

Translate this page into:
Structure-based design, and development of amidinyl, amidoximyl and hydroxamic acid based organic molecules as novel antimalarial drug candidates
⁎Corresponding author at: Covenant University Bioinformatics Research (CUBRe), Covenant University, Ota, Nigeria. ezekiel.adebiyi@covenantuniversity.edu.ng (Ezekiel Adebiyi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Malaria remains a significant global health concern causing numerous fatalities and the emergence of antimalarial drug resistance highlights the urgent need for novel therapeutic options with innovative mechanisms of action and targets. This study aimed to design potential inhibitors of Plasmodium falciparum 6-pyruvoyltetrahydropterin synthase (PfPTPS), synthesize them, and experimentally validate their efficacy as antimalarial agents. A structure-based approach was employed to design a series of novel derivatives, including amidinyl, amidoximyl and hydroxamic acid analogs (1c, 1d, 2b, and 3b), with a focus on their ability to bind to the Zn2+ present in the active site of PfPTPS. The syntheses of these compounds were accomplished through various multi-step synthetic pathways and their structural identities were confirmed using 1H and 13C NMR spectra, mass spectra, and elemental analysis. The compounds were screened for their antiplasmodial activity against the NF54 strain of P. falciparum and in vitro cytotoxicity testing was performed using L-6 cells. The in vivo acute toxicity of the compounds was evaluated in mice. Docking studies of the compounds with the 3D structure of PfPTPS revealed their strong binding affinities, with compound 3b exhibiting notable metal–acceptor interaction with the Zn2+ in the protein binding pocket thereby positioning it as a lead compound for PfPTPS inhibition. The in vitro antiplasmodial studies revealed moderate efficacies against the Pf NF54 strain, particularly compounds 1d and 3b which displayed IC50 < 0.2 μM. No significant cytotoxicity was noted on the L-6 rat cell line. Moreover, in vivo studies suggested that compound 3b exhibited both safety and efficacy in treating rodent malaria. The identified lead compound in this study represents a possible candidate for antimalarial drug development and can be further explored in the search for alternative antifolate drugs to combat the malaria menace.
Keywords
Acute toxicity
Cytotoxicity
Hydroxamic acids
Malaria
Suppression

1 Introduction
Malaria remains a significant global health burden with the World Health Organization (WHO) reporting 247 million cases worldwide, predominantly in Africa (WHO, 2022). Tragically, children under the age of five bear the brunt of this disease, accounting for 76 % of all malaria-related deaths globally. Human malaria is caused by five species of the Plasmodium parasite, with Plasmodium falciparum and P. vivax being the most prevalent in Africa and countries outside of Africa, respectively (Umumararungu et al., 2023). Despite the continuing effort to eradicate malaria, there is an increase in its resistance to the most effective drugs such as artemisinin, mefloquine, and piperaquine leading to the use of artemisinin-based combination therapies (ACTs) (Lin et al., 2023; Mishra et al., 2023). However, the emergence of ACT resistance have also warranted the search for new compound candidates with new modes of action (Hanboonkunupakarn et al., 2022).
The advent of multidisciplinary approaches to drug design is one that is currently being explored. Computer-aided drug design (CADD) techniques such as structure-based drug design (SBDD), quantitative structure activity relationships (QSAR), and molecular docking have proven instrumental in identifying and designing potential pharmacologically active compounds (Rajguru et al., 2022; Sabe et al., 2021). CADD techniques are becoming increasingly important in drug development as they are valuable in identifying potential therapeutic candidates at low cost (Acuna et al., 2020; Brogi et al., 2020). These computational techniques effectively reduce the use of animal models in pharmacological studies, assist in the rational design of novel and safe drug candidates, reposition marketed drugs, and assist medicinal chemists and pharmacologists throughout the drug discovery process (Chen, 2021). Many inhibitors targeting various biological targets have been developed using CADD, including SBDD and ligand-based drug design (LBDD) (Mouchlis et al., 2020). Also, many antimalarial drug candidates obtained from SBDD are in the medicines for malaria venture (MMV) pipeline being considered as future antimalarial drugs (Rajguru et al., 2022). Choosing a specific set of compounds or ligands play a vital role in the early stages of lead discovery. Docking chemical libraries into the binding cavity of the target protein is used to find hits, and this is carried out using virtual screening methods (Batool et al., 2019). Hits are ligands with the lowest binding energy or affinity or strongest binders from the virtual screening of a chemical library (Oduselu et al., 2021). When evaluating a group of compounds to be selected as leads, their chemical accessibility (e.g., number of synthetic steps) and ease of synthesis of their analogues may be essential factors to consider; particularly for those leads that are not fully drug-like in their properties and thus require extensive modifications (Brodniewicz and Grynkiewicz, 2010; Wunberg et al., 2006). The ability to predict the activities of molecules before they are synthesized has helped to reduce cost, time, and resources required in the lead identification process (Iwaloye et al., 2020).
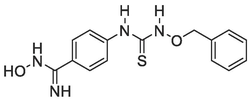
A good number of the P. falciparum targets have been identified, experimentally validated, and their biological roles have been defined (Mishra et al., 2017). For instance, the design and development of targeted inhibitors against P. falciparum dihydrofolate reductase, which disrupts folate production in the parasite, have yielded antifolate malaria medications like pyrimethamine, WR99210, and cycloguanil (Fig. 1) (Adams et al., 2023; Akinnusi et al., 2023; Dasgupta et al., 2009). However, resistance to these antifolate drugs has emerged rapidly, rendering them ineffective against the parasite. Consequently, the identification of alternative antimalarial drug targets in the de novo folate synthesis pathway has become crucial. Among these potential targets is Plasmodium falciparum 6-pyruvoyltetrahydropterin synthase (PfPTPS) (Fatumo et al., 2009). PfPTPS is a promising antimalarial target due to its distinctive structural features and the potential for developing parasite-specific inhibitors (Hyde et al., 2008). This enzyme plays a vital role in the conversion of 7,8-dihydroneopterin (DHNP) to 6-pyruvoyl-tetrahydropterin (PTP) through an internal redox transfer process (Khairallah et al., 2020). In this study, we aimed to employ a structure-based design approach to develop inhibitors of PfPTPS, synthesize these compounds through multi-step synthetic pathways, and evaluate their antimalarial efficacies. The ultimate goal is to identify new antimalarial drug candidates capable of combating drug resistance and improving treatment outcomes for malaria.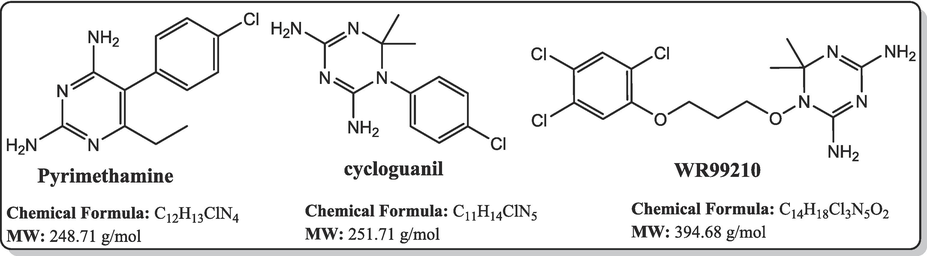
Known antifolate antimalarials that are active-site inhibitors of the P. falciparum dihydrofolate reductase, a common target in the de novo folate synthesis pathway.
2 Materials and methods
2.1 Structure-based drug design (SBDD)
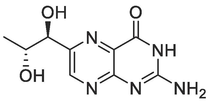
The PfPTPS crystal structure (PDB ID: 1Y13) was downloaded from Protein Data Bank (PDB) and used for the structure-based design. The protein structure possesses three chains with a resolution of 2.20 Å and a sequence length of 181. The active site of PfPTPS contains a co-ligand biopterin and a zinc ion. In the deeper pocket, a negative charged glutamate and aromatic side chains explain the binding mode of the native binder, biopterin (Fig. 2). Due to the small size of the binding pocket and the zinc ion in the active site, the fragment-like subset of the ZINC Database was screened using a graph-matching strategy for zinc-binding groups. The selected ligands were optimized, and a basic functionality has been added to enable the interaction to a negatively charged glutamate 128 at the bottom of the binding pocket. The compounds designed from the structure-based drug design approach include different amidinyl, amidoximyl and hydroxamic acid analogs (1c, 1d, 2b, 3b) and these compounds were taken further for synthesis.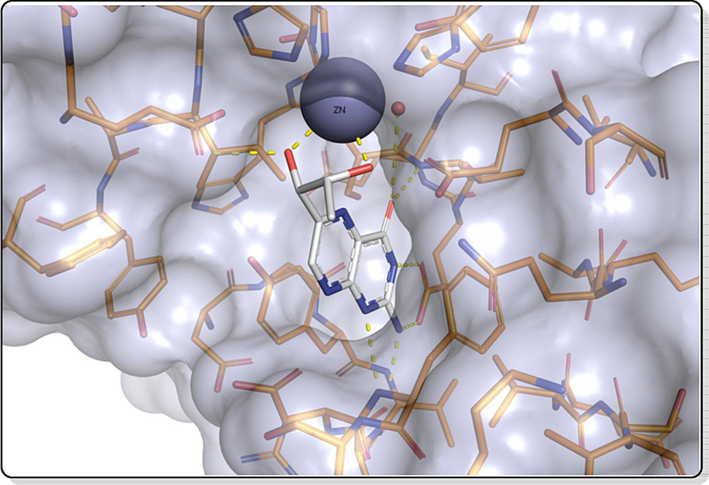
The active site of PfPTPS (PDB 1D: 1Y13) and the native ligand biopterin.
2.2 Chemistry
2.2.1 Instruments
1H NMR and 13C NMR spectra were recorded in DMSO‑d6 using a JEOL Delta NMR ECX 400 spectrometer operating at 400 MHz and 100 MHz, respectively. Mass spectra were obtained using Q-Trap 2000 Spectrometer at 70 eV. Elemental analyses (C, H, N) of the compounds were performed using various MICRO V1.5.1 Elemental Analyzers. The progress of reactions was monitored with TLC using CHCl3/CH3OH solvent system and the developed plates were visualized under UV where necessary. Column chromatographic purifications were carried out on Merck silica gel F (Mesh 200–300) using DCM/MeOH as eluting solvent in various ratios depending on the polarity of the crude compounds. Organic solutions were dried over anhydrous MgSO4 while the concentration and removal of solvents was achieved with an RE-2000B Rotary Evaporator at reduced pressure. All chemicals were obtained from Sigma-Aldrich Chemicals (purity ≥ 90 %) and other manufacturers in Germany.
2.2.2 Synthesis of the designed compounds
2.2.2.1 Synthesis of N-(Benzyloxy)-4-cyanobenzamide, 1a
Dichloromethane DCM (27.00 mL) was added to 4-cyanobenzoyl chloride (9.00 mmol, 1.50 g) with stirring at rt until total dissolution. o-Benzyl hydroxyl amine (13.50 mmol, 0.90 mL, 1.50 eq.) was added to 27 mL of DCM in a dispensing adaptor followed by the addition of triethylamine (13.50 mmol, 1.80 mL, 1.50 eq) and mixed thoroughly. The contents in the adaptor was added dropwise to the flask solution above. The resulting solution was stirred at rt for 5 h first and later overnight until the starting material had been consumed as indicated by TLC. The resulting solution was quenched by diluting with more DCM, transferred into separatory funnel, washed with 1 M HCl, followed by saturated NaHCO3 and finally with distilled water. The organic layer was dried over anhydrous MgSO4, evaporated to get crude solid which was purified by column chromatography (DCM/MeOH → 98:2) and vacuum dried to afford a white-coloured solid N-(benzyloxy)-4-cyanobenzamide, 1a (1.77 g, 78.0 %). 1H NMR (400 MHz, DMSO‑d6) δ: 11.93 (br s, 1H, NH), 7.91–7.89 (d, J = 8.4 Hz, 2H, Ar—H), 7.84–7.82 (d, J = 8.8 Hz, 2H, Ar—H), 7.39–7.32 (m, 5H, ArH), 4.87 (s, 2H, CH2—Ar). 13C NMR (100 MHz, DMSO‑d6) δ: 136.9 (C⚌O), 133.1 (3 × CH), 129.5 (3 × CH), 128.9 (4 × CH), 128.4 (2 × CH), 118.8 (C≡N), 77.5 (CH2) ppm. MS-ESI: in m/z (rel. %): 274.99 (100 %), 270.00 (66.67 %), 253.09 (M+ + 1, 95.2 %), 213.80 (19.9 %), 203.23 (15.1 %), 149.10 (17.8 %), 141.20 (22.6 %), 130.00 (28.8 %). Anal. % Calcd. for C15H12N2O2 (252.27): C, 71.43; H, 4.76; N, 11.11. Found: C, 71.47; H, 4.95; N, 11.27.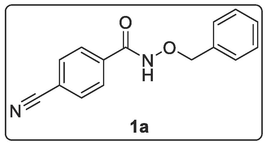
2.2.2.2 Synthesis of N-(Benzyloxy)-4-(N-hydroxycarbamimidoyl)benzamide, 1b
A solution of N-benzyloxy-4-cyanobenzamide, 1a (4.00 mmol, 1.00 g), hydroxylamine hydrochloride (5.40 mmol, 0.37 g), and Na2CO3 (4.00 mmol, 0.42 g) in EtOH (30 mL) was heated under reflux for 7 h. Additional amount of NH2OH.HCl (5.40 mmol, 0.37 g) and Na2CO3 (4.00 mmol, 0.42 g) were added and the mixture was stirred under reflux for 4 h. The resulting mixture was allowed to cool, filtered by suction and the solvent was evaporated to dryness and later vacuum dried to give white-coloured solid N-(benzyloxy)-4-(N-hydroxycarbamimidoyl)benzamide, 1b (1.13 g, 99.12 %). 1H NMR (400 MHz, DMSO‑d6) δ: 11.82 (s-br, 1H, OH), 9.65 (s, 1H, NH), 7.70–7.68 (d, J = 8.4 Hz, 2H, Ar—H), 7.61–7.59 (d, J = 8.4 Hz, 2H, Ar—H), 7.39–7.38 (d, J = 7.2 Hz, 2H, Ar—H), 7.30–7.22 (m, 3H, Ar—H), 5.74 (s, 2H, NH2), 4.83 (s, 2H, CH2—Ar). MS-ESI: in m/z (rel. %): 308.02 (62.2 %), 292.95 (6.1 %), 286.04 (M+ + 1, 100 %), 195.02 (6.2 %), 178.10 (7.5 %), 161.09 (3.0 %). Molecular weight C15H15N3O3 (285.30).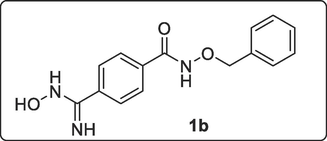
2.2.2.3 Synthesis of N-Hydroxy-4-(N-hydroxycarbamimidoyl)benzamide, 1c
N-(Benzyloxy)-4-(N-hydroxycarbamimidoyl)benzamide, 1b (1.00 mmol, 0.29 g) was dissolved in 40 mL of methanol followed by addition of 10 % Pd/C (0.10 g). Hydrogen gas was continuously bubbled into solution under stirring at room temperature for 4 h 40 min. The reaction was quenched and washed with more MeOH and filtered. The filtrate was evaporated to dryness with rotary evaporator after which the solid was vacuum-dried to give a white-coloured N-hydroxy-4-(N-hydroxycarbamimidoyl)benzamide, 1c (0.16 g, 88.89 %). Melting Point: 181–182 °C. 1H NMR (400 MHz, DMSO‑d6) δ: 7.87–7.81 (m, 2H, 2 × OH), 7.71–7.69 (d, J = 8.0 Hz, 2H, Ar—H), 7.57–7.55 (d, J = 8.0 Hz, 2H, Ar—H), 5.86 (s, 1H, NH), 5.72 (s, 2H, NH2). MS-ESI: in m/z (rel. %): 196.08 (M + 1, 36.4 %), 180.07 (M – NH, 38.6 %), 164.10 (M - NOH, 100 %), 147.06 (M – NOH – OH, 25.01 %), 145.04 (M – NHOH – H2O, 6.2 %), 134.04 (M – (NH2)2COH, 7.5 %), 118.99 (M – (NH2)2COH – NH2, 4.5 %). Anal. % Calcd. for C8H9N3O3 (195.17): C, 49.23; H, 4.65; N, 21.53. Found: C, 48.06; H, 5.11; N, 18.11.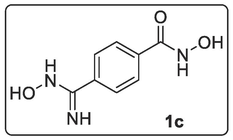
2.2.2.4 Synthesis of 4-Carbamimidoyl-N-hydroxybenzamide Hydrochloride, 1d
Potassium formate was prepared in situ from HCOOH (10.00 mmol, 0.38 mL) and K2CO3 (5 mmol, 0.69 g) in MeOH (1.50 mL). The parent amidoxime N-(benzyloxy)-4-(N-hydroxycarbamimidoyl)benzamide, 1b (1.00 mmol, 0.29 g) was dissolved in acetic acid (1 mL) and acetic anhydride (1.10 mmol, 0.104 mL) was added at room temperature (r.t.). After 5 min, potassium formate solution in MeOH was added followed by 10 % Pd/C (0.15 g). The mixture was stirred at r.t. until reaction was completed (1 h) based on TLC (DCM/MeOH → 90:10). The solids were filtered, washed with MeOH, and the filtrate was evaporated to get crude solid residue (1.83 g). The residue was dissolved in anhydrous EtOH (50 mL) and 5 M HCl in anhydrous EtOH (2 mL/24 mL, 12 eq) was then added and the stirring continues for about 10–15 min. The solids were filtered, washed with anhydrous EtOH and the filtrate was evaporated to afford the pure white-coloured solid amidine hydrochloride, 4-carbamimidoyl-N-hydroxybenzamide, 1d (0.19 g). Melting Point: 252–253 °C. 1H NMR (400 MHz, DMSO‑d6) δ: 9.55 (s, 1H, NH), 9.41 (s, 1H, NH), 7.92–7.90 (d, J = 5.60 Hz, 2H, Ar—H), 7.89–7.88 (d, J = 5.60 Hz, 2H, Ar—H). 13C NMR (100 MHz, DMSO‑d6) δ: 165.8 (C⚌O), 128.8 (2 × CH arom.), 128.7 (2 × C), 128.4 (2 × CH), 127.8 (C⚌N) ppm. MS-ESI, m/z (rel. %): 332.82 (MH + KCl + HCl + H2O + Na, 13.7 %), 316.87 (MH + KCl + HCl + H2O + Na – NH2, 6.9 %), 234.90 (M + 2HCl – H2O, 21.1 %), 220.18 (M + 2HCl – NHOH, 10.5 %), 193.10 (M–N, 16.8 %), 180.11 (M + 1, 56.8 %), 164.11 (M–NH, 100 %), 147.05 (M–NHOH, 26.3 %), 136.9 (M–NH2CN, 61.0 %), 120.96 (M–NHOHCN, 5.3 %). Anal. % Calcd. for C8H9N3O2.HCl + KCl (290.19): C, 33.10; H, 3.47; N, 14.48; Cl 24.43. Found: C, 36.37; H, 4.09; N, 14.22; Cl 25.45.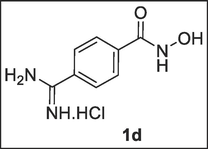
2.2.2.5 Synthesis of 1-(Benzyloxy)-3-(4-cyanophenyl)thiourea, 2a
Toluene (10 mL) was added to 4-cyanophenyl isothiocyanate (3.00 mmol, 0.48 g) and stirred at rt until total dissolution was observed. Then, o-benzylhydroxyamine (3.00 mmol, 0.19 mL) was added dropwise and after a while (7 min), plenty of white precipitate appeared. It stirred at room temperature for 4 h. The precipitate was collected by suction filtration, washed with more toluene and dried to afford yellow-coloured solid 1-(benzyloxy)-3-(4-cyanophenyl)thiourea, 2a (0.75 g, 88.23 %). 1H NMR (400 MHz, DMSO‑d6) δ: 11.28 (s, 1H, NH), 9.90 (s, 1H, NH), 7.78–7.76 (d, J = 8.92 Hz, 2H, Ar—H), 7.72–7.70 (d, J = 8.92 Hz, 2H, Ar—H), 7.46–7.45 (dd, J1 = 1.76 Hz, J2 = 9.36 Hz, 1H, Ar—H), 7.44–7.43 (dd, J1 = 1.32 Hz, J2 = 9.36 Hz, 1H, Ar—H), 7.35–7.29 (m, 3H, Ar—H), 4.88 (s, 2H, CH2—Ar). 13C NMR (100 MHz, DMSO‑d6) δ: 143.9 (C⚌S), 134.8, 132.7 (C≡N), 129.8, 128.7, 128.6 (3 × CH aromatic), 127.1 (CH aromatic), 126.9 (3 × CH aromatic), 116.3 (2 × CH aromatic), 63.4 (CH2) ppm. MS-ESI: in m/z (rel. %): 589.03 (2 M + Na, 19.6 %), 567.12 (2 M + 1, 17.4 %), 305.96 (M + Na, 23.9 %), 283.97 (M+, 100 %), 124.10 (4.3 %), 102.15 (Ph-CN, 47.8 %). Anal. % Calcd. for C15H13N3S (283.34): C, 63.58; H, 4.62; N, 14.83. Found: C, 63.79; H, 4.72; N, 15.11.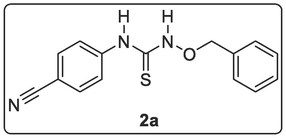
2.2.2.6 Synthesis of 4-(3-(Benzyloxy)thioureido)-N-hydroxybenzimidamide, 2b
A solution of 1-(benzyloxy)-3-(4-cyanophenyl)thiourea, 2a (1.00 mmol, 0.28 g), hydroxylamine hydrochloride, NH2OH.HCl (1.35 mmol, 0.09 g) and Na2CO3 (1.35 mmol, 0.14 g) in EtOH (30 mL) was heated under reflux for 7 h. Additional amount of NH2OH.HCl (1.35 mmol, 0.09 g) and Na2CO3 (1.35 mmol, 0.14 g) were added and the mixture was stirred under reflux for additional 10 h. The resulting solution was allowed to cool, filtered by suction and it was evaporated to dryness and later vacuum-dried to give a solid product (0.30 g) which was purified by using the dichloromethane/methanol (90:10) as eluting solvent to afford pure yellow-coloured solid 4-(3-(benzyloxy)thioureido)-N-hydroxybenzimidamide, 2b (0.11 g, 38.87 %). 1H NMR (400 MHz, DMSO‑d6) δ: 11.17 (s, 1H, NH), 9.01 (s, 1H, OH), 7.63–7.60 (d, J = 8.92 Hz, 2H, Ar—H), 7.55–7.53 (d, J = 8.92 Hz, 2H, Ar—H), 7.37–7.33 (m, 2H, Ar—H), 7.28–7.27 (d, J = 4.4 Hz, 2H, Ar—H), 7.21–7.17 (m, 1H, Ar—H), 6.06 (s, 2H, NH2), 5.11–5.09 (t, J = 5.60 Hz, 1H, CHa of CH2-Ph), 4.46–4.45 (d, J = 5.60 Hz, 1H, CHb of CH2-Ph), 1.14 (s, 1H, SH).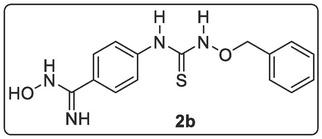
2.2.2.7 Synthesis of N-(Benzyloxy)-4-nitrobenzamide, 3a
4-Nitrobenzoyl chloride (1.00 mmol, 0.19 g, 1 eq) was dissolved in CH2Cl2 DCM (9.00 mL) and cool to 0 °C in an ice bath. A mixture of o-benzylhydroxy amine NH2-O-Bzl (1.50 mmol, 0.095 mL, 1.50 eq.) and triethylamine TEA (1.5 mmol, 0.21 mL, 1.50 eq.) was added to 9.00 mL of DCM in a dropping funnel and mixed thoroughly. The content in the dropping funnel was drop-wisely added to the continuously stirred solution of acid chloride under ice bath in such a way that the temperature was maintained below 5 °C (ca 15 mins.). The stirring continues under ice bath for additional 15 mins. after which the content was warmed up to room temperature and stirred there for 6 h (TLC monitored; DCM/MeOH → 99:1). The resulting solution was quenched, transferred into separatory funnel, washed with 1 M HCl (15.00 mL), followed by saturated NaHCO3 (15 mL) and finally with distilled water (20.00 mL). Organic layer was dried over anhydrous MgSO4, evaporated to get a solid which was vacuum-dried to a crude compound which was purified by column chromatography (DCM/MeOH → 99:1) to afford brown-coloured solid N-(benzyloxy)-4-nitrobenzamide, 3a (0.26 g, 96.30 %). 1H NMR (400 MHz, DMSO‑d6) δ: 12.04 (s-br, 1H, NH), 8.29–8.26 (d, J = 8.64 Hz, 2H, Ar—H), 7.95–7.93 (d, J = 8.64 Hz, 2H, Ar—H), 7.44–7.42 (d, J = 6.96 Hz, 2H, Ar—H), 7.39–7.32 (m, 3H, Ar—H), 4.93 (s, 2H, CH2—Ar). MS-ESI: in m/z (rel. %): 545.11 (2 M + 1, 10.0 %), 290.00 (M + H2O, 52.0 %), 273.03 (M + 1, 40.0 %), 223.17 (8.0 %), 102.13 (100 %).
2.2.2.8 Synthesis of 4-Aminobenzenecarbohydroxamic acid, 3b
Palladium on activated carbon (10 %, 37 mg) was added to a mixture of N-(benzyloxy)-4-nitrobenzamide, 3a (0.73 mmol, 0.20 g) and ammonium formate (3.67 mmol, 0.23 g, 5 eq.) in anhydrous methanol (20 mL). The mixture was heated under reflux under a streaming flow of argon at 60–70 °C for 3 h until all the starting material was totally consumed. The mixture was cooled and filtered to give a filtrate which was evaporated to dryness under reduced pressure while the product was purified by column chromatography (DCM/MeOH → 90:1) to afford brown-coloured solid 4-aminobenzenecarbohydroxamic acid 3b (0.21 g, 88.87 %). Melting Point: 121–122 °C.1H NMR (400 MHz, DMSO‑d6) δ: 8.38 (s, 1H, NH), 7.55–7.53 (d, J = 8.26 Hz, 1H, NH), 7.44–7.42 (d, J = 9.26 Hz, 2H, Ar—H), 7.28–7.26 (d, J = 8.26 Hz, 1H, NH), 6.50–6.47 (d, J = 9.26 Hz, 2H, Ar—H), 5.55 (s-br, 1H, OH). 13C NMR (100 MHz, DMSO‑d6) δ: 166.1 (C⚌O), 152.0, 129.6, 128.8, 119.8, 113.1 (3 × C aromatic) ppm. MS-ESI: in m/z (rel. %): 243.11 (M + CH2Ph, 100 %), 193.11 (28.6 %), 153.09 (M + 1, 60.7 %), 137.1 (M–NH, 50.0 %), 120.1 (M–NHOH, 85.7 %), 102.2 (69.6 %), 91.1 (C6H4NH, 42.9 %).
2.3 Molecular docking studies of selected synthesized compounds
The P. falciparum 6-pyruvoyltetrahydropterin synthase (PfPTPS) crystal structure with the PDB code 1Y13 was used for the docking studies. The protein structure was prepared by the removal of non-amino acid residues (the complexed native binder, biopterin) and water molecules, computation of Gasteiger charges, addition of polar hydrogens and merging of the nonpolar hydrogens using AutoDockTools 1.5.6 (Trott and Olson, 2009). The 2-dimensional (2D) structures of the synthesized compounds, biopterin ligand and pyrimethamine standard, were built using ChemDraw Professional 15.0 by PerkinElmer USA (Cousins, 2005). Pyrimethamine was employed as the standard for the docking studies because it is a known antifolate antimalarial drug (Pathak et al., 2017). Also, ChemDraw was used to generate the simplified molecular-input line-entry system (SMILES) that were converted to their corresponding 3D structures using FRee Online druG 3D conformation generator (FROG) (Leite et al., 2007) and saved in.mol2 format. In addition, PyRx software was used to minimize the structures and the.mol2 files were converted to the AutoDock docking formats (.pdbqt), which were further used for the docking simulation. The grid box was constructed using 35, 35, and 35, pointing in x, y, and z directions, respectively, with a grid point spacing of 0.375 Å. The center grid box was of 73.255 Å, 2.524 Å and 23.775 Å around the binding pocket of biopterin. The molecular docking analysis was performed carried out using Autodock vina and the post docking analysis was carried out using Discovery Studio.
2.4 Biological screening
2.4.1 In vitro antiplasmodial screening using P. falciparum NF54 strain
In vitro activity against erythrocytic stages of P. falciparum was determined using a 3H-hypoxanthine incorporation assay using the drug sensitive NF54 strain and the standard drugs chloroquine (Sigma C6628). Compounds were dissolved in DMSO at 10 mg/ml and further diluted in medium before they were added to parasite cultures incubated in RPMI 1640 medium without hypoxanthine, supplemented with HEPES (5.94 g/L), NaHCO3 (2.1 g/L), neomycin (100 U/mL), AlbumaxR (5 g/L) and washed human red cells A+ at 2.5 % haematocrit (0.3 % parasitaemia). Serial drug dilutions of seven 2-fold dilution steps covering a range from 10 to 0.02 μg/ml were prepared. The 96-well plates were incubated in a humidified atmosphere at 37 °C; 4 % CO2, 3 % O2, 93 % N2. After 48 h, 50 μL of 3H-hypoxanthine (=0.5 μCi) was added to each well of the plate. The plates were incubated for a further 24 h under the same conditions. They were then harvested with a Betaplate™ cell harvester (Wallac, Zurich, Switzerland), and the red blood cells transferred onto a glass fibre filter then washed with distilled water. The dried filters were inserted into a plastic foil with 10 mL of scintillation fluid and counted in a Betaplate™ liquid scintillation counter (Wallac, Zurich, Switzerland). IC50 values were calculated from sigmoidal inhibition curves by linear regression using Microsoft Excel.
2.4.2 In vitro cytotoxicity with L-6 cells
Assays were performed in 96-well microtiter plates, each well containing 100 μL of RPMI 1640 medium supplemented with 1 % l-glutamine (200 mM) and 10 % fetal bovine serum, and 4000 L-6 cells (a primary cell line derived from rat skeletal myoblasts). Serial drug dilutions of eleven 3-fold dilution steps covering a range from 100 to 0.002 μg/ml were prepared. After 70 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterile conditions. 10 μL of Alamar Blue was then added to each well and the plates incubated for another 2 h. Later they were read with a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wavelength of 536 nm and an emission wave length of 588 nm. The IC50 values were calculated by linear regression (Huber and Koella, 1993) from the sigmoidal dose inhibition curves using SoftmaxPro software (Molecular Devices Cooperation, Sunnyvale, CA, USA). Podophyllotoxin (Sigma P4405) as control.
2.4.3 In vivo studies
2.4.3.1 Ethical approval
Ethical clearance was obtained from the Covenant Health Research Ethics Committee (CHREC) with the ethical code NHREC/TR/ 02/06/2007a. Guidelines from the National Code for Health Research Ethics (NCHRE) was employed in all experiments as required.
2.4.3.2 Acute toxicity
The toxicity of compound 3b was determined using 6–8 week old male and female Swiss albino mice according to the Organization for Economic Cooperation and Development (OECD) standard (2001). The compound was dissolved in 10 % DMSO, 10 % Tween 80 and 80 % sterile distilled water. The mice were weighed and categorized into six groups. The compound was administered orally in a single dose to five groups (1–5) each at varying concentrations of 100, 200, 400, 2000, and 5000 mg/kg (body weight) respectively, and a sixth group of negative control given the vehicle. The vehicle was prepared in a solution of 10 % DMSO, 10 % Tween 80, and 80 % sterile distilled water. Physical signs of intoxication were recorded for the first thirty minutes post-dosage, and consistently for a period of 14 d. Certain signs were looked out for, such as behavior, heart rate, tremors, salivation, sleep, coma, injury, diarrhea, and mortality. For groups one to three (100, 200, and 400 mg/kg) weights were obtained 14 d post-treatment, while weights for 2000 and 5000 mg/kg were obtained 7 days post-treatment but monitored for 7 more days afterward. The median lethal dose (LD50) was determined using Lorke’s method as described by (1983). It was calculated as the geometric mean of least lethal dose (A) and the highest dose with no lethality (B) with the formula LD50 = √A × B. Weights of the mice were obtained on the first and final day of evaluation for each group. Histological examination was conducted on the kidney and liver of the test animals.
2.4.3.3 Suppressive studies
This study employed the Peter’s four-day suppressive method (Peters, 1965). Male and female Swiss albino mice weighing 14–31 g were inoculated with Plasmodium berghei NK 65 strain (obtained from the Nigerian Institute of Medical Research, Yaba) via oral administration. The mice were randomly divided into five groups, including three treatment groups of test compounds dissolved in the vehicle at varying concentrations (100 mg/kg, 200 mg/kg, and 400 mg/kg), and two control groups, the positive (chloroquine) and negative (placebo) groups. The stock concentration of the chloroquine used was 10 mg/ml and the dosage concentration by body weight was 100 mg/kg body weight. The negative control consisted of the vehicle minus any drug. Treatment was orally administered as a single dose daily for 5 days (Days 0, 1, 2, 3, and 4). On day 0, treatment was administered 2 h post-inoculation. Blood samples were collected from mice's tail veins on the fifth and final day of treatment administration (Day 4). Blood smears were made (both thick and thin films) and stained using a 3 % Giemsa solution. Microscopy was carried out with the oil immersion 100× magnification power on the thick and thin smears for parasitemia examination (Gebrehiwot et al., 2019). Parasitemia was determined by counting the number of parasitized red blood cells out of at least 200 red blood cells in each field.
2.4.3.4 Mean Survival Time (MST)
Mice mortality was monitored and recorded for a period of thirty days from day 0 of inoculation (Orabueze et al., 2020). Mice from the treatment groups that outlive the untreated mice postinoculation are considered as efficacious (Mulaw et al., 2019).
2.4.3.5 Statistical analysis
A one-way analysis of variance (ANOVA) followed by Tukey’s Post Hoc test at a 95 % confidence level (Cl) using GraphPad prism 9 was used to determine how statistically significant the difference is between the treatment and control groups for the antiplasmodial studies. The difference was considered statistically significant for p values ≤0.05. The paired t-test was used to determine the statistical significance between the groups during the acute toxicity studies with a 95 % CI.
3 Results and discussion
3.1 Chemistry
The pathways to achieve the synthesis of the proposed compounds (1c, 1d, 2b, 3b) from the structure-based drug design are shown in Schemes 1-3.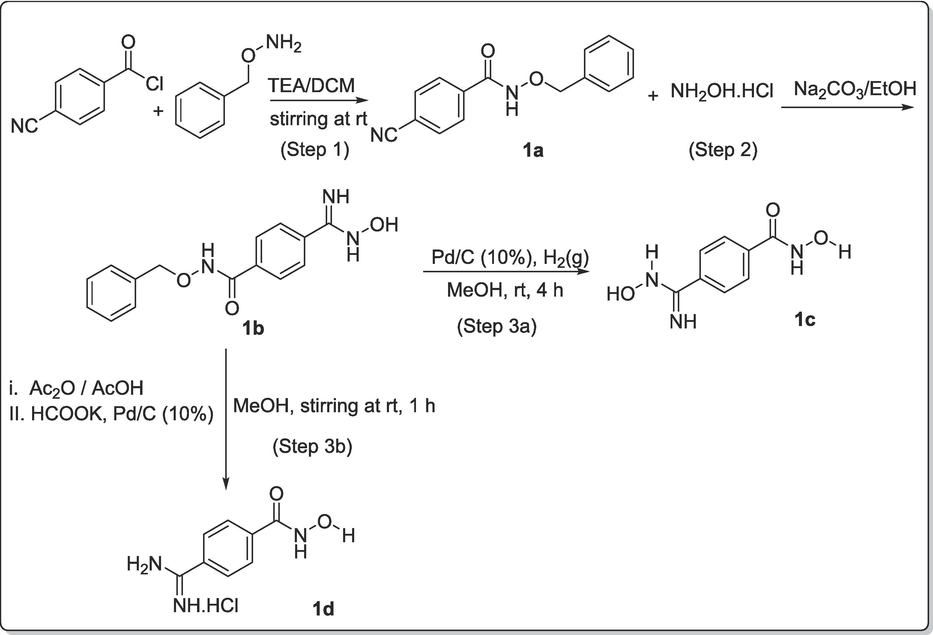
Synthetic pathway for achieving the designed amidinyl- compound (1c) and amidoximyl- compound (1d).
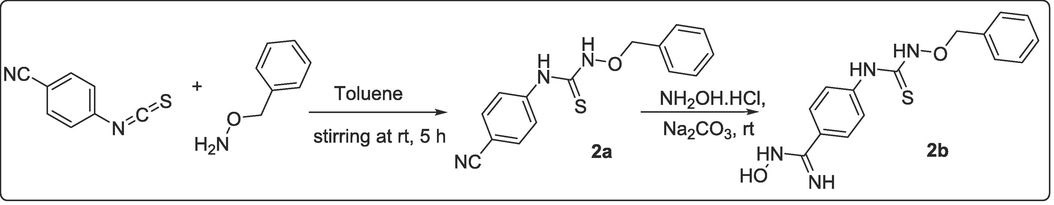
Synthetic pathway for achieving the designed amidoximyl- analog, 2b.
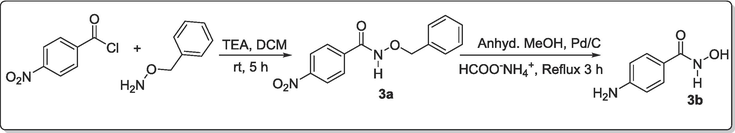
Synthetic pathway for achieving the designed amidoximyl- analogs, 3b.
The designed compounds from the SBDD were taken further for synthesis. The amidine and amidoxime groups were incorporated in the final products (1c, 1d, 2b, 3b) because of the ability of these two groups to act as good lone pair donors, and increase their efficiency as zinc binding group (Xu and Pang, 2010). Therefore, a suitable pathway was constructed for the synthesis of amidine 1c via three steps expeditious synthetic pathway. The overall view was as shown in Scheme 1. In detail, the first step involved the protection of the acid chloride (4-cyano benzoylchloride) by coupling such functionality with o-benzylhydroxyl amine (NH2-O-Bzl) using triethylamine base (TEA). This step afforded the corresponding N-benzyloxy-4-cyanobenzamide, 1a in excellent yield (77.97 %). In the second step, the cyano functional group on the para position of 1a was successfully transformed to produce amidoxime, 1b (Nikitas et al., 2021). This involved the use of Na2CO3/ EtOH and this afforded the product in excellent yield, 99.12 % (Scheme 1). In the third step, we employed two approaches to reduce the amidoxime to amidines (Scheme 1), one under catalytic hydrogenation in the presence of Pd/C (Step 3a of Scheme 1), the second by acylating the amidoximes and reducing them via potassium formate route as reported by Nadrah and Dolenc, 2007 (Nadrah and Dolenc, 2007) (Step 3b of Scheme 1).
The first trial was by bubbling hydrogen gas into a mixture of methanolic solution of amidoxime 1b and palladized charcoal (10 %) for 4 h. Although, there was a change in TLC indicating that the reaction had reached completion. However, this reaction step did not produce the amidine as envisaged, but rather hydroxamic acid 1c of the former amidoxime 1b. Despite the change in TLC, the product 1c obtained still has the amidoxime functionality intact as depicted by its spectroscopic features as well as the elemental analytical data. Thus, the change in TLC was as a result of the cleaving away of the good leaving group (benzylium ion) alone. Therefore, this method might require a longer reaction time to allow for the complete reduction of amidoxime as well. On the contrary, the reduction of amidoxime 1b with potassium formate in acetic acid proceeded quite impressively and resulted in the expected amidine 1d in quantitative yield within a short reaction time. This method has proved to be very efficient; supporting the fact that potassium formate is a more active donor of hydrogen than sodium and ammonium formate (Wiener et al., 1991). The kinetic of the reaction was enhanced by first acylating the amidoxime before subsequent potassium formate reduction (Wiener et al., 1991). The intermediate product 1a and 1b are thermally stable and their chemical properties could not be easily compromised.
Similarly, the method used for the preparation of 1a by coupling of acid chloride with o-benzyl hydroxylamine was applied to the coupling of the 4-cyano phenyl isothiocyanate with o-benzyl hydroxylamine using toluene as the solvent of choice through which an excellent yield was achieved for 2a (Scheme 2). This reaction was kinetically favored because within 5 min of stirring at rt, the cloudy precipitate started appearing. However, it was allowed to stir for 5 h in order to ensure total consumption of the precursor for maximum yield. Afterward, the synthetic manipulation of the cyano group (CN) on the para position of 2a to amidoxime was executed reaction of 2a and hydroxylamine hydrochloride in an ethanolic solution of sodium carbonate (Na2CO3) at rt for 20 h to afford the expected amidoxime of thiourea 2b (38.87 %).
Furthermore, the synthesis of N-(Benzyloxy)-4-nitrobenzamide, 3a was achieved by treatment of 4-nitrophenyl benzoyl chloride with o-benzyl hydroxylamine in the presence of triethylamine at room temperature stirring for 5 h (Scheme 3). The essence of o-hydroxybenzylamine was to also protect the carboxyl group as a more stable benzamide was formed. The second stage involved the synthetic conversion of the nitro compound 3a to the amine counterpart 4-aminobenzenecarbohydroxamic acid, 3b according to a known procedure (Tulevski et al., 2007). This was carried out by refluxing a mixture of 3a and HCOONH4 on Pd/C in the presence of anhydrous methanol for 3 h. This was successfully achieved but did not only produce the amine but also led to the cleaving away of benzylium ion to afford hydroxamic acid 3b (Adebiyi et al., 2021) (Scheme 3).
3.2 Molecular docking
The redocking of the co-ligand of PfPTPS (biopterin) was carried out in its binding pocket to ascertain the accuracy of the docking protocol. It was observed that the redocked biopterin has a similar conformation and occupies a similar position with the ligand binding pocket as compared to the bound co-crystallized biopterin from the original structure. The redocked ligand also formed hydrogen bond interactions in the binding pocket of the protein structure at Glu A:128, Thr A:127, Glu A:161, Glu A:38, His A:43 similar to that of the bound ligand (Fig. 3). The molecular docking studies of the final products (1c, 1d, 2b and 3b) and the standard pyrimethamine in the binding pocket of PfPTPS showed that their binding energies ranged between −6.6 and −5.6 kcal/mol (Table 1). The order of the binding energies is: 1c = Biopterin > 1d > 3b > Pyrimethamine > 2b. The native ligand, biopterin had a binding affinity of −5.6 kcal/mol and pyrimethamine −5.9 kcal/mol. Compound 2b possessed the best binding affinity of −6.6 kcal/mol and predicted Ki of 13.95 µM, better than the native ligand and standard pyrimethamine. The post-docking analyses showed the interactions of the atoms of the compounds in the binding pocket of the protein target. The oxygen of the hydroxyl group of the hydroxamic acid on compound 3b was observed to interact with the Zn2+ present in the protein binding pocket via a metal-acceptor interaction (Fig. 4). This interaction is key in the inhibition of PfPTPS due to the role zinc ion play in the stability of the native ligand, biopterin in the protein binding pocket as observed in Fig. 3A. Also, residue Glu161 is highly conserved at the active site similar to the zinc ion (Khairallah et al., 2020). The interaction of compound 3b with the Glu161 via a conventional hydrogen bond shows that it is a good fit of the docking model. Hence, compound 3b is a good potential inhibitor of the protein, that can be considered as a lead for future antimalarial discovery.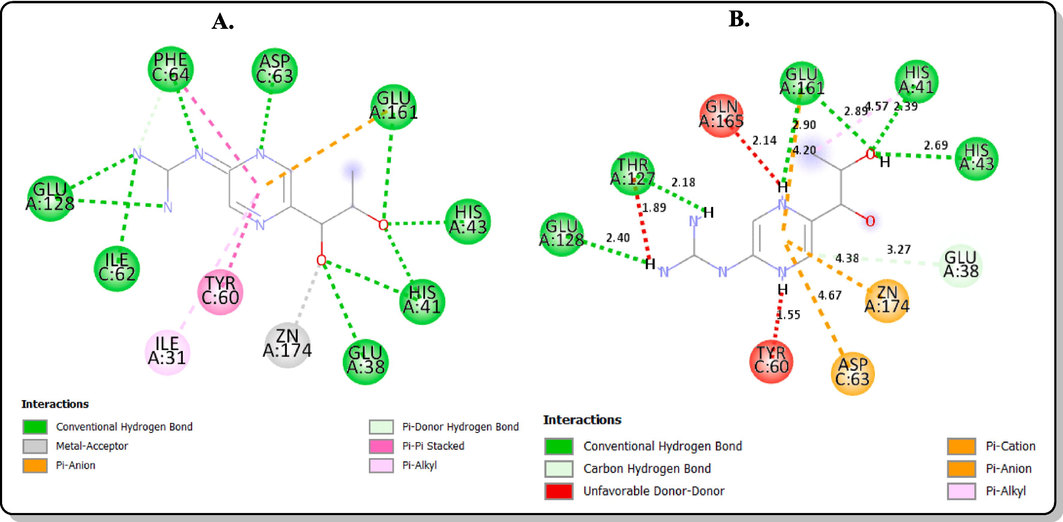
2D interactions with the amino acid residues in the binding site of the protein target (PDB ID: 1Y13) (A) co-crystallized biopterin (B) redocked biopterin.
Cpd ID
Structures
Binding energy (kcal/mol)
Predicted Ki (µM)
Binding Interactions
1c
−5.6
75.90
Conventional hydrogen bond: GluA:128 (3.07 Å), ThrA:127 (1.92 Å), GluA:161 (2.34 Å), GluA:38 (2.09 Å)
Pi-Cation/ Pi-Anion: GluA:38 (3.00 Å), HisA:41 (4.67 Å)
1d
−5.7
64.07
Conventional hydrogen bond: GluA:128 (2.46 Å), ThrA:127 (2.22 Å), AspC:63 (4.39 Å)
Pi-Anion: GluA:161 (4.24 Å), GluA:128 (4.92 Å)
Pi-Pi stacked: PheC:64 (5.41 Å), TyrC:60 (4.95 Å)
Pi-Alkyl: IleA:31 (5.41 Å)
2b
−6.6
13.95
Conventional hydrogen bond: CysA:27 (2.95 Å), HisA:80 (2.20 Å)
Carbon hydrogen bond: AspA:116 (3.59 Å)
Pi-Alkyl: LeuA:40 (5.37 Å), AlaA:28 (3.58 Å)
3b

−5.9
45.66
Conventional hydrogen bond: GluA:128 (2.29 Å), TyrC:60 (2.73 Å), GluA:161 (2.16 Å)
Metal-Acceptor: ZnA:174 (2.21 Å)
Pi-Anion: GluA:161 (4.96 Å)
Pi-Pi stacked: TyrC:60 (3.94 Å), PheC:64 (4.21 Å)
Biopterin
−5.6
75.90
Conventional hydrogen bond: GluA:128 (2.40 Å), ThrA:127 (2.18 Å), GluA:161 (2.90 Å), His A:41 (2.39 Å), HisA:43 (2.69 Å)
Carbon hydrogen bond: GluA:38 (3.27 Å)
Pi-Cation/ Pi-Anion: ZnA:174 (4.38 Å), AspC:63 (4.67 Å)
Unfavourable Donor–Donor: GlnA:165 (2.14 Å), TyrC:60 (1.55 Å)
Pyrimethamine
−5.9
45.66
Conventional hydrogen bond: GluA:38 (2.49 Å), TyrC:60 (2.68 Å)
Pi-Anion: GluA:161 (4.65 Å), AspC:63 (3.97 Å)
Pi-Pi stacked: TyrC:60 (4.73 Å), PheC:64 (5.33 Å)
Alkyl/Pi-alkyl: TyrC:60 (4.79 Å), PheC:64 (5.19 Å), IleA:31(5.33 Å), LysC:68 (5.24 Å)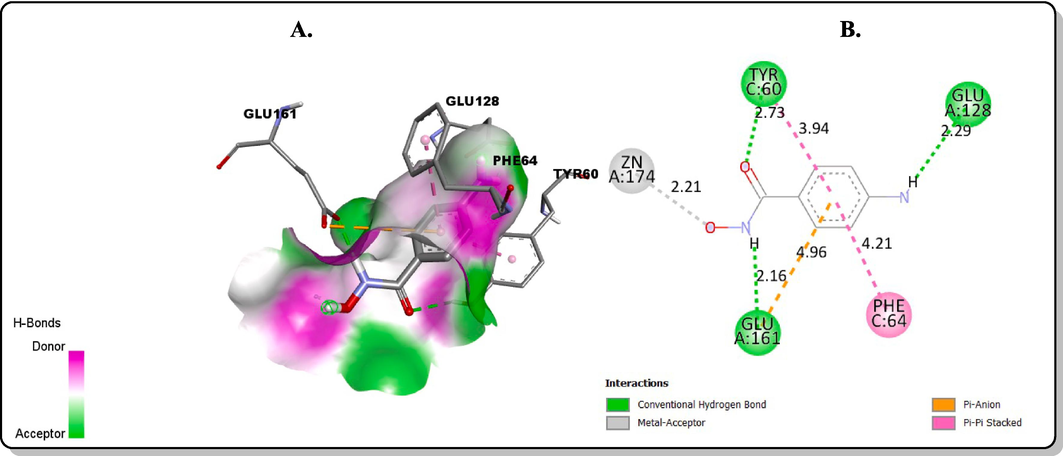
Molecular docking interactions between 3b and the binding sites of PfPTPS: (A) 3D model (B) 2D model.
3.3 Antimalarial activity
3.3.1 In vitro antimalarial screening and cytotoxicity
Antiplasmodial sensitivity and cytotoxicity testing for 1c, 1d, 2b, and 3b were carried out against the PfNF54 using chloroquine and Podophyllotoxin as standard controls respectively. Antimalarial and cytotoxicity were represented as the inhibitory concentration (IC50) for each compound (Table 2).
Compounds
Cytotoxicity (L6) IC50 (μM)
Antiplasmodial (Pf) IC50 (μM)
Selectivity Index
1c
10.950
0.405
27.04
1d
19.645
0.199
98.72
2b
18.239
2.404
7.59
3b
6.603
0.147
44.92
Chloroquine
66.138
0.002
33069.00
Podophyllotoxin
0.002
–
–
Compound 3b demonstrated the most noteworthy antiplasmodial activity (IC50 = 0.147 μM) among the tested compounds, closely resembling chloroquine (IC50 = 0.002 μM) against the P. falciparum NF 54 strain. When comparing the IC50 value of 3b with those of cycloguanil (0.0045 μM) and pyrimethamine (0.017 μM) against the same P. falciparum NF 54 strain (Miglianico et al., 2023), significant similarities were observed. The compounds appear to be selective for the parasite and do not seem to show general toxicity against a mammalian cell line. Also, compound 3b appears to be 45x more active against the parasite in terms of selectivity. Considering SAR properties, 1c and 1d were structurally related and differed only in the functional moieties at the para position of their phenyl ring. Compound 1c was amidoxime-based while 1d was amidine-based at their para positions with 1d being more than two-fold active than 1c. This indicates that the conversion of amidoxime to amidine is a valid route to obtaining antimalarial active compounds, at least in this present study. The availability of a lone-pair donor amino group in 1d made amidine a valuable pharmacophore.
The cytotoxic activities of the compounds on L-6 cells also showed moderate toxicity compared to the reference drug podophyllotoxin (IC50 = 0.002 μM). The cytotoxicity order was 1d < 2b < 1c < 3b, most compounds showed toxicity of IC50 ≥ 10 μM except 3b (IC50 = 6.603 μM). The compounds showed a selective inhibition to the parasite. Hence, these bioactive motifs are potential drug candidates for antimalarial drug investigation.
3.3.2 In vivo screening
3.3.2.1 Acute toxicity
All mice displayed a stretching relaxed position after administration for the first thirty minutes even though active if perturbed by agitating conditions. No other behavioral change was observed as a sign of intoxication in the mice after administration throughout the study. The LD50 was determined to be ≥5000 mg/kg. Mice given the highest dosage concentration was also monitored and found to be healthy after a month of administration. It was also observed that with an increase in the concentration of compound, mice gained additional weight (Fig. 5) which may be due to increase in food intake influenced by the compound, whereas the control didn’t, given the same environmental and feeding conditions (Table 3). *DC = Dosage concentrations, D = Day, No = Number, bdw = body weight.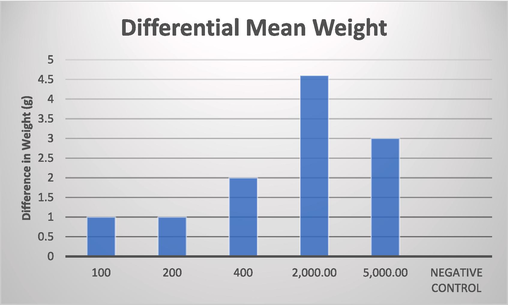
Graph describing the increase in weight based on the concentration.
Groups
No of mice
DC (mg/kg) bdw
Mortality D0
Mortality D1
Mortality D5
Mortality D7
Mortality D14
% Mortality
Lorke' s Phase 1
Group 1
4
100
0
0
1
0
0
25
Group 2
4
200
0
0
0
0
0
0
Group 3
4
400
0
1
0
0
0
25
Group 4
4
−ve Control
0
0
0
0
0
0
Lorke' s Phase 2
Group 5
4
2,000
0
0
0
0
0
0
Group 6
1
5,000
0
0
0
0
0
0
3.3.2.2 Histology
Kidney tubular degeneration was observed at 100 mg/kg dosage of 3b comparable to the control group which showed no lesion (Fig. 6). Renal or kidney tubular degeneration may arise due to several etiologies which could be perturbations that repair themselves as may be in this case. This moderate degeneration did not lead to an injury (Suttie, 2018). Renal congestion arises from increased pressure at the central vein of the kidney, at moderate level, there’s no impairment in the kidney (Komuro et al., 2019). Severe congestion and edema as observed at 400 mg/kg concentration of 3b is a clinical expression of a failing circulation resulting from the presence of accumulated influxes of sodium ion (Na+) in the kidney which could be treated with diuretics (Kuriyama, 2019). At 2000 mg/kg, there were no lesions, therefore, it may be that the volume of dosage between both concentrations might have been the source of the congestion at 400 mg/kg. The control group showed no lesion. In the liver, at 100 mg/kg, 200 mg/kg and 400 mg/kg (Fig. 7), mild to moderate vacuolar degeneration of hepatocytes were observed but did not lead to necrosis which indicates the possibility of regeneration of affected hepatocytes would repair themselves over time (Wolf and Wheeler, 2018). In the control group, the moderate periportal cellular infiltration did not lead to any form of injury.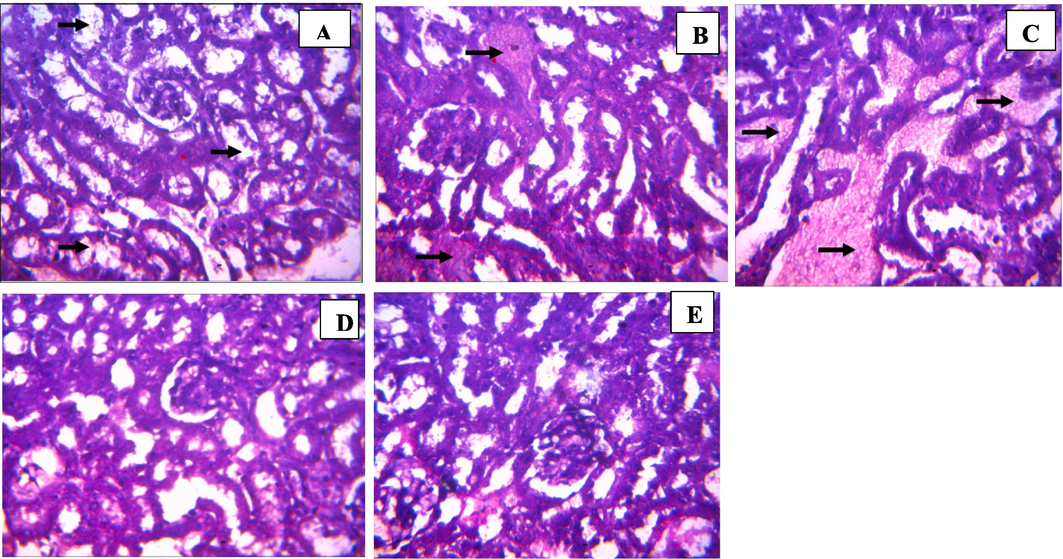
Histology of kidney sections from test animals treated with benzamide compound (9b): (A) 100 mg/kg, (B) 200 mg/kg, (C) 400 mg/kg, (D) 2000 mg/kg, (E) Control (Magnification ×400).
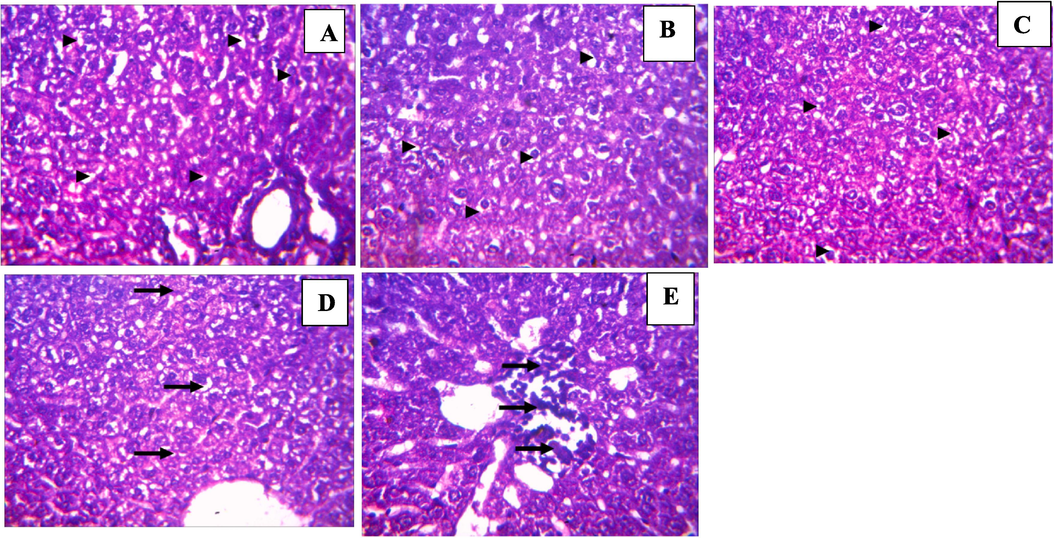
Histology of liver sections from test animals treated with benzamide compound (9b): (A) 100 mg/kg, (B) 200 mg/kg, (C) 400 mg/kg, (D) 2000 mg/kg, (E) Control (Magnification ×400).
3.3.2.3 Peter’s 4-day suppressive test
Significant reduced parasitemia was observed in a dose-dependent fashion, p ≤ 0.0001 (Table 4). Highest chemosuppressive activity (80.53 %) was observed among the mice group that was administered 400 mg/kg of the compound, and MST of > 30 days amongst the three concentrations. This was comparable to the standard drug which exhibited a chemosuppressive activity of 81.71 % and MST of 30 (+) days. The three treatment groups were statistically significant at p values ≤0.0001 compared to the negative control. Data are represented as mean ± SEM. SEM = Standard Error of Mean. (+) = animals survived and didn’t die.
Group
Concentration (mg/kg)
% Parasitemia
% Suppression ± SEM
MST (days)
1
100
6.25 ± 0.95
51.99 ± 10.38
24 ± 1.00
2
200
2.95 ± 1.95
78.55 ± 13.35
29.5 ± 2.25
3
400
2.6 ± 0.6
80.53 ± 3.26
30 (+)
4
Negative Control
13.205 ± 0.875
0
16
5
Chloroquine
2.4 ± 0.08
81.71 ± 1.82
30 (+)
Compound 3b showed no toxicity at the highest concentration limit (5000 mg/kg). Fig. 5 described the weight-gain pattern observed in the mice groups during the acute toxicity study. Recent studies have suggested that compounds with hydroxamate moieties have histone deacetylase (HDACs) inhibitory properties which offer therapeutic advantages in antiplasmodial drug development. HDACs are very important in the regulation of post-translational modification in mammals. The HDAC isoforms in P. falciparum are reported to be vulnerable to these inhibitors except for a clause of non-selectivity between the human and parasite HDACs (Vreese et al., 2017). Suggestions were offered by Vreese et al., (2017) for the modification of HDAC inhibitors which selectively bind to the Plasmodium isoforms other than the human HDACs. The absence of P. berghei in the mice during the acute toxicity experiments may have allowed the compound to inhibit mice HDACs and thereby increasing histone acetylation leading to cell-cycle progression and therefore influences the rapid increase in weight and improvement of mice health. Mahmoud (2022), reported that certain epigenetic factors influence increase in obesity especially histone alterations as may be the case in this study (Mahmoud, 2022). Compounds with ≥50 % suppression are of consideration as at least moderate for antiplasmodial activity. All mice in group 3 which were administered with 400 mg/kg, were treated with no mortality even after the experiment, while group 1 and 2 (200 mg/kg) stayed alive till the 24th and 29th day post-inoculation (Okokon et al., 2022). The outcome of this experiment suggests that this compound is both safe and efficacious in the treatment of rodent malaria.
4 Conclusion
All living organisms require folate cofactors essential to their DNA synthesis. P. falciparum is capable of both de novo folate biosynthesis and salvage of pre-formed folate from the plasma of its human host. Studies show that both pathways are essential to the success of parasite development and the inhibition of key steps in folate metabolism has been the basis of combinatorial advances (such as pyrimethamine and sulfadoxine) against malaria. However, the use of some of the combinatorial drugs have an increased resistance rate. This study addresses the need for new antifolates and novel therapeutic options to combat malaria, considering the emergence and persistence of antimalarial drug resistance. The design and synthesis of potential inhibitors targeting PfPTPS, a protein target in the de novo folate biosynthesis pathway, were successfully accomplished using a structure-based approach. The designed compounds, including amidinyl, amidoximyl and hydroxamic acid analogs (1c, 1d, 2b, and 3b) were synthesized successfully using different synthetic pathways. All of the compounds demonstrated favorable binding affinities to the active site of PfPTPS with 4-(3-(benzyloxy)thioureido)-N-hydroxybenzimidamide 2b being the strongest binder (-6.6 kcal/mol), followed by 4-aminobenzenecarbohydroxamic acid 3b (-5.9 kcal/mol). 4-Carbamimidoyl-N-hydroxybenzamide hydrochloride 1d and compound 3b exhibited significant efficacy with IC50 values of 0.199 μM and 0.147 μM, respectively, indicating their potential as potent antimalarial agents. Moreover, cytotoxicity testing demonstrated that all the compounds showed moderate toxicity compared to the reference drug podophyllotoxin (IC50 = 0.002 μM), further supporting their suitability for drug development. In vivo studies using Swiss albino mice confirmed the safety and efficacy of compound 3b in treating rodent malaria, providing additional evidence of its therapeutic potential. The identified lead compounds from this study hold promise for further exploration and development as alternative antifolate drugs to address the ongoing challenge of malaria. Overall, these findings contribute to the search for new strategies to combat malaria, offering new avenues for the development of effective antimalarial drugs that can overcome drug resistance, and reduce the burden of this global health concern.
Funding
This work was supported by the Fogarty National Institutes of Health [grant number: 1U2RTW010679].
Acknowledgements
First synthesis was performed while OOA worked with Dr. Serghei Glinca, Prof. Gerhard Klebe, Dr. Regina Ortmann, Dr. Armin Reichenberg, and Prof. Martin Schlitzer at the Institute for Pharmaceutical Chemistry, Marburg University using an Institutional grant from Covenant University and DAAD. EA and a part of his group are housed at the IPMB, Heidelberg University and worked on part of this work while been sponsored by the AvH, 2014–2016. The PhD project of DVA was funded by Fogarty NIH, grant number 1U2RTW010679 and she performed the 2nd and 3rd syntheses working with KDK, Dr. Joe Lewis and Dr. David Will at EMBL, Heidelberg, Germany. GOO also worked on the synthesis and carried out the computational studies to predict the binding orientation of the compounds. The preclinical in vitro testing was performed as a service by Prof. Maeser and co-workers of Swiss Tropical and Public Health Institute (TPH), Switzerland. GPA and GIO worked on the in vivo biological screening at Covenant University. EA would also like to thank Dr. Aubry Miller of the Cancer Drug Development group at the DKFZ for assessment of the syntheses and bioactivity results obtained at critical stages and his useful comments.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Computer-Aided Drug Design for the Organic Chemistry Laboratory Using Accessible Molecular Modeling Tools. J. Chem. Educ.. 2020;97:760-763.
- [CrossRef] [Google Scholar]
- In silico screening of phytochemicals from Dissotis rotundifolia against Plasmodium falciparum Dihydrofolate Reductase. Phytomedicine Plus. 2023;3:100447
- [CrossRef] [Google Scholar]
- Structure-based scoring of anthocyanins and molecular modeling of PfLDH, PfDHODH, and PfDHFR reveal novel potential P. falciparum inhibitors. Informatics Med. Unlocked. 2023;38:101206
- [CrossRef] [Google Scholar]
- A structure-based drug discovery paradigm. Int. J. Mol. Sci.. 2019;20:2783.
- [CrossRef] [Google Scholar]
- Editorial: In silico Methods for Drug Design and Discovery. Front. Chem.. 2020;8:1-5.
- [CrossRef] [Google Scholar]
- Development of Stimulator of Interferon Genes Agonists in Silico. J. Phys. Conf. Ser.. 2021;1893
- [CrossRef] [Google Scholar]
- ChemDraw Ultra 9.0. CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. See Web site for pricing options. J. Am. Chem. Soc.. 2005;127:4115-4116.
- [CrossRef] [Google Scholar]
- Dasgupta, T., Chitnumsub, P., Kamchonwongpaisan, S., Maneeruttanarungroj, C., Nichols, S.E., Lyons, T.M., Jorgensen, W.L., Yuthavong, Y., Anderson, K.S., 2009. Exploiting Structural Analysis, in Silico Screening, and Serendipity To Identify Novel Inhibitors of Drug-Resistant Falciparum Malaria Articles Exploiting Structural Analysis, in Silico Screening, and Serendipity To Identify Novel Inhibitors of Drug-R. https://doi.org/10.1021/cb8002804.
- Estimating novel potential drug targets of Plasmodium falciparum by analysing the metabolic network of knock-out strains in silico. Infect. Genet. Evol.. 2009;9:351-358.
- [CrossRef] [Google Scholar]
- Gebrehiwot, S., Shumbahri, M., Eyado, A., Yohannes, T., 2019. Phytochemical-Screening-and-in-Vivo-Antimalarial-Activity-of-Two-Traditionally-Used-Medicinal-Plants-of-Afar-Region-Ethiopia-against-Plasmodium-berghei-in-Swiss-Albino-MiceJournal-of-Parasitology-Research.pdf.
- Artemisinin resistance and malaria elimination: Where are we now? Front. Pharmacol.. 2022;13:1-10.
- [CrossRef] [Google Scholar]
- A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop.. 1993;55:257-261.
- [CrossRef] [Google Scholar]
- Plasmodium falciparum: a paradigm for alternative folate biosynthesis in diverse microorganisms? Trends Parasitol.. 2008;24:502-508.
- [CrossRef] [Google Scholar]
- In silico molecular studies of selected compounds as novel inhibitors for phosphodiesterase-5 (PDE5) in the management of erectile dysfunction. J. Comput. Sci. Syst. Biol.. 2020;13:2.
- [CrossRef] [Google Scholar]
- Probing the Structural Dynamics of the Plasmodium falciparum Tunneling-Fold Enzyme 6-Pyruvoyl Tetrahydropterin Synthase to Reveal Allosteric Drug Targeting Sites. Front. Mol. Biosci.. 2020;7:1-17.
- [CrossRef] [Google Scholar]
- Demonstration of Improved Renal Congestion After Heart Failure Treatment on Renal Perfusion Imaging With Contrast-Enhanced Ultrasonography. Circ. Rep.. 2019;1:593-600.
- [CrossRef] [Google Scholar]
- A Potential Mechanism of Cardio-Renal Protection with Sodium-Glucose Cotransporter 2 Inhibitors: Amelioration of Renal Congestion. Kidney Blood Press. Res.. 2019;44:449-456.
- [CrossRef] [Google Scholar]
- Frog: A FRee Online druG 3D conformation generator. Nucleic Acids Res.. 2007;35:568-572.
- [CrossRef] [Google Scholar]
- Evaluation of an Innovative Point-of-Care Rapid Diagnostic Test for the Identification of Imported Malaria Parasites in China. Trop. Med. Infect. Dis. Artic.. 2023;8:1-10.
- [Google Scholar]
- An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions. Int. J. Mol. Sci.. 2022;23
- [CrossRef] [Google Scholar]
- Assessment of the drugability of initial malaria infection through miniaturized sporozoite assays and high-throughput screening. Commun. Biol.. 2023;6:1-10.
- [CrossRef] [Google Scholar]
- Quebrachitol from Putranjiva roxburghii Wall. (Putranjivaceae) a potent antimalarial: Pre-clinical efficacy and its interaction with PfLDH. Parasitol. Int.. 2023;92:102675
- [CrossRef] [Google Scholar]
- Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem.. 2017;125:1300-1320.
- [CrossRef] [Google Scholar]
- Computer-aided drug design of β-secretase, γ- secretase and anti-tau inhibitors for the discovery of novel alzheimer’s therapeutics. Int. J. Mol. Sci.. 2020;21
- [CrossRef] [Google Scholar]
- Evaluation of Antimalarial Activity of the 80% Methanolic Stem Bark Extract of Combretum molle Against Plasmodium berghei in Mice. J. Evid.-Based Integr. Med.. 2019;24:1-9.
- [CrossRef] [Google Scholar]
- Preparation of amidines by amidoxime reduction with potassium formate. Synlett 2007:1257-1258.
- [CrossRef] [Google Scholar]
- Photochemical Activation of Aromatic Aldehydes: Synthesis of Amides, Hydroxamic Acids and Esters. Chem. - A Eur. J.. 2021;27:7915-7922.
- [CrossRef] [Google Scholar]
- Structure-Based Drug Design in Discovering Target Specific Drugs against Tropical Journal of Natural Product Research Structure-Based Drug Design in Discovering Target Specific Drugs against. Trop. J. Nat. Prod. Res.. 2021;5:739-743.
- [CrossRef] [Google Scholar]
- In vitro and in vivo antimalarial activity and chemical profiling of sugarcane leaves. Sci. Rep.. 2022;12:1-13.
- [CrossRef] [Google Scholar]
- Antimalarial potentials of Stemonocoleus micranthus Harms (leguminoseae) stem bark in Plasmodium berghei infected mice. J. Tradit. Complement. Med.. 2020;10:70-78.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of antimalarial activity of new derivatives of 2,4,6-s-triazine. Chem. Cent. J.. 2017;11:1-11.
- [CrossRef] [Google Scholar]
- Drug Resistance in Multiple Drug Vincke Resistance and Lips, The development of a strain of Plasmodium berghei resistant to chloroquine (RC strain) and another resistant to the triazine metabo- lite of proguanil (B strain) was described in the first. Exp. Parasitol.. 1965;102:97-102.
- [Google Scholar]
- Identification of promising inhibitors for Plasmodium haemoglobinase Falcipain-2, using virtual screening, molecular docking, and MD Simulation. J. Mol. Struct.. 2022;1248:131427
- [CrossRef] [Google Scholar]
- Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem.. 2021;224:113705
- [CrossRef] [Google Scholar]
- Suttie, A.W., 2018. Boorman’s Pathology of the Rat. Elsevier. https://doi.org/10.1016/C2010-0-69040-7.
- AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem.. 2009;31:455-461.
- [CrossRef] [Google Scholar]
- Chemically assisted directed assembly of carbon nanotubes for the fabrication of large-scale device arrays. J. Am. Chem. Soc.. 2007;129:11964-11968.
- [CrossRef] [Google Scholar]
- Recent developments in antimalarial drug discovery. Bioorg. Med. Chem. 2023:88-89.
- [CrossRef] [Google Scholar]
- Exploration of thiaheterocyclic hHDAC6 inhibitors as potential antiplasmodial agents. Chimia (aarau).. 2017;9:357-364.
- [CrossRef] [Google Scholar]
- WHO, 2022. World malaria report 2022.
- Transfer Hydrogenolysis of Aryl Halides and Other Hydrogen Acceptors by Formate Salts in the Presence of Pd/C Catalyst. J. Org. Chem.. 1991;56:6145-6148.
- [CrossRef] [Google Scholar]
- A critical review of histopathological findings associated with endocrine and non-endocrine hepatic toxicity in fish models. Aquat. Toxicol.. 2018;197:60-78.
- [CrossRef] [Google Scholar]
- Improving the hit-to-lead process: Data-driven assessment of drug-like and lead-like screening hits. Drug Discov. Today. 2006;11:175-180.
- [CrossRef] [Google Scholar]
- Zinc binding-induced near-IR emission from excited-state intramolecular proton transfer of a bis(benzoxazole) derivative. Chem. Commun.. 2010;46:4070-4072.
- [CrossRef] [Google Scholar]




