

Translate this page into:
Synthesis, anticancer activity and docking studies of pyrazoline and pyrimidine derivatives as potential epidermal growth factor receptor (EGFR) inhibitors
⁎Corresponding author.at: School of Chemical Sciences, Universiti Sains Malaysia, 11800 Penang, Malaysia. melati@usm.my (Melati Khairuddean),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A search for anticancer agents has prompted the design and synthesis of new chalcone, pyrazoline and pyrimidine derivatives as potential epidermal growth factor receptor (EGFR) kinase inhibitors. These derivatives’ binding affinities were predicted by AutoDock, which showed that chalcone, pyrazoline and pyrimidine derivatives as EGFR-kinase inhibitors have good binding energies, ranging from −10.91 to −7.32 kcal/mol. These compounds were synthesized and characterized using elemental analysis (CHN analysis) and spectroscopic techniques (FTIR and NMR). Among the pyrazoline derivatives, 4Aiii has revealed a superior in vitro activity, inhibiting the EGFR kinase even at a low concentration of 0.19 μM compared to the pyrimidine derivative, 5Bii. In contrast, the cytotoxic effect of these derivatives was studied against hormonal and non-hormonal breast cancer cell lines. Most of the pyrazoline derivatives were able to express their cytotoxic effect efficiently against hormonal breast cancer but only one pyrimidine derivative managed to express its activity against hormonal breast cancer.
Keywords
Pyrazoline
Pyrimidine
EGFR inhibitors
AutoDock
Anticancer

- EGFRs
-
epidermal growth factor receptors
- DEPT
-
distortionless enhancement by polarization transfer
- COSY
-
correlation spectroscopy
- HSQC
-
heteronuclear single quantum correlation
- MTT
-
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- LGA
-
lamarckian genetic algorithm
- ATCC
-
american type culture collection
- RPMI
-
roswell park memorial institute
- DMEM
-
dulbecco’s modified eagle medium
- SI
-
selectivity index
Abbreviations
1 Introduction
Globally, cancer is the second highest cause of death after cardiovascular diseases. Cancer-related deaths have escalated from 8.2 million in 2012 (Ferlay et al., 2015) to 8.7 million in 2015 and 9.5 million in 2018 (Fitzmaurice et al., 2017). In the absence of effective interventions, cancer-related mortality would rise to an estimated 13 million and 16 million deaths among cancer patients by 2030 and 2040, respectively (The International Agency for Research on Cancer, IARC). Cancer is a disorder of abnormal cell growth with the potential of spreading into or invading nearby tissues and organs. Despite many treatment for cancer, chemotherapy is considered the mainstay of cancer therapy. However, chemotherapy has some limitations such as limited efficiency, selectivity, high cast, genotoxicity, and drug resistance. It also produces side effects as a result of damage to normal body cells and organ toxicities which reduced the quality of life in cancer patients (Tang et al., 2017). Despite the advances in chemotherapy, there are no agents that selectively target the cancer cells. This has confirmed the urge to develop new chemotherapeutic agents which are more selective for cancer cells, and thus produce fewer side effects (Al-Anazi et al., 2018; Slaihim et al., 2019; Khairuddean et al., 2020; Salum et al., 2020; Al-Anazi et al., 2021; Mohammad, et al., 2021a; Mohammad, et al., 2021b; Mohammad, et al., 2022; Jumaah et al., 2022). Tyrosine kinase protein-inhibitors are of great interest due to their therapeutic capacity in treating a variety of diseases, especially cancer (Backes et al., 2008a; 2008b; Gschwind et al., 2004).
Among protein tyrosine kinase, the epidermal growth factor receptor (EGFR) has emerged as a key and main target for the development of new anti-cancer agents (Speake et al., 2005; Warnault et al., 2013; Yewale et al., 2013). EGFR kinase inhibition in cancer treatment is performed by blocking this enzyme with small molecules (drugs). To date, over 10 EGFR inhibitors have been approved by the United States Food and Drug Administration (US-FDA) in the past two decades. Some of these inhibitors include Erlotinib (Tarceva®), Gefitinib (Iressa®) and Lapatinib (Tykerb) which is also known as GW-572016, with quinazoline-based derivatives (Dowell et al., 2005; Ganjoo and Wakelee, 2007; Stamos et al., 2002). Although the therapeutic effects of the quinazoline-based agents on different cancers have been well established, many side effects such as skin rashes, diarrhea, vomiting, nausea and hemorrhage were also reported (Sequist and Lynch, 2008). Clearly, as anticancer agents, it is necessary to discover drugs with minimum adverse effects that provide much more hope for mankind. Based on the previously cited studies, the exploration of EGFR with newly synthesized hybrids is a fruitful and fertile research area. It was reported that compounds with chalcone moiety, a class of secondary metabolites of flavonoids exhibit various biological activities, especially as anticancer agents (Karthikeyana et al., 2015; Alman et al., 2020). In addition, nitrogen-containing heterocycles have become important in the development of new effective drugs for the treatment of cancer (Kerru et al., 2020). Nitrogen-based heterocyclic compounds constitute nearly 60% of all drugs approved by the FDA and represent about 73% of the approved anticancer drugs in 2015 (Martins et al., 2015; Hosseinzadeh et al., 2018). Compounds with heterocyclic rings such as pyrazoline (Bhutani et al., 2015; Vincent et al., 2020) and pyrimidine (Kaur et al., 2015; Kumar and Narasimhan, 2018; Mahapatra et al., 2021) have demonstrated a wide variety of promising biological activities especially for anticancer.
Nitrogen-containing compounds, especially pyrimidines, have been screened as potential anti-cancer agents for clinical use because pyrimidine plays an important role in the synthesis of nucleic acid (Parker, 2009). The synthesis of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) depends on several pyrimidine derivatives including adenine, guanine, cytosine, thymine, and uracil. In this study, in continuation of the EGFR inhibitors as anticancer agents (Al-Anazi et al., 2018; Al-Anazi et al., 2021), compounds with pyrimidine rings were proposed. The molecular docking and synthesis of a series of pyrazoline and pyrimidine derivatives as anticancer agents are reported. The characterization of these compounds was confirmed using spectroscopic techniques such as FTIR, 1H NMR, 13C NMR and elemental analysis (CHN analysis). Consequently, the biological activity of the synthesized compounds was also evaluated against the EGFR kinase enzyme, and breast cancer cell lines.
2 Results and discussion
2.1 Molecular docking
Molecular docking is an important tool in the drug discovery toolbox which predicts the orientation, interaction and binding energies of ligands in their targeted binding sites. Currently, AutoDock 4.2 is reported to be the most popular molecular docking program. Its high accuracy and versatility have expanded its application (Sousa et al., 2006; Meng et al., 2011; Al-Najjar, 2018b). Compounds with chalcone, pyrazoline and pyrimidine moieties were successfully docked against the 3POZ crystal structure, and the results are shown in Table 1. TAK-285 is a novel investigational small molecule (N-[2-[4-[3-chloro-4-[3-(trifluoromethyl)phenoxy] anilino]pyrrolo[3,2-d]pyrimidin-5-yl]ethyl]-3-hydroxy-3-methylbutanamide) which inhibits EGFR and specifically targets HER2, such as lapatinib. It has antiproliferative activity in vitro and in vivo cytotoxic activity against cancer cells (Ishikawa et al., 2011) and was used as a control ligand.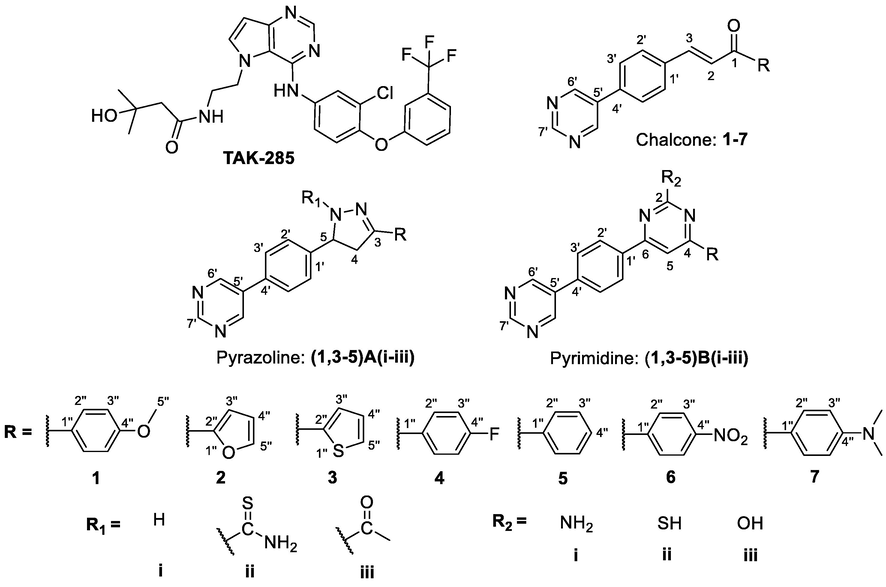
Compound
Binding energy
(Kcal/mol)Interacting amino acids
Compound
Binding energy
(Kcal/mol)Interacting amino acids
TAK-285
−10.15
MET 793, ARG 776, LEU 777, THR790, ARG 841, THR 854,
Chalcone
1
−10.91
MET 793, LYS 745
5
−10.31
MET 793
2
−8.27
MET 793
6
−9.63
MET 793
3
−10.76
MET 793
7
−8.23
LEU 777
4
−10.19
MET 793, LEU 777
Pyrazoline
1Ai
-0.8.49
MET 766, MET 793
4Ai
−7.81
MET 793
1Aii
-0.8.16
MET 793, ASP 855
4Aii
−8.82
LYS 745, MET 793
1Aiii
−9.16
LYS 745, MET 793, ASP 855
4Aiii
−8.80
LYS 745, MET 793, ASP 855
3Ai
−8.26
MET 793, ASP 855
5Ai
−7.66
MET 793
3Aii
−8.80
MET 793, ASN 842, ASP 855
5Aii
−7.32
ASP 855
3Aiii
−8.92
LYS 745, LEU 788, MET 793
5Aiii
−8.92
LYS 745, MET 793, ASN 842
Pyrimidine
1Bi
-0.9.32
MET 793, ASN 842, ASP 855
4Bi
−9.41
MET 793, ASN 842, ASP 855
1Bii
-0.9.22
MET 793, ASN 842, ASP 855
4Bii
−9.43
MET 793, ASN 842, ASP 855
1Biii
−8.96
MET 793, ASN 842, ASP 855
4Biii
−8.40
LYS 745, MET 793, ASP 855
3Bi
−9.23
MET 793, THR 854, ASP 855
5Bi
−7.82
MET 793, ASP 855
3Bii
−7.41
LEU 788, ARG 841
5Bii
−9.71
MET 793, ASN 842, ASP 855
3Biii
−9.42
MET 793, THR 854, ASP 855
5Biii
−9.15
MET 793, ASN 842, ASP 855
Structures of TAK-285 and new chalcone, pyrazoline and pyrimidine derivatives.
The results of the docking score for the chalcone were nearly similar to TAK-285, except for compounds 2 and 7, where the modified extra chains did not affect the affinity of binding inside the pocket. Likewise, compound 6 was not selected for further investigation because of the environmental toxicity, carcinogenicity and mutagenicity of the aromatic nitro compound (Kovacic and Somanathan, 2014). On the other hand, most of the proposed pyrazoline and pyrimidine derivatives have shown good binding energies, ranging from −9.71 to −7.32 kcal/mol. Compound 5Bii revealed the lowest binding energy of −9.71 kcal/mol, while, 5Aii showed the highest binding energy of −7.32 kcal/mol.
Intermolecular interactions of the docked compounds are shown in Supplementary. Fig. 1 shows the binding of TAK-285 within the adenosine triphosphate (ATP) binding pocket of the catalytic tyrosine kinase domain, competing with ATP. Several studies have recently stated that the formation of a hydrogen bond with the ATP binding site of EGFR (MET 793 and/or LYS 745) helps to inhibit its activity (Aertgeerts et al., 2011; Mahajan et al., 2017; Al-Anazi et al., 2021), which can be seen in all of the proposed compounds except in compounds 7, 5Aii and 3Bii. ASN 842 performed conventional hydrogen bonds with TAK-285, matching with compounds 3Aii, 5Aiii, 1B(i-iii), 4B(i-ii) and 4B(ii-iii), by hydrogen bonds interaction. However, amino acid residue LYS 745 has performed a hydrophobic interaction with TAK-285 but formed conventional hydrogen bonds with compounds 1Aiii, 3Aiii, 4Aii, 4Aiii, 5Aiii and 4Biii. Therefore, based on the binding energy study of all the designed compounds, it was decided to synthesize compounds 1, 3, 4 and 5 of chalcone derivatives and some heterocyclic derivatives of pyrazoline (Ai-iii) and pyrimidine (Bi-iii). These compounds have shown good binding energies and strong interactions in the targeted active site.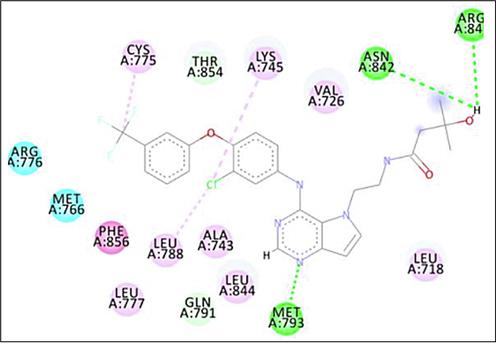
2D intermolecular interactions between docked (TAK-285) and 3POZ protein. Green and pink colored amino acids represent their contribution to hydrogen bonds and hydrophobic interactions.
3 Chemistry
The series of pyrazoline, (1,3–5)A(i-iii) and pyrimidine, (1,3–5)B(i-iii) derivatives were synthesized from cyclization reactions of chalcone compounds 1, 3–5, as shown in Scheme 1. All the synthesized compounds were confirmed using FT-IR, NMR spectrometers and CHN elemental analysis.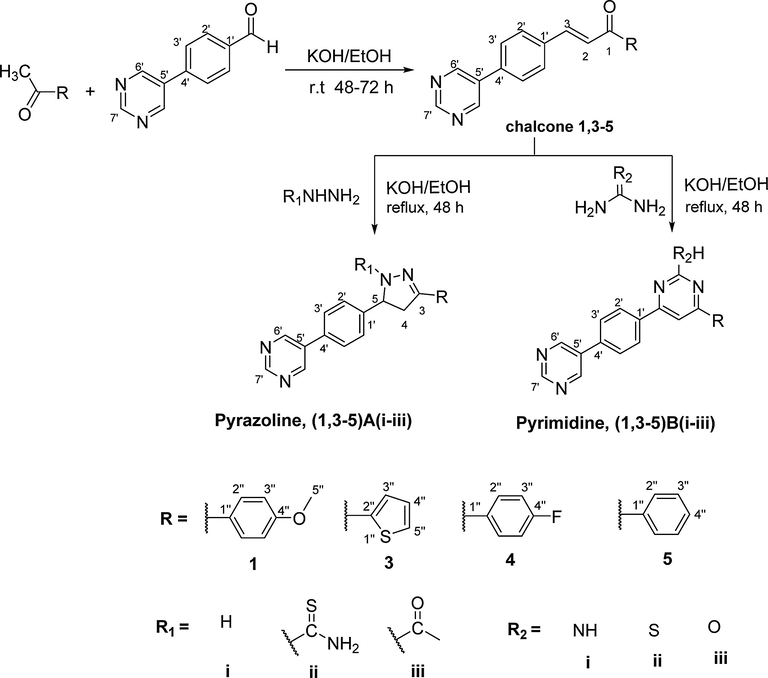
Synthesis pathway of chalcone, pyrazoline and pyrimidine derivatives.
The IR spectra manifested the presence of the diagnostic bands. For example, in compounds 1Aiii and 1Bii, the absorption bands were observed at υ 3070 (Csp2-H str), 2914–2910 and 2847–2840 cm−1 for (Csp3-H of the asymmetrical and symmetrical str), 1608–1605 (C = N str), 1553–1521 (C = C str), and 1250–1230 (C-N str) cm−1, respectively. The absorption band at υ 1637–1644 cm−1 was attributed to C = O stretching of the N-acetyl derivative, Aiii and the band at υ 2590–2530 cm−1 was attributed to the C-SH stretching of the 4,6-diphenyl-pyrimidine-2-thiol derivative, Bii. The IR spectra for derivatives Ai-ii, Bi and Biii showed little differences compared to Aiii and Bii compounds. This is due to the disappearance of the stretching absorption bands of the C = O, C-SH and the appearance of the absorption bands of the NH and NH2 moieties in derivatives Ai-ii and also the appearance of the absorption bands of the NH2 and OH moieties in derivatives Bi and Biii, respectively, as shown in the Supplementary Figure S2.
The 1H NMR spectra supported the diagnostic tools for the positional elucidation of the protons. The assignment of the characteristic signals is based on the chemical shifts and intensity patterns. In all the pyrazoline derivatives, two protons of Ha and Hb at C-4 of the pyrazoline ring, which is geminal, were observed and appeared as two doublets of doublets at δH 2.86–3.35 and 3.47–3.96. A proton at C-5 of the pyrazoline ring also appeared as a doublet of doublets at δH 4.88–6.12 due to the vicinal coupling with two nonequivalent germinal protons of C-4 carbon. However, this proton was observed to be more downfield than those of Ha and Hb in all the products, due to its proximity to a benzene ring. The coupling constants for two nonequivalent geminal protons become smaller (0–3 Hz), as predicted when the HCH angle becomes larger. So, in this study, the small changes in bond angles resulting from stereochemical changes influence the geminal coupling constant (>17 Hz). That means coupling constant become larger when the HCH angle become smaller (a = 120°, 2JHH = 0–3 Hz & a = 107°, 2JHH = 17.5 Hz) (Pavia et al., 2014). A singlet centered at δH 7.24 and δH 7.83 were consistent with the formation of a pyrimidine ring in 2,4,6-trisubstituted pyrimidine derivatives of Bi-iii.
Both the pyrazoline and pyrimidine derivatives exhibited aromatic protons in the region of δH 8.82–9.27 and δH 9.11–9.25. In all the compounds, a singlet was assigned to H6′ and H7′ of the terminal pyrimidine ring. Signals for NH (Ai), NH2 (Aii, Bi), SH (Bii), acetyl-CH3 (Aiii) and OH (Biii) protons resonated as a singlet at δH 7.61–10.23, 7.23–9.82, 6.72–6.83, 9.25–9.29, 2.34–2.46 and 7.36–5.42, respectively. The aromatic protons were observed within the expected region along with the integral values. The 1H NMR spectrum of compound 1Aiii (Supplementary Figure S2.13(b)) showed two signals for acetyl-CH3 and methoxy-H5′' protons which resonated as a singlet at δH 2.36 and δH 3.78, respectively. The presence of the pyrazoline ring was proven by the diastereotopic protons of Ha and Hb on C-4 which appeared as a pair of doublets at δH 3.09 and δH 3.71 while the proton at C-5 was observed as a doublet of doublets at δH 5.61.
The 1H NMR spectrum of 1Bii pyrimidine derivative (Supplementary Figure S2.21(b)) has shown a signal for methoxy-H5′' proton which resonated as a singlet at δH 3.86. The presence of 2,4,6-trisubstituted pyrimidine ring was proven by proton H5 which appeared as a singlet at δH 7.42. In both derivatives, 1Aiii and 1Bii, the signals for protons H6′ and H7′ of the terminal pyrimidine ring appeared as a singlet at δH 8.82–9.16 and δH 9.11–9.18. Lastly, the remaining signals were observed as the aromatic protons which resonated as doublets at δH 7.32–828 (H2′), δH 7.45–7.87 (H3′), δH 7.62–8.11 (H2′') and δH 6.87–7.02 (H3′').
The 13C NMR spectra of all derivatives (Ai-iii and Bi-iii) (Supplementary Figure S2.(13,21)c) have confirmed the absence signals of trans-alkene carbons and the appearing signals of the pyrazoline ring which were observed at δC 37.5–43.0 and δC 54.9–63.8, attributed to C-4 and C-5, respectively. On the other hand, the appearing signal at δC 94.8–106.4 was attributed to C5 on the pyrimidine ring, Other characteristic aromatic carbons have appeared in the expected region.
Furthermore, the carbon skeleton of compound 1Aiii was confirmed by 13C NMR which showed the aliphatic carbons at δC 21.9, 55.4, 42.3, 59.5 for the acetyl-CH3, methoxy-C5′', C-4 and C-5 of pyrazoline, respectively. The carbonyl carbon was observed at δC 168.8 and the aromatic carbons appeared in the range of δC 161.4–114.2. Likewise, the carbon skeleton of compound 1Bii was confirmed by 13C NMR that showed the aliphatic carbon at δC 55.6 for the methoxy-C5′', C-5 for the 2,4,6-trisubstituted pyrimidine ring at δC 101.9 and the aromatic carbon at δC 114.1–189.5. The DEPT-90 and DEPT-135 NMR spectra (Supplementary Figure S2.(13,21)d,e) have confirmed all the methine (CH), methylene (CH2), methyl (CH3) and quaternary carbons (C) in both of 1Aiii and 1Bii. The extreme downfield signals at δC 168.8 and δC 189.5 were assigned to the carbonyl carbon and C2 (C-SH) carbon of 1Aiii and 1Bii, respectively. The extreme upfield signals at δC 59.5, 55.4, 42.3 and 21.9 were assigned to the methine (C5), methoxy carbon (C5′'), methylenic (C4) which appeared as a negative signal in the DEPT-135) and methyl carbon of the acetyl moiety in 1Aiii, respectively. Similarly, for derivative 1Bii, the extreme upfield signal at δC 55.6 and δC 101.9 were assigned to the methoxy (C5′') and the methine (C5) carbons of the pyrimidine ring, respectively. The quaternary carbons (C4′', C1′', C3, C1′, C4′, C5′) and (C4′', C1′', C4, C2, C6, C1′, C4′, C5′) in 1Aiii and 1Bii, respectively, were identified through their absence in the DEPT-90 and DEPT-135 spectra.
The 2D-NMR correlation using 1H–1H COSY (Supplementary Figure S2.29) and 1H–13C HSQC (Supplementary Figure S2.30) spectra were used for unambiguous assignment. The 1H–1H COSY spectrum of pyrazoline derivative 1Aiii showed the correlations of H4a with H4b, H4a with H5, H4b with H4a, H4b with H5, H5 with H4b, H5 with H4a, H3′' with H2′', H2′ with H3′, H3′ with H2′ and H2′' with H3′'. The 1H–1H COSY spectrum of pyrimidine derivative 1Bii has shown the correlations of H3′' with H2′', H3′ with H2′, H2′' with H3′' and H2′ with H3′. On the other hand, the 1H–13C HSQC spectrum has revealed the correlations between the proton and carbon and confirmed the assigned structure of pyrazoline 1Aiii and pyrimidine 1Bii. In pyrazoline 1Aiii, a cross peak was observed between both protons H4a and H4b at δH 3.09 and δH 3.71 with the methylene carbon C4 (CH2) at δC 42.3. In addition to this, two protons at δH 3.78 and δH 2.36 have shown two cross-peaks with the aliphatic carbons of C5′' and CH3-CO at δC 55.4 and δC 21.9, respectively. Also, a cross-peak of the pyrimidine derivative was observed between proton H5 at δH 7.59 with methine carbon C5 (CH) at δC 101.9. In addition to this, a proton at δH 3.83 has shown a cross-peak with the aliphatic carbon (C5′') at δC 55.6. Furthermore, two protons of H6′ and H7′ of the terminal pyrimidine ring in derivatives 1Aiii and 1Bii have appeared at δH 8.82–9.25 and δH 9.11–9.21 which showed two cross-peaks for the methine carbons at δC 155.2–154.8 and δC 157.4–157.8, respectively. The remaining four aromatic protons H3′', H2′, H3′, H2′' appeared at δH 6.87–7.03, δH 7.32–8.33, δH 7.45–7.95 and δH 7.62–8.16, showing a cross-peak with methine carbon at δC 114.1–114.2, δC 126.8–128.0, δC 127.3–127.6 and δC 128.2–128.7, respectively. Finally, All the carbons were determined and figuratively represented in Supplementary Figure S2.
4 In-vitro assays
4.1 Recombinant EGFR kinase assay
To confirm the molecular docking results of the ability of compounds 4Aiii and 5Bii to bind and inhibit EGFR kinase, the luminescence of ATP conversion was measured using ADP-GloTM kinase assay at two different concentrations of 50 and 0.19 μM. Results indicate a significant inhibition of the recombinant kinase when incubated with the inhibitors. Compound 4Aiii has managed to inhibit the enzyme with 83% and 82% of the inhibition rate, respectively for both concentrations. On the other hand, compound 5Bii showed an inhibition rate of 89% at the concentration of 50 μM. However, when the concentration drops to 0.19 μM its inhibition rate falls to 72% which is still considered as a significant inhibitory activity (Fig. 2). This goes consistently with the molecular docking results where both compounds gave a very comparable binding affinity and the same number of interactions with the surrounding amino acids seated in the EGFR binding pocket. Conceptually, this indicates the high potential of these pyrazoline and pyrimidine derivatives to be further investigated for their true cytotoxic activity.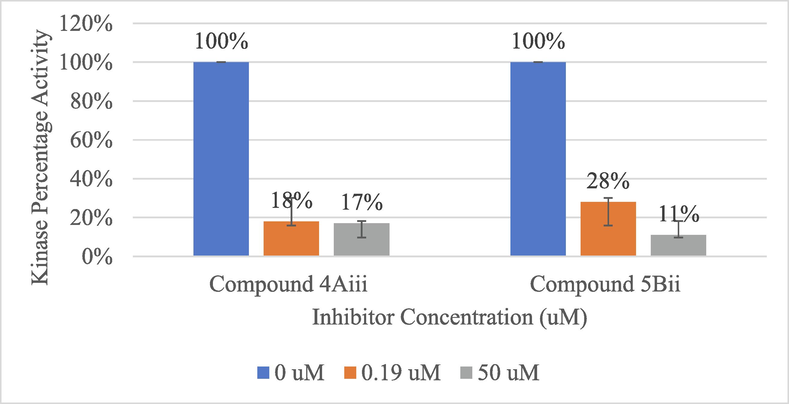
Recombinant kinase activity measured using ADP-GloTM for compounds 4Aiii and 5Bii at three different concentrations (0, 0.19 and 50 μM).
5 Cytotoxicity assay
The newly synthesized chalcone, pyrazoline and pyrimidine derivatives were evaluated for their in-vitro cytotoxic activity (0–100 μg/mL) against two human breast cancer cell lines (MCF7 and MDA-MB-231) and one non-cancerous breast (MCF-10A), with the aid of tamoxifen as reference drugs. The concentration required for 50% inhibition of cell viability (IC50) alongside the selectivity index (SI) was calculated and is presented in Table 2.
Compounds
IC50 (72 h) (μM)
Selective Index
MCF-7
MDA-MB-231
MCF-10A
MCF-7
MDA-MB-231
1
31.2 ± 2.17
100 ± 0.01
6.33 ± 0.58
0.2
0.06
3
9.55 ± 0.22
100 ± 0.01
4.73 ± 0.49
0.5
0.05
4
100 ± 0.01
100 ± 0.01
6.86 ± 1.68
0.07
0.07
5
9 ± 0.94
100 ± 0.01
32.9 ± 1.15
3.66
0.33
1Ai
100 ± 0.01
64.56 ± 7.8
100 ± 0.01
1
1.55
1Aii
53.7 ± 8.9
100 ± 0.01
100 ± 0.01
1.86
1
1Aiii
66.8 3 ± 1.8
51.2 9 ± 6.75
100 ± 0.01
1.5
1.95
3Ai
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
3Aii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
3Aiii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
4Ai
40.27 ± 1.08
100 ± 0.01
100 ± 0.01
2.48
1
4Aii
38.01 ± 2.96
100 ± 0.01
100 ± 0.01
2.63
1
4Aiii
58.88 ± 2.63
100 ± 0.01
100 ± 0.01
1.7
1
5Ai
26.3 ± 0.61
83.17 ± 0.97
100 ± 0.01
3.8
1.2
5Aii
67.61 ± 1.15
93.32 ± 2.15
100 ± 0.01
1.48
1.07
5Aiii
50.12 ± 4.13
69.18 ± 2.46
100 ± 0.01
2
1.45
1Bi
60.25 ± 7.25
100 ± 0.01
100 ± 0.01
1.66
1
1Bii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
1Biii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
3Bi
5.5 ± 0.07
100 ± 0.01
100 ± 0.01
18.18
1
3Bii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
3Biii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
4Bi
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
4Bii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
4Biii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
5Bi
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
5Bii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
5Biii
100 ± 0.01
100 ± 0.01
100 ± 0.01
1
1
Tamoxifen
26.95 ± 3.01
23.36 ± 3.84
–
–
0.3
In general, results showed that pyrazoline derivatives have better anti-cancer potentials compared to the pyrimidine-derived entities. For the chalcone attached to the pyrimidine ring (compounds 1, 3, 4 and 5, Supplementary Figure S3.1), they displayed different cytotoxic activity according to the attached substituent, in the order of C6H5- > thiophene ring > C6H4-OMe > C6H4F against MCF-7. Likewise, for the MDA-MB-231 cell line, all chalcone compounds (1, 3–5) exhibited moderate cytotoxic (IC50: 100 µM) activity. Compounds 5 also displayed a high cytotoxic selectivity of 3.66 compared to tamoxifen. On the other hand, compounds 1, 3, and 4 exhibited a lower degree of cytotoxic selectivity against MCF-7, which shows increased cytotoxicity toward the healthy cell.
Among the pyrazoline derivatives (Supplementary Figure S3.2–3), 5Ai showed the lowest IC50 value (26.30 ± 0.61 μM) which was consistent with tamoxifen IC50 (26.95 ± 3.01 μM), indicating an excellent cytotoxic activity. Also, 5Ai showed a high cytotoxic selectivity index toward MCF-7 avoiding massive toxicity of normal cells (MCF-10A). This enhanced the activity of 5Ai which might be due to the presence of 1H and the 3-phenyl within the pyrazole ring. Another pyrazoline, 4Ai (IC50: 40.27 ± 1.08, SI: 2.48) has 1H and 3-(p-fluorophenyl) while pyrazoline 4Aii (IC50: 38.01 ± 2.96, SI: 2.63) has 1-thioamide and 3-(p-fluorophenyl) within the ring. Remarkably, pyrazoline 1Aiii was the only compound that reasonably appears to be effective against the MDA-MB-231 cell line (IC50: 51.29 ± 6.75 μM, SI: 1.95) with 1-ethanone and 3-(p-methoxyphenyl).
On the other hand, compound 3Bi (Supplementary Figure S3.2–3) was the only pyrimidine derivative that expressed the best cytotoxic activity. This compound has conveyed a superior activity against the MCF-7 cell line (IC50: 5.5 ± 0.07 μM, SI: 18.18) compared to the other tested pyrimidine derivatives including the tamoxifen standard which 3Bi topped it with magnitudes of selectivity and specificity. The superior activity of 3Bi was due to the presence of thiophen-2-yl over position no. 4 of the pyrimidine ring.
6 Conclusion
Several chalcone, pyrazoline and pyrimidine derivatives which may function as EGFR inhibitors were studied. Some selected compounds of pyrazoline (1,3–5)A(i-iii) and pyrimidine (1,3–5)B(i-iii) derivatives with promising EGFR inhibitory activity were synthesized and characterized using spectroscopic techniques such as FTIR, 1H NMR, 13C NMR and elemental analysis (CHN analysis). These synthesized compounds were investigated in vitro for their activity against the EGFR kinase enzyme, then for their anti-proliferative and cytotoxic activities using multiple cell lines of the breast. Interestingly, the molecular docking study revealed that the chalcone, pyrazoline and pyrimidine derivatives have good binding affinities towards EGFR kinase. These results are consistent with their effect on the enzyme where dropping the concentration of the pyrazoline compound 4Aiii did not affect its inhibitory activity. Moreover, the cytotoxic activity of pyrazoline derivatives against breast cancer cell lines revealed that 5Ai is the best-proposed candidate against hormonal breast cancer, while 1Aiii is the most potent against non-hormonal breast cancer. On the other hand, only one compound of the pyrimidine derivatives exhibited excellent activity against hormonal breast cancer while other pyrimidine derivatives appeared to be active against non-hormonal breast cancer. Further investigations and studies are encouraged to test these compounds against other cancer cell lines and other possible modifications can also be done to improve their activity.
7 Materials and methodology
7.1 Overview
Computational modeling has been used to increase the efficiency of the drug design process as well as to reduce the experimental cost and time (Al-Najjar et al., 2017; Al-Najjar, 2018a; 2018b;). EGFR tyrosine kinase was selected as a therapeutic target for novel chalcone, pyrazoline and pyrimidine derivatives since it is known as the main target for the development of new anti-cancer agents. The X-ray crystallographic structure of the EGFR kinase domain with a resolution of 1.5 Å was found from Protein Data Bank (https://www.rcsb.org/pdb) (PDB ID: 3POZ) (Aertgeerts et al., 2011). AutoDock 4.2 (The Scripps Research Institute, San Diego, CA, USA) was used to study the intermolecular interactions and binding energies of pyrazoline and pyrimidine derivatives within the active site of EGFR.
7.2 Molecular docking
The three-dimensional (3D) structure of pyrazoline, pyrimidine derivatives were drawn using ACD/ChemSketch (https://www.acdlabs.com) and saved in mol2 format before being converted to pdb files. The 3D crystal structure of the EGFR domain bound to TAK-285 was retrieved from the protein data bank (PDB ID: 3POZ). Ligand files in the pdb format were set to be prepared by AutoDockTools (mgltools.scripps.edu). Both atomic charges were added, and hydrogen atoms were merged to new pyrazoline, pyrimidine derivatives and the protein. Kollman and Gasteiger charges were added to protein and pyrazoline and also pyrimidine derivatives, respectively. The pdb files of the ligands and receptor were converted to pdbqt formats using the ‘Quick ligand’ and ‘Grid’ options in AutoDockTools. The receptor-binding site to which the ligands were docked was defined by a grid box with the size of 40, 40 and 40 Å. The grid box was centered on the coordinates 16.732, 33.121, and 12.166 (x, y and z, respectively). A set of grid maps have been created by AutoGrid 4.2. The default docking parameters of AutoDock 4.2 were utilized except for the number of Lamarckian Genetic Algorithm (LGA) runs, which was set to 100 runs. Molecular docking simulations were executed using AutoDock 4.2. The scoring function in AutoDock 4.2 was utilized to predict the binding affinity of the receptor-ligand interaction. The most suitable conformation with the lowest binding energy has been chosen. Docking results were visualized using Discovery Studio Visualizer (https://www.3ds.com).
7.3 Instrumentation and chemicals
FT-IR absorption spectra were obtained via a Perkin Elmer Frontier FT-IR Spectrometer with Perkin Elmer Universal ATR Sampling Accessory (ν, cm−1) in a range of 600 to 4000 cm−1. 1D NMR spectra (1H, 13C, DEPT-90, and DEPT-135) and 2D NMR (COSY and HSQC) of all products were recorded by Bruker UltrashieldTM spectrometer operating at 500 MHz at the School of Chemical Sciences, Universiti Sains Malaysia. The chemical shifts (δ, ppm) were expressed in (ppm) downfield from tetramethylsilane (TMS) as internal standard and the coupling constants (J) were expressed in Hertz (Hz). Elemental analyses were obtained utilizing Perkin Elmer II, 2400 CHN analyzer. Likewise, melting points of all products obtained were determined via Stuart Scientific SMP1 melting point apparatus with an open capillary tube that was used utilizing a temperature range of 25–350 °C. All chemicals and reagents were obtained from Aldrich (Sigma-Aldrich, USA) and were utilized without further purification.
7.4 Synthesis
Synthesis of chalcones attached to the pyrimidine ring 1 and 3–5 (Mohammad et al., 2021).
Chalcone 1 and 3–5 with different substituents were synthesized via Claisen-Schmidt condensation reaction between chalcone attached to the pyrimidine ring. Reactant 1-(4-methoxyphenyl)ethan-1-one (6 mmol, 1.000 g) was reacted with 4-(pyrimidin-5-yl)benzaldehyde (6.6 mmol, 1.227 g) to form chalcone 1, with 1-(thiophen-2-yl)ethan-1-one (6.6 mmol, 0.832 g) to form chalcone 3, with 1-(4-fluorophenyl)ethan-1-one (6.6 mmol, 0.920 g) to form chalcone 4, and with acetophenone (6.6 mmol, 0.800 g) to yield chalcone 5. Reactant mixtures were stirred at room temperature in the presence of potassium hydroxide (6.6 mmol, 0.373 g) in 20 mL ethanol hours for 48 h. Thick suspensions were formed, and their precipitates were filtered off, washed several times with water and air-dried. Recrystallization from ethanol gave chalcones 1 and 3–5 (80–90%) as off-white powders, except for compound 4 which gave a yellow powder (Supplementary Material, S4. Characterization data).
Synthesis of pyrazoline derivatives Ai-Aiii (Salum et al., 2020; Mohammad et al., 2021).
A mixture of chalcone (1,3–5) (0.5 mmol) and hydrazine hydrate 99% (0.05 mL, 3 × 0.5 mmol) or substituted hydrazine hydrate (2 × 0.5 mmol) in absolute ethanol (10 mL) containing sodium hydroxide (2 × 0.5 mmol, 0.040 g) or acetic acid (5 mL) was refluxed for 48 h. After cooling, the solid formed was filtered off, air-dried and recrystallized from absolute ethanol or methanol except for pyrazoline from thiosemicarbazide. All the compounds were purified by column chromatography on silica gel using hexane–ethyl acetate (Supplementary Material, S4. Characterization data).
Synthesis of pyrimidine derivatives Bi-Biii (Mohammad et al., 2021).
To a solution of chalcones (1,3–5) (0.5 mmol) in absolute ethanol (10 mL), urea (3 × 0.5 mmol, 0.090 g), or substituted urea (3 × 0.5 mmol) and aqueous sodium hydroxide (3 × 0.5 mmol, 0.060 g) were added. The reaction mixture was heated under reflux for 48 h and poured into ice-cold water. The product obtained was filtered, washed with water and recrystallized from absolute ethanol or methanol except for pyrimidine from guanidine, which was purified by column chromatography on silica gel using hexane–ethyl acetate.
8 Biological assay
8.1 Recombinant EGFR kinase assay
The molecular docking part has shown the consistency of the binding affinity of compounds 4Aiii and 5Bii. It was decided that these synthesized inhibitors were screened for their EGFR inhibitory effect using ADP-Glo™ Kinase Assay (Promega, Madison). The principle of the assay depends on measuring the ADP formed from the kinase reaction. Later, this ADP will transform into ATP that generates light and reflects the kinase activity. The assay was conducted according to the company protocol. Positive control with 5 μM of ATP-substrate was used to calculate 100% kinase activity. Negative control (blank) did not contain the inhibitors nor the enzyme and was used to calculate 0% kinase activity. Inhibitors’ activities were measured thrice at two distinct concentrations (50 and 0.19 μM). The luminescent signals generated from the ADP-Glo™ assay were converted to percentage activity by subtracting the values of the negative control (0% kinase activity) from all points.
8.2 Cytotoxicity assay
In order to confirm the cytotoxic activity of the synthesized pyrazoline and pyrimidine derivatives, this cell viability and cytotoxicity assay was initiated. Two human breast cancer (MCF7, MDA-MB-231) and a non-cancerous breast (MCF-10A) were originally obtained from the American Type Culture Collection (ATCC, USA). MCF-7 cell line was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium. MDA-MB-231 cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with L-alanyl, L-glutamine (2 mM), penicillin/streptomycin antibiotics (0.1%, v/v) and 10% heat-inactivated fetal bovine serum (FBS). MCF-10A cell line was cultured in a mixture of DMEM and Ham’s F-12 (DMEM/F12) medium, containing 5% (v/v) horse serum, 10 μg/mL insulin, 20 ng/mL human epidermal growth factor (hEGF), 0.5 μg/mL hydrocortisone and 0.01% (v/v) penicillin–streptomycin antibiotics. All cell lines were routinely cultured and maintained in a humidified atmosphere with 5% CO2 at 37 °C.
MCF-7 and MDA-MB-231 cancer cell lines were seeded at a density of 1 × 104 cells/well in a 96-well plate. MCF-10A was used as a control cell line to determine the selectivity index (SI) values. The cells were seeded and allowed for attachment overnight. Subsequently, they were treated using a fresh assay medium supplemented with an increasing concentration of chalcone derivatives (0–100 μg/mL) within 24–72 h of incubation at 37 °C. Standard chemotherapeutic drugs including tamoxifen were used as positive controls, while medium alone was used as the negative control (untreated). At each incubation period, 10 μL (5 mg/mL) of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was added into each well, and incubated for 4 h at 37 °C, 5% CO2. Following this, the MTT solution was discarded and replaced with 100 μL of dimethyl sulfoxide (DMSO) into each well for crystal solubilization. The absorbance of each sample was measured in a microplate reader at 570 nm wavelength with a reference wavelength of 620 nm according to the manufacturer’s protocol. Half-maximal inhibitory concentration (IC50) values for all cell lines were determined based on Equation (1). The selectivity index (SI) values were calculated as the ratio of the 50% cytotoxic concentration (IC50 in cancer cell line) to the 50% cytotoxic concentration on the control cell line (IC50 in non-cancerous cell line) (Equation (2)).
where, Abs: Absorbance reading at 570 nm.
sample: Respective compounds or chemotherapeutic drug.
blank: Culture medium alone.
Supporting Information: The following are available online, Figure S1.1 2D & 3D intermolecular interactions between docked chalcone compounds 1, 3, 4 and 5 with 3POZ.PDB. Figure S1.2 2D & 3D intermolecular interactions between docked pyrazoline compounds 1A(i-iii) and (3–5)A(i-iii) with 3POZ protein. Figure S1.3 2D & 3D intermolecular interactions between docked pyrimidine compounds 1B(i-iii) and (3–5)B(i-iii) with 3POZ protein. Green and pink colored amino acids represent their contribution to hydrogen bond and hydrophobic interactions, respectively. Figure S2. (a) IR (b) 1H (C) 13C, (d) DEPT 90 and (e) DEPT135 NMR spectra of chalcone, pyrazoline and pyrimidine derivatives in CDCl3 and DMSO‑d6, 1H–1H COSY NMR spectra of compounds 1, 1Aiii and 1Bii in CDCl3 and DMSO‑d6, respectively, and 1H–13C HSQC NMR spectra of compounds 1, 1Aiii and 1Bii in CDCl3 and DMSO‑d6, respectively. Figure S3.1 Cytotoxic activity of chalcone derivatives at 72 h in the inhibition of MCF-7 and MDA-MB-231 cells based on MTT assay. Data are expressed as mean ± SEM of a representative experiment performed in triplicate (n = 3). *Symbol above the bars indicate significant differences. The significance was considered at P < 0.0001. Figure S3.2 Cytotoxic activity of pyrazoline and pyrimidine derivatives at 72 h in the inhibition of MCF-7 cell based on MTT assay. Figure S3.3 Cytotoxic activity of pyrazoline and pyrimidine derivatives at 72 h in the inhibition of MDA-MB-231 cell based on MTT assay. Figure S4. Characterization data. This material is available free of charge via the Internet at https://xxx.
Availability of data and materials
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Funding
The authors would like to thank the Ministry of Higher Education Malaysia for the Fundamental Research Grant Scheme (FRGS) with the project code FRGS/1/2019/STG01/USM/02/16, which is used to finance this research work.
CRediT authorship contribution statement
Menier Al-Anazi: Conceptualization, Methodology, Software, Validation, Investigation, Writing – original draft. Melati Khairuddean: Conceptualization, Methodology, Software, Investigation, Writing – original draft, Writing – review & editing, Supervision. Belal O. Al-Najjar: Conceptualization, Methodology, Software, Validation, Investigation, Writing – review & editing, Supervision. Mohammad Murwih Alidmat: Methodology. Nik Nur Syazni Nik Mohamed Kamal: Methodology, Validation. Musthahimah Muhamad: Methodology, Validation.
Acknowledgement
Not applicable.
Conflicts of Interest: The authors declare no conflict of interest related to this work.
References
- Structure-Based Drug Design Studies Toward the Discovery of Novel Chalcone Derivatives as Potential Epidermal Growth Factor Receptor (EGFR) Inhibitors. Molecules. 2018;23(12):3203.
- [Google Scholar]
- EGFR Inhibitors and Apoptosis Inducers: Design, Docking, Synthesis, and Anticancer Activity of Novel Tri-Chalcone Derivatives. Systematic Reviews in Pharmacy.. 2021;12(3):809-820.
- [Google Scholar]
- Chalcone – Promising Entity for Anticancer Activity: An Overview. International Journal of Pharmaceutical Scientific Research.. 2020;11(5):2027-2041.
- [Google Scholar]
- Small-molecule inhibitors binding to protein kinase. Part II: the novel pharmacophore approach of type II and type III inhibition. Expert Opinion on Drug Discovery. 2008;3(12):1427-1449.
- [CrossRef] [Google Scholar]
- Small-molecule inhibitors binding to protein kinases. Part I: exceptions from the traditional pharmacophore approach of type I inhibition. Expert Opinion on Drug Discovery. 2008;3(12):1409-1425.
- [CrossRef] [Google Scholar]
- Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-E386.
- [CrossRef] [Google Scholar]
- Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncology. 2017;3(4):524-548.
- [CrossRef] [Google Scholar]
- Review of erlotinib in the treatment of advanced non-small cell lung cancer. Biologics: Targets & Therapy. 2007;1(4):335-346. PMCID: PMC2721286
- [Google Scholar]
- The discovery of receptor tyrosine kinases: targets for cancer therapy. Nature Reviews Cancer. 2004;4(5):361-370.
- [CrossRef] [Google Scholar]
- Anti-cancer nitrogen-containing heterocyclic compounds. Curr. Org. Chem.. 2018;22(23):2256-2279.
- [Google Scholar]
- Design, Synthesis, Characterization and Cytotoxic Activity of New Ortho-hydroxy and Indole Chalcone Derivatives as Anticancer Agents. Med. Chem. Res.. 2022;2022
- [CrossRef] [Google Scholar]
- Advances in Chalcones with Anticancer Activities Recent Patents on Anti-Cancer Drug Discovery.. 2015;10(1):97-115.
- Anti-Cancer Pyrimidines in Diverse Scaffolds: A Review of Patent Literature. Recent Pat. Anti-Cancer Drug Discovery. 2015;10:23-71.
- [Google Scholar]
- A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules. 2020;25(8):1909.
- [Google Scholar]
- Therapeutic potential of heterocyclic pyrimidine scafolds. Chem. Cent. J.. 2018;12:38.
- [Google Scholar]
- Pyrimidine: a review on anticancer activity with key emphasis on SAR. Future J. Pharmaceutical Sciences.. 2021;7:123.
- [Google Scholar]
- Heterocyclic anticancer compounds: recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules. 2015;20(9):16852-16891.
- [Google Scholar]
- Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des.. 2011;7(2):146-157.
- [Google Scholar]
- Docking Studies, Synthesis, Characterisation and Cytotoxicity Activity of New Bis-Chalcones Derivatives. Biomedical Research and Therapy.. 2021;8(4):4294-4306.
- [Google Scholar]
- Synthesis, Characterization, Cytotoxicity Study and Docking Studies of New Fused-pyrazoline Derivatives derived from Bis-Chalcones Against Breast Cancer cells. Egypt. J. Chem.. 2021;64(12):6801-6811.
- [Google Scholar]
- Synthesis. Characterization, Molecular Docking and Cytotoxicity Evaluation of New Thienyl Chalcone Derivatives Agains Breast Cancer Cells, Systematic Reviews in Pharmacy.. 2022;12(10):707-717.
- [Google Scholar]
- Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev.. 2009;109(7):2880-2893.
- [Google Scholar]
- Design, synthesis, characterization, and cytotoxicity activity evaluation of mono-chalcones and new pyrazolines derivatives. J. Applied Pharmaceutical Science.. 2020;10(08):020-036.
- [Google Scholar]
- EGFR tyrosine kinase inhibitors in lung cancer: an evolving story. Annu Rev Med. 2008;59:429-442.
- [CrossRef] [Google Scholar]
- Synthesis, characterisation of new derivatives with mono ring system of 1,2,4-triazole scaffold and their anticancer activities. J. Mol. Struct.. 2019;1196:78-87.
- [Google Scholar]
- Synthesis, Characterisation of Some New Schiff Base for the Piperidinium 4-Amino-5-Substituted-4H-1,2,4-Triazole-3-Thiolate and Their n Vitro Anticancer Activities. Int. J. Natural Sci. Human Sciences.. 2020;1(1):48-58.
- [Google Scholar]
- Recent developments related to the EGFR as a target for cancer chemotherapy. Current Opinion in Pharmacology. 2005;5(4):343-349.
- [CrossRef] [Google Scholar]
- Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 2002;277(48):46265-46272.
- [CrossRef] [Google Scholar]
- Targeted multidrug delivery system to overcome chemoresistance in breast cancer. Int. J. Nanomed.. 2017;12:671.
- [Google Scholar]
- Recent advances in drug design of epidermal growth factor receptor inhibitors. Current Medicinal Chemistry. 2013;20(16):2043-2067.
- [CrossRef] [Google Scholar]
- Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690-8707.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.03.037.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




