

Translate this page into:
Synthesis and greener pastures biological study of bis-thiadiazoles as potential Covid-19 drug candidates
⁎Corresponding authors riyadh1993@hotmail.com (Sayed M. Riyadh), stefan.braese@kit.edu (Stefan Bräse), smgomha@iu.edu.sa (Sobhi M. Gomha)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A novel series of bis- (Abdelhamid et al., 2017; Banerjee et al., 2018; Bharanidharan et al., 2022)thiadiazoles was synthesized from the reaction of precursor dimethyl 2,2′-(1,2-diphenylethane-1,2-diylidene)-bis(hydrazine-1-carbodithioate) and hydrazonyl chlorides in ethanol under ultrasonic irradiation. Spectral tools (IR. NMR, MS, elemental analyses, molecular dynamic simulation, DFT and LUMO and HOMO) were used to elucidate the structure of the isolated products. Molecular docking for the precursor, 3 and ligands 6a-i to two COVID-19 important proteins Mpro and RdRp was compared with two approved drugs, Remdesivir and Ivermectin. The binding affinity varied between the ligands and the drugs. The highest recorded binding affinity of 6c with Mpro was (−9.2 kcal/mol), followed by 6b and 6a, (−8.9 and −8.5 kcal/mol), respectively. The lowest recorded binding affinity was (−7.0 kcal/mol) for 6 g. In comparison, the approved drugs showed binding affinity (−7.4 and −7.7 kcal/mol), for Remdesivir and Ivermectin, respectively, which are within the range of the binding affinity of our ligands. The binding affinity of the approved drug Ivermectin against RdRp recoded the highest (−8.6 kcal/mol), followed by 6a, 6 h, and 6i are the same have (−8.2 kcal/mol). The lowest reading was found for compound 3 ligand (−6.3 kcal/mol). On the other side, the amino acids also differed between the compounds studied in this project for both the viral proteins. The ligand 6a forms three H-bonds with Thr 319(A), Sr 255(A) and Arg 457(A), whereas Ivermectin forms three H-bonds with His 41(A), Gly143(A) and Gln 18(A) for viral Mpro. The RdRp amino acids residues could be divided into four groups based on the amino acids that interact with hydrogen or hydrophobic interactions. The first group contained 6d, 6b, 6 g, and Remdesivir with 1–4 hydrogen bonds and hydrophobic interactions 1 to 10. Group 2 is 6a and 6f exhibited 1 and 3 hydrogen bonds and 15 and 14 hydrophobic interactions. Group 3 has 6e and Ivermectin shows 4 and 3 hydrogen bonds, respectively and 11 hydrophobic interactions for both compounds. The last group contains ligands 3, 6c, 6 h, and 6i gave 1–3 hydrogen bonds and 6c and 3 recorded the highest number of hydrophobic interactions, 14 for both 6c and 6 h. Pro Tox-II estimated compounds’ activities as Hepatoxic, Carcinogenic and Mutagenic, revealing that 6f-h were inactive in all five similar to that found with Remdesivir and Ivermectin. The drug-likeness prediction was carried out by studying physicochemical properties, lipophilicity, size, polarity, insolubility, unsaturation, and flexibility. Generally, some properties of the ligands were comparable to that of the standards used in this study, Remdesivir and Ivermectin.
Keywords
Bis-[1,2,4]thiadiazoles
Molecular docking
Binding energy
Remdesivir
Ivermectin
Drug-likeness prediction

1 Introduction
The manifestation and spreading of Covid-19 virus have orchestrated many researchers to formulate and construct novel bioactive heterocycles as antiviral agents. Hydrazones tethered azoles were considered as template for pharmaceutical drugs that inhibit Epidermal Growth Factor Receptors (EGFR) kinase enzyme as anticancer agents (Senkardes et al., 2021; Labib et al., 2018). Analogously, hydrazono-azines displayed potent growth inhibition activity against lung, leukemia, and ovarian cancer cell lines (Zhang et al., 2014). Also, a diverse class of hydrazones displayed anti-inflammatory (Bharanidharan et al., 2022), anticholinesterase (Cosar et al., 2022), and antimicrobial (Khoramil and Shaterian, 2015) activities. The therapeutic effect of bis-heterocycles has been studied for several pathological conditions including inflammation, cancer, and hypertension. For example, bis-thiadiazoles revealed high potency as an antihypertensive a-blocking (El-Enany et al., 2019), antimicrobial (El-Enany et al., 2021; Mahmoud et al., 2021; Gomha et al., 2018), and anticancer (Gomha et al., 2016) activities. Furthermore, the inhibition corrosion efficiency of bis-thiadiazoles, tethered by alkyl linker, on mild steel was reported (Singh and Quraishi, 2010). Additionally, these compounds with high nitrogen content have high detonation performance and are insensitive to external, which stimuli could be used as promising candidates for high-energy materials (Pu et al., 2020). Thus, the synergism of bis-thiadiazoles with hydrazone moiety in a hybridized molecule may increase its biological potency and industrial applications. Promoted by the above observations, it was aimed to synthesize and evaluate their biological importance.
2 Results and discussions
2.1 Chemistry
The precursor material, namely dimethyl 2,2′-(1,2-diphenylethane-1,2-diylidene)-bis(hydrazine-1-carbodithioate) (3), was readily synthesized from a condensation reaction of benzil (1) with two equivalent of methyl hydrazinecarbodithioate (2) in 2-propanol under stirring condition (Scheme 1). Spectral data and elemental analysis were in favor of the proposed product. The absorption bands in the IR spectrum of compound 3 appeared at 3290, 1626, and 1375 cm−1 due to stretching vibration of (NH), (C⚌N), and (C⚌S) groups, respectively. 1H NMR was characterized by a singlet signal at δ = 2.47 ppm attributed to (SCH3) group in carbodithioate moiety (Abdelhamid et al., 2017).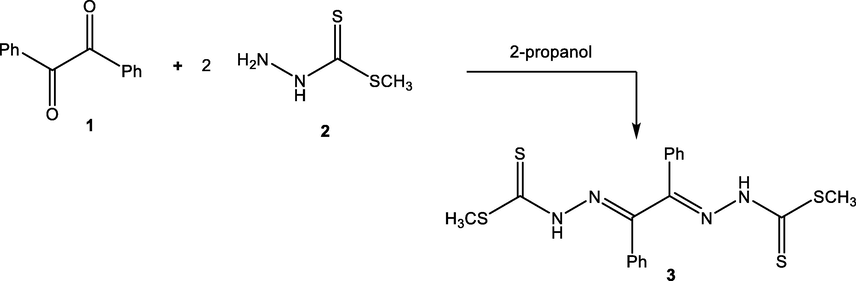
Synthesis of bis(hydrazine-1-carbodithioate).
Continuing our work on bis-heterocycle synthesis (El-Enany et al., 2021; Mahmoud et al., 2021; Gomha et al., 2018; Gomha et al., 2016; Mahmoud et al., 2019; Gomha et al., 2015), the chemical reactivity of compound 3 towards various hydrazonoyl chlorides was studied to prepare new series of bis-thiadiazole derivatives. Thus, the reaction of bis(hydrazine-1-carbodithioate) 3 with various derivatives of hydrazonoyl chlorides 4a-i (two equivalents) (Eweiss and Osman, 1980; Shawali and Abdelhamid, 1971) in ethanol under ultrasonic irradiation (20–60 min) in the presence of triethylamine as a basic catalyst, afforded the respective bis-thiadiazoles 6a-i as depicted in Scheme 1. The development of all reactions was tracked by thin- layer chromatography (TLC). The mechanistic pathway of this reaction was preceded by sequential nucleophilic substitution of thiol groups to give non-isolable intermediate 5 followed by intramolecular cyclization and elimination of methanethiol to give the respective isolable products 6a-i (Scheme 2).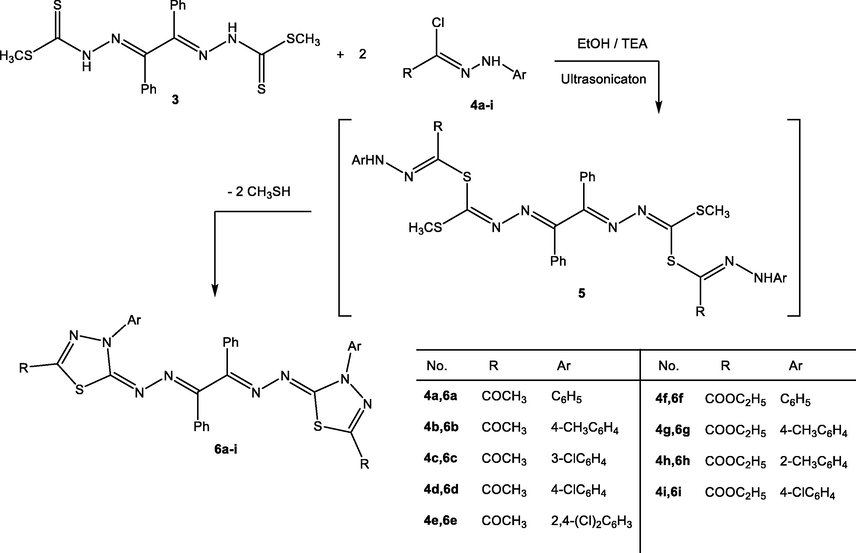
Synthesis of bis-thiadiazole derivatives 6a-i.
The structural assignment for the compounds 6a-i was based on their spectroscopic investigations. IR spectrum revealed the absence of (C⚌S) and (NH) absorption bands and the presence of a new absorption band due to the carbonyl group. 1H NMR spectrum displayed up-field signals attributed to protons of acetyl and ester groups.
The chemical evidence for the assigned structure of compounds 6a-i was achieved through alternative synthesis. Thus, treatment of two equivalents of ethyl 2-hydrazono-3-phenyl-1,3,4-thidiazoline-5-carboxylate (7) with benzil (1) under thermal condition afforded authentic product identical in all respects (mp, mixed mp, and IR) to the isolated product 6f (Scheme 2).
2.2 Molecular modeling with 6LU7 and 6 M71
Covid-19 and its variants made the world experience hard-time at several social and economic levels. Omicron, Delta variants and more continue to emerge; some appear and vanish while others continue (Donnelly and Patrinos, 2021; Gallo Marin et al., 2021; Zhang et al., 2020; Zia et al., 2021). Therefore, we have been stimulated to research how our ligands 3 and 6a-i might interact with the active site of Covid-19 sites. This study includes viral proteases, RNA-dependent RNA polymerase (RdRp, PDB, 6 M71), and a target protein 6LU7. Both M and N viral proteins are significant in various stages of viral replication. Importantly it is an attractive goal for numerous antiviral remedial agents. The present study screened two potential approved drugs, Ivermectin and Remdesivir, alongside our ligands for a comparison. The Mpro, 6LU7, was used in this study because it was molecularly docked with the N3 inhibitor published in Nature journal (Jin et al., 2020). RdRp is used as a target for viral drug design and one example is Remdesivir which was designed against RdRp's Ebola virus (Picarazzi et al., 2020).
Significantly, and toward encouraging greener biological pastures preliminary results, we used free computer-aided software to screen and filter ligands 3 and 6a-i versus the approved medicine before any in vivo or/and other experiments to save energy. Interestingly, computer-aided science techniques are now outstanding in accuracy and accessibility. Computer-assisted drug designs have been used for over 40 years (van Gunsteren and Karplus, 1982). The molecular docking for 6a-i and the approved drugs, Ivermectin and Remdesivir, with 6LU7, are summarized in Fig. 1. The superpositions of all compounds, alongside Ivermectin, and Remdesivir drugs, against the Mpro, 6LU7, gave a glorious insight image of the laydown of all molecules with concerning the approved drugs. For example, 6 h, 6i, and 6 g are parallel to Ivermectin and Remdesivir drugs, whereas 6c, 6e, 6d, and 6f intersect the drugs molecule. The presentations of the ligands before and after docking into 6LU7 are not identical in all docking cases (Fig. 1). Compound 3 is presented separately as a key sample in Fig. 2. The display of compound 3 is shown before and after docking, demonstrating a rearrangement of 3 to fit the cavity.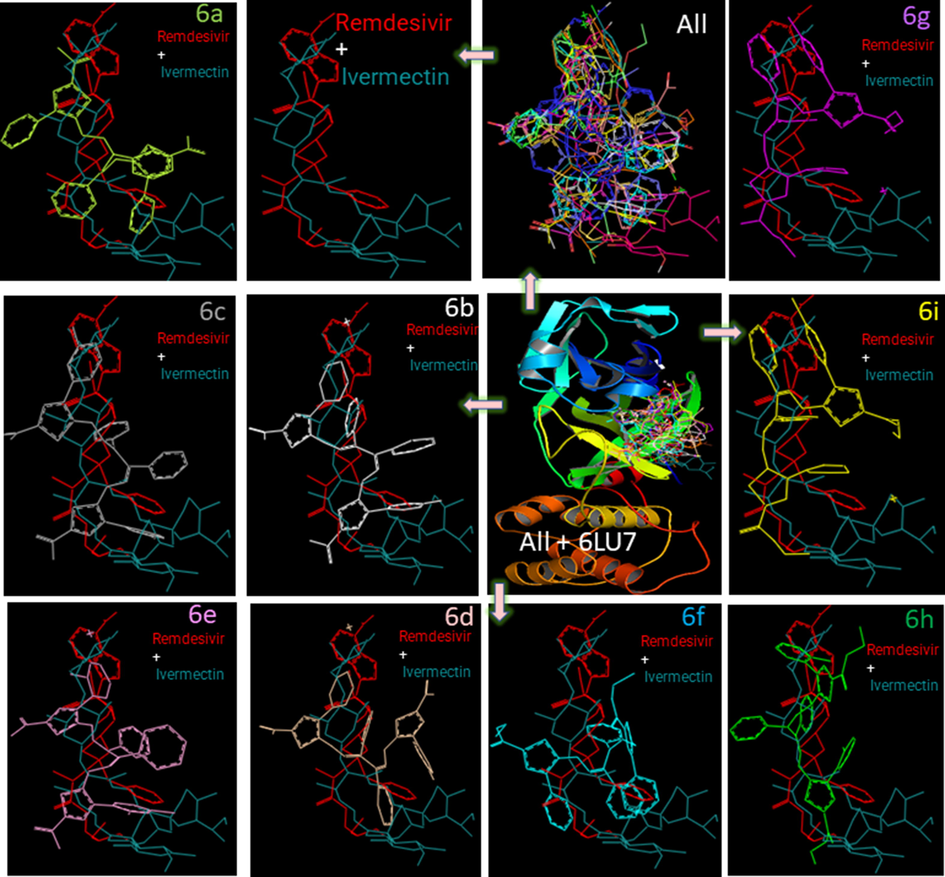
The superpositions of 6a–i, Ivermectin, and Remdesivir docked together into the binding pocket of 6LU7 for comparison. All compounds are color-coded. Each rectangular shows a superimposed ligand over both Remdesivir and Ivermectin for orientation comparison using PyMOL (DeLano, 2004).
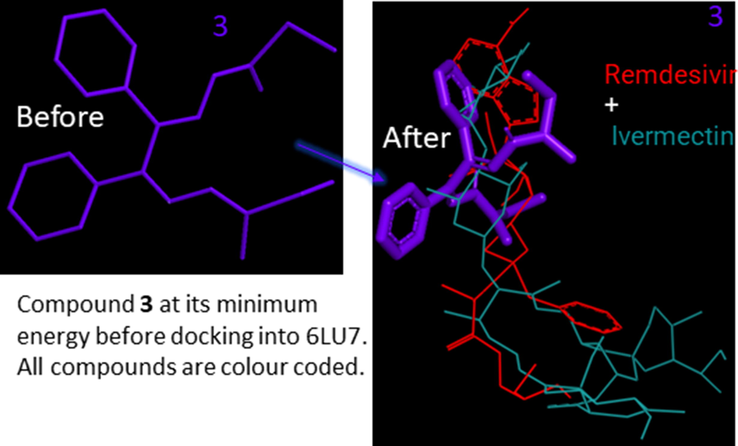
The superpositions of compound 3, Ivermectin and Remdesivir drugs, molecularly docked together into the binding pocket of 6LU7 as a key sample for Fig. 1.
Furthermore, the binding affinity of Remdesivir and Ivermectin with 6LU7 are (−7.7 and −7.4 kcal/mol), respectively. The binding energy of compounds 6a-i is in the range of (−9.2 to −6.6 kcal/mol), suggesting possible similar biological behavior to Remdesivir and Ivermectin drugs (Ottesen and Campbell, 1994; Shah et al., 2020; Rubin et al., 2020). Molecular docking was also done using the same parameters for the same compounds with 6 M71 to show binding energy (−7.7 and −7.4 kcal/mol) for Remdesivir and Ivermectin, respectively. In contrast, the binding energy of compounds 6a-i is in the range of (−9.4 to −6.6 kcal/mol) (Fig. 3). This could encourage a further study of the compounds 6a-i and 3 toward finding a potential inhibitor for COVID-19. The two approved drugs were chosen because they are active against several viral diseases, including influenza (Shah et al., 2020).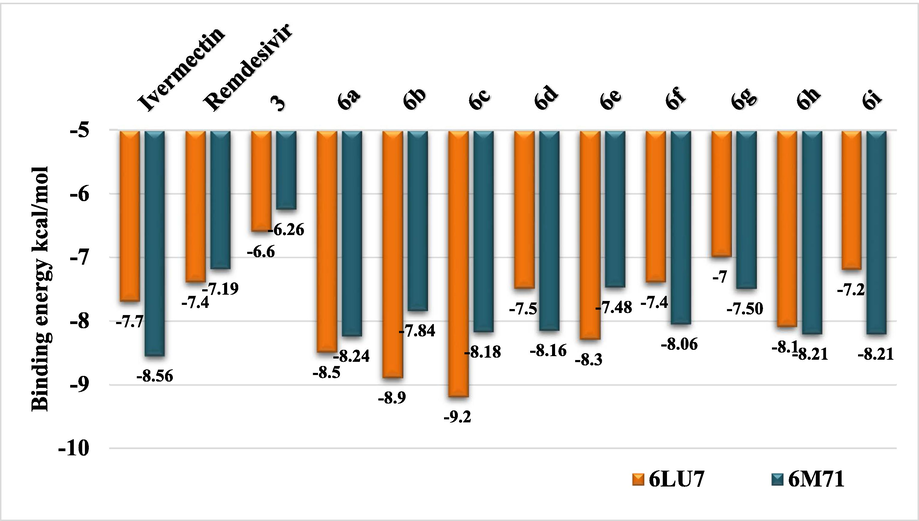
Binding energies of compounds 3 and 6a-i, Ivermectin and Remdesivir with 6LU7 and 6 M71 for comparison. Binding affinities/energies are shown on each compound's top of each column.
The presence of hydrogen bonds and hydrophobic interactions between ligands 3 and 6a-i and the receptor's active amino acid residues of 6LU7 and 6 M71 (representative examples are shown in Fig. 4) are associated with the binding affinity (Williamson and Williams, 1984). An example of hydrogen bonds and hydrophobic interactions between ligand 6a and the receptor's active amino acid residues of 6LU7 and 6 M71 are presented in Fig. 4. It could be seen that 6a forms three H-bonds with Thr 319(A), Sr 255(A) and Arg 457(A) whereas Ivermectin forms three H-bonds with His 41(A), Gly143(A) and Gln 18(A). Key symbols were reported in our previous work (Alsafi et al., 2020).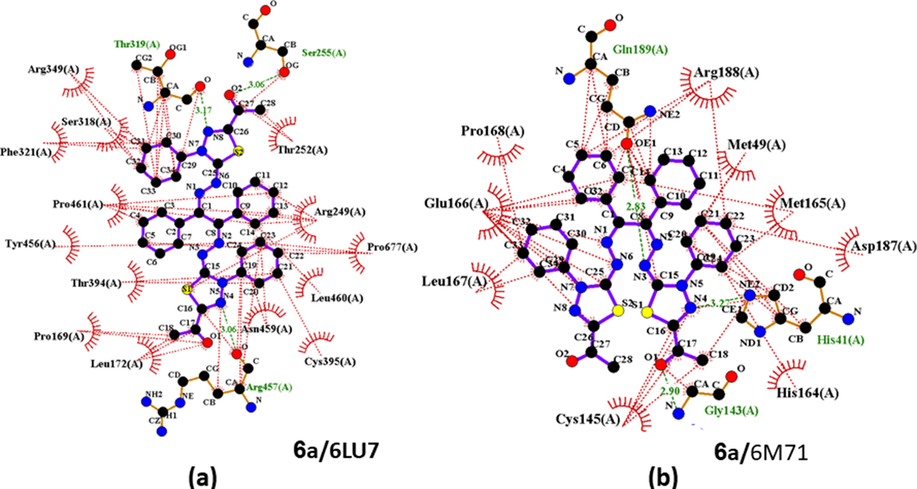
(a) A diagram of 2D LIGPLOT depiction of 6a against 6LU7; (b) 6a with 6 M71. Complex showing the hydrogen bonds (green lines) and hydrophobic interactions (red lines).
All interactions have been summarized for compounds 3, 6a-I, and the approved drugs against 6LU7 in Fig. 5.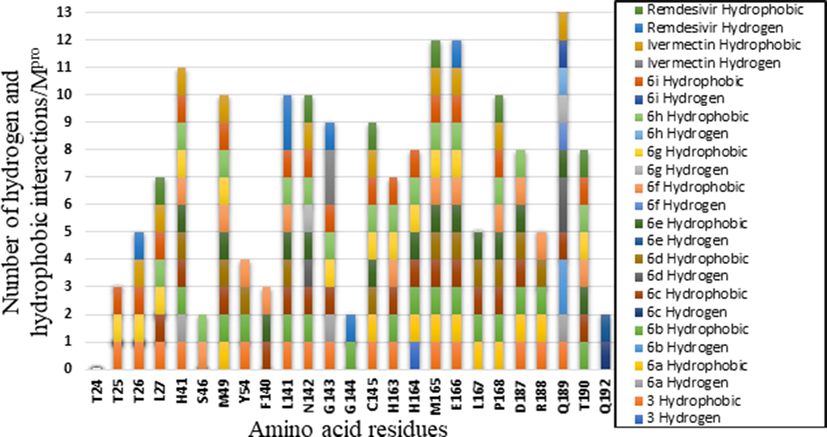
Hydrophobic interactions and H-bonds unveiled upon docking with 6LU7 protease for Ivermectin and Remdesivir, 3 and 6a-i. They are color-coded.
Amino acids H41, M49, L141, N142, M165, E166, P168 and Q189 appeared ten times or more, in Fig. 5, with all ligands and drugs in this study (Shah et al., 2020) The amino acid, P189, displayed 13 interactions forming either a H-bond or hydrophobic interaction with the amino acid residues of the Mpro substrate of COVID-19 (6LU7). The maximum interactions are 4H-bond and hydrophobic interactions. Surprisingly, residues 87 amino acid residues were involved in the case of RdRp.
Also, the interaction of compounds 3, 6a-i, and the approved drugs against 6 M71 is presented in Fig. 6. Amino acid residues Phe35, Lys47, Lys50, Tyr129, His133, Asn138, Thr206, Asn209, Asp218, Lys780 and Asn781 appeared at least four times as a result of the interaction, between all the ligands and 6 M71 (RdRp), in the form of either a H-bond or hydrophobic interactions (Fig. 6). Amino acid residue Lys780 scored the highest interactions (Skariyachan et al., 2020).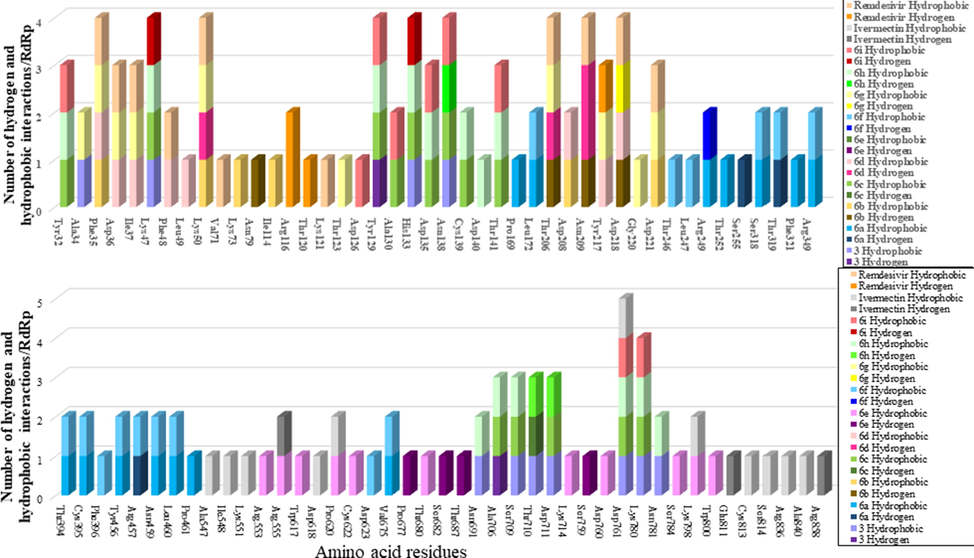
Number of H-bonds and hydrophobic interactions found upon docking 6 M71 RNA-dependent RNA polymerase (RdRp) against our compounds 3, 6a-i, and the two approved medicines Ivermectin and Remdesivir.
COVID-19's RNA-dependent RNA polymerase (RdRp), also called nsp12 structure is complex. It contains different domains; nsp12, nsp7, and two copies of nsp8. Then the enzyme's catalytic site contains several motifs named by the letters A to G (Fig. 7) (Gao et al., 2020; Jiang et al., 2021; Poustforoosh et al., 2021). In this study, the newly synthesized ligands 6a-i, and their precursor 3 were compared to the antiviral approved medicine Remdesivir (Eastman et al., 2020] and the antiparasitic Ivermectin (Conterno et al., 2020). The RdRp amino acids were docked against all different active sites of the protein similar to those identified recently (Poustforoosh et al., 2021; Muratore and Komai, 2020). They divided RdRp protein depending upon druggability. Some sites contain residues essential for RdRp function; Gao et al., 2020; Jiang et al., 2021, Poustforoosh et al., 2021). Docking results of different ligands and approved drugs can be divided into four groups (Fig. 7, Table 1, and Table 1S).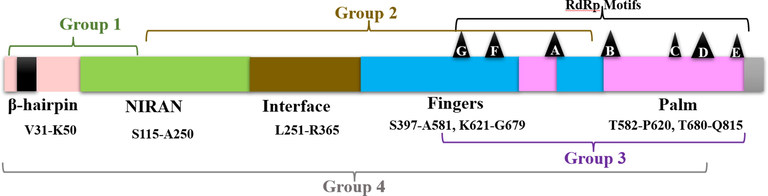
COVID-19 RdRp (nsp12) protein domain organization and groups defined after docking against the ligands, Remdesivir and Ivermectin. Domains organizations are based on Gao and co-workers (Gao et al., 2020).
Groups
Ligands and Drugs
First Amino acid
Last amino acid
Interactions
Min-Max Hydrogen
Min-Max Hydrophobic
1
6b, 6d, 6 g, Remdesivir
Ala34
Asp221
1–4
6–10
2
6a, 6f
Pro169
Val675
1–3
14–15
3
6e, Ivermectin
Ala547
Arg858
3–4
11
4
3, 6c, 6 h, 6i
Tyr32
Asn781
1–3
9–11
The first group 6d, 6b, 6 g, and Remdesivir have a range of 1 to 4 hydrogen bonds and 1 to 10 hydrophobic interactions. These amino acids were at the β-hairpin and NIRAN domains of the enzyme (Fig. 7). β-hairpin domain was identified in the N-terminus of the protein and it was found to stabilize protein structure by being inserted into the groove and held by both the NIRAN domain and the palm subdomain (Gao et al., 2020). The NIRAN domain is identified between Ser115-Ala250. Three out of four of the compounds in this group have four hydrogen bonds, mainly in the domain (Table 1S). Group 2 is 6a and 6f exhibit 1 and 3 hydrogen bonds, respectively, and 15 and 14 hydrophobic interactions (Table 1).
Group 3 is 6e and Ivermectin show 4 and 3 hydrogen bonds, respectively and both in total have 11 hydrophobic interactions. Motif A in the finger's domain is the active site for RdRp in the range of 611–662. It has the classic divalent-cation–binding D618 [Gao et al., 2020]. Interestingly, it was reported that Ivermectin docked into this amino acid. In this group, both compounds docked to the catalytic residues Ser759, Asp760 and D761 [Gao et al., 2020]. However, 6e shows a hydrogen bond with Ser759 and hydrophobic interaction with 760, whereas Ivermectin displays a hydrophobic interaction with 761 (Fig. 7 and Table 1S). These two compounds also interacted in the docking with Arg553 and Arg555, residues in the F motif (Fig. 7). This motif is important in forming clamped RNA template grooves with G and E motifs [Gao et al., 2020]. The 6e has hydrophobic interaction with both residues, while Ivermectin displays a hydrogen bond with 555.
The last group, group 4 contains compounds 3, 6c, 6 h, and 6i. Docking of these compounds resulted in interactions with different domains of the RdRp protein (Fig. 7). Compound 6c displayed the minimum number of hydrogen bonds (1), whereas 6c and 3 demonstrated the highest number of hydrogen bonds (3). The highest hydrophobic interactions found were 14 for 6c and 6 h (Table 1 and Table 1S).
2.3 Ligands toxicity and drug-likeness properties prediction
ProTox-II virtual tool predicted oral toxicity LD50, which is presented as the Lethal Dose (LD) at 50 % milligrams per kilograms tested population weight. Molecule toxicity in ProTox-II is divided into six gradual classes 1–6; one is the highest, whereas six is the lowest. Compound 3 and the nine ligands 6a-i were classified as 4 or 5 and compared to the approved drug Remdesivir 4. Compound 3 and 6e with LD50 (381 and 2000), respectively (Table 2). The ligands 6f, 6 g, and 6 h were also class 4 with LD50 predicted (500 mg/kg). The rest of the ligands were less toxic and belonged to class 5 with LD50 (2580 mg/kg) for 6a, 6b, and 6c and (5000 mg/kg) for 6d and 6i (Table 2). The average similarity for 3 and Remdesivir was (47.75 % and 40.93 %), respectively and predicted accuracy was similar for both compounds (54.26 %) (Table 2). The average similarity for all other ligands from 6a to 6i was close, with a range of (1.97 %) (Table S1). Dark green (strong inactive); light green (moderate inactive); Red colour (strong active), Pink (moderate active).
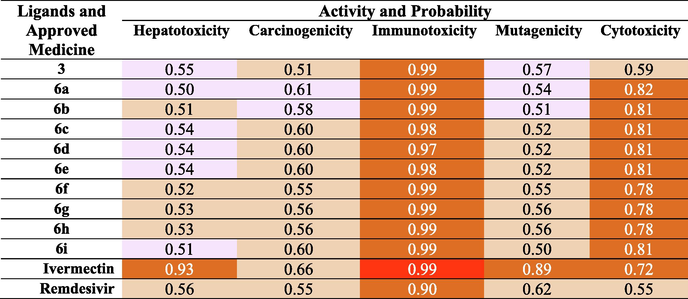
The ProTox-II web server also predicted organ toxicity targeting hepatoxicity estimation of the precursor and its ligands compared to Remdesivir. The ligands (6a −6i), including 3 varied in their hepatoxicity. Some were moderately active 3, 6a, 6c, 6d, 6e, and 6i with a probability between (0.50–0.55). The other ligands 6b, 6f, 6 g, and 6 h were moderately inactive similar to that of Remdesivir with a calculated probability between (0.51–0.53). However, Ivermectin was strongly inactive with a probability of (0.99). Carcinogenicity was predicted and only 6a and 6b were moderately active with a probability of (0.61 and 0.58), respectively (Table 2). All other compounds, including the precursor and both standards, were moderately inactive with a probability between (0.51–0.66). In addition, most compounds in this study were predicted to be strongly inactive except Ivermectin which was strongly active as immunotoxicity. The precursor, 6a and 6b were found to be moderately active as a mutagen. Nevertheless, compounds 6d, 6e-6i and the standard were inactive mutagens (Table 2). The ligands 6a-6i were anticipated to be strongly inactive as cytotoxic compounds with a probability of (0.72–0.82). But compound 3 and the standard Remdesivir were moderately inactive with a probability of (0.59 and 0.55), respectively (Table 2).
The Pro Tox-II estimation of all compounds showed that they vary in their activities as Hepatoxic, carcinogenic and mutagenic. We could conclude that 6f-h were predicted to be inactive in the five activities comparable to Remdesivir (Table 2).
The physicochemical properties also valued by SwissADME included the molecular weight (g/mol), the molecular refractivity, and the topological polar surface area (Å2). The molecular weight (MW) of compound 3 was in the accepted range of 418.62. All other ligands, including Remdesivir, were out of the MW range between 50 and 500 g/mol (Table 4 and Fig. 8). The total surface area polarity (TSAP) was evaluated. It was higher than 20–130 Å2 range, for all compounds, including the standard, Remdesivir. The H-bond acceptor, which indicates molecule solubility, should not exceed six. However, all ligands were more than 6 accept the precursor 3 (Table 3).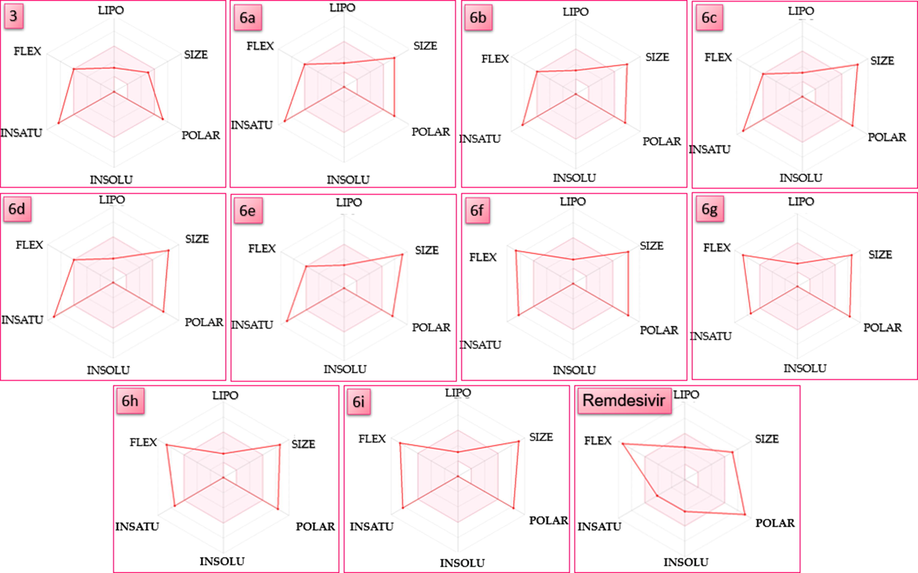
Bioavailability radar for ligands 6a-i, 3 and the Remdesivir drug.
Physicochemical
3
6a
6b
6c
6d
6e
6f
6 g
6 h
6i
Remdesivir
Molecular Weight g/mol
418.62
642.75
670.81
711.64
711.64
780.53
702.80
730.86
730.86
771.69
602.58
Heavy atom
26
46
48
48
48
50
50
52
52
52
42
Arom. heavy atom
12
34
34
34
34
34
34
34
34
34
15
Fraction Csp3
0.11
0.06
0.11
0.06
0.06
0.06
0.11
0.16
0.16
0.11
0.48
Rotatable bond
9
9
9
9
9
9
13
13
13
13
14
H–Bond acceptor
2
8
8
8
8
8
10
10
10
10
12
H–Bond donor
2
0
0
0
0
0
0
0
0
0
4
Molar refractivity
123.67
180.35
190.29
190.37
190.37
200.39
192.14
202.07
202.07
202.16
150.43
Polar surface area Å2
163.56
175.70
175.70
175.70
175.70
175.70
194.16
194.16
194.16
194.16
213.36
Lipophilicity
MLOGP
–
–
–
–
–
–
–
–
–
–
0.18
WLOGP
4.27
5.90
6.51
7.20
7.20
8.51
5.84
6.46
7.15
7.15
1.91
XLOGP3
–
–
–
–
–
–
–
–
–
–
1.91
To estimate drug-likeness, the bioavailability radar of the ligands and standard were drawn based on the physicochemical properties, lipophilicity, size, polarity, insolubility, unsaturation, and flexibility (Fig. 8). The ideal range of these properties is the pink area on the radar (Fig. 8). The red line on the radar plot represents the compounds analyzed (Fig. 8). The drawing of the precursor was similar and had four properties, lipophilicity, size, insolubility, and flexibility. Three properties were drawn in the pink area of the radar for the ligands 6a-6e, whereas ligands 6f-6i had only two properties lipophilicity and insolubility and unsaturation (Fig. 8). Generally, some properties of the ligands were comparable to the standard used in this study.
2.4 Molecular dynamics simulation and SEM map.
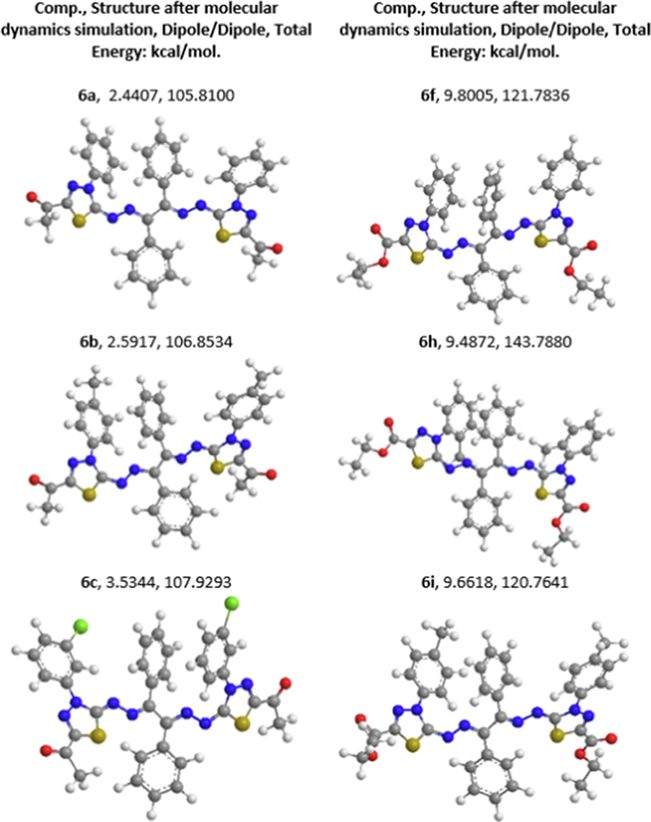
Molecular dynamics simulation (MDS) has been broadly used to better understand any molecule's structure to a function association (Hospital et al.; 2015). MDS is an easy computer-aided tool, time-saving, and hence an environmentally friendly method to gather information about the dynamic properties of the molecules, which is a proper preliminary study of the compounds. MDS with the dipole/dipole and total minimizing energy of selected compounds, using the MM2 method, are provided in Table 4. An increase in the dipole/dipole and total minimized energy by replacing the aldehyde group (6a) with an acetate group (6f) on the thiadiazol ring; however, MDS indicates the mechanical stability in this study of the selected molecules.
To predict and analyze electron-rich areas and electron-deficient for compound 6a as a representative example in this study, MEP was calculated by applying the same method and the basis sets used for geometry optimization. Calculation of frontier molecule orbital density distributions of 6a is also conducted. As displayed in Fig. 9, the negative regions are mainly shown on the aldehyde group on both sides (the energy color-coded scale is provided on the top of the MEP surface for easy comparison).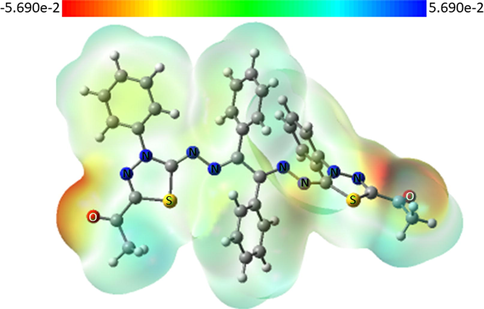
Molecular electrostatic potential (MEP) surfaces of 6a were calculated using the B3LYP/6-311G(D, P) basis set.
One important identification method and helpful information provider of molecular activity is HOMO–LUMO of any molecular structure, e.g., the more negative LUMO energy value is chemically more active molecules (Parlak et al., 2022). It was documented that compounds with higher stabilized LUMO orbitals show more biological activities (Kumar et al., 2018). HOMO–LUMO plots and calculated energy values for 6a, as a representative example, are shown in Fig. 10 The LUMO and HOMO orbitals are mainly located over the thiadiazol ring. The energy difference between the HOMO and LUMO is calculated to be 0.11501 eV (Fig. 10).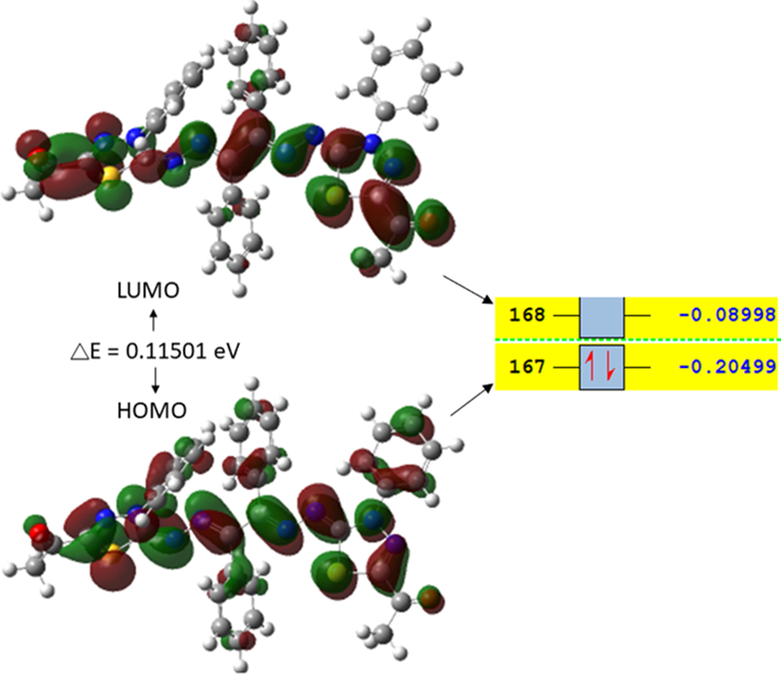
Calculated HOMO and LUMO orbitals of 6a using the B3LYP/6-311G(D, P) basis set.
3 Experimental section
3.1 Materials and methods
3.1.1 Instruments
An electrothermal Gallenkamp apparatus IA 9000 was operated to measure the melting points for the newly synthesized compounds. Pye-Unicam SP300 instrument in potassium bromide discs was used to measure IR spectra. A Varian Mercury VXR-300 spectrometer (300 MHz for 1H NMR and 75 MHz for 13C NMR) was manipulated to measure the 1H NMR and 13C NMR spectra and the chemical shifts were related to that of the solvent. GCMS-Q1000-EX Shimadzu spectrometer was conducted to record the mass spectra of the samples on the ionizing voltage at 70 eV. Elemental analyses were measured by an Elementarvario LIII CHNS analyzer (Germany). Shimadzu TGA-50H Thermal Analyzer was utilized to study the thermal degradation behavior of the samples from room temperature to 500 °C with a heating rate of 10 °C min−1.
3.1.2 Synthesis of dimethyl 2,2′-(1,2-diphenylethane-1,2-diylidene)bis(hydrazinecarbodithioate) (3)
A solution of benzil (1) (2.10 g, l0 mmol) and methyl hydrazinecarbodithioate 2 (2.44 g, 20 mmol) in 20 mL 2-propanol was stirred for 2 h at ordinary temperature. The formed precipitate was isolated via filtration then recrystallized from EtOH to afford compound 3 as yellow solid in 78 %; m.p. 191–193 °C; IR: v = 3290 (NH), 3048, 2914 (CH), 1626 (C⚌N), 1375 (C⚌S) cm−1; 1H NMR: δ = 2.47 (s, 6H, 2SCH3), 7.27–7.78 (m, 10H, Ar-H), 7.97 (br, s, 2H, 2NH); 13C NMR (DMSO‑d6): δ = 25.13 (CH3), 127.23, 128.16, 129.47, 134.11, 144.52 (Ar-C and C⚌N), 196.99 (C⚌S); MS m/z (%): 418 (M+, 38). Anal. Calcd for C18H18N4S4 (418.04): C, 51.65; H, 4.33; N, 13.38; S, 30.63. Found C, 51.51; H, 4.30; N, 13.26; S, 30.42 %.
3.1.3 General procedure for the synthesis of bis-1,3,4-thiadiazole derivatives (6a-i)
An ethanolic solution of bis(hydrazine-1-carbodithioate) 3 (0.418 g, 1 mmol) and the proper hydrazonoyl chlorides 4a-i (2 mmol) containing 1 mL of triethylamine was irradiated in an ultrasonic generator at 50 °C for 20–60 min (Radiation exposure continued until all the starting materials vanished and the product was developed, TLC supervised). The obtained precipitate of TEA / HCl was filtered off, and the mother liquor was evaporated. The formed solid product in each case was filtered off and crystallized from the appropriate solvent to give the respective bis-thiadiazole derivatives 6a-i.
The physical constants and analytical data of synthesized products 6a-i are listed below:
3.1.4 1,1′-(5,5′-((1,2-Diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-phenyl-4,5-dihydro-1,3,4-thiadiazole-2-yl-5-ylidene))diethanone (6a)
Orange solid, mp 216–218 °C; IR (KBr) υ = 3057, 2925 (CH), 1667 (C⚌O), 1596 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 2.45 (s, 6H, 2COCH3), 6.39–7.96 (m, 20H, Ar-H); 13C NMR (DMSO‑d6): δ = 24.93 (CH3), 117.26, 123.46, 127.85, 129.46, 129.96, 130.26, 131.05, 134.28, 137.55, 144.98, 149.91 (Ar-C and C⚌N), 191.99 (C⚌O); MS, m/z (%) 642 (M+, 23). Anal. calcd for C34H26N8O2S2 (642.16): C, 63.53; H, 4.08; N, 17.43; S, 9.98. Found: C, 63.43; H, 4.01; N, 17.35; S, 10.02 %.
3.1.5 1,1′-(5,5′-((1,2-Diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(p-tolyl)-4,5-dihydro-1,3,4-thiadiazole-2-yl-5-ylidene))diethanone (6b)
Yellow solid; mp 219–221 °C; IR (KBr) υ = 3052, 2917 (CH), 1655 (C⚌O), 1598 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 2.19 (s, 6H, 2Ar-CH3), 2.38 (s, 6H, 2COCH3), 6.46–7.97 (m, 18H, Ar-H); 13C NMR (DMSO‑d6): δ = 21.53 (Ar-CH3), 24.36 (CH3), 119.26, 123.42, 127.55, 128.96, 129.76, 130.26, 132.05, 135.28, 138.55, 144.88, 150.41 (Ar-C and C⚌N), 190.39 (C⚌O); MS, m/z (%) 670 (M+, 26). Anal. calcd for C36H30N8O2S2 (670.19): C, 64.46; H, 4.51; N, 16.70; S, 9.56. Found: C, 64.41; H, 4.42; N, 16.58; S, 9.68 %.
3.1.6 1,1′-(5,5′-((1,2-Diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(3-chloro phenyl)-4,5-dihydro-1,3,4-thiadiazole-2-yl-5-ylidene))diethanone (6c)
Orange solid; mp 233–235 °C; IR (KBr) υ = 3054, 2929 (CH), 1672 (C⚌O), 1596 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ 2.39 (s, 6H, 2COCH3), 6.19–7.97 (m, 18H, Ar-H); 13C NMR (DMSO‑d6): δ = 25.13 (CH3), 117.22, 121.46, 123.54, 125.85, 127.68, 128.46, 129.86, 130.66, 131.85, 134.28, 138.55, 145.18, 149.93 (Ar-C and C⚌N), 190.12 (C⚌O); MS, m/z (%) 712 (M++2, 18), 710 (M+, 5). Anal. calcd for C34H24Cl2N8O2S2 (710.08): C, 57.38; H, 3.40; N, 15.75; S, 9.01. Found: C, 57.45; H, 3.27; N, 15.64; S, 8.96 %.
3.1.7 1,1′-(5,5′-((1,2-Diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(4-chloro phenyl)-4,5-dihydro-1,3,4-thiadiazole-2-yl-5-ylidene))diethanone (6d)
Orange solid; mp 252–254 °C; IR (KBr) υ = 3051, 2927 (CH), 1669 (C⚌O), 1596 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 2.40 (s, 6H, 2COCH3), 6.24–7.95 (m, 18H, Ar-H); 13C NMR (DMSO‑d6): δ = 25.13 (CH3), 121.26, 125.46, 127.95, 128.94, 129.76, 130.76, 131.85, 135.18, 137.55, 145.98, 151.11 (Ar-C and C⚌N), 192.39 (C⚌O); MS, m/z (%) 712 (M++2, 50), 710 (M+, 13). Anal. calcd for C34H24Cl2N8O2S2 (710.08): C, 57.38; H, 3.40; N, 15.75; S, 9.01. Found: C, 57.48; H, 3.25; N, 15.59; S, 8.84 %.
3.1.8 1,1′-(5,5′-((1,2-Diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(2,4-dichloro phenyl)-4,5-dihydro-1,3,4-thiadiazole-2-yl-5-ylidene))diethanone (6e)
Brown solid; mp 281–283 °C; IR (KBr) υ = 3047, 2926 (CH), 1666 (C⚌O), 1598 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 2.47 (s, 6H, 2COCH3), 7.26–7.97 (m, 16H, Ar-H); 13C NMR (DMSO‑d6): δ = 25.08 (CH3), 120.22, 121.96, 123.81, 125.85, 127.78, 128.66, 129.83, 130.37, 132.81, 135.08, 138.15, 146.23, 150.43 (Ar-C and C⚌N), 192.85 (C⚌O); MS, m/z (%) 778 (M+, 5). Anal. calcd for C34H22Cl4N8O2S2 (778.01): C, 52.32; H, 2.84; N, 14.36; S, 8.21. Found: C, 52.46; H, 2.81; N, 14.29; S, 8.34 %.
3.1.9 5,5′-Diethyl 5,5′-((1,2-diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-phenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate) (6f)
Dark yellow solid; mp 202–204 °C; IR (KBr) υ = 3048, 2930 (CH), 1734 (C⚌O), 1626 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 1.14–1.32 (t, J = 6.8 Hz, 6H, 2CH2CH3), 4.19–4.27 (q, J = 6.8 Hz, 4H, 2CH2CH3), 7.25–7.71 (m, 20H, Ar-H); 13C NMR (DMSO‑d6): δ = 15.41 (CH3), 56.18 (CH2), 121.26, 122.46, 125.85, 128.46, 130.96, 131.86, 133.65, 135.28, 137.55, 145.18, 149.92 (Ar-C and C⚌N), 171.21 (C⚌O); MS, m/z (%) 702 (M+, 46). Anal. calcd for C36H30N8O4S2 (702.18): C, 61.52; H, 4.30; N, 15.94; S, 9.12. Found: C, 61.39; H, 4.22; N, 15.79; S, 9.24 %.
3.1.10 5,5′-Diethyl 5,5′-((1,2-diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(p-tolyl) −4,5-dihydro-1,3,4-thiadiazole-2-carboxylate) (6 g)
Yellow solid; mp 231–233 °C; IR (KBr) υ = 3059, 2918 (CH), 1723 (C⚌O), 1599 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 1.11–1.31 (t, J = 6.8 Hz, 6H, 2CH2CH3), 2.44 (s, 6H, 2Ar-CH3), 4.17–4.25 (q, J = 6.8 Hz, 4H, 2CH2CH3), 7.25–7.71 (m, 18H, Ar-H); 13C NMR (DMSO‑d6): δ = 15.25 (CH3), 21.85 (Ar-CH3), 55.88 (CH2), 121.16, 122.12, 125.75, 128.46, 129.16, 130.46, 132.65, 135.21, 137.27, 144.98, 150.12 (Ar-C and C⚌N), 170.98 (C⚌O); MS, m/z (%) 730 (M+, 74). Anal. calcd for C38H34N8O4S2 (730.21): C, 62.45; H, 4.69; N, 15.33; S, 8.77. Found: C, 62.33; H, 4.62; N, 15.19; S, 8.65 %.
3.1.11 5,5′-Diethyl 5,5′-((1,2-diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(o-tolyl) −4,5-dihydro-1,3,4-thiadiazole-2-carboxylate) (6 h)
Brown solid; mp 218–220 °C; IR (KBr) υ = 3047, 2927 (CH), 1729 (C⚌O), 1626 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 1.13–1.30 (t, J = 6.8 Hz, 6H, 2CH2CH3), 2.44 (s, 6H, 2Ar-CH3), 4.20–4.27 (q, J = 6.8 Hz, 4H, 2CH2CH3), 7.27–7.97 (m, 18H, Ar-H); 13C NMR (DMSO‑d6): δ = 15.33 (CH3), 21.82 (Ar-CH3), 56.18 (CH2), 120.16, 122.12, 123.98, 124.75, 125.87, 127.46, 128.16, 130.46, 131.53, 134.21, 137.27, 142.98, 149.72 (Ar-C and C⚌N), 170.45 (C⚌O); MS, m/z (%) 730 (M+, 17). Anal. calcd for C38H34N8O4S2 (730.21): C, 62.45; H, 4.69; N, 15.33; S, 8.77. Found: C, 62.49; H, 4.60; N, 15.24; S, 8.58 %.
3.1.12 5,5′-Diethyl 5,5′-((1,2-diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-(4-chlorophenyl)-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate) (6i)
Orange solid; mp 252–254 °C; IR (KBr) υ = 3047, 2922 (CH), 1725 (C⚌O), 1626 (C⚌N) cm−1; 1H NMR (300 MHz, DMSO‑d6) δ = 1.12–1.33 (t, J = 6.8 Hz, 6H, 2CH2CH3), 4.20–4.26 (q, J = 6.8 Hz, 4H, 2CH2CH3), 7.26–7.97 (m, 18H, Ar-H); 13C NMR (DMSO‑d6): δ = 15.42 (CH3), 56.28 (CH2), 121.26, 125.46, 127.85, 128.45, 129.96, 131.86, 133.21, 135.28, 138.55, 146.18, 152.92 (Ar-C and C⚌N), 170.11 (C⚌O); MS, m/z (%) 772 (M++2, 100), 770 (M+, 31). Anal. calcd for C36H28Cl2N8O4S2 (770.11): C, 56.03; H, 3.66; N, 14.52; S, 8.31. Found: C, 55.94; H, 3.52; N, 14.38; S, 8.18 %.
3.1.13 Alternate synthesis of 5,5′-Diethyl 5,5′-((1,2-diphenylethane-1,2-diylidene)bis(hydrazine-2,1-diylidene))bis(4-phenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate) (6f)
An ethanolic solution of benzil 1 (0.210 g, 1 mmol) and ethyl 2-hydrazono-3-phenyl-1,3,4-thidiazoline-5-carboxylate (7) (0.528 g, 2 mmol) was heated under reflux for 4 h. The formed precipitate was isolated via filtration then recrystallized from DMF to give authentic product 6a.
3.2 Docking in silico studies
The docking calculations of compounds 3, 6a-i, Remdesivir and Ivermectin and using 6LU7 and 6 M71(Mpro, RdRp PDB, https://www.rcsb.org/) were accomplished using the Autodock Vina wizard in PyRx 0.8. (Trott and Olson, 2009). Settings are made identical for docking in this research study: Grid box center X = 0, center Y = 0, center Z = 0, size X = 0, size Y = 0 and size Z = 0. The remaining parameters were used as a default setting in the Autodock Vina-PyRx. All drugs and ligands were converted to SDF file type using Chem. Draw program and were used as input to Autodock vina in PyRx. Before docking. The same is used for energy minimization. The PyMOL molecular viewer was used to present the output data (van Gunsteren and Karplus, 1982). Schematic diagrams of protein–ligand interactions were generated using the LIGPLOT program (Wallace et al., 1995).
3.3 In silico prediction
3.3.1 Ligands toxicity and drug-likeness properties prediction
Predict toxicity levels of likely synthesized drugs using In silico methods became a popular way or a few constraints; time, ethical and financial considerations (Rim, 2020; Raies and Bajic, 2016). This study estimated toxicity using ProTox-II platform (Banerjee et al., 2018). The Acute oral toxicity predictions for ligands and approved medicine were classified into different toxicity classes, depending upon the LD50 (mg/kg body weight). These classes were the same as in the Globally Harmonized System (GHS) classification and labeling of chemicals. These classes were ordered as following: class 1-fatal if swallowed (LD50 ≤ 5 mg/kg); class 2-fatal if swallowed (5 mg/kg < LD50 ≤ 50 mg/kg); class 3-toxic if swallowed (50 mg/kg < LD50 ≤ 300 mg/kg); class 4-harmful if swallowed (300 mg/kg < LD50 ≤ 2000 mg/kg); class 5- may be harmful if swallowed (2000 mg/kg < LD50 ≤ 5000 mg/kg). In silico predication for pharmacokinetic and drug-like properties of the Ligndes was carried out using SwissADME (Banerjee et al., 2018). SwissADME is an online server ( https://www.swissadme.ch/, accessed on 22 December 2021 as previously reported (Daina et al., 2017).
3.3.2 DFT, molecular dynamic simulations studies
DFT studies were performed using Gaussian 09, with the B3LYP functional in conjunction with the 6-311G(D,P) basis set for all atoms (Frisch et al.; 2009). MDS and molecules were optimized using the classical MM2 force field (Zare et al., 2016). Parameter Quality: Step Interval: 2.0 fs, Frame Interval: 10 fs, Terminate After: 10,000 steps, Heating/Cooling Rate: 1.000 Kcal/atom/ps, Target Temperature: 300 Kelvin.
4 Conclusions
To evaluate the potentiality of our novel compounds, computer-aided methods were used as a gesture for greener pastures to rank the compounds concerning the approved drugs Remdesivir and Ivermectin against Covid-19 infection. We synthesized a novel series of bis- (Abdelhamid et al., 2017; Banerjee et al., 2018; Bharanidharan et al., 2022)thiadiazoles 6a-i starting with dimethyl 2,2′-(1,2-diphenylethane-1,2-diylidene)-bis(hydrazine-1-carbodithioate) (3) via ultrasonic irradiation and elucidated their structures using spectral and elemental analyses. Molecular docking for precursor 3, ligands 6a-i Remdesivir and Ivermectin to two COVID-19 important proteins Mpro and RdRp was carried out under the same conditions and parameters. The RdRp amino acid residues showed various interactions of hydrogen or hydrophobic interactions. Compounds 6d, 6b, 6 g, and Remdesivir are in one group exhibiting 1–4 hydrogen bonds and 1–10 hydrophobic interactions. Compounds 6a and 6f exhibited fewer hydrogen bonds (1 and 3) and 15 and 14 hydrophobic interactions. Compound 6e and Ivermectin showed 4 and 3 hydrogen bonds and 11 hydrophobic interactions for both compounds. Compounds 3, 6c, 6 h, and 6i displayed 1–3 hydrogen bonds and 6c and 3 recorded the highest number of hydrophobic interactions, 14. The binding affinities with Mpro for compounds in this study were in the range of (−9.2 to 6.3 kcal/mol). The binding affinities for the approved medicines, Ivermectin and Remdesivir, were (−7.7 and −7.4 kcal/mol), respectively. Pro Tox-II estimated compounds' activities as Hepatoxic, Carcinogenic and Mutagenic, revealing that 6f-h were inactive similar to that found with Remdesivir and Ivermectin. The drug-likeness prediction was carried out by studying physicochemical properties, lipophilicity, size, polarity, insolubility, unsaturation, and flexibility. The preliminary results based on the comparative study in this paper suggest further investigation in the context of possible medicinal agents for COVID-19. The dipole/dipole and total minimizing energy increase by adding a chloro or methyl group to the aromatic ring attached to the thiadiazol ring. MEP surface of 6a shows the negative region is mainly shown on the aldehyde group.
Acknowledgements
The authors acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology. Stefan Bräse is grateful for support from the DFG-funded cluster program “3D Matter Made To Order” under Germany's Excellence Strategy -2082/1-390761711. The authors acknowledge grants from Science and Technology Commission of Shanghai Municipality (19440741300).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis of Certain New Thiazole and 1,3,4-Thiadiazole Derivatives via the Utility of 3-Acetylindole. J. Heterocycl. Chem.. 2017;54:1529-1536.
- [CrossRef] [Google Scholar]
- First COVID-19 molecular docking with a chalcone-based compound: synthesis, single-crystal structure and Hirshfeld surface analysis study. Acta Crystallographica Section C Struct. Chem.. 2020;76:1043-1050.
- [CrossRef] [Google Scholar]
- ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.. 2018;46:W257-W263.
- [CrossRef] [Google Scholar]
- Catalytic synthesis, in-vitro anti-inflammatory activity, and moleculardocking studies of novel hydrazone derivatives bearing N and S-heterocycles. Mater. Today Proc.. 2022;48:357-364.
- [CrossRef] [Google Scholar]
- Anticholinesterase activities of novel indole-based hydrazide-hydrazone derivatives: Design, synthesis, biological evaluation, molecular docking study and in silico ADME prediction. J. Mol. Struc.. 2022;1247:131398
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Reports. 2017;7:1-13.
- [CrossRef] [Google Scholar]
- PyMOL Reference Guide. Delano Scientific: San Carlos, CA, US; 2004. p. :1-68.
- Learning loss during Covid-19: An early systematic review. Prospects. 2021;10:1-9.
- [CrossRef] [Google Scholar]
- Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci.. 2020;6:672-683.
- [CrossRef] [Google Scholar]
- Synthesis and molecular docking of some new bis-thiadiazoles as anti-hypertensive α-blocking agents. Synth. Commun. 2019:1-12.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of Some Novel Bis-Thiadiazoles as Antimicrobial and Antitumor Agents. Polycycl. Aromat. Compds.. 2021;41:2071-2082.
- [CrossRef] [Google Scholar]
- Synthesis of heterocycles. Part II. New routes to acetylthia- diazolines and alkylazothiazoles. J. Heterocycl. Chem.. 1980;17:1713-1717.
- [CrossRef] [Google Scholar]
- Gaussian 09, Revision A.1. Wallingford, CT: Gaussian Inc; 2009.
- Predictors of COVID-19 severity: A literature review. Rev. Med. Virol.. 2021;31:1-10.
- [CrossRef] [Google Scholar]
- Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. BioSci. Trends. 2020;14:72-73.
- [CrossRef] [Google Scholar]
- Solvent-Drop Grinding Method: Efficient Synthesis, DPPH Radical Scavenging and Anti-diabetic Activities of Chalcones, bis-chalcones, Azolines, and bis-azolines. Curr. Org. Synth.. 2015;12:220-228.
- [CrossRef] [Google Scholar]
- One Pot Single Step Synthesis and Biological Evaluation of Some Novel Bis(1,3,4-thiadiazole) Derivatives as Potential Cytotoxic Agents. Molecules. 2016;21:1532.
- [CrossRef] [Google Scholar]
- Utility of Bis-Hydrazonoyl Chlorides as Precursors for Synthesis of New Functionalized Bis-Thiadiazoles as Potent Antimicrobial Agents. J. Heterocycl. Chem.. 2018;55:844-851.
- [Google Scholar]
- L Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem.. 2015;19(8):37-47.
- [CrossRef] [Google Scholar]
- RNA-dependent RNA polymerase: Structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun.. 2021;538:47-53.
- [CrossRef] [Google Scholar]
- Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289-293.
- [CrossRef] [Google Scholar]
- Preparation of 2-amino-3-cyano-4-aryl-5,10-dioxo-5,10-dihydro-4H-benzo[g]chromene and hydroxyl naphthalene-1,4-dione derivatives. Res. Chem. Intermed.. 2015;41:3171-3191.
- [CrossRef] [Google Scholar]
- Design, synthesis, DFT, docking studies and ADME prediction of some new coumarinyl linked pyrazolylthiazoles: Potential standalone or adjuvant antimicrobial agents. PLoS ONE. 2018;19:e0196016
- [CrossRef] [Google Scholar]
- Azole-hydrazone derivatives: Design, synthesis, in vitro biological evaluation, dual EGFR/HER2 inhibitory activity, cell cycle analysis and molecular docking study as anticancer agents. Bioorg. Chem.. 2018;76:67-80.
- [CrossRef] [Google Scholar]
- Synthesis and characterization of some novel bis-thiazoles. J. Heterocycl. Chem.. 2019;56:3157-3163.
- [CrossRef] [Google Scholar]
- Synthesis, Antimicrobial Evaluation and Molecular Docking of New Functionalized Bis(1,3,4-Thiadiazole) and Bis(Thiazole) Derivatives. Polycycl. Aromat. Compds.. 2021;41:2029-2041.
- [CrossRef] [Google Scholar]
- Theoretical study of the adiponectin receptors: binding site characterization and molecular dynamics of possible ligands for drug design. SN Appl. Sci.. 2020;2:533.
- [CrossRef] [Google Scholar]
- Ivermectin in human medicine. J. Antimicrob. Chemother.. 1994;34:195-203.
- [CrossRef] [Google Scholar]
- Interaction between favipiravir and hydroxychloroquine and their combined drug assessment: in silico investigations. Chem Zvesti. 2022;76:1471-1478.
- [CrossRef] [Google Scholar]
- Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge. Molecules. 2020;25:5695.
- [CrossRef] [Google Scholar]
- Evaluation of potential anti-RNA-dependent RNA polymerase (RdRP) drugs against the newly emerged model of COVID-19 RdRP using computational methods. Biophys. Chem.. 2021;272:106564
- [CrossRef] [Google Scholar]
- Theoretical design of bis-azole derivatives for energetic compounds. RCS adv.. 2020;10:13185-13195.
- [CrossRef] [Google Scholar]
- In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci.. 2016;6:147-172.
- [CrossRef] [Google Scholar]
- In silico prediction of toxicity and its applications for chemicals at work. Toxicol. Environ. Health Sci.. 2020;14:1-12.
- [CrossRef] [Google Scholar]
- FDA Approval of Remdesivir - A Step in the Right Direction. New England J. Med.. 2020;383:2598-2600.
- [CrossRef] [Google Scholar]
- Novel 2,6-disubstituted pyridine hydrazones: Synthesis, anticancer activity, docking studies and effects on caspase-3-me diate d apoptosis. J. Mol. Struc.. 2021;1223:128962
- [CrossRef] [Google Scholar]
- In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci.. 2020;252:117652
- [CrossRef] [Google Scholar]
- Synthesis and reactions of phenylcarbamoylarylhydrazidic chlorides. Tetrahedron. 1971;27:2517-2528.
- [CrossRef] [Google Scholar]
- The effect of some bis-thiadiazole derivatives on the corrosion of mild steel in hydrochloric acid. Corros. Sci.. 2010;52:1373-1385.
- [CrossRef] [Google Scholar]
- Structural and molecular basis of the interaction mechanism of selected drugs towards multiple targets of SARS-CoV-2 by molecular docking and dynamic simulation studies- deciphering the scope of repurposed drugs. Comp. Biol. Med.. 2020;126:104054
- [CrossRef] [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2009;31:455-461.
- [CrossRef] [Google Scholar]
- Protein Dynamics in Solution and in a Crystalline Environment: A Molecular Dynamics Study. Biochem.. 1982;21:2259-2274.
- [CrossRef] [Google Scholar]
- Ligplot: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng.. 1995;8:127-134.
- [CrossRef] [Google Scholar]
- Hydrophobic interactions affect hydrogen bond strengths in complexes between peptides and vancomycin or ristocetin. Eur. J. Biochem.. 1984;138:345-348.
- [CrossRef] [Google Scholar]
- A comparative QSAR analysis and molecular docking studies of phenyl piperidine derivatives as potent dual NK1R antagonists/serotonin transporter (SERT) inhibitors. Comput. Biol. Chem.. 2016;67:22-37. PMID: 28043073
- [CrossRef] [Google Scholar]
- Synthesis of sulfonylhydrazone and acylhydrazone-substituted 8-ethoxy-3-nitro-2H-chromenes as potent antiproliferative and apoptosis inducing agents full paper synthesis of sulfonylhydrazone and acylhydrazone substituted 8-ethoxy-3-nitro-2H- chromene. Arch. Pharm.. 2014;347:576-588.
- [Google Scholar]
- Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583:437-440.
- [CrossRef] [Google Scholar]
- Exploring the potential of novel phenolic compounds as potential therapeutic candidates against SARS-CoV-2, using quantum chemistry, molecular docking and dynamic studies. Bioorg. Med. Chem. Lett.. 2021;43
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104101.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




