

Translate this page into:
Synthesis of nonionic surfactants with azole ring bearing N-glycosides and their antibacterial activity
⁎Corresponding author. Tel.: +213 771537746; fax: +213 41425763 adelaliothman@gmail.com (Adil A. Othman)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.

Abstract
Six azoles with n-pentyl side chain 6–9, 11 and 12 were synthesized from n-hexanoic acid. Three N-glycosides namely: 5-pentyl-2-(d-amino arabinoside)-1,3,4-oxadiazole (13), 5-pentyl-2-(d-aminoglycoside)-1,3,4-thiadiazole (14), and 3-pentyl-4-(d-amino xyloside)-4H-1,2,4-triazole-5-thiol (15) were prepared from already synthesized n-pentyl azoles 6, 7 and 11, respectively. Surface activity properties of water soluble synthesized compounds 6, 7, and 11–15 were studied in terms of surface tension, cloud point and critical micelle concentration. The antibacterial activities were assessed using the paper disk diffusion and broth dilution methods against gram-positive and gram-negative bacteria. Some of the synthetic compounds showed promising activity against microorganisms under test in comparison to commercially available antibiotics polymixine and oxytetracycline.
Keywords
Hexanoic acid
1,3,4-Oxadiazole
1,3,4-Thiadiazole
1,2,4-Triazole
N-glycosides
Surface activity
Antibacterial activity

1 Introduction
The most important category of fatty nonionic surfactants is synthesis of organic compounds containing active hydrogen atoms such as OH, NH, SH and CO2H in the presence of a base (Crees, 1987).
Sugar-based surfactants are a very interesting class of compounds on account of their low toxicity and their synthesis from renewable resources (Noiret et al., 2002). They are biocompatible and have various applications in personal care and food industry, such as detergents, pharmaceuticals, agrochemicals and explosives (Noiret et al., 2002; Holmberg, 2003; Rybinski and Hill, 1998; Hill and Rhode, 1999). Some non-ionic sugar surfactants such as glycosides have been used for the extraction of biological proteins of the cellular membrane (Garavito and Rosenbusch, 1980). The most frequent types of linkage between the hydrophilic and the hydrophobic moieties are ester, amines, amides and glycosidic in nature (Stabenrrauch, 2001). These kinds of surfactants are sensitive toward hydrolysis, especially in alkaline conditions, while glucamides and glycosides are usually considered as rather stable in water solution (Holmberg, 2001; Söderman and Johansson, 2000). However, little is known about N-glycoside surfactants and their properties. The heterocyclic linkage between hydrophilic and hydrophobic moieties seems to be attractive for two reasons: First, heterocyclic azole rings such as 1,3,4-oxadiazole, 1,3,4-thiadiazole, 1,2,4-triazole and 4-amino-1,2,4-triazole are known to have a wide range of biological activity (Belkadi and Othman, 2006; Benhammadi et al., 2010; Datoussaid et al., 2012; Bradshaw et al., 1986). Second: these heterocyclic rings are readily synthesized from carboxylic acid groups found in naturally occurring fatty acids (El-Sayed and Grasas, 2006).
2 Results and discussion
2.1 Synthesis
Two parent compounds N-thioforamidosemicarbazide hexanoate (3) are considered as a starting material for the synthesis of three azole compounds 6–8. And n-pentyl hydrazide (5) might be considered the parent compound for the preparation of the heterocyclic rings, 9, 11 and 12, from n-hexanoic acid in two separate routes.
Compound 3 was achieved by refluxing acid 1 with thionyl chloride to give n-hexanoyl chloride 2. When 2 was treated with thiosemicarbazide in THF at room temperature it yielded the first parent compound 3 in 95% yields (Furniss et al., 1989). Characterization of 3 was by IR exhibiting bands at 3257 and 3119 cm−1 for NH and NH2, 1619 and 1182 cm−1for N–C⚌O and C⚌S groups respectively (Pretsh et al., 2000a,b, p. 158). 1H NMR showed signals at, 7.40 and 4.66 ppm for O⚌C–NH–N, N–NH–C⚌S and S⚌C–NH2 fragments.
Another route to obtain 3 consisted of three steps: Step 1, preparation of methyl ester 4 by literature method (Furniss et al., 1989). Step 2, refluxing 4 with hydrazine revealed into second parent compound namely n-pentyl hydrazide 5 which by heating with ammoniumthiocyanate yielded the first parent compound 3 in 65% yield, therefore the first route is considered more preferable. Compound 5 was characterized by IR which showed the band at 1626 cm−1 attributed to O⚌C–N bond and supported by 1H NMR which exhibited a signal at 8.90 ppm for hydrogen of O⚌C–NH fragment and signal at 4.13 ppm for NH2 group.
The amino-oxadiazole 6 was obtained by cyclization of 3 in the alkaline ethanolic solution in the presence of catalytic amounts of KI/I2 (George, 2008). Compound 6 was characterized by IR spectrum which showed absorptions at 3490, 1625 and 1112 cm−1 for NH, C⚌N and ⚌COC⚌, respectively. 1H NMR showed signals at 4.73 ppm for NH2 group. 13C NMR exhibited signals at 175.35 and 148.66 ppm attributed to carbons of the ring attached to the side chain.
Synthesis of 2-pentyl-5-amino-1,3,4-thiadiazole (7) was successful by cyclization of 3 preferentially by H3PO4 (Khanum et al., 2003) rather than H2SO4which gave less yield. Compound 7 was characterized by IR, which exhibited the following bands, 3405 and 1637 cm−1 for NH2 group and C⚌N–S of the ring respectively.
The triazole-thiol 8 was separated in 85% yield after refluxing thiosemicarbazide 3 with aqueous NaOH (Khiati et al., 2007). The IR spectrum showed a band at 1637 cm−1 attributed to C⚌N in the ring. 1H NMR exhibited signals at 10.49 ppm attributed to NH of the ring, and 6.09 ppm for SH (Pretsh et al., 2000a,b).
The other heterocyclic ring compounds 9, 11 and 12 were obtained from hydrazide 5. Compound 9 was prepared by refluxing 5 with CS2 in aqueous KOH solution leading to the isolation of 5-pentyl-1,3,4-oxadiazole-2-thione (9) (Khiati et al., 2007). 5-Pentyl-4N-amino-1,2,4-triazole-3-thiol (11) was obtained by refluxing hydrazide 5 and CS2 in n-butanol in the presence of KOH to give first the intermediate salt 10. 1The H NMR spectrum showed correctly all the hydrogen of n-pentyl chain with a particular difference in one H1 which was found in a slightly higher field at 3.20 ppm (see Section 3). The molecular model for compound 10 showed that the hydrogen concerned is situated in an isotropic field of C⚌S group (Othman and Al-Timary, 1980). Treatment of compound 10 with hydrazine yielded 11 in good yields. The IR spectrum showed the characteristic bands at 3378, 2911 and 1637 cm−1 for NH2, SH and C⚌N stretching respectively. 1H NMR, exhibited the proton signals at 5.25 and 3.87 ppm for exocyclic NH2 and SH groups and all protons of the side chain (see Section 3) (see Scheme 1).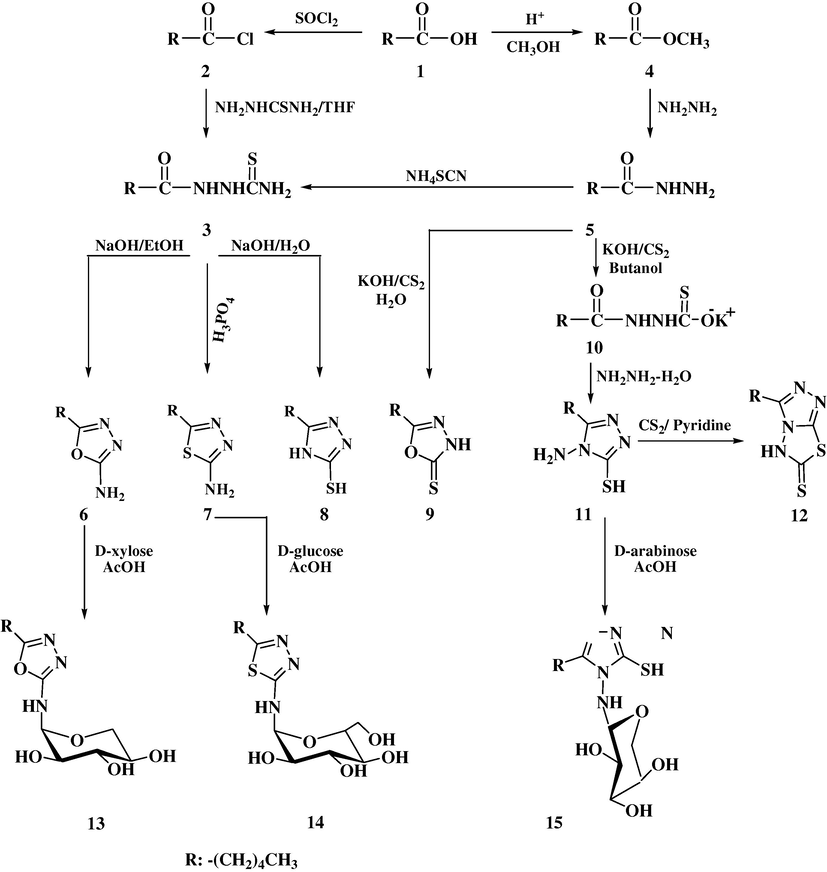
Synthetic pathways to n-pentyl azoles 6–9, 11, 12 and N-glycosides 13–15.
Compound 12 was isolated in 75% yield by heating amino triazole thiol 11 under reflux with CS2 in pyridine (Cacic et al., 2006).
The second target of the synthesis was to obtain the N-glycosides. Those had been achieved by refluxing each of 6, 7 and 11 with the sugars, d-xylose, d-glucose, and d-arabinose in acetic acid to yield the N-glycosides 13, 14 and 15, respectively (Joshi et al., 2008). Theoretically, the amino groups of oxadiazole 6, thiadiazole 7 and triazole 11 preliminarily formed Schiff bases with sugar molecules (El-Essawy and Rady, 2011) leading to ring closure as glyco-hexopyranoside and/or glyco-pentofuranoside(see Scheme 2). 1H and 13C NMR suggested that both possibilities existed.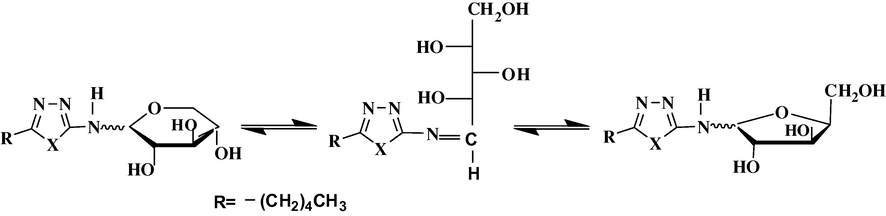
Fischer–Haworth equilibrium systems in sugar molecule.
For example, compound 13 showed an IR spectrum with the following bands, 3312 (broad), 1637 and 1046 cm−1 for bonded OH, C⚌N and C–O–C bonds. 1H NMR exhibited signals at 6.56, 4.77, 4.64 and 4.45 attributed for NH, 3OH groups respectively and two signals at 5.94 and 5.00 ppm attributed to anomeric protons of N-xylo-pentofuranoside and N-xylo-hexopyranoside 13 (Coffey, 1967). 13C NMR revealed the signals at 174.86 for O–C–N (⚌N) of oxadiazole ring, a signal at 92.57 ppm assigned for anomeric carbon of N-xylo-furanoside and at 97.24 ppm for N-xylo-hexopyranoside 13 (Pretsh et al., 2000a,b, p. 152) and all sugar carbons between 97.24 → 72.31 ppm and for side chain carbons between 29.25 → 14 ppm (see Section 3). No further work was done to separate these cyclic isomers and the enantiomers since it was reported that the anomeric configuration has very little effect on the air-water interfacial adsorption properties in sugar enantiomers (Silva et al., 2008). The N-glycoside 14 showed IR absorptions at 3477 (strong and broad), 1638 and 1054 cm−1 assigned for OH free and bonded, C⚌N and C–S–C bonds. 1H NMR exhibited signals at 6.64, 4.85, 4.90, and 4.60 attributed for NH and a multiplet between 6 → 4 ppm for OH groups. A similar phenomenon of a formation of a mixture of cyclic sugar rings had happened in 14. The third N-glycoside 15 was characterized by IR exhibited absorption bands at 3400 (strong and broad) assigned for OH groups and 1624 for C⚌N bond. 13C NMR revealed the following signals 148.86 and 102.00 ppm assigned for heterocyclic carbons, 98.00 → 62.00 ppm for sugar moiety and signals between 34.51 → 14.00 attributed for side chain carbons (see Section 3). However, compound 15 exhibited the same phenomenon of cyclic sugar isomers.
2.2 Surface active properties
The surface activity and related properties of the synthesized compounds have been performed in aqueous solution (1.0 wt.%, pH = 7) at 25 °C are shown in Table 1.
N°comp
Surface tension (dyne/cm)
Cloud point °C
CMC (mmol/l)
3
28
62
4.32
6
27
53
4.22
7
27
52
4.16
9
27
68
4.34
11
26
60
4.12
12
27
59
4.30
13
28
52
3.85
14
28
53
3.90
15
25
60
4.21
2.2.1 Surface and interfacial tension
All prepared compounds have exhibited pronounced surface and interfacial tension (see Table 1). The values are closer to each other since all compounds bear the same hydrophobic n-pentyl chain. The different heterocyclic moieties and n-glycosidic residues were responsible for the slight differences in other surfaces (cloud point and CMC) (El-Sayed and Grasas, 2008).
2.2.2 Cloud point
Since water solubility of nonionic surfactants varies inversely with temperature, all synthesized compounds showed high cloud points which gave performance in hot water (52–68 °C). This is a fact behind all synthesized compounds having a high commercial quality at room temperature (Wang et al., 2008).
2.2.3 Critical micelle concentration (CMC)
CMC had been calculated by Shinoda et al, 1961, while the CMC of the synthesized compounds were evaluated from the break points in the γ versus m experimental curves (Figs. 1 and 2) (Rauter et al., 2005; Boyd et al., 2000; Shinoda et al., 1961). A preliminary analysis of these plots shows that the data obtained from independent dilutions of the compounds tested are in good agreement and also no minimum is observed near the brake point in any of the plots, thus indicating the absence of hydrophobic impurities in the synthesized compounds. Table 1 shows that the surface activity, cloud points and CMC behavior for the tested compounds are close to each other. This overall behavior may be understood due to the effect of the hydrophobic moiety n-pentyl side chain which is present in all tested compounds. However, the slight differences in the values of surface tension, cloud point and CMC are due to the different heterocyclic and sugar residues.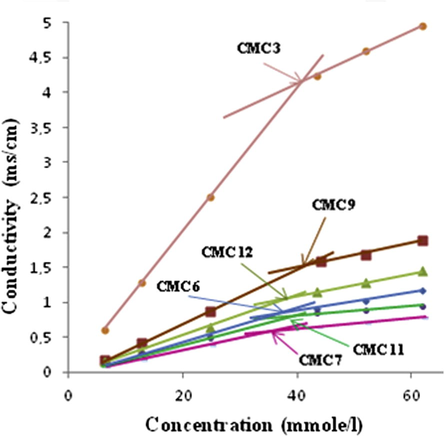
The brake points of thiosemicarbazide 3.
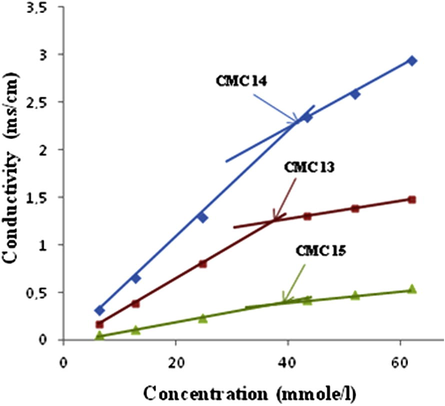
The brake points of N-glycosides 13–15.14 n-pentyl azoles 6, 7, 9, 11 and 12.
2.3 Antibacterial activity
n-Hexanoic acid 1 and its synthesized derivatives 2–15 (except 10) were screened for their activity against Gram-positive Bacilluscereus, Staphylococcus aureus and Gram-negative bacteria, Pseudomonas aeruginosa, Pseudomonas fluorescens and Escherichia coli. The antibacterial activities of the tested compounds were evaluated using the paper disk diffusion method. DMSO which is known as bacterial static in the above mentioned concentration was used as negative control and standard disks (Mast Diagnostics, UK), saturated with known antibiotic polymixine and oxytetracycline as positive control were applied. After incubation at 37 °C for 24 h, the zone of inhibition of growth around each disk was measured in millimeters and zone diameters were interpreted in accordance with CLSI and NCCLS (for Campylobacter spp.) guidelines (CLSI, 2006; NCCLS, 2003, 2005) (Dallal et al., 2010; Clinlcal and Laboratory Standards Institute, 2006; National Committee for Clinical and Laboratory Stand Villanova, 1997). The experiments were performed in duplicates and the average results are summarized in Table 2. The minimal inhibitory concentrations (MIC), 16, 8, 4 and 2 μg/mL data were determined for those compounds which demonstrated to be active in the preliminary paper disk tests. For microorganisms that were not affected by compounds tested, no further dilution tests were conducted. Highly active = (inhibition zone > 20 mm), moderately active = (inhibition zone 11–19 mm), slightly active = (inhibition zone 6–10 mm). Inactive = (inhibition zone < 6 mm); MIC a, b, c, d = 2, 4, 8, 16 μg/mL, ∗10%, v/v = 1/1, †: polymixine, ‡: oxytetracyclin.
Comps
P. aeruginosa
P. fluorescens
E. coli
B. cereus
S. aureus
Inhibition zone (mm)
Relative inhibition%
Inhibition zone (mm)
Relative inhibition%
Inhibition zone (mm)
Relative inhibition%
Inhibition zone (mm)
Relative inhibition%
Inhibition zone (mm)
Relative inhibition%
a
b
c
d
a
b
c
d
a
b
c
d
a
b
c
d
a
b
c
d
1
0
0
0
0
–
0
0
7
7
46.46
0
0
0
9
>100
0
0
0
9
75
0
0
6
10
47.61
2
0
0
8
11
45.83
0
0
0
0
–
0
0
0
0
–
0
0
0
0
–
0
0
0
0
–
3
0
0
16
29
>100
0
0
0
0
–
0
0
0
21
>100
0
0
0
0
–
0
0
0
0
–
4
0
6
7
7
26
0
0
0
0
–
0
6
6
6
85.71
6
7
7
7
58.83
0
0
0
0
–
5
0
0
7
8
33.33
0
6
7
8
53.33
0
0
0
0
–
7
7
7
24
>100
0
0
0
0
–
6
0
0
10
14
58.33
0
0
0
9
60
0
0
0
0
–
0
0
0
0
–
0
0
0
0
–
7
0
0
16
22
91.66
0
0
0
0
–
0
0
0
0
–
0
0
0
7
58.33
6
6
7
11
52.38
8
0
6
7
7
29.16
0
0
0
0
–
0
0
7
7
100
0
0
0
7
58.33
0
0
7
8
38
9
0
0
0
31
100
0
0
0
12
80
0
0
0
16
>100
0
0
7
9
75
0
0
0
0
–
11
0
18
23
32
>100
0
10
12
14
93.93
0
10
12
14
>100
0
0
9
10
83.33
0
14
14
17
81
12
10
12
23
30
>100
6
8
8
13
86.86
0
0
13
14
>100
0
0
9
9
75
9
9
11
16
76
13
0
0
0
0
–
6
6
6
6
40
0
0
0
9
100
0
0
0
9
75
0
0
6
10
47.61
14
0
0
7
7
29.16
18
20
27
32
>100
0
0
6
8
100
0
0
19
22
>100
8
9
18
18
100
15
0
0
0
0
–
17
24
30
36
>100
0
0
0
8
100
9
9
9
9
75
7
11
16
19
90.47
DMSO∗
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
†
24
100
15
100
12
100
7
100
16
100
‡
20
100
15
100
7
100
7
100
21
100
Compounds 11, 12, 14 and 15 have affected all tested Gram-positive and Gram-negative bacteria. The other compounds 1 → 10 and 13 showed selective affection on Gram-positive and Gram-negative microbes. Gram-positive B. cereus is affected by all compounds except 2 and 3 whereas S. aureus is unaffected by 2, 6 and 9.
Gram-negative P. aeruginosa was resistant to compounds 1, 13 and 15. P. fluorescens was unaffected by compounds 2, 3, 4, 7 and 8, while E. coli was resistant to compounds 2, 5, 6 and 7.
3 Experimental
3.1 General
All reactions were monitored by TLC, silica gel F254, made by Merck, Germany. The melting points were measured with a BÜCHI 540 melting point apparatus and are uncorrected. The IR spectra were recorded using KBr disks in a GENESIS II FTIR spectrophotometer, in units of cm−1. The 1H and 13C NMR spectra in 1d were recorded on a Bruker AC 400 MHz spectrometer (Université de Lyon 1, France) in DMSO-d6 and referenced to TMS. Coupling constants, J, are given in Hz, and chemical shifts in ppm. Surface tension was measured by Ring Tensiometer KG (Krusse), (Department of Physics, USTO). Conductivities were performed by digital conductivity meter Phywe 13701.93. Microorganisms in this study were supplied by the university hospital of Oran and identified by the laboratory of phytopathology, the department of Biology, University of Oran Es-Senia. The Mueller Hinton medium was supplied by (Difco).
3.2 Synthesis of compounds
3.2.1 n-Hexanoyl chloride 2
n-Haxanoic acid 1 (4.2 g, 0.036 mol) in chloroform (50 mL), SOCl2 (12 mL) in DMF added to it gradually and refluxed for 22 h. Solvents were removed under vacuum to give a brownish syrup 2 (3.78 g, 90%). IR (CCl4) ν (cm−1): 1780 (C⚌O), 960(C–Cl).
3.2.2 Methyl hexanoate 4
n-Hexanoic acid 1 (12 g, 0.0920 mol) in methanol (30 ml), H2SO4 (4 ml) was added dropwise with stirring for 8 h. Iced water (100 ml) was added at the end of the reaction with care. The aqueous mixture was extracted two times with dichloromethane (25 ml). The combined organic layers were washed with 5% aqueous NaHCO3 until neutralization, then washed with 50 ml of water, dried over anhydrous MgSO4 and filtered. The filtrate was evaporated to dryness to give colorless oil, methyl hexanoate 4 (11.5 g, yield 96%); IR (CCl4), ν (cm−1): 1733.69 (C⚌O), (lit) 1742.
3.3 Hexanoic hydrazide 5
Methyl hexanoate 4 (11 g, 0.0840 mol), ethanol (30 ml) and hydrazine hydrate 64% (12 ml) were refluxed for 10 h. Ethanol and hydrazine were evaporated under reduced pressure, white solid was produced, recrystallized from acetone/cyclohexane to give hexanoic hydrazide 5 (10.5 g, yield 95%). M.p. 69–71 °C, IR νmax/cm−1 (KBr) 3298 (NH, NH2), 1626 (N–C⚌O). 1H NMR, δ (ppm), 8.9 (s, 1H, NH), 4.13 (d, 2H, NH2), 2.0 (t, 2H, H1a, H1b), 1.48 (p, 2H, H2a, H2b), 1.25 (m, 4H, H3a, H3b and H4a, H4b), 0.85 (t, 3H, H5). 13C NMR, δ, ppm, 195.55 (C⚌O), 32.90 (C1, CH2), 32.25 (C2, CH2), 24.15 (C3, CH2), 23.48 (C4, CH2), 14.00 (C5, CH3).
3.4 N-thioforamidosemicarbazide hexanoate 3
3.4.1 First method
n-Hexanoyl chloride 2 (0.7 g, 0.0052 mol) was dissolved in THF (15 ml), thiosemicarbazide (1.80 g) was added and the mixture was stirred for 16 h at room temperature. Aqueous NaHCO3 solution was added dropwise until no further precipitation formed solid was filtered off and washed with cold THF then recrystallized from ethanol to give 3 as bright crystalline (0.66 g, 95%). IR νmax/cm−1 (KBr) νmax (cm−1), 3236 (NH), 1637 (C⚌N), 1402 (C⚌S). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 10.49 (s, 1H, NH); 6.09 (s, 1H, SH); 3.01 (t, 2H, H-1) 2.77 (m, 2H, H-2,); 1.61 (m, 2H, H-3); 1.35 (m, 2H, H-4); 0.90 (t, 3H, H-5). 13C NMR, δ, ppm, 195 (C⚌S), 162.80 (C⚌O), 32.90 (C1, CH2), 32.90 (C2, CH2), 24.65 (C3, CH2), 23.90 (C4, CH2), 14.25 (C5, CH3).
3.4.2 Second method
Hexanoic hydrazide 5 (1 g, 0.0077 mol) was dissolved in n-butanol (20 ml) with stirring. Ammonium thiocyanate (0.58 g) and HCl (30%) were added, reaction mixture was refluxed for 25 h. Excess solvent was evaporated to almost dryness and recrystallized from methanol/petroleum ether to give 3 (1.20 g, 75%), m.p. 180 °C; IR νmax/cm−1 (KBr) 3173 (NH, NH2), 1618 (CO–N), 1118 (C⚌S).
3.5 5-Pentyl-2-amino-1,3,4-oxadiazole 6
Thiosemicarbazide hexanoate 3 (1 g, 0.0035 mol) in (6 ml) of ethanol was added to a solution of (5 N) NaOH with cooling and stirring. To this clear solution was added a solution of KI/I2 until a permanent tinge color of iodine persisted at room temperature. The mixture was immediately refluxed, and more KI/I2 solution was added until the permanent tinge color of iodine remained at the higher temperature. The solution was then cooled and poured into ice-cold water. The solid that separated was collected by filtration and was then washed with distilled water, with dilute thiosulfate solution, and again with distilled water before being dried. The crude compounds were recrystallized from ethanol to give yellowish-brown crystals 6 (1.26 g, 60%), m.p. (218–220), IR (KBr) ν (cm−1): 3490 (NH2, NH); 1625 (C⚌N); 1112 (⚌C–O–C⚌). H1 NMR(400 MHZ, DMSO d6) σ (ppm): 4.73 (d, 2H, NH2); 2.20 (t, 2H, H-1); 1.47 (m, 2H, H-3, H-4); 0.84 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 175.35 (C, NCON); 148.66 (C, CON); 34.51 (C1, CH2); 33.14 (C2, CH2); 25.33 (C3, CH2); 22.94 (C4, CH2); 14.79 (C5, CH3).
3.6 5-Pentyl-2-amino-1,3,4-thiadiazole 7
Thiosemicarbazide hexanoic 3 (1 g, 0.0035 mol) was added gradually under stirring to cooled concentrated sulfuric acid (30 ml) for 1 h. the reaction mixture was further stirred for another 15 h in an ice bath. It was then poured over crushed ice under stirring. The solid precipitated was filtered, washed with water, dried and recrystallized with ethanol to give 7 (0.3 g, 40%), m.p. (202 °C). IR (KBr) ν (cm−1): 3405 (NH2); 1637 (C⚌N–S). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 6.45 (d, 2H, NH2); 2.30 (t, 2H, H-1); 1.30 (m, 2H, H-3, H-4); 0.89 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 132.15 (C, CSN); 31.80 (C1, CH2); 31.70 (C2, CH2); 22.93 (C3, CH2); 22.84 (C4, CH2); 13.60 (C5, CH3).
3.7 5-Pentyl-4H-1,2,4-triazole-3-thiol 8
Thiosemicarbazide 3 (1 g, 0.0035 mol) in 15 ml of water was added to a solution of 10.0% NaOH (20 ml), and the reaction mixture was refluxed immediately for 25 h on a boiling oil bath. The mixture was cooled and acidified with dilute HCl at pH 5–6. The resulting solid was filtered, washed with distilled water, and dried. The crude compounds were recrystallized from ethanol to give 8 (0.85 g, 85%), m.p. (290 °C). IR (KBr) ν max (cm−1): 3236 (NH), 1637 (C⚌N), 1402 (C⚌S). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 10.49 (s, 1H, NH); 6.09 (s, 1H, SH); 3.01 (t, 2H, H-1); 2.77 (m, 2H, H-2); 1.61 (m, 2H, H-3); 1.35 (m, 2H, H-4); 0.90 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 158.25 (C, NCN–S); 142.50 (C, NCN); 31.90 (C1, CH2); 30.15 (C2, CH2); 23.90 (C3, CH2); 23.50 (C4, CH2); 14.25 (C5, CH3).
3.8 5-Pentyl-1,3,4-oxadiazole-2-thione 9
Hexanoic hydrazide 5 (1 g, 0.0077 mol) in water (80 ml) was added to a solution of CS2 (30 ml) and (1.2 g) of KOH, and the mixture was refluxed for 36 h until the release of H2S was ceased. The mixture was then cooled and acidified with dilute HCl. The solid mass that separated was collected and was filtered, washed with ethyl acetate, and dried. The crude compounds were recrystallized from chloroform to give 9 (1.26 g, 60%), m.p. (218–220), IR (KBr) ν (cm−1): 3490 (NH2, NH); 1625 (C⚌N); 1112 (⚌C–O–C⚌). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 4.73 (d, 2H, NH2); 2.20 (t, 2H, H-1); 1.47 (m, 2H, H-3, H-4); 0.84 (t, 3H, H5), C13 NMR (400 MHZ, DMSO d6) σ (ppm): 175.35 (C, NCON); 148.66 (C, CON); 34.51 (C1, CH2); 33.14 (C2, CH2); 25.33 (C3, CH2); 22.94 (C4, CH2); 14.79 (C5, CH3).
3.9 Hexanoyl-potassium thiocarbazinic acid 10
Hexanoic hydrazide 5 (1 g, 0.0077 mol) was dissolved in n-butanol, alcoholic solution of KOH (1.23 g) in n-butanol and CS2 (1.80 g) were added dropwise. The mixture was stirred for 70 h at room temperature. Ether (20 ml) was added and a brownish precipitate was formed. The product was filtered, washed twice with ether, dried at room temperature to give a solid mass which was dissolved partially in warm ethyl acetate and filtered off. The filtrate was evaporated down to almost dryness to give a solid crystalline, recrystallized from chloroform/ethanol (1/1) to give the pure 10 (2.1 g, yield 70%), m.p. 200–208 °C. IR νmax/cm−1 (KBr) 3410, 3227 (NH), 1631 (N–C⚌O), 1590 (C⚌O) and 1261 (C⚌S). 1H NMR, δ, ppm, 4.19 (d, 2H, 2xNH), 3.2 (t, 2H, H1a), 2.28 (t, 2H, H1b), 1.55 (p, 2H, H3), 1.29 (m, 2H, H4), 0.89 (t, 3H, H5). 13C NMR, δ, ppm, 148.55 (N–C⚌O), 71.14 (C⚌S), 42.00 (C1, CH2), 37 (C2, CH2), 31.55 (C3, CH2), 19.48 (C4, CH2), 14.67 (C5, CH3).
3.10 5-Pentyl-4-amino-1,2,4-triazole-3-thiol 11
Hexanoyl- potassium thiocarbazinic acid 10 (0.5 g, 0.0038 mol) dissolved in water (4 ml) and hydrazine hydrate 64% (2 ml) was heated under reflux on an oil bath at 120 °C for 22 h. The reaction mixture was cooled to room temperature, iced-water was added and the solution was made acidic with 10% HCl. A precipitate formed and was filtered off. The filtrate was extracted two times with 20 ml of ethyl acetate. The extracts were combined and dried over anhydrous Na2SO4. Excess solvent was evaporated to almost dryness and recrystallized from ethanol/water to give 11 (0.30 g, 65%), m.p. (270 °C). IR (KBr) ν cm−1: 3376 (NH2, NH), 2911 (SH), 1637 (C⚌N). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 5.25 (d, 2H, NH2); 3.87 (s, 1H, SH); 2.69 (t, 2H, 1); 1.62 (p, 2H, H-2); 1.30 (m, 2H, H-3); 0.95 (p, 2H, H-4); 0.88 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 168.25 (C, NCN-S); 146.30 (C, NCN); 33.90 (C1, CH2); 31.50 (C2, CH2); 24.80 (C3, CH2); 23.90 (C4, CH2); 14.75 (C5, CH3).
3.11 3-Pentyl-1,2,4-triazole (3,4,b)(1,3,4) thiadiazole-5-thione 12
Amino triazole 11 (1.00 g, 0.0048 mol), CS2 (0.36 g, 0.0048 mol) and pyridine (9.0 mL) were refluxed for 21 h on an oil bath at 120 °C. Solid was isolated after cooling, filtered and recrystallized from ethanol to give 12 as crystalline (1.02 g, 75%), m.p. (210 °C). IR νmax/cm−1 (KBr), 3421, 3065 (NH), 1636 (C⚌N), 1271 (C⚌S) and 705 (C–S–C). IR (KBr) ν (cm−1): 3421, 3065 (NH), 2064 (N–C⚌S); 1636 (C⚌N); 1271 (C⚌S); 705 (C–S–C). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 7.61 (s, 1H, NH); 2.79 (t, 2H, 1); 1.67 (p, 2H, H-2); 1.34 (m, 2H, H-3); 1.02 (p, 2H, H-4); 0.87 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 182.15 (C, NCSS); 169.14 (C, NCNS); 129.65 (NCN); 33.94 (C1, CH2); 32.75 (C2, CH2); 23.15 (C3, CH2); 22.89 (C4, CH2); 14.20 (C5, CH3).
3.12 2-Pentyl-5-amino-xyloyl-1,2,4-oxadiazole 13
A solution of 6 (2.37 g, 0.01 mol) in methanol (25 ml) was added to d-xylose (1.8 g, 0.01 mol) in water (2 ml) and few drops of acetic acid. The mixture was heated under reflux for 10 h. Excess solvent was evaporated to almost dryness to give colorless oil 13 (1.06 g, 45%). IR (KBr) ν (cm−1): 3212 (OH); 1637 (C⚌N); 1046 (C–O–C). 1H NMR (400 MHZ, DMSO d6) σ (ppm) 6.56 (s, 2H, NH); 6.20 (s, 1H, H′1); 4.90 (s, 1H, H″-1); 4.91 (s, 1H, H″′-1); 4.77 (m, 1H, OH–C2′); 4.64 (m, 1H, OH–C3′), 4.45 (m, 1H, OH–C4′); 3.02 (m, 1H, OH–C5′), 4.85 (m, 1H, H–C2′); 4.37 (m, 1H, H–C3′); 4.29 (m, 1H, H–C4′); 3.60 (m, 1H, H–C′5); 3.46 (m, 1H, H–C5′); 2.45 (m, 2H, H-2); 2.25 (t, 2H, H-1); 1.50 (m, 4H, H-3,4); 0.89 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 14.31 (C5); 22.45 (C4); 24.84 (C3); 29.09 (C2); 29.25 (C1); 70.93 (C5′); 72.31 (C2′); 73.42 (C4′); 77.09 (C3′); 92.57 (C5f); 97.24 (C5p); 174.86 (C–O–N).
3.13 2-Pentyl-5-amino-d-glycoyl-1,3,4-thiadiazole 14
A solution of 7 (2.37 g, 0.01 mol) in methanol (25 ml) was added to d-glucose (1.8 g, 0.01) in water (2 ml) and few drops of acetic acid. The mixture was heated under reflux for 8 h. Excess solvent was evaporated to almost dryness to give colorless oil 14 (1.06 g, 45%). IR (KBr) ν (cm−1): 3422 (OH); 1638 (C⚌N–S), 1054 (C–S–C). H1 NMR (400 MHZ, DMSO d6) σ (ppm): 6.54 (s, 2H, NH); 6.20 (s, 1H, H′1); 4.85 (s, 1H, H″-1); 4.90 (s, 1H, H′″-1); 4.70 (m, 1H, OH–C2′); 4.60 (m, 1H, OH–C3′), 4.38 (m, 1H, OH–C4′); 3,02 (m, 1H, OH–C5′), 4.80 (m, 1H, H–C2′); 4.25 (m, 1H, H–C3′); 4.20 (m, 1H, H–C4′); 3.58 (m, 1H, H–C′5); 3.44 (m, 1H, H–C5′); 2,45 (m, 2H, H-2); 2.20 (t, 2H, H-1); 1.48 (m, 4H, H-3,4); 0,90 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 14.43 (C5); 22.41 (C4); 24.78 (C3); 29.00 (C2); 29.15 (C1); 70.88 (C5′); 72.38 (C2′); 73.47 (C4′); 77.10 (C3′); 92.00 (C5f); 97.15 (C5p); 134.76 (C–S–N).
3.14 3-Pentyl-4-amino-arabinoyl-1,2,4-triazole-5-thiol 15
A solution of 11 (2.37 g, 0.01 mol) in methanol (25 ml) was added to d-arabinose (1.8 g, 0.01 mol) in water (2 ml) and few drops of acetic acid. The mixture was heated under reflux for 10 h. Excess solvent was evaporated to almost dryness to give colorless oil 15 (1.06 g, 45%).IR (KBr) ν (cm−1): 3400 (OH); 1624 (C⚌N). 1H NMR (400 MHZ, DMSO d6) σ (ppm): 9.00 (s, 2H, NH); 6.00 (s, 1H, H′1); 5.16 (s, 1H, H″-1); 5.07 (s, 1H, H″′-1); 4.99 (m, 1H, OH–C2′); 4.91 (m, 1H, OH–C3′), 4.89 (m, 1H, OH–C4′); 4.72 (m, 1H, OH–C5′), 4.66 (m, 1H, H–C2′); 4.33 (m, 1H, H–C3′); 4.19 (m, 1H, H–C4′); 3.66 (m, 1H, H–C′5); 3.49 (m, 1H, H–C5′); 2.48 (m, 2H, H-2); 1.95 (t, 2H, H-1); 1.34 (m, 4H, H-3,4); 1.09 (t, 3H, H-5). C13 NMR (400 MHZ, DMSO d6) σ (ppm): 14 (C5); 22.94 (C4); 25 (C3); 33 (C2); 34.51 (C1); 62 (C5′); 72.31 (C2′); 73.42 (C4′); 93 (C3′); 98 (C5f); 102 (C5p); 148.86 (C–N–C).
Acknowledgments
Thanks are due to Mr. N. Karkachi and Miss. F. Ghomari of the Laboratory of Phytopathology, Faculty of Science, University of Oran, Essenia, Oran, for antibacterial tests. We also thank Mrs. O. Lantz and Mrs. I. Baunammour of University of Lyon 1, Grenoble, France, for recording 1H and 13C NMR measurements.
References
- Arkivok. 2006;xi:183-195.
- Asian J. Chem.. 2010;22(7):5535-5542.
- Langmuir. 2000;16:7359-7367.
- J. Heterocycl. Chem.. 1986;23:361-368.
- Molecules. 2006;11:134-147.
- Performance Standards for Antimicrobia Susceptibly Testing; Sixteenth International Supplement. 2006;26:11-35.
- Coffey S., ed. Rodd’s Chemistry of Organic Compounds. Vol vol. 1 (F). Amsterdam: Elsevier Publishing Company; 1967. p. :162-164.
- Nonionic Surfactants. Vol vol. 19. New York: Marcel Dekker; 1987.
- Food Control. 2010;21:388-392.
- Afr. J .Chem.. 2012;65:30-35.
- Chem. Heterocycl. Compd.. 2011;47(4):497-506. (Russian original vol. 47, No. 4, April, 2011)
- Grasas Aceites. 2006;57(2):180-188.
- Grasas Aceites. 2008;59(2):110-120.
- Vogel’s Text Book of Practical Organic Chemistry (5th ed.). New York: Longman Scientific & Technical; 1989. p. 647
- J. Cell Biol.. 1980;86:327-329.
- Acta Pharm. 2008:119-129.
- Fett-Lipid. 1999;201:25-33.
- Curr. Opin. Colloid Interface Sci.. 2001;6:148-159.
- Holmberg K., ed. Novel Surfactants. Preparation, Applications and Biodegradability.Holmberg K., ed. Surfactant Science Series. Vol vol. 114. New York: Marcel Dekker; 2003. p. :1-192.
- Eur. J. Med. Chem.. 2008;43:1989-1996.
- Sci. Asia.. 2003;29:383-392.
- S. Afr. J. Chem.. 2007;60:20-24.
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically (4th ed). 1997.
- L’Actualité Chimique. 2002;17(12):70-75.
- Tetrahedron. 1980;36:753-758.
- Structure Determination of Organic Compounds – Tables of Spectral Data. Berlin Heidelberg: Springer-Verlage; 2000. p. 158
- Structure Determination of Organic Compounds – Tables of Spectral Data. Berlin Heidelberg: Springer-Verlage; 2000. p. 152
- Carbohydr. Res.. 2005;340:191-201.
- Angers. Chem. Int. Ed.. 1998;37:1328-1345.
- Bull. Chem. Soc. Jpn.. 1961;34:237-241.
- Bioorg. Med. Chem.. 2008;16:4083-4092.
- Curr. Opin. Colloid Interface Sci.. 2000;4:391-440.
- Curr. Opin. Colloid Interface Sci.. 2001;6:160-170.
- Colloids Surf. B. 2008;61:118-122.




