

Translate this page into:
Synthesis and biological evaluation of 18F-labelled deuterated tropane derivatives as dopamine transporter probes
⁎Corresponding author at: 20 Qianrong Road, Wuxi 214063, China. chenzhengping@jsinm.org (Zhengping Chen)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Purpose
Dopamine transporter (DAT) is a promising target for positron emission tomography (PET) imaging of many neuropsychiatric diseases. 18F-labelled N-alkyl tropane analogues were reported to be useful PET radioligands for DAT. However, the drawback of 18F-labelled tropane analogues is that N-alkyl on tropane is easily metabolized in vivo, which interferes with brain imaging. To develop a more in vivo stable DAT-targeted PET radioligand with high DAT affinity and specificity, in this study, we synthesized and compared a series of 18F-labelled novel deuterated N-fluoropropyl tropane derivatives [18F]4a-e for DAT tracing.
Procedures
Five deuterated N-fluoropropyl-d6 tropane derivatives (4a-e) and corresponding non-deuterated compounds (6a-e) were synthesized, and their semi-inhibitory concentrations (IC50) were measured by competitive binding assay. The radioligands [18F]4a-e and [18F]6a-e were obtained by two-step one-pot radio-labelling reactions. The selectivity and specificity of these radioligands were evaluated by cellular uptake and microPET in normal rats. [18F]4e was selected for further investigation with microPET of the PD model, autoradiography and biodistribution experiments and compared with its non-deuterated [18F]6e. Finally, in vivo metabolic stability was analyzed by radio-HPLC.
Results
Ten tropane compounds had high DAT affinity (IC50 = 2–21 nM), in which FP-CIT-d6 (4e) had the lowest IC50 value of 2.7 nM. Radiolabelled [18F]4a-e and [18F]6a-e were obtained with radiochemical yields ranging from 10.6 ± 2.8% to 35.1 ± 5.4% with molar activities >20 GBq/μmol and the radiochemical purities >99%. [18F]4e showed the highest cell uptake (12%) and CFT inhibition efficacy (∼72%) among [18F]4a-e. MicroPET results showed [18F]4e has the highest target to non-target ratio (striatum/cerebellum). Therefore, [18F]4e was then selected for further biological evaluation. Ex vivo autoradiography experiment confirmed high specific binding of [18F]4e towards DAT. Biodistribution results indicated that [18F]4e has a higher striatum/cerebellum value than [18F]FP-CIT ([18F]6e) at 30–120 min. Furthermore, in vivo metabolism studies in rats revealed improved stability of [18F]4e as compared with that of [18F]6e.
Conclusions
The new probe [18F]4e is a promising candidate with good DAT affinity, specificity and metabolic stability for PET imaging, and might provide reliable diagnosis, treatment and prognostic detection of DAT-related neuropsychiatric diseases.
Keywords
Dopamine transporter
PET
Deuterium
18F-labelling
Tropane
In vivo metabolism

1 Introduction
Dopamine transporter (DAT) plays an important role in neuronal transmission and becomes a potential target of many neurological diseases, such as Parkinson’s disease (PD) (Akdemir et al., 2021), Schizophrenia (Amato et al., 2020), depression (Moriya et al., 2020), and others involving impaired dopaminergic neuron activity (Nikolaus et al., 2022). The changes in DAT density and availability are important indicators of the function of the dopamine transmission system. To visualize DAT, a variety of imaging techniques such as Positron emission tomography (PET) and single photon emission tomography (SPECT) have been developed. A SPECT imaging probe [123I]FP-CIT (Ioflupane I-123 injection, DaTscanTM) has been approved by Food and Drug Administration (FDA) for clinical applications (Hauser and Grosset, 2012). However, PET is able to detect the concentrations of specific biomolecules in the picomolar range and provides measurement of function and metabolism of the brain lesions, with higher sensitivity, better specificity than SPECT (Rahmim and Zaidi, 2008). Therefore, PET imaging with the DAT probes may better aid diagnosis and treatment of PD by estimating the density and distribution of DAT.
A plethora of 11C or 18F-labelled radiotracers have been developed for PET imaging (Riss et al., 2013). Compared with the 11C-labelled probes, 18F-labelled tracers show lower positron energy and more suitable half-life, has been identified as the most widely reported imaging tools. As of today, the most clinically used DAT PET probes are 18F-labelled N-fluoroalkyl tropane derivatives (Nye et al., 2014; Ribeiro et al., 2020; Verger et al., 2021). As reported, these tropane radiotracers are prone to produce N-dealkylation metabolites catalyzed by cytochrome P450 (Shetty et al., 2007; Peyronneau et al., 2012; Guengerich, 2017). For instance, the major radio-metabolites of [18F]PE2I were observed as a result of N-dealkylation (Amini et al., 2013) (Fig. 1). A closely related analogue, [18F]FECNT which has been used in humans, was reported to produce [18F]fluoroacetaldehyde, [18F]fluoroacetic acid and [18F]fluoroethanol as N-dealkylated metabolites in the periphery (Fig. 1). These metabolites may penetrate the blood–brain barrier (BBB) and interfere with the quantitative analysis of DAT in the rat, primate, and human brain (Zoghbi et al., 2006). Additionally, [18F]LBT999, a relatively new DAT PET probe, has been applied in clinical trials. The optimal time window of [18F]LBT999 PET scanning was assessed based on the radio-metabolites analysis (Ribeiro et al., 2020; Arlicot et al., 2022). However, it is reported that N-dealkylation is also one of the metabolism pathways of in vivo [18F]LBT999 (Peyronneau et al., 2012) (Fig. 1).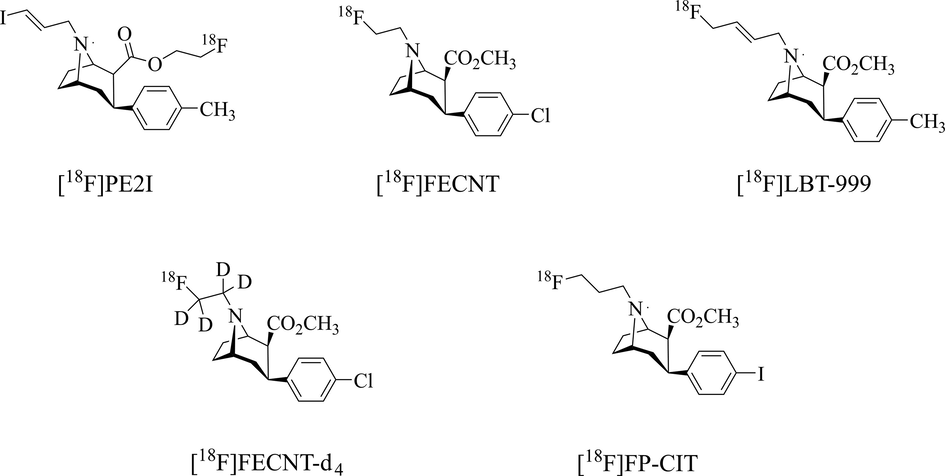
Examples of 18F-labelled tropane probes.
Therefore, it is desired to develop DAT probes with reduced in vivo metabolites and increased intact probe in the target and reference regions, which could contribute to better images and quantitative analysis of PET. Direct substitution of deuterated on easily-metabolite site has been used to improve pharmacokinetics, increase the target uptake, and reduce background activity of radiopharmaceuticals. Several successful deuterated ligands have been reported (Jahan et al., 2011; Kuchar and Mamat 2015; Haider et al., 2020; Nag et al., 2021; Liu et al., 2023). For instance, [18F]FLUDA, a deuterated analogue of [18F]FESCH, revealed improved stability and was employed for mapping A2A receptor activity in the brain (Lai et al., 2021).
We previously reported a deuterated N-[18F]fluoroethyl tropane analogue, [18F]FECNT-d4 (Fig. 1), for DAT imaging (Cao et al., 2021). Biological results showed that [18F]FECNT-d4 retained the original affinity and specificity, and improved in vivo stability compared with non-deuterated [18F]FECNT and provide a higher target/nontarget ratio and lower metabolic rate. Nevertheless, [18F]FP-CIT (Fig. 1), a [18F]fluoropropyl tropane analogue, is a widely clinically used 18F-labelled DAT PET probe for imaging PD and heterogenous dopaminergic neurodegeneration patterns (Suh et al., 2020; Lee et al., 2021). This [18F]fluoropropyl tropane tracer has the advantages of rapid pharmacokinetics, good in vivo half-life, high affinity and specificity, and it is developed for PET quantification of the DAT (Peng et al., 2021). However, the radio-metabolites of [18F]FP-CIT in vivo include the free carboxylic acid form of [18F]FP-CIT decomposed by esterase, [18F]fluoride ion generated by defluorination and [18F]fluoropropyl compounds produced by N-dealkylation (Shin et al., 2012).
Herein, we report the synthesis and biological evaluation of a series of deuterated [18F]fluoropropyl tropanes analogues as DAT probes. To validate that deuterated N-fluoropropyl ligands give fewer N-dealylated metabolites and improve imaging quality, we conducted a series of preclinical studies, including in vivo metabolism and dynamic PET studies in rats. The pharmacokinetic properties of deuterated 18F-labelled ligands were compared, and the results showed that deuterated [18F]N-fluopropyl tropane analogues have improved pharmacokinetics and in vivo stability in comparison with non-deuterated compounds. Among these deuterated [18F]fluoropropyl tropanes, [18F]FP-CIT-d6 ([18F]4e) showed the best selectivity and affinity. More importantly, this probe exhibited higher stability and better striatum/cerebellum (ST/CB) ratio as comparison with [18F]FP-CIT ([18F]6e), thus supporting the clinical application for DAT PET imaging.
2 Materials and methods
2.1 General information
Unless otherwise stated, all reagents were purchased from Energy Chemical (Shanghai, China) or J&K Scientific (Shanghai, China). Semi-preparative high-performance liquid chromatography (HPLC) consists of Waters 1525 HPLC pump, UV-2489 Detector (Waters, USA), and flow-count radioactivity detector (BioScan, USA). An analytical HPLC system was used, consisting of Waters 1525 pump, UV-2998 detector (Waters, USA), radioactive GABI Nova detector (ELYSIA RAYTEST, Germany) and an analytical C18 column (5 μm, 4.6 mm × 150 mm) was purchased from Phenomenex. Kryptfix®222 (K2.2.2, 98.0%) was purchased from ABX (Germany). Solid-phase extraction (SPE) cartridges SepPak QMA light and SepPak C18 Plus were bought from Waters (Milford, USA). The C18 cartridge was activated using ethyl alcohol (EtOH, 20 mL), followed by sterile water (20 mL). Radioactivity was counted with a γ-counter (2480 Wizard2, PerkinElmer, USA).
Compounds were purified on flash chromatography performed on silica gel (200–300 or 300–400 mesh) and detected by analytical thin-layer chromatography (TLC) using an ultraviolet (UV) lamp at 254 nm. Chemical compounds were characterized by proton and carbon nuclear magnetic resonance (1H NMR, 13C NMR and 19F NMR) and mass spectrometry (MS) or gas chromatograph-mass spectrometry (GC-MS) data. NMR spectra were recorded on a Bruker spectrometer operating at 400 or 600 MHz for 1H, 100 or 150 MHz for 13C, and 376 or 565 MHz for 19F acquisitions. Samples were dissolved in chloroform-d (CDCl3) with tetramethylsilane as an internal standard and chemical shifts were expressed as ppm. J was expressed as Hz, and its splitting patterns were reported as s = singlet, d = doublet, t = triplet, m = multiple, or dd = doublet of doublets. The purities of all new compounds determined by analytical HPLC were confirmed to be more than 95% pure.
Animal studies were carried out in accordance with the guidelines put forth by the Animal Care and Ethics Committee of Jiangsu Institute of Nuclear Medicine. Male Sprague-Dawley (SD, 220–260 g) rats were provided by Cavens Laboratory Animal Technology Co. Ltd. (Jiangsu, China). Unilateral-lesioned PD model rats were induced by stereotaxic injection of 16 µg 6-hydroxydopamine (6-OHDA) (Truong et al., 2006).
2.2 Chemical synthesis
General Procedure for the Preparation of 1 and 3a-e. Compound 1 was synthesized according to the reported method (Zhao et al., 2019). Compound 3a-e was prepared according to previously reported methods (Zheng and Mulholland 1996; Gu et al., 2001). All NMR spectra and data are presented in the supporting materials.
2.2.1 3-fluoropropyl-1,1,2,2,3,3-d6 4-methylbenzenesulfonate (2)
The reaction of propane-1,3-diyl-d6 bis(4-methylbenzenesulfonate) (666 mg, 1.70 mmol) with 2.5 mL tetrabutylammonium fluoride (TBAF) 1.0 mol/mL solution in THF was refluxed under nitrogen for 2 h, condensed under reduced pressure and purified by flash chromatography (ethyl acetate/hexanes = 1/4, v/v) to give 2 (0.85 mmol, 50%) as a colorless oil.
General Procedure for the Synthesis of 4a-e. A mixture of 2 (1.0 mmol, 1.0 equiv), 3 (1.0 mmol, 1.0 equiv) and 0.8 mL triethylamine in 30 mL toluene, was refluxed at 115 °C for 4 h and then the mixture was concentrated under reduced pressure, the compound 3 was separated by flash chromatography (ethyl acetate/hexane = 1/4, v/v), then eluted with flash chromatography (hexane/ether/triethylamine = 1/1/0.01, v/v/v) to give 4a-e as a white solid.
2.3 Radiochemistry
[18F]4a-e and [18F]6a-e were prepared by a two-step one-pot automatic radiosynthesis of the tosylate precursor according to the previously reported method (Koivula et al., 2005) on a commercially available PET-MF-2 V-IT-1 (Beijing PET Technology, China) [18F]fluoride multifunction synthesis module (Scheme 2) with slight modification. [18F]fluoride was produced by the cyclotron via the 18O (p, n)18F nuclear reaction and trapped on a SepPak QMA light cartridge. [18F]F- trapped in the cartridge was eluted by a stock solution (15 mg of K222 in 800 µL of acetonitrile (CH3CN)/3.0 mg of K2CO3 in 100 µL of water). The [18F]F- elution solution was evaporated at 105 °C for 3 min in a stream of nitrogen. Afterward, CH3CN (1.0 mL) was added to the reaction tube and the reaction mixture was dried twice at 105 °C under N2. Precursor 1 or 7 (10.0 mg), in CH3CN (500 µL) was added, and the mixture was heated at 90 °C for 15 min. After cooling to room temperature, dry DMF solution containing 3 (12.0 mg) was added, the mixture was heated to 135 °C for 25 min, and then cooled to room temperature. Next, the reaction was quenched with 1.0 mL HPLC solvent, and the crude product was purified by HPLC. HPLC purification was performed on a Phenomenex Luna C18 column (10 mm × 250 mm, 5 µm) with a mobile phase of CH3CN/H2O (60/40, v/v) (0.1% triethylamine, TEA) at a flow rate of 4.0 mL/min. The retention time of [18F]4a-e or [18F]6a-e was ∼ 20.0 min, the required radioactive product was collected and diluted with 30 mL H2O, transferred to a C18 cartridge, and then the cartridge was washed with 10 mL H2O. The trapped [18F]4a-e or [18F]6a-e was eluted with 2.0 mL EtOH and the final product was formulated in a solution containing 10% EtOH in 0.9% saline (0.5 mL, 10% ethanol/normal saline) prior to administration. The total radiosynthesis time was in the range of 90–110 min. The product was identified by co-injection of the corresponding non-radioactive ligand and its radiochemical purity was measured by an analytical HPLC containing a Gemini NX-C18 column (3 mm × 150 mm, 5 µm) with a mobile phase of methanol/water/trifluoroacetic acid (MeOH/H2O/TFA, 40/60/0.1, v/v/v) at 1.0 mL/min flowing rate. The molar activity was calculated by a calibration curve of the non-radioactive compound through HPLC with a UV detector.
2.4 In vitro stability
The in vitro stability assay of all 18F-labelled probes was performed by evaluating their radiochemical purity using a radio-HPLC system. The [18F]4a-e or [18F]6a-e (37 MBq) were added to fetal bovine serum (FBS, 500 µL) and phosphate-buffered saline (PBS, pH 7.4, 500 µL), respectively. At indicated time points (0, 1, 2, 4, and 6 h), the serum samples (100 µL) were mixed with MeOH (100 µL) at each time point and centrifuged at 14,000×g in an Eppendorf 5415R centrifuge (Eppendorf, Germany) for 5 min. The radiochemical purity (RCP) of 18F-labelled probes in supernatant and the PBS solution was determined by radio-HPLC.
2.5 Distribution coefficient (logD7.4)
The values of logD7.4 were determined by the conventional shaking-flask method with n-octanol and PBS as a fractionation system (n = 3). [18F]4a-e or [18F]6a-e (18.5 KBq) was added into a two-phase mixture of PBS (pH 7.4, 3.0 mL) and n-octanol (3.0 mL) and vortexed for 5 min. The PBS and the n-octanol phase were separated via centrifugation (5 min, 1000 r/min). Then, the PBS (1.0 mL) and n-octanol (1.0 mL) solutions were pipetted into γ-counting tubes and determined by a γ-counter, respectively. The logD values were calculated by the formula: logD = log (CPMn-octanol/CPMPBS). The above experiment was repeated 5 times to obtain an average logD value.
2.6 In vitro competitive binding assay
The SD rats were decapitated under ether anesthesia and striata were removed immediately and homogenized through a high throughput group grinder in 50 mM Tris-HCl buffer (pH 7.4) containing 120 mM NaCl, 2 mM KCl, 1 mM CaCl2, and 1 mM MgCl2. After centrifuging at 14,000 g at 4 °C for 5 min, the supernatant was discarded. The precipitate was diluted into 100 mg/mL of striatum homogenate in Tris-HCl buffer, which was divided into multiple tissue samples with a volume of 20 µL (2 mg of striatum homogenate). A competitive binding assay was performed with 20 µL striatal homogenate, 20 µL 99mTc-TRCDAT-1 (∼18.5 KBq, 925KBq/mL), a known DAT binding ligand (Bao et al., 2000), and 20 µL of non-radioactivity competing ligands (non-radioactivity compound 4a-e and 6a-e, 10-6 to 10-12 M) in 440 µL Tris-HCl buffer. The non-specific binding was determined in the presence of DAT inhibitor (CFT, 20 µM) (Milius et al., 1991). After incubating at 37 °C for 60 min, these mixtures were centrifuged for 5 min (14,000×g/min), and the supernatant and precipitates were separated respectively. Tris-HCl buffer (400 µL) was added to wash the striatum precipitates and centrifugation for 5 min (14,000×g/min) again. The bound and the free radioactivity were separated by centrifuging and measured with a γ-counter separately. Data of competition experiments were analyzed using the nonlinear least-square curve fitting program to determine semi-inhibitory concentrations (IC50) with GraphPad Prism.
2.7 Cell uptake and blocking assays
The human neuroblastoma (SH-SY5Y) cells (1 × 105 cells/100 mL) were grown in 24-well plates and assigned to three groups: control, CFT blocking, and DTBZ pretreated group. The control group was added with [18F]4a-e (37 KBq), CFT blocking group was added with [18F]4a-e and CFT (20 μM), and DTBZ (Kilbourn et al., 1996) (VMAT2 inhibitor) pretreated group was added with [18F]4a-e and DTBZ (20 μM). The cells were preincubated with 20 µM DTBZ for 10 min. Then the cells were incubated at 37 °C for 15, 30, 60, 90 and 120 min, respectively. After reaching the expected incubation time, the cells were washed twice with 1.0 mL of ice-cold PBS. Subsequently, the cells were lysed and collected with sodium hydroxide (1.0 mol/mL). The radioactivity in the cell lysates and supernatants were measured via a γ-counter. The percentage of the cell counts was calculated by the cell counts and the total counts.
2.8 In vivo PET imaging studies
A microPET system (Inveon 5000, Germany) was used for real-time scanning of rat brain. The SD rats were fixed to a small animal PET scanner bed under 2% isoflurane anesthesia. After adjusting the location of the brain, dynamic PET scans were acquired from 0 to 120 min after tail vein injection of [18F]4a-e or [18F]6a-e (∼10 MBq, 0.5 mL, 10 % ethanol/saline) respectively. After the scanning, the obtained imaging data was segmented into a sequence of 43 frames: 10 × 0.5 min, 5 × 1 min, 10 × 2 min and 18 × 5 min. The dynamic images were obtained by reconstruction with the filtered back projection (FBP) algorithm. Image analyses were performed using ASIPro VM software (Concorde Microsystems) and time-activity curves (TACs) of the regions of interest (ROIs, striatum and cerebellum) were plotted from the dynamic images.
To verify the specific binding of [18F]4e to DAT in vivo, DAT inhibitor CFT (1.0 mg/kg) was injected into normal rats at 40 min after [18F]4e injection and dynamic imaging was performed for 120 min.
Additionally, to further verify the binding specificity of [18F]4e, microPET scans of DAT blocking and semi-PD model rats were performed. The time window of these PET scans was set at 40–70 min post tracer injection since a relatively high and stable ST/CB values were observed from the TACs of [18F]4e in normal rats. The control and unilateral-lesioned PD model rats were injected with [18F]14e (∼10 MBq). The blocking rats were co-injected with [18F]14e (∼10 MBq) and CFT (1.0 mg/kg). All rats were scanned at 40 min after injection and continued for 30 min (40–70 min). Brain imaging images were collected and analyzed using ASIPro VM software.
2.9 Ex vivo autoradiography with [18F]4e
The specific binding ability of [18F]4e to DAT was further confirmed with ex vivo autoradiography. The control male rat was injected with [18F]4e (∼185 MBq) and sacrificed 40 min later, the brains were immediately dissected and frozen at −25 °C and sliced into 20 µm coronal sections with a cryostat microtome (HM525, Thermo, Germany). The slices were exposed to the imaging plate for 1 h and scanned with a phosphine screen imager. Then images were attained using Cyclone Plus Storage Phosphor System (Perkin Elmer, USA), and ROIs (striatum and cerebellum) were drawn manually. Ex vivo autoradiography was quantified as ratios of the striatum to the cerebellum. For blocking studies, the rat was co-injected with [18F]4e and CFT (1 mg/kg), and the exposure and imaging procedures were the same as described above.
2.10 Biodistribution with [18F]6e and [18F]4e
All SD rats were injected with a solution of [18F]6e or [18F]4e (∼18.5 MBq/rat) to examine the biodistribution. These rats were sacrificed at 5, 15, 30, 60 and 120 min post injection (n = 5). The blood (200 μL) was obtained from the carotid artery. The brain tissues including the striatum, cerebellum, hippocampus and cortex were dissected and other organs of interest (heart, liver, spleen, lung, kidney, stomach, small intestines, testis, bone, muscle, pancreas) were quickly removed and weighed. Then radioactivity of these samples was measured with a γ-counter. The percentage of the injected dose per gram (%ID/g) of brain tissues and the percentage of the injected dose per organ (%ID) of the interested organs were calculated with sample counts and decay-corrected 1% predose counts (100-fold dilution of injection).
2.11 Metabolite analyses with [18F]6e and [18F]4e
Radio-metabolites were analyzed using HPLC slightly modified from previously reported methods (Jin et al., 2018). Two-months-old rats were injected with [18F]4e or [18F]6e (∼185 MBq, 0.5 mL, 10% ethanol/normal saline) intravenously and sacrificed at 5, 15, 30, 60 and 120 min (n = 3) post injection. Plasma, striatum and cerebellum samples were rapidly collected. The samples were homogenized in 0.5 mL MeOH containing 1.0 mg/ml FP-CIT (20 µM), the homogenates were centrifuged at 14,000 g at 4 °C in an Eppendorf 5415R centrifuge for 5 min and the supernatant and the precipitate were collected. The sample extraction efficiency was calculated by measuring the radioactivity counts of the supernatant and the precipitate. The supernatant of the centrifuged samples was injected into the analytic HPLC system for the analysis of in vivo stability. The mobile phase was composed of MeOH/H2O/TFA (40/60/0.1, v/v/v), and the flow rate was 1.0 mL/min. The HPLC fractions at 0.5 min intervals were collected with an automatic collector for 20 min. Then, each fraction tube was counted separately using a well γ-counter. The percentages of the parent tracer were calculated as the ratio of the radioactivity of the parent compound fractions to the total amount of radioactivity collected.
2.12 Statistical analysis
Unless otherwise stated, all data were presented as mean ± standard deviation (sd) as indicated in the legends of the figures. To determine the statistical significance of the experiments, t-test was used and p-values of <0.05 were considered significant.
3 Results
3.1 Chemical synthesis
Syntheses of the non-radioactive ligands 4a-e and 6a-e are shown in Scheme 1. Briefly, 3-fluoropropyl-1,1,2,2,3,3-d6 4-methylbenzenesulfonate (2) was synthesized from propane-1,3-diyl-d6 bis (4-methylbenzenesulfonate) (1) using nucleophilic fluorination with TBAF in 50% yields. Subsequent alkylation of compound 3a-e with 2 or 5 gave compounds 4a-e and 6a-e respectively in 40–60% yields.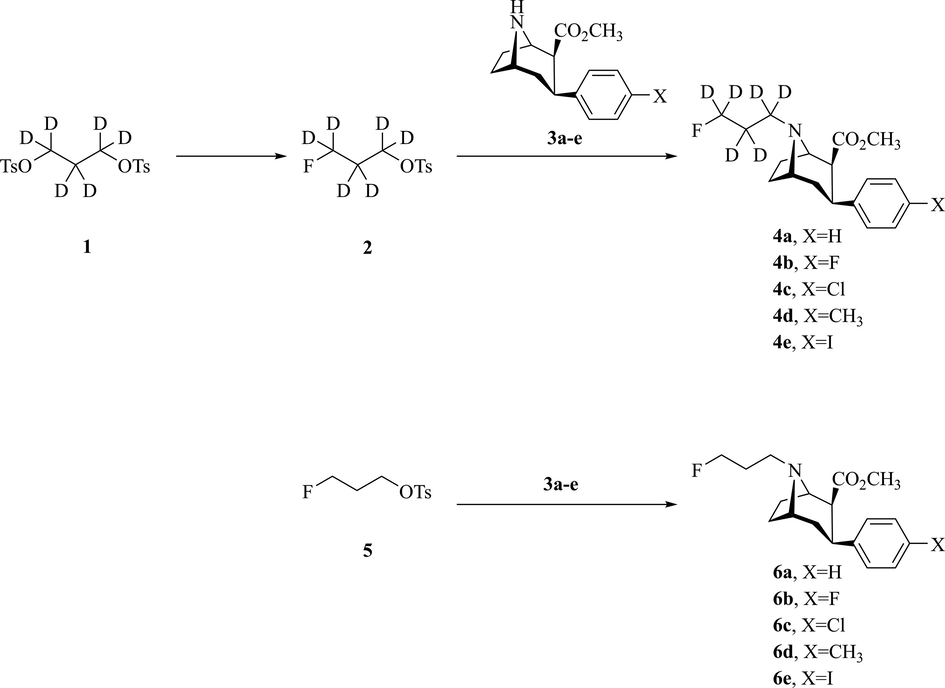
Design and synthesis of non-radioactive fluorinated standards.
![Radiosynthesis of [18F]4a-e and [18F]6a-e.](/content/184/2023/16/11/img/10.1016_j.arabjc.2023.105278-fig3.png)
Radiosynthesis of [18F]4a-e and [18F]6a-e.
3.2 Radiosynthesis, lipophilicity, and in vitro stability
All 18F-labelled ligands ([18F]4a-e and [18F]6a-e) were radio-synthesized by a two-step one-pot approach with nortropane precursors reacting with [18F]fluoropropyl tosylate ([18F]5) or deuterated [18F]fluoropropyl tosylate ([18F]2) (Scheme 2). The decay-corrected radiochemical yields (RCYs) of ligands were obtained between 10.6 ± 2.8% and 35.1 ± 5.4% (Table S1), for an averaged synthesis time of 90–110 min. All the RCPs were >99% (Fig. S2) and the molar activities were >20 GBq/μmol (Table S1). As a final step, a sterile 0.9% saline solution of 18F-labelled ligands was prepared for in vitro and in vivo experiments.
The radiochemical purity of all 18F-labelled ligands remained at least 96% after being incubated in FBS and PBS for 6 h at 37 °C (Fig. S3). The LogD values of all 18F-labelled probes were measured to be 0.60–2.50 (n = 3) (Table S2).
3.3 In vitro binding assay
Upon synthesis, compounds 4a-e and 6a-e were compared for their in vitro binding activity toward DAT. For this purpose, we determined their ability to inhibit DAT activity using of IC50 values against 99mTc-TRODAT-1, a verified DAT tracer (Bao et al., 2000). As shown in Table 1, compounds 4a-e exhibited good potency with IC50 values between 2 and 17 nM. The compounds FP-CIT-d6 (4e) and FP-CIT (6e) bearing an iodophenyl on tropane demonstrated the highest potency among these compounds, with an IC50 value of 2.7 nM and 3.2 nM respectively. The competition curves of the 10 compounds are shown in Fig. S1.
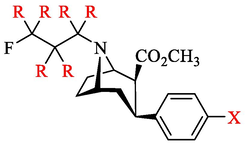
compound
R
X
IC50 (nM)
4a
D
H
17.56
4b
D
F
14.56
4c
D
Cl
5.89
4d
D
CH3
7.60
4e
D
I
2.67
6a
H
H
20.53
6b
H
F
15.87
6c
H
Cl
5.59
6d
H
CH3
7.26
6e
H
I
3.16
3.4 Cellular uptake and blocking assays
The cell uptake of [18F]4a-e was assessed in SH-SY5Y cells. The binding specificity of [18F]4a-e was preliminarily evaluated in cells treated with DAT inhibitor CFT and VMAT2 inhibitor DTBZ. We observed that [18F]4e showed the highest cell uptake (12%) and CFT inhibition efficacy (∼72%) among [18F]4a-e. However, the differences in cell uptake among [18F]4a-e were not significant (P > 0.05). Contrary to the CFT inhibition effect, little difference of [18F]4a-e uptake was found between the DTBZ group and the control group within 120 min (Fig. S4).
3.5 MicroPET
After the synthesis of 18F-labelled ligands, dynamic PET acquisition was carried out in SD rats (n = 3). Fig. 2a-d and Fig. S5 show TACs of PET imaging with [18F]4a-e and [18F]6a-e. Among them, [18F]4c and [18F]4e showed greater average standardized uptake values (SUV) (4.06 ± 1.45 and 3.76 ± 1.73, respectively) of striatum and target-to-background (striatum/cerebellum) standardized uptake value ratios (SUVr) (3.16 ± 1.62 and 3.20 ± 1.73) (Table 2). However, [18F]4e showed the highest maximum SUVr of 6.84. Furthermore, the TACs of [18F]4e since 20 min showed higher striatum/cerebellum (ST/CB) than those of other ligands until the end of the PET scan (Fig. S5).![Time-activity curves of PET imaging with [18F]6e or [18F]4e in rat brain. The TACs of brain SUV (a-b) and SUVr (c-d) of [18F]6e and [18F]4e in the rat. The TACs of brain SUV (e) and SUVr (f) of [18F]4e in control (non-blocking) or blocking (injected CFT at 40 min) rat. Data are expressed as mean ± sd (n = 3).](/content/184/2023/16/11/img/10.1016_j.arabjc.2023.105278-fig4.png)
Time-activity curves of PET imaging with [18F]6e or [18F]4e in rat brain. The TACs of brain SUV (a-b) and SUVr (c-d) of [18F]6e and [18F]4e in the rat. The TACs of brain SUV (e) and SUVr (f) of [18F]4e in control (non-blocking) or blocking (injected CFT at 40 min) rat. Data are expressed as mean ± sd (n = 3).
Radioactive ligand
SUV
SUVr
SUVr max
[18F]4a
1.80 ± 1.31
1.67 ± 0.42
2.29
[18F]4b
2.56 ± 1.26
1.82 ± 0.75
3.47
[18F]4c
3.76 ± 1.73
3.16 ± 1.62
6.33
[18F]4d
2.30 ± 1.28
3.00 ± 1.17
5.53
[18F]4e
4.06 ± 1.45
3.20 ± 1.73
6.84
[18F]6a
1.27 ± 1.07
1.40 ± 0.47
2.29
[18F]6b
3.38 ± 2.44
1.74 ± 0.71
3.31
[18F]6c
3.56 ± 1.68
2.75 ± 1.40
5.10
[18F]6d
2.24 ± 1.46
2.63 ± 1.11
5.94
[18F]6e
3.83 ± 1.30
2.78 ± 1.16
5.01
Considering the lowest IC50 and highest SUVr (ST/CB) maximum of PET imaging results of [18F]4e, we selected [18F]4e for further in vivo PET study and compared with its nondeuterated compound [18F]6e. Fig. 3a-b illustrated representative PET images of [18F]4e and [18F]6e in rat brains. Summation images between 0 and 120 min after the radioligand injection showed a high signal in the striatum, a region with a high DAT concentration. Low radioactivity was observed in the cerebellum, a region with low DAT expression. As shown in the TACs of brain regions (Fig. 2b), SUV of the striatum gradually increased after [18F]4e injection, peaked at 5 min (SUV = 6.68) and then reduced steadily. The SUVr of [18F]4e reached 6.84 at around 90 min (Fig. 2d). The average SUV and SUVr of [18F]4e were 4.06 ± 1.45, and 3.20 ± 1.73 within 120 min, respectively (Table 2).![Coronal microPET images obtained from SD rats injected with [18F]4e (a) or [18F]6e (b) at 0–120 min. (c) Representative PET images of the control group, CFT blocking group and semi-PD model group at 40–70 min post injection. The striatum and lateral skull regions were highlighted with ellipses and arrows, respectively. (d) Quantification of PET of ST/CB. Data are expressed as mean ± sd (n = 3). ****P < 0.0001.](/content/184/2023/16/11/img/10.1016_j.arabjc.2023.105278-fig5.png)
Coronal microPET images obtained from SD rats injected with [18F]4e (a) or [18F]6e (b) at 0–120 min. (c) Representative PET images of the control group, CFT blocking group and semi-PD model group at 40–70 min post injection. The striatum and lateral skull regions were highlighted with ellipses and arrows, respectively. (d) Quantification of PET of ST/CB. Data are expressed as mean ± sd (n = 3). ****P < 0.0001.
To further demonstrate the targeting specificity of [18F]4e in vivo, a blocking test was performed with DAT inhibitor CFT (Milius et al., 1991) (1 mg/kg) at 40 min after the probe injection, and the result showed that the striatum uptake and TAC ratio reduced dramatically after CFT injection (Fig. 2e-f). The specificity of the probe to DAT was thus proved. Representative images and quantitative analysis of the control group, CFT blocking group were shown in Fig. 3c-d.
To investigate the ability of [18F]4e to detect the DAT deficit, the semi-PD model rats (lesioned in the right ST with 6-OHDA) were used for microPET imaging. As shown in Fig. 3c, the radioactivity of the un-lesioned side of the striatum was similar with control rats (STunlesioned/CB = 3.21 ± 0.46), while the lesioned side showed dramatically reduced radioactivity similar to background (STlesioned/CB = 1.04 ± 0.09) (Fig. 3d).
3.6 Ex vivo autoradiography
Fig. 4 showed representative ex vivo autoradiograms of [18F]4e on the rat brain slices. Control rat slice images showed high uptake in the DAT-rich striatum. A low signal was seen in the DAT non-rich regions such as the cerebellum and cortex (CX). When rats were co-injected with CFT, the radioactivity of [18F]4e in the striatum of these slices declined dramatically. In the control rat, the ST/CB and ST/CX value of the control group was 11.28 ± 0.83 and 7.18 ± 0.57, respectively. Contrarily, no visual difference in radioactivity levels among the striatum, cortex and cerebellum of brain slices in the blocking rat was observed. The ST/CB and ST/CX of the blocking group significantly decrease to 1.22 ± 0.09 and 1.04 ± 0.07, respectively (****P < 0.0001). These results indicated that the binding pattern of [18F]4e in rat brain was specific to DAT.![Ex vivo autoradiography of [18F]4e in control or blocking rat brain. ST: striatum, CB: cerebellum, CX: cortex. ****P < 0.0001 (n = 4).](/content/184/2023/16/11/img/10.1016_j.arabjc.2023.105278-fig6.png)
Ex vivo autoradiography of [18F]4e in control or blocking rat brain. ST: striatum, CB: cerebellum, CX: cortex. ****P < 0.0001 (n = 4).
3.7 Biodistribution
Biodistribution studies quantify tracer accumulation in organs and determine metabolic pathways. Fig. 5 shows the radioactivity distribution over 120 min after injection of [18F]6e and [18F]4e in rats.![Biodistribution of [18F]6e (a) and [18F]4e (b) in SD rats. The ratio of uptake in the striatum to the cerebellum for [18F]6e and [18F]4e (c). Data were expressed as % injected dose/organ and expressed as mean ± sd (n = 5).](/content/184/2023/16/11/img/10.1016_j.arabjc.2023.105278-fig7.png)
Biodistribution of [18F]6e (a) and [18F]4e (b) in SD rats. The ratio of uptake in the striatum to the cerebellum for [18F]6e and [18F]4e (c). Data were expressed as % injected dose/organ and expressed as mean ± sd (n = 5).
These studies demonstrated [18F]4e has reversible binding, as well as [18F]6e, in the peripheral main organs, with a maximum uptake between 5 and 15 min. The liver had the highest uptake with 12.85 ± 2.02 %ID at 15 min, similar to [18F]6e. Initial accumulation in the kidneys of [18F]4e was 2.05 ± 0.33 %ID, which was compatible with [18F]6e (Table S3, S4). It was observed that radioactivity in other organs decreased gradually after the initial uptake (Fig. 5a-b). Furthermore, the radioactivity of bone (<0.09 %ID) were quite low for both radiotracers within 120 min.
In brain tissues, High radioactivity was shown in the striatum since 5 min, striatum uptake was the highest after injection, while low radioactivity was observed in the cerebellum and cortex. The radioactivity of [18F]4e in the striatum and cerebellum respectively reached 0.95 ± 0.16 and 0.75 ± 0.32 % ID/g at 5 min and decreased to 0.21 ± 0.15 and 0.04 ± 0.01 % ID/g at 120 min. [18F]4e displayed a higher ST/CB ratio of 5.89 ± 1.23 at 60 min, 5.84 ± 1.36 at 120 min (Fig. 5c, Table S5, S6), while the ST/CB of [18F]6e were 4.42 ± 0.86 and 4.54 ± 0.07 at 60 min and 120 min respectively. These results showed that the ratio of ST/CB of [18F]4e was higher than that of [18F]6e between 60 min and 120 min.
3.8 Metabolite analysis
The extraction efficiency of the total radioactivity was 85 ± 5% (Table S7). Radio-metabolites of [18F]6e or [18F]4e in rat brain and plasma were analyzed at 5, 15, 30, 60 and 120 min after tracer injection through HPLC (Fig. S7). The percentage of unmetabolized parental [18F]4e in plasma decreased throughout the time course, from 85 ± 9% at 5 min to 15 ± 5% at 120 min. Meanwhile, intact [18F]6e in plasma showed lower concentration from 5 to 120 min as compared with [18F]4e, from 48 ± 7% at 5 min to 6 ± 5% at 120 min post injection. In the brain striatum, the parent compound of [18F]4e accounted for about 84 ± 4% at 120 min, whereas the percentage of [18F]6e was only 65 ± 3% at the same time point post injection. In the cerebellum, [18F]4e also possessed higher intact tracer concentration than [18F]6e, for example, 56 ± 6% vs 15 ± 5% at 120 min post injection (Fig. S7 and Table S8). The results demonstrated that metabolic rates of [18F]4e in plasma, striatum and cerebellum were slower than [18F]6e (Fig. 6).![Metabolism analysis of intact [18F]4e (red) and [18F]6e (blue) in rat plasma, striatum and cerebellum (% of total radioactivity, mean ± sd, n = 3).](/content/184/2023/16/11/img/10.1016_j.arabjc.2023.105278-fig8.png)
Metabolism analysis of intact [18F]4e (red) and [18F]6e (blue) in rat plasma, striatum and cerebellum (% of total radioactivity, mean ± sd, n = 3).
4 Discussion
Deuteration may have substantial benefits to the overall pharmacological properties of the resulting compounds if the corresponding C–H bonds are involved in critical steps of the in vivo metabolism. In this study, to increase the tropane probes’ in vivo stability via the limitation of N-dealkylation, we successfully designed, synthesized and characterized a series of novel deuterated [18F]fluoropropyl tropane analogues as DAT probes. Preliminary studies showed that the probes selectively bind to DAT with high affinity, appropriate lipophilicity, and in vitro stability. Our initial experiments showed that [18F]4e possesses the best biological prosperity among all probes. Furthermore, we applied [18F]4e in the 6-OHDA-induced PD rat model for in vivo imaging and the results showed high tracer uptake in regions of DAT-rich striata. Our systematic experimental results demonstrated that the deuterated [18F]fluoropropyl tropane derivative [18F]4e is a promising candidate for PET imaging of DAT, which may be better than the clinically widely used [18F]6e.
Firstly, we synthesized non-radioactive deuterated compounds 4a-e and 6a-e. All compounds were characterized with HPLC, MS and NMR to determine their structure and purity. We found all compounds showed a nanomolar inhibitory activity on DAT, with IC50 values ranging from 2 to 21 nM. No significant change was observed in binding affinity with deuterium substitution in comparison with the original non-deuterated compounds (Table 1). The competitive binding experiments showed that the deuterium strategy retained DAT affinity without significant alteration of IC50 value. Notably, the IC50 values of fluoropropyl-d6 derivatives 4e were lower than those of 4a-d and 6a-d. We speculated that the function of 4e was related to the electronegativity and size of substituent according to relevant literature reported (Carroll et al., 1991).
The radiotracers of [18F]4a-e and [18F]6a-e were radio-synthesized by a two-step one-pot approach from nortropane precursor 3a-e with high RCP (>99%). For these deuterated radiotracers, the characteristics of lipophilicity, in vitro stability, and BBB permeability were retained. The in vitro lipid-water distribution confirmed that the LogD values of 18F-labelled probes (LogD = 0.6–2.5, Table S2) fall in the range of good brain permeability (Dishino et al., 1983; Lipinski et al., 2001; Vermeulen et al., 2019), which suggested the probes were able to cross the BBB. LogD values of pH7.4 were greater than pH7.0, probably due to that these compounds contain a nitrogen atom. The in vitro stability tests at 37 °C in PBS and FBS showed no observable decomposition and defluorination up to 6 h (Fig. S3). Good in vitro stability, and suitable logD values for BBB penetration implied that these tracers could be used for subsequent biological experiments.
To preliminarily evaluate the biological activity of [18F]4a-e in vitro, we applied [18F]4a-e to the SH-SY5Y cell uptake assay (Fig. S4). The cell uptake of [18F]4a-e showed a significant upward trend within 120 min, and the uptake of [18F]4e showed better binding inhibition of ∼ 72% (12% uptake) than that of [18F]4a-d after pretreatment with DAT inhibitor. However, in the cells treated with DTBZ, the radioactivity of [18F]4e did not decrease because DTBZ was a VMAT2 inhibitor and did not bind to DAT specifically. The bioactivity and high selectivity of [18F]4e to DAT were initially proved at the cell level.
After radiosynthesis of [18F]4a-e and [18F]6a-e, we performed in vivo PET imaging with rats. Consistent with expectations, the results of non-deuterated [18F]6a-e microPET were compatible with those as reported (Goodman et al., 1997; Marjamaki et al., 2010; Choi et al., 2012, Cumming et al., 2014). The TACs showed that the SUV of deuterated probes decreased more slowly than those of the non-deuterated probes in the DAT-rich striatum. The ratios of ST/CB confirmed this property, with a continuous increase after 20 min and a slow decrease after reaching a maximum at 40 min (Fig. 2a-d and Fig. S5). [18F]4c-e showed higher ST/CB than the reported [18F]FECNT-d4 after 40 min (Cao et al., 2021). The maximum SUV (SUVmax) of [18F]4e was higher than that of [18F]FECIT-d4, a deuterated [18F]fluoroethyl tropane analog reported by Li et al. (2023). In addition, the TACs of [18F]4e had the maximal mean SUV and SUVr in rat brain.
Considering [18F]4e has a good binding affinity with the lowest IC50, high cellular uptake (Fig. S4) and excellent microPET time-activity data. [18F]4e was selected as the optimal candidate probe for subsequent biological investigation. To demonstrate the targeting specificity of [18F]4e to DAT, we performed a 30-min PET scan in normal and PD model rats. The semi-PD model caused by 6-OHDA is one of the frequently-used PD models, with the un-lesioned-side striatum often used as an internal control for comparative study with the lesioned side (Choi et al., 2012). The signal of the lesioned-side striatum of PD model rats was significantly reduced to cerebellum level, while the un-lesioned side of the striatum retained high radioactivity. The microPET results of normal and semi-PD model rats showed that [18F]4e binds to DAT with high specificity and can be used for the evaluation of dopaminergic degeneration (Fig. 3c-d). To further confirm the DAT specificity of [18F]4e, this probe was co-injected with CFT in normal rats. In the striatum, the concentration of [18F]4e dramatically decreased in the blocking group than in the control group, resulting in a similar signal between the target and non-target regions.
Autoradiography was performed to further prove the selective and specific binding of [18F]4e to DAT. In this study, ex vivo autoradiography of brain slices was performed at 40 min post injection of [18F]4e in rats (Fig. 4). A good radio-specific binding of [18F]4e was found in the DAT-rich striatum in the brain slices. And the high radioactivity of the striatum could be blocked by DAT inhibitor (CFT), giving a dramatic decrease of the target/non-target ratio (ST/CB). These results were consistent with the microPET imaging and further demonstrated that [18F]4e had a specific binding to DAT.
To investigate the pharmacokinetics of [18F]4e and [18F]6e in vivo, a biodistribution experiment was performed. Initial accumulation in the brains of [18F]4e and [18F]6e were shown to be 0.89 ± 0.09 and 1.09 ± 0.12 %ID at 5 min, respectively. The results showed high uptake in the striatum, followed by the hippocampus, with the lowest radioactivity in the cerebellum and cortex among these brain regions at 30 min post injection. The results of [18F]4e and [18F]6e were similar to those reported by Li et al. (2023). The ST/CB of [18F]4e after 30 min was higher than that of [18F]6e (Fig. 5c), which was consistent with TACs of PET imaging. Moreover, the maximum ST/CB value of [18F]4e (5.89 ± 1.23) was higher than [18F]FECIT-d4 (3.45 ± 0.23) reported by Li et al. (2023). These results indicated [18F]4e possessed appropriate pharmacokinetics in the target region. In the body, a similar accumulation of [18F]4e and [18F]6e was observed in most organs. Both tracers were distributed primarily to the liver (>12 %ID), lung (>2.2 %ID), kidney (>1.5 %ID) and lower accumulation in the heart, spleen, stomach, genital, pancreas, bone and muscle (Fig. 5a-b). This phenomenon showed that the main metabolic pathway of probes was predominant through the hepatobiliary system. The bone radioactive accumulation of [18F]6e and [18F]4e at 120 min were 0.50 ± 0.13 and 0.43 ± 0.10 %ID/g, respectively (Fig. S6 and Table S5, S6), indicating that the bone radioactivity of [18F]4e was decreased but not significant (P = 0.37).
In many cases, N-dealkylation starts when the C–H bond splits. Limiting N-dealkylation may increase the stability of the probes in vivo. N-dealkylation is one of the major pathways of metabolism of [18F]6e (Shin et al., 2012). The fraction of intact [18F]6e was l0 − 15% after 140 min (Lundkvist et al., 1997). In our study, metabolite experiments of [18F]4e revealed a remarkably reduced dealkylation in rats compared with [18F]6e (Fig. 6, Fig. S7 and Table S8). The metabolic components of [18F]4e in the striatum, cerebellum and plasma were less than those of [18F]6e at each time point post injection (Fig. 6) while parent tracer fractions of [18F]4e in ST showed a higher percentage than [18F]FECIT-d4 described by Li et al. (2023), indicating that [18F]4e give better in vivo stability. The metabolic analysis was supported by the above-mentioned microPET and biodistribution results. These results implied that [18F]4e had better stability in vivo.
Based on the above experiments, it was reasonable to speculate that the deuterium substitution of hydrogen on N-fluoropropyl might lead to a reduced metabolism in vivo. However, it is noticed that the deuterated probe [18F]FP-CMT-d6 did not give a PET improvement. It was possible that the p-toluene group of [18F]FP-CMT might be the main metabolic site in addition to the N-fluoroalkyl (Cumming et al., 2014) and deuterium substitution at the N-fluoropropyl had a limited effect on its metabolism rate.
In summary, the nanomolar affinity, outstanding selectivity and good in vivo stability of the deuterated DAT probe [18F]4e were confirmed. Nevertheless, the improved effects of [18F]4e had yet to be validated in primates and humans. This PET ligand is warranted for further clinical trials with the improvement of in vivo DAT measurement of related disorders such as PD.
5 Conclusion
In the present study, to develop in vivo stable 18F-labelled tracers for DAT, we successfully synthesized a series of 18F-labelled novel deuterated N-fluoropropyl tropane derivatives [18F]4a-e. These radio-labelled compounds showed good DAT affinity and suitable lipophilicity to penetrate the BBB. Among these radiotracers, [18F]4e showed a higher binding affinity to DAT than other radioligands as evaluated at IC50 value, cellular uptake and microPET imaging study. Further autoradiography and biodistribution experiments demonstrated good specific accumulation of [18F]4e in the target, while the in vivo metabolism study indicated a lower metabolic rate than clinically used [18F]6e. In general, [18F]4e might be a promising candidate for DAT PET measurement, which could be an excellent entry point for further clinical applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82172054), the Natural Science Foundation of Jiangsu Province (BK20201133, BK20210062), the Jiangsu Health Commission (M2022047) and the Science and Technology Project of Wuxi Administration of Traditional Chinese Medicine (ZYKJ202115).
Compliance with ethical standards
All animal experiments were approved by the Animal Care and Ethics Committee of Jiangsu Institute of Nuclear Medicine.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Dopamine transporter SPECT imaging in Parkinson’s disease and parkinsonian disorders. Turk. J. Med. Sci.. 2021;51:400-410.
- [CrossRef] [Google Scholar]
- A dopaminergic mechanism of antipsychotic drug efficacy, failure, and failure reversal: the role of the dopamine transporter. Mol. Psychiatry. 2020;25:2101-2118.
- [CrossRef] [Google Scholar]
- Identification of PET radiometabolites by cytochrome P450, UHPLC/Q-ToF-MS and fast radio-LC: applied to the PET radioligands [11C]flumazenil, [18F]FE-PE2I, and [11C]PBR28. Anal. Bioanal. Chem.. 2013;405:1303-1310.
- [CrossRef] [Google Scholar]
- Imaging of dopamine transporter with [18F]LBT-999: initial evaluation in healthy volunteers. Q. J. Nucl. Med. Mol. Imaging. 2022;66:148-155.
- [Google Scholar]
- Imaging of dopamine transporters with technetium-99m TRODAT-1 and single photon emission computed tomography. J. Neuroimaging. 2000;10:200-203.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of [18F]FECNT-d4 as a Novel PET Agent for Dopamine Transporter Imaging. Mol. Imag. Biol.. 2021;23:733-744.
- [CrossRef] [Google Scholar]
- Synthesis, ligand binding, QSAR, and CoMFA study of 3 beta-(p-substituted phenyl)tropane-2 beta-carboxylic acid methyl esters. J. Med. Chem.. 1991;34:2719-2725.
- [CrossRef] [Google Scholar]
- Evaluation of dopamine transporters and D2 receptors in hemiparkinsonian rat brains in vivo using consecutive PET scans of [18F]FPCIT and [18F]fallypride. Appl. Radiat. Isot.. 2012;70:2689-2694.
- [CrossRef] [Google Scholar]
- Radiosynthesis and validation of [18F]FP-CMT, a phenyltropane with superior properties for imaging the dopamine transporter in living brain. J. Cereb. Blood Flow Metab.. 2014;34:1148-1156.
- [CrossRef] [Google Scholar]
- Relationship between lipophilicity and brain extraction of 11C-labelled radiopharmaceuticals. J. Nucl. Med.. 1983;24:1030-1038.
- [Google Scholar]
- Fluorine-18-FPCT: a PET radiotracer for imaging dopamine transporters. J. Nucl. Med.. 1997;38:119-126.
- [Google Scholar]
- Synthesis and biological evaluation of a series of novel N- or O-fluoroalkyl derivatives of tropane: potential positron emission tomography (PET) imaging agents for the dopamine transporter. Bioorg. Med. Chem. Lett.. 2001;11:3049-3053.
- [CrossRef] [Google Scholar]
- Kinetic deuterium isotope effects in cytochrome P450 reactions. Methods Enzymol.. 2017;596:217-238.
- [CrossRef] [Google Scholar]
- Identification and preclinical development of a 2,5,6-trisubstituted fluorinated pyridine derivative as a radioligand for the positron emission tomography imaging of cannabinoid type 2 receptors. J. Med. Chem.. 2020;63:10287-10306.
- [CrossRef] [Google Scholar]
- [123I]FP-CIT (DaTscan) SPECT brain imaging in patients with suspected parkinsonian syndromes. J. Neuroimaging. 2012;22:225-230.
- [CrossRef] [Google Scholar]
- Decreased defluorination using the novel beta-cell imaging agent [18F]FE-DTBZ-d4 in pigs examined by PET. EJNMMI Res.. 2011;1:33.
- [CrossRef] [Google Scholar]
- Kinetic modeling of [18F]VAT, a novel radioligand for positron emission tomography imaging vesicular acetylcholine transporter in non-human primate brain. J. Neurochem.. 2018;144:791-804.
- [CrossRef] [Google Scholar]
- Effects of dopaminergic drug treatments on in vivo radioligand binding to brain vesicular monoamine transporters. Nucl. Med. Biol.. 1996;23:467-471.
- [CrossRef] [Google Scholar]
- Simplified synthesis ofN-(3-[18F]fluoropropyl)-2β-carbomethoxy-3β-(4-fluorophenyl)nortropane ([18F]β-CFT-FP) using [18F]fluoropropyl tosylate as the labelling reagent. J. Labelled Comp. Radiopharm.. 2005;48:463-471.
- [CrossRef] [Google Scholar]
- Methods to increase the metabolic stability of 18F-radiotracers. Molecules. 2015;20:16186-16220.
- [CrossRef] [Google Scholar]
- Improved in vivo PET imaging of the adenosine A2A receptor in the brain using [18F]FLUDA, a deuterated radiotracer with high metabolic stability. Eur. J. Nucl. Med. Mol. Imaging. 2021;48(9):2727-2736.
- [CrossRef] [Google Scholar]
- Variability of FP-CIT PET patterns associated with clinical features of multiple system atrophy. Neurology. 2021;96:1663-1671.
- [CrossRef] [Google Scholar]
- Deuterated [18F]fluoroethyl tropane analogs as dopamine transporter probes: Synthesis and biological evaluation. Nucl. Med. Biol.. 2023;108334
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 2001;46:3-26.
- [CrossRef] [Google Scholar]
- Design and evaluation of [18F]CHDI-650 as a positron emission tomography ligand to image mutant huntingtin aggregates. J. Med. Chem.. 2023;66:641-656.
- [CrossRef] [Google Scholar]
- [18F] beta-CIT-FP is superior to [11C] beta-CIT-FP for quantitation of the dopamine transporter. Nucl. Med. Biol.. 1997;24:621-627.
- [CrossRef] [Google Scholar]
- Comparison of 2beta-carbomethoxy-3beta-(4-[18F]fluorophenyl)tropane and N-(3-[18F]fluoropropyl)-2beta-carbomethoxy-3beta-(4-fluorophenyl)nortropane, tracers for imaging dopamine transporter in rat. Mol. Imag. Biol.. 2010;12:269-277.
- [CrossRef] [Google Scholar]
- Synthesis and receptor binding of N-substituted tropane derivatives. High-affinity ligands for the cocaine receptor. J. Med. Chem.. 1991;34:1728-1731.
- [CrossRef] [Google Scholar]
- Low dopamine transporter binding in the nucleus accumbens in geriatric patients with severe depression. Psychiatry Clin. Neurosci.. 2020;74:424-430.
- [CrossRef] [Google Scholar]
- PET imaging of VMAT2 with the novel radioligand [18F]FE-DTBZ-d4 in nonhuman primates: comparison with [11C]DTBZ and [18F]FE-DTBZ. ACS Chem. Nerosci.. 2021;12:4580-4586.
- [CrossRef] [Google Scholar]
- Boosted dopamine and blunted serotonin in Tourette syndrome - evidence from in vivo imaging studies. Rev. Neurosci.. 2022;33:859-876.
- [CrossRef] [Google Scholar]
- Quantification of dopamine transporter density with [18F]FECNT PET in healthy humans. Nucl. Med. Biol.. 2014;41:217-222.
- [CrossRef] [Google Scholar]
- Dynamic [18F]FPCIT PET: quantification of Parkinson's disease metabolic networks and nigrostriatal dopaminergic dysfunction in a single imaging session. J. Nucl. Med.. 2021;62:1775-1782.
- [CrossRef] [Google Scholar]
- Difficulties in dopamine transporter radioligand PET analysis: the example of LBT-999 using [18F] and [11C] labelling: part II: Metabolism studies. Nucl. Med. Biol.. 2012;39:347-359.
- [CrossRef] [Google Scholar]
- PET versus SPECT: strengths, limitations and challenges. Nucl. Med. Commun.. 2008;29:193-207.
- [CrossRef] [Google Scholar]
- Usefulness of PET with [18F]LBT-999 for the evaluation of presynaptic dopaminergic neuronal loss in a clinical environment. Front. Neurol.. 2020;11:754.
- [CrossRef] [Google Scholar]
- Tropane-derived 11C-labelled and 18F-labelled DAT ligands. J. Labelled Comp. Radiopharm.. 2013;56:149-158.
- [CrossRef] [Google Scholar]
- Identification and regional distribution in rat brain of radiometabolites of the dopamine transporter PET radioligand [11C]PE2I. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:667-678.
- [CrossRef] [Google Scholar]
- Effect of animal condition and fluvoxamine on the result of [18F]N-3-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl) nortropane ([18F]FP-CIT) PET study in mice. Nucl. Med. Mol. Imaging. 2012;46:27-33.
- [CrossRef] [Google Scholar]
- Unsupervised clustering of dopamine transporter PET imaging discovers heterogeneity of parkinsonism. Hum. Brain Mapp.. 2020;41:4744-4752.
- [CrossRef] [Google Scholar]
- Developing a preclinical model of Parkinson's disease: a study of behaviour in rats with graded 6-OHDA lesions. Behav. Brain Res.. 2006;169:1-9.
- [CrossRef] [Google Scholar]
- Single photon emission computed tomography/positron emission tomography molecular imaging for parkinsonism: a fast-developing field. Ann. Neurol.. 2021;90:711-719.
- [CrossRef] [Google Scholar]
- Design and challenges of radiopharmaceuticals. Semin. Nucl. Med.. 2019;49:339-356.
- [CrossRef] [Google Scholar]
- VMAT2 imaging agent, D6-[18F]FP-(+)-DTBZ: Improved radiosynthesis, purification by solid-phase extraction and characterization. Nucl. Med. Biol.. 2019;72–3:26-35.
- [CrossRef] [Google Scholar]
- Improved synthesis of beta-CIT and [11C]beta-CIT labeled at nitrogen or oxygen positions. Nucl. Med. Biol.. 1996;23:981-986.
- [CrossRef] [Google Scholar]
- PET imaging of the dopamine transporter with [18F]FECNT: a polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med.. 2006;47:520-527.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105278.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




