

Translate this page into:
Targeting tumor cells with pyrazolo[3,4-d]pyrimidine scaffold: A literature review on synthetic approaches, structure activity relationship, structural and target-based mechanisms
⁎Corresponding author at: College of Pharmacy, Jouf University, Sakaka, Aljouf 72341, Saudi Arabia. mhmdgwd@ju.edu.sa (Mohamed A. Abdelgawad)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Pyrazolo[3,4-d]pyrimidine had been attracted awesome interest due to its pharmacological potential especially as an anticancer. Several mechanisms of action were accounted for the anticancer potential of this privileged scaffold. Previous researches explained its role in binding with many receptors as cyclin-dependent kinases, epidermal growth factor receptor, Src kinase, m-TOR, and JAK kinase. Nevertheless, there is an incredible demand for the discovery of target-oriented compounds. In this review, we shed the light on the antitumor potential of this important fused heterocyclic system, different mechanisms of actions of this ring, and SAR studies towards many targets in a trial to pave the way for medicinal chemists to optimize this ring system in order to discover new anticancer agents with better selectivity and increased anticancer potential.
Keywords
Pyrazolopyrimidines
Anticancer agents
Kinases
EGFR
SAR

1 . Introduction
Cancer is among the most dangerous diseases characterized by limitless proliferative potential, growth signals, self-sufficiency, in addition to apoptotic and antiproliferative cues resistance (Ruiz-Torres et al., 2017; González et al., 2019; Caon et al., 2020; Rossari et al., 2020; El Azab et al., 2021). It was detected that many risk elements including smoking, consuming alcoholic beverages, insufficient consumption of vegetables and fruits and obesity were the most important causes of cancer (Alsamarrai et al., 2014; Gul et al., 2016; Onvani et al., 2017; Salem and Mackenzie, 2018; Hur et al., 2019; Barrea et al., 2021; Entwistle et al., 2021; Feng et al., 2021). In addition, in low-income nations, cervical cancer in women occurred due to infection with papilloma virus through sexual transmission (Randall and Ghebre, 2016; Habtu et al., 2018; Cohen et al., 2019; Hull et al., 2020). Cancer receives assistance from the neighboring cells, forming new blood vessels to obtain oxygen and nutrients, metastasizes by spreading via blood vessels and lymphatic system to other organs especially lung, liver, and bones (Popper, 2016; Abdelgawad et al., 2017; Fares et al., 2020; Follain et al., 2020; Steinbichler et al., 2020). It has been established that neither radiation nor surgery nor the combination of both strategies can effectively manage metastatic cancer (Garbe et al., 2020; Cornford et al., 2021; Witjes et al., 2021). Furthermore, conventional chemotherapy is being used to cure cancer, but this kind of therapy does not distinguish between cancer cells and normal cells causing many adverse effects (Egeblad and Werb, 2002; Kerbel and Kamen, 2004; Jackson and Bartek, 2009; Abdellatif et al., 2014b). In an attempt to avoid these side effects, new strategies using molecular targeting agents had been developed to make treatments more tumor-specific (Tietze et al., 2015; Chester et al., 2018; Mooney et al., 2018; Park et al., 2019). This strategy uses medications that inhibit the growth of tumor cell through interfering with specific receptors and signaling pathways, so this strategy is more selective against cancer cells (Ho et al., 2014; Fan et al., 2015; Wang et al., 2019; Pottier et al., 2020; Zhang et al., 2020).
Literature survey recorded that pyrazolo[3,4-d]pyrimidine fused ring drew great attention as a result of its diverse and versatile pharmacological potential due to the chemical similarity between this scaffold and purines (Schenone et al., 2004; Akbari, 2008; Bakr et al., 2012b; Abdellatif et al., 2014a; Bakr and Mehany, 2016; Falsini et al., 2017; Li et al., 2019). It exhibited many pharmacological activities including antiviral (Wang et al., 2018), anti-inflammatory (Yewale et al., 2012; Yadava et al., 2013; Bakr et al., 2016), antimicrobial (Abdel-Megid, 2020; Greco et al., 2020; Hassaneen et al., 2020), antimycobacterial (Kulkarni et al., 2021), antihypertensive (Xia et al., 1997; El-Hamouly et al., 2006), radioprotective (Ghorab et al., 2009; Ghorab et al., 2010; Ghorab et al., 2012), antioxidant (Hassan et al., 2011; Rashad et al., 2011; Kostić et al., 2021).
This scaffold had been documented in literature to exhibit anticancer activity through many different mechanisms (Abdelall et al., 2014; Abdellatif et al., 2014a; Bakr and Mehany, 2016; Bakr et al., 2017; Abdellatif and Bakr, 2021). This review discusses the efforts of researchers in medicinal chemistry in the discovery of novel pyrazolopyrimidines with anticancer potential focusing on the mechanism of action of these candidates with particular emphasis on their structure activity relationship (SAR) and the synthetic procedures for the preparation of the major pyrazolo[3,4-d]pyrimidines.
2 Synthetic approaches
A lot of strategies had been used for the synthesis of pyrazolo[3,4-d]pyrimidines depending on the starting materials.
2.1 From pyrazole derivatives
o-Aminoamide pyrazole I was documented as starting material for preparing many pyrazolopyrimidine derivatives through different pathways (Fig. 1). Treating the aminoamide pyrazole I with diethyl oxalate afforded pyrazolylaminoxoacetate intermediate, which cyclized to the corresponding pyrazolopyrimidine in 73% yield using reflux in glacial acetic acid (Pathway A) (Harb et al., 2005). Also, refluxing o-aminoamide I with the appropriate aromatic aldehydes employing acidic catalyst as Keggin type heteropolyacid H3PW12O40 and Pressler type, H14[NaP5W30O110] for 0.5–3 h afforded novel set of pyrazolopyrimidines (Pathway B) (Heravi et al., 2006). Davoodnia et al. (2006) reported the construction of some pyrazolopyrimidines utilizing one-pot two components reaction of orthoesters and o-aminoamide pyrazole I utilizing silica gel under microwave irradiation [Pathway C]. In addition, cyclizing o-aminoamide I with the appropriate aldehydes using equimolar amount of iodine as oxidizing agent in acetonitrile yielded the corresponding pyrazolopyrimidines (Pathway D) (Bakavoli et al., 2010). On the other hand, reacting o-aminocyanopyrazole II with trimethyl orthoformate in acetic anhydride gave pyrazolylimidoformate derivative. Reacting the latter with hydrazine hydrate followed by heating the product with dry benzene afforded the corresponding pyrazolopyrimidines (Pathway E) (Harb et al., 2005). Besides, refluxing o-aminocyanopyrazole II for 6 h with formic acid produced the corresponding pyrazolopyrimidine (Pathway F) (Yang et al., 2008). In 2019, a group of coworkers designed novel series of pyrazolopyrimidines employing [3 + 2]cycloaddition reaction (Rahmouni et al., 2019). First, refluxing o-aminocyanopyrazole II with acetic anhydride for 6 h followed by thermal intramolecular cyclization of the intermediate with piperidine in ethanol for 4 h (Pathway G). Reaction of o-aminocyanopyrazole II with p-nitrobenzoyl chloride followed by refluxing the pyrazole intermediate with H2O2/NaOH resulted in the pyrazolopyrimidine (Pathway H) (Kaplan et al., 2010). In addition, pyrazolopyrimidine ring could be obtained from o-aminoesterpyrazole III either by reaction with hydrazine hydrate and trimethyl orthoformate (Pathway I) (Ghorab et al., 2010) or chloroacetonitrile in dioxane (Pathway J) (Ogurtsov and Rakitin, 2021) and/or formamide (Pathway K) (Bakr et al., 2012a).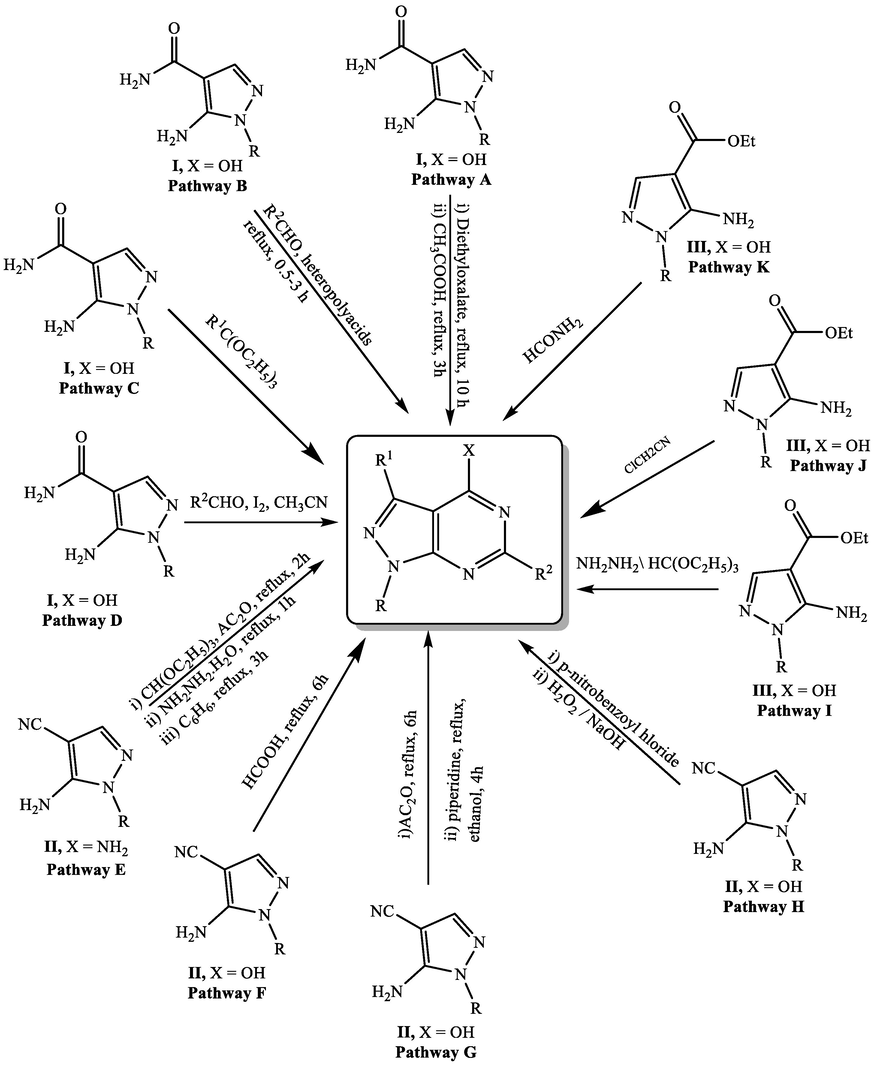
Synthetic strategies for pyrazolopyrimidines starting from substituted pyrazoles.
The mechanism of pyrazolopyrimidine formation from o-aminocyanopyrazole II using acetic anhydride (Pathway G) is demonstrated in Scheme 1 (Abdellatif and Bakr, 2018).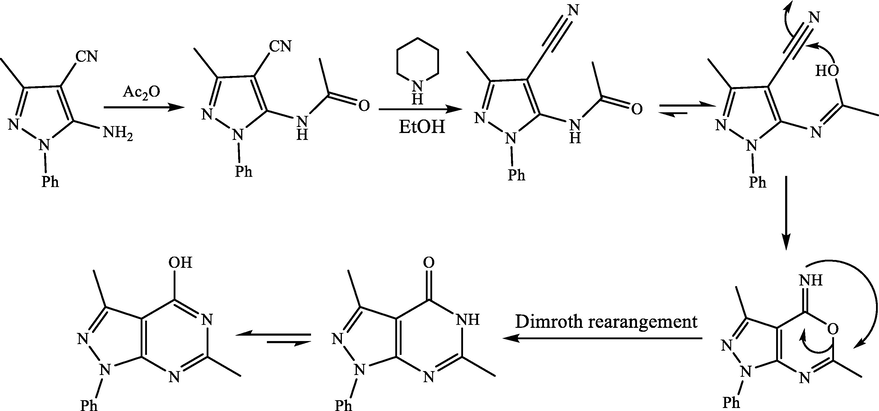
Mechanism of pyrazolopyrimidine formation.
2.2 From pyrimidine derivatives
Pyrimidine derivatives were reported in many publications as starting material for the construction of pyrazolopyrimidines. Refluxing 1,3-dimethyl-6-hydrazinouracil (III) with α-ketoalkynes in ethanol yielded the corresponding pyrazolopyrimidine (Pathway L) (Prajapati et al., 2006) (Fig. 2). In 2017, novel pyrazolopyrimidines were constructed from cyclizing 4-amino-6-chloropyrimidine-5-carbonitrile IV with isopropylhydrazine hydrochloride (Wang et al., 2017) (Pathway M).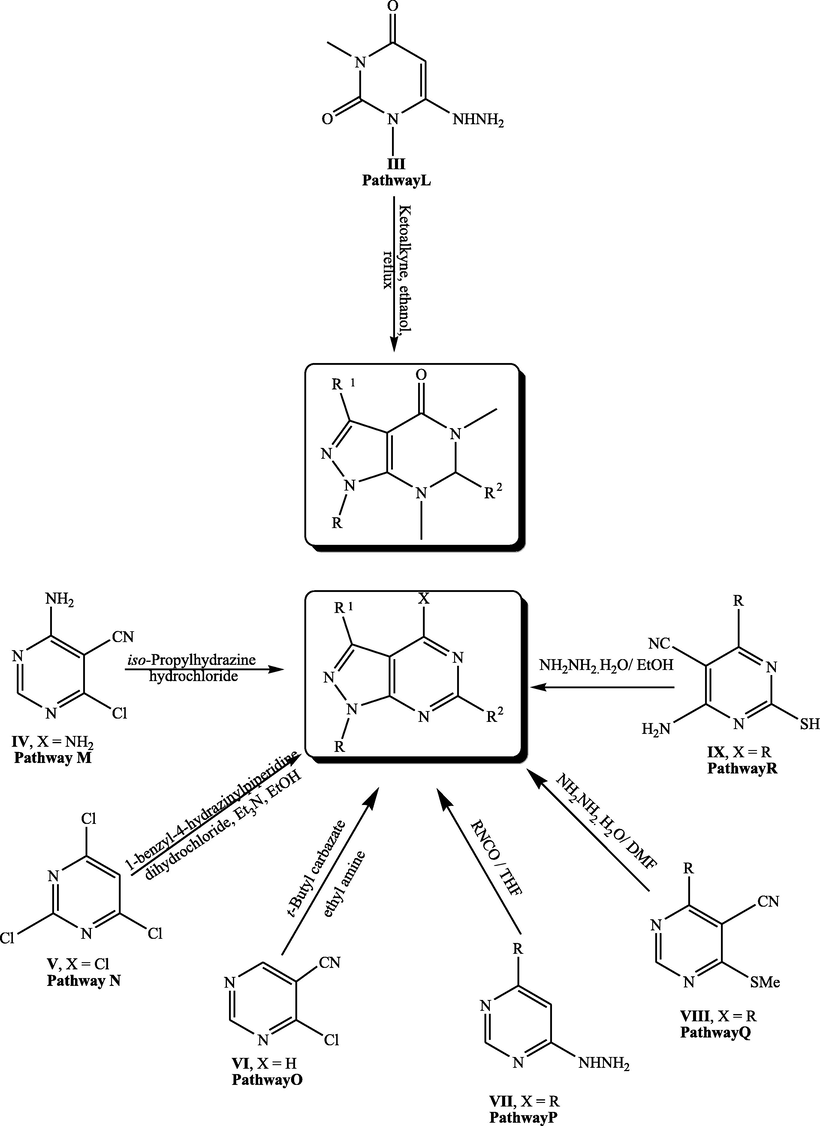
Synthetic strategies for construction of pyrazolopyrimidine starting from substituted pyrimidines.
In addition, pyrazolopyrimidines could be obtained from reacting trichloropyrimidine V with 1-benzyl-4-hydrazinylpiperidine dihydrochloride at 78 °C in presence of trimethyl amine (Pathway N) (Gilbert et al., 2010). Moreover, treating 4-chloro-5-cyanopyrimidine VI with tert-butyl carbazate in ethyl amine produced pyrazolopyrimidine (Pathway O) (Soth et al., 2011). Reacting o-aminocyanopyrazole VII with isocyanates in THF yielded pyrazolopyrimidines in one pot synthesis (Pathway P) (Chauhan and Kumar, 2013). In addition, refluxing 3-cyano-2-methylthiopyrimidine derivative VIII with hydrazine hydrate in dry DMF for 4 h afforded the pyrazolopyrimidine (Pathway Q) (Khobragade et al., 2010). Finally, treating pyrimidine derivatives IX with hydrazine hydrate in ethanol yielded the target pyrazolopyrimidines (Pathway R).
3 Mechanism of action of pyrazolopyrimidines as antitumor agents
3.1 Cyclin dependent kinase (CDK) suppressors
CDKs are kinases belonging to serine threonine kinases, which are responsible for transcription and cell cycle regulation, and the differentiation of cell (Kreis et al., 2019; Ding et al., 2020; Leal-Esteban and Fajas, 2020). The regulatory protein known as cyclin is bound by CDK and the human cdk2 structure has an altered ATP binding region, which modified by cyclin binding (Alexander et al., 2015; Henriques and Lindorff-Larsen, 2020). In absence of cyclin, CDK has little activity as T-loop (activation loop) blocks the cleft, and the position of key amino acid residues is not optimal for binding with ATP (Lu and Schulze-Gahmen, 2006; Liao, 2007). These CDK enzymes are overexpressed in cancer, and targeting CDK by many selective inhibitors like pyrazolopyrimidine derivatives is one of the strategies in curing cancer (Collins and Garrett, 2005; Malumbres and Barbacid, 2007; Musgrove et al., 2011).
Kim et al. (2003) prepared two pyrazolo[3,4-d]pyrimidine series 1a-d and 2a-g and evaluated the activity of these derivatives as CDK2 inhibitors. Structure activity study has been performed on these compounds, which showed that derivatives bearing anilino moiety as in compounds 2a-g showed better CDK2 inhibitory activity than the corresponding benzyl analogues as shown in derivatives 1a-d. The compounds 2d and 2e incorporating 3-fluoroanilino moiety at C-4 of pyrazolopyrimidine were as active as roscovitine (3) that served as the reference. Moreover, the unsubstituted compounds (2a, 2d, 2e, 2f, and 2 g) at N-1 exhibited higher CDK2 inhibitory potential compared to the substituted derivatives (2b, and 2c) (Fig. 3, Table 1).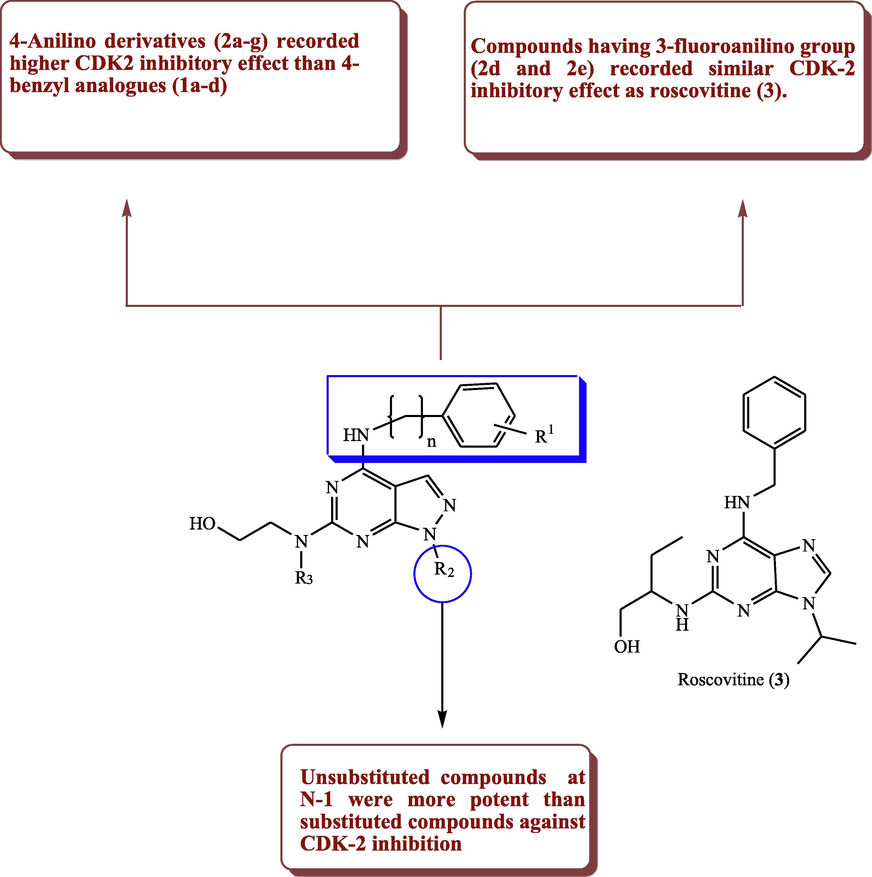
Substitution effect and SAR of some pyrazolopyrimidines with CDK-2 inhibitory potential.
C.N
R1
R2
R3
n (number of carbons)
CDK2 (IC50, µM)
C.N
R1
R2
R3
n (number of carbons)
CDK2 (IC50, µM)
1a
H
H
H
1
29.5
2c
3-Br
CH3
(CH2)2OH
0
>100
1b
H
CH3
(CH2)2OH
1
>100
2d
3-F
H
H
0
0.5
1c
H
CH(CH3)2
H
1
>100
2e
3-F
H
(CH2)2OH
0
0.9
1d
4-NO2
CH(CH3)2
H
1
46.8
2f
3-F
CH3
H
0
21.4
2a
3-Br
H
(CH2)2OH
0
3.8
2 g
3-F
CH3
(CH2)2OH
0
36.3
2b
3-Br
CH3
H
0
>100
3
–
–
–
–
0.5
Le Brazidec et al. (2012) prepared a new set of 1,6-disubstitutedpyrazolopyrimidines as kinase inhibitors, from this set, the target compound 4 (Fig. 4, Table 2) combined strong potency against Aurora-B kinases (AKA/AKB) and CDK1. Aurora family belongs to serine/threonine kinases, which are essential for mitotic cell division and for controlling the precise partition of the duplicated genome into two daughter cells (Al-Sanea et al., 2020). Aurora kinases overexpression has been implicated in many cancer types so, Aurora kinases are targeted for cancer therapy (Tang et al., 2017). SAR study displayed the influence of substituent lipophilicity at N-1 on potency and metabolic stability as explained in (Fig. 4). Furthermore, this study recorded that replacing the primary amide attached to the pyrrole ring at C-6 with disubstituted pyrrolidine amide moiety afforded the compound 5 with increased AKA/CDK1 inhibitory activity (Fig. 4). Furthermore, compound 6 (Fig. 4) had been designed as a roscovitine bioisostere and the anticancer potential of this compound 6 was screened on a panel of human cell lines (Jorda et al., 2011). The results revealed that this compound (6) recorded higher potential over roscovitine towards cancer cell lines, in addition, X-ray crystal structure of this pyrazolopyrimidine 6 binded with CDK2 recorded similar binding mode to that shown by roscovitine.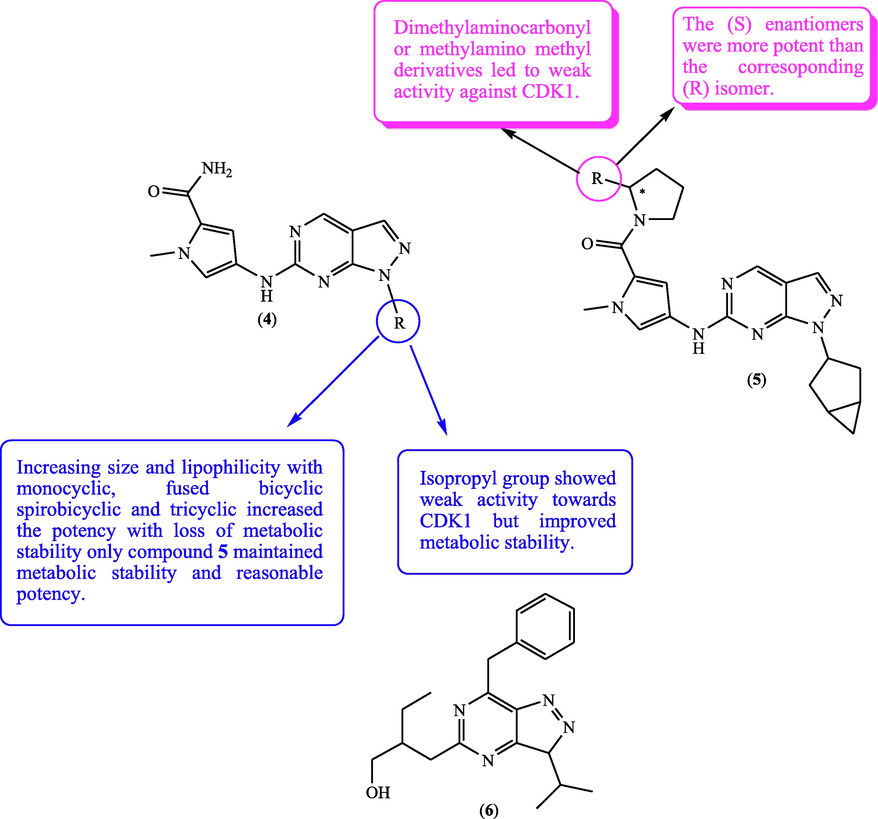
SAR and substitution effect of pyrazolopyrimidines on CDK1 inhibitory potential.
C.N.
R
CDK1 IC50 (µM)
HCT116 IC50 (µM)
cLogP
4

0.024
0.079
1.6
5a
(S)CH2OCH3
0.022
0.01
–
5b
(R)CH2OCH3
0.15
0.06
–
In addition, 4-amino-6-hydroxy-pyrazolopyrimidinisoxazolyl benzenesulphonamide derivative (7) (Fig. 5, Table 3) was documented to exhibit CDK2 inhibitory activity with IC50 = 0.24 µM and displayed excellent docking score (Ibrahim et al., 2009).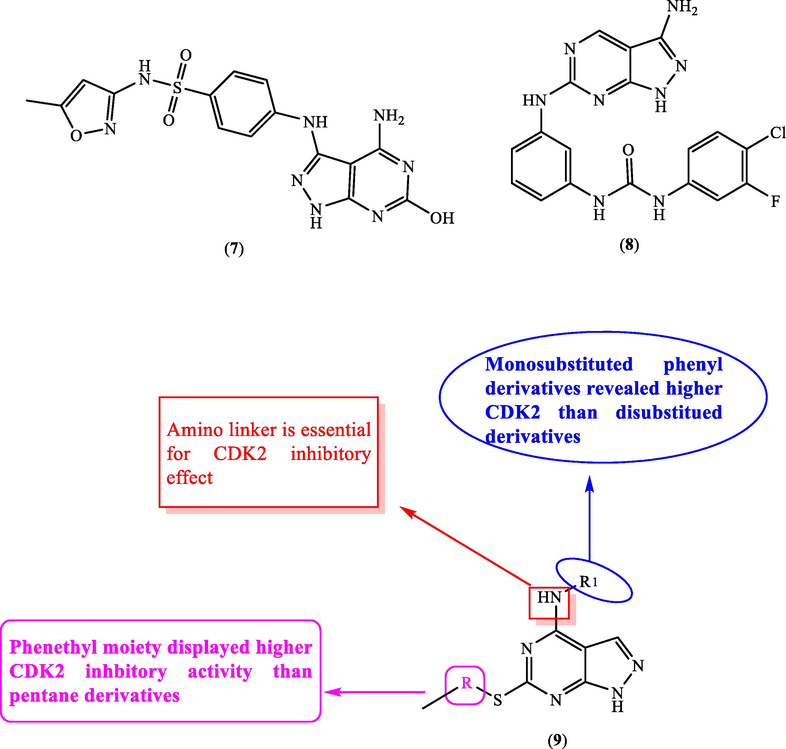
SAR and substitution effect on CDK2 inhibition.
C. N
R
R1
CDK2 IC50
Ab1 (µM)
K-526 (µM)
MCF-7 (µM)
7
–
–
0.24 µM
–
–
–
8
–
–
78 nM
–
–
–
9a
SCH2CH2Ph
3-NO2-Ph
5.1 µM
>25
>12.5
>12.5
9b
SCH2CH2Ph
4-Cl-3F-Ph
13.4 µM
>25
>6.25
>6.25
9c
S(CH2)3
3-NO2-Ph
17.7 µM
>25
>12.5
>12.5
Roscovitine
–
–
0.1 µM
>100
42
11
In 2012, Yang et al. (2012) prepared 3-aminopyrazolopyrimidinylaminophenylurea derivative (8) (Fig. 5, Table 3) which exhibited CDK1 inhibitory activity with IC50 = 78 nM. Recently, in 2018, new derivatives of 4,6-disubstituted pyrazolopyrimidines were constructed and screened for their inhibitory potential against CDK2 (Cherukupalli et al., 2018). SAR displayed that introducing thiophenethyl group at C6 as in 9a (IC50 = 5.1 µM) recorded better inhibitory activity than thiopentane group as in 9c (IC50 = 17.7 µM). In addition, monosubstitutedphenyl moiety at C-4 exhibited higher CDK2 inhibitory effect than the disubstituted phenyl group which is obvious upon comparing target candidate 9a (IC50 = 5.1 µM) with 9b (IC50 = 13.4 µM) (Fig. 5, Table 3).
Moreover, a set of 5-substituted-3-isopropylpyrazolo[3,4-d]pyrimidine derivative 10 substituted at C-5 was prepared and screened for their inhibitory effect towards CDK2 and CDK5. From the obtained outcomes, most of the prepared compounds were more potent (IC50 < 1 µM) than the standard purine compound CR8 (11) towards CDK2/CDK5 inhibition (Vymětalová et al., 2016). The optimized SAR reveled that substitution at C-5 markedly improved the activity as depicted in Fig. 6, Table 4.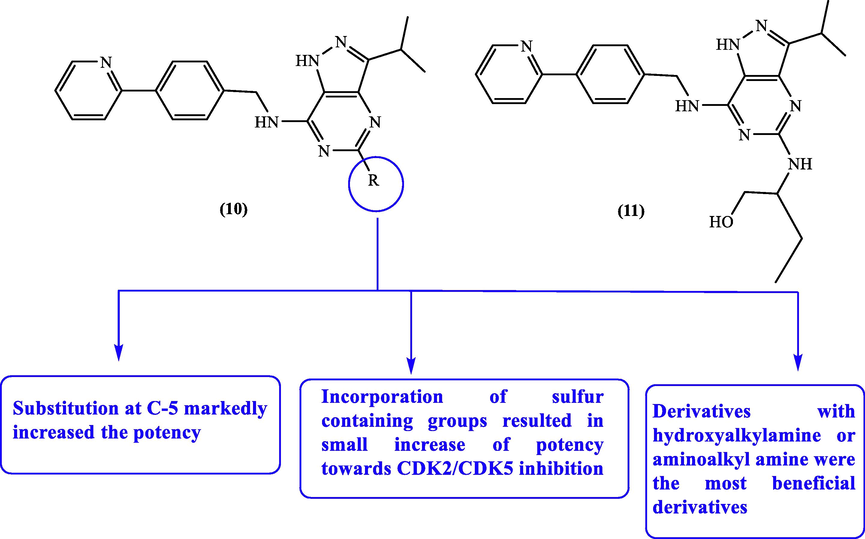
SAR studies of some pyrazolopyrimidines with CDK2/CDK5 inhibitory potential.
C.N
R
CDK2 IC50 (µM)
CDK5 IC50 (µM)
K-562 IC50 (µM)
MCF-7 IC50 (µM)
G361 IC50 (µM)
HCT-116 IC50 (µM)
10a
H
0.197
0.183
1.000
1.190
0.875
1.237
10b
SO2CH3
0.070
0.165
0.640
0.460
0.793
1.140
10c
NHCH2C(CH3)2
0.009
0.001
0.029
0.024
0.048
0.085
11 (CR8)
–
0.062
0.225
0.175
0.160
0.503
0.350
In addition, novel pyrazolo[3,4-d]pyrimidines incorporating benzenesulfonamide moiety were constructed and screened for their antiproliferative potential against MCF-7 and HepG2 cell lines (Hassan et al., 2017). Candidate 12a was the most active cytotoxic agent on MCF-7 cell lines with IC50 = 1.4 µM, while 12b was the most active on HepG2 with IC50 = 0.4 µM, respectively. Moreover, the mechanism of action of compound 12b was postulated to inhibit CDK2 enzyme with IC50 = 0.19 µM. SAR study showed that substitution at positions 4 and 6 was essential for the cytotoxic potential as illustrated in Fig. 7, Table 5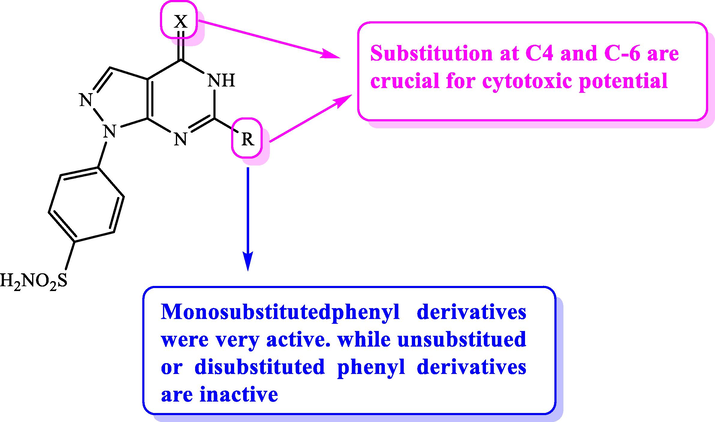
SAR analysis of some pyrazolopyrimidines.
C.N.
R
X
MCF-7
IC50 (µM)HepG2 IC50 (µM)
CDK2
IC50 (µM)
12a
4-OCH3—C6H4
O
1.4
>100
–
12b
C⚌O
NH
>100
0.4
0.19
DOX
–
–
1.02
0.90
–
3.2 Epidermal growth factor receptor (EGFR) inhibitors
EGFR belongs to trans-membrane growth factor receptor protein tyrosine kinases. EGFR has four members HER2 (known also as erbB2), HER1, HER3, and HER4. EGFR overexpression has been implicated in many cancer of epithelial origin (Rayego-Mateos et al., 2018).
In 2005, three series of pyridopyrazolopyrimidine 13a-c, 14, and 15a-f (Fig. 8, Table 6) were constructed and tested for their activity against EGFR, erbB2 kinases (Alberti et al., 2005). SAR study of these derivatives demonstrated that the primary amino-substituted derivative 14 showed no activity against all the tested kinases. While, introducing an anilino moiety instead of the free amino group afforded potent inhibition towards erbB2 and/or EGFR. Substitution on the anilino moiety determines the potency towards erbB2 and/or EGFR for example, 13a having a halogen disubstituted anilino group, showed more selectivity for EGFR than erbB2. While compound 13b incorporating benzyl ether substituted anilino moiety exhibited potent dual EGFR/erbB kinase inhibitory activity. In addition, replacing the anilino moiety with aryl urea diminished the activity against erbB2 or EGFR as in 15a-f. This diminished activity is attributed to inability of the 4-urea derivatives to bind with Thr830 as a result of the forming intramolecular H-bond between the tricyclic core nitrogen and the urea NH (Alberti et al., 2005).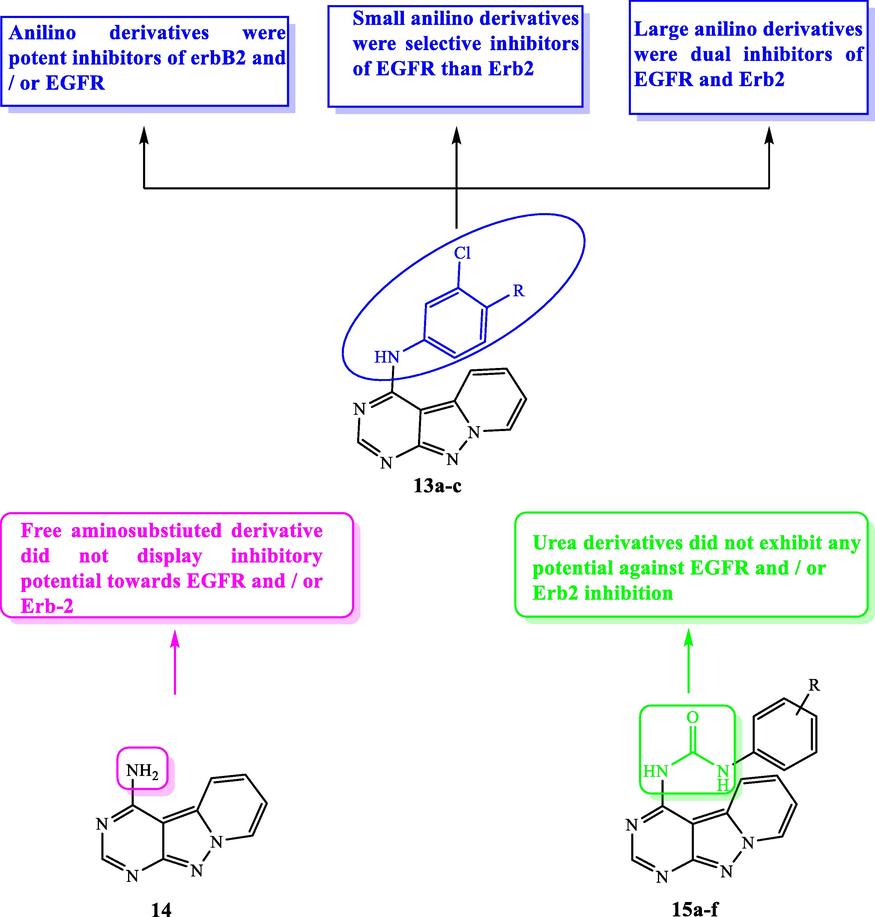
SAR and substitution effect of pyrazolopyrimidines as EGFR inhibitors.
C.N.
R
ErbB-2 IC50 (µM)
EGFR IC50 (µM)
C.N.
R
ErbB-2 IC50 (µM)
EGFR IC50 (µM)
13a
F
>10
0.20
15c
3-NO2
>10
>10
13b
OCH2-3-F—C6H4
0.063
0.032
15d
4-Br
>10
>10
13c
OCH2—C6H5
0.200
0.050
15e
4—CF3
>10
>10
15a
3—CN
>10
>10
15f
4F
>10
>10
15b
3—CF3
>10
>10
In 2006, N-(1,3-benzodioxolylmethylphenylpyrazolopyrimidin-4-amine (16) (Fig. 9, Table 7) was reported by Cavasotto et al. (2006) as a low-micromolar EGFR suppressor. Furthermore, a novel set of pyrazolo[3,4-d]pyrimidin-yloxy-4-substitutedbenzylidene acetohydrazide were prepared and assayed for antitumor activity towards breast carcinoma (MCF-7), non-small cell lung cancer (A549) and human colorectal adenocarcinoma (HT-29) cell lines (Abdelgawad et al., 2016). This study showed that compound 17 recorded the highest anticancer potential with (IC50) between 5.36 and 9.09 µM. Exposing this candidate to a small panel of kinases displayed that this candidate 17 was the most potent suppressor against EGFR with IC50 = 4.18 µM.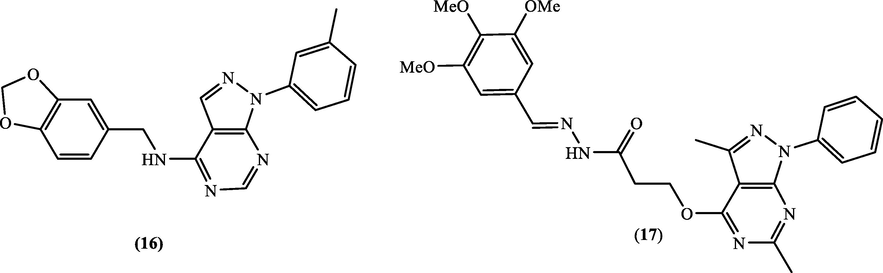
Chemical structures of pyrazolopyrimidines 16 and 17.
C.N.
MCF-7 IC50 (µM)
A549 IC50 (µM)
HT-29
IC50 (µM)EGFR IC50 (µM)
16
–
–
–
15
17
6.14 ± 2.13
9.09 ± 2.36
5.36 ± 1.98
4.18
DOX
2.01 ± 0.87
1.17 ± 0.35
ND
ND
In 2016, Bakr et al. (2017) prepared hybrids of pyrazolopyrimidine incorporating pyrazole scaffold 18a-f (Fig. 10, Table 8). These candidates were tested for their anticancer potential towards HCT-116, MCF-7 and HEPG-2 cell lines. All the tested compounds demonstrated good anticancer potential, especially 18d was the most potent with IC50 = 3.65, 1.45, and 2.00 μM towards HEPG-2, MCF-7 and HCT-116 cell lines, respectively. These compounds (18a-f) were screened for their inhibitory activity against EGFR and this study detected that all the tested derivatives exhibited inhibitory activity with IC50 = 8.27–18.82 µM.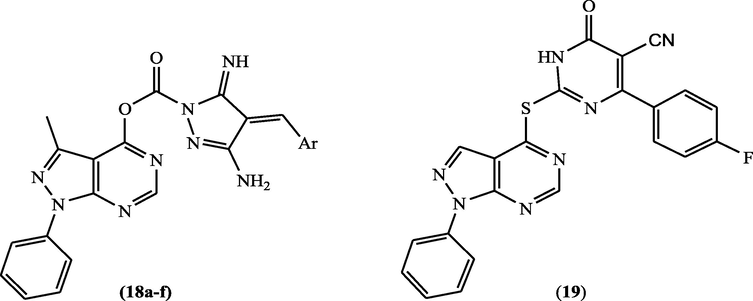
Structure of pyrazolopyrimidine derivatives 18a-f and 19 as EGFR inhibitors.
C.N.
Ar
HEPG-2 IC50 (µM)
MCF-7 IC50 (µM)
HCT-116 IC50 (µM)
EGFR IC50 (µM)
18a
3-OCH3—C6H4
6.86 ± 0.08
8.17 ± 0.04
7.96 ± 0.62
10.91
18b
4-OCH3—C6H4
5.52 ± 0.05
6.80 ± 0.02
5.87 ± 0.57
13.12
18c
4—Cl—C6H4
39.98 ± 0.42
54.19 ± 1.00
50.6 ± 1.52
18.82
18d
4-NO2—C6H4
3.65 ± 0.02
1.45 ± 0.03
2.00 ± 0.32
14.34
18e
2,3-(OCH3)2—C6H3
10.88 ± 0.09
16.81 ± 1.01
17.27 ± 0.91
8.27
18f
2,3,5-(OCH3)3—C6H2
11.01 ± 0.10
15.69 ± 0.07
11.58 ± 0.81
3.71
DOX
–
5.66 ± 0.12
2.60 ± 0.02
8.48 ± 0.32
–
Furthermore, in-silico docking study had been done on compound 18e which detected that this compound exhibited the energy score = -29.50 kcal/mol and demonstrated five H-bonds interactions between OCH3 with Thr766, NH2 with Asp831 and NH2 with Arg817 (Fig. 11) (Bakr et al., 2017).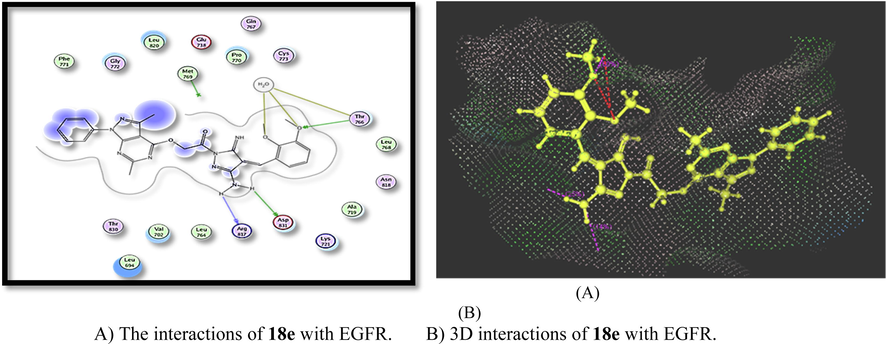
A) The interactions of 18e with EGFR. B) 3D interactions of 18e with EGFR.
Furthermore, in 2015, pyrazolo[3,4-d]pyrimidin-4-yl)thio]dihydropyrimidine-5-carbonitrile (19) (Fig. 10) was found to inhibit EGFR (91%) comparing to the standard drug gefitinb (100%) (Abbas et al., 2015).
In 2021, novel set of pyrazolopyrimidine was designed as mutant-EGFR inhibitors (Lamie et al., 2021). These derivatives were assessed for their anticancer activity against MCF-7, HCT-116 and W138 cell lines. It was found that compound 20d (Fig. 12, Table 9) was the most potent deivative towards MCF-7 (IC50 = 6.50 nM) while, 20c derivative was the most potent aginst HCT-116 (IC50 = 4.80 nM) comparing with the standard drug, lapatinib (IC50 = 21 nM, 12 nM, on MCF-7 and HCT-116, respectively). In addition, derivatives 20a-d were subjected to assessment towards EGFRT790M/HER2 inhibitory potential.the obtained data displayed that these candidates exhibited 81.81–65.70% and 86.66–54.49% inhibition to EGFRT790M and HER2, sequentially (Fig. 12, Table 9).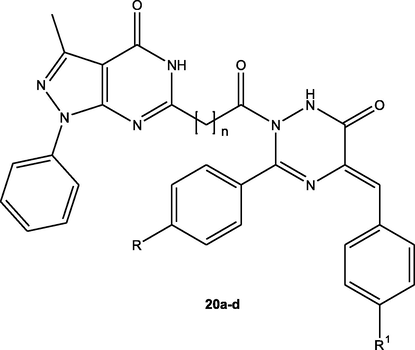
Chemical structure of pyrazolopyrimidines 20a-d as mutant EGFR inhibitors.
C.N.
n
R
R1
HCT-116 IC50 (nM)
MCF-7 IC50 (nM)
W138 IC50 (nM)
EGFR-T790M (% inhibition)
HER2 (% inhibition)
20a
0
CH3
Cl
5.60 ± 0.25
59.30 ± 2.20
26.40 ± 0.59
65.70%
54.49%
20b
0
Cl
Cl
68.90 ± 2.18
6.90 ± 1.40
47.20 ± 1.87
76.35%
83.61%
20c
1
CH3
Cl
4.80 ± 0.10
49.80 ± 1.70
29.40 ± 1.13
76.97%
72.61%
20d
1
Cl
OCH3
12.90 ± 0.54
6.50 ± 0.10
54.90 ± 2.16
81.81%
86.66%
Lapatinib
–
–
–
12.00 ± 0.48
21.00 ± 0.54
45.20 ± 0.96
77%
94%
3.3 Src kinase suppressors
Src kinases family represents a major set of nonreceptor tyrosine kinases, and its members play important roles in signal transduction and so are of basic importance for many biological functions, including proliferation, differentiation, migration, apoptosis, and angiogenesis (Sridhar and Miranti, 2006; Guarino, 2010). Literature survey studies afforded evidence that c-Src enzymes are involved in the generation and metastasis in many tumors, including prostate, head, colon, breast, neck, lung, and pancreatic cancer (Coleman et al., 2005).
4-Amino-5-(4-chlorophenyl)‑7‑(dimethylethyl)pyrazolo[3,4‑d]pyrimidine (PP2) (21) (Fig. 13) is one of the most prominent compounds used to suppress c-Src (Saito et al., 2010). Inhibiting Src by PP2 caused over-expression of markers representative of both epithelial polarization and intestinal terminal differentiation events, leading to up-regulation of Cdx2 and HNF1a and the reduction of polycomb PRC2-related epigenetic repressing activity, so Src family exert a critical role in regulating intestinal epithelial cell terminal differentiation (Seltana et al., 2013).![Structure of pyrazolo[3,4-d]pyrimidines 21 and 22 as Src kinase suppressors.](/content/184/2022/15/5/img/10.1016_j.arabjc.2022.103781-fig14.png)
Structure of pyrazolo[3,4-d]pyrimidines 21 and 22 as Src kinase suppressors.
Preparation of novel pyrazolo[3,4-d]pyrimidines along with evaluating their Src and cell line proliferation inhibitory activity (A431 and 8701-BC cells) had been carried out by Carraro et al (Carraro et al., 2006). From these derivatives, compound 22 (IC50 = 31.2 µM) showed Src inhibitory activity higher than the reference compound (21) (IC50 = 61.8 µM) by two-folds (Fig. 13). Unfortunately, compound 22 showed very low solubility in aqueous media and to enhance solubility Dreassi and coworkers solubilized this derivative with 30% w/v 2-hydroxypropyl-b-cyclodextrin solution. This method increased solubility of pyrazolopyrimidine derivative 22 and improved the antiproliferative potential against osteosarcoma (SaOS-2) and leukemic (K-652,KU-812 and HL-60) cell lines in comparison with the not complexed compounds (Dreassi et al., 2010).
In addition, a novel set of 4-amino substituted pyrazolopyrimidines 23a-d (Fig. 14, Table 10) having a 2-chloro-2-phenylethylamino at N1 and thio-methyl or -propyl groups at C6 was synthesized and screened for their Src kinase inhibitory activity (Schenone et al., 2008). All of the screened derivatives demonstrated potent inhibitory activity with Ki values = 0.21–0.51 µM.![SAR and substitution effect of pyrazolo[3,4-d]pyrimidines on Src inhibitory effect.](/content/184/2022/15/5/img/10.1016_j.arabjc.2022.103781-fig15.png)
SAR and substitution effect of pyrazolo[3,4-d]pyrimidines on Src inhibitory effect.
C.N.
R
R1
R2
Src
Ki (µM)
23a
F
CH3
CH2-4-F—C6H4
0.31 ± 0.07
23b
F
CH3
CH2-2-F—C6H4
0.30 ± 0.06
23c
F
CH3
3-F—C6H4
0.21 ± 0.02
23d
H
C3H7
CH2—C6H5
0.51 ± 0.1
In addition, to improve pharmacokinetic properties of pyrazolopyrimidine fused ring, Vignaroli et al (Vignaroli et al., 2013) designed pyrazolo[3,4-d]pyrimidine derivatives as prodrugs (24a,b) (Fig. 15), with high water-solubility. In vitro studies recorded a significant enhancement of water solubility and plasma stability, proposing predominant in vivo bioavailability. These prodrugs 24a,b did not exhibit any potential against Src and Ab1 but displayed significant antitumor effect in myeloid cell lines, as a result of hydrolysis of solubilizing moiety and release of the active compounds 25a,b (Fig. 15, Table 11).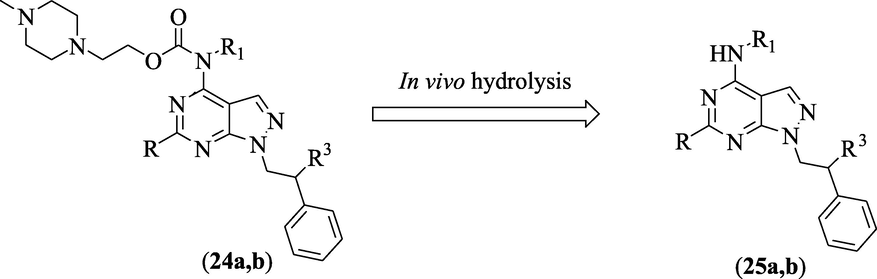
Structure of pyrazolopyrimidines 24a,b and 25a,b.
C.N.
R
R1
R2
Ki (µM)
IC50 (µM)
c-Src
C-Ab1
32D-p210
32D-T315I
24a
SC2H5
nBu
Cl
0.60
0.32
3.5
6.7
24b
SCH3
mBrC6H4
CH3
0.02
1.07
6.2
5.8
25a
SC2H5
nBu
Cl
NA
NA
1.2
2.4
25b
SCH3
mBrC6H4
CH3
NA
NA
2.8
2.6
Radi et al (Radi et al., 2011) prepared novel pyrazolo[3,4-d]pyrimidines 26 and these compounds were able to suppress the growth of SH-SY5Y neuroblastoma cell lines, in particular compound 26a was the most potent derivative (IC50 = 80 μM). In addition, these novel compounds were assayed for their Src inhibitory potential as a mechanism for their antiproliferative potential. SAR analysis described the importance of methylthio group at C-6 and anilino moiety at C-4 of the pyrazolopyrimidine ring as depicted in Fig. 16 and Table 12.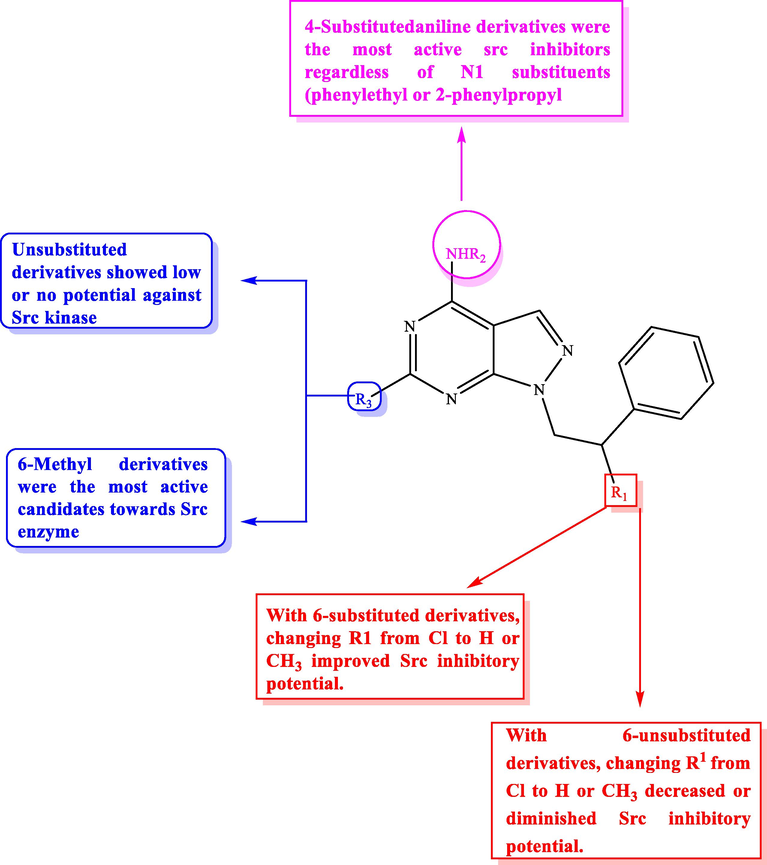
SAR and substitution effect of pyrazolopyrimidines to Src inhibition.
C.N
R1
R2
R3
c-Src (Ki) µM
C.N
R1
R2
R3
c-Src (Ki) µM
26a
H
3—Cl—C6H4-
SCH3
0.09
40 g
CH3
3—Cl—C6H4-
SCH3
0.025
26b
CH3
CH2C6H5
SCH3
3.7
40 h
CH3
3-Br—C6H4-
SCH3
0.018
26c
CH3
CH2-2-FC6H4
SCH3
2.16
40i
H
CH2-2-FC6H4
H
0.51
26d
CH3
CH2-4-FC6H4
SCH3
2.81
40j
H
3—Cl—C6H4-
H
0.48
26e
CH3
CH2-2—ClC6H4
SCH3
2
40 k
CH3
CH2-3-FC6H4
H
0.081
26f
CH3
C6H5
SCH3
0.025
40 l
CH3
CH2-2-FC6H4
H
0.151
Recently, in 2018, Molinari et al. (2018) designed and constructed novel pyrazolopyrimidines 27 incorporating 3-hydroxyphenylamino moiety at C-4 and added different substituents at C-6 in an attempt to modulate ADME and expand SAR. All the designed derivatives had been assayed for their Src inhibitory activity and cellular viability on SH SY5Y cells. The outcomes obtained displayed that compounds 27d and 27f recorded the highest inhibitory potential against Src enzyme with Ki value equal to 3.5 nM. SAR study showed that compounds incorporating 3-amino-4-chlorophenolic moiety are more active than C-4 non halogenated derivatives. This was in accordance with the computational study which recorded a favourable ortho substitution of a hydrogen with a chlorine atom (ΔΔG = 4.8 Kcal/mol) as depicted in Fig. 17 and Table 13.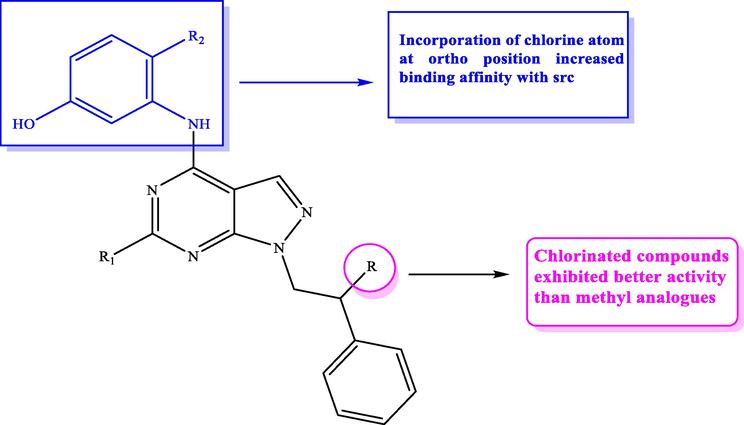
SAR studies of pyrazolopyrimidines 27a-f as Src Kinase suppressors.
C.N.
R
R1
R2
c-Src (Ki) µM
SH SY5Y (IC50) µM
27a
CH3
H
Cl
0.12
8.63 ± 0.7
27b
Cl
H
Cl
0.017
8.63 ± 0.7
27c
CH3
SCH3
Cl
0.05
6.7 ± 1.9
27d
Cl
SCH3
Cl
0.0035
6.7 ± 1.9
27e
CH3
NHCH2CH2OH
H
0.3
3.1 ± 0.3
27f
Cl
SCH2CH2-4-morpholino
Cl
0.0035
6.6 ± 1.8
3.4 m-TOR Inhibitors
Mammalian target of rapamycin (mTOR) is seine/threonine kinase, which acts as the catalytic subunits the catalytic subunit of two essential protein complexes called mTORC1 and mTORC2. These complexes exert fundamental role in signal transduction to regulate proliferation, metabolism, migration, and survival (Chang et al., 2015). Overexpression of mTOR is reported in many cancers and neurodegenerative disorders (Guo et al., 2011). In 2009, Verheijen and co-workers (Verheijen et al., 2009) hybridized 4-morpholino-1H-pyrazolopyrimidines with alkylureidophenyl groups at C-6 of pyrazolo[3,4-d]pyrimidine ring to give target compounds 28 (Fig. 18). These targets 28 demonstrated high mTOR inhibitory activity with (IC50 < 1 μM).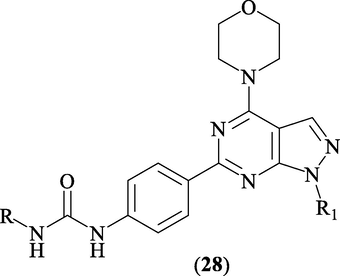
Structure of pyrazolopyrimidines 28.
Fraser et al (Fraser et al., 2016) discovered the lead pyrazolopyrimidine compound 29 (Fig. 19) as m-TOR inhibitory candidate with EC50 = 320 μM. Optimization of the hit compound 29 by introducing 2-amino-1,3-benzoxazole ring at C-3 and adding different acetal groups at N-1 afforded the target compounds 30–32. These compounds are tested for their antiproliferative potential towards MCF-7 and for their kinase inhibitory potential towards panel of kinases. All the new compounds (30–32) possessed superior antiproliferative effect towards MCF-7 cell line than lead compound 29 with EC50 = 8.4, 7.6 and 6.7 μM, respectively. Moreover, these candidates revealed higher inhibition to mTOR than exhibited by lead compound 29 (Fig. 19) (see Table 14).![Structure of pyrazolo[3,4-d]pyrimidines 29–32 with m-TOR inhibitory potential.](/content/184/2022/15/5/img/10.1016_j.arabjc.2022.103781-fig20.png)
Structure of pyrazolo[3,4-d]pyrimidines 29–32 with m-TOR inhibitory potential.
C.N
m-TOR IC50 (µM)
MCF (EC50, µM)
29
328
320
30
15
8.4
31
59
7.6
32
25
6.7
3.5 JAK kinase suppressor
The Janus kinases (JAKs) and their downstream effectors, signal transducer and activator of transcription proteins (STATs) form a critical cell signalling circuit, which control survival, proliferation, and differentiation of a variety of cell. JAK signalling is of fundamental importance in innate immunity, inflammation, and haematopoiesis, and dysregulation is frequently observed in immune disease and cancer (Yin et al., 2018).
In 2018, Yin et al. (2018) designed new pyrazolo[3,4-d]pyrimidines using structure based drug design strategy. This study afforded compound 33 (Fig. 20), which revealed potent JAK3 inhibitory potential (IC50 = 6.2 µM) and antiproliferative activity on T-cell with IC50 = 9.4 µM. One year later, the same group of investigators prepared novel pyrazolo[3,4-d]pyrimidine-4-amino compounds 34 (Fig. 20) trying to develop more active JAK-3 inhibitors (Yin et al., 2019). Unfortunately, these novel candidates exhibited weak Janus inhibitory potential (IC50 > 20 µM) but fortunately they revealed exciting inhibitory potential towards JAK-2. From these compounds, 34a-d were the most active and selective JAK-2 kinase inhibitors revealing IC50 range 6.5–9.7 µM. SAR study displayed that 3,5-disubstituted-1-methylpyrazole moiety improved the potency and selectivity of 3-(1-methyl-1H-pyrazol-3-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-ylamine as discussed in Fig. 20 and Table 15.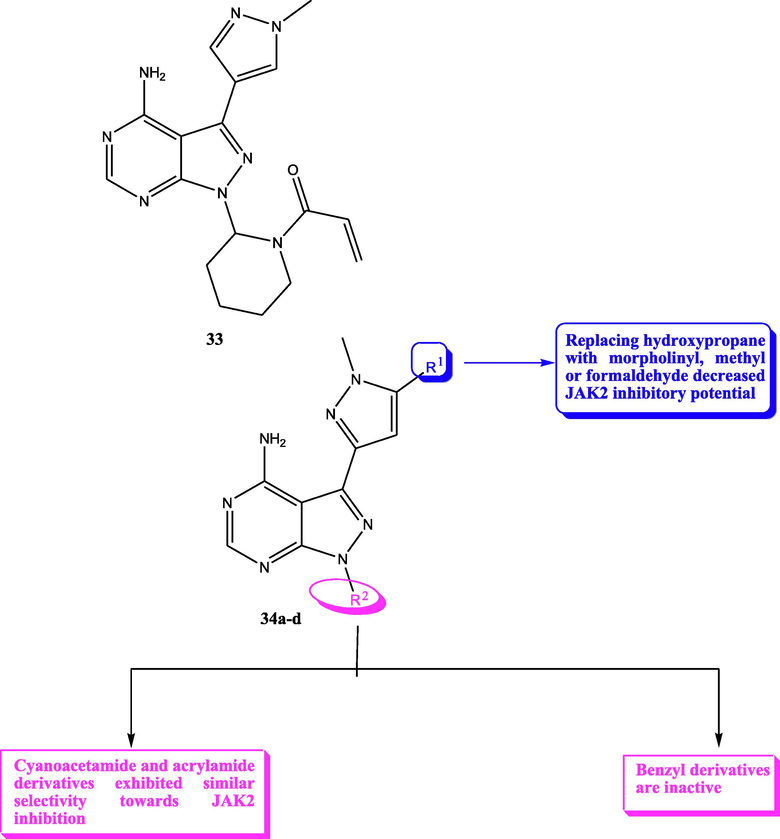
Structure of pyrazolopyrimidines 33, 34a-d as JAK2 inhibitors.
C.N
R1
R2
JAK2 IC50 (µM)
HEL (JAK2V617F)
IC50 (µM)TF-1 (JAK2)
IC50 (µM)THP-1 (JAK1/3)
IC50 (µM)NK-2 (JAK1/3)
IC50 (µM)
34a
7.2
4.7
2.0
18.5
22.1
34b
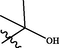
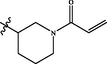
6.5
5.6
2.3
19.8
14.7
34c
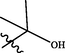
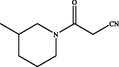
8
–
–
–
–
34d
9.7
4.3
5.4
16.0
23.6
Tofacitinib
–
–
4.3
5.2
2.6
0.9
0.6
3.6 Reactive oxygen species formation
Reactive oxygen species (ROS) are molecules having oxygen that are chemically reactive (Dickinson and Chang, 2011). ROS play critical roles in homeostasis and cell signalling and formed naturally as by-products of the abnormal oxygen metabolism (Brieger et al., 2012; Bystrom et al., 2014). Cancer cells exhibit greater ROS stress than normal cells (Pelicano et al., 2004), (Boonstra and Post, 2004). Rashad et al. (2011) designed and prepared novel sets of pyrazolopyrimidines (35–41) and screened the anticancer potential of these candidates towards MCF-7 using MTT assay. All the screened pyrazolopyrimidines (35–41) exerted excellent anticancer effect towards MCF-7 cell lines, especially compound 38, which recorded higher anticancer effect than the standard drug cisplatin. To elucidate the mode by which the tested derivatives exhibit their anticancer potential, they determined the activities of free radical metabolizing enzymes superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSH-Px), also the levels of glutathione and hydrogen peroxide were also determined. Treating the cells with theses pyrazolo[3,4-d]pyrimidines (35–41) resulted in an increase of SOD activity and H2O2 level, in addition a decrease in both CAT and GSH-Px. These changes were in the following order: 38 > 39 > 35 > 40 > 36 > 37 > 41, which are in accordance with the anticancer potential so these compounds can act as anticancer through the production of ROS.
SAR study displayed that the substituent at C-4 of the pyrazolo[3,4-d]pyrimidine derivatives greatly affects the anticancer activity as explained in Fig. 21, Table 16.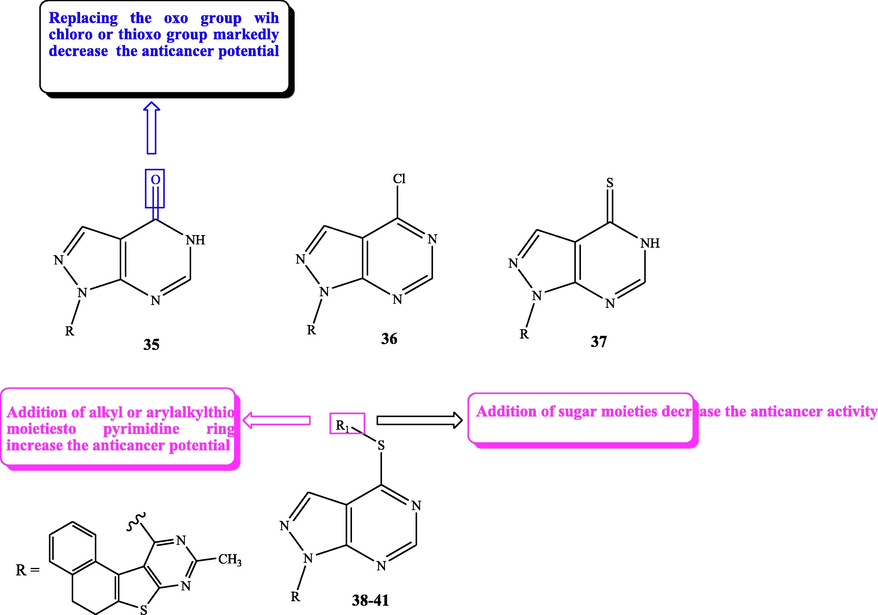
Structure and SAR of pyrazolopyrimidines 35–41.
C.N
R1
SOD U/mg protein
CAT U/mg protein
GSH nmol/mg protein
H2O2 nmol/mg protein
DNA (µg/106 cells)
RNA (µg/106 cells)
35
–
290 ± 31
2.30 ± 0.27
2.65 ± 0.25
60.30 ± 6.50
2.70 ± 0.28
5.25 ± 0.55
36
–
200 ± 24
3.65 ± 0.25
6.70 ± 0.72
52.33 ± 6.11
3.85 ± 0.41
6.50 ± 0.18
37
–
180 ± 19
6.70 ± 0.72
6.82 ± 0.78
46.37 ± 5.12
4.00 ± 0.41
6.80 ± 0.65
38
CH2CH3
385 ± 39
6.82 ± 0.78
2.20 ± 0.24
80 ± 8.60
1.82 ± 0.21
3.61 ± 0.35
39
CH2CH2OCH3
310 ± 35
2.10 ± 0.26
2.90 ± 0.42
66.80 ± 7.50
2.50 ± 0.28
4.80 ± 0.50
40
CH2CH2OCH2CH3
250 ± 29
3.20 ± 0.35
5.25 ± 0.60
55 ± 6.25
3.00 ± 0.33
6.20 ± 0.70
41
Ribose
160 ± 18
5.35 ± 0.63
5.40 ± 0.60
40.33 ± 4.13
4.45 ± 0.45
7.22 ± 0.74
Cisplatin
–
470.60 ± 55
1.20 ± 0.11
1.40 ± 0.18
84.80 ± 9.60
1.11 ± 0.14
2.30 ± 0.26
Furthermore, in 2019, novel pyrazolo[3,4-d]pyrimidine 42 (Fig. 22) was found to suppress the growth of non-small cell lung cancer (NC-H460), renal cancer (786–0), non-small cell lung cancer (A549), and renal cancer (ACHN). The anticancer effect of this compound was exerted by generation of ROS (Gaonkar et al., 2020). Recently, in 2021, Si306 (43) and pro-Si306 (44) (Fig. 22) were reported to stimulate ROS production and expression of antioxidant enzymes in primary Glioblastoma cells (Kostić et al., 2021).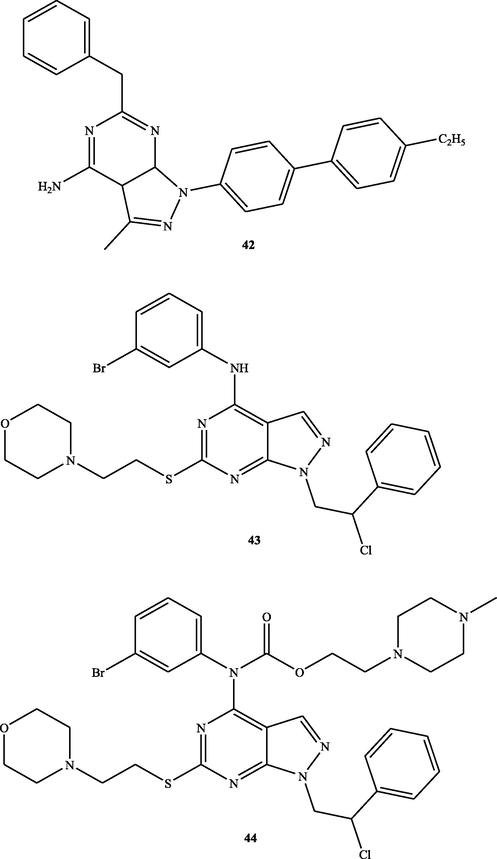
Structure of pyrazolopyrimidines 42–44.
3.7 Bruton’s tyrosine kinase inhibitors
BTK is a nonreceptor tyrosine kinase that has a vital role in B cell receptor signaling and controls B-lymphocyte differentiation, signaling and survival (Corneth et al., 2015; Singh et al., 2018). Abnormal BTK activity is a crucial feature of B-cell malignancies, so targetting BTK is a selective strategy to cure B-cell malignancies. Ibrutinib 45 is the first discovered irreversible BTK inhibitor that approved for treating chronic lymphatic leukemia, mantle cell lymphoma and Waldenstrom's macroglobulinemia (Smith, 2015; Ruella et al., 2016; Papanota et al., 2019). In 2019, Ran et al. (2019) performed some modifications on the chemical structure of ibrutinib to yield novel derivatives of pyrazolopyrimidines 46a-g (Fig. 23, Table 17). These modifications included the replacement of the piperidine moiety with phenyl ring which was linked to N-1 of pyrazolopyrimidine through a methylene group. From these target candidates, 46 g recorded the highest BTK inhibitory potential (IC50 = 27 nM) comparing to the standard drug ibrutinib 45 (IC50 = 8 nM).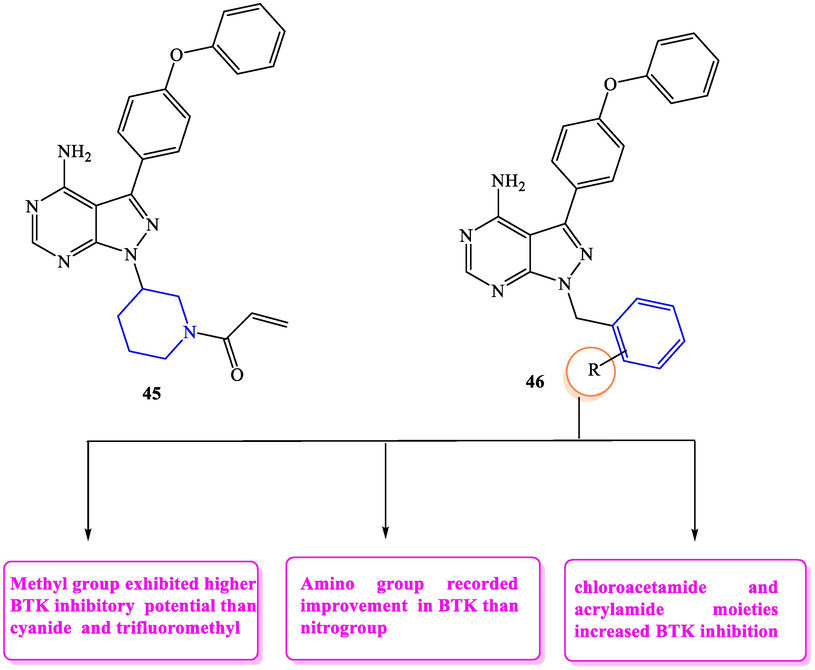
SAR of pyrazolopyrimidines as BTK inhibitors.
C.N.
R
BTK inhibitory % at 1 µM
Cell viability assay, IC50 µM
Mino
Jeko-1
ZI38
Maver-1
46a
4—CH3
66
–
–
–
–
46b
4—CN
12
–
–
–
–
46c
4—CF3
27
–
–
–
–
46d
4-NO2
67
–
–
–
–
46e
4-NH2
93
13.9
13.2
18.4
18.7
46f
3-NHCOCH2Cl
98
0.9
0.4
0.8
0.4
46 g
3-NHCOCH⚌CH2
97
1.3
1.3
2.8
2.6
IBN (45)
–
100
15.7
1.1
9.7
7.8
3.8 Phosphoinositide-3-kinase (PI3-K) suppressors
PI3Ks are lipid kinases that are included in phosphorylation and regulation of cellular metabolism, survival and proliferation (Hoxhaj and Manning, 2020). Abnormal PI3K pathway activation causes PI3Kα isoform overexpression which may suppress PTEN that is essential for converting PIP3 to PIP2 (Jung et al., 2018). Although all PI3K isoforms are included in PTEN-null tumors, PI3K is primarily responsible for malignant cancer transformation (Thorpe et al., 2015).
Parsaclisib (47) (Fig. 24) is a highly selective PI3Kδ (IC50 < 1 nM) pyrazolopyrimidine derivative with drug like ADME properties that recorded an excellent in vivo profile as shown by pharmacokinetic studies in monkeys, dogs and rats (Yue et al., 2019). Furthermore, the pyrazolopyrimidine 50 was designed depending on some chemical modifications on pan-PI3K inhibitor, ZSTK474 (48) (Fig. 24, Table 18) (Gamage et al., 2017). Modelling study of compound 48 displayed that one of the two morpholino moieties was not essential for binding with p110δ active region (Berndt et al., 2010). This point encouraged Gamage et al. (2017) to replace one of morphlino ring with piperazinyl sulphonamide group to afford comound 49 that showed dual PI3Kα/δ inhibition. Optimization of compound 49 was done through replacing the pyrimidine ring with pyrazolopyrimidine to produce compound 50 which displayed potent and selective inhibition towards PI3Kα (IC50 = 2.6 nM) (Table 18).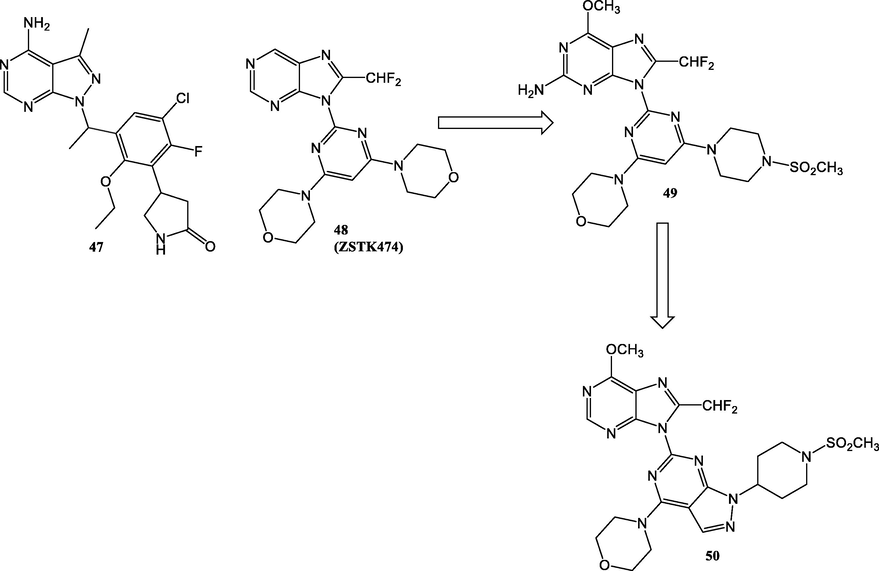
Some examples of some PIK3 inhibitors 47–50.
C.N.
Cell (IC50) nM
Enzyme IC50 (nM)
NZB5
NZOV9
P110α
P110β
P110δ
48
220
287
8.9
58
38
49
74
5
22
116
13
50
114
24
2.6
768
58
3.9 MKK7 inhibitors
Mitogen-activated protein kinase 7 (MKK7) is a part of c-JUN N-terminal kinase (JNK) signaling pathway that is included in many cellular regulation process (Ying et al., 2021). It is associated with many diseases as leukemia, lung cancer, myeloma and Alzheimer’s diseases (Ooshio et al., 2021). Development of MKK7 suppressors received little attention and no MKK7 suppressors have included in clinical trials. Wolle et al. (2019a) prepared novel series of pyrazolopyrimidines 51a-d with MKK7 inhibitory potential. SAR study of these compounds is explained in Fig. 25 (see Table 19).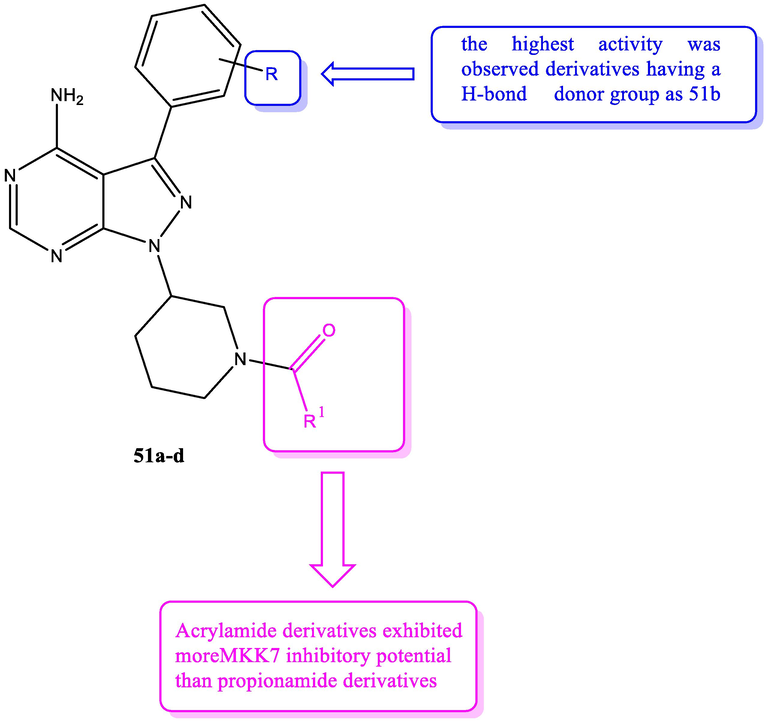
SAR study of pyrazolopyrimidines 51a-d with MKK7 inhibitory effect.
C.N
R
R1
MKK7 (IC50, nM)
51a
3—CF3
—CH⚌CH
159 ± 117
51b
4-OH
—CH⚌CH
8.6 ± 2.9
51c
3—CF3
—CH⚌CH—CH2-N(CH3)2
2611 ± 1503
51d
3—CF3
—CH2—CH3
>10,000
Ibrutinib
–
–
78 ± 21
3.10 BRAF and VEGFR-2 suppressors
RAF kinases are essential regulators in RAS-RAF-MEK-ERK signaling pathway that exert a pivotal role in cell proliferation, survival and differentiation (Roberts and Der, 2007). ARAF, BRAF and CRAF are members of RAF family (Pedersen and Plotkin, 2010). RAF kinases are stimulated via dimerization of their kinase domains (Lavoie and Therrien, 2015). Dysregulation of RAF kinases activation is a powerful cause of cancer (Durrant and Morrison, 2018). The cancer tissues secrete vascular endothelial growth factor (VEGF) to enhance angiogenesis from neighboring blood vessels (Pandya et al., 2006). Suppressing VEGFR-2 is considered as a rational approach for cancer curing. A novel series of pyrazolopyrimidines 52a-d had been constructed and assessed for their inhibitory potential against VEGFR-2 and BRAFV600E (Wolle et al., 2019b). From the obtained results, some SAR was concluded as represented in Fig. 26, Table 20. It is noted that compounds substituted at terminal phenyl ring enhanced the inhibitory potential comparing with the unsubstituted effect, implying that the substituted aryl ring was required for effective BRAFV600E and VEGFR2 suppression.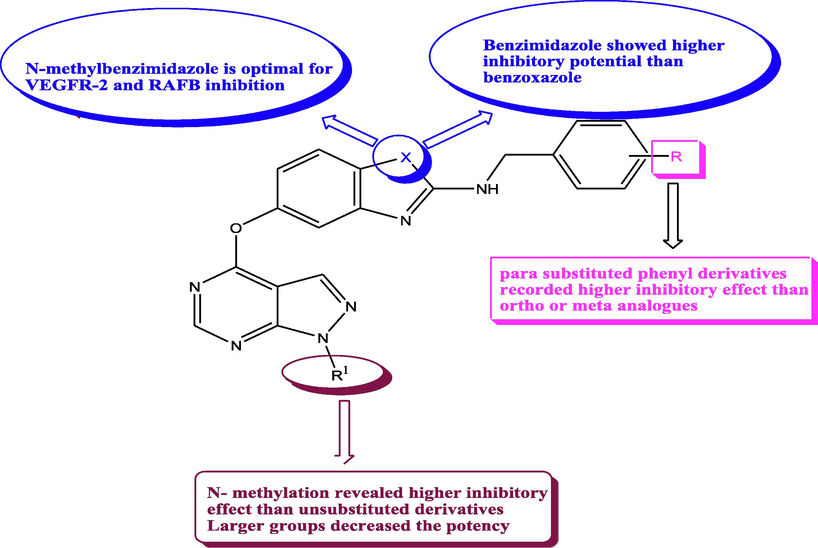
SAR and substitution effects of pyrazolopyrimidines with RAFB and VEGFR-2 inhibitory potential.
C.N
X
R1
R
Kinase inhibitory potential (IC50, µM)
In vitro anticancer activity
BRAFV600E
VEGFR-2
A375
HT-29
HUVEC
52a
NH
CH3
4—Cl
82.0
14.6
>100
22.87
>100
52b
NMe
CH3
4—CH3
90.1
96.6
25.36
6.13
15.71
52c
NMe
CH3
2—Cl
65.4
84.2
>100
80.36
78.62
52d
O
CH3
4-OCF3
56.1
22.3
>100
>100
61.62
4 Conclusion
Pyrazolo[3,4-d]pyrimidine is one of the most important heterocyclic systems due to its wide applications in medicinal chemistry mainly as anticancer. Many chemistry researches used this ring as a template for the design and preparation of many anticancer agents. In this review, we have compiled a list of recent studies on pyrazolopyrimidines and their target specific activities in cancer treatment. Moreover, we displayed the new advances in the synthesis of this scaffold and structure–activity relationship to assist in the generation of novel potential candidates in a short time.
Acknowledgement
This work was funded by the Deanship of Scientific Research at Jouf University under grant No (DSR-2021-01-0308).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding:
Juof University, Saudi Arabia (DSR-2021-01-0308).
References
- 4-Substituted-1-phenyl-1H-pyrazolo [3, 4-d] pyrimidine Derivatives: Design, Synthesis, Antitumor and EGFR Tyrosine Kinase Inhibitory Activity. Chem. Biol. Drug Des.. 2015;85:608-622.
- [Google Scholar]
- Part—II: Utilities of active methylene compounds and heterocycles bearing active methyl or having an active methine in the formation of bioactive pyrazoles and pyrazolopyrimidines. Synth. Commun.. 2020;50:3563-3591.
- [Google Scholar]
- Enhancement to synthesize, design and dock of novel EGFR inhibitors containing pyrazolo [3, 4-d] pyrimidine cores of expected anticancer activity. OCAIJ. 2014;10:470-483.
- [Google Scholar]
- Design, synthesis and antitumor activity of novel pyrazolo [3, 4-d] pyrimidine derivatives as EGFR-TK inhibitors. Bioorg. Chem.. 2016;66:88-96.
- [Google Scholar]
- Design, synthesis and biological evaluation of some novel benzothiazole/benzoxazole and/or benzimidazole derivatives incorporating a pyrazole scaffold as antiproliferative agents. Bioorg. Chem.. 2017;74:82-90.
- [Google Scholar]
- Synthesis, docking study and antitumor evaluation of certain newly synthesized pyrazolo [3, 4-d] pyrimidine derivatives. Organ. Chem. Ind. J.. 2014;10:157-167.
- [Google Scholar]
- Synthesis and anticancer activity of some new pyrazolo [3, 4-d] pyrimidin-4-one derivatives. Molecules. 2014;19:3297-3309.
- [Google Scholar]
- New advances in synthesis and clinical aspects of pyrazolo [3, 4-d] pyrimidine scaffolds. Bioorg. Chem.. 2018;78:341-357.
- [Google Scholar]
- Pyrimidine and fused pyrimidine derivatives as promising protein kinase inhibitors for cancer treatment. Med. Chem. Res.. 2021;30:31-49.
- [Google Scholar]
- Studies on Bioactive Heterocyclic Compounds. Saurashtra University; 2008.
- Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorg. Med. Chem.. 2020;28:115525
- [Google Scholar]
- Discovery and in vitro evaluation of potent kinase inhibitors: Pyrido [1′, 2′: 1, 5] pyrazolo [3, 4-d] pyrimidines. Bioorg. Med. Chem. Lett.. 2005;15:3778-3781.
- [Google Scholar]
- Factors that affect risk for pancreatic disease in the general population: a systematic review and meta-analysis of prospective cohort studies. Clin. Gastroenterol. Hepatol.. 2014;12(1635–1644):e1635
- [Google Scholar]
- Synthesis, EGFR Inhibition and Anti-cancer Activity of New 3, 6-dimethyl-1-phenyl-4-(substituted-methoxy) pyrazolo [3, 4-d] pyrimidine Derivatives. Anti-Canc. Age. Med. Chem. (Forme. Curr. Med. Chem.-Anti-Canc. Age.). 2017;17:1389-1400.
- [Google Scholar]
- Molecular iodine promoted synthesis of new pyrazolo [3, 4-d] pyrimidine derivatives as potential antibacterial agents. Eur. J. Med. Chem.. 2010;45:647-650.
- [Google Scholar]
- Design and synthesis of new EGFR-tyrosine kinase inhibitors containing pyrazolo [3, 4-d] pyrimidine cores as anticancer agents. Bull. Pharm. Sci. Assiut. 2012;35:27-42.
- [Google Scholar]
- Design and synthesis of new EGFR-tyrosine kinase inhibitors containing pyrazolo [3, 4-d] pyrimidine cores as anticancer agents. Bull. Pharm. Sci. Assiut. Univ.. 2012;35:1-16.
- [Google Scholar]
- Synthesis, cyclooxygenase inhibition, anti-inflammatory evaluation and ulcerogenic liability of new 1-phenylpyrazolo [3, 4-d] pyrimidine derivatives. J. Enzyme Inhib. Med. Chem.. 2016;31:6-12.
- [Google Scholar]
- (3, 5-Dimethylpyrazol-1-yl)-[4-(1-phenyl-1H-pyrazolo [3, 4-d] pyrimidin-4-ylamino) phenyl] methanone. Molbank. 2016;2016:M915.
- [Google Scholar]
- Nutritional status and follicular-derived thyroid cancer: An update. Crit. Rev. Food Sci. Nutr.. 2021;61:25-59.
- [Google Scholar]
- The p110δ structure: mechanisms for selectivity and potency of new PI (3) K inhibitors. Nat. Chem. Biol.. 2010;6:117-124.
- [Google Scholar]
- Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1-13.
- [Google Scholar]
- Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid. Redox Signal.. 2014;20:1917-1924.
- [Google Scholar]
- Revisiting the hallmarks of cancer: The role of hyaluronan. In: Seminars in cancer biology. Elsevier; 2020. p. :9-19.
- [Google Scholar]
- Pyrazolo [3, 4-d] pyrimidines as potent antiproliferative and proapoptotic agents toward A431 and 8701-BC cells in culture via inhibition of c-Src phosphorylation. J. Med. Chem.. 2006;49:1549-1561.
- [Google Scholar]
- In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells. Bioorg. Med. Chem. Lett.. 2006;16:1969-1974.
- [Google Scholar]
- Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit. Rev. Oncol./Hematol.. 2015;96:507-517.
- [Google Scholar]
- Medicinal attributes of pyrazolo [3, 4-d] pyrimidines: A review. Bioorg. Med. Chem.. 2013;21:5657-5668.
- [Google Scholar]
- Synthesis, anticancer evaluation, and molecular docking studies of some novel 4, 6-disubstituted pyrazolo [3, 4-d] pyrimidines as cyclin dependent kinase 2 (CDK2) inhibitors. Bioorg. Chem. 2018
- [Google Scholar]
- Immunotherapy targeting 4–1BB: mechanistic rationale, clinical results, and future strategies. Blood J. Am. Soc. Hematol.. 2018;131:49-57.
- [Google Scholar]
- Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem.. 2005;48:6661-6670.
- [Google Scholar]
- Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr. Opin. Pharmacol.. 2005;5:366-373.
- [Google Scholar]
- BTK signaling in B cell differentiation and autoimmunity. B Cell Recep. Signal. 2015:67-105.
- [Google Scholar]
- EAU-EANM-ESTRO-ESUR-SIOG guidelines on prostate cancer. Part II—2020 update: treatment of relapsing and metastatic prostate cancer. Eur. Urol.. 2021;79:263-282.
- [Google Scholar]
- Synthesis of new substituted pyrazolo [3, 4-d] pyrimidin-4-ones under microwave irradiation. Indian J. Heterocycl. Chem.. 2006;16:151-154.
- [Google Scholar]
- Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol.. 2011;7:504-511.
- [Google Scholar]
- The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci.. 2020;21:1960.
- [Google Scholar]
- 2-Hydroxypropyl-β-cyclodextrin strongly improves water solubility and anti-proliferative activity of pyrazolo [3, 4-d] pyrimidines Src-Abl dual inhibitors. Eur. J. Med. Chem.. 2010;45:5958-5964.
- [Google Scholar]
- Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br. J. Cancer. 2018;118:3-8.
- [Google Scholar]
- New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002;2:161.
- [Google Scholar]
- El-Hamouly, W.S., El-Khamry, A.-M.A., Abbas, E.M., 2006. Synthesis of new 4-aryl-isoxazolo [5, 4-d] pyrimidin-6-one (thione) and 4-aryl-pyrazolo [3, 4-d]-pyrimidin-6-one derivatives of potential antihypertensive activity.
- Facile One-Pot Multicomponent Synthesis of Pyrazolo-Thiazole Substituted Pyridines with Potential Anti-Proliferative Activity: Synthesis, In Vitro and In Silico Studies. Molecules. 2021;26:3103.
- [Google Scholar]
- Dietary patterns related to total mortality and cancer mortality in the United States. Cancer Causes Control. 2021;32:1279-1288.
- [Google Scholar]
- The 1, 2, 4-Triazolo [4, 3-a] pyrazin-3-one as a Versatile Scaffold for the Design of Potent Adenosine Human Receptor Antagonists. Structural Investigations to Target the A2A Receptor Subtype. J. Med. Chem.. 2017;60:5772-5790.
- [Google Scholar]
- Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med. Chem.. 2015;7:1511-1519.
- [Google Scholar]
- Molecular principles of metastasis: a hallmark of cancer revisited. Sign. Trans. Target. Therapy. 2020;5:1-17.
- [Google Scholar]
- Prevalence and coprevalence of modifiable risk factors for upper digestive tract cancer among residents aged 40–69 years in Yangzhong city, China: a cross-sectional study. BMJ open. 2021;11:e042006
- [Google Scholar]
- Fluids and their mechanics in tumour transit: shaping metastasis. Nat. Rev. Cancer. 2020;20:107-124.
- [Google Scholar]
- eCF309: a potent, selective and cell-permeable mTOR inhibitor. MedChemComm. 2016;7:471-477.
- [Google Scholar]
- Synthesis and biological evaluation of sulfonamide analogues of the phosphatidylinositol 3-kinase inhibitor ZSTK474. Bioorg. Med. Chem.. 2017;25:5859-5874.
- [Google Scholar]
- Novel pyrazolo [3, 4-d] pyrimidine derivatives inhibit human cancer cell proliferation and induce apoptosis by ROS generation. Arch. Pharm.. 2020;353:1900296.
- [Google Scholar]
- European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment-Update 2019. Eur. J. Cancer. 2020;126:159-177.
- [Google Scholar]
- Synthesis of some new pyrazolo [3, 4-d] pyrimidine derivatives of expected anticancer and radioprotective activity. Eur. J. Med. Chem.. 2010;45:171-178.
- [Google Scholar]
- Synthesis of novel pyrazole and pyrimidine derivatives bearing sulfonamide moiety as antitumor and radiosensitizing agents. Med. Chem. Res.. 2012;21:1376-1383.
- [Google Scholar]
- Synthesis, anticancer and radioprotective activities of some new pyrazolo [3, 4-d] pyrimidines containing amino acid moieties. Arzneimittelforschung. 2009;59:96-103.
- [Google Scholar]
- Novel purine and pyrazolo [3, 4-d] pyrimidine inhibitors of PI3 kinase-α: Hit to lead studies. Bioorg. Med. Chem. Lett.. 2010;20:636-639.
- [Google Scholar]
- González, J.A., Acanda, M., Akhtar, Z., Andrews, D., Azqueta, J., Bass, E., Bellorín, A., Couso, J., García-Ñustes, M.A., Infante, Y., 2019. New combinational therapies for cancer using modern statistical mechanics. arXiv preprint arXiv:190200728.
- Synthesis and Antibacterial Evaluation of New Pyrazolo [3, 4-d] pyrimidines Kinase Inhibitors. Molecules. 2020;25:5354.
- [Google Scholar]
- Nutraceuticals and functional foods: The foods for the future world. Crit. Rev. Food Sci. Nutr.. 2016;56:2617-2627.
- [Google Scholar]
- Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res.. 2011;89:1723-1736.
- [Google Scholar]
- Health seeking behavior and its determinants for cervical cancer among women of childbearing age in Hossana Town, Hadiya zone, Southern Ethiopia: community based cross sectional study. BMC Cancer. 2018;18:1-9.
- [Google Scholar]
- Pyrazoles as building blocks in heterocyclic synthesis: Synthesis of pyrazolo [3, 4-d] pyrimidine, pyrazolo [3, 4-e][1, 4] diazepine, pyrazolo [3, 4-d][1, 2, 3] triazine and pyrolo [4, 3-e][1, 2, 4] triazolo [1, 5-c] pyrimidine derivatives. J. Iran. Chem. Soc.. 2005;2:115-123.
- [Google Scholar]
- Synthesis and in vitro cytotoxic activity of novel pyrazolo [3, 4-d] pyrimidines and related pyrazole hydrazones toward breast adenocarcinoma MCF-7 cell line. Bioorg. Med. Chem.. 2011;19:6808-6817.
- [Google Scholar]
- Novel pyrazolopyrimidines: Synthesis, in vitro cytotoxic activity and mechanistic investigation. Eur. J. Med. Chem.. 2017;138:565-576.
- [Google Scholar]
- Synthesis, reactions, and antimicrobial activity of some novel pyrazolo [3, 4-d] pyrimidine, pyrazolo [4, 3-e][1, 2, 4] triazolo [1, 5-c] pyrimidine, and pyrazolo [4, 3-e][1, 2, 4] triazolo [3, 4-c] pyrimidine derivatives. J. Heterocycl. Chem.. 2020;57:892-912.
- [Google Scholar]
- Protein Dynamics Enables Phosphorylation of Buried Residues in Cdk2/Cyclin-A-Bound p27. Biophys. J.. 2020;119:2010-2018.
- [Google Scholar]
- Catalytic synthesis of 6-aryl-1H-pyrazolo [3, 4-d] pyrimidin-4 [5H]-ones by heteropolyacid: H14 [NaP5W30O110] and H3PW12O40. J. Mol. Catal. A: Chem.. 2006;249:1-3.
- [Google Scholar]
- Current strategies for inhibiting FGFR activities in clinical applications: opportunities, challenges and toxicological considerations. Drug Discov. Today. 2014;19:51-62.
- [Google Scholar]
- The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer. 2020;20:74-88.
- [Google Scholar]
- Cervical cancer in low and middle-income countries. Oncol. Lett.. 2020;20:2058-2074.
- [Google Scholar]
- Effect of dietary red meat on colorectal cancer risk—a review. Compr. Rev. Food Sci. Food Saf.. 2019;18:1812-1824.
- [Google Scholar]
- Structure-based design of a new class of highly selective pyrazolo [3, 4-d] pyrimidines based inhibitors of cyclin dependent kinases. Arkivoc. 2009;7:12-25.
- [Google Scholar]
- Pyrazolo [4, 3-d] pyrimidine bioisostere of roscovitine: evaluation of a novel selective inhibitor of cyclin-dependent kinases with antiproliferative activity. J. Med. Chem.. 2011;54:2980-2993.
- [Google Scholar]
- Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC) Canc. Head Neck. 2018;3:1-13.
- [Google Scholar]
- Discovery of 3, 6-dihydro-2H-pyran as a morpholine replacement in 6-aryl-1H-pyrazolo [3, 4-d] pyrimidines and 2-arylthieno [3, 2-d] pyrimidines: ATP-competitive inhibitors of the mammalian target of rapamycin (mTOR) Bioorg. Med. Chem. Lett.. 2010;20:640-643.
- [Google Scholar]
- The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer. 2004;4:423.
- [Google Scholar]
- Synthesis and antimicrobial activity of novel pyrazolo [3, 4-d] pyrimidin derivatives. Eur. J. Med. Chem.. 2010;45:1635-1638.
- [Google Scholar]
- Synthesis and biological evaluations of pyrazolo [3, 4-d] pyrimidines as cyclin-dependent kinase 2 inhibitors. Eur. J. Med. Chem.. 2003;38:525-532.
- [Google Scholar]
- Pyrazolo [3, 4-d] pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sci.. 2021;11:884.
- [Google Scholar]
- The multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation, migration and cancer therapy. Cancers. 2019;11:1220.
- [Google Scholar]
- Synthesis, characterization, antitubercular and anti-inflammatory activity of new pyrazolo [3, 4-d] pyrimidines. Comb. Chem. High Through. Screen.. 2021;24:1300-1308.
- [Google Scholar]
- Pyrazolo [3, 4-d] pyrimidine-based dual EGFR T790M/HER2 inhibitors: Design, synthesis, structure–activity relationship and biological activity as potential antitumor and anticonvulsant agents. Eur. J. Med. Chem.. 2021;214:113222
- [Google Scholar]
- Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol.. 2015;16:281-298.
- [Google Scholar]
- Synthesis, SAR and biological evaluation of 1, 6-disubstituted-1H-pyrazolo [3, 4-d] pyrimidines as dual inhibitors of Aurora kinases and CDK1. Bioorg. Med. Chem. Lett.. 2012;22:2070-2074.
- [Google Scholar]
- Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis.. 2020;1866:165715.
- [Google Scholar]
- Deep learning enhancing kinome-wide polypharmacology profiling: model construction and experiment validation. J. Med. Chem.. 2019;63:8723-8737.
- [Google Scholar]
- Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem.. 2007;50:409-424.
- [Google Scholar]
- Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J. Med. Chem.. 2006;49:3826-3831.
- [Google Scholar]
- Efficient optimization of pyrazolo [3, 4-d] pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment. Bioorg. Med. Chem. Lett.. 2018;28:3454-3457.
- [Google Scholar]
- Concise review: Neural stem cell-mediated targeted Cancer therapies. Stem Cells Transl. Med.. 2018;7:740-747.
- [Google Scholar]
- 4-Chloro-6-(chloromethyl)-1-methyl-1H-pyrazolo [3, 4-d] pyrimidine. Molbank. 2021;2021:M1253.
- [Google Scholar]
- Adherence to the Healthy Eating Index and Alternative Healthy Eating Index dietary patterns and mortality from all causes, cardiovascular disease and cancer: a meta-analysis of observational studies. J. Hum. Nutr. Diet.. 2017;30:216-226.
- [Google Scholar]
- Hepatocyte Mitogen-Activated Protein Kinase Kinase 7 Contributes to Restoration of the Liver Parenchyma Following Injury in Mice. Hepatology. 2021;73:2510-2526.
- [Google Scholar]
- Evaluating ibrutinib in the treatment of symptomatic Waldenstrom’s macroglobulinemia. J. Blood Med.. 2019;10:291.
- [Google Scholar]
- Alliance with EPR effect: Combined strategies to improve the EPR effect in the tumor microenvironment. Theranostics. 2019;9:8073.
- [Google Scholar]
- A language for biochemical systems: Design and formal specification. In: Transactions on computational systems biology XII. Springer; 2010. p. :77-145.
- [Google Scholar]
- ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates. 2004;7:97-110.
- [Google Scholar]
- Tyrosine kinase inhibitors in cancer: breakthrough and challenges of targeted therapy. Cancers. 2020;12:731.
- [Google Scholar]
- One-pot synthesis of novel 1H-pyrimido [4, 5-c][1, 2] diazepines and pyrazolo [3, 4-d] pyrimidines. Beilstein J. Org. Chem.. 2006;2:5.
- [Google Scholar]
- Identification of potent c-Src inhibitors strongly affecting the proliferation of human neuroblastoma cells. Bioorg. Med. Chem. Lett.. 2011;21:5928-5933.
- [Google Scholar]
- Synthesis of novel isoxazolines and isoxazoles of N-substituted pyrazolo [3, 4-d] pyrimidin-4 (5H)-one derivatives through [3+ 2] cycloaddition. Arabian J. Chem.. 2019;12:1974-1982.
- [Google Scholar]
- Discovery of pyrazolopyrimidine derivatives as potent BTK inhibitors with effective anticancer activity in MCL. Bioorg. Chem.. 2019;89:102943
- [Google Scholar]
- Challenges in prevention and care delivery for women with cervical cancer in sub-Saharan Africa. Front. Oncol.. 2016;6:160.
- [Google Scholar]
- Synthesis and anticancer effects of some novel pyrazolo [3, 4-d] pyrimidine derivatives by generating reactive oxygen species in human breast adenocarcinoma cells. Eur. J. Med. Chem.. 2011;46:1019-1026.
- [Google Scholar]
- Role of epidermal growth factor receptor (EGFR) and its ligands in kidney inflammation and damage. Mediat. Inflamm.. 2018;2018
- [Google Scholar]
- Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-3310.
- [Google Scholar]
- Tumor dormancy as an alternative step in the development of chemoresistance and metastasis-clinical implications. Cell. Oncol.. 2020;43:155-176.
- [Google Scholar]
- The Addition of the BTK inhibitor Ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin. Cancer Res.. 2016;22:2684-2696.
- [Google Scholar]
- An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules. 2017;22:1037.
- [Google Scholar]
- Fyn: a novel molecular target in cancer. Canc. Interdiscip. Int. J. Am. Canc. Soc.. 2010;116:1629-1637.
- [Google Scholar]
- Synthesis, biological evaluation and docking studies of 4-amino substituted 1H-pyrazolo [3, 4-d] pyrimidines. Eur. J. Med. Chem.. 2008;43:2665-2676.
- [Google Scholar]
- Antiproliferative activity of new 1-aryl-4-amino-1H-pyrazolo [3, 4-d] pyrimidine derivatives toward the human epidermoid carcinoma A431 cell line. Eur. J. Med. Chem.. 2004;39:939-946.
- [Google Scholar]
- Src family kinase inhibitor PP2 accelerates differentiation in human intestinal epithelial cells. Biochem. Biophys. Res. Commun.. 2013;430:1195-1200.
- [Google Scholar]
- Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer. 2018;17:1-23.
- [Google Scholar]
- Ibrutinib in B lymphoid malignancies. Expert Opin. Pharmacother.. 2015;16:1879-1887.
- [Google Scholar]
- 3-Amino-pyrazolo [3, 4-d] pyrimidines as p38α kinase inhibitors: design and development to a highly selective lead. Bioorg. Med. Chem. Lett.. 2011;21:3452-3456.
- [Google Scholar]
- Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006;25:2367.
- [Google Scholar]
- Cancer stem cells and their unique role in metastatic spread. In: Seminars in cancer biology. Elsevier; 2020. p. :148-156.
- [Google Scholar]
- PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer. 2015;15:7-24.
- [Google Scholar]
- Magnetic nanoparticle-based drug delivery for cancer therapy. Biochem. Biophys. Res. Commun.. 2015;468:463-470.
- [Google Scholar]
- Discovery of 4-morpholino-6-aryl-1 H-pyrazolo [3, 4-d] pyrimidines as highly potent and selective ATP-competitive inhibitors of the mammalian target of rapamycin (mTOR): optimization of the 6-aryl substituent. J. Med. Chem.. 2009;52:8010-8024.
- [Google Scholar]
- Pyrazolo [3, 4-d] pyrimidine prodrugs: strategic optimization of the aqueous solubility of dual Src/Abl inhibitors. ACS Med. Chem. Lett.. 2013;4:622-626.
- [Google Scholar]
- 5-Substituted 3-isopropyl-7-[4-(2-pyridyl) benzyl] amino-1 (2) H-pyrazolo [4, 3-d] pyrimidines with anti-proliferative activity as potent and selective inhibitors of cyclin-dependent kinases. Eur. J. Med. Chem.. 2016;110:291-301.
- [Google Scholar]
- Synthesis and structure–activity relationship study of pyrazolo [3, 4-d] pyrimidines as tyrosine kinase RET inhibitors. Bioorg. Med. Chem. Lett.. 2017;27:2544-2548.
- [Google Scholar]
- Drug resistance and combating drug resistance in cancer. Cancer Drug Resis.. 2019;2:141-160.
- [Google Scholar]
- Pyrazolo [3, 4-d] pyrimidine derivatives containing a Schiff base moiety as potential antiviral agents. Bioorg. Med. Chem. Lett.. 2018;28:2979-2984.
- [Google Scholar]
- European association of urology guidelines on muscle-invasive and metastatic bladder cancer: summary of the 2020 guidelines. Eur. Urol.. 2021;79:82-104.
- [Google Scholar]
- Characterization of Covalent Pyrazolopyrimidine–MKK7 Complexes and a Report on a Unique DFG-in/Leu-in Conformation of Mitogen-Activated Protein Kinase Kinase 7 (MKK7) J. Med. Chem.. 2019;62:5541-5546.
- [Google Scholar]
- Targeting the MKK7–JNK (mitogen-activated protein kinase kinase 7–c-Jun N-terminal kinase) pathway with covalent inhibitors. J. Med. Chem.. 2019;62:2843-2848.
- [Google Scholar]
- Synthesis and evaluation of polycyclic pyrazolo [3, 4-d] pyrimidines as PDE1 and PDE5 cGMP phosphodiesterase inhibitors. J. Med. Chem.. 1997;40:4372-4377.
- [Google Scholar]
- Pyrazolo [3, 4-d] pyrimidines as inhibitor of anti-coagulation and inflammation activities of phospholipase A 2: insight from molecular docking studies. J. Biol. Phys.. 2013;39:419-438.
- [Google Scholar]
- Highly efficient synthesis of fused bicyclic 2, 3-diaryl-pyrimidin-4 (3H)-ones via Lewis acid assisted cyclization reaction. Tetrahedron Lett.. 2008;49:1725-1728.
- [Google Scholar]
- Discovery of N6-phenyl-1H-pyrazolo [3, 4-d] pyrimidine-3, 6-diamine derivatives as novel CK1 inhibitors using common-feature pharmacophore model based virtual screening and hit-to-lead optimization. Eur. J. Med. Chem.. 2012;56:30-38.
- [Google Scholar]
- Novel 3-substituted-1-aryl-5-phenyl-6-anilinopyrazolo [3, 4-d] pyrimidin-4-ones: Docking, synthesis and pharmacological evaluation as a potential anti-inflammatory agents. Bioorg. Med. Chem. Lett.. 2012;22:6616-6620.
- [Google Scholar]
- Discovery of novel selective Janus kinase 2 (JAK2) inhibitors bearing a 1H-pyrazolo [3, 4-d] pyrimidin-4-amino scaffold. Bioorg. Med. Chem.. 2019;27:1562-1576.
- [Google Scholar]
- Structure-based design and synthesis of 1H-pyrazolo [3, 4-d] pyrimidin-4-amino derivatives as Janus kinase 3 inhibitors. Bioorg. Med. Chem.. 2018;26:4774-4786.
- [Google Scholar]
- Ras-related C3 botulinum toxin substrate 1 combining with the mixed lineage kinase 3-mitogen-activated protein kinase 7-c-Jun N-terminal kinase signaling module accelerates diabetic nephropathy. Front. Physiol.. 2021;12
- [Google Scholar]
- INCB050465 (Parsaclisib), a novel next-generation inhibitor of phosphoinositide 3-kinase delta (PI3Kδ) ACS Med. Chem. Lett.. 2019;10:1554-1560.
- [Google Scholar]
- Overcoming cancer therapeutic bottleneck by drug repurposing. Sign. Trans. Target. Ther.. 2020;5:1-25.
- [Google Scholar]




