

Translate this page into:
Synthesis, in-silico, and in-vitro study of novel chloro methylquinazolinones as PI3K-δ inhibitors, cytotoxic agents
⁎Corresponding author. sherin_el_feky@mans.edu.eg (Sherin M. Elfeky)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The PI3k-δ enzyme is of the key enzymes for cellular growth and proliferation. One of the key mechanisms for cytotoxic agents is to target the PI3k-δ enzyme. 5c and 5d were able to bind at ATP binding pocket of PI3k-δ enzyme in an inhibitory mode forming H-bond interaction with key amino acid Val828. 5c and 5d showed in-vitro inhibition of PI3k-δ enzyme(IC50=8.27±0.19μM), (IC50=1.24±0.03μM) respectively. 5c and 5d showed moderated to good in-vitro cytotoxicity against three cell lines HCT (IC50=8.00 ±0.12 μM), (IC50=47.56 ±0.67 μM), MCF-7 (IC50=21.22 ±0.33 μM), (IC50=41.07 ±0.58 μM) and HePG-2(IC50=58.98 ±0.33 μM), (IC50=17.78 ±0.58 μM), respectively. 5c and 5d showed in-vitro cytotoxicity against the WI-38 cell line (IC50=177.00±3.66μM), (IC50=148.49±1.37μM), respectively.

Abstract
Through a two-step procedure, 3-amino-7-chloro-2-methylquinazolin-4(3H)-one was synthesized from 2-amino-4-chlorobenzoic acid as a starting material. The latter reacted with chloro acetylchloride, then nucleophilically substituted with various secondary amines to produce acetamide derivatives (5a-e), or underwent condensation reaction with various aldehydes to produce arylidine derivatives (6a-e). In-silico study of drug-likeness and ADME descriptors was conducted for all compounds. Compounds showed good oral bioavailability, as well as good gastrointestinal absorption potential and no symptoms of liver or CNS adverse effects. In-vitro cytotoxic activity of the compounds was moderate to good when compared to staurosporine in three cell lines: HCT, MCF-7, and HepG-2. Compound 5c showed the highest cytotoxic activity against the HCT cell line (IC50 = 8.00 ± 0.33 μM), Compound 5d showed the highest cytotoxic activity against the HepG-2 cell line (IC50 = 17.78 ± 0.58 μM). Acetamide derivatives revealed higher cytotoxic activity compared to arylidine derivatives. Compound 5d had the highest enzyme inhibition activity in the in-vitro PI3k-δ enzyme inhibition assay (IC50 = 1.24 ± 0.03 μM) followed by 5c (IC50 = 8.27 ± 0.19 μM). Both 5c and 5d were able to bind at the ATP binding site of the PI3k-δ enzyme in a mode similar to the native ligand where they formed H-bond interactions with the hinge region amino acid Val828 and hydrophobic interactions with other amino acids indicating an agreement between molecular docking simulation study and the biological screening.
Keywords
7-chloro-2-methylquinazolin-4(3H)-ones
PI3k-δ enzyme
Cytotoxic Activity
Molecular docking
Enzyme inhibition
ADME studies

1 Introduction
Phosphoinositide 3-kinase PI3K is a lipid kinase that phosphorylates the 3 hydroxy group of the inositol ring of phosphoinositide. PI3K is a critical part of the cellular signaling pathway responsible for the survival, growth, and proliferation of cells (McNamara and Degterev, 2011). Deregulation of this pathway is manifested in almost all human cancers including; breast cancer, colorectal carcinoma, and others. Hence, targeting this pathway is considered to be a highly effective therapeutic strategy against tumor progression where the selective inhibition of PI3K decreases cellular proliferation and increases cellular death(Cheng et al. 2010, Yuan et al. 2011). There are three classes of PI3K in human cells (I, II, III) (Jean and Kiger, 2014). Class I is responsible for the phosphorylation of membrane-bound phosphatidylinositol-(4,5)-biphosphate (PIP2) to Phosphotidyl inositol - (3,4,5)-triphosphate (PIP3). PIP3 is a secondary messenger that activates the Pleckstrin homology (PH) domain-containing the downstream protein kinase (ATK) that activates cellular proliferation (Lien et al., 2017). Class I of PI3K can be subdivided according to the activating receptor into group A and group B. Group B include PI3K-δ that is activated by G protein-coupled receptors (GPCRs)(Yang et al. 2019). Inhibitors of PI3K-δ have been reported to be safe, highly effective antitumor agents (Akinleye et al., 2013). Different PI3K-δ inhibitors are reported including 4-methylpyridopyrimidinones (Cheng et al., 2013), Thiazolopyrimidinones (Lin et al., 2012), 4-acrylamido-quinolines (Ma et al., 2019), and quinoxalines (El Newahie et al., 2019). Different publications report quinazolines to be highly effective PI3K- δ inhibitors as shown in Fig. 1. Idelalisib (I) is a quinazoline containing a specific PI3K-δ inhibitor and it is the first PI3K inhibitor approved by the American food and drug administration FDA (Zeid et al., 2019). 4-aryl quinazoline (II) is a reported PI3K-δ inhibitor that showed high efficacy in-vivo (Hoegenauer et al., 2016). Quinazolin-4(3H)-one derivative (III) showed cytotoxic activities against HePG-2, MCF-7, and HCT-116 cancer cell lines and showed in-vitro inhibitory activity against PI3K-δ. In the molecular docking study, (III) was able to bind at the PI3K-δ binding site at an inhibitory mode forming hydrogen bonding to the key amino acids at the ATP hinge region at the pocket (Zeid, Mohamed et al., 2019). Resistance to known PI3K inhibitors is caused by a variety of factors, including mutation, drug-related toxicity, and feedback upregulation of PI3K levels as compensatory mechanisms (Mishra et al., 2021). Small molecules that inhibit PIK3 have been linked to a variety of side effects, the most common of which are gastrointestinal toxicity (Hanker et al., 2019). This, in turn, necessitates a special intermittent dose schedule of suboptimal doses to ensure medication safety, posing a challenge to the efficacy of known PIK3-inhibitors(Hudson et al., 2016). This is why researchers are constantly looking for more potent PIK3 inhibitors that can be highly effective at low doses.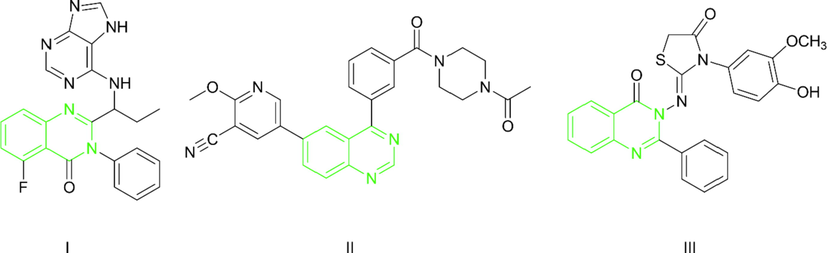
Different quinazoline PI3K-δ inhibitors.
In the present work, we aim to design novel quinazolin-4(3H)-one derivatives to form key interaction at the ATP hinge region at PI3K-δ pocket. Different structural features were proposed to reinforce interaction at the binding site through additional hydrophobic interactions and hydrogen bonding. Structural features to support hydrophobic interaction include the introduction of chlorine at C-7, the Methyl group at C-2 to overcome the steric hindrance of the bulky phenyl group, and different arylidines at N-3. Quinazolin-4(3H)-one bearing substituted acetamide at N-3 to support hydrogen bond interaction features through free NH and carbonyl moiety as shown in Fig. 2. Physicochemical properties, pharmacokinetics profiles of the designed compounds will be studied in-silico to study their drug-likeness before molecular docking simulation. In the molecular docking simulation study, the compounds will be docked into the binding site of the enzyme PI3K-δ to study their modes of interaction and binding scores compared to the native ligand. To support the claim that the designed quinazoline derivatives have potential antitumor activity all the synthesized compounds will be screened for in-vitro cytotoxic activity against three cell lines human colon cancer cell line (HCT-116), breast cancer cell line MCF-7 and, human liver cancer cell line (HepG-2). To support the claim that the designed compounds are inhibitors of enzyme PI3K-δ the compounds that will show highest cytotoxic activity will be further evaluated for in-vitro enzyme inhibition activity. The most active compounds will be further evaluated for selective cytotoxicity against fibroblast-like fetal lung cell lines (WI-38).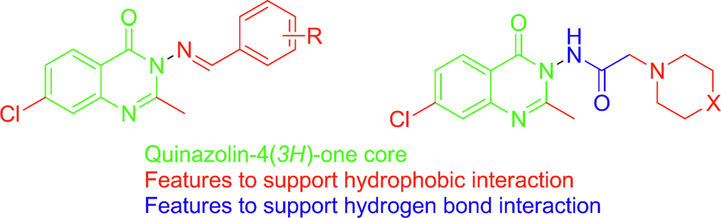
Designed quinazolin-4(3H)-one with different structural features.
2 Results and discussion
The most known synthetic route for quinazolin-4(3H)-ones is the Niementowski reaction which involves the fusion of anthranilic acid derivatives with amides at (130–150 °C). The reaction takes place through the o-amidobenzamide intermediate. The reaction is of limited yield due to the impurities related to conditions, thus it requires further complicated purifications (Alexandre et al., 2002). Advances to Niementowski reaction took place to improve yield and purity of products including; microwave-assisted conditions (de Fatima Pereira et al., 2005, de Fatima Pereira et al., 2007), use of ionic liquid as catalyst (Kathiravan et al., 2011). An improvement to the known Niementowski reaction is synthesis through benzoxazin-4-one intermediate this approach has become one of the most popular approaches for the synthesis of quinazolin-4(3H)-ones (Welch et al., 2001). In which, first anthranilic acid is converted to benzoxazinone through reaction with acetic anhydride. benzoxazin-4-one acts as a source of N-1 and carbonyl carbon in the quinazoline ring system. In the second step, benzoxazinone reacts with an amine as a source of N-3 in a condensation reaction (Kamal et al. 2010, Alagarsamy and Saravanan, 2013). This approach is considered an upgrade from the Niementowski reaction in terms of yield, reaction time, and purity of products. In the present investigation, as shown by (Sheme 1), 2-amino-4-chlorobenzoic acid (1) was converted to the corresponding benzoxazinone through reflux with acetic anhydride till no starting material could be detected on TLC, then the collected 7-chloro-2-methyl-4H-benzo[d][1,3]oxazin-4-one(2) was washed with petroleum ether and allowed to dry. 2 was then refluxed with hydrazine hydrate in ethanol to yield 3-amino-7-chloro-2-methylquinazolin-4(3H)-one (3). (Osarumwense and Iyekowa, 2017). 3-amino-7-chloro-2-methylquinazolin-4(3H)-one (3) was dissolved in dry toluene and cooled to 15 °C then chloroacetyl chloride was added dropwise with stirring, the reaction mixture was refluxed till no starting material was detectable on TLC. The precipitate 2-chloro-N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)acetamide (4) formed after cooling was collected by filtration then washed and recrystallized from ethanol. The 1HNMR for compound 4 showed a singlet at 2.15 ppm corresponding to three protons and another singlet at 4.04 ppm corresponding to two protons of CH2-Cl. Compound 4 showed two carbonyl peaks at 171.74 ppm and 159.44 ppm in 13CNMR. N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-aminoacetamides (5a-e) were prepared through the nucleophilic displacement of chloride of 4 with a variety of secondary amines. This reaction was reported to proceed using potassium carbonate or trimethylamine as a base and using dioxane, or toluene, or a combination of both (Raghavendra et al., 2008, Alagarsamy et al., 2015). In the present work, 2-chloro-N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)acetamide reacted with different piperazines or morpholine in presence of potassium carbonate as a base using dry toluene as solvent under refluxing conditions. The reaction was monitored by TLC till no starting materials could be detectable. Then reaction mixtures were filtered off, excess toluene distilled off, and precipitates collected were recrystallized from ethanol. The 1HNMR for compounds 5a-e showed the characteristic peaks of piperazines or morpholine upfield as two multiplets in the region (3.49–3.06 ppm) and (2.74–2.47 ppm) corresponding to four protons each. The 13CNMR for compounds 5a-e showed characteristic piperazine carbons at 56–60 ppm. The amino group of 3-amino-7-chloro-2-methylquinazolin-4(3H)-one (3) went through a condensation reaction with different aldehydes to yield the corresponding imine derivatives. The reaction proceeded simply by treating the amine derivatives with aldehydes in ethanol in presence of glacial acetic acid (Cordeiro and Kachroo 2020). The reaction was monitored by TLC till no starting materials were detected. (E)-7-chloro-3-((arylidine)amino)-2-methyl-quinazolin-4(3H)-ones (6a-e) were collected in good yield by pouring on ice filtration then recrystallization from ethyl acetate. The 1HNMR for compounds 6a-e showed the characteristic arylidine peak at 8.90–9.06 ppm as a singlet corresponding to one proton. The stability of E and Z diastereoisomers was the subject of in-silico studies (Mansour et al., 2021). Total energies of optimized geometries of E and Z diastereoisomers of target arylidine compounds (6a-6e) were calculated using the MOE 2009. program at the MMFF94x force field. E diastereoisomer of the 6a-6e compounds had energies of 86.18Kcal/mol, 72.54 Kcal/mol, 91.68 Kcal/mol, 70.87 Kcal/mol, and 96.46 Kcal/mol, respectively. While the Z diastereoisomer had higher energies of 97.66 Kcal/mol, 82.81 Kcal/mol, 104.76 Kcal/mol, 81.42 Kcal/mol, and 111.37 Kcal/mol. E diastereoisomers were more stable than the Z diastereoisomers by (10.2–14.9Kcal/mol).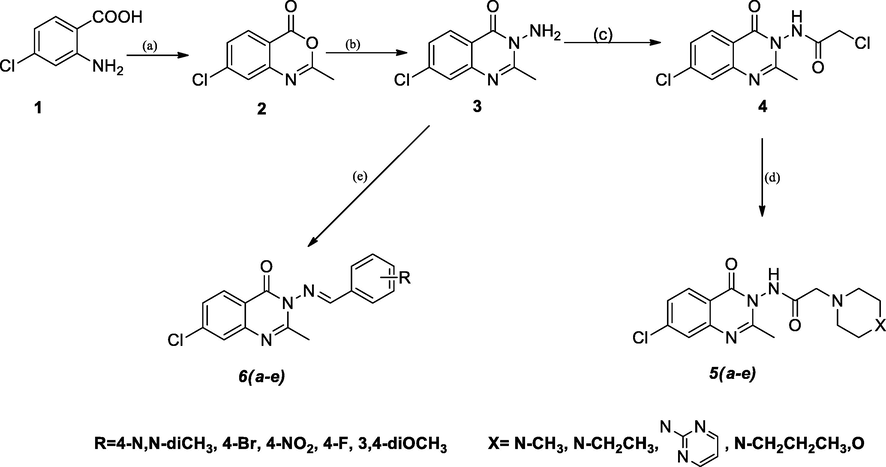
synthesis of N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-aminoacetamides (5a-e) and (E)-7-chloro-3-((arylidine)amino)-2-methylquinazolin-4(3H)-ones (6a-e). (a)acetic anhydride, reflux, 1 h; (b) hydrazine hydrate, ethanol, reflux, 3 h; (c)chloroacetyl chloride, dry toluene, reflux, (d) K2CO3, toluene, reflux; (e) ethanol, glacial acetic acid, reflux.
2.1 In-silico studies
2.1.1 Prediction of biological activities
Chloroquinazolines support a wide range of biological activity; this is why it was important to predict this range of activity using available tools. Swiss Target Prediction is an online tool that can predict possible target macromolecules for a given small molecule. The online tool has access to a large number of known actives and uses the similarity principle to predict targets with the highest similarity to the searched molecule structures and determines the activity probability (Daina et al., 2019). Structures of compounds 5a-5e and 6a-6e were generated as smiles then a query tool in the Swiss Target prediction was used to determine the range of the biological activity of compounds. The query returned results showing that according to structures of compounds, compounds have a high probability to target the PI3k enzyme. The query returned by the Swiss Target Prediction online tool for compound 5a showed that 40% of known actives of high similarity were targets of PI3k enzyme and 20% of them targeted CREB binding protein, 13.3% targeted G-protein coupled receptor and 13.3% targeted protease enzymes.
2.1.1.1 Molecular docking simulation
PI3K- δ was extensively studied as well as its mode of interaction with different inhibitors. From the previous literature, it was revealed that four main regions within the ATP binding pocket are significant for interaction with inhibitors; Adenine pocket (hinge region), Specificity pocket, Affinity pocket, and a Hydrophobic region II at the mouth of the active site. Val828 and Ile910 were the key amino acids involved in H-bond interaction with all inhibitors, Glu826 formed H-bond interaction in most inhibitors (Knight et al., 2006, Williams et al., 2009). The crystallographic structure of PI3K- δ (PDB ID: 2WXG)(Berndt et al., 2010) in complex with reference ligand (SW13) 2-[[4-amino-3-(3-fluoro-5-hydroxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]methyl]-5-methyl-3-(2-methylphenyl) quinazolin-4(3H)-one which exhibited propeller shape conformation where two orthogonally oriented aromatic rings of the inhibitor opened the hydrophobic pocket in the active site of the receptor and the quinazolinone moiety was embedded into the hydrophobic specificity pocket between Trp760 and Ile777 on one side and Met752 and Pro758 on the other side. The reference ligand formed H- bond interaction with Val828, Glu826, Tyr813, Asp911, and Asp787. The ligand also formed hydrophobic interaction with Met752, Thr833, Ile777, Ile910, Met900, and Trp760. Fig. 3 shows SW13 in the binding site of PI3K-δ enzyme in 2D and 3D representation. The newly synthesized compounds were docked using MOE 2009. Program (Elfeky et al., 2020), PDB ID: 2WXG The root mean square differences (RMSD) between the docking poses of the newly synthesized compounds and the native ligand crystallographic geometry of co-crystalized ligand SW13 were <2 Å. Mode of interactions, key amino acids involved in the interactions, energy scores of compounds at the active site of ATP binding site of PI3K- δ enzyme were compared to the native ligand SW13. Compound 5c was able to bind in a similar mode to that of the reference ligand where it was able to form H-bond interaction with hinge region amino acid Val828 the key amino acids at the binding site at the N-1 of the quinazoline ring it also formed additional H-bond interaction to Lys779. It formed several hydrophobic interactions with many key amino acids at the binding site including; Met752, Met900, Trp760, Ile777, Asp911, and Lys755 as shown in Fig. 4. Compound 5d interacted at the ATP binding site of PI3K- δ enzyme in a similar fashion to the SW13 where it formed H-bond interaction to Val828 at N-1 of quinazoline ring, it formed hydrophobic interaction to the key amino acid Ile910 together with other amino acids at the pocket including; Trp760, Ile777, Met900, Asp911, and Lys755, as shown in Fig. 5. As for 6a, the more stable E diastereoisomer was used for docking and nor the Z diastereoisomers. 6a failed to form H-bond interaction to Val828 while it formed H-bond interaction to Lys779 and maintained hydrophobic interaction with key amino acids at the binding site including; Asp911, Met752, Ile910, and Lys755, as shown in Fig. 6. Table 1. shows docking scores, amino acids involved in interactions for compounds 5c, 5d, and 6a compared to reference ligand SW13.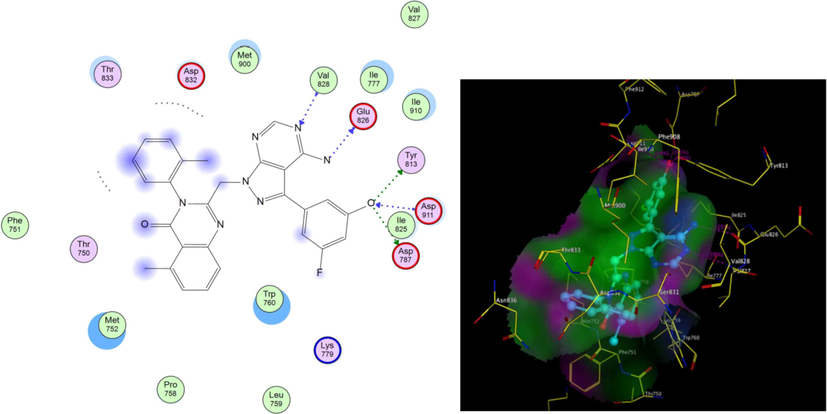
2D,3D representation of SW13 at ATP binding site of PI3K- δ.
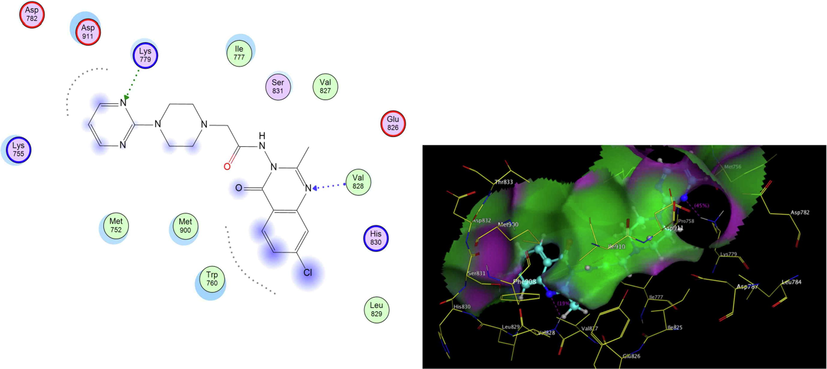
2D,3D representation of 5c at ATP binding site of PI3K- δ.
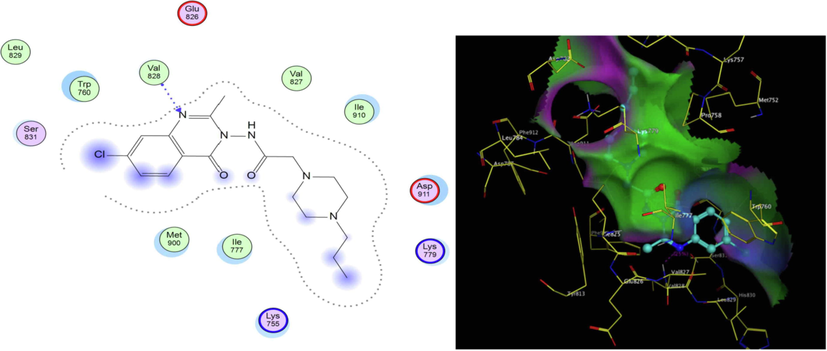
2D, 3D representation of 5d at ATP binding site of PI3K- δ.
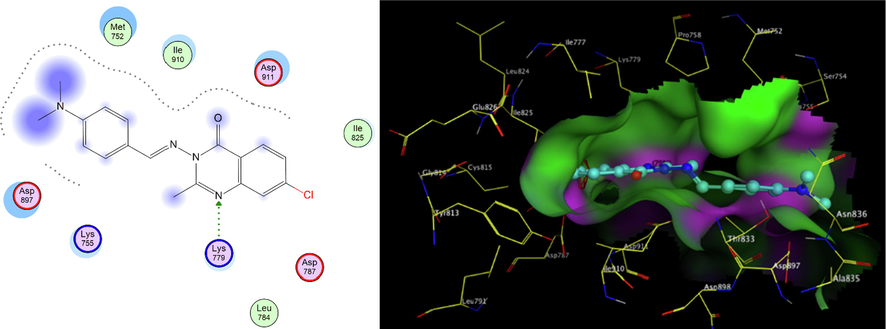
2D,3D representation of 6a at Figure Scheme 1. ATP binding site of PI3K- δ.
Compound
Docking score(kcal/mol)
Amino acids (bond length Å)
SW13
−11.90
Val828(3.01)
Gln826(2.78)
Tyr813(2.55)
Asp911(3.00)
Asp787(2.40)
5c
−10.68
Val828(1.93)
Lys779(1.88)
5d
−10.47
Val828(1.71)
6a
−9.84
Lys779 (1.91)
2.1.1.2 Drug likeness and ADME properties
Drug likeness and ADME properties studies were performed for all compounds using the SwissADME tool online (Daina et al., 2017). All compounds showed physicochemical properties suitable for oral bioavailability where lipophilicity was in the range of 2.40 to 3.49 (Daina et al., 2014). Molecular weight(MM) ranged 314–414 g/mol. Topological polar surface area(TPSA) (Ertl et al., 2000) for compounds was in the range of 40 to 100 Å2 ideally 20–130 Å2. The number of rotatable bonds (RB) ranged from 2 to 6 bonds ideally < 9 bonds. Solubility of compounds varied where compounds 5a and 5e were very soluble, compounds 5b,5c,5d,6a, and 6d were soluble while compounds 6b, 6c, and 6e showed moderate solubility applying topological method. (Ali et al., 2012) according to the applied method. insoluble < -10 < poorly < -6 < moderately < -4 < soluble < -2 < very < 0 < highly. As for the drug-likeness compounds showed no violations of Lipinski s rules. (Lipinski et al., 1997) including molecular weight (MM) ≤ 500, logP (ilogp) ≤ 4.15, number of hydrogen bond donors (HBD) ≤ 5, and acceptors (HBA) ≤ 10. Table 2. shows the physicochemical properties and drug-likeness for compounds 5a-5e and 6a-6e. *MM: molecular weight, ilogp: n-octanol/water partition coefficient, TPSA: topological polar surface area HBD: Hydrogen bond donner, HBA: Hydrogen Bond Acceptor, RB; Rotatable Bonds.
MM* (g/mol)
ilogp*
TPSA(Å)*
violations
HBA*
HBD*
RB*
5a
349.82
2.40
70.47
0
5
1
4
5b
363.84
2.72
70.47
0
5
1
5
5c
413.86
2.75
96.25
0
6
1
5
5d
377.87
3.08
70.47
0
5
1
6
5e
336.77
2.22
76.46
0
5
1
4
6a
340.81
3.34
50.49
0
3
0
3
6b
376.64
3.47
47.25
0
3
0
2
6c
342.74
2.70
93.07
0
5
0
3
6d
315.73
3.21
47.25
0
4
0
2
6e
357.79
3.49
65.71
0
5
0
4
As for the descriptors used for the ADME study, gastrointestinal absorption refers to how well the compounds can penetrate the bloodstream passively through the gut wall, ADME study of this descriptor (G.I absorption) showed that all the compounds have high gastrointestinal absorption using white of the boiled egg model (Daina and Zoete 2016). Except for compounds 6a,6b,6d, and 6e. All compounds showed no blood–brain barrier penetration using the yolk of the boiled egg model (Daina and Zoete 2016), which means these compounds are expected to be safe to CNS. All Compounds were non-inhibitors of cytochrome P450; 2C9, 2C19, and 1A2 except 6a-6e which suggests no expected side effects to the liver. Compounds 5a-5e showed good binding to plasma glycoprotein(p-gp) suggesting compounds can bind to the carrier protein in the blood. Table 3. shows the ADME study results for compounds 5a-5e and 6a-6e. *BBB permeation: Blood-Brain Barrier Permeation, P-gp Substrate: Plasma Glycoprotein Substrate, GI Absorption: Gastrointestinal Absorption.
Solubility
BBB* Permeant
P-gp* Substrate
GI* Absorption
Cytochrome P450
Cyp2C9 inhibitor
Cyp2C19 inhibitor
Cyp1A2 inhibitor
5a
−1.95
no
yes
high
no
no
no
5b
−2.34
no
yes
high
no
no
no
5c
−2.91
no
yes
high
yes
no
no
5d
−2.89
no
yes
high
no
no
no
5e
−1.89
no
yes
high
no
no
no
6a
−3.99
yes
no
high
yes
yes
yes
6b
−4.52
yes
no
high
yes
yes
yes
6c
−4.58
no
no
high
yes
yes
yes
6d
−3.91
yes
no
high
yes
yes
yes
6e
−4.13
yes
no
high
yes
yes
yes
2.2 In-vitro studies
2.2.1 In-vitro cytotoxic activity against HCT, MCF-7, and HepG-2 cell lines
The cytotoxic activity of compounds 5a-5e and 6a-6e was screened against human colon carcinoma (HCT116), Caucasian breast adenocarcinoma (MCF-7), and hepatocellular carcinoma (HepG-2), at different concentrations using MTT assay in comparison to staurosporine as reference. Table 4. shows IC50 in μM of compounds 5a-5e and 6a-6e against HCT, MCF-7, and HepG-2 compared to staurosporine. Among the tested compounds, compounds 5c, 5d, and 6a exhibited good cytotoxicity to the three screened cell lines. Compound 5c exhibited the highest cytotoxicity against the HCT cell line. Compound 5d exhibited the highest cytotoxicity against the HepG-2 cell line. Compound 6a exhibited high cytotoxicity against the MCF-7 cell line. Compound 5a showed the least cytotoxicity to all three cell lines as shown in Fig. 7. According to cytotoxicity, compounds could be divided into three groups: those with low activity (5a, 5b, 6b, 6d, and 6b), those with moderate activity (5e and 6e), and those with high activity (5c, 5d, and 6a). Except for 6a, which demonstrated high cytotoxic activity against MCF-7 cell lines, the majority of compounds with low to moderate activity were arylidine derivatives. The majority of acetamide derivatives demonstrated moderate to high activity.
Compound
Cytotoxicity
IC50(mean ± SD)
μM
HCT
MCF-7
HepG-2
5a
388.60 ± 5.1
176.49 ± 2.32
137.37 ± 2.32
5b
38.82 ± 0.53
90.12 ± 1.23
25.87 ± 1.23
5c
8.00 ± 0.12
21.22 ± 0.33
58.98 ± 0.33
5d
47.56 ± 0.67
41.07 ± 0.58
17.78 ± 0.58
5e
42.24 ± 0.53
13.91 ± 0.18
53.49 ± 0.18
6a
36.95 ± 1.75
11.92 ± 0.15
29.63 ± 0.15
6b
75.23 ± 1.06
112.72 ± 1.59
18.38 ± 1.59
6c
48.38 ± 0.62
53.58 ± 0.69
160.41 ± 0.69
6d
63.16 ± 0.75
42.97 ± 0.68
163.74 ± 0.68
6e
36.62 ± 0.22
48.28 ± 0.38
72.30 ± 0.38
staurosporine
22.44 ± 0.39
29.02 ± 0.51
15.23 ± 0.51
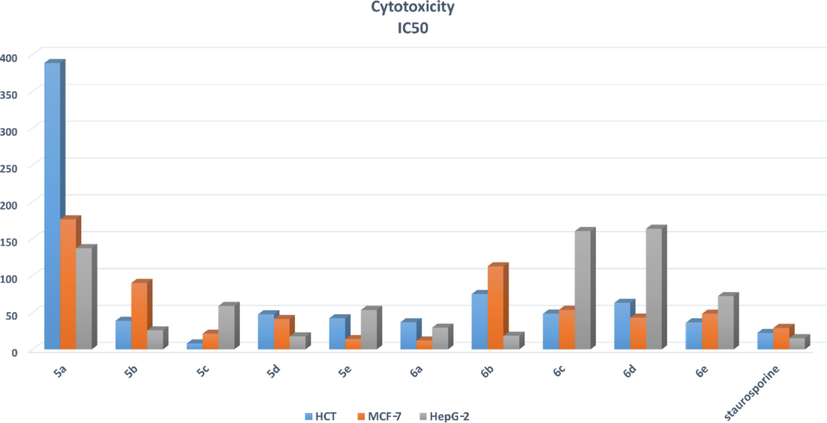
IC50 in μM of compounds 5a-5e and 6a-6e against HCT, MCF-7, and HepG-2 compared to staurosporine.
2.2.2 In-vitro PI3k-δ enzyme inhibition assay
Compounds 5c, 5d, and 6a that displayed the most promising cytotoxic properties compared to staurosporine were further evaluated for inhibitory activity against PI3K-δ enzyme at different concentrations adopting ADP-GLO-assay method and Ly294002 as reference (IC50 = 16.13 ± 0.27 μM). The results revealed that compound 5d showed the highest inhibitory activity (IC50 = 1.24 ± 0.03 μM), compound 5c showed high inhibitory activity (IC50 = 8.27 ± 0.19 μM) and compound 6a showed moderate inhibitory activity against enzyme PI3K-δ (IC50 = 29.10 ± 0.54 μM). Table 5 shows the half-maximal in-vitro inhibitory concentration of PI3k-δ enzyme for compounds 5c, 5d, and 6a compared to reference Ly294002.
Compound
PI3K-δ
IC50(mean ± SD)
μM
5d
1.24 ± 0.03
5c
8.27 ± 0.19
6a
29.10 ± 0.54
LY294002
16.13 ± 0.27
2.2.3 In-vitro cytotoxic activity against WI-38 cell line
Compounds 5c, 5d, and 6a that showed the highest activity both in the cytotoxic screening and the enzyme inhibition assay were further studied for cytotoxicity using Caucasian fibroblast-like fetal lung cell line (WI-38), and staurosporine as reference compound. Results showed that compounds 5c, 5d showed the highest selective cytotoxic activity with (IC50 = 177.00 ± 3.66 μM) and (IC50 = 148.49 ± 1.37 μM) respectively. Compound 6a showed moderate selectivity with (IC50 = 73.41 ± 3.376 μM). Table 6. shows the IC50 of compounds 5c, 5d, and 6a against WI-38 compared to staurosporine.
Compound
Cytotoxicity
IC50(mean ± SD)
μM
WI-38
5c
177.00 ± 3.66
5d
148.49 ± 1.37
6a
73.41 ± 3.37
staurosporine
67.33 ± 1.72
3 Conclusion
In summary, novel chloro methylquinazolinone derivatives were designed and synthesized by introducing different moieties. The activities of the new derivatives were preliminary predicted using online prediction tools. Drug likeness and physicochemical properties of the compounds were studied using in-silico tools as well as descriptors of absorption, distribution, and metabolism. Compounds showed good oral bioavailability characteristics together with good potential for gastrointestinal absorption. The new derivatives showed no signs of side effects to the liver or CNS. All the compounds were screened for cytotoxic activity compared to staurosporine against three cell lines HCT(IC50 = 22.44 ± 0.51 μM), MCF7(IC50 = 29.02 ± 0.51 μM), and HepG2(IC50 = 15.23 ± 0.51 μM). Compound 5c showed the highest cytotoxic activity against the HCT cell line (IC50 = 8.00 ± 0.33 μM), compound 5d showed the highest cytotoxic activity against HepG-2 cell line (IC50 = 17.78 ± 0.58 μM), while compound 6a showed the highest cytotoxic activity against MCF-7 cell line (IC50 = 11.92 ± 0.15 μM). There was an agreement between the molecular docking study and the in-vitro PI3k-δ enzyme inhibition assay, where compound 5d showed enzyme inhibition activity (IC50 = 1.24 ± 0.03 μM) and was able to bind to the ATP binding site of PI3k-δ enzyme where it formed H-bond interaction to the key amino acid Val828 and hydrophobic interaction to the key amino acid ILe910 and other amino acids at the pocket, compound 5c showed enzyme inhibition activity (IC50 = 8.27 ± 0.19 μM) and was able to bind to the ATP binding site of PI3k-δ enzyme as well where it formed H-bond to the key amino acid Val828 and additional H-bond Lys779 and hydrophobic interactions to many amino acids in the pocket. Compound 6a, on the other hand, lacked the key H-bond interaction with the hinge region amino acid Val828 while maintaining hydrophobic interactions with other amino acids at the affinity pocket, specificity pocket, and hydrophobic region II of the PI3k- enzyme's ATP binding site. This could explain 6a's decreased in-vitro enzyme inhibition activity (IC50 = 29.10 ± 0.54 μM). Compounds 5c and 5d showed high cytotoxicity when screened against the WI-38 cell line.
4 Materials & methods
Melting points were recorded on the Stuart melting apparatus. IR. spectra (KBr) were recorded on FT-IR. spectrometer (ν Cm−1). Nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on Bruker 400 MHz spectrometer using DMSO‑d6 as a solvent; the chemical shifts are expressed in δ ppm using TMS as an internal standard in NMR unit Faculty of Pharmacy, Mansoura University. Mass spectra were recorded on Shimadzu QP-GC/MS mass spectrometers. The elemental microanalyses results were within ± 0.4% from the theoretical values. Solvent evaporation was performed under reduced pressure using Buchi R-3000 Rotacool Rotatory Evaporator, thin layer chromatography was performed on pre-coated (0.25 mm) silica gel GF254 plates (E. Merck, Germany); compounds were detected with 254 nm UV lamp. All chemicals and starting materials were commercial chemicals obtained from Sigma-Aldrich.
Synthesis of 7-chloro-2-methyl-4H-benzo[d][1,3]oxazin-4-one (2)
A mixture of 2-amino-4-chlorobenzoic acid (1) (0.01 mol) and acetic anhydride (0.1 mol) was refluxed for 1 h. The reaction was monitored by TLC till no traces of starting material were found. Excess acetic anhydride was evaporated under vacuum. The resulting white precipitate was collected by filtration and washed with petroleum ether. (yield 95%) mp 179–180 °C. mp182°C (Alagarsamy 2004).
Synthesis of 3-amino-7-chloro-2-methylquinazolin-4(3H)-one (3)
Equimolar amounts of 7-chloro-2-methyl-4H-benzo[d][1,3]oxazin-4-one (2) (0.01 mol) and hydrazine hydrate were refluxed in 30 mL ethanol for 3 h. The end of the reaction was monitored by TLC. The reaction mixture was then evaporated under vacuum; a white precipitate was obtained, washed with distilled water, and recrystallized from dimethylformamide (DMF). (Yield = 97%) mp 142–144 °C. mp 138–140 °C (Osarumwense and Iyekowa 2017).
Synthesis of 2-chloro-N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl) acetamide (4)
3-amino-7-chloro-2-methylquinazolin-4(3H)-one (3) (0.01 mol) was dissolved in dry toluene and cooled to 15 °C. Chloroacetyl chloride (0.01 mol) was added dropwise with stirring. The reaction mixture was then refluxed and monitored by TLC till no starting material was present (6 h). The reaction mixture was then cooled; the formed precipitate was collected by filtration, washed and recrystallized from ethanol. The desired intermediate was obtained in good yield (Yield = 95%) mp174-176 °C; IR. (KBr) 3280 Cm−1 (NH), 1689 Cm−1 (quinazoline C = O), 1643 Cm−1 (amide C = O), 700 Cm−1 (C-Cl); 1HNMR (400 MHz, DMSO‑d6) δ 8.05 (d, J = 9.6 Hz, 1H), 7.59(s, 1H), 7.42(m, 1H), 4.04(s,2H), 2.15(s,3H). 13CNMR (100 MHz, DMSO‑d6) δ 171.74, 159.44, 159.01, 137.48, 128.48, 125.48, 125.26, 120.75, 63.15, 22.47; MS m/z (%): 288 (M+2,0.5), Anal. calcd. for C11H19CL2N3O2 (286.11) C,46.18; H, 3.17; N,14.69. Found: C,46.17; H, 3.15; N,14.67.
General procedure for the synthesis of compounds 5a–e
2-chloro-N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)acetamide (4) (0.005 mol) was dissolved in 30 mL dry toluene, to this freshly dried anhydrous potassium carbonate (0.005 mol) and secondary amine (0.005 mol) were added and refluxed for 12 h. The completion of the reaction was monitored by TLC. Excess toluene was then distilled off; precipitate obtained was washed with petroleum ether, dried, and recrystallized from ethanol. The following compounds were prepared:
4.1 N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-(4-methylpiperazin-1-yl) Acetamide (5a)
(Yield: 96%) mp 293–295 °C; IR. (KBr) 3380 Cm−1(NH), 1726 Cm−1(quinazoline C = O), 1700 Cm−1(amide C = O); 1HNMR (400 MHz, DMSO‑d6) δ 8.05(d, J = 9.6 Hz,1H), 7.59 (s,1H), 7.39(m. 1H), 3.83(s,2H), 3.14(m, 4H), 2.96 (m, 4H), 2.36 (s, 3H), 2.15 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 171.04, 165.71, 158.43, 148.48, 137.48, 128.53, 125.63, 125.28, 120.62, 62.63, 55.32, 55.22, 53.45, 53.38, 46.32, 22.30; MS m/z (%): 351 (M+2,11), Anal. calcd. for C16H20CLN5O2 (349.82) C,54.94; H, 5.76; N,20.02. Found: C,54.90; H, 5.77; N,20.03.
4.2 N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-(4-ethylpiperazin-1-yl) Acetamide (5b)
(Yield: 94%) mp 280–282 °C; IR. (KBr) 3360 Cm−1(NH), 1710 Cm−1(quinazoline C = O), 1631 Cm−1(amide C = O); 1HNMR (400 MHz, DMSO‑d6) δ 8.01 (d, J = 8.4 Hz, 1H), 7.55 (s,1H), 7.39 (m, 1H), 3.80 (s, 2H), 2.91 (m, 8H), 2.37 (m, 2H), 2.28 (s, 3H), 1.01 (t, J = 7.2 Hz, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 171.74, 159.44, 159.01, 148.46, 137.48, 128.48, 125.48, 125.26, 120.75, 63.15, 53.54, 53.30, 52.94, 52.19, 48.74, 22.47.12.57; MS m/z (%): 365(M+2,4), 363 (M+,11), Anal. calcd. for C17H22CLN5O2 (363.84) C,56.12; H, 6.09; N,19.25. Found: C,56.11; H, 6.07; N,19.22.
4.3 N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-(4-(pyrimidin-2-yl)piperazin-1-yl)acetamide (5c)
(Yield: 97%) mp 297–298 °C; IR. (KBr) 3402 Cm−1(NH), 1705 Cm−1(quinazoline C = O), 1639 Cm−1(amide C = O); 1HNMR (400 MHz, DMSO‑d6) δ8.47 (d, J = 4.4 Hz, 1H), 8.37 (d, J = 4.4 Hz, 1H), 8.04(m,1H), 7.60 (m,1H), 7.42 (m,1H), 6.63 (m,1H), 3.77 (s,2H), 3.12 (m, 4H), 2.64 (m, 4H), 2.37 (s,3H). 13CNMR (100 MHz, DMSO‑d6) δ 171.08, 166.64, 164.96, 159.46, 158.20, 156.72, 146.79, 136.44, 133.91, 120.18, 118.91,115.11, 64.84, 55.76, 54.28, 53.44, 51.96, 21.54; MS m/z (%): 413 (M+,0.94), Anal. calcd. for C19H20CLN7O2 (413.84) C,55.14; H, 4.87; N,23.69. Found: C, C,55.12; H, 4.84; N,23.66.
4.4 N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-(4-propylpiperazin-1-yl) Acetamide (5d)
(Yield: 92%) mp 253–255 °C; IR. (KBr) 3402 Cm−1(NH), 1705 Cm−1(quinazoline C = O), 1639 Cm−1(amide C = O); 1HNMR (400 MHz, DMSO‑d6) δ 8.20 (d, J = 7.5 Hz, 1H), 7.69 (d, J = 1.5 Hz, 1H), 7.46 (dd, J = 7.5, 1.5 Hz, 1H), 3.24 (s, 2H), 2.66 – 2.62 (m, 4H), 2.62 – 2.58 (m, 4H), 2.56 (s, 3H), 2.44 (t, J = 7.1 Hz, 2H), 1.57 (m, 2H), 0.90 (t, J = 8.0 Hz, 3H). 13C NMR (100 MHz, DMSO‑d6) δ171.29, 160.10, 157.77, 148.48, 137.28, 130.58, 121.32, 118.00, 60.24, 58.32, 57.59, 57.24, 56.35, 56.20, 21,53, 19.27, 11.30; MS m/z (%): 338 (M+2,0.32), Anal. calcd. for C18H24CLN5O2 (377.84) C,57.21; H, 6.40; N,18.53. Found: C,57.20; H, 6.41; N,18.50.
N-(7-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-morpholinoacetamide(5e)
(Yield: 95%) mp 243–245 °C; IR. (KBr) 3240 Cm−1(NH), 1700 Cm−1(quinazoline C = O), 1660 Cm−1(amide C = O); 1HNMR (400 MHz, DMSO‑d6) δ 8.11 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.59 (dd, J = 8.5, 2.1 Hz, 1H), 3.49 – 3.06 (m, 4H), 3.19 (s, 2H), 2.74 – 2.47 (m, 4H), 2.42 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ169.62, 158.69, 158.44, 148.09, 140.13, 129.37, 128.97, 128.68, 127.61, 126.61, 125.78, 119.84, 67.18, 66.48, 60.73, 53.74, 46.00, 21.72; MS m/z (%): 338(M+2,23), 336(M+,66), Anal. calcd. for C15H17CLN4O3 (336.77) C,53.50; H, 5.09; N,16.64. Found: C, 53.53; H, 5.11; N,16.60.
4.5 General procedure for the synthesis of compounds 6a–e
3-amino-7-chloro-2-methylquinazolin-4(3H)-one (3) (0.005 mol) was dissolved in 30 mL ethanol. To this mixture, a few drops of acetic acid and aromatic aldehyde (0.005 mol) were added and refluxed for 12–24 h. The completion of the reaction was monitored by TLC. The reaction mixture was allowed to cool then poured on crushed ice. The obtained precipitate was collected by filtration, dried then recrystallized from ethyl acetate. The following compounds were prepared:
(E)-7-chloro-3-((4-(dimethylamino)benzylidene)amino)-2-methylquinazolin-4(3H)-one (6a)
(Yield: 92%) mp 242–245 °C; IR. (KBr) 3410 Cm−1(NH), 1705 Cm−1(C = O), 1590 Cm−1(C = N); 1HNMR (400 MHz, DMSO‑d6) δ 9.06 (s, 1H), 8.21 (d, J = 7.5 Hz, 1H), 7.68 (d, J = 1.5 Hz, 1H), 7.54 – 7.50 (m, 2H), 7.49 (dd, J = 7.5, 1.5 Hz, 1H), 6.86 – 6.51 (m, 2H), 3.03 (s, 6H), 2.60 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 160.64, 156.41, 153.63, 151.47, 146.67, 136.52, 129.44, 127.42, 126.98, 126.32, 122.79, 121.46, 111.72, 40.28, 21.45; MS m/z (%): 342(M+2,23), 340(M+,69), Anal. calcd. for C18H17CLN4O (340.81) C,63.44; H, 5.03; N,16.44. Found: C, 63.40; H, 5.107; N,16. 40.
(E)-3-((3-bromobenzylidene)amino)-7-chloro-2-methylquinazolin-4(3H)-one (6b)
(Yield: 95%) mp 193–195 °C; 1HNMR (400 MHz, DMSO‑d6) δ 8.94 (d, J = 0.7 Hz, 1H), 8.25 (d, J = 7.5 Hz, 1H), 7.93 – 7.85 (m, 1H), 7.70 (m, 1H), 7.66 – 7.56 (m, 2H), 7.50 (dd, J = 7.5, 1.5 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 2.60 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 160.33, 155.58, 152.09, 146.83, 136.83, 136.02, 131.35, 130.33, 129.07, 127.34, 127.19, 126.94, 122.29, 121.28, 120.75, 21.41; MS m/z (%):376(M+2,0.33), Anal. calcd. for C16H11BrCLN3O (376.64) C,51.02; H, 2.94; N,11.16. Found: C,51.03; H, 2.93; N,11.17.
(E)-7-chloro-2-methyl-3-((4-nitrobenzylidene)amino)quinazolin-4(3H)-one (6c)
(Yield: 97%) mp 172–174 °C; 1HNMR (400 MHz, DMSO‑d6) δ 9.04 (s, 1H), 8.22 (dd, J = 3.0, 1.7 Hz, 1H), 8.21 – 8.19 (m, 2H), 8.07 (d, J = 1.3 Hz, 1H), 8.05 (t, J = 0.8 Hz, 1H), 7.68 (d, J = 1.5 Hz, 1H), 7.49 (dd, J = 7.5, 1.5 Hz, 1H), 2.60 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 160.36, 155.98, 153.33, 147.84, 146.58, 139.69, 136.73, 127.78, 127.43, 127.18, 123.94, 122.48, 120.86, 21.37; MS m/z (%):342(M+,18), Anal. calcd. for C16H11CLN4O3 (342.74) C,56.07; H, 3.23; N,16.35. Found: C,56.05; H, 3.20; N,16.33.
(E)-7-chloro-3-((4-fluorobenzylidene)amino)-2-methylquinazolin-4(3H)-one (6d)
(Yield: 94%) mp 223–225 °C; IR. (KBr) 3295 Cm−1(NH), 1700 Cm−1(C = O), 1599 Cm−1(C = N); 1HNMR (400 MHz, DMSO‑d6) δ 9.07 (s, ,1H), 8.18 (d, J = 7.5 Hz, 1H), 7.88 – 7.75 (m, 2H), 7.64 (d, J = 1.5 Hz, 1H), 7.53 (dd, J = 7.5, 1.5 Hz, 1H), 7.28 – 7.13 (m, 2H), 2.59 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 162.77, 160.32, 160.25, 155.68, 153.52, 146.76, 136.23, 131.61, 131.58, 129.79, 129.71, 127.38, 127.18, 122.16, 120.66, 116.47, 116.27, 21.37; MS m/z (%):317(M+2,0.31), Anal. calcd. for C16H11FCLN3O (315.73) C,60.87; H, 3.51; N,11.23. Found: C,60.88; H, 3.50; N,11.24.
(E)-7-chloro-3-((3,4-dimethoxybenzylidene)amino)-2-methylquinazolin-4(3H) -one (6e)
(Yield: 96%) mp189-192 °C; 1HNMR (400 MHz, DMSO‑d6) δ 8.90 (s, 1H), 8.23 (d, J = 7.5 Hz, 1H), 7.69 (d, J = 1.5 Hz, 1H), 7.48 (dd, J = 7.5, 1.4 Hz, 1H), 7.41 (dd, J = 1.5, 0.5 Hz, 1H), 7.30 – 7.19 (m, 1H), 6.99 (d, J = 7.5 Hz, 1H), 3.86 (s, 6H), 2.60 (s, 3H). 13CNMR (100 MHz, DMSO‑d6) δ 160.40, 155.98, 152.16, 150.91, 149.79, 146.66, 136.96, 127.83, 127.34, 127.19, 125.58, 122.43, 120.86, 112.31, 110.33, 55.89, 55.87, 21.37; MS m/z (%):359 (M+2, 33), 357(M+,100), Anal. calcd. for C18H16FCLN3O3 (357.79) C,60.42; H, 4.51; N,11.74. Found: C,60.40; H, 4.55; N,11.70.
4.6 Molecular docking studies
Docking was performed following the literature, (Elfeky et al., 2018). The crystallographic structure of PI3K- δ (PDB ID: 2WXG) was downloaded from PDB and prepared for molecular docking by eliminating ligand, the addition of hydrogens, and energy minimization by using MOE software 2009. The energy minimized structure was further used as a receptor for docking. The catalytic site of PI3K- δ was obtained using the site finder algorithm in MOE. The two‐dimensional structures of the synthesized compounds were generated using ChemBioOffice, then constructed from fragment libraries in the MOE 2009. and energy minimized using MMFF94x force field in MOE. The docking was performed with selected parameters (Rescoring function 1 and Rescoring function 2: London dG, Placement: Triangle matcher, retain: 2, and Refinement: Force field) to find and evaluate the interaction between ligands and The ATP site of PI3K- δ. The most efficient hits were selected based on the S-score of SW13 inhibitor and root-mean-square deviation (RMSD) values. The retrieved compounds that have higher S-value and lower RMSD were recorded.
4.7 In-vitro cytotoxic activity against HCT, MCF7, and HepG2 cell lines
The standard MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5- diphenyl-2H-tetrazolium bromide) method (Letafat et al., 2013), was used to evaluate the cytotoxic activity of the synthesized compounds against three cancer cell lines including human colon carcinoma (HCT116), human breast adenocarcinoma (MCF-7), and human hepatocellular carcinoma (HepG-2), The quantitative assay depends on the ability of the mitochondrial dehydrogenase of viable cells to cleave the tetrazolium ring of MTT. The produced purple color is measured spectrophotometrically, and thus the increase or decrease in the cell number can indicate the antitumor activity of tested compounds. the antitumor activity was conveyed as the concentration of the compound that caused 50% growth inhibition (IC50, mean ± SD) in comparison to the growth of untreated cells. Cells for cell lines were obtained from American Type Culture Collection and cultured using DMEM (Invitrogen) supplemented with 10% FBS (Hyclone), 10 μg/mL of insulin (Sigma), and 1% penicillin–streptomycin. A 96-well plate was used for the test. Cells were treated with serial concentrations of test compounds and incubated for 48 h at 37°c, and then the plate was examined under an inverted microscope before the MTT assay. the cultures were removed from the incubator to the laminar flow hood, and MTT was added as 10% of the culture medium volume and then incubated for 2–4 h. After removal from the incubator, the formed formazan crystals were dissolved; using MTT solubilizing solution, the absorbance was measured at a wavelength of 570 nm.
4.8 In-vitro PI3k-δ enzyme inhibition assay
The PI3 Kinase Inhibitor Assay (Yanamandra et al., 2015, Davidson et al., 2021) is a competitive assay used for the fast and sensitive quantitation of activity of PI3 kinases δ. The assay works on the principle that PI3 Kinase phosphorylates phosphatidylinositol-(4,5)-bisphosphate (PIP2) and converts it into phosphatidylinositol-(3,4,5)- trisphosphate (PIP3). The pleckstrin homology (PH) domain of the protein GRP-1 binds to PIP3 with high affinity and specificity. The kit includes this recombinant protein that is used as the capture protein. This protein binds to the glutathione plate and captures either the PIP3 generated as part of the kinase reaction or the biotinylated-PIP3 tracer included in the kit. The captured biotinylated-PIP3 is detected using streptavidin-HRP conjugate and a colorimetric read-out (OD 450). The lower the signal, the higher the PI3 Kinase activity.
4.9 In-vitro cytotoxic activity against WI-38 cell line
The cytotoxic effect of the highly active compounds was determined using a normal Caucasian fibroblast-like fetal lung cell line (WI-38). cells were cultured using DMEM (Invitrogen/Life Technologies, Waltham, MA, USA) supplemented with 10% FBS (Hyclone, Waltham, MA, USA), 10 µg/mL of insulin (Sigma), and 1% penicillin–streptomycin. All of the other chemicals and reagents were from Sigma or Invitrogen. Plate cells (cells density 1.2–1.8 × 10,000 cells/well) in a volume of 100 µL complete growth medium + 100 µL of the tested compound per well in a 96-well plate for 24 h before the MTT assay (Sofan et al., 2021).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J. Hematol. Oncol.. 2013;6(1):1-17.
- [Google Scholar]
- Synthesis and pharmacological investigation of some novel 2-methyl-3-(substituted methylamino)-(3H)-quinazolin-4-ones as histamine H1-receptor blockers. Die Pharm.-Int. J. Pharm. Sci.. 2004;59(10):753-755.
- [Google Scholar]
- Synthesis and anticonvulsant activity of novel quinazolin-4 (3 H)-one derived pyrazole analogs. Med. Chem. Res.. 2013;22(4):1711-1722.
- [Google Scholar]
- Design and synthesis of quinazolinyl acetamides for their analgesic and anti-inflammatory activities. Zeitschrift für Naturforschung B. 2015;70(8):597-604.
- [Google Scholar]
- Microwave-assisted Niementowski reaction. Back to the roots. Tetrahedron Lett.. 2002;43(21):3911-3913.
- [Google Scholar]
- In silico prediction of aqueous solubility using simple QSPR models: the importance of phenol and phenol-like moieties. J. Chem. Inf. Model.. 2012;52(11):2950-2957.
- [Google Scholar]
- The p110 delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol.. 2010;6(2):117-124.
- [Google Scholar]
- Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04691502 through structure based drug design. MedChemComm. 2010;1(2):139-144.
- [Google Scholar]
- Structure-based design, SAR analysis and antitumor activity of PI3K/mTOR dual inhibitors from 4-methylpyridopyrimidinone series. Bioorg. Med. Chem. Lett.. 2013;23(9):2787-2792.
- [Google Scholar]
- Synthesis and biological evaluation of anti-tubercular activity of Schiff bases of 2-Amino thiazoles. Bioorg. Med. Chem. Lett.. 2020;30(24):127655
- [Google Scholar]
- iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model.. 2014;54(12):3284-3301.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7(1):1-13.
- [Google Scholar]
- SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res.. 2019;47(W1):W357-W364.
- [Google Scholar]
- A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117.
- [Google Scholar]
- Thyroid hormone receptor beta inhibits PI3K-Akt-mTOR signaling axis in anaplastic thyroid cancer via genomic mechanisms. J. Endocr. Soc.. 2021;5(8):bvab102.
- [Google Scholar]
- Efficient synthesis of novel pentacyclic 6, 7-dihydro-5a, 7a, 13, 14-tetraaza-pentaphene-5, 8-diones. Tetrahedron Lett.. 2005;46(20):3445-3447.
- [Google Scholar]
- Synthesis of novel 2, 3-substituted quinazolin-4-ones by condensation of alkyl or aromatic diamines with 5-(N-arylimino)-4-chloro-5H-1, 2, 3-dithiazoles. Tetrahedron. 2007;63(4):847-854.
- [Google Scholar]
- Design and synthesis of new quinoxaline derivatives as anticancer agents and apoptotic inducers. Molecules. 2019;24(6):1175.
- [Google Scholar]
- Elfeky, s., T. Sobahi, M. Geninah and N. Ahmed (2018). Ultrasound one-pot synthesis of fused Quinazolinones and Quinazolinediones, screening and molecular modeling study as phosphodiesterase 7A inhibitors.
- Synthesis, biological screening, and molecular docking of quinazolinone and quinazolinethione as phosphodiesterase 7 inhibitors. Arch. Pharm.. 2020;353(1):1900211.
- [Google Scholar]
- Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem.. 2000;43(20):3714-3717.
- [Google Scholar]
- Challenges for the clinical development of PI3K inhibitors: strategies to improve their impact in solid tumors. Cancer Discovery. 2019;9(4):482-491.
- [Google Scholar]
- Discovery and pharmacological characterization of novel quinazoline-based PI3K delta-selective inhibitors. ACS Med. Chem. Lett.. 2016;7(8):762-767.
- [Google Scholar]
- Intermittent high-dose scheduling of AZD8835, a novel selective inhibitor of PI3Kα and PI3Kδ, demonstrates treatment strategies for PIK3CA-dependent breast cancers. Mol. Cancer Ther.. 2016;15(5):877-889.
- [Google Scholar]
- Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci.. 2014;127(5):923-928.
- [Google Scholar]
- “A. viswanath, and F.” Sultana, SNCvL Pushpavalli, M. Pal-Bhadra, HK Srivastava, GN Sastry, A. Juvekar, S. Sen, and S. Zingde, Quinazolinone linked pyrrolo [2, 1-c][1, 4] benzodiazepine (PBD) conjugates: Design, synthesis and biological evaluation as potential anticancer agents. Bioorg. Med. Chem. 2010;18:526-542.
- [Google Scholar]
- The Synergy of Combined Use of DMSO and Bronsted Acid (Ionic Liquid) as a Catalyst Part I: Efficient Niementowski Synthesis of Modified Quinazolinones. Green Sustain. Chem.. 2011;1(01):12.
- [Google Scholar]
- A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125(4):733-747.
- [Google Scholar]
- Synthesis and in vitro cytotoxic activity of novel chalcone-like agents. Iranian J. Basic Med. Sci.. 2013;16(11):1155.
- [Google Scholar]
- Rational design, synthesis, and SAR of a novel thiazolopyrimidinone series of selective PI3K-beta inhibitors. ACS Med. Chem. Lett.. 2012;3(7):524-529.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23(1–3):3-25.
- [Google Scholar]
- Novel 4-acrylamido-quinoline derivatives as potent PI3K/mTOR dual inhibitors: the design, synthesis, and in vitro and in vivo biological evaluation. Front. Chem.. 2019;7:236.
- [Google Scholar]
- Inversion kinetics of some E/Z 3-(benzylidene)-2-oxo-indoline derivatives and their in silico CDK2 docking studies. RSC Adv.. 2021;11(14):7839-7850.
- [Google Scholar]
- Small-molecule inhibitors of the PI3K signaling network. Future Med. Chem.. 2011;3(5):549-565.
- [Google Scholar]
- PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci.. 2021;22(7):3464.
- [Google Scholar]
- Synthesis and antibacterial activity of 7-chloro-2-methyl-4H-benzo [d][1, 3]–oxazin-4-one and 3-amino-7-chloro-2-methyl-quinazolin-4 (3H)-one. Tropical J. Natural Prod. Res.. 2017;1:173-175.
- [Google Scholar]
- Synthesis and antimicrobial activity of some novel substituted piperazinyl-quinazolin-3 (4H)-ones. E-J. Chem.. 2008;5(1):23-33.
- [Google Scholar]
- Synthesis, Cytotoxicity Assessment and Antioxidant Activity of Some New Thiazol-2-yl carboxamides. J. Heterocycl. Chem. 2021
- [Google Scholar]
- Atropisomeric quinazolin-4-one derivatives are potent noncompetitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonists. Bioorg. Med. Chem. Lett.. 2001;11(2):177-181.
- [Google Scholar]
- Form and flexibility in phosphoinositide 3-kinases. Biochem. Soc. Trans.. 2009;37(4):615-626.
- [Google Scholar]
- Development and application of PI3K assays for novel drug discovery. Expert Opin. Drug Discov.. 2015;10(2):171-186.
- [Google Scholar]
- Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer. 2019;18(1):1-28.
- [Google Scholar]
- PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol. Cancer Ther.. 2011;10(11):2189-2199.
- [Google Scholar]
- Zeid, I. F., N. A. Mohamed, N. M. Khalifa, E. M. Kassem, E. S. Nossier, A. A. Salman, K. Mahmoud and M. A. Al-Omar (2019). “PI3K inhibitors of novel hydrazide analogues linked 2-pyridinyl quinazolone scaffold as anticancer agents.” Journal of Chemistry 2019.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103614.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




