-3139.
- [Google Scholar]
- Asymmetric synthesis and DNA intercalation of (-)-6-[[(aminoalkyl)oxy]methyl]-4-demethoxy-6,7-dideoxydaunomycinones. J. Org. Chem.. 1996;61:6958-6970.
- [Google Scholar]
- Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem.. 2008;73:4615-4624.
- [Google Scholar]
- Understanding the mechanism of non-polar Diels-Alder reactions. A comparative ELF analysis of concerted and stepwise diradical mechanisms. Org. Biomol. Chem.. 2010;8:5495-5504.
- [Google Scholar]
- ORTEP-3 for Windows – a version of ORTEP-III with a graphical user interface (GUI) J. Appl. Cryst.. 1997;30:565.
- [Google Scholar]
- WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst.. 1999;32:837-838.
- [Google Scholar]
- The Demjanov and Tiffeneau-Demjanov one-carbon ring enlargements of 2-aminomethyl-7-oxabicyclo[2.2.1]heptane derivatives. The stereo- and regioselective additions of 8-oxabicyclo[3.2.1]oct-6-en-2-one to soft electrophiles. Tetrahedron. 1993;49:1649-1664.
- [Google Scholar]
- The Diels-Alder Reaction: Selected Practical Methods. Chichester: J. Wiley & Sons; 2002.
- Lewis acid-promoted stereoselective Diels-Alder cycloadditions of captodative olefins acetylvinyl carboxylates and NMR structural study of their cyclopentadiene adducts. Anal. Quím., Int. Ed.. 1996;92:108-117.
- [Google Scholar]
- An efficient total synthesis of (±)-brefeldin-A. J. Am. Chem. Soc.. 1980;102:7583-7584.
- [Google Scholar]
- Pseudoguaianolides. 1. Stereospecific total synthesis of the ambrosanolides dl-ambrosin and dl-damsin. J. Am. Chem. Soc.. 1982;104:4226-4233.
- [Google Scholar]
- Enantioselective Diels-Alder reactions with anomalous endo/exo selectivities using conformationally flexible chiral supramolecular catalysts. Angew. Chem. Int. Ed.. 2011;50:12189-12192.
- [Google Scholar]
- Magnetic properties of aromatic transition states: the Diels-Alder reactions. Angew. Chem. Int. Ed. Engl.. 1994;33:1376-1378.
- [Google Scholar]
- Regio- and stereoselectivity of captodative olefins in 1,3-dipolar cycloadditions. A DFT/HSAB theory rationale for the observed regiochemistry of nitrones. J. Org. Chem.. 2001;66:1252-1263.
- [Google Scholar]
- Synthesis of new captodative alkenes: alkyl 2-aroyloxy acrylates – structure, and reactivity in Diels-Alder cycloadditions. Eur. J. Org. Chem. 2001:4657-4669.
- [Google Scholar]
- 1-Acetylvinyl acrylates: new captodative olefins bearing an internal probe for the evaluation of the relative reactivity of captodative against electron-deficient double bonds in Diels-Alder and Friedel-Crafts reactions. J. Braz. Chem. Soc.. 2005;16:456-466.
- [Google Scholar]
- Transition states of epoxidations: diradical character, spiro geometries, transition state flexibility, and the origins of stereoselectivity. J. Am. Chem. Soc.. 1997;119:10147-10152.
- [Google Scholar]
- Hudlicky, M., 1990. Oxidations in Organic Chemistry. American Chemical Society. Washington, p. 130.
- Stable 5-substituted cyclopentadienes for the Diels-Alder cycloaddition and their application to the synthesis of palau’amine. Angew. Chem. Int. Ed.. 2008;47:8885-8888.
- [Google Scholar]
- Mechanistic studies of a CAB-catalyzed asymmetric Diels-Alder reaction. J. Am. Chem. Soc.. 1993;115:10412-10413.
- [Google Scholar]
- Ishihara, K., Sakakura, A., 2014. Intermolecular Diels-Alder reactions, in: Knochel, P., Molander, G.A. (Eds.), Comprehensive Organic Synthesis. Elsevier: Amsterdam, vol. 5, Chap. 5.09, pp. 351–408.
- Searching for zwitterionic intermediates in Hetero Diels-Alder reactions between methyl α,p-dinitrocinnamate and vinyl-alkyl ethers. Comp. Theor. Chem.. 2014;1046:93-98.
- [Google Scholar]
- An experimental and theoretical study of the hetero Diels-Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: one-step or zwitterionic, two-step mechanism? Reac. Kinet. Mech. Cat.. 2014;113:333-345.
- [Google Scholar]
- A reexamination of the molecular mechanism of the Diels-Alder reaction between tetrafluoroethene and cyclopentadiene. Reac. Kinet. Mech. Cat.. 2016;119:49-57.
- [Google Scholar]
- First example of stepwise, zwitterionic mechanism for bicyclo[2.2.1]hept-5-ene (norbornene) formation process catalyzed by the 1-butyl-3-methylimidazolium cations. Monatsh. Chem.. 2016;147:1207-1213.
- [Google Scholar]
- Structural, spectroscopic, and theoretical study of 1-acetylvinyl p-nitrobenzoate, a highly reactive and selective captodative olefin in cycloaddition reactions. J. Phys. Chem. A. 1997;101:10082-10089.
- [Google Scholar]
- Synthesis of polypropionate fragments containing tertiary-alcohol moieties. Cross-aldolisations with lithium enolates of 7-oxabicyclo[2.2.1]heptan-2-one derivatives. Helv. Chim. Acta. 1995;78:301-324.
- [Google Scholar]
- Diversity-oriented approach to unusual amino acid derivatives and heterocycles via methyl 2-acetamidoacrylate and its congeners. Tetrahedron. 2014;70:5361-5384.
- [Google Scholar]
- An efficient approach to Aspidosperma alkaloids via [4 + 2] cycloadditions of aminosiloxydienes: stereocontrolled total synthesis of (±)-tabersonine. Gram-scale catalytic asymmetric syntheses of (+)-tabersonine and (+)-16-methoxytabersonine. Asymmetric syntheses of (+)-aspidospermidine and (-)-quebrachamine. J. Am. Chem. Soc.. 2002;124:4628-4641.
- [Google Scholar]
- The Baeyer-Villiger oxidation of ketones and aldehydes. Org. React.. 1993;43:251-798.
- [Google Scholar]
- Synthesis of cyclopentitols by ring-closing approaches. Chem. Rev.. 2009;109:6809-6857.
- [Google Scholar]
- Theoretical studies of conformations of acrolein, acrylic acid, methyl acrylate, and their Lewis acid complexes. J. Am. Chem. Soc.. 1987;109:14-23.
- [Google Scholar]
- Synthesis of all stereoisomeric carbapentofuranoses. J. Org. Chem.. 1995;60:5224-5235.
- [Google Scholar]
- Exo-selective Diels-Alder reaction based on a molecular recognition approach. J. Am. Chem. Soc.. 1994;116:12115-12116.
- [Google Scholar]
- An approach to seco-prezizaane sesquiterpenoids: enantioselective total synthesis of (+)-1S-minwanenone. J. Org. Chem.. 2012;77:8056-8070.
- [Google Scholar]
- 1,3-Dipolar cycloaddition reactions: a DFT and HSAB principle theoretical model. J. Phys. Chem. A. 1998;102:6292-6296.
- [Google Scholar]
- Effect of aryl substituents on the reactivity of the captodative olefins 1-acetylvinyl arenecarboxylates. J. Mex. Chem. Soc.. 2006;50:47-56.
- [Google Scholar]
- Biosynthesis inspired Diels-Alder route to pyridines: synthesis of the 2,3-dithiazolylpyridine core of the thiopeptide antibiotics. Chem. Commun. 2002:1760-1761.
- [Google Scholar]
- Strain-promoted retro-Dieckmann-type condensation of [2.2.2]- and [2.2.1]bicyclic systems: a fragmentation reaction for the preparation of functionalized heterocycles and carbocycles. Org. Biomol. Chem.. 2013;11:7016-7025.
- [Google Scholar]
- Highly enantioselective Diels-Alder reactions of maleimides catalyzed by activated chiral oxazaborolidines. Org. Lett.. 2010;12:632-635.
- [Google Scholar]
- Highly selective 1,3-dipolar cycloadditions of captodative olefins 1-acetylvinyl carboxylates to diverse dipoles. Tetrahedron Lett.. 1996;37:6835-6838.
- [Google Scholar]
- The Diels-Alder reaction in total synthesis. Angew. Chem. Int. Ed.. 2002;41:1668-1698. and references cited therein
- [Google Scholar]
- Captodative olefin 3-p-nitrobenzoyloxy-3-buten-2-one as a Diels-Alder ketene equivalent for the synthesis of γ-hydroxycyclohexenones. Tetrahedron. 1999;55:14535-14546.
- [Google Scholar]
- Orbital unsymmetrization of olefins arising from non-equivalent orbital interactions. σ-π Coupling in bicyclo[2.2.2]octenes. Chem. Pharm. Bull.. 1996;44:296-306.
- [Google Scholar]
- Orbital-controlled stereoselections in sterically unbiased cyclic systems. Chem. Rev.. 1999;99:1337-1375.
- [Google Scholar]
- Highly efficient synthesis of the natural spiroterpenoid (±)-andirolactone. Synthesis 1993:375-377.
- [Google Scholar]
- Electronic control of stereoselectivity. 3. Stereoselection operative in [4+2] π cycloadditions to cyclopentadiene rings fused at C2, C3 to bicyclic frameworks. J. Am. Chem. Soc.. 1980;102:1186-1188.
- [Google Scholar]
- The α-hydroxyl ketone (α-ketol) and related rearrangements. Org. React.. 2003;62:477-567. and references cited therein
- [Google Scholar]
- Acceleration and selectivity enhancement of Diels-Alder reactions by special and catalytic methods. Chem. Rev.. 1993;93:741-761.
- [Google Scholar]
- The role of supramolecular intermediates in the potential energy surface of the Diels-Alder reaction. J. Mex. Chem. Soc.. 2013;57:267-275.
- [Google Scholar]
- Asymmetric Diels-Alder reaction catalyzed by some chiral Lewis acids. Tetrahedron: Asymmetry. 1990;1:199-214.
- [Google Scholar]
- Highly selective Diels-Alder cycloadditions of captodative dienophiles 1-acetylvinyl arenecarboxylates to unsymmetrically substituted butadienes. J. Org. Chem.. 1990;55:1024-1034.
- [Google Scholar]
- New chiral auxiliaries and new optically pure ketene equivalents derived from tartaric acids. Improved synthesis of (-)-7-oxabicyclo[2.2.1]hept-5-en-2-one. Tetrahedron: Asymmetry. 1990;1:729-736.
- [Google Scholar]
- Origin of the endo selectivity in the Diels-Alder reaction between cyclopentadiene and maleic anhydride. Eur. J. Org. Chem. 2016:4275-4278.
- [Google Scholar]
- New captodative olefins: 3-(2-furoyloxy)-3-buten-2-one and alkyl 2-(2-furoyloxy)-2-propenoates, and their reactivity in addition reactions. J. Mex. Chem. Soc.. 2007;51:198-208.
- [Google Scholar]
- Mechanistic aspects of Diels-Alder reactions: a critical survey. Angew. Chem Int. Ed. Engl.. 1980;19:779-807.
- [Google Scholar]
- Isotope effects and the distinction between synchronous, asynchronous, and stepwise Diels-Alder reactions. Tetrahedron. 2001;57:5149-5160.
- [Google Scholar]
- Stereochemistry and mechanism of thermal and base-catalyzed rearrangements of α-hydroxy ketones. J. Org. Chem.. 1972;37:2091-2097.
- [Google Scholar]
- On the origin of the endo/exo selectivity in Diels-Alder reactions. Chem. Commun. 1998:385-386.
- [Google Scholar]
- Gold-catalyzed oxidative reactions of propargylic carbonates involving 1,2-carbonate migration: stereoselective synthesis of functionalized alkenes. J. Org. Chem.. 2014;79:4055-4067.
- [Google Scholar]
- New dienophiles: 1-acetylvinyl arenecarboxylates. Reactivity towards cyclopentadiene and exocyclic dienes. Helv. Chim. Acta. 1981;64:188-197.
- [Google Scholar]
- Origins of stereoselectivity in intramolecular Diels-Alder cycloadditions of dienes and dienophiles linked by ester and amide tethers. J. Org. Chem.. 2001;66:1938-1940.
- [Google Scholar]
- A general approach to 1-hydroxymethylquinolizidine and 8-hydroxymethylindolizidine stereoisomers: synthesis of (+)-epitashiromine and formal syntheses of (+)-epilupinine and (+)-tashiromine. Eur. J. Org. Chem. 2013:7282-7285.
- [Google Scholar]
- New synthetic reactions. Oxidative decarboxylation of α-methylthiocarboxylic acids, new approach to acyl anion and ketene synthons. J. Am. Chem. Soc.. 1977;99:3101-3113.
- [Google Scholar]
- Stereodivergent synthesis of piperidine alkaloids by ring-rearrangement metathesis/reductive lactam alkylation of nitroso Diels-Alder cycloadducts. Chem. Eur. J.. 2013;19:9358-9365.
- [Google Scholar]
- An asymmetric allylic alkylation-Smiles rearrangement-sulfinate addition sequence to construct chiral cyclic sulfones. Org. Lett.. 2013;15:4786-4789.
- [Google Scholar]
- Recrossing and dynamic matching effects on selectivity in a Diels-Alder reaction. Angew. Chem. Int. Ed.. 2009;2009(48):9156-9159.
- [Google Scholar]
- The existence of secondary orbital interactions. J. Comput. Chem.. 2007;28:344-361.
- [Google Scholar]
- Synthesis of α-ketoles by functionalization of captodative alkenes and divergent preparation of heterocycles and natural products. Tetrahedron. 2015;71:6961-6978.
- [Google Scholar]
- Furan-based o-quinodimethanes by gold-catalyzed dehydrogenative heterocyclization of 2-(1-alkynyl)-2-alken-1-ones: a modular entry to 2,3-furan-fused carbocycles. Angew. Chem. Int. Ed.. 2014;53:6542-6545.
- [Google Scholar]
- Lewis base catalyzed asymmetric substitution/Diels-Alder cascade reaction: a rapid and efficient construction of enantioenriched diverse tricyclic heterocycles. Org. Biomol. Chem.. 2013;11:7080-7083.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data (1H and 13C NMR spectra of compounds 11a-b, 12a-b, 15, 16, 19b, 22a, 23, 26-29, and 31a-b) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.arabjc.2017.08.008.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




