

Translate this page into:
Synthesis of novel xanthone and acridone carboxamides with potent antiproliferative activities
⁎Corresponding author. ikkostakis@pharm.uoa.gr (Ioannis K. Kostakis)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Several new amino-substituted acridone and xanthone derivatives have been designed and synthesized, using an efficient methodology from suitable acridone- or xanthone-carboxylic acid intermediates. The antiproliferative activity of the target compounds has been evaluated against four cancer cell lines, namely breast adenocarcinoma MCF-7, acute lymphocytic leukemia CCRF-CEM, and its doxorubicin-resistant variant CEM/ADR5000 and prostate cancer PC-3 cell lines. Selected derivatives have also been tested against the urinary bladder T24 and metastatic melanoma WM266-4 cancer cell lines. Two nitro substituted acridones, bearing a basic side chain as well, were endowed with a remarkable profile against the majority of the cell lines tested, with IC50 values in the low micromolar range. Both compounds cause accumulation at G0/G1 phase, induce apoptosis, and act as potent autophagy inhibitors in PC-3 cells, suggesting their further evaluation in various pathophysiological environments, conditions, and regimens.
Keywords
Acridone
Xanthone
Antiproliferative activity
Cell cycle arrest
Autophagy

1 Introduction
Among the many different classes of chemotherapeutic agents used against malignant diseases, acridone and xanthone derivatives have been extensively studied as anticancer compounds. Furthermore, both scaffolds, xanthone and its aza-analogue acridone, have been used as lead compounds for the synthesis of various analogues, which are endowed with a number of interesting, albeit diverse, biological properties, such as anti-inflammatory (Chen et al., 2002; Feng et al., 2020), anti-viral (Tonelli et al., 2011; Bernal and Coy-Barrera, 2015), anti-allergic (Chukaew et al., 2008; Shagufta and Ahmad, 2016), anti-malarial (Kumar et al., 2009; Tomar et al., 2010; Yu et al., 2012) and anti-parasitic (Shagufta and Ahmad, 2016) functions. They are characterized by a π-conjugated planar structure, which makes them considerably hydrophobic and allows their interaction with different bio-molecular targets. Due to this unique planar system, they intercalate in between base pairs in the double-stranded DNA, thereby inhibiting DNA replication in the rapidly growing cancer cells (Chiron and Galy, 2004; Demeunynck, 2004; Belmont et al., 2007; Belmont and Dorange, 2008; Kaur and Singh, 2011; Zhang et al., 2014; Shagufta and Ahmad, 2016). It is well established that various acridone and xanthone analogues exert their potent anticancer activities, through inhibition, among others, of topoisomerases, telomerase and cyclin-dependent kinases. The effectiveness of bioactive acridone derivatives is affected by their ability to exist in two tautomeric forms. The heterocyclic nitrogen adopts either an acceptor or a donor conformation, which has a radical effect on the binding properties of the molecule.
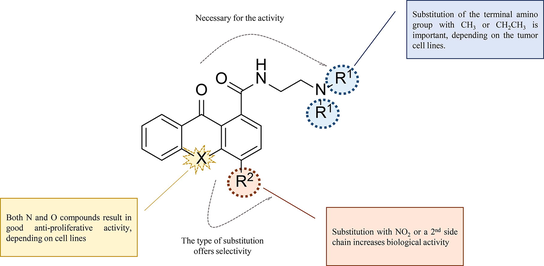
The most representative examples in this field include pyrazoloacridines (Zalupski et al., 1998; Bu et al., 2002; Ramaswamy et al., 2011), thiadiazinoacridines (Antonini et al., 2003), triazoloacridones (Lemke et al., 2004, 2005), imidazoloacridones (Mazerska et al., 2003; den Brok et al., 2005), pyrazoloxanthones (Kostakis et al., 2005, 2006), and aminoxanthones (Kostakis et al., 2005). Among them, PZA, an amino substituted 5-nitropyrazolo[3,4,5-kl]acridine derivative (Zalupski et al., 1998; Ramaswamy et al., 2011), imidazoloacridine C-1311 (Cholody et al., 1990; Wiśniewska et al., 2007), acridine carboxamide DACA (Atwell et al., 1987; Wolf et al., 2009) and acronycine (Elomri et al., 1996; Guilbaud et al., 2002) are of great biological interest (Fig. 1); hence, many analogues of them have been synthesized. Following extensive SAR studies in this class of compounds, it has been proposed that the substitution with one or two basic side chains, a 7-methoxyl, or an electron-deficient nitro-group, play important role in their activity and selectivity.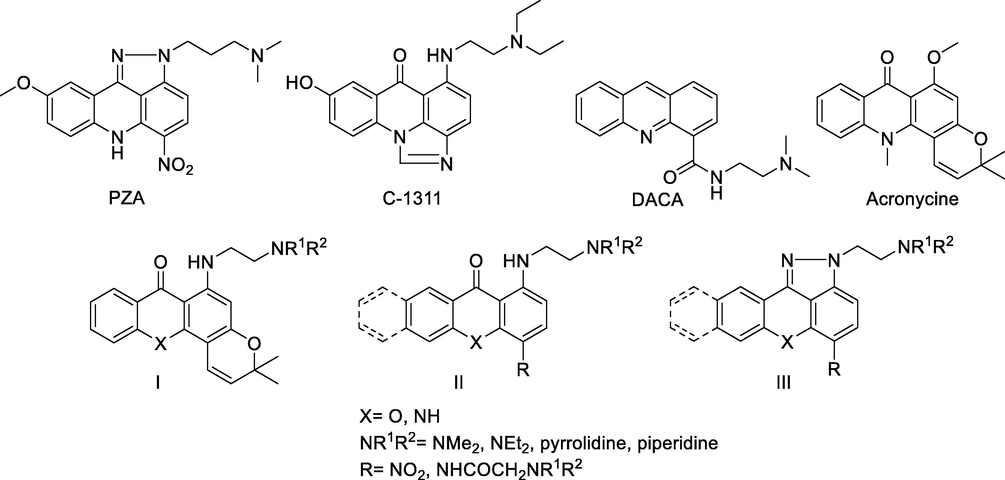
Structures of PZA, C-1311, DACA, acronycine, and compounds prepared by our group (I – III).
As part of an extensive research program concerning the synthesis and pharmacological evaluation of acridone and xanthone analogues (Kostakis et al., 2002, 2005, 2006, 2008; Giannouli et al., 2007, 2015), we have recently reported the synthesis of several amino-substituted xanthones and acridones (general formulas I–III, Fig. 1). These compounds share common structural features with the biologically active derivatives described above, bearing at least one basic side chain together with a nitro group, or a second amino substituted side chain. They exhibit strong cytotoxic activities against a panel of cancer cell lines, probably due to DNA binding and intercalation.
Prompted by these results, we, next, decided to examine these two scaffolds further and we present herein the synthesis and biological evaluation of two series of novel xanthone and acridone carboxamides, with direct similarity to PZA and other amino substituted acridones. In these derivatives, a carbonyl group has been inserted between the amino side chain and the chromophore, in order to gain a better insight of the structure–activity relationships regarding: a) the influence of the altered flexibility and spatial arrangement of the amide side chain, b) the effect of an additional substitution with either a nitro group, or a second basic side chain, as well as an α,β-unsaturated side chain, which could probably be susceptible to nucleophilic attack from DNA bases (i.e. guanine’s N-7) and c) the contribution of the closely related acridone or xanthone core to the biological activity.
2 Results and discussion
2.1 Chemistry
The synthesis of the amino-substituted xanthones and acridones started from 4-nitro-9-oxo-9,10-dihydroacridine-1-carboxylic acid (8) (Scheme 1), and 4-nitro-9-oxo-9H-xanthene-1-carboxylic acid (17) (Scheme 2). For the synthesis of acridone carboxylic acid 8, we have used the nitro derivative 1 which was prepared from the commercially available methyl 4-aminobenzoate following a previously described procedure (Decodts et al., 1983; Knepper et al., 2006). Bromide 1 was then reacted with methyl anthranilate (3), in the presence of Pd[P(C6H5)3]4 as catalyst and Cs2CO3 as base, to yield the intermediate methyl ester 4. Saponification of compound 4 and ring closure of the intermediate dicarboxylic acid 5, upon treatment with concentrated H2SO4, provided the acridone carboxylic acid 8 (Burdeska et al., 1972). Unfortunately, the overall yield of this procedure was only 35%, taking also under consideration the difficult purification of the last step (42%); therefore, a slightly different synthetic pathway was examined. This involves the initial preparation of methyl ester 6, by reaction of bromide 1 with anthranilic acid (2). Acid 6 was then ring closed, upon treatment with a mixture of TFAA and TFA, and the resulting methyl ester 7 was saponified to afford the desired acid 8 (85%) in 65% overall yield.![Reagents and conditions: a) c. H2SO4, abs. EtOH, reflux; b) Cs2CO3, Pd[P(C6H5)3]4, toluene (or toluene-DMA (3/1) for 6), reflux; c) NaOH 15%, MeOH, rt; d) c. H2SO4, 90 °C; e) TFAA-TFA.](/content/184/2020/13/11/img/10.1016_j.arabjc.2020.09.025-fig3.png)
Reagents and conditions: a) c. H2SO4, abs. EtOH, reflux; b) Cs2CO3, Pd[P(C6H5)3]4, toluene (or toluene-DMA (3/1) for 6), reflux; c) NaOH 15%, MeOH, rt; d) c. H2SO4, 90 °C; e) TFAA-TFA.
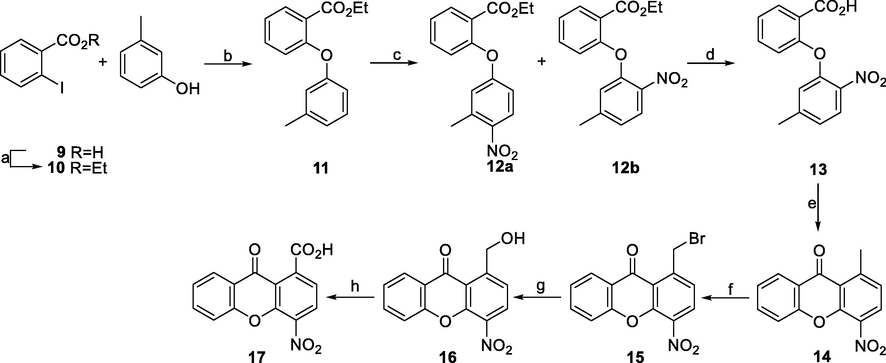
Reagents and conditions: a) c. H2SO4, abs. EtOH, reflux; b) CuCl, K2CO3, Pyr, reflux; c) fum. HNO3, 0 °C, rt; d) NaOH 40%, EtOH, rt; e) PPA, 110 °C; f) NBS, dibenzoyl peroxide, CCl4, UV (140 Watt); g) AgNO3, (CH3)2CO-H2O, rt; h) Jones reagent, (CH3)2CO, rt.
For the preparation of xanthone carboxylic acid 17, we followed a slightly different approach from the methodology previously reported from our team (Hadjipavlou et al., 2006). Thus, commercially available 2-iodobenzoic acid was first esterified to the corresponding ethyl ester 10 (Baker et al., 1965; Hadjipavlou et al., 2006) and then reacted with m-cresol, in the presence of cuprous chloride and potassium carbonate, to provide the ether 11 (Hadjipavlou et al., 2006) (Scheme 2). Treatment of compound 11 with fuming nitric acid, in the presence of acetic anhydride, resulted in a mixture of isomeric nitro compounds 12a and 12b. Structural discrimination of compounds 12a and 12b was unambiguously established by NMR spectroscopy using both direct and long-range experiments. The nitration site resulted from the observation that for compound 12b, two aromatic protons (namely H-4′ and H-6′, exhibit 3J coupling with the methyl group, while in the case of compound 12a, only H-2′ exhibits 3J coupling with the methyl group. Then, ester 12b was saponified under mild conditions, and the resulting carboxylic acid 13 was ring closed upon treatment with PPA providing xanthone 14 (Recanatini et al., 2001). Xanthone 14 was then brominated with NBS, in the presence of a catalytic amount of dibenzoyl peroxide applying UV irradiation, to give the bromomethyl analogue 15 (Recanatini et al., 2001), which upon hydrolysis in the presence of AgNO3, provided the benzyl alcohol 16. The target carboxylic acid 17 was then obtained from xanthone 16 upon oxidation with Jones reagent.
The nitro derivatives 18a-b and 24a-b were prepared upon amidation of acridone acid 8 and xanthone acid 17 respectively, with the appropriately substituted amines (Scheme 3). The 1H NMR spectra of compounds 18a-b display a characteristic doublet in the range of 8.15–8.28 ppm, integrating for 1 proton, attributed to H-8. On the other hand, H-3 appears downfield as a doublet at 8.44–8.60 ppm, while H-2 shifts upfield at 7.04–7.14 ppm. Next, the nitro group was reduced by hydrogenation over palladium on activated carbon, to afford the anilines 19a-b and 25a-b, which upon treatment with chloracetyl chloride afforded the amides 20a-b and 26a-b. Finally, reaction of these amides with the suitable amines resulted in the target diamines 21a-b and 27a-b. By analogy to compounds 18a-b, the 1H NMR spectrum of 21a-b shows a doublet at 7.82–7.90 ppm for H-8. The most characteristic H-3 appears as a doublet at 7.30–7.36 ppm while the proton at position 2 observed as a doublet at 6.68–6.70 ppm.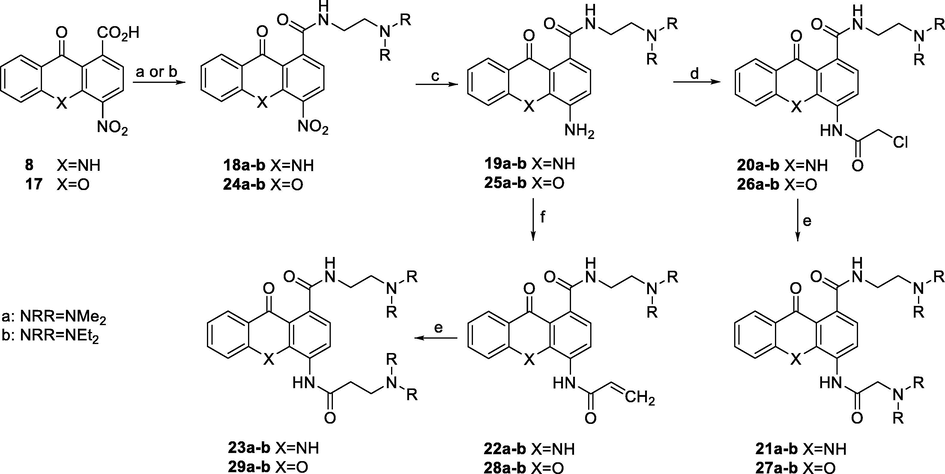
Reagents and conditions: a) 1. CDI, dry DMF, rt; 2. suitable diamine, 50 °C; b) 1. NBS, PPh3, dry CH2Cl2, 0 °C; 2. suitable diamine, rt; c) H2, Pd/C, 50 psi, EtOH abs., rt; d) K2CO3, chloroacetyl chloride, dry CH2Cl2; e) suitable secondary amine, EtOH abs., 60 °C; f) 3-chloropropionyl chloride, K2CO3, dry CH2Cl2, rt.
For the synthesis of the acrylamides 22a-b and 28a-b, and the diamines 23a-b and 29a-b, we followed a similar strategy. Therefore, anilines 19a-b and 25a-b were converted to the corresponding acrylamides 22a-b and 28a-b respectively, by treatment with 3-chloropropionyl chloride, in the presence of potassium carbonate. Finally, the aforementioned acrylamides, underwent 1,4-Michael’s addition using suitable secondary amines to provide the target diamines 23a-b and 29a-b.
2.2 Biological activity
All acridone and xanthone analogues were evaluated for their antiproliferative activities against four human cancer cell lines, namely, MCF-7 (breast adenocarcinoma) (Seo et al., 2015), CCRF-CEM and its variant CEM/ADR5000, a doxorubicin-resistant sub-clone (acute lymphocytic leukemia: ALL) (Kimmig et al., 1990; Efferth et al., 2008; Kadioglu et al., 2016) and PC-3 (prostate cancer) (Vistica et al., 1991). For comparative reasons and in order to further evaluate the activity of the new compounds, four derivatives which emerged from the initial screening, namely 18a, 18b, 28b and 29b, were also examined against two additional human cancer cell lines, the T24 (urinary bladder cancer) (Giannopoulou et al., 2019b) and the MW266-4 (metastatic melanoma / skin cancer) (Giannopoulou et al., 2019a), which are being typified by high malignancy grades and strong metastatic capacities (Table 1). Furthermore, for compounds 18a and 18b flow cytometric analysis of DNA content was also conducted.
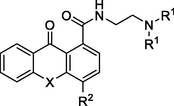
Cmp
X
R1
R2
MCF-7 %MCVa (30 µM)
CCRF/ CEM %MCVa (10 µM)
CEM/ ADR5000 %MCVa (10 µM)
PC3 IC50b (µM)
T24 %MCVc
MW266-4 %MCVc
18a
NH
CH3
NO2
82.08 ± 0.83
58.92 ± 3.42
86.63 ± 3.38
1.20 ± 0.06
50.80 ± 1.30 (50 µM) 37.40 ± 2.37 (100 µM)
52.00 ± 2.14 (50 µM) 26.50 ± 0.82 (100 µM)
18b
NH
CH2CH3
NO2
73.84 ± 1.40
56.67 ± 2.32
87.94 ± 1.85
2.00 ± 0.05
56.00 ± 0.90 (50 µM) 40.50 ± 1.03 (100 µM)
54.60 ± 0.77 (50 µM) 29.20 ± 1.09 (100 µM)
21a
NH
CH3
NHCOCH2N(CH3)2
86.42 ± 1.44
84.32 ± 3.48
84.27 ± 2.91
>40
21b
NH
CH2CH3
NHCOCH2N(CH2CH3)2
86.03 ± 1.19
86.71 ± 1.79
103.09 ± 1.82
40.00 ± 1.72
22a
NH
CH3
NHCOCH = CH2
85.06 ± 2.04
80.40 ± 2.18
92.68 ± 2.86
>40
22b
NH
CH2CH3
NHCOCH = CH2
82.87 ± 1.17
84.56 ± 2.53
92.42 ± 2.36
>40
23a
NH
CH3
NHCOCH2CH2N(CH3)2
84.60 ± 1.64
87.77 ± 2.07
96.29 ± 3.14
>40
23b
NH
CH2CH3
NHCOCH2CH2N(CH2CH3)2
85.43 ± 1.50
87.94 ± 1.61
94.20 ± 2.95
>40
24a
O
CH3
NO2
84.56 ± 1.23
75.79 ± 2.32
88.84 ± 2.31
24.20 ± 0.91
24b
O
CH2CH3
NO2
86.41 ± 1.35
75.37 ± 1.92
89.46 ± 0.78
33.30 ± 1.20
27a
O
CH3
NHCOCH2N(CH3)2
83.76 ± 2.31
75.72 ± 3.84
86.17 ± 3.03
14.30 ± 0.41
27b
O
CH2CH3
NHCOCH2N(CH2CH3)2
81.33 ± 1.16
73.54 ± 2.85
83.45 ± 3.80
15.30 ± 0.52
28a
O
CH3
NHCOCH = CH2
83.84 ± 1.17
75.07 ± 2.57
82.02 ± 2.33
31.00 ± 0.94
28b
O
CH2CH3
NHCOCH = CH2
N/T
61.09 ± 3.38
59.43 ± 4.01
7.20 ± 0.23
60.60 ± 1.54 (50 µM) 14.40 ± 0.87 (100 µM)
2.20 ± 0.83 (50 µM) 3.00 ± 0.72 (100 µM)
29a
O
CH3
NHCOCH2CH2N(CH3)2
85.85 ± 0.83
66.58 ± 3.73
86.77 ± 2.32
18.30 ± 0.65
29b
O
CH2CH3
NHCOCH2CH2N(CH2CH3)2
85.10 ± 0.93
67.81 ± 1.95
85.72 ± 3.00
7.40 ± 0.23
82.40 ± 1.95 (50 µM) 77.90 ± 3.35 (100 µM)
96.20 ± 0.57 (50 µM) 38.60 ± 1.33 (100 µM)
5-FU (Fluorouracil)
3.10 ± 0.09
2.2.1 In vitro antiproliferative activity against MCF-7 cell line
The cytotoxicity of target compounds was tested using the Resazurin assay (O'Brien et al., 2000). The results, expressed as cell viability at 30 µM, can be visualized in Supplementary Figure S1. All the new compounds showed low activity and only the nitro-acridone 18b, displayed an interesting activity against MCF-7 cell line with 73.84%±1.40 cell viability. It has been reported that certain quinone type derivatives are also endowed with cytotoxic effects against this cell line and this could be marginally related to the activity of the compounds presented herein (Mollica et al., 2014).
2.2.2 In vitro antiproliferative activity against CCRF-CEM and CEM/ADR5000 cell lines
The new compounds were assayed in vitro for their antiproliferative activities against CCRF-CEM and CEM/ADR5000 cell lines using the Resazurin assay (O'Brien et al., 2000). Results, expressed as % mean cell viability at 10 µM, are summarized in Supplementary Figure S2. The majority of the new compounds show improved cytotoxic properties against the two ALL cell lines, compared to the MCF-7 one. The cytotoxic potency of xanthone analogues is slightly higher than the one of corresponding acridones. However, it is noticeable that acridones 18a and 18b, bearing a nitro substitution, are the most potent derivatives in this series and display mean cell viabilities of 58.92%±3.42 and 56.67%±2.32 respectively, in CCRF-CEM cell line. This is consistent with the observations concerning the MCF-7 cell line and unveils the beneficial effect of the nitro group in terms of cytotoxicity. Notably, from a direct comparison between the CCRF-CEM and CEM/ADR5000 respective responses to the administered compounds, it is clearly observed that the derivatives show comparable anticancer activities against both cell lines, with the exception of the nitro acridones 18a and 18b, which are less effective against the doxorubicin-resistant sub-clone CEM/ADR5000. These results strongly suggest the importance of the acridone core and the nitro substitution, thus, providing evidence regarding the favorable scaffold and substitution for the maximization of cytotoxic capacity of these classes of compounds.
It must be underlined that although the antiproliferative activity of the di-substituted xanthones 28a-b and 29a-b is moderate, their potency is enhanced, compared to the di-substituted acridones 22a-b and 23a-b. Compound 28b exhibits the highest cytotoxicity among the xanthone analogues, with mean cell viability 61.09%±3.38, followed by compounds 29a-b. Besides, compound 28b showed high cytotoxicity against the doxorubicin-resistant CEM/ADR5000 cell sub-clone, with an inhibitory activity of approximately 40%, at 10 µM. On the contrary, the acridone analogue 22b possesses negligible activity, whereas the most active derivative among the acridone analogues, 18b, possesses a remarkable inhibitory activity of approximately 44% against the parental CCRF/CEM cells and only 13% against the resistant sub-clone. This could suggest that the di-substituted xanthones possessing an additional side chain could partly overcome multi-drug resistance in an ALL cellular setting.
2.2.3 In vitro antiproliferative activity against PC-3 cell line
The antiproliferative activity of the new compounds was evaluated in vitro against the PC-3 human prostate cancer cell line. The results of the MTT dye reduction assay, expressed as 50% inhibitory concentrations (IC50) in µM, are depicted in Table 1. The majority of xanthone analogues show interesting anticancer activities, with IC50 values varying within the range of 7.20 to 33.3 µM. In general, the di-substituted compounds 27a-b, 28a-b, and 29a-b seem considerably more active when compared with the nitro substituted analogues 24a-b. These data indicate that the replacement of the nitro group with a second basic side chain affords higher cytotoxicity against prostate cancer cells.
On the contrary, we observe that the di-substituted acridone analogues 21a-b, 22a-b, and 23a-b, appear to be considerably less active when compared with the corresponding di-substituted xanthones 27a-b, 28a-b, and 29a-b. In consistence with the previous results, the nitro acridones 18a and 18b emerge as the most potent compounds, showing strong cytotoxicity with IC50 values of 1.20 and 2.00 µM, respectively. The data indicate that in the case of the acridone analogues, in contrast to the xanthone counterparts, the replacement of the nitro group significantly reduces the activity. This could probably be attributed to a different mechanism of action; nevertheless, the existence of the two tautomeric forms, which the acridone core can adopt, could have a radical effect on the activity. This issue remains to be clarified.
2.2.4 In vitro antiproliferative activity against T24 human urinary bladder cancer cell line and WM266-4 human metastatic melanoma cancer cell line
Through employment of an MTT-based protocol, a cancer-cell type-specific sensitivity to the four herein tested compounds is clearly revealed (see Supplementary Figures S3 and S4). An overall assessment typifies T24 (human) urinary bladder cancer cells (Stravopodis et al., 2009; Giannopoulou et al., 2019b) more refractory to each one of the four examined compounds, as compared to the WM266-4 (human) metastatic melanoma (skin cancer) cells (Giannopoulou et al., 2019a). Specifically, compounds 18a and 18b seem capable to significantly kill both T24 and WM266-4 cancer-cell types, at 50 and 100 µM compound doses, with T24 and WM266-4 retaining survival-percentage values of approximately 40% and 30%, respectively, at the highest compound concentration being added. Similarly, administration of compound 28b, with a dose of 50 or 100 µM, results in complete eradication of WM266-4 cell populations, with the highest compound concentration remarkably reducing T24 viability, albeit rather incompletely, since an approximate 15% of T24 cells remains unaffected in the presence of 100 µM of 28b. In contrast to 28b, compound 29b was effective against T24 or WM266-4 cells only in the highest concentration tested. Interestingly, although T24 are rather refractory to 100 µM of 29b, WM266-4 cells are presented prominently vulnerable to the cytotoxic power of 29b compound.
Since compound 29b carries surprisingly decreased killing activity against T24 and WM266-4 cancer cells, when compared to the vinyl substituted compound 28b, it could be suggested that the electrophilic character of 28b is in favor of enhanced cytotoxicity regarding the specific cell lines.
2.2.5 Flow cytometric analysis of DNA content
Cell-cycle perturbation and arrest of exponentially growing PC-3 cells with compounds 18a and 18b for 24, 48 and 72 h are given in Table 2. Both compounds caused accumulation of PC-3 cells at G0/G1 phase, reducing in parallel the percentage of cells at G2/M and S phase of the cycle (though the reduction of S phase was marginally non-significant).
Sample
(Compound)
Time
(h)
G0/G1
(%)
S
(%)
G2/M
(%)
Control
24
56.4
17.1
26.5
18a
24
60.3
16.1
23.6
18b
24
61.1
18
20.9
Control
48
59
17.2
23.8
18a
48
67.9
15.3
16.8
18b
48
70
13.3
16.7
Control
72
54.1
15
30.9
18a
72
60.5
12.3
27.2
18b
72
60.6
17.3
22.1
Nevertheless, both compounds were found to significantly induce apoptosis of PC-3 cells at 72 h, with 18b being comparatively the most potent. More specifically, 18b induced at 72 h treatment the mobilization of late apoptosis at an elevated 40% ratio, as compared to control cells, whereas 18a increased late apoptosis almost 10% at 72 h of exposure (Fig. 2).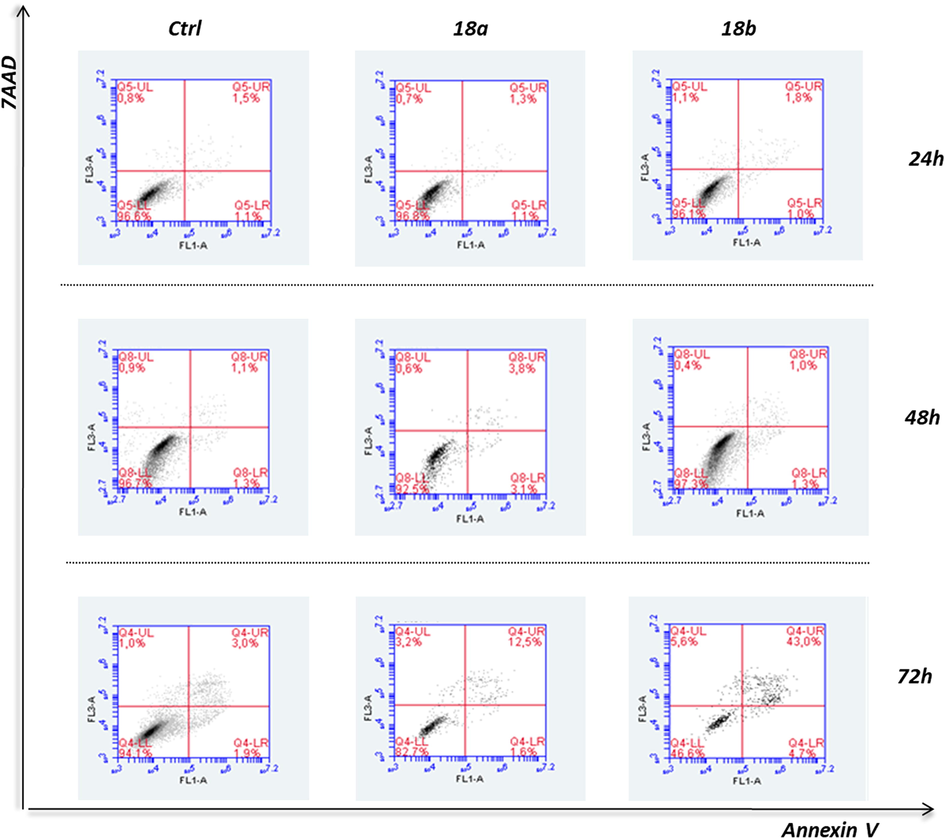
Flow cytometric analysis of DNA content. Early or late apoptosis and necrosis were estimated in PC-3 cells after 24-, 48- and 72-h exposure to the compounds 18a and 18b, versus control cells (Ctrl), based on the AnnexinV − 7AAD staining. Representative graphs illustrate the overall apoptosis activated upon each treatment, on an incubation-time basis.
The semi-quantification of autophagy engagement revealed that 18a and 18b showed a similar pattern compared to control cells, by decreasing the levels of CYTO ID fluorescence signal (ΔMFI = -0.230) and (ΔMFI = -0.235), respectively (Fig. 3). ΔMFI values for rapamycin (potent autophagy inducer) and chloroquine (potent autophagic flux inhibitor) were calculated as 0.800 and −0.420, respectively (Guo et al., 2011; Guo and White, 2013). This indicates that both 18a and 18b likely act as potent autophagy inhibitors in a PC-3 cellular environment (Fig. 3).![Representative flow cytometry graphs showing ΔMFI Cyto-ID after 48-hour treatment of the compounds 18a and 18b in PC-3 cells, in concentration equal to the corresponding IC50 values. [ΔMFI Cyto-ID = MFI Cyto-ID (treated) - MFI Cyto-ID (control)].](/content/184/2020/13/11/img/10.1016_j.arabjc.2020.09.025-fig7.png)
Representative flow cytometry graphs showing ΔMFI Cyto-ID after 48-hour treatment of the compounds 18a and 18b in PC-3 cells, in concentration equal to the corresponding IC50 values. [ΔMFI Cyto-ID = MFI Cyto-ID (treated) - MFI Cyto-ID (control)].
Since autophagy seems to facilitate the survival of hypoxic cells and hypoxia has been associated with resistance to anticancer therapy (Tan et al., 2016), novel compounds 18a and 18b (by inhibiting autophagy) may hold strong promise in the prompt development of new drug cocktail-based regimens for the successful management of human prostate cancer in the clinic.
3 Conclusions
Two series of novel compounds possessing the acridone and xanthone scaffold were synthesized and evaluated for their antiproliferative activities against a panel of four tumor cell lines. Selected derivatives have been subsequently examined against two additional highly metastatic cancer cell lines. Two analogues with the most interesting cytotoxic properties have also been studied in terms of cell-cycle perturbation and arrest of exponentially growing PC-3 cells, as well as for their ability to affect apoptosis and autophagy. All new compounds possess moderate to good antiproliferative properties, with the most interesting results - among all inhibitors - being extracted from their profile against the human prostate cancer cells. Apart from the nitro substituted analogues, 18a and 18b, no other acridone-containing derivative possesses a considerably interesting profile against the tested cell lines, in contrast to the synthesized compounds belonging to the xanthone series. Compounds 18a and 18b, which also bear a basic side chain, emerged as the most efficient of the acridone series, with the best activities being demonstrated against the PC-3 (prostate cancer) cells. Interestingly, both compounds were found to act as potent autophagy inhibitors in a PC-3 cellular environment, causing accumulation of PC-3 cells at G0/G1 phase and significantly induce apoptosis. Regarding xanthone analogues, all of them showed interesting cytotoxicity profiles, although, in this case the nitro xanthones were not among the noteworthy analogues. Compounds 28b and 29b, bearing an additional side chain, appear very potent against the PC-3 cell line, with IC50 values estimated at the low micromolar range, especially the vinyl-substituted derivative 28b, which in parallel showed substantial cytotoxic activity against T24 and mainly WM266-4 cancer-cells. When compared to the previously reported xanthone analogues which possess a direct amino substituent on the chromophore (Kostakis et. al. 2006), we could state that the carboxamides presented in this work appear overall less cytotoxic. This could indicate that in this scaffold the insertion of the carbonyl group is not beneficial for the biological activity.
In terms of their clinical relevance and therapeutic potential, 18a, 18b, 28b and 29b are suggested to be further optimized and investigated for prostate cancer treatment. Although the mechanisms for the tumor-suppressing functions of the herein described novel compounds are still unclear, our results indicate that in the case of xanthone analogues, the replacement of nitro group with a second side chain significantly improves the anticancer activity. Furthermore, the nitro acridones appear considerably more potent, when compared with the corresponding nitro xanthones, whereas the di-substituted compounds are inactive. These results strongly suggest the pivotal role of these “structural transformations” in the mechanism of action for the herein presented compounds. Since a number of tumors seem to require autophagy for their survival and growth, the 18a- and 18b- mediated suppression of autophagic flux may compel prostate cancer cells (PC-3) to apoptotic death; yet, more studies are needed to further clarify how these alterations of the autophagic potential may serve as death signals. On the basis of these results, the establishment of a preliminary Structure-Activity-Relationship (SAR) is feasible, in order to determine the structural features that possess a crucial role in enhanced and targeted inhibition of cancer cell survival, and growth
4 Experimental section
4.1 Chemistry
All commercially available chemicals were purchased from Alfa Aesar. Melting points were determined on a Büchi apparatus and were uncorrected. 1H NMR, 13C NMR and 2D spectra were recorded on a Bruker Avance III 600, 400 and 200 spectrometer (Bruker GmbH, Germany) using dimethyl sulfoxide (DMSO‑d6), methanol (CD3OD) and chloroform (CDCl3) as deuterated solvents and were referenced to TMS (δ scale). The signals of 1H and 13C spectra were unambiguously assigned by using 2D NMR techniques: 1H1H COSY, HMQC, and HMBC. 1H NMR and 13C NMR of compounds 1 (Decodts et al., 1983; Knepper et al., 2006), 8 (Burdeska et al., 1972), 11 (Hadjipavlou et al., 2006), 14 (Recanatini et al., 2001), 15 (Recanatini et al., 2001) and 17 (Burdeska et al., 1972) were confirmed with those reported in the literature (See Supplementary Material). Flash chromatography was performed on Merck silica gel 60 (0.040–0.063 mm). Analytical thin layer chromatography (TLC) was carried out on pre-coated (0.25 mm) Merck KgaA (Darmstadt, Germany) silica gel F-254 plates. HRMS were obtained on an LTQ-Orbitrap Discovery Mass Spectrometer (Thermo Scientific, Brehmen, Germany).
4.1.1 Methyl 3-{[2-(methoxycarbonyl)phenyl]amino}-4-nitrobenzoate (4)
A suspension of methyl anthranilate (617 mg, 4.08 mmol, 2), 3-bromo-4-nitrobenzoate (1.06 g, 4.08 mmol, 1), Cs2CO3 (6.64 g, 18 mmol) and tetrakis(triphenylphosphine)palladium (0) (235 mg, 0.20 mmol) in dry toluene (100 mL), was heated at 110 °C, under argon, for 24 h. After completion of the reaction, the volatiles were removed under reduced pressure and the residue was extracted with CH2Cl2, dried over Na2SO4, and evaporated to dryness. Flash chromatography on silica gel, using a mixture of cyclohexane / EtOAc 10 / 1, as the eluent, afforded 1.2 g (89%) of the title compound 4. M.p. 160–161 °C (CH2Cl2 - n-Pentane); 1H NMR (600 MHz, CDCl3) δ 11.16 (s, D2O exch., 1H, NH), 8.28 (d, J = 1.5 Hz, 1H, H-2), 8.21 (d, J = 8.7 Hz, 1H, H-5), 8.06 (d, J = 8.7, 1H, H-3΄), 7.55–7.47 (m, 3H, H-6΄, H-5΄, H-6), 7.10 (td, J = 8.2 Hz, 2.0 Hz, 1H, H-4΄), 3.97 (s, 3H, CH3), 3.91 (s, 3H, CH3΄); 13C NMR (151 MHz, CDCl3) δ 167.5 (COOCH3΄), 165.4 (COOCH3΄), 141.9 (C-1΄), 139.4 (C-4), 138.9 (C-3), 135.5 (C-1), 133.7 (C-5΄), 132.2 (C-3΄), 126.9 (C-5), 122.4 (C-4΄), 120.3 (C-2), 120.0 (C-6), 118.9 (C-6΄), 118.8 (C-2΄), 52.8 (COOCH3), 52.4 (COOCH3΄); HRMS (ESI-) m/z 329.0779 (calcd for C16H13N2O6-, 329.0771).
4.1.2 3-[(2-Carboxyphenyl)amino]-4-nitrobenzoic acid (5)
To a solution of the diester 4 (1.2 g, 3.6 mmol) in methanol (150 mL), at room temperature, was added dropwise a cold 15% NaOH solution (10 mL) and the mixture was stirred at room temperature for 12 h. After completion of the reaction, the mixture was poured into water, acidified with 18% HCl solution (pH ∼ 2) and the resulting solid was filtered and dried over P2O5 to afford 1.05 g (96%) of the title compound 5, which was used for the next step without any further purification. M.p. > 270 °C (MeOH - Et2O); 1H NMR (600 MHz, DMSO‑d6) δ 11.12 (s, D2O exch., 1H, NH), 8.26 (d, J = 7.6 Hz, 1H, H-5), 8.16 (s, 1H, H-2), 8.05 (d, J = 7.6 Hz, 1H, H-3΄), 7.61 (m, 2H, H-5΄, H-6΄), 7.55 (d, J = 7.6 Hz, 1H, H-6), 7.19 (t, J = 8.7 Hz, 1H, H-4΄); 13C NMR (151 MHz, DMSO‑d6) δ 168.6 (COOH), 165.7 (COOH), 141.6 (C-1΄), 138.3 (C-3), 136.4 (C-1), 133.7 (C-5΄), 131.9 (C-3΄), 130.0 (C-4), 127.0 (C-5), 122.3 (C-4΄), 120.2 (C-2), 119.8 (C-6), 119.0 (C-6΄), 118.9 (C-2΄).
4.1.3 2-{[5-(Methoxycarbonyl)-2-nitrophenyl]amino}benzoic acid (6)
A suspension of methyl 3-bromo-4-nitrobenzoate (1 g, 3.80 mmol, 1), anthranilic acid (615 mg, 4.07 mmol, 3), Cs2CO3 (7.2 g, 20.40 mmol) and tetrakis(triphenylphosphine)palladium (0) (235 mg, 0.20 mmol) in a 3 / 1 mixture of toluene / N,N-dimethylacetamide (100 mL), was stirred under reflux, for 24 h, under argon. After completion of the reaction, the volatiles were removed under reduced pressure and the residue was poured into water, acidified with 18% HCl solution (pH ∼ 2) and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and evaporated to dryness. Flash chromatography on silica gel, using a mixture of cyclohexane / EtOAc 1 / 3, as the eluent, afforded 1.1 g (92%) of the title compound 6. M.p. 230–231 °C (THF - n-Pentane); 1H NMR (400 MHz, DMSO‑d6) δ 11.10 (s, D2O exch., 1H, NH), 8.23 (d, J = 7.6 Hz, 1H, H-5), 8.11 (d, J = 1.7 Hz, 1H, H-2), 7.99 (d, J = 7.6 Hz, 1H, H-3΄), 7.58–7.49 (m, 3H, H-5΄, H-6΄, H-6), 7.15 (t, J = 8.7 Hz, 1H, H-4΄), 3.84 (s, 3H, COOCH3); 13C NMR (151 MHz, DMSO‑d6) δ 168.6 (COOH), 164.7 (COOCH3), 141.6 (C-1΄), 139.2 (C-4), 138.3 (C-3), 135.0 (C-1), 133.7 (C-5΄), 131.9 (C-3΄), 127.1 (C-5), 122.4 (C-4΄), 120.0 (C-2), 119.8 (C-6), 119.0 (C-2΄), 118.9 (C-6΄), 53.3 (COOCH3); HRMS (ESI-) m/z 329.0623 (calcd for C15H11N2O6-, 329.0627).
4.1.4 Methyl 4-nitro-9-oxo-9,10-dihydroacridine-1-carboxylate (7)
A suspension of acid 6 (3.25 g, 10.28 mmol) in a 2 / 1 mixture of trifluoroacetic acid - trifluoroacetic anhydride (12 mL) was stirred at room temperature for 14 h. After completion of the reaction, the mixture was poured into crushed ice, the precipitate was filtered, washed with water and air dried, to afford 2.54 g (83%) of the title compound, which was used for the next step without any further purification. M.p. > 270 °C (THF - Et2O). 1H NMR (600 MHz, DMSO‑d6) δ 11.64 (s, D2O exch., 1H, NH), 8.71 (d, J = 8.2 Hz, 1H, H-3), 8.20 (d, J = 7.4 Hz, 1H, H-8), 8.11 (d, J = 8.4 Hz, 1H, H-5), 7.84 (t, J = 7.5 Hz, 1H, H-6), 7.44 (t, J = 7.5 Hz, 1H, H-7), 7.36 (d, J = 8.4 Hz, 1H, H-2), 3.93 (s, 3H, CH3); 13C NMR (151 MHz, DMSO‑d6) δ 175.0 (C-9), 168.7 (COCH3), 140.6 (C-4a), 139.8 (C-10a), 135.9 (C-4), 135.1 (C-1), 134.8 (C-6), 131.4 (C-3), 125.7 (C-8), 123.7 (C-7), 120.6 (C-8a), 119.4 (C-9a), 119.3 (C-2), 118.2 (C-5), 52.6 (COCH3); HRMS (ESI-) m/z 297.0517 (calcd for C15H9N2O5-, 297.0522).
4.1.5 4-Nitro-9-oxo-9,10-dihydroacridine-1-carboxylic acid (8)
Method A: A solution of acid 5 (1 g, 3.31 mmol) in c. sulfuric acid (10 mL) was stirred at 110 °C for 2 h. After cooling, the mixture was poured into crushed ice, the precipitate was filtered, washed with water and dried (P2O5). Flash chromatography on silica gel, using a mixture of CH2Cl2 / MeOH 6 / 1, afforded the title compound 8 (400 mg, 42%). Method B: To a solution of ester 7 (1.5 g, 5.03 mmol) in methanol (60 mL), at room temperature, was added dropwise a cold 40% NaOH solution (2.5 mL) and the mixture was stirred at room temperature for 12 h. After completion of the reaction, the mixture was poured into water, acidified with 18% HCl solution (pH ∼ 2) and the resulting solid was filtered and dried over P2O5 to afford 1.35 g (85%) of the title compound 8, which was used for the next step without any further purification.
4.1.6 Ethyl 2-(5-methyl-2-nitrophenoxy)benzoate (12b)
To a suspension of ester 11 (4.1 g, 16 mmol) in acetic anhydride (9.34 mL, 99 mmol) at 0 °C was added dropwise a solution of fuming HNO3 (0.68 mL, 16 mmol) in acetic anhydride (2.36 mL, 25 mmol) and the mixture was stirred at room temperature for 24 h. After completion of the reaction, the mixture was poured into ice - water, basified with 15% NaOH solution (pH ∼ 9) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over anh. Na2SO4 and evaporated to dryness. The residue was purified by column chromatography (silica gel), using a mixture of petroleum ether / EtOAc (100 / 0 – 30 / 1), as the eluent, to afford 1.01 g (21%) of ester 12a and 3.03 g (63%) of the ester 12b.
Physicochemical data of ethyl 2-(5-methyl-2-nitrophenoxy)benzoate (12a): oil; 1H NMR (400 MHz, CDCl3) δ 8.03 (dd, J = 8.0 Hz, 2 Hz, 1H, H-6), 7.92 (d, J = 8.0 Hz, 1H, H-3΄), 7.58 (td, J = 8.0 Hz, 2 Hz, 1H, H-4), 7.33 (td, J = 8.0 Hz, 2 Hz, 1H, H-5), 7.11 (d, J = 8.0 Hz, 1H, H-3), 6.95 (d, J = 8.0 Hz, 1H, H-4΄), 6.58 (s, 1H, H-6΄), 4.22 (q, J = 7.0 Hz, 2H, CH2CH3), 2.30 (s, 3H, CH3) 1.16 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (50 MHz, CDCl3) δ 164.9 (CO), 153.8 (C-2), 151.7 (C-1΄), 145.9 (C-5΄), 137.8 (C-2΄), 133.9 (C-4), 132.3 (C-6), 125.8 (C-3΄),125.2 (C-5), 124.0 (C-1), 123.1 (C-4΄), 122.1 (C-3), 118.5 (C-6΄), 61.2 (CH2CH3), 21.6 (CH3), 13.8 (CH2CH3).
Physicochemical data of ethyl 2-(3-methyl-4-nitrophenoxy)benzoate (12b): M.p. 63–64 °C (Et2O - n-Hexane); 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.0 Hz, 1H, H-5΄), 8.03 (d, J = 8.0 Hz, 1H, H-6), 7.62 (t, J = 8.0 Hz, 1H, H-4), 7.36 (t, J = 8.0 Hz, 1H, H-5), 7.15 (d, J = 8.0 Hz, 1H, H-3), 6.80 (s, 1H, H-2΄), 6.75 (d, J = 8.0 Hz, 1H, H-6΄), 4.20 (q, J = 7.0 Hz, 2H, CH2CH3), 2.56 (s, 3H, CH3), 1.18 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (50 MHz, CDCl3) δ 164.7 (CO), 162.0 (C-1΄), 153.4 (C-2), 143.1 (C-4΄), 137.0 (C-3΄), 134.1 (C-4), 132.3 (C-6), 127.4 (C-5΄), 125.7 (C-5), 124.5 (C-1), 123.1 (C-3), 119.4 (C-2΄), 113.9 (C-6΄), 61.17 (CH2CH3), 21.5 (CH3), 14.0 (CH2CH3).
4.1.7 2-(5-Methyl-2-nitrophenoxy)benzoic acid (13)
To a solution of ester 12b (3.01 g, 10 mmol) in ethanol (40 mL) was added a 20% sodium hydroxide solution (8 mL, 40 mmol) and the mixture was stirred at room temperature for 3 h. After completion of the reaction, the mixture was poured into water and acidified with a 36% HCl solution (pH ∼ 2). The precipitate was filtered and dried over P2O5, to afford the title compound 13 (2.62 g, 96%), which was used without any further purification for the next reaction. M.p. 144–145 °C (EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.22 (dd, J = 8.0 Hz, 2 Hz, H-6), 8.04 (d, J = 8.0 Hz, 1H, H-3΄), 7.57 (td, J = 8.0 Hz, 2 Hz, H-4), 7.26 (t, J = 8.0 Hz, 1H, H-5), 7.15 (d, J = 8.0 Hz, 1H, H-3), 6.94 (d, J = 8.0 Hz, 1H, H-4΄), 6.90 (s, 1H, H-6΄), 2.43 (s, 3H, CH3); 13C NMR (50 MHz, CDCl3) δ 168.9 (CO), 155.6 (C-2), 150.1 (C-1΄), 146.6 (C-5΄), 138.6 (C-2΄), 135.0 (C-4), 133.3 (C-6), 126.1 (C-3΄), 124.8 (C-5), 124.7 (C-1), 121.4 (C-4΄), 120.7 (C-3), 120.2 (C-6΄), 21.6 (CH3).
4.1.8 1-(Hydroxymethyl)-4-nitro-9H-xanthen-9-one (16)
A suspension of bromide 15 (1.67 g, 5 mmol) and AgNO3 (3.57 g, 21 mmol) in a 2 / 1 mixture of acetone / water (100 mL) was stirred in the dark, at room temperature, for 36 h. After completion of the reaction, most of the volatiles were removed under reduced pressure and the residue was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over anh. Na2SO4 and evaporated to dryness. Flash chromatography on silica gel, using a mixture of cyclohexane / EtOAc 1 / 1 as the eluent, afforded 1.23 g (91%) of the title compound 16. M.p. 229–230 °C (EtOH); 1H NMR (600 MHz, DMSO‑d6) δ 8.50 (d, J = 8.0 Hz, 1H, H-3), 8.14 (dd, J = 8.0 Hz, 1 Hz, 1H, H-8), 7.89 (d, J = 8.0 Hz, 1H, H-2), 7.88 (td, J = 8.0 Hz, 1 Hz, 1H, H-6), 7.62 (d, J = 8.0 Hz, 1H, H-5), 7.51 (t, J = 8.0 Hz, 1H, H-7), 5.68 (t, J = 8.0 Hz, D2O exch, 1H, OH), 5.21 (d, J = 5.0 Hz, 2H, CH2OH); 13C NMR (151 MHz, DMSO‑d6) δ 177.0 (C-9), 154.3 (C-10a), 153.5 (C-1), 149.1 (C-4a), 137.6 (C-4), 136.3 (C-6), 130.2 (C-3), 126.4 (C-8), 125.7 (C-7), 122.1 (C-8a), 120.2 (C-2), 119.6 (C-9a), 118.2 (C-5), 62.6 (CH2OH); HRMS (ESI-) m/z 271.0481 (calcd for C14H9NO5-, 271.0488).
4.1.9 4-Nitro-9-oxo-9H-xanthene-1-carboxylic acid (17)
A solution of Jones reagent (1 mmol / mL) was added dropwise at room temperature, to a solution of alcohol 16 (100 mg, 0.36 mmol) in acetone (8 mL). The addition is continued until the characteristic orange - red color persists for about 5 min. After completion of the reaction, isopropanol was added until color disappearance and the residual green salts were filtered off. The volatiles were removed under reduced pressure and the residue was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anh. Na2SO4 and evaporated to dryness. Flash chromatography on silica gel, using a mixture of CH2Cl2 / MeOH (9 / 2) as the eluent, afforded 92.5 mg (88%) of the title compound 17 (Burdeska et al., 1972).
4.1.10 N-[2-(Dimethylamino)ethyl]-4-nitro-9-oxo-9,10-dihydroacridine-1-carboxamide (18a)
To a solution of acid 8 (300 mg, 1.05 mmol) in dry DMF (10 mL) was added 1,1′-carbonyldiimidazole (300 mg, 1.05 mmol) and the mixture was stirred under argon, at room temperature, for 25 min. N,N-dimethylethylenodiamine (572 µL, 5.25 mmol) was then added and the reaction mixture was heated at 50 °C for 12 h. After completion of the reaction, DMF was removed under reduced pressure and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anh. Na2SO4 and evaporated to dryness. Flash chromatography on silica gel, using a mixture of CH2Cl2 / MeOH 10 / 1–4 / 1 as the eluent, afforded 240 mg (63%) of the title compound 18a. M.p. 224–225 °C (CH2Cl2 - n-Pentane); 1H NMR (600 MHz CDCl3) δ 11.37 (s, D2O exch., 1H, NH), 8.60 (d, J = 8.3 Hz, 1H, H-3), 8.28 (dd, J = 8.0 Hz, 1.2 Hz, 1H, H-8), 7.68 (td, J = 8.0 Hz, 1.2 Hz, 1H, H-6), 7.38 (d, J = 8.0 Hz, 1H, H-5), 7.30 (t, J = 8.0 Hz, 1H, H-7), 7.14 (d, J = 8.3 Hz, 1H, H-2), 6.57 (brs, D2O exch., 1H, CONH), 3.62 (q, J = 8.7 Hz, 2H, CONHCH2), 2.67 (t, J = 8.7 Hz, 2H, CH2CH2N(CH3)2), 2.27 (s, 6H, N(CH3)2); 13C NMR (151 MHz, CDCl3) δ 176.1 (C-9), 169.4 (CONH), 146.6 (C-1), 138.9 (C 10a), 136.6 (C-4a), 134.8 (C-6), 134.2 (C-4), 131.2 (C-3), 127.3 (C-8), 124.0 (C-7), 122.1 (C-8a), 120.6 (C-9a), 119.3 (C-2), 117.7 (C-5), 57.4 (CH2N(CH3)2), 45.0 (N(CH3)2), 37.12(CONHCH2); HRMS (ESI+) m/z 355.1401 (calcd for C18H19N4O4+, 355.1406).
4.1.11 N-[2-(Diethylamino)ethyl]-4-nitro-9-oxo-9,10-dihydroacridine-1-carboxamide (18b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 18a, using N,N-diethylethylenodiamine. Yield: 70%. M.p. 193–194 °C (CH2Cl2 - n-Pentane); 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 8.2 Hz, 1H, H-3), 8.15 (d, J = 7.9 Hz, 1H, H-8), 7.64 (t, J = 7.9 Hz, 1H, H-6), 7.37 (d, J = 7.9 Hz, 1H, H-5), 7.25 (t, J = 7.9 Hz, 1H, H-7), 7.04 (d, J = 8.2 Hz, 1H, H-2), 6.83 (brs, D2O exch., 1H, CONH), 3.61 (q, J = 8.7 Hz, 2H, CONHCH2), 2.78 (t, J = 8.7 Hz, 2H, CH2N(CH2CH3)2), 2.60 (t, J = 8.4 Hz, 4H, N(CH2CH3)2), 0.99 (t, J = 8.4 Hz, 6H, N(CH2CH3)2); 13C NMR (151 MHz, CDCl3) δ 175.7 (C-9), 169.1 (CONH), 146.5 (C-1), 138.7 (C-10a), 136.1 (C-4a), 134.7 (C-6), 134.0 (C-4), 131.0 (C-3), 126.9 (C-8), 123.8 (C-7), 121.6 (C-8a), 120.1 (C-9a), 119.2 (C-2), 117.7 (C-5), 51.3 (CH2N(CH2CH3)2), 46.6 (N(CH2CH3)2), 37.4 (CONHCH2), 11.5 (N(CH2CH3)2); HRMS (ESI+) m/z 383.1714 (calcd for C20H23N4O4+, 383.1718).
4.1.12 4-[(Chloroacetyl)amino]-N-[2-(dimethylamino)ethyl]-9-oxo-9,10-dihydroacridine-1-carboxamide (20a)
A suspension of 18a (240 mg, 0.67 mmol) in absolute ethanol (50 mL) was hydrogenated in the presence of 10% Pd / C (30 mg) under a pressure of 50 psi, at room temperature, for 7 h. The resulting mixture was filtered through a Celite pad, and the filtrate was evaporated to dryness, to afford amine 19a. Without any further purification, the amine was dissolved under argon in dry THF (5 mL) and to this solution were added K2CO3 (166 mg, 1.20 mmol) and chloracetyl chloride (34 µL, 0.40 mmol), at 0 °C and the mixture was stirred at room temperature for 8 h. After completion of the reaction, THF was vacuum evaporated and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anh. Na2SO4 and evaporated to dryness. Flash chromatography on silica gel, using a mixture of CH2Cl2 / MeOH 10 / 1 to 4 / 1 as the eluent, afforded 70 mg (43%) of the title compound 20a. M.p. > 270 °C (MeOH - Et2O); 1H NMR (600 MHz, CD3OD) δ 8.35 (d, J = 8.4 Hz, 1H, H-8), 7.85 (d, J = 8.4 Hz, 1H, H-5), 7.83–7.77 (m, 2H, H-3, H-6), 7.37 (t, J = 8.4 Hz, 1H, H-7), 7.22 (d, J = 7.9 Hz, 1H, H-2), 4.50 (s, 2H, COCH2), 3.89 (t, J = 8.7 Hz, 2H, CONHCH2), 3.56 (t, J = 8.7 Hz, 2H, CH2N(CH3)2), 3.27 (s, 6H, N(CH3)2); 13C NMR (151 MHz, CD3OD) δ 179.1 (C-9), 175.2 (CONHCH2), 169.7 (NHCOCH2), 142.0 (C-10a), 138.9 (C-1), 136.5 (C-4a), 135.8 (C-6), 132.3 (C-3), 127.6 (C-4), 127.3 (C-8), 123.8 (C-7), 122.1 (C-8a), 121.0 (C-2), 119.0 (C-5), 118.9 (C-9a), 60.4 (CH2N(CH3)2), 44.6 (N(CH3)2), 44.0 (COCH2), 35.8 (CONHCH2); HRMS (ESI+) m/z 401.1375 (calcd for C20H22ClN4O3+, 401.1367).
4.1.13 4-[(Chloroacetyl)amino]-N-[2-(diethylamino)ethyl]-9-oxo-9,10-dihydroacridine-1-carboxamide (20b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 20a, starting from compound 18b. Yield: 45%. M.p. 210–211 °C (MeOH - Et2O); 1H NMR (600 MHz, CD3OD) δ 8.28 (dd, J = 8.1 Hz, 1.8 Hz, 1H, H-8), 7.90 (d, J = 8.1 Hz, 1H, H-5), 7.83–7.78 (m, 2H, H-3, H-6), 7.36 (t, J = 8.1 Hz, 1H, H-7), 7.20 (d, J = 7.9 Hz, 1H, H-2), 4.53 (s, 2H, COCH2), 3.91 (t, J = 8.7 Hz, 2H, CONHCH2), 3.60 (q, J = 7.3 Hz, 4H, N(CH2CH3)2), 3.55 (t, J = 8.7 Hz, 2H, CH2N(CH2CH3)2), 1.49 (t, J = 7.3 Hz, 6H, N(CH2CH3)2); 13C NMR (151 MHz, CD3OD) δ 178.7 (C-9), 174.8 (CONHCH2), 169.5 (NHCOCH2), 142.0 (C-10a), 138.5 (C-1), 136.2 (C-4a), 135.7 (C-6), 132.0 (C-3), 127.6 (C-4), 127.0 (C-8), 123.8 (C-7), 122.0 (C-8a), 121.1 (C-2), 119.0 (C-5), 118.8 (C-9a), 54.2 (CH2N(CH2CH3)2), 48.8 (N(CH2CH3)2), 44.1 (COCH2), 35.3 (CONHCH2), 9.2 (N(CH2CH3)2); HRMS (ESI+) m/z 429.1688 (calcd for C22H26ClN4O3+, 429.1693).
4.1.14 4-[2-(Dimethylamino)acetamido]-N-[2-(dimethylamino)ethyl]-9-oxo-9,10-dihydroacridine-1-carboxamide (21a)
To a solution of compound 20a (70 mg, 0.17 mmol) in absolute ethanol (25 mL) was added dimethylamine (0.32 mL, 1.80 mmol, 5.6 M in ethanol) and the mixture was refluxed for 8 h. Upon cooling, the mixture was vacuum-evaporated, extracted with EtOAc / water, the organic layer was dried (anh. Na2SO4) and evaporated to dryness. The residue was purified by column chromatography (silica gel, CH2Cl2 / MeOH 10 / 1 – 4 /1) to afford the title compound (40 mg, 57%). M.p. 220–222 °C (MeOH - Et2O); 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.0 Hz, 1H, H-8), 7.59 (d, J = 8.0 Hz, 1H, H-5), 7.41 (t, J = 8.0 Hz, 1H, H-6), 7.36 (d, J = 7.9 Hz, 1H, H-3), 6.94 (t, J = 8.0 Hz, 1H, H-7), 6.68 (d, J = 7.9 Hz, 1H, H-2), 3.67 (q, J = 8.3 Hz, 2H, CONHCH2), 3.45 (s, 2H, NHCOCH2), 3.06 (t, J = 8.3 Hz, 2H, CONHCH2CH2), 2.68 (s, 6H, COCH2N(CH3)2), 2.49 (s, 6H, CH2CH2N(CH3)2); 13C NMR (151 MHz, CDCl3) δ 177.02 (C-9), 172.28 (CONHCH2), 170.40 (NHCOCH2), 140.1 (C-10a), 135.0 (C-1), 133.5 (C-4a), 133.2 (C-6), 127.9 (C-4), 126.1 (C-3), 125.5 (C-8), 121.7 (C-7), 120.3 (C-8a), 119.6 (C-2), 118.0 (C-5), 117.5 (C-9a), 63.5 (NHCOCH2), 60.4 (CONHCH2CH2), 45.8 (COCH2N(CH3)2), 44.7 (CH2CH2N(CH3)2), 35.9 (CONHCH2); HRMS (ESI+) m/z 410.2187 (calcd for C22H28N5O3+, 410.2189).
4.1.15 4-[2-(Diethylamino)acetamido]-N-[2-(diethylamino)ethyl]-9-oxo-9,10-dihydroacridine-1-carboxamide (21b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 21a, using diethylamine. Yield: 62%. M.p. 219–221 °C (MeOH - Et2O); 1H NMR (400 MHz, CDCl3) δ 7.90 (dd, J = 8.1, 1.4 Hz, 1H, H-8), 7.45 (m, 2H, H-5, H-6), 7.30 (d, J = 8.1 Hz, 1H, H-3), 6.90 (td, J = 8.1 Hz, 1.4 Hz, 1H, H-7), 6.70 (d, J = 8.1 Hz, 1H, H-2), 3.60 (t, J = 7.0 Hz, 2H, CONHCH2), 3.45 (s, 1H, NHCOCH2), 2.90 (t, J = 7.0 Hz, 2H, CONHCH2CH2), 2.80 (m, 8H, N(CH2CH3)2, N(CH2CH3)2), 1.06 (t, J = 7.0 Hz, 6H, CH2CH2N(CH2CH3)2), 1.19 (t, J = 7.3 Hz, 6H, COCH2N(CH2CH3)2); 13C NMR (151 MHz, CDCl3) δ 176.6 (C-9), 172.1 (CONHCH2), 171.7 (NHCOCH2), 140.2 (C-10a), 135.1 (C-1), 134.0 (C-4a) 133.1 (C-6), 126.5 (C-4), 125.9 (C-3), 125.9 (C-8), 121.7 (C-7), 120.1 (C-8a), 119.7 (C-2), 118.1 (C-5), 118.1 (C-9a), 57.7 (NHCOCH2), 51.9 (CONHCH2CH2), 48.3 (COCH2N(CH2CH3)2), 45.6 (CH2CH2N(CH2CH3)2), 36.6 (CONHCH2), 12.0 (COCH2N(CH2CH3)2), 10.6 (CH2CH2N(CH2CH3)2); HRMS (ESI+) m/z 466.2813 (calcd for C26H36N5O3+, 466.2816).
4.1.16 4-(Acryloylamino)-N-[2-(dimethylamino)ethyl]-9-oxo-9,10-dihydroacridine-1-carboxamide (22a)
This compound was synthesized by an analogous procedure as described for the preparation of compound 20a, using 3-chloropropionyl chloride. Yield: 75%. M.p. 206–207 °C (MeOH - Et2O); 1H NMR (600 MHz, CD3OD) δ 8.22 (d, J = 8.1 Hz, 1H, H-8), 7.74–7.68 (m, 3H, H-3, H-5, H-6), 7.27 (t, J = 8.1 Hz, 1H, H-7), 7.10 (d, J = 7.9 Hz, 1H, H-2), 6.69 (dd, J = 16.0 Hz, 10.1 Hz, 1H, CH = CH2), 6.47 (dd, J = 16.0, 1.0 Hz, 1H, CH = CH2), 5.92 (d, J = 10.1 Hz, 1H, CH = CH2), 3.73 (t, J = 7.1 Hz, 2H, CONHCH2), 3.03 (t, J = 7.1 Hz, 2H, CONHCH2CH2), 2.70 (s, 6H, N(CH3)2); 13C NMR (151 MHz, CD3OD) δ 178.7 (C-9), 174.5 (CONHCH2), 167.8 (NHCOCH = CH2), 141.9 (C-10a), 138.2 (C-1), 136.5 (C-4a), 135.2 (C-6), 132.2 (CH = CH2), 131.2 (C-3), 128.4 (CH = CH2), 127.6 (C-4), 127.1 (C-8), 123.4 (C-7), 122.1 (C-8a), 121.1 (C-2), 119.2 (C-9a), 118.9 (C-5), 59.4 (CONHCH2CH2), 45.2 (N(CH3)2), 37.7 (CONHCH2); HRMS (ESI+) m/z 379.1765 (calcd for C21H23N4O3+, 379.1768).
4.1.17 4-(Acryloylamino)-N-[2-(diethylamino)ethyl]-9-oxo-9,10-dihydroacridine-1-carboxamide (22b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 20a, using 3-chloropropionyl chloride. Yield: 70%. M.p. 160–162 °C (MeOH - Et2O); 1H NMR (600 MHz, CD3OD) δ 8.31 (d, J = 8.0 Hz, 1H, H-8), 7.87–7.76 (m, 3H, H-3, H-5, H-6), 7.35 (t, J = 8.0 Hz, 1H, H-7), 7.19 (d, J = 7.9 Hz, 1H, H-2), 6.68 (dd, J = 16.0, 10.1 Hz, 1H, CH = CH2), 6.48 (dd, J = 16.0, 1.1 Hz, 1H, CH = CH2), 5.93 (d, J = 10.1 Hz, 1H, CH = CH2), 3.84 (t, J = 7.6 Hz, 2H, CONHCH2), 3.39 (m, 6H, CONHCH2CH2), 1.42 (t, J = 7.7 Hz, 6H, N(CH2CH3)2); 13C NMR (151 MHz, CD3OD) δ 178.7 (C-9), 174.8 (CONHCH2), 167.8 (NHCOCH = CH2), 142.1 (C-10a), 138.5 (C-1), 136.2 (C-4a), 135.5 (C-6), 132.2 (CH = CH2), 131.4 (C-3), 128.5 (CH = CH2), 128.0 (C-4), 127.1 (C-8), 123.7 (C-7), 122.1 (C-8a), 121.1(C-2), 119.1 (C-5), 119.0 (C-9a), 53.9 (CONHCH2CH2), 48.6 (N(CH2CH3)2), 36.0 (CONHCH2), 9.8 (N(CH2CH3)2); HRMS (ESI+) m/z 407.2078 (calcd for C23H27N4O3+, 407.2081).
4.1.18 N-[2-(Dimethylamino)ethyl]-4-[3-(dimethylamino)propanamido]-9-oxo-9,10-dihydroacridine-1-carboxamide (23a)
To a solution of compound 22a (60 mg, 0.16 mmol) in absolute ethanol (25 mL) was added dimethylamine (0.30 mL, 1.68 mmol, 5.6 M in ethanol) and the mixture was refluxed for 8 h. Upon cooling, the mixture was vacuum - evaporated, extracted with EtOAc - water, the organic layer was dried (anh. Na2SO4) and evaporated to dryness. The residue was purified by column chromatography (silica gel, CH2Cl2 - MeOH 10 / 1– 4 /1) to afford the title compound (55 mg, 81%). M.p. 183–184 °C (CH2Cl2 - Et2O); 1H NMR (400 MHz, CDCl3) δ 10.38 (s, D2O exch., 1H, NH), 7.85 (d, J = 8.0 Hz, 1H, H-8), 7.44 (d, J = 8.0 Hz, 1H, H-5), 7.39 (t, J = 8.0 Hz, 1H, H-6), 7.29 (d, J = 7.5 Hz, 1H, H-3), 6.92 (t, J = 8.0 Hz, 1H, H-7), 6.66 (d, J = 7.5 Hz, 1H, H-2), 3.64 (q, J = 8.0 Hz, 2H, CONHCH2), 2.90 (m, 6H, CONHCH2CH2, NHCOCH2CH2), 2.55 (s, 6H, COCH2CH2N(CH3)2), 2.49 (s, 6H, NHCH2CH2N(CH3)2); 13C NMR (151 MHz, CDCl3) δ 176.8 (C-9), 172.6 (CONHCH2), 172.1 (NHCOCH2), 139.8 (C-10a), 135.2 (C-1), 133.4 (C-4a), 133.1 (C-6), 128.2 (C-4), 126.6 (C-3), 125.7 (C-8), 121.6 (C-7), 120.4 (C-8a), 119.6 (C-2), 117.7 (C-5), 117.5 (C-9a), 65.8 (CONHCH2CH2), 57.8 (NHCOCH2CH2), 45.5 (COCH2CH2N(CH3)2), 44.9 (NHCH2CH2N(CH3)2), 36.6 (CONHCH2CH2), 34.7 (NHCOCH2CH2); HRMS (ESI+) m/z 424.2343 (calcd for C23H30N5O3+, 424.2346).
4.1.19 N-[2-(Diethylamino)ethyl]-4-[3-(diethylamino)propanamido]-9-oxo-9,10-dihydroacridine-1-carboxamide (23b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 23a, using diethylamine. Yield: 85%. M.p. 188–190 °C (CH2Cl2 - Et2O); 1H NMR (600 MHz, CDCl3) δ 10.22 (s, D2O exch., 1H, NH), 7.84 (d, J = 8.0 Hz, 1H, H-8), 7.62 (d, J = 8.0 Hz, 1H, H-5), 7.45 (t, J = 8.0 Hz, 1H, H-6), 7.38 (d, J = 7.9 Hz, 1H, H-3), 6.99 (t, J = 8.0 Hz, 1H, H-7), 6.69 (d, J = 7.9 Hz, 1H, H-2), 3.59 (q, J = 8.2 Hz, 2H, CONHCH2), 3.10 (m, 8H, CONHCH2CH2, NHCOCH2CH2, NHCH2CH2N(CH2CH3)2), 2.98 (t, J = 8.2 Hz, 2H, NHCOCH2), 2.81 (t, J = 7.8 Hz, 4H, COCH2CH2N(CH2CH3)2), 1.08 (m, 12H, N(CH2CH3)2, N(CH2CH3)2); 13C NMR (151 MHz, CDCl3) δ 176.8 (C-9), 172.4 (CONHCH2), 172.0 (NHCOCH2), 140.0 (C-10a), 134.6 (C-1), 133.2 (C-6), 133.0 (C-4a), 127.5 (C-4), 126.6 (C-3), 125.6 (C-8), 121.7 (C-7), 120.4 (C-8a), 119.6 (C-2), 117.9 (C-5), 117.3 (C-9a), 52.2 (CONHCH2CH2), 48.5 (NHCOCH2CH2), 46.9 (COCH2CH2N(CH2CH3)2), 46.6 (NHCH2CH2N(CH2CH3)2, 33.9 (CONHCH2), 29.7 (NHCOCH2), 11.3 (COCH2CH2N(CH2CH3)2), 10.1 (NHCH2CH2N(CH2CH3)2); HRMS (ESI+) m/z 480.2969 (calcd for C27H38N5O3+, 480.2973).
4.1.20 N-[2-(Dimethylamino)ethyl]-4-nitro-9-oxo-9H-xanthene-1-carboxamide (24a)
A mixture of acid 17 (500 mg, 1.75 mmol), triphenylphosphine (917 mg, 3.5 mmol) and NBS (717 mg, 4.03 mmol) in dry CH2Cl2 (50 mL) was stirred under argon, at 0 °C. After 25 min, N,N-dimethylethylenediamine (668 µL, 6.13 mmol) was added and stirring was continued at room temperature for 25 more minutes. Upon completion of the reaction, the mixture was washed with 5% sodium bicarbonate solution and water, dried over anh. Na2SO4 and evaporated to dryness. Flash chromatography on silica gel, using a mixture of CH2Cl2 / MeOH (10 / 1–7 / 1) as the eluent, afforded 570 mg (91%) of the title compound 24a. M.p. 116 – 118 °C (EtOAc); 1H NMR (400 MHz, CD3OD) δ 8.45 (d, J = 8.0 Hz, 1H, H-3), 8.25 (dd, J = 8.0 Hz, 1.3 Hz, 1H, H-8), 7.91 (td, J = 8.0 Hz, 1.3 Hz, 1H, H-6), 7.65 (d, J = 8.0 Hz, 1H, H-5), 7.54 (t, J = 8.0 Hz, 1H, H-7), 7.49 (d, J = 8.0 Hz, 1H, H-2), 3.66 (t, J = 8.0 Hz, 2H, CONHCH2), 2.86 (t, J = 8.0 Hz, 2H, CH2N(CH3)2), 2.50 (s, 6H, N(CH3)2); 13C NMR (50 MHz, CD3OD) δ 174.6 (C-9), 169.7 (CONH), 155.0 (C-10a), 148.4 (C-4a), 141.8 (C-1, C-4), 136.1 (C-6), 130.0 (C-3), 126.0 (C-8), 125.3 (C-7), 122.3 (C-2), 121.3 (C-8a), 120.0 (C-9a), 117.8 (C-5), 57.5 (CH2CH2N(CH3)2), 44.0 (N(CH3)2), 36.9 (CONHCH2); HRMS (ESI+) m/z 356.1241 (calcd for C18H18N3O5+, 356.1246).
4.1.21 N-[2-(Diethylamino)ethyl]-4-nitro-9-oxo-9H-xanthene-1-carboxamide (24b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 24a, using N,N-diethylethylenediamine. Yield: 94%. M.p. 175 – 177 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 8.30 (d, J = 8.4 Hz, 1H, H-3), 8.25 (dd, J = 8.4 Hz, 1.7 Hz, 1H, H-8), 7.82 (td, J = 8.4 Hz, 1.7 Hz, 1H, H-6), 7.60 (d, J = 8.4 Hz, 1H, H-5), 7.47 (td, J = 8.4 Hz, 1.7 Hz, 1H, H-7), 7.40 (d, J = 8.4 Hz, 1H, H-2), 6.93 (s, D2O exch., 1H, CONH), 3.68 (t, J = 7.1 Hz, 2H, CONHCH2), 2.87 (t, J = 7.1 Hz, 2H, CH2N(CH2CH3)2), 2.67 (q, J = 7.0 Hz, 4H, N(CH2CH3)2), 1.04 (t, J = 7.1 Hz, 6H, N(CH2CH3)2); 13C NMR (151 MHz, CDCl3) δ 174.5 (C-9), 168.0 (CONHCH2), 154.9 (C-10a), 148.8 (C-4a), 143.0 (C-1), 139.3 (C-4), 135.9 (C-6), 130.0 (C-3), 126.7 (C-8), 125.5 (C-7), 122.3 (C-2), 121.6 (C-8a), 120.5 (C-9a), 118.3 (C-5), 51.3 (CH2N(CH2CH3)2), 46.5 (N(CH2CH3)2), 37.1 (CONHCH2), 11.1 (N(CH2CH3)2); HRMS (ESI+) m/z 384.1554 (calcd for C20H22N3O5+, 384.1558).
4.1.22 4-[(Chloroacetyl)amino]-N-[2-(dimethylamino)ethyl]-9-oxo-9H-xanthene-1-carboxamide (26a)
This compound was synthesized by an analogous procedure as described for the preparation of compound 20a, starting from 24a. Yield: 25%. M.p. > 270 °C (EtOH); 1H NMR (600 MHz, CD3OD) δ 8.55 (d, J = 8.1 Hz, 1H, H-3), 8.25 (d, J = 8.0 Hz, 1H, H-8), 7.92 (t, J = 8.0 Hz, 1H, H-6), 7.76 (d, J = 8.0 Hz, 1H, H-5), 7.51 (t, J = 8.0 Hz, 1H, H-7), 7.35 (d, J = 8.1 Hz, 1H, H-2), 4.47 (s, 2H, NHCOCH2) 3.85 (t, J = 7.4 Hz, 2H, CONHCH2), 3.56 (t, J = 7.4 Hz, 2H, CH2N(CH3)2), 3.20 (s, 6H, N(CH3)2); 13C NMR (151 MHz, CD3OD) δ 178.4 (C-9), 173.7 (CONHCH2), 168.0 (NHCOCH2), 156.7 (C-10a), 148.7 (C-1), 137.5 (C-6), 132.1 (C-4a), 129.6 (C-4), 128.0 (C-3), 127.5 (C-8), 126.3 (C-7), 123.7 (C-2), 122.4 (C-8a), 119.4 (C-9a), 119.3 (C-5), 60.1 (CH2N(CH3)2), 44.6 (N(CH3)2), 44.0 (NHCOCH2), 36.1 (CONHCH2); HRMS (ESI+) m/z 402.1215 (calcd for C20H21ClN3O4+, 402.1219).
4.1.23 4-[(Chloroacetyl)amino]-N-[2-(diethylamino)ethyl]-9-oxo-9H-xanthene-1-carboxamide (26b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 20a, starting from 24b. Yield: 25%. M.p. 260–262 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 9.46 (s, D2O exch., 1H, NHCO), 8.52 (s, D2O exch., 1H, CONHCH2), 8.41 (d, J = 8.1 Hz, 1H, H-3), 8.06 (d, J = 8.1 Hz, 1H, H-8), 7.66 (t, J = 8.1 Hz, 1H, H-6), 7.50 (d, J = 8.1 Hz, 1H, H-5), 7.32 (t, J = 8.1 Hz, 1H, H-7), 7.09 (d, J = 8.1 Hz, 1H, H-2), 4.40 (s, 2H, NHCOCH2), 3.82 (q, J = 7.0 Hz, 2H, CONHCH2), 3.41 (t, J = 7.1 Hz, 2H, CH2N(CH2CH3)2), 3.24 (q, J = 8.1 Hz, 4H, N(CH2CH3)2), 1.34 (t, J = 8.1 Hz, 6H, N(CH2CH3)2); 13C NMR (151 MHz, CDCl3) δ 176.0 (C-9), 170.6 (CONHCH2), 164.8 (NHCOCH2), 154.6 (C-10a), 145.6 (C-1), 135.4 (C-6), 131.7 (C-4a), 128.7 (C-4), 126.3 (C-8), 124.7 (C-7), 124.2 (C-3), 123.0 (C-2), 121.3 (C-8a), 118.1 (C-9a), 117.8 (C-5), 52.3 (CH2N(CH2CH3)2), 46.9 (N(CH2CH3)2), 43.4 (NHCOCH2), 35.3 (CONHCH2), 8.9 (N(CH2CH3)2); HRMS (ESI+) m/z 430.1528 (calcd for C22H25ClN3O4+, 430.1533).
4.1.24 N-[2-(Dimethylamino)ethyl]-4-[2-(dimethylamino)acetamido]-9-oxo-9H-xanthene-1-carboxamide (27a)
This compound was synthesized by an analogous procedure as described for the preparation of compound 21a, starting from 26a. Yield: 84%. M.p. 149–150 °C (EtOAc); 1H NMR (600 MHz, CDCl3) δ 8.75 (d, J = 8.1 Hz, 1H, H-3), 8.22 (dd, J = 8.1 Hz, 1.5 Hz, 1H, H-8), 7.74 (td, J = 8.1 Hz, 1.5 Hz, 1H, H-6), 7.46 (d, J = 8.1 Hz, 1H, H-5), 7.39 (td, J = 8.1 Hz, 1.5 Hz, 1H, H-7), 7.29 (d, J = 8.1 Hz, 1H, H-2), 3.75 (q, J = 7.0 Hz, 2H, CONHCH2), 3.22–3.16 (m, 4H, CH2CH2N(CH3)2, NHCOCH2), 2.68 (s, 6H, CH2CH2N(CH3)2), 2.51 (s, 6H, COCH2N(CH3)2); 13C NMR (151 MHz, CDCl3) δ 176.5 (C-9), 170.4 (CONHCH2), 169.3 (NHCOCH2), 154.9 (C-10a), 145.8 (C-1), 135.1 (C-6), 131.8 (C-4a), 128.3 (C-4), 126.1 (C-8), 124.8(C-7), 123.9 (C-3), 123.4 (C-2), 122.0 (C-8a), 118.7 (C-9a), 117.6 (C-5), 63.8 (NHCOCH2), 58.0 (CH2CH2N(CH3)2), 46.3 (COCH2N(CH3)2), 44.7 (CH2CH2N(CH3)2), 36.2 (CONHCH2); HRMS (ESI+) m/z 411.2027 (calcd for C22H27N4O4+, 411.2029).
4.1.25 N-[2-(Diethylamino)ethyl]-4-[2-(diethylamino)acetamido]-9-oxo-9H-xanthene-1-carboxamide (27b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 27a, using diethylamine. Yield: 89%. M.p. 123–125 °C (EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.85 (d, J = 8.1 Hz, 1H, H-3), 8.25 (d, J = 8.1, 1.5 Hz, 1H, H-8), 7.77 (t, J = 8.1 Hz, 1.5 Hz, 1H, H-6), 7.49 (d, J = 8.1 Hz, 1H, H-5), 7.42 (t, J = 8.1 Hz, 1.5 Hz, 1H, H-7), 7.33 (d, J = 8.1 Hz, 1H, H-2), 3.82 (q, J = 7.1 Hz, 2H, CONHCH2), 3.30 (m, 8H, CH2CH2N(CH2CH3)2, NHCOCH2, CH2CH2N(CH2CH3)2), 2.78 (q, J = 7.0 Hz, 4H, COCH2N(CH2CH3)2), 1.29 (t, J = 7.1 Hz, 6H, CH2CH2N(CH2CH3)2), 1.23 (t, J = 7.1 Hz, 6H, COCH2N(CH2CH3)2); 13C NMR (50 MHz, CDCl3) δ 176.6 (C-9), 171.9 (CONHCH2), 171.8 (NHCOCH2), 155.1 (C-10a), 146.2 (C-1), 136.0 (C-6), 130.8 (C-4a), 128.4 (C-4), 126.2 (C-8), 125.0 (C-7), 123.8 (C-3), 122.8 (C-2), 121.2 (C-8a), 118.0 (C-9a), 117.4 (C-5), 58.2 (NHCOCH2), 52.3 (CH2CH2N(CH2CH3)2), 48.6 (COCH2N(CH2CH3)2), 47.4 (CH2CH2N(CH2CH3)2), 35.1 (CONHCH2), 11.8 (COCH2N(CH2CH3)2), 8.5 (CH2CH2N(CH2CH3)2); HRMS (ESI+) m/z 467.2653 (calcd for C26H35N4O4+, 467.2658).
4.1.26 4-(Acryloylamino)-N-[2-(dimethylamino)ethyl]-9-oxo-9H-xanthene-1-carboxamide (28a)
This compound was synthesized by an analogous procedure as described for the preparation of compound 22a, starting from 24a. Yield: 80%. M.p. 155–157 °C (EtOH); 1H NMR (600 MHz, CD3OD) δ 8.54 (d, J = 8.0 Hz, 1H, H-3), 8.18 (dd, J = 8.1 Hz, 1.3 Hz, 1H, H-8), 7.86 (td, J = 8.1 Hz, 1.3 Hz, 1H, H-6), 7.73 (d, J = 8.1 Hz, 1H, H-5), 7.46 (t, J = 8.1 Hz, 1H, H-7), 7.28 (d, J = 8.0 Hz, 1H, H-2), 6.76 (dd, J = 16.1, 10.0 Hz, 1H, CH = CH2), 6.51 (dd, J = 16.1, 1.3 Hz, 1H, CH = CH2), 5.92 (dd, J = 10.0 Hz, 1.3 Hz, 1H, CH = CH2), 3.67 (t, J = 7.6 Hz, 2H, CONHCH2), 2.92 (t, J = 7.6 Hz, 2H, CH2N(CH3)2), 2.56 (s, 6H, N(CH3)2); 13C NMR (151 MHz, CD3OD) δ 175.9 (C-9), 171.3 (CONHCH2), 165.1 (NHCOCH = CH2), 155.0 (C-10a), 147.1 (C-4a), 135.3 (C-6), 132.1 (C-1), 130.6 (CH = CH2), 128.3 (C-9a), 127.6 (CH = CH2), 126.3 (C-3), 125.8 (C-8), 124.5 (C-7), 122.4 (C-2), 121.2 (C-8a), 118.2 (C-4), 117.8 (C-5), 57.6 (CH2N(CH3)2), 43.9 (N(CH3)2), 36.7 (CONHCH2); HRMS (ESI+) m/z 380.1605 (calcd for C21H22N3O4+, 380.1608).
4.1.27 4-(Acryloylamino)-N-[2-(diethylamino)ethyl]-9-oxo-9H-xanthene-1-carboxamide (28b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 28a, starting from 24b. Yield: 83%. M.p. 265–267 °C (EtOH); 1H NMR (400 MHz, CD3OD) δ 8.68 (d, J = 8.1 Hz, 1H, H-3), 8.26 (d, J = 8.1 Hz, 1H, H-8), 7.96 (t, J = 8.1 Hz, 1H, H-6), 7.86 (d, J = 8.1 Hz, 1H, H-5), 7.55 (t, J = 8.1 Hz, 1H, H-7), 7.39 (d, J = 8.1 Hz, 1H, H-2), 6.83 (dd, J = 16.0, 10.1 Hz, 1H, CH = CH2), 6.51 (d, J = 16.0 Hz, 1H, CH = CH2), 5.93 (d, J = 10.1 Hz, 1H, CH = CH2), 3.90 (t, J = 8.1 Hz, 2H, CONHCH2), 3.65–3.51 (m, 6H, CH2N(CH2CH3)2, N(CH2CH3)2), 1.49 (t, J = 8.1 Hz, 6H, N(CH2CH3)2); 13C NMR (50 MHz, CD3OD) δ 177.0 (C-9), 172.2 (CONHCH2), 165.2 (NHCOCH = CH2), 155.3 (C-10a), 147.3 (C-4a), 136.0 (C-6), 131.2 (C-1), 130.6 (CH = CH2), 128.8 (C-9a), 127.8 (CH = CH2), 126.7 (C-3), 125.8 (C-8), 124.9 (C-7), 122.4 (C-2), 120.9 (C-8a), 118.2 (C-5), 117.9 (C-4), 52.8 (CH2N(CH2CH3)2), 46.6 (N(CH2CH3)2) 34.3 (CONHCH2), 7.9 (N(CH2CH3)2); HRMS (ESI+) m/z 408.1918 (calcd for C23H26N3O4+, 408.1920).
4.1.28 N-[2-(Dimethylamino)ethyl]-4-[3-(dimethylamino)propanamido]-9-oxo-9H-xanthene-1-carboxamide (29a)
This compound was synthesized by an analogous procedure as described for the preparation of compound 23a, starting from 28a. Yield: 91%. M.p. 213–215 °C (MeOH - Et2O); 1H NMR (600 MHz, CD3OD) δ 8.64 (d, J = 8.1 Hz, 1H, H-3), 8.25 (d, J = 8.1 Hz, 1H, H-8), 7.93 (t, J = 8.1 Hz, 1H, H-6), 7.84 (d, J = 8.1 Hz, 1H, H-5), 7.51 (t, J = 8.1 Hz, 1H, H-7), 7.34 (d, J = 8.1 Hz, 1H, H-2), 3.85 (t, J = 7.4 Hz, 2H, CONHCH2), 3.52 (t, J = 7.4 Hz, 2H, NHCH2CH2N(CH3)2), 3.35 (t, J = 7.4 Hz, 2H, NHCOCH2CH2N(CH3)2), 3.14 (s, 6H, NHCH2CH2N(CH3)2), 3.12 (t, J = 7.4 Hz, 2H, COCH2CH2N(CH3)2), 2.84 (s, 6H, COCH2CH2N(CH3)2); 13C NMR (151 MHz, CD3OD) δ 178.4 (C-9), 173.6 (CONHCH2), 171.9 (NHCOCH2), 156.7 (C-10a), 148.3 (C-1), 137.3 (C-6), 132.5 (C-4a), 130.2 (C-4), 127.5 (C-8), 127.5 (C-3), 126.2 (C-7), 123.8 (C-2), 122.5 (C-8a), 119.4 (C-9a), 119.4 (C-5) 59.8 (NHCH2CH2N(CH3)2), 55.3 (COCH2CH2N(CH3)2), 44.7 (NHCH2CH2N(CH3)2), 44.4 (COCH2CH2N(CH3)2), 36.4 (CONHCH2), 33.0 (NHCOCH2); HRMS (ESI+) m/z 425.2183 (calcd for C23H29N4O4+, 425.2185).
4.1.29 N-[2-(Diethylamino)ethyl]-4-[3-(diethylamino)propanamido]-9-oxo-9H-xanthene-1-carboxamide (29b)
This compound was synthesized by an analogous procedure as described for the preparation of compound 29a, using diethylamine. Yield: 85%. M.p. 167–169 °C (MeOH - Et2O); 1H NMR (400 MHz, CD3OD) δ 8.61 (d, J = 8.1 Hz, 1H, H-3), 8.21 (d, J = 8.1 Hz, 1H, H-8), 7.87 (t, J = 8.1 Hz 1H, H-6), 7.67 (d, J = 8.1 Hz, 1H, H-5), 7.47 (t, J = 8.1 Hz, 1H, H-7), 7.25 (d, J = 8.1 Hz, 1H, H-2), 3.57 (t, J = 7.5 Hz, 2H, CONHCH2), 2.94 (t, J = 7.5 Hz, 2H, COCH2CH2N(CH2CH3)2), 2.88 (t, J = 7.5 Hz, 2H, NHCH2CH2N(CH2CH3)2), 2.74 (m, 10H, NHCH2CH2N(CH2CH3)2), NHCOCH2CH2N(CH2CH3)2), 1.16 (s, 6H, COCH2CH2N(CH2CH3)2), 1.14 (s, 6H, NHCH2CH2N(CH2CH3)2); 13C NMR (50 MHz, CD3OD) δ 175.7 (C-9), 172.6 (CONHCH2), 171.2 (NHCOCH2), 155.1 (C-10a), 146.5 (C-1), 135.3 (C-6), 131.9 (C-4a), 128.6 (C-4), 126.0 (C-8), 125.4 (C-3), 124.5 (C-7), 122.8 (C-2), 121.4 (C-8a), 118.3 (C-9a), 117.5 (C-5) 50.7 (NHCH2CH2N(CH2CH3)2), 47.9 (COCH2CH2N(CH2CH3)2), 47.3 (NHCH2CH2N(CH2CH3)2), 46.9 (COCH2CH2N(CH2CH3)2), 36.8 (NHCH2CH2N(CH2CH3)2), 33.0 (COCH2CH2N(CH2CH3)2), 10.2 (COCH2CH2N(CH2CH3)2); HRMS (ESI+) m/z 481.2809 (calcd for C27H37N4O4+, 481.2813).
4.2 Biological assays and experiments
4.2.1 Cell viability Resazurin assays
For the maintenance and culturing of the MCF-7 human breast cancer cells, which were provided by ATCC-LGC Standards GmbH (Wesel, Germany), EMEM, containing EBSS supplemented with 2 mM Glutamine, 1% Non-Essential Amino Acids (NEAA) and 10% Fetal Bovine Serum (FBS), was used as growth medium. Sub-culturing routine was performed via splitting of sub-confluent cultures (70–80%) 1:2 to 1:6 times, for succeeding an optimal seeding of 2–4 × 104 cells / cm2, using 0.25% Trypsin (or Trypsin / EDTA), as detachment agent. Cells were grown in constant chamber conditions of 5% CO2, 37 °C and ≥ 95% relative humidity.
The drug-sensitive CCRF-CEM and doxorubicin-resistant CEM/ADR5000 ALL cells were grown in RPMI medium supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 µg/ml). CCRF-CEM and CEM/ADR5000 cells being obtained from exponential-phase cultures were counted and seeded into 96-well plates. The seeding density was ∼ 100 cells per well for both cell lines. Cells were, then, exposed to tested agents at a single 30 µM concentration. After a 72 h incubation period in a 5% CO2 incubator, under>95% humidified environment, 20 µL of Resazurin (0.01% w/v) were added to each well and the plates were further incubated at 37 °C for 4 h. Fluorescence was measured on an Infinite M2000 Pro plate reader (Tecan, Crailsheim, Germany). Compounds were defined as active if the mean cell viability was<32% in one or both cell lines. Viability was evaluated based on comparison with untreated cells. For active compounds, the same procedure was repeated using 0.03, 0.1, 0.3, 1, 3, 10 and 30 µM dose against the sensitive and resistant cell line. Dose-response curves were generated by plotting the mean cell viability (%) against the concentration of each examined compound (µM). IC50 values were calculated from a calibration curve by linear regression using Microsoft Excel.
4.2.2 Cell viability MTT assays
PC-3 is an androgen insensitive, p53-negative and K-Ras mutated human prostate cancer cell line, which was obtained from ATCC-LGC Standards GmbH (Wesel, Germany). PC-3 cells were cultured in DMEM media containing 10% FBS. To test the inhibitory activities of compounds using a cell-based protocol, MTT assay was performed for cell viability. Briefly, cells (750 / well) were seeded in 96-well plates, incubated overnight at 37 °C in 5% CO2 and treated with the compounds in a dose-dependent manner for 96 h. Dimethyl sulfoxide (DMSO) was used as the vehicle control. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was added at a concentration of 5 mg/ml directly to each well for 4 h at 37 °C. The medium was aspirated and the blue MTT formazan precipitate was dissolved in DMSO. Absorbance was determined in a Powerwave microplate spectrophotometer (Biotek Instruments, Inc., Vermont, USA) at 540 nm. Viable cell numbers were determined by tetrazolium conversion to its formazan dye. Each experiment was performed in triplicate.
The T24 human urinary bladder cancer cell line (major mutation signature: HRASG12V; p53ΔY126) was provided by ATCC-LGC Standards GmbH (Wesel, Germany), while the WM2664 human metastatic melanoma (skin cancer) cell line (major mutation signature: BRAFV600D) was purchased from ECACC-Sigma-Aldrich (Missouri, USA). Cells were cultured and exponentially grown in complete DMEM medium supplemented with 10% FBS, in a 5% CO2 environment and at 37 °C. For the MTT assay, cells were seeded onto 48-well plates at approximately 80% confluency and subsequently treated with different doses of the herein examined four compounds, for 24 h. Next, cells were incubated with MTT solution for 4 h and the formazan crystals being produced were carefully dissolved in isopropanol. Spectrophotometric absorbance was measured via engagement of a Dynatech MR5000 Elisa microtiter-plate reader (Dynatech Laboratories, Virginia, USA), at 550 nm, using 630 nm as the wavelength of reference. Each MTT assay was repeated three times, using 3 wells per tested compound and cell line. All four compounds were dissolved in DMSO. Statistical significance of differences being observed in compound-treated versus control (DMSO) cell-survival values was determined using the unpaired, two-sided Student’s t-test. Data are herein being reported as mean ± SD (standard deviation) of the mean value. P < 0.001 was considered statistically significant.
4.2.3 DNA staining and flow cytometric analysis of apoptosis and autophagy
Exponentially growing PC-3 human prostate cancer cells were treated with the IC50 values of the compounds 18a and 18b or the corresponding DMSO concentration (vehicle) for 24, 48 and 72 h. For cell cycle analysis, cell culture supernatants and attached cells were collected, centrifuged, washed in PBS, fixed in 50% ethanol and stained with an RNase-containing propidium iodide solution (50 µg/ml). For cell apoptosis assay, cells were harvested and stained with Annexin V binding buffer, Annexin V-FITC and PI (Annexin V-FITC Apoptosis Detection Kit, BD Systems) and were kept in the dark at room temperature for 15 min before being analyzed. DNA content was analyzed on a BD Accuri C6 Flow Cytometer, using the BD CSampler software (BD Biosciences, USA). Non-apoptotic events were used to calculate the percentage of cells distributed in each phase. A P value < 0.05 was considered to be statistically significant (Student’s t-test). Finally, quantification of autophagy was conducted using Cyto-ID Autophagy Detection Kit (Enzo Life Sciences, USA), according to manufacturer’s instructions. ΔMFI Cyto-ID values were calculated for each treatment relative to control, as values > 0 indicate autophagy activation, whereas values < 0 indicate inhibition of autophagic flux.
Acknowledgements
The research work was funded by the Hellenic Foundation for Research and Innovation (HFRI) under the HFRI PhD Fellowship grant (Fellowship Number: 1559/27.04.2018). This work was supported by the Special Account for Research Grants and the National and Kapodistrian University of Athens.
Declaration of Competing Interest
The authors declared that there is no conflict of interest.
References
- Rational design, synthesis and biological evaluation of thiadiazinoacridines: a new class of antitumor agents. Bioorg. Med. Chem.. 2003;11:399-405.
- [CrossRef] [Google Scholar]
- Potential antitumor agents. 50. In vivo solid-tumor activity of derivatives of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J. Med. Chem.. 1987;30:664-669.
- [CrossRef] [Google Scholar]
- Baker, G.P., Mann, F.G., Sheppard, N., Tetlow, A.J., 1965. 681. The structure of o-iodosobenzoic acid and of certain derivatives. J. Chem. Soc. 3721–3728. 10.1039/JR9650003721.
- Acridine and acridone derivatives, anticancer properties and synthetic methods: where are we now? Anticancer agents. Med. Chem.. 2007;7:139-169.
- [CrossRef] [Google Scholar]
- Acridine/acridone: a simple scaffold with a wide range of application in oncology. Expert Opin. Ther. Pat.. 2008;18:1211-1224.
- [CrossRef] [Google Scholar]
- Searching for antimicrobial and antiviral xanthones: a molecular docking study coupled to multivariate analysis. Planta Med.. 2015;81:PM_58.
- [CrossRef] [Google Scholar]
- Synthesis and cytotoxicity of potential anticancer derivatives of pyrazolo[3,4,5-kl]acridine and indolo[2,3-a]acridine. Tetrahedron. 2002;58:175-181.
- [CrossRef] [Google Scholar]
- Über die synthese von in 1-stellung substituierten 4-nitro. Helv. Chim. Acta. 1972;55:1948-1958.
- [CrossRef] [Google Scholar]
- Synthesis and antiinflammatory evaluation of 9-Anilinoacridine and 9-Phenoxyacridine derivatives. J. Med. Chem.. 2002;45:4689-4694.
- [CrossRef] [Google Scholar]
- Reactivity of the acridine ring: A review. Synthesis. 2004;2004:313-325.
- [CrossRef] [Google Scholar]
- 5-[(Aminoalkyl)amino]imidazo[4,5,1-de]acridin-6-ones as a novel class of antineoplastic agents. Synthesis and biological activity. J. Med. Chem.. 1990;33:49-52.
- [CrossRef] [Google Scholar]
- Potential anti-allergic acridone alkaloids from the roots of Atalantia monophylla. Phytochemistry. 2008;69:2616-2620.
- [CrossRef] [Google Scholar]
- Suicide inhibitors of proteases. 1983. Lack of activity of halomethyl derivatives of some aromatic lactams. Eur. J. Med. Chem.. 1983;18:107-111.
- [Google Scholar]
- Development and validation of an LC–UV method for the quantification and purity determination of the novel anticancer agent C1311 and its pharmaceutical dosage form. J. Pharm. Biomed. Anal.. 2005;39:46-53.
- [CrossRef] [Google Scholar]
- Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res.. 2008;14:2405-2412.
- [CrossRef] [Google Scholar]
- Synthesis and cytotoxic activity of acronycine derivatives modified at the pyran ring. Chem. Pharm. Bull.. 1996;44:2165-2168.
- [CrossRef] [Google Scholar]
- Xanthones, a promising anti-inflammatory scaffold: structure, activity, and drug likeness analysis. Molecules. 2020;25:598.
- [CrossRef] [Google Scholar]
- Gene-specific intron retention serves as molecular signature that distinguishes melanoma from non-melanoma cancer cells in Greek patients. Int. J. Mol. Sci.. 2019;20:937.
- [CrossRef] [Google Scholar]
- Revisiting histone deacetylases in human tumorigenesis: the paradigm of urothelial bladder cancer. Int. J. Mol. Sci.. 2019;20:1291.
- [CrossRef] [Google Scholar]
- Design, synthesis, and evaluation of the antiproliferative activity of a series of novel fused xanthenone aminoderivatives in human breast cancer cells. J. Med. Chem.. 2007;50:1716-1719.
- [CrossRef] [Google Scholar]
- Synthesis and antiproliferative activity of some novel benzo-fused imidazo[1,8]naphthyridinones. Bioorg. Med. Chem. Lett.. 2015;25:2621-2623.
- [CrossRef] [Google Scholar]
- Acronycine derivatives as promising antitumor agents. Anticancer Drugs. 2002;13:445-449.
- [CrossRef] [Google Scholar]
- Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev.. 2011;25:460-470.
- [CrossRef] [Google Scholar]
- Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRAS(G12D)-driven lung tumors. Autophagy. 2013;9:1636-1638.
- [Google Scholar]
- 1-Ethyl-1H-3-nitrobenzopyrano[4,3,2-cd]isoindole: a novel heterocyclic ring system bearing an unusually labile deuterium-exchangeable aromatic proton. Tetrahedron Lett.. 2006;47:3681-3684.
- [CrossRef] [Google Scholar]
- Genomic and transcriptomic profiling of resistant CEM/ADR-5000 and sensitive CCRF-CEM leukaemia cells for unravelling the full complexity of multi-factorial multidrug resistance. Sci. Rep.. 2016;6:36754.
- [CrossRef] [Google Scholar]
- Acridine derivatives: a patent review (2009–2010) Expert Opin. Ther. Pat.. 2011;21:437-454.
- [CrossRef] [Google Scholar]
- Susceptibility of multidrug-resistant human leukemia cell lines to human interleukin 2-activated killer cells. Cancer Res.. 1990;50:6793-6799.
- [Google Scholar]
- Nitrogen functionalities in palladium-catalyzed reactions on solid supports: a case study. Eur. J. Org. Chem.. 2006;2006:1886-1898.
- [CrossRef] [Google Scholar]
- Design, synthesis, and antiproliferative activity of some new pyrazole-fused amino derivatives of the pyranoxanthenone, pyranothioxanthenone, and pyranoacridone ring systems: a new class of cytotoxic agents. J. Med. Chem.. 2002;45:2599-2609.
- [CrossRef] [Google Scholar]
- Design, synthesis, and antiproliferative activity of some novel aminosubstituted xanthenones, able to overcome multidrug resistance toward MES-SA/Dx5 cells. Bioorg. Med. Chem. Lett.. 2005;15:5057-5060.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel amino-substituted xanthenones and benzo[b]xanthenones: evaluation of their antiproliferative activity and their ability to overcome multidrug resistance toward MES-SA/D x 5 cells. Bioorg. Med. Chem.. 2006;14:2910-2934.
- [CrossRef] [Google Scholar]
- Design, synthesis and cell growth inhibitory activity of a series of novel aminosubstituted xantheno[1,2-d]imidazoles in breast cancer cells. Bioorg. Med. Chem.. 2008;16:3445-3455.
- [CrossRef] [Google Scholar]
- Synthesis of 9-anilinoacridine triazines as new class of hybrid antimalarial agents. Bioorg. Med. Chem. Lett.. 2009;10:6996-6999.
- [CrossRef] [Google Scholar]
- The antitumor triazoloacridone C-1305 is a topoisomerase II poison with unusual properties. Pharmacol. Mol. 2004
- [CrossRef] [Google Scholar]
- Induction of unique structural changes in guanine-rich DNA regions by the triazoloacridone C-1305, a topoisomerase II inhibitor with antitumor activities. Nucleic Acids Res.. 2005;33:6034-6047.
- [CrossRef] [Google Scholar]
- Molecular mechanism of the enzymatic oxidation investigated for imidazoacridinone antitumor drug, C-1311. Biochem. Pharmacol.. 2003;66:1727-1736.
- [CrossRef] [Google Scholar]
- Synthesis and anti-cancer activity of naturally occurring 2,5-diketopiperazines. Fitoterapia. 2014;98:91-97.
- [CrossRef] [Google Scholar]
- Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem.. 2000;267:5421-5426.
- [CrossRef] [Google Scholar]
- Phase II trial of pyrazoloacridine (NSC#366140) in patients with metastatic breast cancer. Invest. New Drugs. 2011;29:347-351.
- [CrossRef] [Google Scholar]
- A new class of nonsteroidal aromatase inhibitors: design and synthesis of chromone and xanthone derivatives and inhibition of the P450 enzymes aromatase and 17α-Hydroxylase/C17,20-Lyase. J. Med. Chem.. 2001;44:672-680.
- [CrossRef] [Google Scholar]
- Cytotoxicity of natural products and derivatives toward MCF-7 cell monolayers and cancer stem-like mammospheres. Phytomedicine. 2015;22:438-443.
- [CrossRef] [Google Scholar]
- Recent insight into the biological activities of synthetic xanthone derivatives. Eur. J. Med. Chem.. 2016;116:267-280.
- [CrossRef] [Google Scholar]
- Grade-dependent effects on cell cycle progression and apoptosis in response to doxorubicin in human bladder cancer cell lines. Int. J. Oncol.. 2009;34:137-160.
- [CrossRef] [Google Scholar]
- Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia. 2016;18:347-355.
- [CrossRef] [Google Scholar]
- Synthesis of new chalcone derivatives containing acridinyl moiety with potential antimalarial activity. Eur. J. Med. Chem.. 2010;45:745-751.
- [CrossRef] [Google Scholar]
- Acridine derivatives as anti-BVDV agents. Antiviral Res.. 2011;91:133-141.
- [CrossRef] [Google Scholar]
- Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production. Cancer Res.. 1991;51:2515-2520.
- [Google Scholar]
- Metabolic transformations of antitumor imidazoacridinone, C-1311, with microsomal fractions of rat and human liver. Acta Biochim. Pol.. 2007;54 831–838. 10.18388/abp.2007_3179
- [Google Scholar]
- In vitro assessment of novel transcription inhibitors and topoisomerase poisons in rhabdomyosarcoma cell lines. Cancer Chemother. Pharmacol.. 2009;64:1059-1069.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of Acridine derivatives as antimalarial agents. Chem. Med. Chem.. 2012;7:587-605.
- [CrossRef] [Google Scholar]
- Evaluation of pyrazoloacridine in patients with advanced pancreatic carcinoma. Invest. New Drugs. 1998;16:93-96.
- [CrossRef] [Google Scholar]
- Acridine and its derivatives: a patent review (2009–2013) Expert Opin. Ther. Pat.. 2014;24:647-664.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Synthetic procedures for compounds 1, 8, 10, 11, 14 and 15, 1H and 13C NMR spectra can be found online at https://doi.org/10.1016/j.arabjc.2020.09.025.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




