

Translate this page into:
Synthesis of functionalized aminopyrazole and pyrazolopyrimidine derivatives: Molecular modeling and docking as anticancer agents
⁎Corresponding author. nmmohamed@uqu.edu.sa (Nashwa M. El-Metwaly)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Three new functionalized 4-aminopyrazole derivatives were synthesized and cyclized with phenyl isothiocyanate to yield the corresponding three pyrazolo[4,3-d]pyrimidine analogues. The DFT quantum chemical calculations were utilized in the determination of the frontier molecular orbital energies and Fukui’s indices. The data showed that they have a low HOMO-LUMO energy gap, ranging from 1.16 to 2.35 eV for 5 and 6, respectively. The newly created analogues' cytotoxic qualities were evaluated in comparison to the reference 5-florouracil (5-Fu) using an in vitro MTT cytotoxicity screening investigation toward four different cell lines, including HCT-116, HepG2, MCF-7, and WI38. The results showed variable potency against human cell lines, with MCF-7 and HepG-2 showing cytotoxic selectivity. The most potent agent against MCF-7 and HCT-116 human cancer cells were found to be aminopyrazole and pyrazolopyrimidine derivatives 4–9. The structure–activity relationships (SAR) for the synthesized compounds were discussed. The examined compounds had superior cytotoxic properties; the most potent derivative 7, had an IC50 ranged from 11.51 ± 0.35 to 21.25 ± 0.37 µM. Meanwhile, quantum chemical computation used independent variables EH, EL, ΔEH-L, χ and η were applied to determine the best way to describe activity. As a result, an increase in the HOMO-LUMO gap and hardness will result in an increase in the anticancer activity. While the EH, EL, and showed negative coefficients, increasing them will decrease the anticancer activity. Furthermore, 5IVE protein's crystal structure for KDM5A was docked with the newly created aminopyrazole and pyrazolopyrimidine derivatives to afford the theoretical prediction on the KDM5A enzyme.
Keywords
4-Aminopyrazole
Pyrazolo[4,3-d]pyrimidines
Fukui’s indices
Cytotoxicity
Molecular docking

1 Introduction
Cancer remains one of the most menacing diseases in the world (Akram et al., 2017). It is considered the second cause of death after cardiac disease (Harding et al., 2018). Liver cancer is the second leading cause of cancer death worldwide (Cao et al., 2021), followed by colorectal cancer (Rawla et al., 2019). Abnormalities in the genetic material are the causes of the various cancer types (Thiagalingam et al., 2002). One important approach to treating cancer cells' uncontrolled cell division and growth is to use drugs that interfere with the synthesis of the nucleic acids DNA and RNA and inhibit their normal function (Shai et al., 2022, Demény and Virág, 2021). In the development of cancer treatments and cellular apoptosis, the pyrazole scaffold is a highly adaptable drug-like template (Elbastawesy et al., 2019, Lolli et al., 2018, Zhong et al., 2020, Kerru et al., 2020). These substances have the ability to block a variety of enzymes, proteins, and receptors that are essential for cell proliferation, resulting in impressive anticancer effects (Kumar et al., 2013). Similarly, pyrimidine has served as a fruitful source of inspiration for medicinal chemists and has drawn their interest due to a variety of activities (Bakavoli et al., 2010). Meanwhile, one of most valuable category in heterocyclic chemistry is the pyrazolo-pyrimidine (PPs) class, have dissimilar attachments between pyrazole and pyrimidine rings: [3,4-d], [4,3-d], [5,1-b], and [1,5-a] (Asati et al., 2021). Where, several modified chemical structures of PPs have been linked to pharmacological effects that include antimicrobial, antiviral, anti-fungal, anti-inflammatory, anti-tumor, and herbicidal properties throughout the past few decades (Yewale et al., 2012, He et al., 2011, Liao et al., 2013). The main effective point in their pharmaceutical activity of PPs, PPs and both of purine and adenosine receptors have almost identical fundamental structures instead of the imidazole ring found in purines and adenosine, the pyrazolopyrimidine have a pyrazole moiety fused with the pyrimidine unit (Chauhan and Kumar, 2013, Kumar et al., 2021). Many PPs and their derivatives have recently been found to have anticancer potential through interactions with various enzymes and receptors (Gaber et al., 2021, Lamie et al., 2021). For instance, SAR of PPs has been presented as a prospective anticancer agent with (IC50 = 13.91, 22.37, 6.56, 8.72, 4.17, and 5.57 μM/L) for two cancer MCF-7 and hela cell lines, in addition to PIM1 inhibitors (Fig. 1) (Asati et al., 2021, Metwally et al., 2019).
Pyrazolopyrimidines as PIM1 inhibitors and anti-cancer agents.
Recently, many researchers are interested in computational DFT calculations to support and get understanding of the experimental results where such calculations have been employed widely to determine several parameters, e.g., frontier molecular orbitals (HOMO and LUMO) and Mulliken charges of pyrazolopyrimidine derivatives (Hossan et al., 2023). Where, their HOMO-LUMO energies and gap have been strongly affected on the bioactivity, i.e., anticancer activity which is based on mainly the intramolecular charge transfer occurred within the molecule and to some extent on its other properties like electronegativity, hardness and softness where the reactive sites to nucleophilic and electrophilic attacks were correlated with the biologically reactive regions(Issa et al., 2020, Kamel et al., 2017).
Our work in this research paper is aimed to synthesis of new functionalized 4-aminopyrazole and their corresponding pyrazolo[4,3-d]pyrimidine derivatives, conformation their corrected structures through different spectroscopic analysis, evaluate their anticancer activates. Also, the theoretical modeling DFT studies to stand on the reactivity of these derivatives according to HOMO-LUMO energy gap correlate the relation energy gab between the quantum chemical computation and anticancer activity, and theoretical docking study to predict the activity of the synthesized derivative toward KDM5A enzyme to help the biological specialists to examined it in the future.
2 Experimental
2.1 Chemistry
2.1.1 General remarks
All melting points were determined with the assistance of the Gallenkamp electric apparatus. The IR spectra (KBr discs) were obtained with the assistance of a ThermoScientific Nicolet iS10 FTIR spectrometer. The JEOL’s spectrometer was utilized in order to record the NMR spectra in DMSO‑d6 at frequencies of 500 MHz (1H NMR) and 125 MHz (13C NMR). On a Quadrupole ThermoScientific mass spectrometer DSQII, the mass analyses were carried out with an energy setting of 70 eV. The elemental analyses were carried out using a Perkin-Elmer 2400 analyzer (C, H, and N).
2.1.2 Synthesis of 4-amino-1-(4-chlorophenyl)-N-phenyl-1H-pyrazole-3-carboxamides 4, 6, and 8
To a solution of N-(4-chlorophenyl)-2-oxo-2-(phenylamino)acetohydrazonoyl cyanide (3) (1.19 g, 4 mmol) in 30 mL dioxane, 4 mmol of the appropriate alpha-halogenated reagent (namely; chloroacetone, ethyl bromoacetate and/or chloroacetonitrile) and 0.2 mL triethylamine were added. The mixture was refluxed for 4 h and then allowed to cool. The solid that formed was collected and dried to pick up the corresponding 4-amino-5-substituted-pyrazole analogues 4, 6 and 8, respectively.
2.1.2.1 5-Acetyl-4-amino-1-(4-chlorophenyl)-N-phenyl-1H-pyrazole-3-carboxamide (4)
Yield = 77 %, m.p. = 207–208 °C. IR (ν/cm−1): 3419, 3319, 3268 (NH2 and N—H), 1661 (C⚌O, amide), 1597 (C⚌O, acetyl). 1H NMR (δ/ppm): 2.10 (s, 3H, COCH3), 7.09 (t, J = 7.00 Hz, 1H), 7.35–7.39 (m, 6H), 7.48 (br. s, 2H, NH2), 7.65 (d, J = 7.00 Hz, 2H), 9.78 (s, 1H, N—H). 13C NMR (δ/ppm): 28.04 (–CH3), 118.08 (pyrazole-C5), 120.32 (2 Ar-C), 121.89 (2 Ar-C), 126.94 (Ar-C), 128.29 (2 Ar-C), 129.40 (2 Ar-C), 131.27 (pyrazole-C3), 132.18 (Ar-C), 133.62 (pyrazole-C4), 136.84 (Ar-C), 137.91 (Ar-C), 162.47 (C⚌O, amide), 186.68 (C⚌O, acetyl). MS m/z (%): 356 (M+ + 2, 18.26), 354 (M+, 57.43). Anal. calcd.: C18H15ClN4O2 (354.09): C, 60.94; H, 4.26; N, 15.79 %. Found: C, 60.81; H, 4.20; N, 15.70 %.
2.1.2.2 Ethyl 4-amino-1-(4-chlorophenyl)-3-(phenylcarbamoyl)-1H-pyrazole-5-carboxylate (6)
Yield = 81 %, m.p. = 241–242 °C. IR (ν/cm−1): 3361, 3321, 3262 (NH2 and N—H), 1665 (C⚌O, amide), 1636 (C⚌O, ester). 1H NMR (δ/ppm): 1.20 (t, J = 7.00 Hz, 3H, CH3), 4.13 (q, J = 7.00 Hz, 2H, OCH2), 6.67 (br. s, 2H, NH2), 7.07 (t, J = 7.00 Hz, 1H), 7.30–7.37 (m, 6H), 7.64 (d, J = 8.50 Hz, 2H), 9.68 (s, 1H, N—H). 13C NMR (δ/ppm): 14.57 (–CH3), 59.04 (–OCH2), 120.08 (2 Ar-C), 121.13 (pyrazole-C5), 121.82 (2 Ar-C), 126.79 (Ar-C), 128.34 (2 Ar-C), 128.71 (pyrazole-C4), 129.46 (2 Ar-C), 131.33 (pyrazole-C3), 132.23 (Ar-C), 137.00 (Ar-C), 138.11 (Ar-C), 162.58 (C⚌O, amide), 163.50 (C⚌O, ester). MS m/z (%): 384 (M+, 17.09). Anal. calcd.: C19H17ClN4O3 (384.10): C, 59.30; H, 4.45; N, 14.56 %. Found: C, 59.41; H, 4.48; N, 14.47 %.
2.1.2.3 4-Amino-1-(4-chlorophenyl)-5-cyano-N-phenyl-1H-pyrazole-3-carboxamide (8)
Yield = 73 %, m.p. = 228–229 °C. IR (ν/cm−1): 3267, 3200 (NH2 and N—H), 2256 (C≡N), 1668 (C⚌O). 1H NMR (δ/ppm): 6.06 (s, 2H, NH2), 6.76 (t, J = 7.00 Hz, 1H), 7.18 (t, J = 7.50 Hz, 2H), 7.26 (d, J = 7.50 Hz, 2H), 7.32 (d, J = 8.50 Hz, 2H), 7.53 (d, J = 8.50 Hz, 2H), 8.83 (s, 1H, N—H). 13C NMR (δ/ppm): 99.56 (pyrazole-C5), 115.27 (C≡N), 120.64 (2 Ar-C), 122.05 (2 Ar-C), 126.77 (Ar-C), 127.16 (pyrazole-C4), 128.38 (2 Ar-C), 129.44 (2 Ar-C), 130.98 (pyrazole-C3), 132.40 (Ar-C), 137.16 (Ar-C), 137.85 (Ar-C), 162.53 (C⚌O, amide). MS m/z (%): 337 (M+, 21.79). Anal. calcd.: C17H12ClN5O (337.07): C, 60.45; H, 3.58; N, 20.73 %. Found: C, 60.53; H, 3.64; N, 20.80 %.
2.1.3 Synthesis of 1-(4-chlorophenyl)-N,6-diphenyl-5-thioxo-1H-pyrazolo[4,3-d]-pyrimidine-3-carboxamides 5 and 7
In a 100 mL RBF, a mixture of each 4-amino-1-(4-chlorophenyl)-pyrazole analogue 4 or 6 (3 mmol) and phenyl isothiocyanate (0.36 mL, 3 mmol) was dissolved in 30 mL pyridine. After six hours of refluxing, the mixture was cooled, and then poured into ice-water. The resulting solid was collected and subjected to recrystallization from ethanol to provide the desired pyrazolo[4,3-d]pyrimidine derivatives 5 and 7, respectively.
2.1.3.1 1-(4-Chlorophenyl)-7-methyl-N,6-diphenyl-5-thioxo-5,6-dihydro-1H-pyrazolo[4,3-d]pyrimidine-3-carboxamide (5)
Yield = 67 %, m.p. = 287–288 °C. IR (ν/cm−1): 3217 (N—H), 1661 (C⚌O). 1H NMR (δ/ppm): 2.37 (s, 3H, CH3), 7.04 (d, J = 9.00 Hz, 2H), 7.34 (d, J = 7.50 Hz, 2H), 7.41–7.54 (m, 8H), 7.74 (d, J = 9.00 Hz, 2H), 9.83 (s, 1H, N—H). 13C NMR (δ/ppm): 11.17 (–CH3), 109.55 (pyrazole-C5), 121.70 (2 Ar-C), 122.25 (2 Ar-C), 125.17 (pyrimidine-C), 126.88 (Ar-C), 128.40 (Ar-C), 128.58 (2 Ar-C), 129.06 (2 Ar-C), 129.54 (2 Ar-C), 130.33 (Ar-C), 132.42 (2 Ar-C), 134.38 (pyrazole-C3), 135.06 (Ar-C), 137.13 (Ar-C), 141.79 (Ar-C), 154.48 (pyrazole-C4), 161.63 (C⚌O, amide), 172.93 (C⚌S, ring). MS m/z (%): 473 (M+ + 2, 16.28). 471 (M+, 51.43). Anal. calcd.: C25H18ClN5OS (471.09): C, 63.62; H, 3.84; N, 14.84 %. Found: C, 63.41; H, 3.76; N, 14.96 %.
2.1.3.2 1-(4-Chlorophenyl)-7-oxo-N,6-diphenyl-5-thioxo-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-d]pyrimidine-3-carboxamide (7)
Yield = 61 %, m.p. > 300 °C. IR (ν/cm−1): 3221, 3183 (N—H), broad at 1664 (C⚌O). 1H NMR (δ/ppm): 7.07 (t, J = 7.50 Hz, 1H), 7.28–7.35 (m, 4H), 7.42–7.47 (m, 5H), 7.57 (d, J = 8.00 Hz, 2H), 7.77 (d, J = 8.00 Hz, 2H), 9.76 (s, 1H, N—H), 13.02 (s, 1H, N—H). 13C NMR (δ/ppm): 117.22 (pyrazole-C5), 121.61 (2 Ar-C), 122.05 (2 Ar-C), 125.91 (pyrazole-C4), 126.73 (Ar-C), 128.25 (2 Ar-C), 128.69 (Ar-C), 128.95 (2 Ar-C), 129.37 (2 Ar-C), 129.74 (2 Ar-C), 132.16 (Ar-C), 134.69 (Ar-C), 135.08 (pyrazole-C3), 137.27 (Ar-C), 138.36 (Ar-C), 162.51 (C⚌O, outer ring), 164.72 (C⚌O, lactam group), 174.60 (C⚌S, ring). MS m/z (%): 473 (M+, 23.55). Anal. calcd.: C24H16ClN5O2S (473.07): C, 60.82; H, 3.40; N, 14.78 %. Found: C, 60.97; H, 3.47; N, 14.90 %.
2.1.4 Synthesis of 1-(4-chlorophenyl)-7-imino-N,6-diphenyl-5-thioxo-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-d]pyrimidine-3-carboxamide (9):
In a 100 mL RBF, a mixture of 4-amino-1-(4-chlorophenyl)-pyrazole analogue 8 (1.01 g, 3 mmol) and phenyl isothiocyanate (0.36 mL, 3 mmol) was dissolved in 30 mL dimethylformamide. After eight hours of heating at 85–90 °C, the mixture was cooled to 25 °C, and then poured into ice-water. The resulting solid was collected and subjected to recrystallization from ethanol to provide the desired pyrazolo[4,3-d]pyrimidine derivative 9. Yield = 58 %, m.p. = 271–272 °C. IR (ν/cm−1): 3363, 3197 (N—H), 1654 (C⚌O), 1633 (C⚌N). 1H NMR (δ/ppm): 7.11 (t, J = 7.50 Hz, 1H), 7.19–7.24 (m, 5H), 7.34 (t, J = 8.00 Hz, 2H), 7.40–7.44 (m, 4H), 7.64 (d, J = 7.50 Hz, 2H), 10.28 (s, 1H, N—H), 11.17 (s, 1H, N—H, imine), 12.96 (s, 1H, N—H, thioamide). 13C NMR (δ/ppm): 117.15 (pyrazole-C5), 121.64 (2 Ar-C), 122.29 (2 Ar-C), 125.79 (pyrazole-C4), 127.04 (Ar-C), 128.44 (Ar-C), 128.71 (2 Ar-C), 129.00 (2 Ar-C), 129.37 (2 Ar-C), 129.82 (2 Ar-C), 132.13 (Ar-C), 133.99 (pyrazole-C3), 136.37 (Ar-C), 137.10 (Ar-C), 138.08 (Ar-C), 155.45 (C = NH), 162.41 (C⚌O, amide), 174.56 (C⚌S). MS m/z (%): 472 (M+, 16.98). Anal. calcd.: C24H17ClN6OS (472.09): C, 60.95; H, 3.62; N, 17.77 %. Found: C, 60.81; H, 3.67; N, 17.87 %.
2.2 Computational studies
The obtained aminopyrazole and pyrazolopyrimidine derivatives were studied using the DFT/B3LYP/6–311++G (Becke, 1993, Lee et al., 1988, Perdew and Wang, 1992) incorporated in Gaussian 09 W program (Frisch et al., 2009). The optimized geometries of all derivatives exhibited positive frequencies that cleared its stability. The Fukui indices were determined by Materials studio package DMol3 module (BIOVIA, 2017) utilizing the GGA and B3LYP functional with DNP (version 3.5) (Delley, 2006).
2.3 Cytotoxicity assay
Three individual cancer cell lines HepG2, MCF-7, and HCT-116, were examined toward in vitro cytotoxicity over the established MTT technique (Rahmouni et al., 2016) (as shown in the supplementary file).
2.4 Molecular docking (MD)
Using MOE, the most stable conformers of the synthetic analogues were constructed in 2D, 3D, and surface maps (software 2015.10). Lowest energy conformers of the synthetic analogues were docked into PDB: 5IVE. Protein's binding pocket from the Data Bank (Metwally et al., 2019). After the hydrogens were introduced, the enzyme structure was enhanced using a technique that gradually reduced and raised the restrictions on the enzyme until the RMSD gradient was lower than 1.5.
3 Results and discussion
3.1 Chemistry
N-(4-Chlorophenyl)-2-oxo-2-(phenylamino)acetohydrazonoyl cyanide (3) has been prepared in the light of the reported literature by diazo-coupling of diazotized 4-chloroaniline with 2-cyanoacetanilide (Shawali and Abd El-Galil, 1971). Our synthetic strategy for producing many functionalized 4-aminopyrazole derivatives is based on the cyclization of N-(4-chlorophenyl)-2-oxo-2-(phenylamino)acetohydrazonoyl cyanide (3) with various alpha-halogenated reagents. Thus, heating in dioxane in the presence of triethylamine is required for the reaction of N-(4-chlorophenyl)-2-oxo-2-(phenylamino)-acetohydrazonoyl cyanide (3) with chloroacetone. The strong non-classical acidity of hydrazone-hydrogen (C⚌N-NH-) has been shown to be reactive in replacing chlorine from chloroacetone. The produced intermediate (A) undergoes intramolecular cyclization through nucleophilic addition of a methylene group on the nitrile function to produce the corresponding 5-acetyl-4-amino-pyrazole analogue 4 (Scheme 1). This analogue was utilized as a building block for the production of 7-methyl-pyrazolo[4,3-d]pyrimidine derivative 5 upon refluxing with phenyl isothiocyanate in pyridine. The compatible spectral and elemental analyses support the structures of the 4-aminopyrazole analogue 4 and the pyrazolo[4,3-d]pyrimidine derivative 5. The IR spectrum of 4-aminopyrazole 4 revealed the absorption frequencies of the –NH2 and N—H functions at 3419, 3319, 3268 cm−1. The absorption frequencies of the amidic carbonyl and the conjugated acetyl-carbonyl (intramolecular hydrogen bonded with the OH group) were observed at 1661 and 1597 cm−1, respectively. The 1H NMR signals of compound 4 are observed as a singlet at δ 2.10 ppm for the protons of methyl group (COCH3), a singlet at δ 7.48 ppm for the protons of amino group (–NH2), and a singlet at δ 9.78 ppm for the proton of the N—H group. The nine aromatic protons are observed as triplet, multiplet and doublet signals in the region δ 7.09–7.65 ppm.![Synthesis of 5-acetyl-4-aminopyrazole and pyrazolo[4,3-d]pyrimidine derivatives 4 and 5.](/content/184/2023/16/4/img/10.1016_j.arabjc.2023.104645-fig2.png)
Synthesis of 5-acetyl-4-aminopyrazole and pyrazolo[4,3-d]pyrimidine derivatives 4 and 5.
In addition, treatment of N-(4-chlorophenyl)-2-oxo-2-(phenylamino)acetohydrazonoyl cyanide (3) with ethyl bromoacetate proceeded in refluxing dioxane and triethylamine to furnish the conforming 5-ethoxycarbonyl-4-amino-pyrazole analogue 6, which undergoes heating with phenyl isothiocyanate in pyridine to yield the corresponding 7-oxo-pyrazolo[4,3-d]pyrimidine derivative 7 (Scheme 2). The chemical structures of 4-aminopyrazole analogue 6 and its corresponding pyrazolo[4,3-d]pyrimidine derivative 7 were secured based on the compatible spectral and elemental analyses. The IR spectrum of pyrazolo[4,3-d]pyrimidine derivative 7 indicated the absorptions N—H groups at 3221 and 3183 cm−1 in addition to a broad absorption for the carbonyl group at 1664 cm−1. The 1H NMR spectrum of compound 7 resonated as triplet, multiplet and doublet signals in the region δ 7.07–7.77 ppm for the aromatic protons and two singlet signals at δ 9.76 and 13.02 ppm for the protons of N—H groups. Furthermore, the reaction of N-(4-chlorophenyl)-2-oxo-2-(phenylamino)acetohydrazonoyl cyanide (3) with chloroacetonitrile in boiling dioxane and triethylamine afforded the targeting 4-amino-5-cyano-pyrazole analogue 8. The production of 7-imino-pyrazolo[4,3-d]pyrimidine derivative 9 has been achieved upon heating 4-amino-5-cyanopyrazole analogue 8 with phenyl isothiocyanate in dimethylformamide (DMF) at 85–90 °C for eight hours. The spectral and elemental analyses are used to elucidate the structure of aminopyrazole analogue 8 and its conforming pyrazolo[4,3-d]pyrimidine derivative 9.![Synthesis of 5-functionalized-4-aminopyrazoles 6 and 8, and their corresponding pyrazolo[4,3-d]pyrimidines 7 and 9.](/content/184/2023/16/4/img/10.1016_j.arabjc.2023.104645-fig3.png)
Synthesis of 5-functionalized-4-aminopyrazoles 6 and 8, and their corresponding pyrazolo[4,3-d]pyrimidines 7 and 9.
3.2 Molecular modeling features
The optimized structure of the 4-amino-1-(4-chlorophenyl)-N-phenyl-1H-pyrazole-3-carboxamide derivatives, 4 and 6, have a non-planar configuration in which the 4-chlorophenyl group was slanted on the pyrazolyl ring plane where the dihedral angle data disclosed that the C3(Pyz)–N2(Pyz)-N1(Pyz)-C1(PhPyz) and N2(Pyz)-N1(Pyz)-C1(PhPyz)–C2(PhPyz) were 173.0° and 45.2°, in the former, and −177.3° and 138.3°, in the latter, respectively. In contrary, the cyano analogue, derivative 8, has planar configuration, i.e., the corresponding angles were 180.0°. Furthermore, the phenyl carboxamide and amino groups in all derivatives was positioned in the pyrazole plane, where CO(Carb)–C3(Pyz)–N2(Pyz)-N1(Pyz) = 179.5–180.0°, N2(Pyz)–C3(Pyz)–CO(Carb)–NH(Carb) = 0.0–1.1°, N2(Pyz)–C3(Pyz)–CO(Carb)–OC(Carb) = 178.8–180.0°, C3(Pyz)–CO(Carb)–NH(Carb)-C1(PhCarb) = 179.8–180.0° and NH2(Pyz)-C4(Pyz)-C5(Pyz)-N1(Pyz) = 178.7–180.0° (Fig. 2) (Table S1).
DFT Optimized structures of the 4, 6 and 8 derivatives.
On the other hand, the 1-(4-chlorophenyl)-N,6-diphenyl-5-thioxo-5,6-dihydro-1H-pyrazolo[4,3-d]pyrimidine-3-carboxamide derivatives, 5, 7 and 8, have also a non-planar configuration where the former was more distorted than the others. The dihedral angle exhibited that each of pyrazolyl and pyrimidinyl rings, individually, were deviated from the planar structure, e.g., the C3(Pyp)-C3a(Pyp)-C7a(Pyp)-N1(Pyp) and C3a(Pyp)-C7a(Pyp)-C7(Pyp)-N6(Pyp), in average, were 1.3° and 7.3°, while the joint between exhibited more distortion, e.g., the C7(Pyp)-C7a(Pyp)-N1(Pyp)–N2(Pyp) and C3(Pyp)-C3a(Pyp)-C7a(Pyp)-C7(Pyp), average, were 171.0° and 172.5°, respectively. The chlorophenyl moiety was differentially tilted on the pyrazolopyrimidine plane, e.g., in derivative 4, the C7(Pyp)-C7a(Pyp)-N1(Pyp)-C1(PhPyp) and N2(Pyp)-N1(Pyp)-C1(PhPyp)–C2(PhPyp) were 22.7° and 37.0°, respectively, while the corresponding angles in derivative 7 were 0.6° and 174.1°, and in 9 were 13.5° and 137.6°, respectively. Similarly, the other phenyl ring, in position 6, was nearly vertical on the fused ring plane in all derivatives, i.e., C7(Pyp)-N6(Pyp)-C1(Ph)–C2(Ph) has 92.2° average value. Moreover, both of the carbonyl carbon and phenyl ring of the phenyl carboxamide group were almost coplanar with the fused ring, e.g., CO(Carb)–C3(Pyp)–N2(Pyp)-N1(Pyp) ≈ 178.8° and CO(Carb)–NH(Carb)-C1(PhCarb)–C2(PhCarb) ≈ 179.4°, while both their amidic nitrogen and oxygen atoms were shifted out, e.g., N2(Pyp)–C3(Pyp)–CO(Carb)–NH(Carb) ≈ 157.5° and N2(Pyp)–C3(Pyp)–CO(Carb)–OC(Carb) ≈ 24.3°, respectively. Likewise, the thioxo sulfur atom were sited out the pyrazolopyrimidine plane by 5.0°, i.e., C7(Pyp)-N6(Pyp)-C5(Pyp)-S(Thio) ≈ 174.8°. Finally, the methyl, oxo and imino substituents in derivatives 5, 7 and 9, were roughly in plane with the fused ring, i.e., N1(Pyp)-C7a(Pyp)-C7(Pyp)-Me(Pyp), N1(Pyp)-C7a(Pyp)-C7(Pyp)-O(Oxo) and N1(Pyp)-C7a(Pyp)-C7(Pyp)–NH(Pyp) were 0.8°, −2.6° and 1.6°, respectively (Fig. 3) (Table S1).
DFT Optimized structures of the 5, 7 and 9 derivatives.
Additionally, the DFT calculated bond lengths and angles were nearly coincided with those found in single crystal X-ray of similar compounds (Zhang et al., 2016, Şener et al., 2017, Wu et al., 2010) where lengths were longer by 0.11 Å maximum with 0.044–0.050 Å RMSD while the angles were deviated by 10.3° maximum with 5.34–6.91° RMSD, which could be attributed to the absence of intermolecular columbic interactions in the quantum chemical calculations as it carried out for isolated molecule in gaseous state, whilst the practical values were gotten for solid crystal lattice interacting molecules (Sajan et al., 2011) (Table S2-S3).
3.2.1 Frontier molecular orbitals
The molecule HOMO-LUMO construction and energetic values illuminate its electrons donation or receiving behavior (Bulat et al., 2004) as small HOMO-LUMO gap leads to ease intramolecular charge transfer (Xavier et al., 2015, Makhlouf et al., 2018) that may affect the molecule’s bioactivity (Bouchoucha et al., 2018). The 3D illustrations of the examined compounds FMO are presented in Figs. 3-4. The graph cleared that compound 4, 6 and 8 have HOMO spread over the whole molecule and consisted of the phenyl and pyrazole π-orbital with secondary participation of the heteroatom’s lone pairs. However, their LUMO was primarily built from the π*-orbitals of the whole molecule with very low contribution of the carboxamide phenyl ring. Thus, the HOMO-LUMO charge transfer may be mainly described as π → π* transition (Fig. 4).
The frontier molecular orbital of the compounds 4, 6 and 8.
On contrary, the HOMO of 5, 7 and 9 derivatives were built mainly of the lone pair of electrons belong to the thioxo sulfur atom with minimum involvement of the π-orbital of the phenyl substituent on pyrazolopyrimidine moiety whereas their LUMO were constructed from the π*-orbital of the fused pyrazolopyrimidine ring. Therefore, the HOMO-LUMO charge transfer may be mainly described as n → π* transition (Fig. 5).
The frontier molecular orbital of the compounds 5, 7 and 9.
The HOMO-LUMO energy, EH and EL, were influenced by the aforementioned details. Hence, the data revealed that the all derivatives have close values of the EH and EL, range −5.66 - −6.21 and −3.58 - −4.49 eV, respectively, where 5 and 6 exhibited lowest EH and EL, respectively. Additionally, the ΔEH-L gap data indicated that 5 and both of 6 and 8 possessed the lowest and highest values, 1.16 and 2.35 eV, respectively. Accordingly, the studied derivatives could be ordered according to energy gap as 5 < 9 < 7 < 4 < 6 = 8 which clear that the pyrazolopyrimidine derivatives have lower energy gap than the corresponding pyrazoles (Table 1).
Compound
EH
EL
ΔEH-L
χ
η
δ
ω
4
−6.00
−3.76
2.25
4.88
1.12
0.89
10.60
5
−5.66
−4.49
1.16
5.08
0.58
1.72
22.13
6
−5.93
−3.58
2.35
4.75
1.18
0.85
9.60
7
−6.21
−4.07
2.13
5.14
1.07
0.94
12.39
8
−6.19
−3.84
2.35
5.01
1.17
0.85
10.71
9
−5.91
−3.80
2.10
4.85
1.05
0.95
11.22
Respectively, chemical reactivity descriptors, such as electronegativity (χ), global hardness (η), softness (δ) and electrophilicity (ω), were determined using the values of EH and EL by the following formulae (Xavier et al., 2015).
The data revealed that, according to electronegativity (χ), derivative 6 and 7 exhibited the lowest and highest Lewis’s acid character, 4.75 and 5.14 eV, respectively. Whereas, the global softness (δ) and hardness (η) values designated that compound 5 possessed the maximum electrons receiving and charge transfer ability, 1.72 and 0.58 eV, respectively. Hence, in consistent with hardness, the studied derivatives may be ordered as 5 < 9 < 7 < 4 < 8 > 6, in agreement with that of ΔEH-L gap (Table 1).
3.2.2 Atomic Mulliken’s charges and Fukui’s indices
The molecules electronegativity and charge transfer processes could be related to the Mulliken’s atomic charges (Bhagyasree et al., 2013). In the pyrazole derivatives, the nitrogen atom, N1(Pyz), has positive charge in 4 and 6 derivatives, acetyl and carboxylate, respectively, while acquire a negative charge in the cyano derivative, 8. On contrary, the second pyrazolyl nitrogen atom, N1(Pyz), has negative charge in the former two derivative and positive in the last one. Furthermore, the pyrazolyl carbon atoms N4(Pyz) and N5(Pyz), possessed negative and positive charges, respectively, where both increased from 4 to 8 which may be attributed to the electron withdrawing or donating ability of the substituent’s acetyl, carboxylate and cyano, respectively (Table 2).
Atom
4
6
8
Atom
5
7
9
N1(Pyz)
0.535
0.195
−0.679
N1(Pyp)
0.231
0.267
0.203
N2(Pyz)
−0.582
−0.410
0.447
N2(Pyp)
−0.614
−0.637
−0.599
C3(Pyz)
0.173
−0.313
−0.103
C3(Pyp)
−0.252
−0.247
−0.320
C4(Pyz)
−0.460
−0.655
−0.883
C3a(Pyp)
−0.431
−0.103
0.111
C5(Pyz)
0.125
0.527
0.464
N4(Pyp)
0.600
−0.169
−0.146
C1(PhPyz)
−0.594
−0.652
−0.637
C5(Pyp)
−0.179
−0.166
−0.155
C4(PhPyz)
0.263
0.441
0.845
N6(Pyp)
0.582
0.635
0.863
Cl(PhPyz)
0.835
0.824
0.863
C7(Pyp)
−0.236
0.132
0.136
CO(Carb)
0.414
0.462
0.849
C7a(Pyp)
−0.284
0.177
0.152
OC(Carb)
−0.366
−0.364
−0.371
C1(PhPyp)
−0.786
−0.089
−0.554
NH(Carb)
−0.328
−0.284
−0.164
C4(PhPyp)
0.341
0.929
0.518
C1(PhCarb)
−0.526
−0.249
−0.418
Cl(PhPyp)
0.877
0.862
0.858
C4(PhCarb)
−0.443
−0.517
−0.498
CO(Carb)
0.087
0.097
0.009
NH2(Pyz)
−0.711
−0.749
−0.754
OC(Carb)
−0.259
−0.227
−0.252
CO(Acet)
0.199
NH(Carb)
−0.330
−0.264
−0.221
OC(Acet)
−0.384
C1(PhCarb)
−0.514
−0.670
−0.727
Me(Acet)
−0.209
C4(PhCarb)
−0.356
−0.242
−0.279
CO(Est)
−0.360
S(Thio)
−0.264
−0.439
−0.457
OC(Est)
−0.316
C1(Ph)
−0.821
−0.335
−0.278
OEt(Est)
0.186
C4(Ph)
−0.349
−0.236
−0.218
Et(Est)
−0.598
Me(Pyp)
−0.008
CN(Cyno)
−0.278
O(Oxo)
−0.035
NC(Cyno)
−0.100
NH(Pyp)
−0.090
In pyrazolopyrimidine derivatives, nitrogen atoms N1(Pyp) and N2(Pyp) have close positive and negative charges, respectively, however, the other nitrogen atom, N4(Pyp), acquired positive in the methyl derivative, 5, and negative in both oxo and imino derivatives, 7 and 9, which may be ascribed to their subscription in the pyrazolopyrimidine moiety resonating structure. Noteworthy, the increase in the positive charge of the pyrazolopyrimidine nitrogen N6(Pyp) from 0.582 in 5 to 0.863 in 9, in addition to turning of the negative charge of the carbon atoms C7(Pyp) and C7a(Pyp) in the methyl derivative, 5, to be positively charged in oxo and imino derivatives, 7 and 9, could be ascribed to the oxo and imino groups electron withdrawing effect in the latter derivatives. Furthermore, the other heteroatoms in all derivatives, sulfur, oxygen and nitrogen atoms were acquired negative charge (Table 2).
The atomic Fukui’s reactivity indices,
,
and
, toward nucleophilic, electrophilic and radical attacks, respectively, were evaluated (Olasunkanmi et al., 2016, El Adnani et al., 2013, Mi et al., 2015, Messali et al., 2018). The pyrazole derivatives 4, 6 and 8 toward nucleophilic attack Fukui’s indices (
) revealed that the pyrazole nitrogen N2(Pyz) was the most labile atom, except in 8, it resided in the second place after the cyano nitrogen NC(Cyano). However, the pyrazolopyrimidine derivatives 5, 7 and 9 showed different pattern in which the thioxo sulfur and pyrazolopyrimidine, S(Thio) and N2(Pyp), were the most susceptible two sites, respectively (Table 3). Otherwise, the electrophilic attack Fukui’s indices (
) of the pyrazole derivatives 4, 6 and 8, offered comparable patterns for the most labile atoms in whom the amino nitrogen atom was in the first position followed by the chloro atom in 4 and 6 while the cyano nitrogen appeared in 8 as the second active site followed by the chloro atom. Likewise, the pyrazolopyrimidine 5, 7 and 9 exhibited close order where the thioxo sulfur atom S(Thio) was the most active and the pyrazolopyrimidine carbon C5(Pyp) occupied the second place except in compound 7 in which the oxo oxygen atom was in second position and followed by the pyrazolopyrimidine carbon C5(Pyp). Eventually, the radical attack (
) indices of the pyrazolopyrimidine 5, 7 and 9 exhibited close orderliness to those obtained for the nucleophilic attack Fukui’s indices (
) where the thioxo sulfur and pyrazolopyrimidine, S(Thio) and N2(Pyp), were the most susceptible two sites, respectively. In contrast, the derivatives 4, 6 and 8 different activity arrangements where the acetyl oxygen OC(Acet) was on the top in the first compound, 4, while the chloro Cl(PhPyz) and cyano nitrogen NC(Cyano), were for the other derivatives, respectively. However, the amino nitrogen NH2(Pyz) was occupied the second place in both 4 and 6 whilst in compound 8, the chloro Cl(PhPyz) appeared in the second position (Table 3).
4
6
8
5
7
9
atom
atom
atom
atom
atom
atom
NH2(Pyz)
0.095
NH2(Pyz)
0.099
NH2(Pyz)
0.094
S(Thio)
0.397
S(Thio)
0.407
S(Thio)
0.408
Cl(PhPyz)
0.059
Cl(PhPyz)
0.063
NC(Cyno)
0.078
C5(Pyp)
0.040
O(Oxo)
0.048
C5(Pyp)
0.044
OC(Acet)
0.057
C4(PhCarb)
0.045
Cl(PhPyz)
0.076
Cl(PhPyp)
0.037
C5(Pyp)
0.044
NH(Pyp)
0.040
C4(PhCarb)
0.047
C5(Pyz)
0.043
C4(PhCarb)
0.047
C4(Ph)
0.033
Cl(PhPyp)
0.037
C4(Ph)
0.037
C5(Pyz)
0.043
C3(Pyz)
0.043
C3(Pyz)
0.040
N2(Pyp)
0.032
C4(Ph)
0.036
Cl(PhPyp)
0.034
4
6
8
5
7
9
atom
atom
atom
atom
atom
atom
N2(Pyz)
0.094
N2(Pyz)
0.091
NC(Cyno)
0.095
S(Thio)
0.116
S(Thio)
0.099
S(Thio)
0.085
OC(Acet)
0.085
Cl(PhPyz)
0.068
N2(Pyz)
0.085
N2(Pyp)
0.086
N2(Pyp)
0.092
N2(Pyp)
0.085
Cl(PhPyz)
0.068
OC(Est)
0.065
Cl(PhPyz)
0.075
C7(Pyp)
0.082
O(Oxo)
0.070
Cl(PhPyp)
0.073
CO(Acet)
0.060
OC(Carb)
0.050
OC(Carb)
0.054
N4(Pyp)
0.053
Cl(PhPyp)
0.066
NH(Pyp)
0.058
C3(Pyz)
0.047
C3(Pyz)
0.048
C3(Pyz)
0.049
Cl(PhPyp)
0.049
C7(Pyp)
0.057
C3(Pyp)
0.046
4
6
8
5
7
9
atom
atom
atom
atom
atom
atom
OC(Acet)
0.071
Cl(PhPyz)
0.065
NC(Cyno)
0.086
S(Thio)
0.257
S(Thio)
0.253
S(Thio)
0.247
NH2(Pyz)
0.066
NH2(Pyz)
0.064
Cl(PhPyz)
0.076
N2(Pyp)
0.059
N2(Pyp)
0.061
N2(Pyp)
0.058
N2(Pyz)
0.063
N2(Pyz)
0.060
NH2(Pyz)
0.061
C7(Pyp)
0.053
O(Oxo)
0.059
Cl(PhPyp)
0.053
Cl(PhPyz)
0.063
OC(Est)
0.053
N2(Pyz)
0.055
Cl(PhPyp)
0.043
Cl(PhPyp)
0.052
NH(Pyp)
0.049
CO(Acet)
0.045
C3(Pyz)
0.045
C3(Pyz)
0.045
N4(Pyp)
0.036
C7(Pyp)
0.036
C3(Pyp)
0.031
4
6
8
5
7
9
atom
S+/S-
atom
S+/S-
atom
S+/S-
atom
S+/S-
atom
S+/S-
atom
S+/S-
N2(Pyz)
3.03
C1(PhPyz)
6.00
CO(Carb)
3.40
NH(Carb)
5.50
C3a(Pyp)
15.00
CO(Carb)
20.00
CO(Carb)
2.30
N2(Pyz)
3.14
N2(Pyz)
3.27
N6(Pyp)
3.42
CO(Carb)
11.00
C3a(Pyp)
13.00
CO(Acet)
2.07
CO(Carb)
2.90
OC(Carb)
2.45
C7(Pyp)
3.28
C2(PhPyp)
8.00
C2(PhPyp)
11.50
C1(PhPyz)
2.00
OC(Carb)
2.27
CN(Cyno)
2.00
C2(PhPyp)
3.00
NH(Carb)
5.50
NH(Carb)
7.50
OC(Carb)
1.91
CO(Est)
2.14
C1(PhPyz)
1.63
N4(Pyp)
2.79
C7(Pyp)
3.56
C5(PhPyp)
3.57
4
6
8
5
7
9
atom
S-/S+
atom
S-/S+
atom
S-/S+
atom
S-/S+
atom
S-/S+
atom
S-/S+
C1(PhCarb)
8.50
C1(PhCarb)
7.50
C1(PhCarb)
17.00
C5(Pyp)
4.44
C2(Ph)
9.00
C5(Pyp)
11.00
C2(PhCarb)
2.67
NH2(Pyz)
3.30
NH2(Pyz)
3.24
S(Thio)
3.42
C6(Ph)
8.00
C2(Ph)
10.00
NH2(Pyz)
2.57
C2(PhCarb)
2.30
C2(PhCarb)
2.00
C1(PhCarb)
3.00
C5(Pyp)
7.33
C6(Ph)
9.00
C6(PhCarb)
2.27
C6(PhCarb)
1.92
C6(PhCarb)
1.85
C2(Ph)
2.67
S(Thio)
4.11
S(Thio)
4.80
NH(Carb)
2.12
NH(Carb)
1.84
C4(Pyz)
1.60
C6(Ph)
2.33
C4(Ph)
2.00
C1(PhCarb)
3.00
As the Fukui’s indices may be imprecisely predicted the active site for nucleophilic and electrophilic attack, the relative electrophilicity and nucleophilicity descriptors, and , respectively, were determined (Roy et al., 1998b, Roy et al., 1998a, Roy et al., 1999), where , and δ is global softness. The relative nucleophilicity descriptors data, , of derivative 4 showed that pyrazole nitrogen N2(Pyz) and carboxamide carbon CO(Carb) were the first and second active atoms, respectively, while in 6 and 8, the former appeared in second place after the phenyl carbon C1(PhPyz) and carboxamide carbon CO(Carb), respectively. Moreover, the data indicated that the carboxamide and pyrazolopyrimidine nitrogen atoms, NH(Carb) and N6(Pyp), were the first two susceptible in compound 5, respectively, whereas in 7 and 9, the pyrazolopyrimidine and carboxamide carbon atoms, C3a(Pyp) and CO(Carb), were occupied were the most active sties, respectively. In contrast, the relative electrophilicity descriptors, , data displayed almost coincided patterns in case of the pyrazole derivatives 4, 6 and 8, wherein the carboxamide phenyl carbon C1(PhCarb) was on the top followed by the carbon C2(PhCarb) in the former and amino nitrogen NH(Pyz) atom in the other two derivatives. Furthermore, the pyrazolopyrimidine derivatives 5 and 9 data revealed that the pyrazolopyrimidine carbon C5(Pyp) was the most active atom while the phenyl carbon C2(Ph) was in 7 (Table 3).
3.3 Cytotoxicity assay
Six 4-aminopyrazole and pyrazolopyrimidine analogues were examined over MTT assay toward several cancer cell lines such as HCT-116 (colon carcinoma), HepG2 (liver carcinoma), MCF-7(breast carcinoma), and WI38 (normal cell) at the National Cancer Institute, (Egypt). Table 4 and Fig. 6 revealed the cytotoxic results in standings of IC50 values and contrasted them to the extensively used anticancer reference (5-Fu). Some of the synthesized aminopyrazoles and pyrazolopyrimidines presented good efficiency to inhibit the growth of (MCF-7) and (HCT-116) rather than HepG2 (Fayed et al., 2019). Among the deliberate analogues, pyrazolopyrimidine analogues 5, 7, and 9 were presented extra cytotoxic activities especially against both of (MCF-7) and (HCT-116). The examined results of pyrazolopyrimidine analogues deliver understanding into the structural features related to anticancer activity. Initially, aminopyrazole analogue 4 have acetyl moiety in 5-position demonstrated reasonable activity in general with IC50 = 23.42 ± 0.26, 27.25 ± 0.14, and 33.75 ± 0.42 μM against MCF-7, HCT-116, and HepG2, respectively. While, analogue 6 with ethyl ester group in 5-position offered weak activities with IC50 = 34.51 ± 0.47, 39.76 ± 0.19, and 37.51 ± 0.32 μM against the same sequence of cell lines MCF-7, HCT-116, and HepG2, correspondingly. Further analogue 8 through nitrile group in 5-position presented enhancement of cytotoxic activity with IC50 = 19.33 ± 0.18, 22.25 ± 0.53, and 18.42 ± 0.10 μM. However, pyrazolopyrimidine analogues such as analogue 5 with methyl group in 7-position displayed better antitumor activities linked to their substituents with IC50 = 13.47 ± 0.25, 19.35 ± 0.27, and 14.82 ± 0.38 μM. Moreover, analogue 7 with a carbonyl moiety in 7-position exhibited eminent cytotoxic activity with IC50 = 11.51 ± 0.35, 15.08 ± 0.27, and 21.25 ± 0.37 μM. Furthermore, analogue 9 with an imino moiety in 7-position revealed superior activities with IC50 values 16.48 ± 0.36, 16.38 ± 0.27, and 17.39 ± 0.46 μM toward the same order of cell lines MCF-7, HCT-116, and HepG2, respectively. All of the synthesized analogues were studied in comparison to reference (5-Fu).
Compound
HepG2
HCT-116
MCF-7
WI38
4
33.75 ± 0.42
27.25 ± 0.14
23.42 ± 0.26
43.87 ± 0.21
5
14.82 ± 0.38
19.35 ± 0.27
13.47 ± 0.25
47.64 ± 0.12
6
37.51 ± 0.32
39.76 ± 0.19
34.51 ± 0.47
39.37 ± 0.23
8
18.42 ± 0.10
22.25 ± 0.53
19.33 ± 0.18
46.28 ± 0.45
7
21.25 ± 0.37
19.33 ± 0.27
11.51 ± 0.35
52.49 ± 0.54
8
18.42 ± 0.10
22.25 ± 0.53
19.33 ± 0.18
46.28 ± 0.45
9
17.39 ± 0.46
16.38 ± 0.27
16.48 ± 0.36
48.57 ± 0.39
5-Fua
7.15 ± 0.44
5.22 ± 0.36
10.26 ± 0.38
10.16 ± 0.24

In vitro cytotoxicity of the prepared analogues.
3.4 Structural activity relationship
Inspired the above results of the cytotoxic effectiveness, the six aminopyrazole and pyrazolopyrimidine analogues showed potent activities against (MCF-7, HCT-116) and less potent against HepG2 as it was reported before (Hassan et al., 2021). Among the amino-pyrazole analogues 4, 6, and 8 substituted with different acetyl, ester, and cyano moieties in 5-position, respectively, displayed various cytotoxic activities, analogue 4 have acetyl moiety demonstrated reasonable activity, analogue 6 with ethyl ester group offered weak activities, analogue 8 through nitrile group presented enhancement of cytotoxic activity. These results presented that, the cytotoxic activities follow the following order CN substituent > acetyl substituent > ester substituent which agrees with pervious literature (Farag et al., 2010). Meanwhile, pyrazolopyrimidine analogues were own broadly powerful than aminopyrazole as antitumor activity towards certain cancer cell lines (Hassan et al., 2020). Pyrazolopyrimidine analogues 5, 7, and 9 displayed significant anticancer activities toward MCF-7 (IC50 = 13.47 ± 0.25, 11.51 ± 0.35, 16.48 ± 0.36), respectively, which is presented a potent activities than pervious Pyrazolopyrimidine derivatives that was recorded in the literature with (IC50 = 13.91 ± 1.4 and 22.37 ± 1.8 μM/L) (Metwally et al., 2019). As analogue 7 have methyl group in 7-position showed high IC50 value may attribute to the existence of hydrophobic group which increases the dual property lipophilic/hydrophilic part in the structure rather than both of 5 and 9 analogues with carbonyl and imino groups that possess a higher hydrophilic property (Wang et al., 2021).
Finally, the Multiple Linear Regression (MLR) (Worachartcheewan et al., 2011) incorporated in OriginPro software (OriginLab, 2018) was applied to correlate the anticancer activity (IC50), dependent variable, with the descriptors of the quantum chemical calculation, EH, EL, ΔEH-L, χ and η, independent variables, to find out the most effective descriptor on the anticancer activity (Table 5). The data indicated that all the cell lines, except WI38, exhibited close coefficients in which the most effective descriptor were the ΔEH-L gap and hardness (η), both have positive values, and thus the increase in HOMO-LUMO gap and hardness will lead to increase in the activity. While the EH, EL, and χ displayed negative coefficients and consequently their increase will result in reduction of the anticancer activity. Moreover, these cell lines showed close intercept values, 263.96–284.46, and good regression coefficients, R2 = 0.8593–0.9999 with standard deviation, SD = 0.14–6.75. On contrary, the normal cell, WI38, exhibited opposite behavior where the ΔEH-L gap and hardness (η) were negative whereas the others have positive coefficients with regression coefficient, R2 = 0.9854.
Intercept
EH (×1015)
EL (×1016)
ΔEH-L (×1015)
χ (×1016)
η (×1015)
R2
SD
HepG2
284.46
−6.40
−1.75
4.43
−2.39
2.21
0.8593
6.75
HCT-116
263.96
−5.06
−2.68
8.69
−3.19
4.35
0.9772
2.88
MCF-7
282.57
−3.05
−1.94
6.55
−2.25
3.27
0.9999
0.14
WI38
−107.39
0.615
1.27
−4.83
1.33
−2.41
0.9854
1.20
3.5 Molecular docking
A molecular docking simulation via utilizing MOE v10.2015.10 program was carried out to determine the compound's preferred manner of interacting with the enzyme's active site. Energy score (S, kcal/mol) was used to calculate the binding affinity of the synthesized derivatives with the enzyme active site; a low energy score implies a good affinity. The interaction types that are presented in molecular docking are hydrogen bonds, π-H, H-π, and π- π interactions, and arene cation interactions. By using several tested ligand and proteins as a target in a molecular docking simulation, the target-ligand interaction was determined. According to literature survey, it was noticed that PPs recorded a potent activity toward histone lysine demethylases (KDM) inhibitors (Metwally et al., 2019). The crystal structure of the KDM5A protein, which was determined from the protein data bank (PDB- ID: 5IVE), and the most stable conformation of certain investigated drugs were used to perform molecular docking to introduce a theoretical prediction for the reactivity of the synthesized derivatives towards (KDM) to help the biological scientists to investigate these test experimentally. The docking results (Table 6) displayed binding energy score (S), route main square (RMSD), types and site of interactions between ligands and 5IVE amino-acids, and the intermolecular distances (Å) for the synthesized analogues. 5-Acetyl-4-amino-1-(4-chlorophenyl)-N-phenyl-1H-pyrazole-3-carboxamide (4) displayed binding energy value S = −7.1126 kcal/mol, standing on RMSD = 1.4975 through two π-H interaction bindings, phenyl ring with both of His 483 and His 571 through 4.47 Å, 4.28 Å, respectively (Figure S1). Whereas, 1-(4-Chlorophenyl)-7-methyl-pyrazolo[4,3-d]pyrimidine-3-carboxamide analogue 5 revealed best RMSD = 0.8213 from one π- π interaction phenyl ring with His 483 through 3.91, with binding energy value S = -7.5571 kcal/mol (Fig. 7).
Code
S (energy score)
(Kcal/mol)RMSD
(refine unit)Interaction with ligand
Types of Interactions
Distance (Å)
4
−7.1126
1.4975
6‐ring with His 483
6‐ring with His 571π-H
π-H4.47
4.28
5
−7.5571
0.8213
6‐ring with His 483
π- π
3.91
6
−7.5158
1.4039
O 24 with His 483
6-ring with His 483H-acceptor
π- π2.95
3.77
7
−8.3527
1.0311
N 16 with His 483
H-π
3.65
8
−7.0005
1.0765
O 9 with Lys 501
N 24 with His 571
6‐ring with His 483H-acceptor
H-acceptor
π- π3.46
3.18
3.79
9
−7.2823
0.8924
S 7 with Leu 536
6‐ring with His 571H-acceptor
π-H3.95
4.88
5-Fu
−4.3533
0.9599
O 3 with Lys 501
6-ring with Phe 480H-acceptor
π-π3.07
3.70
Docking interactions over pyrazolopyrimidine compound 5 with 5IVE.
While, analogue 6 with amino and ester substituents on the pyrazole moiety showed RMSD = 1.4039 with binding energy value S = -7.5158 kcal/mol through H-acceptor between O24 of carbonyl ester with His 483 which form another π- π interaction between the phenyl-pyrazole ring through distances 2.95 and 3.77 Å (Figure S2). Likewise, analogue 7 revealed better binding score, S = -8.3527 kcal/mol with RMSD value = 1.0311 over H-π interaction between N 16 of amide linkage with His 483 through 3.65 Å (Fig. 8).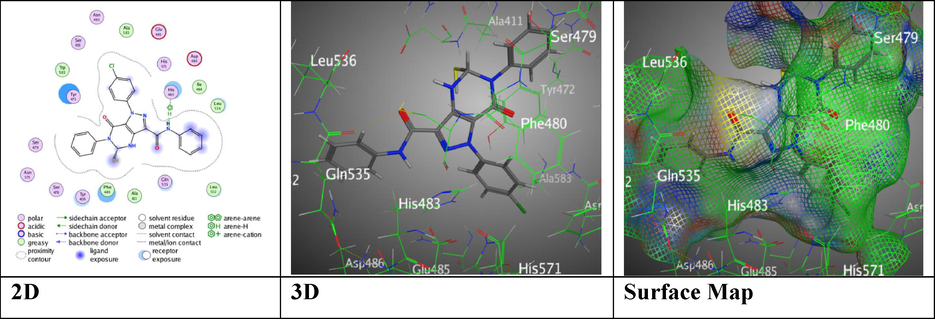
Docking interactions over pyrazolopyrimidine compound 7 with 5IVE.
Also, analogue 8 showed lower value, S = -7.0005 kcal/mol with RMSD value = 1.0765 from H-acceptor raised between O9 of amide linkage with Lys 501, N 24 of nitrile moiety with His 571, and π- π interaction between the phenyl-pyrazole ring with His 483 through 3.46, 3.18, and 3.79 Å (Figure S3). Meanwhile, analogue 9 disclosed one H-acceptor between S7 of thio-emino moiety with Leu 536 through 3.25 Å, π-H interaction between the phenyl-pyrazole ring with His 571 through 4.88 Å, binding energy value S = -7.2823 kcal/mol with RMSD = 0.8924 (Figure S4). Similarly, 5-fluorouracil one H-acceptor between O3 of one of carbonyl group in the pyrimidinone ring with LYS 501 through 3.07 Å, π- π interaction between the pyrimidinone ring with Phe 480 through 3.70 Å, binding energy value S = -4.3533 kcal/mol with RMSD = 0.9599 (Figure S5).
Finally, the stimulating reasonable docking experiment is used to confirm how much the synthesized analogues interactions change as they interact with many analogues. A network of hydrogen bonding with the chemically aromatic moiety that interacts differently with the amino acids of 5IVE supports each of the six synthesized analogues that were created. The 5IVE pocket, which can be visualized as a fork enclosed by polar and nonpolar residues of amino-acids (His 483, His 571, Lys 501, Leu 536, Gly287) with the majority of the analogues that were prepared. Moreover, most of different derivatives are related to the same amino acid (s is considered as strong evidence for success docking procedure.
4 Conclusion
Six new functionalized 4-aminopyrazole and their corresponding pyrazolo[4,3-d]pyrimidine derivatives were synthesized and confirmed. The DFT optimized structure of the investigated compounds indicated that all were deviated from planar structure and have low HOMO-LUMO energy gap but the pyrazolopyrimidine derivatives exhibited lower values than the corresponding pyrazoles following the order 5 < 9 < 7 < 4 < 6 = 8. Moreover, the newly produced analogues' cytotoxic efficacy was investigated over the reference (5-Fu). The cytotoxic results exhibited good potency contrary to human cell lines, with MCF-7 and HCT-116 presenting cytotoxic effectiveness. Midst these analogues, the pyrazolopyrimidine derivatives 5, 7, and 9 were revealed better efficiency toward MCF-7 and HCT-116 cancer cells. Studies on the structure–activity relationship (SAR) were contributed to present the source of the variable activities. Meanwhile, the inspected derivatives were docked toward 5IVE, which was acquired from PDB.
Acknowledgement
Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R22), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Pyrazolopyrimidines as anticancer agents: a review on structural and target-based approaches. Eur. J. Med. Chem.. 2021;225:113781
- [Google Scholar]
- Molecular iodine promoted synthesis of new pyrazolo[3,4-d]pyrimidine derivatives as potential antibacterial agents. Eur. J. Med. Chem.. 2010;45:647-650.
- [Google Scholar]
- Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys.. 1993;98:5648-5652.
- [Google Scholar]
- Vibrational spectroscopic (FT-IR, FT-Raman, (1)H NMR and UV) investigations and computational study of 5-nitro-2-(4-nitrobenzyl) benzoxazole. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2013;102:99-113.
- [Google Scholar]
- Materials Studio. San Diego: Dassault Systèmes; 2017.
- Synthesis and characterization of new complexes of nickel (II), palladium (II) and platinum(II) with derived sulfonamide ligand: Structure, DFT study, antibacterial and cytotoxicity activities. J. Mol. Struct.. 2018;1161:345-355.
- [Google Scholar]
- Condensation of frontier molecular orbital Fukui functions. J. Phys. Chem.. 2004;108:342-349.
- [Google Scholar]
- Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chinese Med. J.. 2021;134:783-791.
- [Google Scholar]
- Medicinal attributes of pyrazolo[3,4-d]pyrimidines: a review. Bioorg. Med. Chem.. 2013;21:5657-5668.
- [Google Scholar]
- Ground-state enthalpies: evaluation of electronic structure approaches with emphasis on the density functional method. J. Phys. Chem.. 2006;110:13632-13639.
- [Google Scholar]
- The PARP enzyme family and the hallmarks of cancer part 1. cell intrinsic hallmarks. Cancers. 2021;13:2042.
- [Google Scholar]
- DFT theoretical study of 7-R-3methylquinoxalin-2 (1H)-thiones (RH;CH3;Cl) as corrosion inhibitors in hydrochloric acid. Corros. Sci.. 2013;68:223-230.
- [Google Scholar]
- Novel Pyrazoloquinolin-2-ones: Design, synthesis, docking studies, and biological evaluation as antiproliferative EGFR-TK inhibitors. Bioorg. chem.. 2019;90:103045
- [Google Scholar]
- Design, synthesis and structure–activity relationship study of novel pyrazole-based heterocycles as potential antitumor agents. Eur. J. Med. Chem.. 2010;45:5887-5898.
- [Google Scholar]
- Design, synthesis, cytotoxicity and molecular modeling studies of some novel fluorinated pyrazole-based heterocycles as anticancer and apoptosis-inducing agents. Mol. Diver.. 2019;23:165-181.
- [Google Scholar]
- Frisch, M., Trucks, G., Schlegel, H., Scuseria, G., Robb, M., Cheeseman, J., Scalmani, G., Barone, V., Mennucci, B., Petersson, G., 2009. Gaussian 09, Revision A. 1. Wallingford, CT, USA: Gaussian. .
- Pharmacophore-linked pyrazolo[3,4-d]pyrimidines as EGFR-TK inhibitors: synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies. Arch. Pharm. 2021:e2100258.
- [Google Scholar]
- Peer reviewed: transitions from heart disease to cancer as the leading cause of death in US States, 1999–2016. Prev. Chronic Dis. 2018:e158.
- [Google Scholar]
- Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids. ACS Omega. 2021;6:12361-12374.
- [Google Scholar]
- New fused pyrazolopyrimidine derivatives; heterocyclic styling, synthesis, molecular docking and anticancer evaluation. J. Heterocycl. Chem.. 2020;57:2704-2721.
- [Google Scholar]
- Novel pyrazolo[3,4-d]pyrimidine derivatives as potential antitumor agents: exploratory synthesis, preliminary structure-activity relationships, and in vitro biological evaluation. Molecules. 2011;16:10685-10694.
- [Google Scholar]
- Synthesis, anticancer activity, and molecular docking of new pyrazolo[1,5-a]pyrimidine derivatives. J. Saudi Chem. Soc.. 2023;27:101599
- [Google Scholar]
- Computational study of 3-thiophene acetic acid: Molecular docking, electronic and intermolecular interactions investigations. Comput. Biol. Chem.. 2020;86:107268
- [Google Scholar]
- Theoretical study of solvent and co-solvent effects on the interaction of Flutamide anticancer drug with Carbon nanotube as a drug delivery system. J. Mole. Liq.. 2017;248:490-500.
- [Google Scholar]
- A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules. 2020;25:1909.
- [Google Scholar]
- Pyrazole scaffold: A remarkable tool in the development of anticancer agents. Eur. J. Med. Chem.. 2013;70:248-258.
- [Google Scholar]
- Efficient green protocols for the preparation of pyrazolopyrimidines. ChemistrySelect. 2021;6:5807-5837.
- [Google Scholar]
- Pyrazolo[3,4-d]pyrimidine-based dual EGFR T790M/HER2 inhibitors: design, synthesis, structure–activity relationship and biological activity as potential antitumor and anticonvulsant agents. Eur. J. Med. Chem.. 2021;214:113-222.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev.. 1988;37:785-789.
- [Google Scholar]
- Synthesis of 1H-pyrazolo[3,4-d]pyrimidines via solid-phase Aza-Wittig/electrocyclic ring closure reaction. Tetrahedron Lett.. 2013;54:6855-6857.
- [Google Scholar]
- Use of human dihydroorotate dehydrogenase (hDHODH) inhibitors in autoimmune diseases and new perspectives in cancer therapy. Recent Patents on Anti-Cancer Drug Discov.. 2018;13:86-105.
- [Google Scholar]
- Experimental and DFT insights into molecular structure and optical properties of new chalcones as promising photosensitizers towards solar cell applications. Appl. Surf. Sci.. 2018;452:337-351.
- [Google Scholar]
- A new schiff base derivative as an effective corrosion inhibitor for mild steel in acidic media: Experimental and computer simulations studies. J. Mol. Struct.. 2018;1168:39-48.
- [Google Scholar]
- Design, synthesis, anticancer evaluation, molecular docking and cell cycle analysis of 3-methyl-4,7-dihydropyrazolo[1,5-a]pyrimidine derivatives as potent histone lysine demethylases (KDM) inhibitors and apoptosis inducers. Bioorg. Chem.. 2019;88:102929
- [Google Scholar]
- Theoretical evaluation of corrosion inhibition performance of three antipyrine compounds. Comput. Theor. Chem.. 2015;1072:7-14.
- [Google Scholar]
- Adsorption and corrosion inhibition properties of N-{n-[1-R-5-(quinoxalin-6-yl)-4,5-dihydropyrazol-3-yl]phenyl} methanesulfonamides on mild steel in 1 M HCl: experimental and theoretical studies. RSC Adv.. 2016;6:86782-86797.
- [Google Scholar]
- Originlab, 2018. Origin Pro 2018 ed.: OriginLab Corporation Northampton, MA.
- Pair-distribution function and its coupling-constant average for the spin-polarized electron gas. Phys. Rev.. 1992;46:12947-12954.
- [Google Scholar]
- Synthesis and biological evaluation of novel pyrazolopyrimidines derivatives as anticancer and anti-5-lipoxygenase agents. Bioorg. Chem.. 2016;66:160-168.
- [Google Scholar]
- Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz. Gastroenterol.. 2019;14:89-103.
- [Google Scholar]
- Site of protonation in aniline and substituted anilines in the gas phase: a study via the local hard and soft acids and bases concept. The J. Phys. Chem.. 1998;102:7035-7040.
- [Google Scholar]
- Local softness and hardness based reactivity descriptors for predicting intra-and intermolecular reactivity sequences: carbonyl compounds. J. Phys. Chem.. 1998;102:3746-3755.
- [Google Scholar]
- Natural bond orbital analysis, electronic structure, non-linear properties and vibrational spectral analysis of L-histidinium bromide monohydrate: A density functional theory. Spectrochim. Acta A Mole. Biomole. Spectrosc.. 2011;81:85-98.
- [Google Scholar]
- A combined experimental and DFT investigation of disazo dye having pyrazole skeleton. J. Mol. Struct.. 2017;1129:222-230.
- [Google Scholar]
- Inhibiting mutant KRAS G12D gene expression using novel peptide nucleic acid-based antisense: A potential new drug candidate for pancreatic cancer. Oncol. Lett.. 2022;23:130.
- [Google Scholar]
- Cyanoacetarylamides—I: Preparation and reactions of their arylazo derivatives with diazonium ion and Grignard reagents. Tetrahedron. 1971;27:4305-4316.
- [Google Scholar]
- Loss of heterozygosity as a predictor to map tumor suppressor genes in cancer: molecular basis of its occurrence. Cur. Opin. Oncol.. 2002;14:65-72.
- [Google Scholar]
- Bioengineered dual-targeting protein nanocage for Stereoscopical loading of synergistic hydrophilic/hydrophobic drugs to enhance anticancer efficacy. Adv. Funct. Mater.. 2021;31:2102004.
- [Google Scholar]
- Predicting the free radical scavenging activity of curcumin derivatives. Chemom. Intell. Lab. Syst.. 2011;109:207-216.
- [Google Scholar]
- Regiospecific synthesis of 6-aryl-3-cyano-5-alkylamino/arylamino-1-p-tolyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-ones via iminophosphorane-mediated annulation. Tetrahedron. 2010;66:5112-5120.
- [Google Scholar]
- NBO, conformational, NLO, HOMO–LUMO, NMR and electronic spectral study on 1-phenyl-1-propanol by quantum computational methods. Spectrochim. Acta A Mole. Biomole. Spectrosc.. 2015;137:306-320.
- [Google Scholar]
- Novel 3-substituted-1-aryl-5-phenyl-6-anilinopyrazolo[3,4-d]pyrimidin-4-ones: Docking, synthesis and pharmacological evaluation as a potential anti-inflammatory agents. Bioorg. Med. Chem. Lett.. 2012;22:6616-6620.
- [Google Scholar]
- Formal [4+1] Annulation of α-arylhydrazonoketones and dimethylsulfoxonium methylide: one-pot synthesis of substituted pyrazoles and dihydropyrazoles. J. Org. Chem.. 2016;81:6036-6041.
- [Google Scholar]
- Small-molecule fms-like tyrosine kinase 3 inhibitors: an attractive and efficient method for the treatment of acute myeloid leukemia. J. Med. Chem.. 2020;63:12403-12428.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104645.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




