

Translate this page into:
Synthesis of benzimidazole derivatives and their antiglycation, antioxidant, antiurease and molecular docking study
⁎Corresponding author. mtaha@iau.edu.sa (Muhammad Taha)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Diabetes and ulcer are the major health problems all over the world. The present study reports synthesis and bio-evaluation of 19 benzimidazole analogs in search of antiglycation, antioxidant and antiulcer agents. The synthetic analogs were characterized using 1H NMR, 13C NMR and HR-EIMS. All compounds were checked for their antiglycation, antiurease and antioxidant activities. The fluorophenyl benzimidazole analogs 12–14 strongly inhibited glycation with IC50 values ranging from 142 µM to 193 µM. The same fluorophenyl analogs (12–14) were also found to exhibit the highest antioxidant activity with IC50 values ranging from 1.2 µM to 6.6 µM which further highlights the significance of these bioactive analogs. The dihydroxyphenyl analogs 6–9 demonstrated the most potent enzyme inhibitory activity with IC50 values ranging from 3.10 µM to 5.90 µM. Molecular docking studies were performed on the active analogs to investigate their interactions with the urease enzyme and provide a plausible explanation for their observed urease inhibitory activity.
Keywords
Novel Benzimidazole derivatives
In silico study
Glycation
Urease

1 Introduction
In the human body, interaction of protein with sugar occurs via an enzymatic and non-enzymatic pathway (Dil et al., 2019). The process by which proteins and sugars interact enzymatically to create glycoproteins is known as glycosylation (Aebi, 2013), while the non-enzymatic interaction is called glycation and does not often occur under normal circumstances. Glycation is usually more prevalent in aging and hyperglycemia (Yao et al., 2018). In the initial step of glycation, D-glucose, D-ribose, and D-fructose combine with a protein's free amino group to form the unstable molecule fructosamine in a Schiff base reaction. These compounds on rearrangement forms a stable Amadori product, which, on further oxidation, gives advanced glycation end-products (AGE) (Marcial and Graves, 2019, Twarda-Clapa et al., 2022). Additionally, AGEs have the ability to crosslink proteins in the mitochondrial respiratory chain, inhibiting ATP synthesis and increasing the formation of reactive oxygen species (ROS) (Abdallah et al., 2022). In this context, looking for potent AGEs inhibitors is a viable tactic to stop or slow down the development and accumulation of AGEs which will aid in reducing the chance of developing several health disorders, such as those related to diabetes (Vasarri et al., 2020). Other medications that have already received FDA approval (such as aspirin, metformin, diclofenac, etc.) are insufficient to stop the glycation process when chronic hyperglycemia is present (Rasheed et al., 2018).Fig. 1..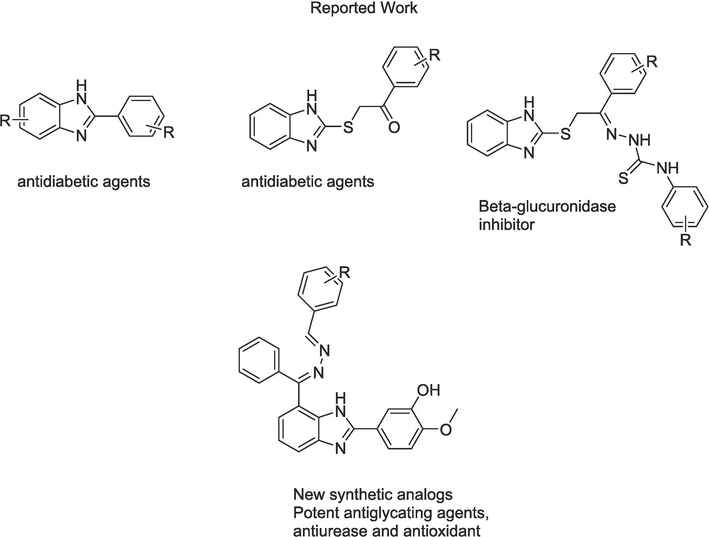
Rational of the current study.
There are also health complications that may arise due to urease activity. It is found in a wide range of organisms including various plants, microorganisms like algae, bacteria, fungus and in soil as soil enzymes (Singh et al., 2020). The urease enzyme catalyzes the rapid transformation of urea into ammonia and carbamic acid, and the carbamate then quickly and spontaneously decomposes to produce a second molecule of ammonia and one of carbon dioxide (Sohrabi et al., 2022). The enzymatic activity of urease causes an elevation in pH because the concentration of ammonia rises, which has a number of detrimental impacts on both agriculture and medicine (Matczuk and Siczek, 2021, Maz et al., 2023). Urease is a key virulence component of most fatal bacterial infections, including Mycobacterium TB, Proteus mirabilis, and Helicobacter pylori. These bacterial complications may cause peptic ulcer, stomach cancer, kidney stones, catheters blocking urolithiasis, urinary catheter encrustation, pyelonephritis and hepatic coma which eventually causes cancer in human body (Chaudhry et al., 2020). Therefore, inhibiting urease in humans is a valuable tactic for preserving human health.
The cellular redox process produces reactive oxygen species (ROS) and reactive nitrogen species (RNS), which have been shown to have a dual function as harmful and advantageous species (Sharpe et al., 2011). The overproduction of these species causes oxidative stress, a condition in which the biological system is unable to maintain the oxidant-antioxidant equilibrium (Singh et al., 2020, Brahimi et al., 2023). These reactive species can attack nucleic acids, lipids, proteins, fatty acids, carbohydrates and induce their oxidation, which may lead to oxidative damage such as protein change, membrane dysfunction, enzymatic inactivation and break of DNA strains (Dai et al., 2017, Lozynskyi et al., 2017). It is crucial to render the free radicals ineffective in order to safeguard the body from these harms (Bentz et al., 2017).
Benzimidazole is a significant pharmacophore of numerous physiologically active heterocyclic compounds having a wide range of biological activities (Satija et al., 2021). Benzimidazole derivatives has been reported as anti-inflammatory (Sharma et al., 2020), antihistaminic (Aroua et al., 2021), antiprotozoal (Patel et al., 2020), antitubercular (Patel et al., 2020), anti-HIV (Pan et al., 2015), and anticonvulsant (Tsay et al., 2013). Keeping in view the importance of benzimidazole, our research group has previously synthesized various benzimidazole scaffolds as potent inhibitors of various biological issues such antiurease (Mumtaz et al., 2022), anti-Alzheimer (Hussain et al., 2022, Adalat et al., 2023), anti-diabetic (Hayatullah et al., 2022a, Khan et al., 2022, Hayat et al., 2023) and β-glucuronidase (Hayat et al., 2022b, Taha et al., 2022). Therefore, in this study we aim to further utilize the extensive pharmacological properties of the benzimidazole nucleus by designing a new series of synthetic benzimidazole analogs and testing them for anti-glycation, anti-urease, and antioxidant properties.
2 Results and discussion
2.1 Chemistry
The benzimidazole analogs have been synthesized according to Scheme 1. Nineteen benzimidazole analogs (5–23) were synthesized followed reaction procedure (Taha et al., 2020) by refluxing 5–6 h of (2,3-diaminophenyl)(phenyl)methanone (1) with 3-hydroxy-4-methoxybenzaldehyde (2) in DMF as a solvent and sodium meta bisulfate as a catalyst to give product 3. Compound 3 was further refluxed with a methanolic hydrazine hydrate to afford intermediate 4 which was reacted with various aldehydes to get the target benzimidazole analogs (5–23) with adequate yields. The reaction completion at all steps of the reactions were checked by thin layer chromatography (TLC). The structure of all synthetic benzimidazole analogs (5–23) were confirmed by different spectroscopic techniques such as 1H NMR, 13C NMR and HR-EIMS.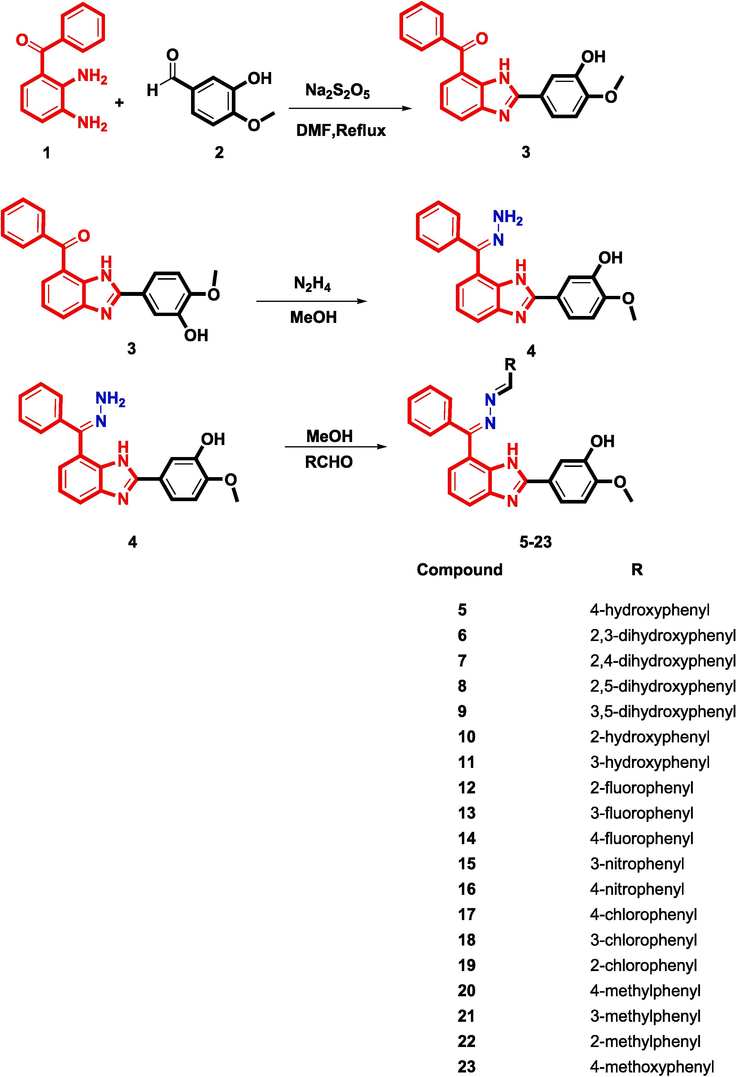
Synthesis of Schiff base derived benzimidazole analogs.
1H NMR spectra for the compounds showed a singlet peak at around 8.5 ppm corresponding the imine group proton (CH = N), and this proves the formation of the intended Schiff bases. The singlet at around 10–12 ppm was assigned to the amino proton (NH) of the benzimidazole nucleus, while the singlets around 9 ppm and 3–3.8 ppm were attributed to the hydroxyl and methoxy protons, respectively. Aromatic proton peaks were observed at around 6.6–8 ppm and their total integral values were in agreement with our proposed structures which further proves the successful synthesis of the intended analogs. There were also certain peaks that were characteristic to some analogs, for instance compounds 6–11 showed more singlet peaks that appeared around 9–13 ppm corresponding to hydroxyl protons, while 20–22 showed peaks at around 2.4 ppm that were assigned to the extra methyl group protons of the structures and 23 showed a singlet peak at 3.81 ppm corresponding to the structure’s extra methoxy protons.
13C NMR spectra showed peaks around 110–140 ppm that were assigned to aromatic carbon atoms, while the three (C = N) groups that are present in all compounds showed peaks at about 150–170 ppm. The peaks at 40–55 ppm were attributed to OCH3 and this was more apparent in 23 whereby a further peak at about 40 ppm was observed for the extra methoxy group present in the structure. Compounds 20–22 showed peaks at around 18–20 ppm corresponding to the methyl group present in their structures. Finally, HR-EIMS showed the relevant peaks for all the synthetic compounds, which proves the formation of the intended synthetic compounds.
2.2 Biological activity
2.2.1 Antiglycation activity
The synthetic analogs were evaluated for their antiglycation activity using Rutin (IC50 = 289.60 µM) as a standard drug (Table 1). All analogs displayed varying degree of antiglycation activity with IC50 values ranging from 140.50 µM to 325.20 µM. The most active analogs among the series were those that contained a fluorophenyl group, namely, compounds 12, 13 and 14, with 14 demonstrating the highest bioactivity with an IC50 value of 140.50 µM, followed by 12 and 13. Analogs possessing the dihydroxyphenyl group (6–9) were also found to be active with compound 6 exhibiting the highest activity with an IC50 value of 174.50 µM, followed by 7, 8 and 9 which showed moderate activity. Activity associated with these analogs may be a result of acetal production from the reaction of dihydroxyphenyl analogs with the carbonyl group of methylglyoxal. Moreover, hydroxyphenyl analogs (5, 10 and 11) also exhibited good bioactivity with IC50 values of 255.20 µM, 240.70 µM and 286.70 µM, respectively. The antiglycation activity of these analogs might be due to hemiacetal formation with glyoxal (Khan et al., 2009). The chlorophenyl analogs (17–19) were also found to exhibit moderate activity with IC50 values of 211.40 µM, 266.30 µM and 251.70 µM, respectively. Finally, the addition of a methoxy group as in 23, resulted in antiglycation activity that is very similar to that of the standard drug Rutin. Nitrophenyl analogs 15 and 16, in addition to methyl analogs 20, 21 and 22 showed weak bioactivity that was less than the standard drug.
Compound
R
IC50 Antiglycation
(µM ± SEM)IC50 Antiurease
(µM ± SEM)IC50 Antioxidant
(µM ± SEM)
5
255.20 ± 0.60
7.30 ± 0.10
15.20 ± 0.60
6
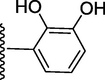
174.50 ± 0.90
3.10 ± 0.10
20.10 ± 0.90
7
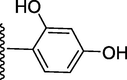
228.90 ± 0.70
4.30 ± 0.10
18.10 ± 0.70
8
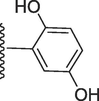
254.30 ± 0.90
5.70 ± 0.10
22.20 ± 0.90
9
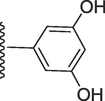
271.80 ± 0.10
5.90 ± 0.10
2.40 ± 0.10
10
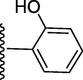
240.70 ± 0.4
8.70 ± 0.20
10.40 ± 0.4
11

286.70 ± 0.10
11.20 ± 0.20
1.60 ± 0.10
12
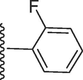
158.50 ± 0.10
7.40 ± 0.20
6.60 ± 0.10
13
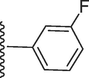
192.60 ± 0.10
11.10 ± 0.20
1.20 ± 0.10
14
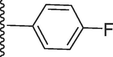
140.50 ± 0.10
4.20 ± 0.10
1.80 ± 0.10
15
319.10 ± 0.70
15.60 ± 0.30
19.20 ± 0.70
16
292.60 ± 0.80
17.40 ± 0.30
21.20 ± 0.80
17
211.40 ± 0.10
18.80 ± 0.40
11.60 ± 0.10
18
266.30 ± 0.10
20.50 ± 0.40
6.10 ± 0.10
19
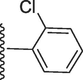
251.70 ± 0.80
17.80 ± 0.30
21.70 ± 0.80
20
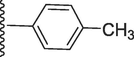
317.60 ± 0.70
20.40 ± 0.40
19.60 ± 0.70
21
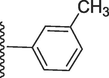
325.20 ± 0.60
21.20 ± 0.40
15.20 ± 0.60
22
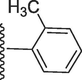
310.10 ± 0.90
22.10 ± 0.50
20.10 ± 0.90
23
288.10 ± 0.70
18.10 ± 0.70
18.10 ± 0.70
Standard drug
–
Rutin
289.60 ± 0.80
Thiourea
21.00 ± 0.80
n-propyl gallate 289.60 ± 0.80
Therefore, it can be deduced that the type of substituents and their position on the benzene ring affects antiglycation activity for the benzimidazole derivatives, with highest bioactivity being observed with the 4-fluorophenyl analog.
2.2.2 Antiurease activity
The synthetic analogs were also screened for their anti-urease potential. All analogs exhibited good to moderate inhibition potential when compared to the standard drug thiourea which has having IC50 value of 21 µM. It is clear from the IC50 values given in Table 1 that the most active analogs in the series are those possessing the dihydroxyphenyl group (6–9) with compound 6 exhibiting the inhibitory activity against urease enzyme with an IC50 value of 3.10 µM. The high inhibition potential of these analogs might be due to the electron donating effect of the hydroxyl group and also due to its involvement in hydrogen bonding with the active site of the enzyme (Taha et al., 2019). It can also be observed that there is a slight difference in activity across compounds 6–9 and this could be due to difference in position of the hydroxyl groups on the phenyl ring which would in turn affect certain potential interactions with the enzyme’s active site. Furthermore, it was observed that even analogs with a hydroxyphenyl group (5, 10 and 11) were found to exhibit antiurease activity with 5 being the most active and possessing an IC50 value of 7.30 µM indicating the ability of its para-OH to form important interactions with the active sites through hydrogen bonding and block the enzyme’s activity (Hamad et al., 2020). This shows that a substituent with a lower number of hydroxyl groups will still result in an active analog but the activity will be relatively lower than analogs containing a dihydroxyphenyl group.
The fluorinated analogs 12, 13 and 14 possessed good inhibitory activity against urease with 14 being the most active fluorinated analog and the second most active compound after 6, with an IC50 value of 4.20 µM. These results indicate the significance of having the fluorine atom at the para position of the benzene ring for optimal activity, in addition to demonstrating that the moderate electronic effects of halogenated groups enhance enzyme inhibition (Chaudhry et al., 2020). The case was different with the chlorinated analogs (17–19), whereby the compounds’ inhibitory activity was higher than the standard drug, but relatively less than that of the fluorinated analogs, which indicates the superiority of the fluorophenyl group.
The nitrophenyl analogs 15 and 16 also exhibited inhibitory activity against urease with 15 being the more active analog with an IC50 value of 15.60 µM. The methylphenyl analogs (20–22) possessed relatively lower activity than the other analogs, but they still exhibited similar activity to that of the standard drug thiourea. However, replacing the methyl group by a methoxy group as in analog 23 resulted in increased urease inhibition with an IC50 value of 18.10 µM.
In conclusion, SAR analysis showed that the introduction of electron withdrawing/donating groups in the structural framework of the benzimidazole moiety, as well as other electronic variables, have significant effects on the inhibitory potentials of the compounds.
2.2.3 Antioxidant activity
The capacity of synthetic compounds to scavenge free radicals was evaluated by using DPPH free radical absorbance assay (Table 1). The majority of synthetic derivatives were found to be good to excellent scavengers. Among all the examined derivatives, compound 13, a 3-fluorophenyl analogue, showed a substantially greater activity with an IC50 value of 1.20 µM, while isomers of 13, such as analogs 12 and 14, also showed good activity with IC50 values of 6.60 µM and 1.80 µM, respectively. The high activity of these analogs might be due to the small size and electron withdrawing nature of fluorine atom. Increasing the size of halogen atoms often results in decreased inductive effect and also reduces radical scavenging activity, and these effects were observed from the IC50 values of analog 18 (IC50 = 6.10 µM) and 19 (IC50 = 21.70 µM) which had a chlorine atom instead of fluorine (Kanwal et al., 2021). It is also clear from the values that halogen atom at 3 position of phenyl ring played a crucial role in inhibition.
Hydroxyphenyl analogs 5, 10 and 11 were another group of compounds that demonstrated excellent radical scavenging activities. Among these analogs, analog 11, a 3-hydroxyphenyl derivative, was superior in activity having a value of 1.60 µM. Analog 5 (IC50 = 15.20 µM) and analog 10 (IC50 = 10.40 µM) were less potent than 11 which indicates that the substituent’s position on the phenyl ring plays an important role in activity. Increasing the number of hydroxyl groups on the phenyl ring resulted in a decrease of antioxidant activity as shown by the IC50 values of analogs 6, 7 and 8, however, the dihydroxyphenyl analogue 9 was an exception as an increase in antioxidant activity was observed (IC50 = 2.40 µM). This shows that activity depends not only on the number of substituents but also on their position on the phenyl ring (Taha et al., 2014).
Nitrophenyl analog 15 and 16 demonstrated moderate antioxidant activity with IC50 values of 19.20 µM and 21.20 µM, respectively, and activity might be due to the electron withdrawing effect of the nitro group. The case was similar with the methylphenyl analogs 20, 21 and 22, whereby moderate activity was also observed with analog 21 being superior in activity and possessing an IC50 value of 15.20 µM. Moderate activity was observed with analog 23 which had an IC50 value of 18.10 µM, and it is suggested that this analog’s activity was limited due to unfavorable steric hindrance which also increases lipophilicity and membrane partitioning (Jeong et al., 2007).
2.3 Molecular docking studies on urease enzyme
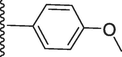
In silico molecular docking studies against the urease enzyme were conducted for the synthetic compounds in order to attempt to provide an explanation for their observed in vitro inhibitory activity. Prior to docking and analysis of the binding mode of the two most active compounds 6 and 14, the docking method was validated through control docking of native inhibitor (acetohydroxamic acid). The acetohydroxamic acid was docked into urease (PDB code: 4UBP) and compared by superimposing with the native ligand in the protein. The rmsd value between docked and native pose of acetohydroxamic acid was found to be 0.92A. The molecular docking results for the native ligand (hydroxamic acid) binding within the active site of urease displayed binding energy of −13.08 kcal/mol and formation a rather stable conventional hydrogen bond at the distance of 3.02A with the backbone (O) of Gly280. It was also observed that hydroxamic acid, through its oxygen atom, could possibly form a metal-acceptor interaction with one of the catalytic nickel atoms (Ni799) at the distance of 2.01A while forming stable hydrophobic alkyl interactions with residues Ala366 and Met367.The molecular docking result for compound 6, identified as the most active compound, which has a binding energy of −44.39 kcal/mol, presents a comprehensive understanding of its potential interactions with the urease enzyme. Compound 6 engages in diverse types of interactions, including hydrogen bond, carbon hydrogen bond, and other interactions, with various residues in the protein's binding site (Fig. 2). The presence of several stable hydrogen bonds, particularly taking place between the hydroxyl groups and His249 (HD1), His222 (NE2), and Asp363 (OD2), suggests robust ligand–protein interactions. The relatively short distances of these hydrogen bonds indicate strong binding affinity, potentially stabilizing compound 6 within the binding pocket and influencing its orientation for optimal interactions with the protein. The hydrogen bond interaction between the hydroxyl groups with the sidechain (HD1) of His249 (2.57 Å and 2.70 Å) may be crucial for the ligand's specificity and recognition within the active site. In addition, the hydroxyl group at ortho and meta position displayed the ability to form interactions with metal ions Ni798I and Ni799I present, intriguing possibilities for metal-binding ligands with specific biological activities. Additionally, carbon hydrogen bond interactions were observed between the aromatic rings with backbone of Ala170 (4.20 Å) and sidechain (OD2) of Asp224 (2.82 Å) offer alternative means of ligand binding and stabilization. These less common interactions could contribute to the ligand's conformational preferences and influence its overall binding mode. Understanding these interactions aids in deciphering the ligand's precise binding orientation and its potential effects on protein function.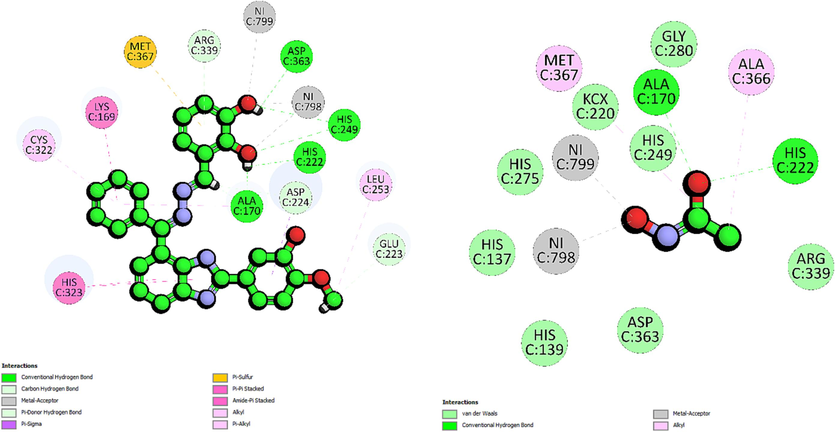
The 2D-interaction diagram of compound 6 and acetohydroxamic acid interacting with active site of urease enzyme.
The second most active analogue was the 4-fluorophenyl derivative, compound 14, recorded binding energy of −54.11 kcal/mol. Molecular docking of 14 showed that it forms a hydrogen bond with the residue His249 (2.77 Å). The presence of fluorine atom at ortho position enhances the stability of the ligand–protein complex, as indicated by the relatively short distance between the fluorine atom and the sidechain (HD1) of His249, suggesting strong binding affinity (Fig. 3). This hydrogen bond plays a significant role in stabilizing compound 14 within the binding pocket, influencing its binding orientation and specificity for His249. Additionally, Compound 14, through its fluorine atom, establishes carbon-hydrogen bond interactions with the sidechain (HD2) of His275 (3.02 Å). Meanwhile on the other hand, the imidazole ring forms another carbon-hydrogen bond with the sidechain (NE2) of His323 (2.38 Å). Compound 14 also participates in halogen interactions with metal ions Ni798 and Ni799, both involving interactions with the fluorine atoms at 2.05 Å and 2.71 Å. These interactions introduce favorable polar interactions that may influence the ligand binding mode and function within the protein. Furthermore, halogen interactions with other residues, GLY280 (3.31 Å) and Asp363 (3.38 Å) could have further enhanced compound 14 specificity for the target residues through van der Waals forces and polar contacts. An electrostatic π-anion interaction is observed between compound 14 and the sidechain (OD2) of Asp224 at the distance of 4.67 Å. Compound 14 also engages in several hydrophobic interactions, including a π-Sulphur interaction with backbone (SG) of Cys322 (4.18 Å), π-π T-shaped interaction with His323 (4.78 Å) and π-alkyl interaction with Lys169 (5.18 Å).
The 2D-interaction diagram of 14 interacting with active site of urease enzyme.
The molecular docking results complement the experimental findings and provide mechanistic insights on the observed SAR trends. The results revealed key structures that facilitated the interactions between the active compounds and critical amino acid residues like His249, Leu253, His323 and Met367 in the protein binding pocket. It was observed that the active compounds bind to urease by having their benzimidazole ring position in such a way that it could form electrostatic interactions with His323. Meanwhile on the other end of the compounds, the extended ring displayed ability to form hydrophobic interactions with Met367 while having substituents to form metal interactions with nickel atom (NIC798 and NIC799).
2.4 Molecular dynamics
Molecular dynamics studies had been conducted using the most active compound 6 in complex with urease enzyme. The molecular dynamics simulation for compound 6 in complex with the urease enzyme reveals insights on the structural dynamics of the simulated system over time. Analysis on RMSD fluctuation of the protein showed that initially during the equilibration phase between 1 and 5 ns, the RMSD values exhibit stability at relatively high levels, suggesting the system is adjusting to the simulation conditions (Fig. 4). Around 5 to 35 ns, the RMSD values fluctuate within a moderate range, indicating a steady state where structural fluctuations are balanced which implies a relatively stable conformation. Around 35–45 ns, there is a notable spike in RMSD values, indicating a perturbation or significant structural change, possibly triggered by external forces or molecular events like ligand binding interaction. The RMSD values then gradually decrease, signifying a recovery phase where the system returns to its equilibrium state, with slightly altered structural characteristics. Around 60–65 ns, another increase in RMSD suggests a structural rearrangement, possibly reflecting that the system was exploring different conformational states. Towards the end of the simulation, the RMSD values stabilize once again, at a slightly elevated level compared to the initial steady state, indicating a final equilibrium with potentially modified structural features. In order to further confirm the stability of the complex, MM-GBSA analysis was conducted to observe any major fluctuation in the binding energy occurring during the simulation. Initially the binding energy is recorded at −26.74 kcal/mol, indicating a stable initial interaction. This suggests that compound 6 formed a favorable binding conformation with the urease protein. At 5 ns and 15 ns, the binding energy becomes more negative, suggesting stronger binding interactions or a more favorable binding conformation adopted by compound 6 within the urease active site. Conversely, at 7.5 ns, 12.5 ns, and 22.5 ns, the binding energy values are close to zero, indicating weaker or less favorable binding interactions, potentially due to structural rearrangements within the complex or fluctuations in the solvent environment. Moreover, significant perturbations in the binding energy are noted at specific time points, such as 32.5 ns, 35 ns, and 40 ns, where the binding energy becomes more negative. This suggests a sudden strengthening of the binding interaction, possibly due to conformational changes in either compound 6 or the urease protein, or due to the formation of additional favorable interactions within the binding interface. Conversely, at 47.5 ns, 55 ns, and 62.5 ns, the binding energy values become more significantly negative, indicating a potential stabilization in the binding interaction, possibly due to favorable interactions. There are also transient unbinding events evident throughout the simulation, as reflected by positive binding energy values at several time points. These positive values suggest a partial or complete dissociation of compound 6 from the urease complex, followed by subsequent re-association. Towards the end of the simulation, the binding energy values stabilize around −30 kcal/mol, indicating a relatively steady state in the interaction between compound 6 and urease. This stabilization suggests that, despite fluctuations and transient unbinding events, compound 6 maintains a generally stable binding interaction with the urease protein over the course of the simulation.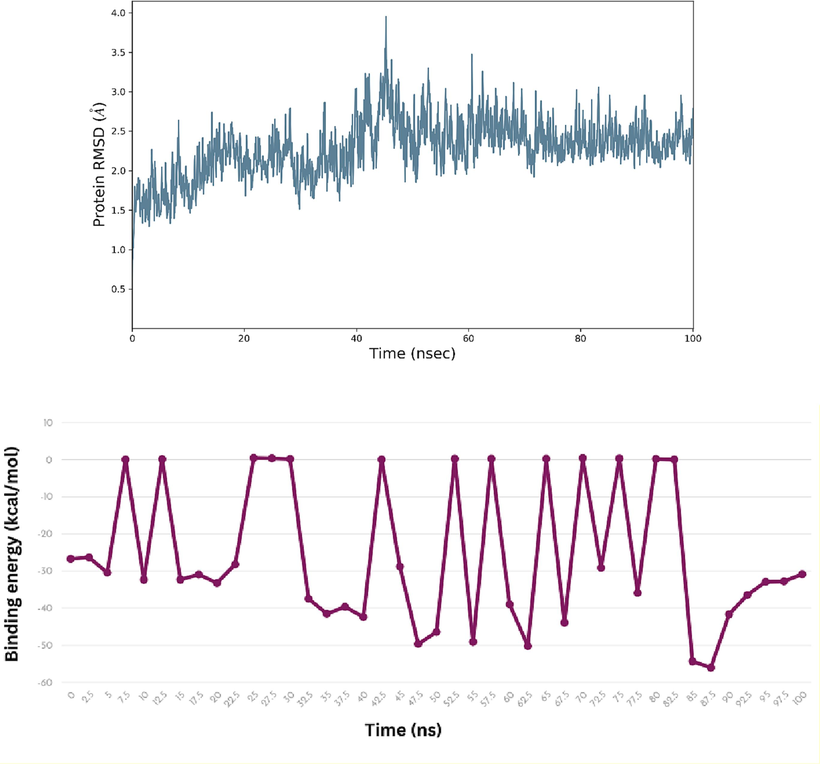
The Root Mean Square Deviation (RMSD) and MM-GBSA analysis for compound 6 in complex with urease enzyme.
3 Materials and methods
3.1 General Experimental
Melting points were determined on a Büchi 434 melting point apparatus. NMR Experiments were performed on Avance Bruker AM 600 MHz. instruments. HR Electron impact mass spectra (HREI MS) were recorded on a Finnigan MAT-311A, Germany. Thin layer chromatography (TLC) was performed on precoated silica gel aluminum plates (Kieselgel 60, 254, E. Merck, Germany). Chromatograms were viewed under UV light at 254 and 365 nm.
3.2 Synthesis of 5-(7-(hydrazono(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (4)
(2,3-diaminophenyl)(phenyl)methanone (1) (10 mmol) were refluxed with 10 mmol of 3-hydroxy-4-methoxybenzaldehyde (2) in DMF (50 mL) and sodium meta bisulfate (20 mmol) as a catalyst for 5–6 h to give intermediate product 3. The intermediate product 3 after workup was further refluxed with 50 mL of methanolic hydrazine hydrate in a 1:1 ratio to afford intermediate product 4 (Taha et al., 2020a).
3.3 Synthesis of benzimidazole analogues 5–23
The intermediate 4 (0.5 mmol) was reacted with various aldehydes (0.5 mmol) in methanol to get the target benzimidazole analogs Schiff bases (5–23) with suitable yields. The reaction completion at all steps of the reactions were optimized by thin layer chromatography (TLC).
5-(7-((E)-((4-hydroxybenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (5).
Rf value 0.59 % (30 % ethyl acetate); Light brown; Yield: 0.196 g (85 %); M.p.: 278 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.08 (s, 1H, NH), 10.03 (s, 1H, OH), 10.01 (s, 1H, OH), 8.55 (s, 1H, CH), 8.04–7.99 (m, 3H, Ar-H), 7.68–7.64 (m, 2H, Ar-H), 7.58–7.32 (m, 5H, Ar-H), 6.90–6.84 (m, 4H, Ar-H), 6.78 (d, J = 7.4 Hz, 1H, Ar-H), 3.80 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 160.4, 152.7, 149.2, 147.3, 147.0, 138.7, 132.7, 129.9, 129.5, 129.3, 128.6, 128.4, 128.3, 128.2, 128.1, 127.9, 127.6, 125.5, 115.8, 115.7, 115.6, 115.6, 117.0, 116.0, 111.3, 110.6, 55.5.; HREI-MS: m/z calcd for C28H22N4O3 [M]+462.1692; Found; 462.1676.
3-((((E)-(2-(3-hydroxy-4-methoxyphenyl)-1H-benzo[d]imidazol-7-yl)(phenyl)methylene) hydrazono)methyl)benzene-1,2-diol (6).
Rf value 0.54 % (30 % ethyl acetate); brown; Yield: 0.197 g (82 %); M.p.: 292 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.78 (s, 1H, OH), 10.51 (s, 1H, NH), 9.48 (s, 2H, 2OH), 8.49 (s, 1H, CH), 7.78 (d, J = 7.8 Hz, 1H, Ar-H), 7.71 (d, J = 7.5 Hz, 2H, Ar-H), 7.67 (d, J = 7.5HZ, 1H, Ar-H), 7.64 (d, J = 7.4 Hz, 1H, Ar-H), 7.46 (t, J = 6.8 Hz, 3H, Ar-H), 7.43 (t, J = 6.5 Hz, 1H, Ar-H), 7.21 (d, J = 7.5 Hz, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 6.83 (d, J = 7.5 Hz, 1H, Ar-H), 6.75 (d, J = 7.3 Hz, 1H, Ar-H), 6.70 (t, J = 6.4 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.2, 157.0, 152.3, 151.6, 147.3, 147.0, 146.0, 138.7, 132.7, 131.9, 129.0, 129.0, 128.2, 128.2, 128.1, 124.7, 124.3, 124.1, 123.6, 122.6, 122.3, 119.6, 119.3, 118.2, 117.6, 113.6, 111.2, 56.0.; HREI-MS: m/z calcd for C28H22N4O4 [M]+478.1641; Found; 478.1611.
4-((((E)-(2-(3-hydroxy-4-methoxyphenyl)-1H-benzo[d]imidazol-7-yl)(phenyl)methylene) hydrazono)methyl)benzene-1,3-diol (7).
Rf value 0.50 % (30 % ethyl acetate); brown; Yield: 0.201 g (84 %); M.p.: 296 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.10 (s, 1H, NH), 10.09 (s, 1H, OH), 10.01 (s, 1H, OH), 9.69 (s, 1H, OH), 8.57 (s, 1H, CH), 8.05 (d, J = 7.6 Hz, 1H, Ar-H), 8.00 (d, J = 7.6 Hz, 2H, Ar-H), 7.57–765 (m, 3H, Ar-H), 7.43–750 (m, 4H, Ar-H), 6.95–7.01 (m, 4H, Ar-H), 3.83 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 162.0, 161.8, 157.5, 152.6, 147.3, 147.0, 138.7, 134.3, 133.7, 132.0, 131.4, 130.6, 130.3, 128.7, 128.6, 127.92, 127.90, 127.6, 126.9, 125.7, 125.0, 124.7, 123.9, 115.7, 111.1, 108.6, 55.0.; HREI-MS: m/z calcd for C28H22N4O4 [M]+478.1641; Found; 478.1617.
2-((((E)-(2-(3-hydroxy-4-methoxyphenyl)-1H-benzo[d]imidazol-7-yl)(phenyl)methylene) hydrazono) methyl)benzene-1,4-diol (8).
Rf value 0.48 % (30 % ethyl acetate); brown; Yield: 0.206 g (86 %); M.p.: 297 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.21(s, 1H, NH), 11.11 (s, 1H, OH), 9.48 (s, 1H, OH), 9.45 (s, 1H, OH), 8.41 (s, 1H, CH), 7.78 (d, J = 7.6 Hz, 1H, Ar-H), 7.71 (d, J = 7.6 Hz, 2H, Ar-H), 7.67 (d, J = 7.6 Hz, 1H, Ar-H), 7.64 (d, J = 7,5Hz, 1H, Ar-H), 7.46 (t, J = 6.5 Hz, 3H, Ar-H), 7.43 (t, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 7.08 (s, 1H, Ar-H), 6.83 (d, J = 7.4 Hz, 1H, Ar-H), 6.75 (d, J = 7.4 Hz, 1H, Ar-H), 6.66 (d, J = 7.4 Hz, 1H, Ar-H), 3.83 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.3, 157.1, 153.7, 151.7, 147.1, 147.0, 139.0, 132.4, 131.0, 129.1, 129.1, 128.4, 128.4, 128.0, 124.2, 124.1, 123.0, 122.6, 120.2, 119.6, 119.3, 118.0, 117.3, 116.1, 111.2, 113.6, 56.0.; HREI-MS: m/z calcd for C28H22N4O4 [M]+478.1641; Found; 478.1609.
5-((((E)-(2-(3-hydroxy-4-methoxyphenyl)-1H-benzo[d]imidazol-7-yl)(phenyl)methylene) hydrazono)methyl)benzene-1,3-diol (9).
Rf value 0.51 % (30 % ethyl acetate); brown; Yield: 0.191 g (80 %); M.p.: 302 °C; 1H NMR (600 MHz, DMSO‑d6): δ 9.65 (s, 1H, NH), 9.60 (s, 1H, OH), 8.63 (s, 2H, 2OH), 8.57 (s, 1H, CH), 8.02 (d, J = 7.6 Hz, 1H, Ar-H), 7.94–7.98 (m, 3H, Ar-H), 7.62–7.70 (m, 4H, Ar-H), 7.00 (d, J = 7.5 Hz,1H, Ar-H), 6.77–6.84 (t, J = 6.8 Hz, 3H, Ar-H), 6.47 (t, J = 6.8 Hz, 1H, Ar-H), 6.39 (s, 1H, Ar-H), 3.77 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 159.8, 159.6, 152.7, 149.2, 147.3, 147.0, 140.8, 138.7, 132.7, 130.5, 129.0, 129.0, 128.6, 128.6, 128.0, 124.2, 124.0, 122.7, 122.6, 118.0, 117.2, 113.5, 111.1, 107.0, 107.0, 105.5, 55.5.; HREI-MS: m/z calcd for C28H22N4O4 [M]+478.1641; Found; 478.1599.
5-(7-((E)-((2-hydroxybenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (10).
Rf value 0.60 % (30 % ethyl acetate); light brown; Yield: 0.192 g (83 %); M.p.: 279 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.21 (s, 1H, NH), 11.11 (s, 1H, OH), 9.48 (s, 1H, OH), 8.43 (s, 1H, CH), 7.78 (d, J = 7.6 Hz, 1H, Ar-H), 7.71 (d, J = 7.5 Hz, 2H, Ar-H), 7.67 (d, J = 7.4 Hz, 1H, Ar-H), 7.66 (d, J = 7.4 Hz, 1H, Ar-H), 7.64 (d, J = 7.5 Hz, 1H, Ar-H), 7.46 (t, J = 6.8 Hz, 3H, Ar-H), 7.43 (t, J = 6.5 Hz, 1H, Ar-H), 7.32 (t, J = 7.3 Hz, 1H, Ar-H), 7.16 (t, J = 6.4 Hz, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 6.93 (d, J = 7.3 Hz, 1H. Ar-H), 6.83 (d, J = 7.4 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.6, 161.0, 157.3, 152.9, 147.4, 147.5, 139.0, 132.4, 132.1, 132.0, 131.6, 129.0, 129.0, 128.4, 128.4, 128.2, 124.3, 124.1, 123.1, 122.09, 121.4, 118.3, 118.1, 117.6, 117.5, 113.9, 111.4, 56.0.; HREI-MS: m/z calcd for C28H22N4O3 [M]+462.1692; Found; 462.1677.
5-(7-((E)-((3-hydroxybenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (11).
Rf value 0.61 % (30 % ethyl acetate); light brown; Yield: 0.196 g (85 %); M.p.: 284 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.48 (s, 1H, NH), 9.48 (s, 1H, OH), 9.45 (s, 1H, OH), 8.47 (s, 1H, CH), 7.78 (d, J = 7.6 Hz, 1H, Ar-H), 7.67 (d, J = 7.5 Hz, 1H, Ar-H), 7.64 (d, J = 7.4 Hz, 1H, Ar-H), 7.46 (t, J = 6.8 Hz, 3H, Ar-H), 7.43 (t, J = 6.5 Hz, 1H, Ar-H), 7.32 (d, J = 7.6 Hz, 2H, Ar-H), 7.25 (s, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 7.13 (t, J = 6.5 Hz, 1H, Ar-H), 6.93 (d, J = 7.3 Hz, 1H, Ar-H), 6.82 (d, J = 7.2 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.6, 158.0, 152.7, 149.0, 147.6, 147.4, 139.0, 136.0, 132.9, 131.0, 130.2, 129.2, 129.2, 128.8, 128.8, 128.4, 124.5, 124.3, 123.1, 122.9, 121.8, 118.2, 118.1, 117.5, 114.5, 113.9, 111.4, 56.1.; HREI-MS: m/z calcd for C28H22N4O3 [M]+ 462.1692; Found; 462.1677.
5-(7-((E)-((2-fluorobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (12).
Rf value 0.71 % (30 % ethyl acetate); dark brown; Yield: 0.187 g (80 %); M.p.: 277 °C; 1H NMR (600 MHz, DMSO‑d6): δ 9.58 (s, 1H, NH), 9.51 (s, 1H, OH), 8.51 (s, 1H, CH), 8.50 (m, 1H, Ar-H), 7.99–7.90 (m, 4H, Ar-H), 7.85 (d, J = 7.5 Hz, 1H, Ar-H), 7.58 (d, J = 7.4 Hz, 2H, Ar-H), 7.44–7.49 (m, 4H, Ar-H), 6.94–6.88 (m, 3H, Ar-H), 3.82 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 159.0, 152.7, 149.2, 147.3, 147.0, 138.7, 132.7, 132.2, 130.5, 130.5, 129.0, 129.0, 128.6, 128.6, 128.0, 124.5, 124.2, 124.0, 122.7, 122.6, 118.0, 118.0, 117.2, 115.4, 113.5, 111.1, 56.5.; HREI-MS: m/z calcd for C28H21FN4O2 [M]+464.1649; Found; 464.1620.
5-(7-((E)-((3-fluorobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (13).
Rf value 0.72 % (30 % ethyl acetate);Grey; Yield: 0.1995 g (86 %); M.p.: 282 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.61 (s, 1H, NH), 9.48 (s, 1H, OH), 8.49 (s, 1H, CH), 7.80 (s, 1H, Ar-H), 7.78 (d, J = 7.8 Hz, 1H, Ar-H), 7.71 (d, J = 7.6 Hz, 2H, Ar-H), 7.67 (d, J = 7.5 Hz, 1H, Ar-H), 7.64 (d, J = 7.5 Hz, 1H, Ar-H), 7.63 (t, J = 7.5 Hz, 1H, Ar-H), 7.53 (t, J = 6.8 Hz, 1H, Ar-H), 7.46 (t, J = 6.8 Hz, 3H, Ar-H), 7.44 (t, J = 6.5 Hz, 1H, Ar-H), 7.43 (d, J = 7.5 Hz, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 6.83 (d, J = 7.3 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.6, 163.0, 152.9, 149.4, 147.5, 147.4, 139.0, 135.3, 132.09, 131.0, 130.4, 129.3, 129.2, 128.8, 128.8, 128.4, 124.5, 124.3, 123.1, 122.9, 118.1, 117.8, 117.5, 114.0, 113.9, 111.4, 56.1.; HREI-MS: m/z calcd for C28H21FN4O2 [M]+ 464.1649; Found; 464.1611.
5-(7-((E)-((4-fluorobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (14).
Rf value 0.70 % (30 % ethyl acetate); Grey; Yield: 0.195 g (84 %); M.p.: 286 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.01 (s, 1H, NH), 8.58 (s, 1H, OH), 8.02 (s, 1H, CH), 7.99 (d, J = 7.6 Hz, 1H, Ar-H), 7.92 (m,1H, Ar-H), 7.71–7.75 (m, 2H, Ar-H), 7.57.48 (m, 5H, Ar-H), 7.36 (t, J = 6.8 Hz, 1H, Ar-H), 7.23 (t, J = 6.6 Hz, 3H, Ar-H), 6.93 (m, 2H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 165.0, 164.2, 152.7, 149.3, 147.5, 147.4, 139.0, 132.9, 131.3, 131.0, 130.6, 130.6, 129.2, 129.2, 128.8, 128.8, 128.4, 124.5, 122.3, 123.1, 122.9, 118.1, 117.5, 115.6, 115.6, 113.9, 111.4, 55.8.; HREI-MS: m/z calcd for C28H21FN4O2 [M]+ 464.1649; Found; 464.1611.
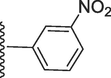
2-Methoxy-5-(7-((E)-((3-nitrobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)phenol (15).
Rf value 0.65 % (30 % ethyl acetate);Orange; Yield: 0.201 g (82 %); M.p.: 297 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.02 (s, 1H, NH), 8.52 (s, 1H, OH), 8.01 (s, 1H, CH), 7.99 (m, 2H, Ar-H), 7.74–7.79 (m, 4H, Ar-H), 7.52–7.56 (m, 2H, Ar-H), 7.77 (d, J = 7.5 Hz, 2H, Ar-H), 7.69 (t, J = 6.8 Hz, 2H, Ar-H), 7.32 (t, J = 6.8 Hz, 3H, Ar-H), 6.95 (m, 3H, Ar-H), 3.58 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 152.7, 149.2, 147.5, 147.3, 147.0, 138.7, 135.0, 134.2, 132.7, 130.5, 129.5, 129.0, 129.0, 128.6, 128.6, 128.0, 126.0, 124.2, 124.0, 122.7, 122.6, 121.0, 118.0, 117.2, 113.5, 111.1, 55.5.; HREI-MS: m/z calcd for C28H21N5O4 [M]+491.1594; Found; 491.1577.
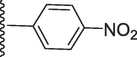
2-Methoxy-5-(7-((E)-((4-nitrobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)phenol (16).
Rf value 0.61 % (30 % ethyl acetate); Orange; Yield: 0.206 g (84 %); M.p.: 298 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.61 (s, 1H, NH), 9.48 (s, 1H, OH), 8.49 (s, 1H, CH), 8.33 (d, J = 7.6 Hz, 2H, Ar-H), 8.13 (d, J = 7.6 Hz, 2H, Ar-H), 7.78 (d, J = 7.5 Hz, 1H, Ar-H), 7.71 (d, J = 7.5 Hz, 2H, Ar-H), 7.67 (d, J = 7.4 Hz, 1H, Ar-H), 7.64 (d, J = 7.3 Hz, 1H, Ar-H), 7.48 (t, J = 6.8 Hz, 3H, Ar-H), 7.46 (t, J = 6.5 Hz, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 6.83 (d, J = 7.4 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.2, 152.7, 150.2, 149.2, 147.3, 147.1, 141.6, 139.0, 132.6, 131.0, 129.1, 129.1, 128.4, 128.4, 128.2, 127.4, 124.3, 124.1, 124.0, 124.0, 123.0, 122.6, 118.0, 117.3, 113.6, 111.4, 55.8.; HREI-MS: m/z calcd for C28H21N5O4 [M]+ 491.1594; Found; 491.1561.
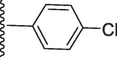
5-(7-((E)-((4-chlorobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (17).
Rf value 0.68 % (30 % ethyl acetate); pale yellow; Yield: 0.204 g (85 %); M.p.: 305 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.81 (s, 1H, NH), 10.03 (s, 1H, NH), 8.92 (s, 1H, CH), 8.76 (d, J = 7.6 Hz, 2H, Ar-H), 8.72 (d, J = 7.5 Hz, 1H, Ar-H), 8.45 (d, J = 7.5 Hz, 2H, Ar-H), 8.36 (d, J = 7.5 Hz, 1H, Ar-H), 8.28 (d, J = 7.4 Hz, 1H, Ar-H), 8.05 (d, J = 7.6 Hz, 2H, Ar-H), 7.96 (t, J = 6.8 Hz, 3H, Ar-H), 7.83 (t, J = 6.5 Hz, 1H, Ar-H), 7.75 (s, 1H, Ar-H), 7.58 (d, J = 7.5 Hz, 1H, Ar-H), 3.38 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 152.7, 149.2, 147.3, 147.0, 138.7, 136.0, 133.4, 132.7, 130.5, 129.0, 129.0, 128.6, 128.6, 128.5, 128.5, 128.0, 127.7, 127.7, 124.2, 124.0, 122.7, 122.6, 118.0, 117.2, 113.5, 111.1, 55.5.; HREI-MS: m/z calcd for C28H21ClN4O2 [M]+480.1353; Found; 480.1322.
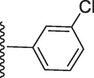
5-(7-((E)-((3-chlorobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (18).
Rf value 0.70 % (30 % ethyl acetate); yellow; Yield: 0.197 g (82 %); M.p.: 296 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.09 (s, 1H, NH), 9.98 (s, 1H, OH), 8.72 (s, 1H, CH), 8.35 (m, 2H, Ar-H), 7.98 (m, 2H, Ar-H), 7.71 (m, 2H, Ar-H), 7.57 (m, 2H, Ar-H), 7.44 (m, 3H, Ar-H), 7.14 (s, 1H, Ar-H), 6.83 (m, 3H, Ar-H), 3.88 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.3, 152.9, 149.4, 147.5, 147.4, 139.0, 135.1, 134.4, 132.9, 131.1, 131.0, 130.2, 129.2, 129.2, 128.8, 128.8, 128.3, 127.1, 128.0, 124.4, 124.2, 122.9, 118.1 117.5, 113.9, 111.4, 56.0.; HREI-MS: m/z calcd for C28H21ClN4O2 [M]+480.1353; Found; 480.1320.
5-(7-((E)-((2-chlorobenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)-2-methoxyphenol (19).
Rf value 0.73 % (30 % ethyl acetate); light yelow; Yield: 0.206 g (86 %); M.p.: 292 °C; 1H NMR (600 MHz, DMSO‑d6): δ 11.11 (s, 1H, NH), 8.65 (s, 1H, OH), 8.01 (s, 1H, CH), 7.98 (d, J = 7.6 Hz, 2H, Ar-H), 7.89 (d, J = 7.5 Hz, 1H, Ar-H), 7.62 (m, 3H, Ar-H), 7.33–7.40 (m, 5H, Ar-H), 7.28 (t, J = 6.8 Hz, 1H, Ar-H), 6.94 (t, J = 6.5 Hz, 3H, Ar-H), 3.86 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 152.7, 150.2, 147.3, 147.0, 138.7, 136.0, 133.5, 132.7, 132.0, 130.5, 130.0, 129.0, 129.0, 128.6, 128.6, 128.0, 127.0, 126.6, 124.2, 124.0, 122.7, 122.6, 118.0, 117.2, 113.5, 111.1, 55.5.; HREI-MS: m/z calcd for C28H21ClN4O2 [M]+480.1353; Found; 480.1319.
2-Methoxy-5-(7-((E)-((4-methylbenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)phenol (20).
Rf value 0.73 % (30 % ethyl acetate); off white; Yield: 0.200 g (87 %); M.p.: 287 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.61 (s, 1H, NH), 9.48 (s, 1H, OH), 8.49 (s, 1H, CH), 7.78 (s, 1H, Ar-H), 7.77 (d, J = 7.6 Hz, 2H, Ar-H), 7.71 (d, J = 7.5 Hz, 2H, Ar-H), 7.67 (d, J = 7.5 Hz, 1H, Ar-H), 7.64 (d, J = 7.4 Hz, 1H, Ar-H), 7.48 (t, J = 6.8 Hz, 3H, Ar-H), 7.46 (t, J = 6.5 Hz, 2H, Ar-H), 7.23 (d, J = 7.5 Hz, 2H, Ar-H), 6.83 (d, J = 7.6 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3) 2.41 (s, 3H, CH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.6, 152.9, 149.4, 147.5, 147.4, 147.0, 139.0, 132.9, 132.7, 131.0, 129.2, 129.2, 129.1, 129.1, 129.0, 129.0, 128.6, 128.2, 124.5, 124.5, 123.1, 122.9, 118.1, 117.5, 113.9, 111.4, 56.0, 21.0.; HREI-MS: m/z calcd for C29H24N4O2 [M]+460.1899; Found; 460.1870.
2-Methoxy-5-(7-((E)-((3-methylbenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)phenol (21).
Rf value 0.74 % (30 % ethyl acetate); Off white; Yield: 0.184 g (80 %); M.p.: 282 °C; 1H NMR (600 MHz, DMSO‑d6): δ 10.09 (s, 1H, NH), 8.48 (s, 1H, OH), 8.01 (s, 1H, CH), 7.98 (d, J = 7.5 Hz, 1H, Ar-H), 7.84 (d, J = 7.5 Hz, 2H, Ar-H), 7.60–7.67 (m, 4H, Ar-H), 7.40–7.46 (m, 2H, Ar-H), 7.33–7.38 (m, 2H, Ar-H), 6.83–6.89 (m, 4H, Ar-H), 3.86 (s, 3H, OCH3), 1.92 (s, 3H, CH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.3, 152.6, 149.2, 147.3, 147.1, 139.0, 138.2, 133.2, 132.6, 131.1, 131.0, 129.2, 129.0, 129.0, 128.4, 128.4, 128.3, 128.3, 128.0, 127.6, 124.5, 124.5, 124.3, 123.1, 122.9, 118.1, 117.3, 111.2, 56.1, 21.1; HREI-MS: m/z calcd for C29H24N4O2 [M]+460.1899; Found; 460.1879.
2-Methoxy-5-(7-((E)-((2-methylbenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)phenol (22).
Rf value 0.75 % (30 % ethyl acetate);white; Yield: 0.1887 g (82 %); M.p.: 277 °C; 1H NMR (600 MHz, DMSO‑d6): δ 12.61 (s, 1H, NH), 9.48 (s, 1H, OH), 8.49 (s, 1H, CH), 7.78 (d, J = 7.6 Hz, 1H, Ar-H), 7.71 (t, J = 6.6 Hz, 3H, Ar-H), 7.67 (d, J = 7.4 Hz, 1H, Ar-H), 7.64 (d, J = 7.3 Hz, 1H, Ar-H), 7.46 (t, J = 6.5 Hz, 3H, Ar-H), 7.43 (t, J = 6.5 Hz, 1H, At-H), 7.23 (t, J = 6.5 Hz, 2H, Ar-H), 7.21 (d, J = 7.6 Hz, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 6.83 (d, J = 7.0 Hz, 1H, Ar-H), 3.86 (s, 3H, OCH3), 2.43 (s, 3H, CH3); 13C NMR (150 MHz, DMSO‑d6): δ 164.0, 152.9, 149.7, 147.3, 147.2, 139.2, 135.1, 132.6, 131.2, 131.0, 130.9, 129.1, 129.1, 129.0, 128.4, 128.4, 128.2, 126.3, 125.1, 124.1, 123.4, 122.6, 118.1, 117.5, 113.9,111.4, 56.1, 18.9.; HREI-MS: m/z calcd for C29H24N4O2 [M]+460.1899; Found; 460.1865.
2-Methoxy-5-(7-((E)-((4-methoxybenzylidene)hydrazono)(phenyl)methyl)-1H-benzo[d]imidazol-2-yl)phenol (23).
Rf value 0.72 % (30 % ethyl acetate); pale yellow; Yield: 0.210 g (88 %); M.p.: 294 °C; 1H NMR (600 MHz, DMSO‑d6): δ 9.70 (s, 1H, NH), 9.60 (s, 1H, OH), 8.59 (s, 1H, CH), 8.52 (d, J = 7.6 Hz, 2H, Ar-H), 8.04 (d, J = 7.4 Hz, 1H, Ar-H), 7.99 (d, J = 7.7 Hz, 2H, Ar-H), 7.75 (d, J = 7.5 Hz, 1H, Ar-H), 7.66 (t, J = 6.8 Hz, 3H, Ar-H), 7.52 (t, J = 6.5 Hz, 1H, Ar-H), 7.32 (s, 1H, Ar-H), 7.22 (d, J = 7.8 Hz, 2H, Ar-H), 6.94 (d, J = 7.3 Hz, 2H, Ar-H), 3.86 (s, 3H, OCH3), 3.81 (s, 3H, OCH3);13C NMR (150 MHz, DMSO‑d6): δ 162.9, 162.0, 160.3, 155.0, 150.8, 138.2, 135.7, 133.7, 132.8, 131.9, 130.3, 129.4, 129.3, 128.9, 127.9, 127.8, 127.7, 126.2, 125.3, 125.0, 124.8, 121.8, 120.6, 116.3, 115.7, 115.7, 115.7, 55.9, 55.0; HREI-MS: m/z calcd for C29H24N4O3 [M]+476.476.1848; Found; 476.1826.
3.4 Docking studies
All compound structures were drawn using Chemdraw and minimized using CHARMM forcefield. The crystal structure of urease (PDB: 4UBP) had been obtained from RCSB database and the structure was optimized using macromolecule module in Discovery Studio. Molecular docking had been carried out using CDocker protocol by defining the binding site based on the native ligand coordinate within 4UBP (x: 30.746, y: 68.623, z: 75.780). Molecular docking results were prepared and analysed using Discovery Studio visualizer 2016.
3.5 Biological evaluation
3.5.1 Antioxidant assay protocol
Using published procedures, 1,1-diphenyl-2-picrylhydrazil (DPPH) was used to assess the free radical scavenging activity (Taha et al., 2020). Reaction mixture is made by adding five liters of the test sample (1 mM in DMSO) and ninety-five liters of DPPH (Sigma, 300 M) in ethanol. For 30 min, the reaction mixture was heated to 37 °C. On a microtiter plate reader (made by Molecular Devices, CA, USA), the absorbance was measured at 515 nm. In comparison to a control that included DMSO, the percentage of radical scavenging activity was calculated by following formula.
% inhibition = 1-absorbance of analyte/absorbance of control group x100.
The IC50 value is the amount of a chemical needed to scavenge 50 % of DPPH radicals. A positive control was propyl gallate and the substances utilized were all of analytical quality (Sigma, USA).
3.5.2 Anti-glycation assay protocol
Rutin and methylglyoxal (MG) (40 % aqueous solution) were purchased from Sigma Aldrich (Japan), Bovine Serum Albumin (BSA) was purchased from Merck Marker Pvt. Ltd. (Germany), sodium dihydrogen phosphate (NaH2PO4), disodium hydrogen phosphate (Na2HPO4), and sodium azide (NaN3) were purchased from Scharlau Chemie, S.A. while DMSO, was bought from Fischer Scientific (UK) (Taha et al., 2020b).
Bovine Serum Albumin (10 mg/mL), methyl glyoxal (14 mM), a range of chemical concentrations (made in DMSO, 10 % final concentration), and 0.1 M phosphate buffer (pH 7.4) containing sodium azide (30 mM) were incubated for nine days at 37 °C under sterilized conditions. Each sample was tested against a sample blank after 9 days to see if any unique fluorescence had developed (excitation: 330 nm; emission: 440 nm). The positive control was rutin (Sattarahmady, N. et al 2007). For each inhibitor drug, the percent inhibition of AGE formation in the test sample compared to the control was computed using the formula below:
% inhibition= (1-test sample fluorescence/control group fluorescence) x 100.
3.5.3 Anti-urease assay protocol
In urease assay protocol 25 mL of the synthesized chemical were combined with 25 mL of urease solution and incubated at 30° for 15 min. A further 30 min were spent incubating after the addition of 55 L of urea substrate at a concentration of 100 mM. Each well received 45 L of carbolic acid (0.005 % w/w and 1 % w/v Na2[Fe(CN)5NO)) and 70 L of basic reagent (0.1 % NaOCl and 0.5 % NaOH), which were added and incubated for 45 min at 30° C. The urease inhibitory concentration was determined by the weather burn method utilizing an ELISA plate reader (Spectra Max M2, Molecular Devices, CA, USA) at 630 nm (Weatherburn, 1967). Thiourea was employed as the standard in the test. Each assay was run in triplicate.
3.5.4 Molecular docking studies
The 3D structure of active compoundswas prepared using Chem3D by CambridgeSoft and were being optimized using MM2 molecular forcefield. The active compounds were then docked into the active site of urease enzyme (PDB: 4UBP) at the coordinate of x: 30.948823, y: 65.986721, and z: 76.458516. The molecular docking experiment was performed using the CDOCKER (CHARMm-based molecular docking) module in Discovery Studio 2016. The default docking parameters were composed of CHARMm (chemistry at Harvard macromolecular mechanics) forcefield, 10 diverse top poses, 0.1 post cluster radius, 10 random conformations, 10,000 dynamics steps, 1000 °C dynamics target temperature, electrostatic interaction, 10 orientations to refine bad maximum orientations and simulated annealing. The docking was performed by maintaining the protein rigidness and retaining the ligand in flexible mode. The protein–ligand interactions were visualized using Discovery studio. The molecular docking protocol has been validated by redocking the native ligand (acetohydroxamic acid) that demonstrated root mean square deviation (RMSD) values of less than 2 Å.
4 Conclusion
The current attempt is based on logical research and includes the synthesis of 19 analogues that contained benzimidazole moiety. All the synthetic compounds were characterized using several spectroscopic techniques, such as NMR and HR-EIMS. These compounds were investigated for a range of biological activities after the targeted moieties have been verified, including antiurease, antiglycation, and antioxidant activities. Among the benzimidazole derivatives, the dihydroxyphenyl analogs 6, 7, 8 and 9 displayed excellent inhibition against urease enzyme with IC50 values ranging from 3.10 µM to 5.90 µM. However, in case of antiglycation and antioxidant activities, it was found that fluorophenyl analogs 12, 13 and 14 demonstrated the highest activity. Structure activity relationship has been established for all compounds which reveals the effects of substituents, their number and position on biological activity discussed in detail. Finally, the analogs’ binding with the urease enzyme was investigated via molecular docking in order to study the potential binding interactions between the enzyme and the synthetic analogs and utilize that to provide an explanation behind the analogs’ observed bioactivity.
Acknowledgement
This research has been funded by Scientific Research Deanship at University of Ha'il - Saudi Arabia through project number RG-23 102
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Phenolics from Chrozophora oblongifolia aerial parts as inhibitors of α-glucosidases and advanced glycation end products: in-vitro assessment, molecular docking and dynamics studies. Biology (basel).. 2022;11:762.
- [CrossRef] [Google Scholar]
- Biologically potent benzimidazole-based-substituted benzaldehyde derivatives as potent inhibitors for alzheimer’s disease along with molecular docking study. Pharmaceuticals (basel).. 2023;16:208.
- [CrossRef] [Google Scholar]
- N-linked protein glycosylation in the ER. Biochim. Biophys. Acta - Mol. Cell Res.. 2013;1833:2430-2437.
- [CrossRef] [Google Scholar]
- A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity. Bioorg. Chem.. 2021;114:105073
- [CrossRef] [Google Scholar]
- Donor-acceptor interactions as descriptors of the free radical scavenging ability of flavans and catechin. Comput. Theor. Chem.. 2017;1110:14-24.
- [CrossRef] [Google Scholar]
- Antioxidant activities exhibited by Benzofuran-1, 3-Thiazolidin-4- one derivative: a theoretical study. J. Med. Chem. Drug Design. 2023;1:103.
- [Google Scholar]
- Identification of imidazolylpyrazole ligands as potent urease inhibitors: synthesis, antiurease activity and in silico docking studies. ChemistrySelect. 2020;5:11817-11821.
- [CrossRef] [Google Scholar]
- The mechanism for cleavage of three typical glucosidic bonds induced by hydroxyl free radical. Carbohydr. Polym.. 2017;178:34-40.
- [CrossRef] [Google Scholar]
- Development of sulfonamide-based schiff bases targeting urease inhibition: synthesis, characterization, inhibitory activity assessment, molecular docking and ADME studies. Bioorg. Chem.. 2020;102:104057
- [CrossRef] [Google Scholar]
- Benzimidazole bearing thiourea analogues: synthesis, β-glucuronidase inhibitory potential and their molecular docking study. J. Mol. Struct.. 2022;1270:133941
- [CrossRef] [Google Scholar]
- Benzimidazole bearing thiosemicarbazone derivatives act as potent α-amylase and α-glucosidase inhibitors; synthesis, bioactivity screening and molecular docking study. Molecules. 2022;27:6921.
- [CrossRef] [Google Scholar]
- Synthesis of benzimidazole-thiosemicarbazone hybrid derivatives, in vitro.-glucosidase and.-amylase activities, and an in silico molecular docking study. Chem. Data Collect.. 2023;45:101027
- [CrossRef] [Google Scholar]
- Multipotent cholinesterase inhibitors for the treatment of alzheimer’s disease: synthesis, biological analysis and molecular docking study of benzimidazole-based thiazole derivatives. Molecules. 2022;27:6087.
- [CrossRef] [Google Scholar]
- Antioxidant and chemosensitizing effects of flavonoids with hydroxy and/or methoxy groups and structure-activity relationship. J. Pharm. & Pharm. Sci.. 2007;10:537.
- [Google Scholar]
- Indole-3-acetamides: as potential antihyperglycemic and antioxidant agents; synthesis, in vitro α-amylase inhibitory activity, structure-activity relationship, and in silico studies. ACS Omega. 2021;6:2264-2275.
- [CrossRef] [Google Scholar]
- Synthesis of bis-schiff bases of isatins and their antiglycation activity. Bioorganic & Med. Chem.. 2009;17:7795-7801.
- [CrossRef] [Google Scholar]
- Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct.. 2022;1269:133812
- [CrossRef] [Google Scholar]
- Synthesis, antioxidant and antimicrobial activities of novel thiopyrano[2,3-d]thiazoles based on aroylacrylic acids. Mol. Divers.. 2017;21:427-436.
- [CrossRef] [Google Scholar]
- Implementation and evaluation of diabetes clinical practice guidelines in a primary care clinic serving a hispanic community. Worldviews Evidence-Based Nurs.. 2019;16:142-150.
- [CrossRef] [Google Scholar]
- Design, synthesis and evaluation of aryl-tailored oxadiazole-thiones as new urease inhibitors. ChemistrySelect. 2023;8
- [CrossRef] [Google Scholar]
- New triazinoindole bearing benzimidazole/benzoxazole hybrids analogs as potent inhibitors of urease: synthesis, in vitro analysis and molecular docking studies. Molecules. 2022;27:6580.
- [CrossRef] [Google Scholar]
- Development of benzimidazole derivatives to inhibit HIV-1 replication through protecting APOBEC3G protein. Eur. J. Med. Chem.. 2015;95:500-513.
- [CrossRef] [Google Scholar]
- N-mannich bases of benzimidazole as a potent antitubercular and antiprotozoal agents: their synthesis and computational studies. Synth. Commun.. 2020;50:858-878.
- [CrossRef] [Google Scholar]
- Drug repurposing: in-vitro anti-glycation properties of 18 common drugs. PLoS One. 2018;13:e0190509-e.
- [CrossRef] [Google Scholar]
- Benzimidazole based derivatives as anticancer agents: structure activity relationship analysis for various targets. J. Heterocycl. Chem.. 2021;59:22-66.
- [CrossRef] [Google Scholar]
- Recent advancements in the development of heterocyclic anti-inflammatory agents. Eur. J. Med. Chem.. 2020;200:112438
- [CrossRef] [Google Scholar]
- Synthesis, molecular docking, α-glucosidase inhibition, and antioxidant activity studies of novel benzimidazole derivatives. Med. Chem. Res.. 2020;29:1846-1866.
- [CrossRef] [Google Scholar]
- Synthesis of novel derivatives of 4-methylbenzimidazole and evaluation of their biological activities. Eur. J. Med. Chem.. 2014;84:731-738.
- [CrossRef] [Google Scholar]
- Synthesis, α-amylase inhibition and molecular docking study of bisindolylmethane sulfonamide derivatives. Med. Chem. Res.. 2019;28:2010-2022.
- [CrossRef] [Google Scholar]
- Synthesis, α-glycosidase inhibitory potential and molecular docking study of benzimidazole derivatives Muhammad. Bioorg. Chem.. 2020;95:103555
- [CrossRef] [Google Scholar]
- Synthesis, antiglycation and antioxidant potentials of benzimidazole derivatives. J. King Saud Univ. - Sci.. 2020;32:191-194.
- [CrossRef] [Google Scholar]
- Synthesis of new 1,2-disubstituted benzimidazole analogs as potent inhibitors of β-glucuronidase and in silico study. Arab. J. Chem.. 2022;15:103505
- [CrossRef] [Google Scholar]
- Coumarins hinged directly on benzimidazoles and their ribofuranosides to inhibit hepatitis C virus. Eur. J. Med. Chem.. 2013;63:290-298.
- [CrossRef] [Google Scholar]
- Advanced glycation end-products (AGEs): formation, chemistry, classification, receptors, and diseases related to AGEs. Cells. 2022;11:1312.
- [CrossRef] [Google Scholar]
- Annona cherimola miller fruit as a promising candidate against diabetic complications: an in vitro study and preliminary clinical results. Foods (basel, Switzerland). 2020;9:1350.
- [CrossRef] [Google Scholar]
- Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem.. 1967;39:971-974.
- [CrossRef] [Google Scholar]
- Advanced glycation end product concentrations in follicular fluid of women undergoing IVF/ICSI with a GnRH agonist protocol. Reprod. Biomed. Online. 2018;36:20-25.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105700.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




