

Translate this page into:
Synthesis and antibacterial activity of 1-N-(β-d-glucopyranosyl)-4-((1-substituted-1H-1,2,3-triazol-4-yl)ethoxymethyl)-1,2,3-triazoles
⁎Corresponding author. Tel.: +964 7906487332, +964 61422117596. adnanimchem@yahoo.com (Adnan Ibrahim Mohammed)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.

Abstract
Abstract
Four gluco-triazole conjugates were synthesized employing Cu(I) catalyzed click reactions between peracetylated glucosyl azide and alkyl-(propynoxy)ethyl-triazoles which in turn were prepared under similar reaction conditions using the appropriate alkyl azide (C7, C8, C10, and C12) and but-3-yn-1-ol. Removal of the acetyl protecting groups afforded additional four compounds. All eight conjugates were assessed toward inhibition of bacterial growth (Escherichia coli, Staphylococcus aureus). Slight growth inhibition compared to kanamycin was observed at a concentration of 20 μg/mL, while no growth inhibition was observed at the lowest concentration (5 μg/mL).
Keywords
Sugar 1,2,3-triazoles
Dipolar cycloaddition
Triazolyl alcohols
Alkynyl 1,2,3-triazoles
Click chemistry

1 Introduction
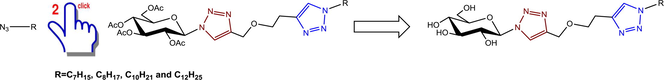
Cu(I) catalyzed cycloaddition reactions are called click reactions (Hein and Fokin, 2010). Forming substituted triazole system has created a wide range of applications (Binder and Sachsenhofer, 2007; Kolb and Sharpless, 2003; Evans, 2007; Francis et al., 2011; Nenajdenko et al., 2010; Rathwell et al., 2010; Wan et al., 2006; Whiting et al., 2006). As it has been shown in many works, significant sugar based 1,2,3-triazole derivatives have been synthesized (Mohammed et al., 2012; Jwad, 2011; Ali et al., 2009). As a result, a number of 1,2,3-triazoles have been synthesized regarding click conditions using microwave irradiation and the biological activities of the prepared compounds were monitored against different types of microorganisms (Mohammed et al., 2010) and were observed for their important behavior toward bacteria and fungi. Recently, biologically interesting tetrakis-1,2,3-triazoles starting from d-mannitol utilizing the copper catalyzed ‘click’ protocol were synthesized and the antibacterial activity was measured in vitro (Mohammed et al., 2013), in addition; these triazole derivatives showed a significant antibacterial activity. Many other examples showed the importance of the synthesized 1,2,3-triazole derivatives as antimicrobial agents (Dharshan et al., 2012; Gautam and Chourasia, 2012). 1,2,3-Triazoles containing quinoline moiety presented a broad spectrum of antifungal activity (Sumangala et al., 2010). Moreover, a series of synthesized 1,2,3-triazolphanes exhibited both antifungal and antibacterial activities (Lal et al., 2013). Adding to this, glycal-derived 1,2,3-triazoles appeared to resemble antimicrobial activity (Reddy et al., 2010). This paper describes the synthesis of new ether-linked bis-1,2,3-triazole derivatives based on d-glucose and the measurement of the antibacterial activity of these derivatives in vitro against the Escherichia coli (−) and Staphylococcus aureus (+). (See Fig. 1 and Scheme 1.)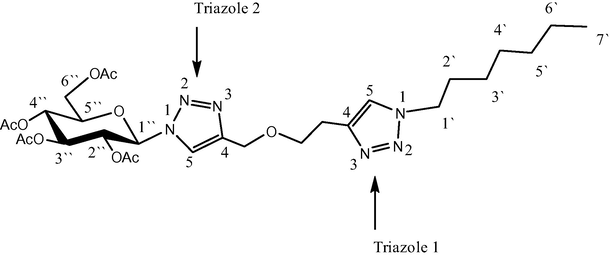
Numbering of compound (4a).
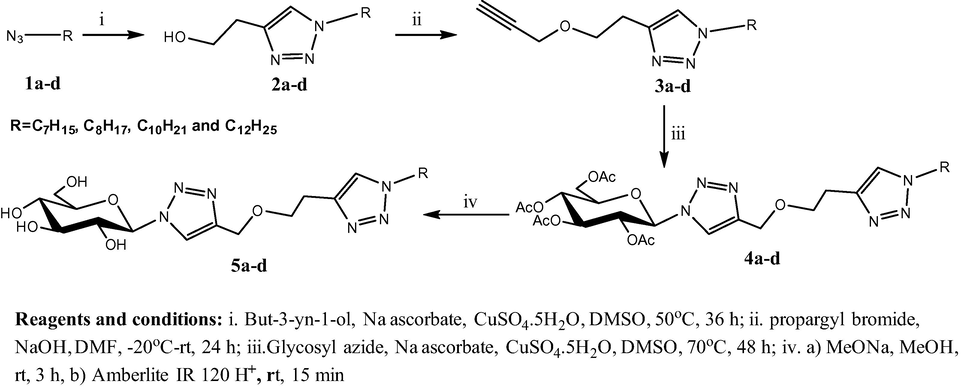
The synthetic route of d-glucose based bis-1,2,3-triazoles.
2 Experimental section
2.1 General experimental information
All chemicals and solvents were obtained from commercial sources and were used without further purification. Optical rotations were determined at the sodium D line at 25 °C. Infrared spectra were recorded using SHIMADZU 2001 FT-IR. Routine 1H and 13C NMR spectra were obtained on a Bruker spectrometer DPX 300 (300 MHz for 1H NMR and 75 MHz for 13C NMR, respectively) or on a Bruker Avance III 400 spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR, respectively). Chemical shifts were recorded in parts per million (ppm) relative to solvent nuclei as an internal reference. HRMS spectra were recorded on a Thermo LTQ FT mass spectrometer. Purification of the crude products by column chromatography was performed on silica gel 60 (230–400 mesh, 0.040–0.063 mm). Silica TLC plates were used with an aluminum backing (0.2 mm, 60 F254). The reactions were monitored by TLC and visualized by the development of the TLC plates with an alkaline potassium permanganate dip. The antibacterial activity was determined using the agar well diffusion method.
2.2 Synthesis
2.2.1 General procedure for synthesis of 1,2,3-triazol-4-yl alcohols (2a–d)
A solution of but-3-yn-1-ol (7.0 mmol) in DMSO (3 mL) was added dropwise to the suspension of sodium ascorbate (0.13 g, 0.63 mmol) and CuSO4.5H2O (0.077 g, 0.35 mmol) in DMSO (2 mL). The mixture was stirred for few minutes and to this was added the suitable n-alkyl azide (7.0 mmol) then it was stirred at 50 °C for 36 h. The reaction mixture was diluted with distilled water (70 mL), extracted with EtOAc (3 × 60 mL), the combined organic layers were washed with sat. NaCl (2 × 20 mL), dried over Na2SO4, and evaporated to dryness under reduced pressure. The residue was flash chromatographed (silica gel, 3:1 to 1:2 n-hexane: Et2O) to yield the desired 1,2,3-triazol-4-yl alcohols 2a–d.
2.2.1.1 2-(1-Heptyl-1H-1,2,3,-triazol-4-yl)ethanol (2a)
White solid (1.36 g, 92%), mp 50–52 °C; (CAS number 1249705–13-2), Rf = 0.17 (n-hexane/EtOAc 1:1), IR (KBr): 3433, 3257, 3134, 3080, 2954, 2926, 2852, 1462, 1373, 1215, 1145, 1058, 1020, 831, 729, 675 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.84 (t, J 6.8 Hz, 3H, H7′), 1.26 (m, 8H, H3′-H6′), 1.85 (quin, J 7.4 Hz, 2H, H2′), 2.91 (t, J 6.0 Hz, 2H, C4triazole—CH2—CH2—OH), 3.13 (broad s, 1H, —OH), 3.90 (t, J 6.0 Hz, C4triazole—CH2—CH2—OH), 4.28 (t, J 7.3 Hz, 2H, H1′), 7.37 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.1, 22.6, 26.5, 28.7, 28.8, 30.4, 31.6, 50.4, 61.6, 121.5, 145.5; HRMS (ESI) calcd for C11H22N3O 212.1756 [M + H]+, found 212.1757.
2.2.1.2 2-(1-Octyl-1H-1,2,3,-triazol-4-yl)ethanol (2b)
White solid (1.48 g, 94%), mp 53–54 °C (waxy solid, Nulwala et al., 2009); Rf = 0.16 (n-hexane/EtOAc 1:1), IR (KBr): 3261, 3138, 3084, 2951, 2924, 2854, 1458, 1371, 1217, 1145, 1056, 1022, 829, 732, 678 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.86 (t, J 6.7 Hz, 3H, H8′), 1.27 (m, 10H, H3′—H7′), 1.87 (quin, J 7.4 Hz, 2H, H2′), 2.67 (broad s, 1H, —OH), 2.94 (t, J 5.8 Hz, 2H, C4triazole—CH2—CH2—OH), 3.93 (t, J 5.9 Hz, C4triazole—CH2—CH2—OH), 4.30 (t, J 7.3 Hz, 2H, H1′), 7.37 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 22.7, 26.6, 28.8, 29.1, 29.14, 30.4, 31.8, 50.4, 61.8, 121.5, 145.6; HRMS (ESI) calcd for C12H23N3ONa 248.1733 [M + Na]+, found 248.1731.
2.2.1.3 2-(1-Decyl-1H-1,2,3,-triazol-4-yl)ethanol (2c)
White solid (1.60 g, 90%), mp 56–58 °C; (CAS number 1457635–78-7) Rf = 0.15 (n-hexane/EtOAc 1:1), IR (KBr): 3258, 3138, 3084, 2949, 2922, 2850, 1458, 1371, 1215, 1145, 1066, 1020, 833, 729, 678 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.85 (t, J 6.8 Hz, 3H, H10′), 1.29 (m, 14H, H3′—H9′), 1.86 (quin, J 7.4 Hz, 2H, H2′), 2.92 (t, J 6.0 Hz, 2H, C4triazole—CH2—CH2—OH), 3.00 (broad s, 1H, —OH), 3.91 (t, J 6.0 Hz, C4triazole —CH2—CH2—OH), 4.28 (t, J 7.3 Hz, 2H, H1′), 7.37 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 22.7, 26.6, 28.8, 29.1, 29.3, 29.5, 29.53, 30.4, 31.9, 50.4, 61.7, 121.5, 145.5; HRMS (ESI) calcd for C14H27N3ONa 276.2046 [M + Na]+, found 276.2044.
2.2.1.4 2-(1-Dodecyl-1H-1,2,3,-triazol-4-yl)ethanol (2d)
White solid (1.73 g, 88%), mp 67–69 °C (67–68 °C, Sabbah et al., 2012); Rf = 0.15 (n-hexane/EtOAc 1:1), IR (KBr): 3257, 3136, 3066, 2920, 2850, 1458, 1371, 1215, 1145, 1058, 1022, 833, 727, 678 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.85 (t, J 6.7 Hz, 3H, H12′), 1.27 (m, 18H, H3′—H11′), 1.87 (quin, J 7.2 Hz, 2H, H2′), 2.92 (t, J 6.0 Hz, 2H, C4triazole—CH2—CH2—OH), 3.01 (broad s, 1H, —OH), 3.91 (t, J 6.0 Hz, C4triazole—CH2—CH2—OH), 4.28 (t, J 7.3 Hz, 2H, H1′), 7.37 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 22.8, 26.6, 28.8, 29.1, 29.4, 29.5, 29.6, 29.67, 29.7, 30.4, 32.0, 50.4, 61.7, 121.5, 145.5; HRMS (ESI) calcd for C16H31N3ONa 304.2359 [M + Na]+, found 304.2358.
2.2.2 General procedure for synthesis of alkynyl triazoles (3a–d)
Triazolyl alcohol 2 (4 mmol) was dissolved in DMF (15 mL) and NaOH pellets (0.6 g, 15 mmol) were added. The mixture was cooled in ice-salt bath to −20 °C and the contents were stirred vigorously for 10 min and then propargyl bromide (0.4 mL, 4.3 mmol) was added drop wise. The heterogeneous reaction mixture was stirred vigorously for 24 h, slowly warming to room temperature. The mixture was filtered and H2O (50 mL) was added and the product was extracted with Et2O (4 × 50 mL). The organic phases were combined and washed sequentially with 5% HCl (2 × 50 mL) and distilled water (50 mL). The organic phase was dried over Na2SO4, filtered and the solvent was evaporated to dryness under reduced pressure to yield a residue which was flash chromatographed (silica gel, 9:1 to 1:1 n-hexane: Et2O) to afford the alkynyl triazoles 3a–d.
2.2.2.1 1-Heptyl-4-(2-(prop-2-yn-1-yloxy)ethyl)-1H-1,2,3-triazole (3a)
Pale yellow oil (0.86 g, 86%); Rf = 0.25 (n-hexane/EtOAc 1:1), IR (neat): 3226, 3082, 2954, 2929, 2860, 2113, 1456, 1361, 1217, 1101, 1053, 950, 912, 802, 715, 667 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.81 (t, J 6.8 z 3H, H7′), 1.24 (m, 8H, H3′—H6′), 1.83 (quin, J 7.2 Hz, 2H, H2′), 2.38 (t, J 2.4 Hz, 1H, O—CH2—C≡CH), 2.97 (t, J 6.5 Hz, 2H, C4triazole—CH2—CH2—O), 3.75 (t, J 6.5 HZ, 2H, C4triazole —CH2—CH2—O), 4.11 (d, J 2.4 Hz, 2H, O—CH2—C≡CH), 4.25 (t, J 7.2 Hz, 2H, H1′), 7.36 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.0, 22.5, 26.4, 26.43, 28.7, 30.3, 31.6, 50.2, 58.1, 68.9, 74.5, 79.7, 121.6, 144.8; HRMS (ESI) calcd for C14H23N3ONa 272.1733 [M + Na]+, found 272.1730.
2.2.2.2 1-Octyl-4-(2-(prop-2-yn-1-yloxy)ethyl)-1H-1,2,3-triazole (3b)
Pale yellow oil (0.90 g, 85%); Rf = 0.25 (n-hexane/EtOAc 1:1), IR (neat): 3292, 3078, 2956, 2922, 2850, 2115, 1462, 1369, 1145, 1107, 1058, 952, 912, 825, 725, 671 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.87 (t, J 6.8 z 3H, H8′), 1.27 (m, 10H, H3′—H7′), 1.88 (quin, J 7.3 Hz, 2H, H2′), 2.42 (t, J 2.4 Hz, 1H, O—CH2—C≡CH), 3.02 (t, J 6.5 Hz, 2H, C4triazole —CH2—CH2—O), 3.80 (t, J 6.5 HZ, 2H, C4triazole—CH2—CH2—O), 4.20 (d, J 2.4 Hz, 2H, O—CH2—C≡CH), 4.30 (t, J 7.2 Hz, 2H, H1′), 7.39 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 22.7, 26.5, 26.6, 29.1, 29.2, 30.5, 31.8, 50.4, 58.3, 69.1, 74.6, 79.8, 121.7, 145.0; HRMS (ESI) calcd for C15H25N3ONa 282.1889 [M + Na]+, found 282.1885.
2.2.2.3 1-Decyl-4-(2-(prop-2-yn-1-yloxy)ethyl)-1H-1,2,3-triazole (3c)
Waxy solid (0.99 g, 85%), mp 40–42 °C; Rf = 0.24 (n-hexane/EtOAc 1:1), IR (KBr): 3292, 3076, 2922, 2854, 2115, 1552, 1458, 1365, 1145, 1217, 1105, 1043, 952, 910, 825, 725, 669, 634 cm−1. 1H NMR (300 MHz, CDCl3) δ ppm: 0.86 (t, J 6.5 z 3H, H10′), 1.29 (m, 14H, H3′—H9′), 1.87 (broad quin, J 7.1 Hz, 2H, H2′), 2.41 (t, J 2.5 Hz, 1H, O—CH2—C≡CH), 3.02 (t, J 6.5 Hz, 2H, C4triazole—CH2—CH2—O), 3.80 (t, J 6.5 HZ, 2H, C4triazole—CH2—CH2—O), 4.16 (d, J 2.4 Hz, 2H, O—CH2—C≡CH), 4.29 (t, J 7.2 Hz, 2H, H1′), 7.38 (s, 1H, H5) .13C NMR (75 MHz, CDCl3) δ ppm: 14.2, 22.8, 26.5, 26.6, 29.1, 29.4, 29.5, 29.6, 30.4, 32.0, 50.3, 58.3, 69.1, 74.6, 79.9, 121.6, 145.0; HRMS (ESI) calcd for C17H29N3ONa 314.2203 [M + Na]+, found 314.2198.
2.2.2.4 1-Dodecyl-4-(2-(prop-2-yn-1-yloxy)ethyl)-1H-1,2,3-triazole (3d)
Pale yellow oil (1.0 g, 80%), mp 47–49 °C; Rf = 0.22 (n-hexane/EtOAc 1:1), IR (KBr): 3292, 3078, 2954, 2922, 2850, 2115, 1554, 1462, 1365, 1215, 1145, 1107, 1060, 952, 912, 825, 725, 669, 632 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.86 (t, J 6.8 z 3H, H12′), 1.29 (m, 18H, H3′—H11′), 1.86 (broad quin, J 6.9 Hz, 2H, H2′), 2.41 (t, J 2.4 Hz, 1H, O—CH2—C≡CH), 3.01 (t, J 6.5 Hz, 2H, C4triazole—CH2—CH2—O), 3.80 (t, J 6.5 HZ, 2H, C4triazole—CH2—CH2—O), 4.20 (d, J 2.4 Hz, 2H, O—CH2—C≡CH), 4.29 (t, J 7.2 Hz, 2H, H1′), 7.38 (s, 1H, H5) .13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 22.8, 26.5, 26.6, 29.1, 29.4, 29.5, 29.6, 29.8, 30.5, 32.0, 50.4, 58.3, 69.1, 74.6, 79.8, 121.7, 145.0; HRMS (ESI) calcd for C19H33N3ONa 342.2516 [M + Na]+, found 342.2511.
2.2.3 General procedure for synthesis of bistriazoles (4a–d)
Alkynyl triazole 3 (1.0 mmol) and 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl azide were added to the suspension of sodium ascorbate (0.018 g, 0.09 mmol) and CuSO4.5H2O (0.011 g, 0.045 mmol) in DMSO (5 mL). The mixture was stirred at 70 °C for 48 h. After which, it was diluted with water (30 mL), extracted with EtOAc (3 × 30 mL) and the combined organic layers were washed with brine (2 × 20 mL), dried over Na2SO4 and evaporated to dryness under reduced pressure. The residue was flash chromatographed (silica gel, 3:1 to 1:2 EtOAc/n-hexane) to yield the desired compounds 4a–d.
2.2.3.1 1-N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-4-(((1-n-heptyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (4a)
White solid (0.50 g, 80%); mp 121–122 °C, Rf = 0.21 (n-hexane/EtOAc 1:2), [α]d = +15.4 (c 0.5, DCM), IR (KBr): 3120, 3088, 2958, 2929, 2860, 1741, 1556, 1458, 1373, 1226, 1219, 1107, 1041, 947, 852, 669, 605 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.84 (t, J 6.8 Hz, 3H, H7′), 1.25(m, 8H, H3′—H6′), 1.84, 2.01, 2.04, 2.05 (s,12H, —CH3 acetate), 1.87 (m, 2H, H2′), 3.00 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.75 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.99 (ddd, J 10.2, 5.0, 2.1 Hz, 1H, H5′′), 4.13 (dd, J 12.6, 2.1 Hz, 1H, Ha6″), 4.28 (t, J 7.4 Hz, 2H, H1′), 4.30 (dd, J 12.5, 2.7 Hz, 1H, Hb6′′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.22 (m, 1H, H4′′), 5.41 (m, 2H, H2′′, H3′′), 5.86 (m, 1H, H1′′), 7.39, 7.75 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, CDCl3) δ ppm: 14.1, 20.3, 20.6, 20.62, 20.8, 22.6, 26.5, 28.7, 30.4, 31.6, 50.3, 61.6, 64.2, 67.8, 69.6, 70.4, 72.7, 75.2, 85.8, 121.0, 121.7, 144.9, 145.9, 169.0, 169.4, 170.0, 170.6; HRMS (ESI) calcd for C28H42N6O10Na 645.2855 [M + Na]+, found 645.2847.
2.2.3.2 1-N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-4-(((1-n-octyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (4b)
White solid (0.52 g, 82%); mp 123–124 °C, Rf = 0.21 (n-hexane/EtOAc 1:3), [α]d = +20.1 (c 0.5, DCM), IR (KBr): 3120, 3084, 2926, 2858, 1740, 1554, 1444, 1371, 1224, 1105, 1041, 923, 846, 682, 603 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.85 (t, J 6.7 Hz, 3H, H8′), 1.25 (m, 10H, H3′—H7′), 1.86, 2.01, 2.05, 2.06 (s, 12H, —CH3 acetate), 1.87 (m, 2H, H2′), 3.00 (t, J 6.6 Hz, 2H, C4triazole1—CH2CH2—O), 3.76 (t, J 6.6 Hz, 2H, C4triazole1—CH2CH2—O), 3.99 (ddd, J 10.1, 5.0, 2.1 Hz, 1H, H5′′), 4.14 (dd, J 12.6, 2.1 Hz, 1H, Ha6′′), 4.27 (t, J 7.4 Hz, 2H, H1′), 4.31 (dd, J 12.5, 2.7 Hz, 1H, Hb6′′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.22 (m, 1H, H4′′), 5.40 (m, 2H, H2′′, H3′′), 5.87 (m, 1H, H1′′), 7.39, 7.75 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, CDCl3) δ ppm: 14.1, 20.3, 20.6, 20.62, 20.8, 22.7, 26.56, 26.6, 29.1, 29.12, 30.4, 31.8, 50.3, 61.6, 64.2, 67.8, 69.6, 70.4, 72.7, 75.2, 85.8, 121.0, 121.7, 144.9, 145.9, 169.0, 169.4, 170.0, 170.6; HRMS (ESI) calcd for C29H45N6O10 637.3192 [M + H]+, found 637.3187.
2.2.3.3 1-N-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-4-(((1-n-Decyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (4c)
White solid (0.55 g, 82%); mp 131–133 °C, Rf = 0.20 (n-hexane/EtOAc 1:2), [α]d = +27.7 (c 0.5, DCM), IR (KBr): 3473, 3120, 3076, 2926, 2858, 1741, 1554, 1456, 1375, 1217, 1105, 1041, 921, 846, 721, 675, 605 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.84 (t, J 6.7 Hz, 3H, H7′), 1.24 (m, 14H, H3′—H9′), 1.84, 2.00, 2.04, 2.06 (s,12H, —CH3 acetate), 1.87 (m, 2H, H2′), 3.00 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.76 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.99 (ddd, J 10.2, 5.0, 2.1 Hz, 1H, H5′′), 4.14 (dd, J 12.6, 2.1 Hz, 1H, Ha6′′), 4.27 (t, J 7.2 Hz, 2H, H1′), 4.30 (dd, J 12.5, 3.8 Hz, 1H, Hb6′′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.22 (m, 1H, H4′′), 5.41 (m, 2H, H2′′, H3′′), 5.86 (m, 1H, H1′′), 7.39, 7.75 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 20.3, 20.6, 20.63, 20.8, 22.7, 26.6, 26.61, 29.1, 29.3, 29.5, 29.6, 30.4, 31.9, 50.3, 61.6, 64.2, 67.8, 69.6, 70.4, 72.7, 75.2, 85.8, 121.0, 121.7, 144.9, 145.9, 169.0, 169.5, 170.0, 170.6; HRMS (ESI) calcd for C31H48N6O10Na 687.3324 [M + Na]+, found 687.3316.
2.2.3.4 1-N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-4-(((1-n-dodecyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (4d)
White solid (0.58 g, 84%); mp 134–135 °C, Rf = 0.19 (n-hexane/EtOAc 1:2), [α]d = +9.6 (c 0.5, DCM), IR (KBr): 3477, 3120, 3082, 2924, 2854, 1741, 1552, 1458, 1375, 1224, 1103, 1039, 921, 848, 723, 682, 601 cm−1. 1H NMR (400 MHz, CDCl3) δ ppm: 0.85 (t, J 6.7 Hz, 3H, H7′), 1.25 (m, 18H, H3′—H11′), 1.84, 2.01, 2.05, 2.06 (s,12H, —CH3 acetate), 1.85 (m, 2H, H2′), 3.00 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.76 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.99 (ddd, J 10.2, 5.0, 2.1 Hz, 1H, H5′′), 4.14 (dd, J 12.6, 2.1 Hz, 1H, Ha6′′), 4.27 (t, J 7.2 Hz, 2H, H1′), 4.30 (dd, J 12.5, 4.2 Hz, 1H, Hb6′′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.22 (m, 1H, H4′′), 5.41 (m, 2H, H2′′, H3′′), 5.86 (m, 1H, H1′′), 7.39, 7.75 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, CDCl3) δ ppm: 14.2, 20.3, 20.6, 20.63, 20.8, 22.8, 26.6, 26.61, 29.1, 29.4, 29.5, 29.6, 29.7, 30.4, 40.0, 50.3, 61.6, 64.2, 67.8, 69.6, 70.4, 72.7, 75.2, 85.8, 121.0, 121.7, 144.9, 145.9, 169.0, 169.5, 170.0, 170.6; HRMS (ESI) calcd for C33H52N6O10Na 715.3637 [M + Na]+, found 715.3628.
2.2.4 General procedure for deprotection of bistriazoles 4a–d. Synthesis of compounds (5a–d)
Methanolic NaOMe (0.20 mL, 0.1 mmol, 0.5 M) was added to a solution of protected bistriazoles 4a–d (0.5 mmol) in anhyd. MeOH (3 mL). The mixture was stirred for 3 h at rt then Amberlite IR 120 (H+) resin (0.5 g) was added. The mixture was allowed to stir until the neutralization occurred (15–20 min), the resin was filtered off. The filtrate was concentrated in vacuo to yellow syrup then the residue was dissolved in small amount of EtOH and triturated with n-pentane to afford the deprotected bistriazoles 5a–d.
2.2.4.1 1-N-(β-d-Glucopyranosyl)-4-(((1-n-heptyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (5a)
White solid (0.22 g, 95%); mp 139–141 °C, Rf = 0.19 (DCM/MeOH 9:1), [α]d = +39.2 (c 0.1, MeOH), IR (KBr): 3446, 3381, 2954, 2858, 1641, 1554, 1462, 1371, 1224, 1047, 900, 827, 765, 584 cm−1. 1H NMR (400 MHz, MeOH-d4) δ ppm: 0.89 (t, J 6.7 Hz, 3H, H7′), 1.32 (m, 8H, H3′—H6′), 1.88 (quin, J 7.3 Hz, 2H, H2′), 2.96 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.77 (t, J 6.6 Hz, 2H, C4triazole1—CH2CH2—O), 3.48–3.92 (m, 6H, H5′′, Ha6′′, Hb6′′, H4′′, H3′′, H2′′), 4.32 (t, J 7.2 Hz, 2H, H1′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.62 (d, J 9.2 Hz, 1H, H1′′), 7.76, 8.17 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, MeOH-d4) δ ppm: 14.4, 23.6, 27.1, 27.4, 29.7, 31.3, 32.8, 51.3, 62.4, 64.6, 70.2, 70.9, 74.0, 78.4, 81.1, 89.6, 124.0, 124.3, 145.9, 146.1; HRMS (ESI) calcd for C20H35N6O6 455.2612 [M + H]+, found 455.2612.
2.2.4.2 1-N-(β-d-Glucopyranosyl)-4-(((1-n-octyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (5b)
White solid (0.22 g, 93%); mp 141–143 °C, Rf = 0.18 (DCM/MeOH 9:1), [α]d = +44.5 (c 0.1, MeOH), IR (KBr): 3441, 3356, 3142, 2926, 2854, 1641, 1552, 1462, 1373, 1222, 1099, 1049, 900, 827, 765, 732, 603 cm−1. 1H NMR (400 MHz, MeOH-d4) δ ppm: 0.89 (t, J 6.8 Hz, 3H, H8′), 1.32 (m, 10H, H3′-H7′), 1.87 (quin, J 7.4 Hz, 2H, H2′), 2.96 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.77 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.48–3.92 (m, 6H, H5′′, Ha6′′, Hb6′′, H4′′, H3′′, H2′′), 4.34 (t, J 7.2 Hz, 2H, H1′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.62 (d, J 9.2 Hz, 1H, H1′′), 7.76, 8.16 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, MeOH-d4) δ ppm: 14.4, 23.7, 27.1, 27.5, 30.0, 30.2, 31.3, 32.9, 51.3, 62.4, 64.6, 70.2, 70.9, 74.0, 78.4, 81.1, 89.6, 124.0, 124.3, 145.9, 146.1; HRMS (ESI) calcd for C21H37N6O6 469.2769 [M + H]+, found 469.2765.
2.2.4.3 1-N-(β-d-Glucopyranosyl)-4-(((1-n-decyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (5c)
White solid (0.24 g, 96%); mp 150–152 °C, Rf = 0.16 (DCM/MeOH 9:1), [α]d = +12.3 (c 0.1, MeOH), IR (KBr): 3414, 3383, 3142, 2926, 2855, 1637, 1554, 1462, 1371, 1224, 1099, 1049, 900, 825, 765, 721, 599 cm−1. 1H NMR (400 MHz, MeOH-d4) δ ppm: 0.90 (t, J 6.6 Hz, 3H, H10′), 1.32 (m, 14H, H3′—H9′), 1.87 (quin, J 7.3 Hz, 2H, H2′), 2.96 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.77 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.49–3.92 (m, 6H, H5′′, Ha6′′, Hb6′′, H4′′, H3′′, H2′′), 4.34 (t, J 7.2 Hz, 2H, H1′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.62 (d, J 9.2 Hz, 1H, H1′′), 7.76, 8.16 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, MeOH-d4) δ ppm: 14.5, 23.7, 27.1, 27.5, 30.1, 30.4, 30.55, 30.6, 31.3, 33.0, 51.3, 62.4, 64.6, 70.2, 70.8, 74.0, 78.4, 81.1, 89.6, 124.0, 124.3, 145.9, 146.0; HRMS (ESI) calcd for C23H41N6O6 497.3082 [M + H]+, found 497.3080.
2.2.4.4 1-N-(β-d-Glucopyranosyl)-4-(((1-n-dodecyl-1H-1,2,3-triazol-4-yl)ethoxy)methyl)-1H-1,2,3-triazole (5d)
White solid (0.24 g, 91%); mp 155–156 °C, Rf = 0.15 (DCM/MeOH 9:1), [α]d = +19.8 (c 0.1, MeOH), IR (KBr): 3506, 3387, 3134, 2922, 2854, 1556, 1463, 1373, 1321, 1228, 1103, 1043, 900, 833, 721, 621, 588 cm−1. 1H NMR (400 MHz, MeOH-d4) δ ppm: 0.90 (t, J 6.6 Hz, 3H, H12′), 1.32 (m, 18H, H3′—H11′), 1.87 (quin, J 7.3 Hz, 2H, H2′), 2.96 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.77 (t, J 6.5 Hz, 2H, C4triazole1—CH2CH2—O), 3.48–3.92 (m, 6H, H5′′, Ha6′′, Hb6′′, H4′′, H3′′, H2′′), 4.34 (t, J 7.2 Hz, 2H, H1′), 4.63 (s, 2H, C4triazole2—CH2—O), 5.62 (d, J 9.2 Hz, 1H, H1′′), 7.75, 8.16 (s, 2H, H5triazole1, H5triazole2). 13C NMR (100 MHz, MeOH-d4) δ ppm: 14.5, 23.7, 27.1, 27.5, 30.1, 30.5, 30.6, 30.7, 30.74, 31.3, 33.1, 51.3, 62.4, 64.6, 70.2, 70.9, 74.0, 78.4, 81.1, 89.6, 124.0, 124.3, 145.9, 146.1; HRMS (ESI) calcd for C25H45N6O6 525.3395 [M + H]+, found 525.3393.
2.3 Biological screening (Jayanna et al., 2012)
The antibacterial efficacy of the synthesized compounds was tested against Gram positive bacteria S. aureus and Gram negative bacteria E. coli by the agar well diffusion method.
Twenty four old Muller–Hinton broth cultures of test bacteria were swabbed on sterile Muller–Hinton agar plates using sterile cotton swab followed by punching wells of 6 mm with the help of sterile cork borer. 20 mg/mL in 10% DMSO and control (10% DMSO) were added to respectively labeled wells. The plates are allowed to stand for 30 min and were incubated at 37 °C for 24 h in upright position and the zone of inhibition was recorded.
3 Results and discussion
3.1 Synthesis
Alkyl azides 1a–d were converted to the corresponding triazolyl alcohols 2a–d in approximately excellent yields via click protocol. Proton NMR spectra of compounds 2a–d showed a clear signal at 7.37 ppm which attributed to the H5 of the triazole ring. In addition, the signals at 121.5 ppm C5 and 146.5 ppm C4 in 13C NMR spectra provided an excellent evidence for triazoles formation. The following scheme describes the overall synthetic route of the targeted compounds:
The subsequent step was Williamson etherification of compounds 2a–d using propargyl bromide and NaOH as heterogeneous catalysts. Compounds 3a–d were isolated in very good yields 80–86% and the formation of alkynyl triazoles was investigated by the appearance of the triplet acetylenic signals between 2.38 and 2.42 ppm and the doublet between 4.11 and 4.20 ppm that attributed to the methylene group O—CH2—C≡CH. Furthermore, three important signals that appeared in the 13C NMR spectra of compounds 3a–d supported the formation of the alkynyl triazoles. The first one appeared around 50.3 ppm referred to the methylene carbon O—CH2—C≡CH. The second signal at 74.5 ppm assigned the terminal acetylenic carbon while the third signal allocated the quaternary acetylenic carbon at 79.8 ppm.
The readily available 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl azide has been used as a precursor of bis-1,2,3-triazoles 4a–d. Cu(I) catalyzed 1,3-dipolarcycloaddition reaction between alkynyl triazoles 3a–d and 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl azide afforded compounds 4a–d in very good yields 80–84%. The structures of these compounds were also determined by NMR spectroscopy in addition to the other techniques. Owing to facilitate the explanation of NMR spectra, numbering of compound 4a is shown below:
Beside the sugar signals, there was an important singlet at 7.75 ppm which clearly referred to the H5 of triazole 2. In the same way, 13C NMR spectra supported the structures of bistriazoles. The signals at 121.0 and 145.9 ppm, for example; were attributed to the C5 and C4 of triazole heterocycle 2, respectively. All assignments of proton and carbon NMR were based on COSY and HSQC. The acetate groups of glucose moiety were removed readily by treating compounds 4a–d with sodium methoxide in order to give the corresponding compounds 5a–d. The disappearance of the methyl group signals in 1H and 13C NMR spectra and the other signals of the C⚌O group are evident proof for the deprotection.
3.2 Antibacterial activity
The antibacterial activity of the synthesized bistriazoles was tested against Gram positive bacteria S. aureus and Gram negative bacteria E. coli by the agar well diffusion method. DMSO was considered as control while kanamycin was used as standard. It is shown from Table 1 that both protected and deprotected bistriazoles did not give any activity at the concentration 5 μg/mL compared to kanamycin. In contrast, all the mentioned compounds exhibited low to moderate activity at the concentrations 10 and 20 μg/mL respectively. On the other hand, the protected and deprotected compounds with C8 and C10 in the aliphatic chain exhibited maximum antibacterial activity compared to the other measured compounds. The antibacterial activity of bistriazoles can be attributed to their behavior as glycolipids mimics (Treyvaud et al., 2011), biosurfactants analogs (Ahimou et al. 2000), the asymmetrical structure of the whole molecule or the protein-binding properties (Banday et al., 2012).
Compound
Zone of inhibition in (mm), concentration (μg/mL)
G+ Staphylococcus aureus
G− Escherichia coli
5
10
20
5
10
20
DMSO
–
–
–
–
–
–
Kanamycin
27
28
28
28
27
28
4a
–
03
05
–
02
05
4b
–
05
11
–
06
11
4c
–
05
11
–
06
12
4d
–
04
08
–
03
05
5a
–
05
11
–
04
07
5b
–
06
13
–
05
11
5c
–
06
12
–
05
10
5d
–
05
07
–
04
09
4 Conclusion
In conclusion, this research has developed a convenient strategy for the synthesis of novel biologically important bis-1,2,3-triazoles based on d-glucose using copper catalyzed ‘click’ protocol. The obtained products thus were found to be slight antibacterial agents in two concentrations.
Acknowledgments
We gratefully acknowledge Dr. James Hook and the staff of NMR Facility at the University of New South Wales, Sydney, Australia for their assistance in NMR analysis. Our thanks are also extended to Dr. Lewis Adler and the staff of Bioanalytical Mass Spectrometry Facility at the same university for the HRMS analysis.
References
- Chem. Soc. Rev.. 2010;39:1302-1315.
- Macromol. Rapid Commun.. 2007;28:15-54.
- Drug Disc. Today. 2003;8:1128-1137.
- Aust. J. Chem.. 2007;60:384-395.
- J. Fluorine Chem.. 2011;132:898-906.
- Eur. J. Org. Chem.. 2010;8:1445-1449.
- Tetrahedron. 2010;66:4002-4009.
- J. Org. Chem.. 2006;71:8244-8249.
- J. Med. Chem.. 2006;49:7697-7710.
- Tetrahedron Lett.. 2012;53:5081-5083.
- J. AL-Nahrain Univ.. 2011;14:44-54.
- Ali, Y., Mohammed, A.I., Jwad, R.S., 2009. Proceedings of 3rd Scientific Conference of the College of Science, University of Baghdad, Baghdad. pp. 1370–1374.
- Asian J. Chem.. 2010;12:5592-5596.
- Int. J. Chem. Sci.. 2013;11:1-11.
- Der Pharma Chem.. 2012;4:272-281.
- Ind. J. Chem.. 2012;51 B:1020-1026.
- Arch. Pharm. Res.. 2010;33:1911-1918.
- J. Chem. Pharm. Res.. 2013;5:261-264.
- Carbohydr. Res.. 2010;345:1515-1521.
- Macromolecules. 2009;42:6068-6074.
- Bioorg. Med. Chem.. 2012;20:4727-4736.
- Int. J. Chem. Sci.. 2012;10:1529-1540.
- Can. J. Microbiol.. 2011;57:745-749.
- Enzyme Microb. Technol.. 2000;27:749-754.
- Organ. Med. Chem. Lett.. 2012;2:13-19.
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.arabjc.2014.02.016.
Appendix A
Supplementary data
Supplementary data 1
Supplementary data 1
This document file contains Supplementary Materials.




