

Translate this page into:
The role of commonly used transition metals in total synthesis of indole alkaloids
⁎Corresponding authors. nasirrasool@gcuf.edu.pk (Nasir Rasool), syedadnan@uitm.edu.my (Syed Adnan Ali Shah),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
This Paper reviews the most recent advances in synthesizing indole alkaloids using transition metal-catalyzed methods. The essential class of natural substances known as indole alkaloids has several biological effects, including anti-inflammatory, anti-cancer, and anti-bacterial properties. Although there are numerous known natural sources of indole alkaloids, due to their scarcity and complexity, total synthesis has emerged as a promising technique of manufacture. Transition metal-catalyzed methods have become efficient tools for synthesizing indole alkaloids due to their high selectivity and efficiency. The various metal-catalyzed reactions that are used to synthesize indole alkaloids are discussed in this paper, including cross-coupling, cycloaddition, and oxidative cyclization reactions in last five years.
Keywords
Indole
Alkaloids
Transition metals
Natural product
Biological activities

1 Introduction
Amino acids are converted into alkaloids, a varied family of substances with a nitrogen atom in a heterocyclic ring. The Arabic name “al-qali,” associated with the plant from which soda was first extracted, is where the word “alkaloid” originates. The German scientist Carl F. W. Meissner used the phrase for the first time in 1819 (Buchanan, Gruissem, & Jones, 2015). Alkaloids are naturally occurring organic compounds that contains nitrogen atoms and can be basic, neutral, or weakly acidic. Alkaloids may also contain carbon, hydrogen, sulfur, oxygen, bromine, phosphorus, or chlorine atoms in addition to nitrogen. Historically, alkaloids were considered plants' end products of nitrogen metabolism. However, it is now known that plants produce alkaloids for various purposes, including defense against herbivores and diseases, pollinator attraction, and plant growth and development regulation (Sundberg, 1988).
Since the discovery of the first alkaloid, morphine, in the nineteenth century, alkaloids have been employed in medicine. Anti-analgesics (e.g., morphine), antiarrhythmics (e.g., ajmaline), cough suppressants (e.g., codeine), antimalarials (e.g., quinine), anti-tumors (e.g., vinblastine), and anti-hypertensive drugs (e.g., vincamine) are all examples of alkaloids. Alkaloids' various activities are owing to their complex structures and capacity to interact with biological targets in distinct ways (Babbar, 2015). Because of their broad physiological effects on animals and pharmacological qualities, such as anticancer and antibiotic activity, as well as their potential use as medications, stimulants, and poisons, alkaloids have considerably impacted human history. Alkaloids have been utilized in traditional medicine for millennia and have played a part in developing several medicines. Simultaneously, several alkaloids have been employed for their euphoric effects, leading to addiction and abuse (Ziegler & Facchini, 2008). An alkaloid with antiparalytic effects is D-tubocurarine. It blocks acetylcholine receptor sites at neuromuscular junctions, preventing muscles from relaxing (Das, Vedasiromoni, Chauhan, & Ganguly, 1997; Kaur & Arora, 2015). Several alkaloids, including serotonin, histamine, and dopamine, act as animal neurotransmitters. These chemicals serve critical roles in regulating physiological processes like mood, sleep, and appetite.
Furthermore, alkaloids containing indole are well-known bioactive compounds employed in the pharmaceutical and agrochemical sectors (Song, Chen, & Gong, 2017). The indole ring system is a key structural component with numerous pharmacological applications. This contains, among other things, antifungal, antihistaminic, antibacterial, plant growth regulating, antioxidant, anti-HIV, anti-inflammatory, analgesic, and anticonvulsant properties (Singh & Singh, 2018). Indoles are a crucial component in drug development and are considered one of the most crucial heterocycles (Smith, Stevenson, Swain, & Castro, 1998). Indole alkaloids are the most complex class of marine alkaloids and are common in marine creatures. Sponges, tunicates, acorn worms, red algae, and symbiotic bacteria are among the marine creatures that create these chemicals. Indole alkaloids account for one-quarter of all marine alkaloids (Gul & Hamann, 2005). Ergot alkaloids are indole alkaloids generated from the fungus Claviceps purpurea. These chemicals have been used in traditional medicine for millennia for various purposes, including stimulating uterine contractions during childbirth.
On the other hand, Ergot alkaloids can be exceedingly dangerous and result in symptoms like gangrene, hallucinations, and convulsions. On the other hand, indole alkaloids like tryptophan are essential nutrients for animals and people (Plemenkov, 2001). For almost a century, traditional synthetic techniques have been employed to produce indole alkaloids, including the Fischer indole synthesis and the Paal-Knorr pyrrole synthesis. However, these procedures necessitate pre-functionalized starting materials, which can be expensive and potentially harmful. For example, the Fischer indole synthesis and the Larock/Castro methods necessitate hazardous aryl hydrazines and costly 2-iodoaniline derivatives(Kasahara, 1981). Transition metal-catalyzed techniques have been developed as an alternative solution for synthesizing and transforming heterocycles, including indole alkaloids. For example, the synthesis of indoles has been achieved through intra- or intermolecular reactions of 2-alkynyl anilides with aryl- or alkyl halides and through palladium-induced cycloadditions of 2-haloanilines with terminal or internal alkynes. These techniques have shown promising results and offer advantages such as high efficiency, mild reaction conditions, and the ability to use readily available starting materials (Cacchi, Fabrizi, Lamba, Marinelli, & Parisi, 2003; Djakovitch, Batail, & Genelot, 2011; Yoshikai & Wei, 2013; Zeni & Larock, 2006). This study will offer an overview of current advancements in the synthesis and total synthesis of indole alkaloids over the last five years, highlighting their origins and biological potential. We focus on using transition metal-catalyzed methods, such as Heck and Suzuki cross coupling, for the total synthesis of indole alkaloids.
2 Role of gold in total synthesis of indole alkaloids
2.1 Rhazinilam
The advantage of this Cycloisomerization –sulfonyl migration catalyzed by Gold that they discovered proved a very efficient method for synthesis of rhazinilam and other natural compounds.(Miaskiewicz et al., 2016) After successfully completed the first two critical steps of the proposed synthesis of rhazinilam Gold-catalyzed cycloisomerization − sulfonyl migration and Suzuki-Miyaura type coupling reaction using sample molecules, the total synthesis was initiated. Two precursors (1, 2) were prepared using N-tosyl- or N-4-methoxybenzenesulfonyl glycine methyl esters and tert-butyl (E)-4-ethylidene-7-iodoheptanoates in three steps. The gold-catalyzed cycloisomerization sulfonyl migration cascade was carried out for each derivative, producing the predicted N-alkylated pyrrolyl sulfonates (3, 4) in high yields. The total synthesis of rhazinilam was then accomplished through the trans-annular cyclization of a halogenated 13-membered lactam, followed by the reverse procedure using (3,4). The presence of the alkene moiety was attributed to the macrolactam structure. The unprotected 2-aminophenylboronic acid (5) was coupled with (3, 4), resulting in 2-aminophenyl pyrrole in lower yield from (4) and high yield from (3). The reaction was completed through an intramolecular oxidative Heck-type reaction, macrolactamization, and hydrogenation of the vinyl side chain. The intra-chain Heck-type reaction at high temperature produced compound (6, 7), allowing for the isolation of the cyclized product in a standard to yield Rhazilium (9){Kim, 2017 #141} (J. H. Kim et al., 2017)(Scheme 1).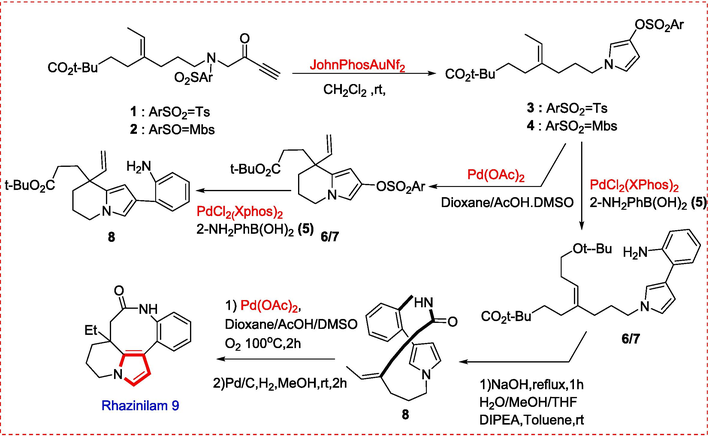
Synthesis of Rhazinilam alkaloid.
2.2 Strictamine
Strictamine is an alkaloid that was first isolated from Rhazya stricta by Ganguli and colleagues in 1966. It belongs to the akuammiline alkaloid family, which are characterized by their cage-like structures made up of indolenine. Strictamine has been found to have inhibitory effects on nuclear factor-κB, a protein that plays a crucial role in controlling the expression of genes involved in inflammation and immune response. In the synthesis of congested ring system of strictamine alkaloid different research groups have employed different methods like Garg's approach focused on completing the D-ring by selecting the C5–N4 bond for intramolecular substitution. Intermediate 6 was formed through Fischer indolization of ketone, which carried the C and E rings,(Moreno, Picazo, Morrill, Smith, & Garg, 2016) The Zhu group took a different route by disconnecting the C15–C20 bond in the E-ring, simplifying strictamine to vinyl iodide intermediate. The D-ring was constructed through intramolecular nucleophilic substitution of brominated compound, which bore the A and C rings.(Ren, Wang, & Zhu, 2016) The first total enantioselective synthesis of (+)-strictamine was reported by the Garg group from UCLA used an incomplete Fischer indolization approach which is better than Zhu group synthesis of Strictamine due to poor yield of last step involving Nickel Catalyzed reaction. The synthesis involved several key steps. Firstly, desymmetrization, oxidation, saponification, and enolization were used to create silyl enol ether 11 from dibenzoate 10. Subsequently, an epoxidation/Wittig olefination sequence was performed to convert [3.3.1]-azabicyclic enone 12 to enal 13 vi Toste cycloisomerization catalyzed by Au(I). The carbon structure of keto lactone 14 was built up over seven stages. The intermediate imine 15 was reduced by a hydride source, leading to smooth Fischer indolization, which resulted in intermediate 16. During this phase, the challenging C7 quaternary stereocenter was formed. The practical synthesis of hydroxyestewas challenging due to the epimerization of the C16 stereocenter when subjected to various direct lactone ring-opening conditions. Eventually, a five-step work-around procedure involved reducing the lactone, selectively silylating the main alcohol and oxidizing and esterifying the material. Mesylation and chlorination were then used to activate the hydroxyl group. The total synthesis of strictamine 18 was completed by denosylation and ring-closing by the released amine group after oxidation by PCC to expose a ketimine functional group. Overall, this total synthesis of strictamine provides a powerful tool for exploring its biological properties and therapeutic potential (Scheme 2).(Ren et al., 2016).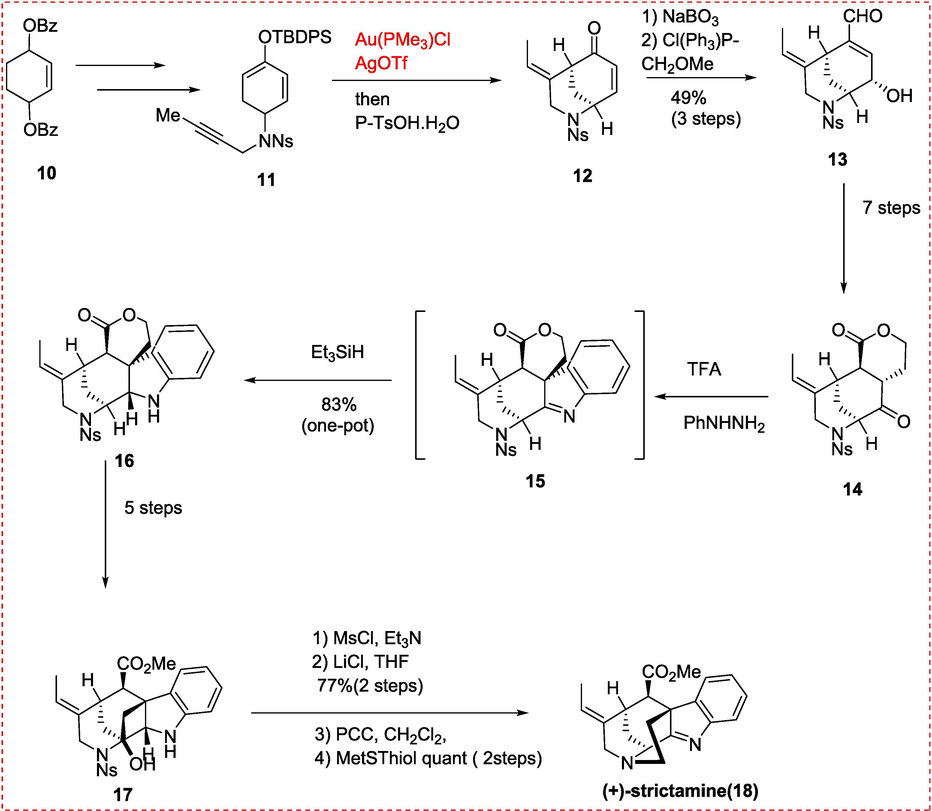
Gold Catalyzed Synthesis of Strictamine.
3 Role of palladium in total synthesis of indole alkaloids
3.1 Tetracyclic indole alkaloid ht-13-A
Kamiguchi and Yasui discovered two tetracyclic indole alkaloids from the bacterium Streptomyces species in 2000(Lounasmaa & Tolvanen, 2000). The compounds ht-13-A have 3,4-oxepino-fused indoles that bind to serotonin receptors to cause better sleep patterns. Previously ht-13-B was synthesized by using the same methodology.(Y. Zhang, Hubbard, Akhmedov, Petersen, & Söderberg, 2015). The synthesis of ht-13-A was first time reported by Zhang et al. from 4-amino-2-hydroxybutyric acid by acyliminium ion allylation followed by Mitsunobo reaction.Then palladium-catalyzed stille-kelly intramolecular cross-coupling to acquire the oxepane ring, and carbon monoxide stimulated reaction catalyzed by reductive palladium N-heterocyclization to acquire pyrrole.
Pyrrolidine precursor 19 was synthesized and was used to make compound 21 by reacting it with 2-bromo-3-nitrophenol (20) in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosphine. The tricyclic molecule 22 was produced by treating compound 21 with hexamethylditin in the presence of bis(dibenzylidenacetone)palladium-triphenylphosphine. In the presence of carbon monoxide, reductive palladium-catalyzed N-heterocyclization yielded the desired tetracyclic indole 23. At last, employing sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al) with toluene as solvent, the methoxycarbonyl was converted to a methyl group, yielding ht-13-A (24) in high yield (Scheme 3, Scheme 4) (Y. Zhang, McArdle, Hubbard, Akhmedov, & Söderberg, 2016).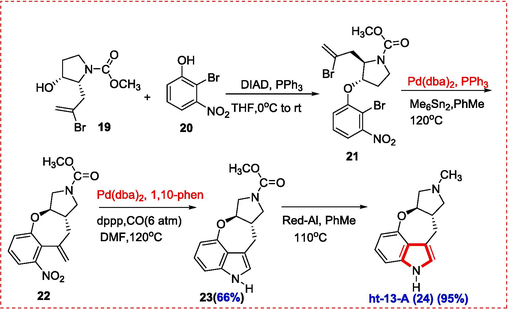
Synthesis of ht-13-A alkaloid.
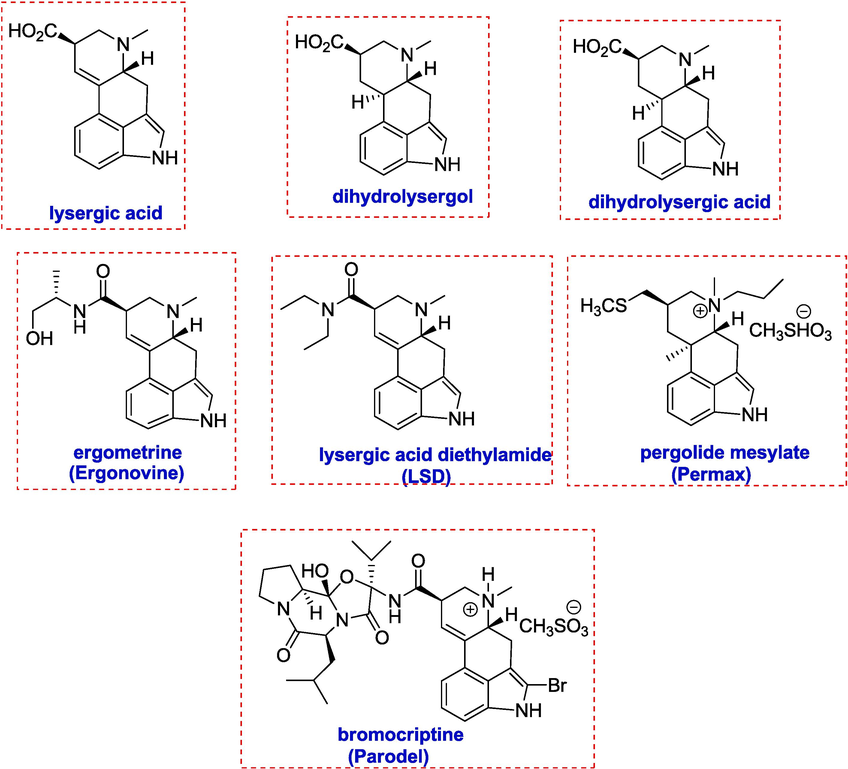
Various ergot alkaloids.
3.2 Dihydrolysergic acid and dihydrolysergol alkaloids
The fungus Claviceps purpurae produces ergot alkaloids, a class of indole alkaloids with pharmacological effects. LSD (lysergic acid diethylamide), a potent psychoactive and hallucinogen, is one of them. The pharmacophores of phenethylamine, dopamine, related biogenic amines, and serotonin are present in conformationally constrained forms in the structures of ergot alkaloids.
Lysergic acid was synthesized by Lee et al. by coupling reaction of compound 30 in the presence of Pd2(dba)3, Pd(PPh3)4, and PdCl2(PPh3)2 in an inert environment. This reaction occurs under soluble organic bases like Et3N, which exceeds inorganic bases like K2CO3. This product 31 is then reacted under inverse Diels Alder type reactions to form compound 32 possessing a pyridine ring, which is converted to N-methylated by reacting it with CH3I (80 °C, 10 h), yielding the quantitative salts of pyridinium 33. This crude salt was then subjected to various reduction conditions. A stepwise reduction (CH3OH, NaCNBH3 65 °C, 18 h; then HOAc, NaCNBH3, 1 h, 25 °C) produced a single diastereoisomer. Concomitant oxidation that converts indoline to indole also causes N-benzoyl amide removal Dihydrolysergic acid (36) was obtained by first treating 34 with HCl in MeOH(0.5 M,95 °C,72 h) to give 35, which is further hydrolyzed (1 N NaOH/CH3OH, 40 °C, 3 h) in the end. Dihydrolysergol 37 was also obtained by ester reduction(THF, LiAlH4, 30 min, 0 °C, 92 %) of the same compound 38 (Scheme 5) (Lee, Poudel, Glinkerman, & Boger, 2015).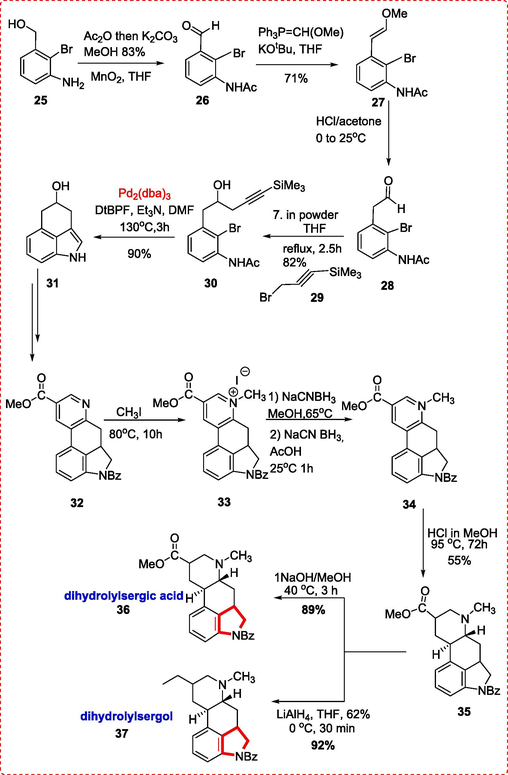
Synthesis of dihydrolylsergic acid and dihydrolylsergol alkaloids.
One significant and notable aspect of the method is the use of a particular advanced ketone-derived enamine. This allows for a divergent preparation process in conjunction with a series of inverse electron demand [4 + 2] cycloaddition processes of heterocyclic azadienes.(Boger, 1987) This method makes it possible to create heterocyclic derivatives that are isomeric and alternate while yet significantly altering the ergot skeleton structurally. These modifications are more difficult to do with traditional techniques, emphasizing the unique and creative nature of this approach.(Boger & Mullican, 1984).
3.3 Unsymmetrical 3,3′-diindolylmethanes indole alkaloids
Diindolylmethanes (DIMs) are distinct indole alkaloids with numerous therapeutic effects, including anti-proliferative, anti-hyperlipidemic, and anti-cancer qualities. DIMs are also used in chemosensor research as a building component for creating oxidized-bisindole chromophores and receptors for conjugated tris(indolyl) methane anions. Unsymmetrical DIMs have been synthesized in recent years by a number of research groups employing a leaving group strategy,(de la Herran, Segura, & Csaky, 2007; Kaiser et al., 2006; Lin et al., 2015) which is supposed to work by creating stable alkylidene ion intermediates from 3-substituted indoles in situ. However, in order to build the symmetrical DIMs system, most current routes used intricately pre-functionalized indoles as reactants. Investigating a feasible and efficient method to quickly access diversely asymmetrical DIMs is therefore still very desirable but difficult.(Guo, Yuan, Gu, Lin, & Yao, 2016) In light of our ongoing research into the synthesis of indole-containing polycyclic compounds, we will reveal an unprecedented palladium-catalyzed cascade here. Allenamides and o-ethynylanilines undergo a Heck/cyclization process to produce unsymmetrically substituted 3,3′-diindolylmethane frameworks. It is possible to produce two C–C bonds and one C-N bond in a single pot(C.-C. Chen, Chin, Yang, & Wu, 2010). Using the cascade Heck/cyclization reaction catalyzed by palladium, an efficient synthesis of unsymmetrical 3,3′-diindolylmethanes (44–50) with various substituents has been devised. N-allenyl-2-iodoaniline 38 and 4-methyl-N-(2-(phenylethynyl)phenyl)benzenesulfonamide 39 were initially employed in the study. The yield increased when acetonitrile was employed as a solvent; however, when other solvents, such as MeOH, DCE, toluene, and THF, were used, the yield fell. This protocol exhibited great functional group tolerance and scalability. Several N-protected groups were discovered to be compatible with N-aryl-N-allenamide 38 and the resulting compounds could be separated in moderate to good yields. High tolerance products, such as methoxy, halogens (Cl, F, Br), methyl, and cyano, yielded good yields ranging from 70 to 90 % (Scheme 6).
Synthesis of 3,3′-diindolylmethanes via Pd-catalyzed Heck cyclization.
Allenamide and o-ethynylaniline demonstrated a feasible catalytic cycle for this reaction. The oxidative addition of allenamide 38 to the active Pd(0) catalyst produce Pd(II) species 40, which then experiences a Heck reaction (intramolecular) to produce the allylpalladium 41. By ligand exchange, the triple bond of o-ethynylaniline 39 coordinates to intermediate, resulting in intermediate 42. Species 42 could easily switch to species 43 by base. Finally, species 43 undergoes C–C reductive elimination, yielding diindolylmethane 44–50 (Scheme 8) and regenerating the Pd(0) species for another catalytic cycle (Scheme 7) (Guo et al., 2021).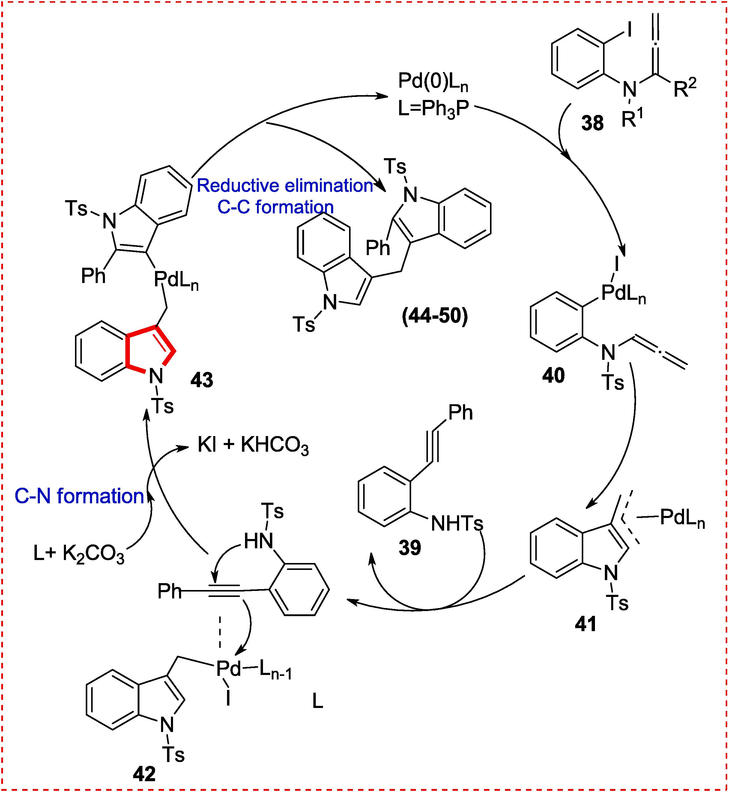
Mechanistic study to synthesize diindolylmethanes.

Chippiine-dippinine type indole alkaloid.
3.4 (+)-tronocarpine
More than 30 years after the discovery of chippiine, a secondary metabolite of the post-iboga alkaloid Tabernaemontana chippii, structurally related compounds were discovered, leading to the discovery of the dippinine-chippiine type indole alkaloids family. Because of the different oxidation states at C-3, C-10, C-11, C-19, and C-20, this family has structural variety. Tronocarpine is a special member of this family since it underwent an N4-C21 rearrangement followed by lactamization with ester functionality at the C16 bridgehead.
The earlier method of producing (+)-tronocarpine used a 15-step linear synthetic sequence (20 steps from commercially available tryptamine), which was a bit laborious for easily providing the samples for upcoming biological analysis. In this article, we offer an enhanced method that can synthesize (+)-tronocarpine using the longest linear sequence possible 11 stages.Tan et al. reported the synthesis of tronocarpine entails multiple processes. They begin with azepanone (51), which undergoes a Dessmartin oxidation in the presence of a Pd(OAc)2 catalyst to generate compound 52 in 88 % yield. Compound 52 is subsequently transformed to compound 53 (enone) with a 75 % yield by treating it with AgCO3 base in DMSO in an oxygen atmosphere. The enone 53 is then alpha-iodinated to obtain compound 54 with 92 % yield. Decarbonylation of compound 54 at C-15, combined with palladium-catalyzed Stille cross-coupling, enables ethyoxycinytannane 55 to integrate a side chain into compound 54, resulting in compound 56 (diene). Compound 56 is subsequently converted to compound 57 with an 86 % yield using Luche reduction. Compound 58 is obtained in 50 % yield by radical deoxygenation of compound 57 utilizing Barton-McCombie. Finally, compound 58 is reduced with LiBHEt3, and quenching occurs to generate tronocarpine 59 in 74 % yield with 1 M HCl (Scheme 9) (D.-X. Tan, Zhou, & Han, 2020).
Synthesis of tronocarpine.
3.5 (+)-minfiensine
The large group of natural products consists of Monoterpene indole alkaloids having an array of akuammiline and strychnos alkaloids having various biological properties and cage like structure. For achieving the total synthesis of Minfiensine previously two methods are employed i.e. Asymmetric Heck-type reaction(Dounay, Overman, & Wrobleski, 2005) and Asymmetric Diels –Alder Cycloaddition(Jones, Simmons, & MacMillan, 2009). In this paper they have introduced new approach for synthesis of (+)-Minfiensine which is based on asymmetric dearomatization of indole. The advantage of this approach is that C = C bond which facilitates creation of hydroxymethyl functionality in (+)-minfiensine(Z. X. Zhang, Chen, & Jiao, 2016)(Liu, He, Dai, & You, 2008).
The conversion of the intermediate (+)-59 to (+)-minfiensine 65 was rather simple. TMSOTf/2,6-lutidine was used to remove the N-Boc group, resulting in the intermediate (+)-60, (Bastiaans, van der Baan, & Ottenheijm, 1997) in which the hydroxy group was at same transformed into the TMS ether. The iodoallyl side chain was introduced by further N-alkylation of crude (+)-60 with tosylate 61, resulting in intermediate (+)-62. Second, a reaction of the Heck type created the final ring by joining the side chain to the tetracyclic core's C2, resulting in the intermediate 63. The intermediate 64 carrying an endocyclic olefin was then created as a result of b-hydride elimination. In order to create (+)-minfiensine 65 in a high yield, the crude 64 was treated with NaOH in MeOH/H2O to remove the CO2CH3 protective group[3e] and the TMS ether. (Scheme 10 (Z. X. Zhang et al., 2016).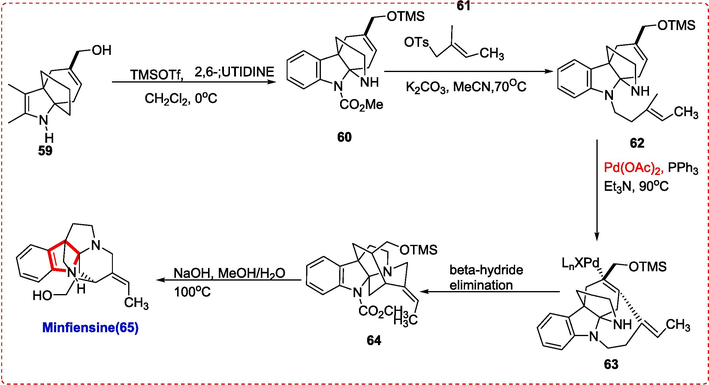
Synthesis of Minfiensine.
3.6 Pyroclavine, festuclavine, costaclavine, pibocin A, epiCostaclavine, 9-deacetoxyfumigaclavine C, dihydrosetoclavine and fumigaclavine G
Claviceps purpurae was discovered to have a wide variety of indole alkaloids called ergot alkaloids.(de Groot, van Dongen, Vree, Hekster, & van Roosmalen, 1998; Rigbers & Li, 2008) They have a wide range of therapeutic benefits. Throughout human history, their significance as natural poisons has been well recognized. Two ergot alkaloids, pergolide, and cabergoline, are known to treat Parkinson's disease and migraines, respectively. Inspite of several synthetic approaches have been done in order to synthesize C/D ring or B/C ring in one step but no approach could acheve synthesis of B/C/D ring in one single step(Dounay et al., 2005; Inuki, Iwata, Oishi, Fujii, & Ohno, 2011). The synthesis of the advanced common intermediate and the B, C, and D rings of ergot alkaloids may be synthesized in a single step by using an intramolecular palladium-catalyzed Tsuji Trost allylation cascade/Larock indole annulationhad been reported by Liu et al..(Shan, Gao, & Jia, 2013a) With some tweaks at the end, this method might be used for a divergent practical synthesis of compounds like deacetoxyfumifclavin, Fumiaclavine G, Pyroclavin, Costaclavin, Dihydrosetoclavine, and Pibocin A. These alkaloids can be synthesized from 66. Under our optimizedcatalyst combination of Me-phos and Pd(OAc)2 at 100 °C, we performed the intramolecular Larock indole annulation of 66 catalyzed by palladium, resulting in the desired tricyclic indole 67 with a yield of 92 %.(Trost & Crawley, 2003) However, when the process was carried out on a gram scale, a small quantity of a surprising tetracyclic molecule 68 was isolated. Tricyclic indole 67 was subjected to Tsuji Trost allylation, resulting in compound 68. The reaction using 0.9 equivalent of Me-phos and 0.3 equivalent of Pd(OAc)2 yielded the necessary 67 with 25 % of 66 in 65 % yield. Carbamate is obtained in 80 % yield when the tert-butanesulfinyl group is removed, and the resulting secondary amine is N-protected with ClCO2CH3. Tricyclic indole 68 might be converted to 69 by removing the silyl and tertbutanesulfinyl groups with HCl, then chlorinating the resulting free hydroxyl group with SOCl2, then treating the resulting compound with NaOH and protecting the secondary amine with N. LiAlH4 was used to decrease the carbamate 69, and olefin 70 was produced in a 93 % yield. Lastly, olefin 69 was selectively reduced with 5 % Pd/C, yielding 71 and 72 in an 8:1 ratio and a 95 % overall yield. Furthermore, the olefin 70 was reduced by Raney Ni to produce the desired molecules 71 and 72 with a 90 % yield and a 5:3 ratio. The physical attributes of synthesized compounds 71 & 72 are similar to those of the originals described in the literature data. We have done the first total synthesis of Pyroclavin(71) and festuclavine(72) (Matuschek, Wallwey, Wollinsky, Xie, & Li, 2012)(Scheme 11).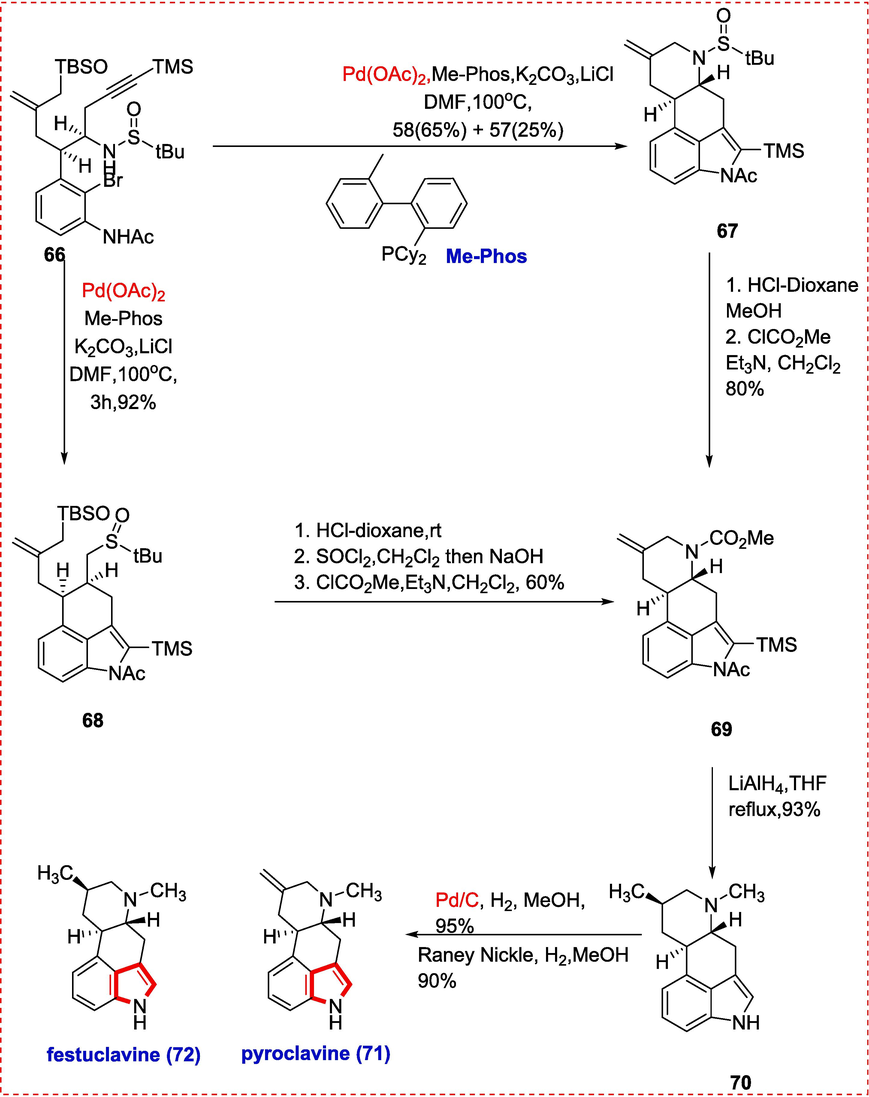
Pyroclavin & Festuclavine total synthesis.
After synthesizing compounds 71 and 72, we investigated their direct functionalization to synthesize fumigaclavine G 73, pibocin A 74, 9-deacetoxyfumigaclavine C 75 and Di-hydrosetoclavine 76,.(Yan Gao, Shan, & Jia, 2014; Larock & Yum, 1991; L. Li, Yang, Wang, & Jia, 2015; Shan, Gao, & Jia, 2013b).
It was straightforward to transform compound 77 into the desired teracycle 78. In conclusion, under different circumstances, the exocyclic C–C bond hydrogenation with Raney Ni or Palladium yielded only epi-costclavine (83). In contrast, reduction with Crabtree's catalyst 79 yielded both costaclavine 82 and epi-costaclavine 83 in a ratio of 1:1.2 (Scheme 12).
Synthesis of costaclavine and epi-costaclavine.
3.7 Herbindole B and cis-trikentrin A
The polyalkylated cyclopentindoles alkaloids include herbindole and trikentrin. Herbindole alkaloids were discovered from the sponge Axinella species present in western Australia. They were shown to be poisonous to KB cells and effective fish repellents. Trikentron flabelliforme sea sponges contain trikentrin alkaloids, which have been extracted and shown to have antibacterial effects against Gram-positive bacteria. Many research groups have targeted the non-asymmetric and enantioselective synthesis of trikentrin and herbindile like Macleod, Kanemastu, Funk, Boger, Saito, silva and Kerr.(Macleod & Monahan, 1988; Muratake, Mikawa, & Natsume, 1992; Silva Jr, Craveiro, & Tebeka, 2010).The synthesis of both cis-trikentrin A (89) and herb-indole B (93) was reported by Leal et al. by using the C–H amination process. To simplify the enantiodivergent synthesis of herbindole B and cis-trikentrin A, an enantioselective diol desymmetrization of the meso-hydroquinone was designed. Thanks to the adaptable aryl triflate functional handle, we created a comprehensive synthesis of cis-trikentrin A and employed 88 as a typical intermediate. Aryl triflate 84 was hydrogenolyzed with H2 under increased pressure to produce 85 in 89 % yield, which was then used to make 86. By Buchwald-Hartwig amination of aryl tosylate 85 and triflation of the resultant aniline progressed to aniline triflamide 86. Triflamide 86 was treated to our newly discovered indolization process, which isolated the corresponding indole 87 in 61 % yield (1C-N bond and 1C = C produced; average 78 % yield per activity). By converting N-triflyl indole 87 into N-tosyl indole 88, an intermediate in the formation of cis-trikentrin A (89), Jackson and Kerr completed the formal synthesis of the compound (Scheme 13) (Leal et al., 2016).
Palladium-catalyzed synthesis of cis-trikentrin A.
From compound 90 triflyl group was removed, and further, tosylation gave compound 91, which acts as an intermediate for the synthesis of Herbindole B (93) (Scheme 14).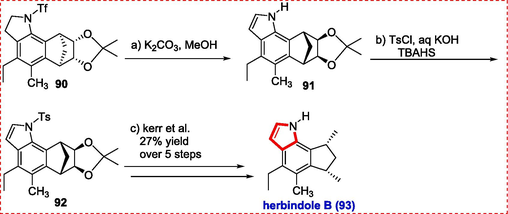
Palladium-catalyzed synthesis of herbindole B.
3.8 Scalaridine A, alocasin A and hyrtinadine B
A group of bisindole alkaloid metabolites derived from tunicates and marine sponges showed strong anti-fungal, anti-viral, anti-bacterial, and anti-plasmodial properties. The presence of an imidazole or pyazinone ring immediately connected to the third indole position is a trait shared by all of these alkaloids. In contrast, Alocasin A, a unique alkaloid extracted from the rhizomes of the plant Alocasia macrorrhiza, has hydroxylation at two odd 5-positions. The marine sponge Hytrios sp. also contains scalaridine A, another pyridine or pyridine-linked bisindole alkaloid with unique behavior. Previously Scalaridine A,(S. H. Kim & Sperry, 2015b) Alocasin A(S. H. Kim & Sperry, 2015a) and Hytinadine A(Tasch, Merkul, & Müller, 2011) was prepared by using cross coupling reaction catalyzed by palladium but Hyrtinadine B was not synthesized. Nurul and Bjorn reported using a unique method for to synthesizing all four alkaloids. Carbon monoxide is used as the last reducing agent in a N-heterocyclization of 2-nitroaryl-substituted alkenes catalyzed by palladium to generate indoles. This strategy is quickly becoming a go-to for synthesizing various substituted indoles.
From 4-benzyloxy-2-bromo-1-nitrobenzene (94), alocasin A was made in six stages, five of which used a palladium catalyst. The anticipated products 95 and 96 were produced in high isolated yields by a deprotection process driven by tetrabutylammonium fluoride (TBAF) after the first Sonogashira coupling of 94 with trimethylsilylethyne. Alami et al have reported on highly -regioselective hydrostannylations of ortho-substituted aryl alkynes (Alami, Liron, Gervais, Peyrat, & Brion, 2002; Hamze, Veau, Provot, Brion, & Alami, 2009; Provot, Hamze, Peyrat, Brion, & Alami, 2013). Regioselective hydrostannylation of 96 using their method produced the anticipated vinylstannane 97 in good yield. In the present synthesis of alocasin A, the core pyrazine ring was created via a one-pot double Kosugi-Migita-Stille cross-coupling of vinylstannane 97 with 2,5- dibromopyrazine 98, producing 99 in a relatively low isolated yield. A 40 % yield homo-coupling product 100 was also identified along with the expected coupling product. A high yielding di-O-benzyl-protected alocasin A 102 was generated by a dual palladium-catalyzed reductive N-heterocyclization of pure 100 in the presence of CO (120 °C, pCO = 6 atm) and a 1,3-bis(diphenyl)propane-1,10-phenanthroline catalyst system. After the benzyl groups were smoothly removed, the naturally occurring bisindolopyrazine alocasin A 102 was created by hydrogenolysis using Pd/C-H2 (H2 = 4 atm 10 % Pd,) in ethanol solvent at 60 °C. (Scheme 15).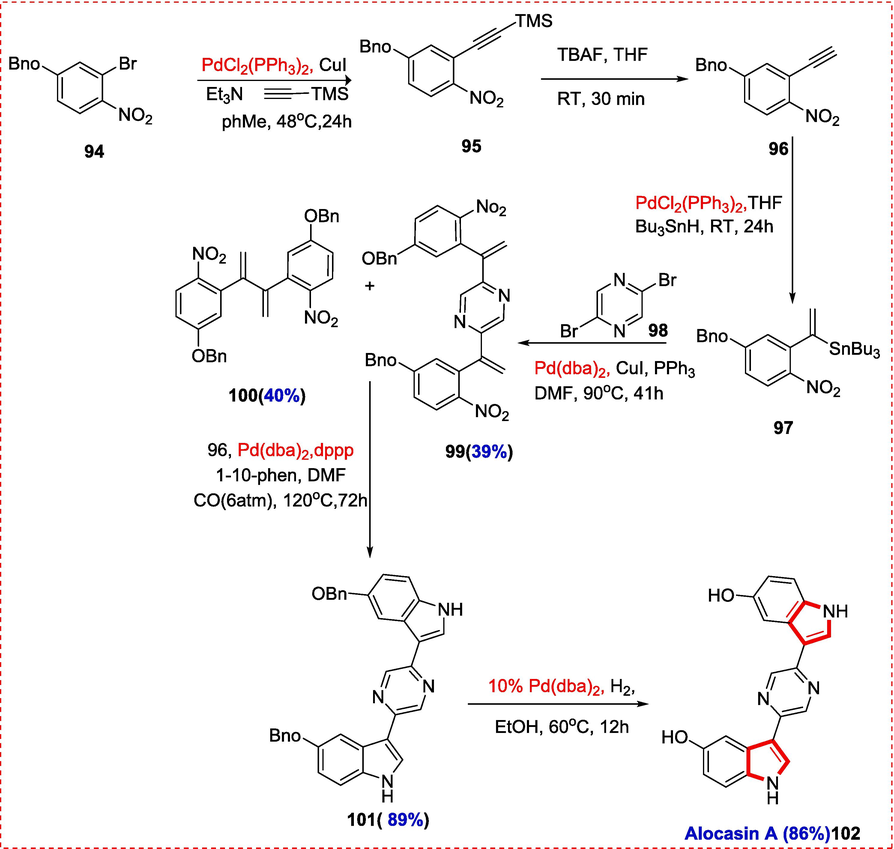
Short synthesis of Alocasin A.
Hydrostannylation of 3,5-diethynylpyridine (1 0 3) catalyzed by palladium in our instance provided 104 with purely α-selectivity, which was satisfying for the production of scalaridine A. The desired product 106 was produced by further cross-coupling 104 with 4-benzyloxy-2-iodo-1-nitrobenzene (1 0 5). Scalaridine A 108 was produced by debenzylation after a reductive N-heterocyclization produced the pyridine bisindole 107 as the last step in the synthesis process. Starting with 103, four synthetic transformations produced a total yield of 13 % (Scheme 16).
Palladium-catalyzed synthesis of Scalaridine A.
2,5-dibromopyrimidine(1 0 9) is coupled with trimethylsilylethyne by double sonogashira coupling to produce product 110, desilylated with K2CO3 in CH3OH 2,5-diethynylpyrimidine (1 1 1). The divinyltiN-substituted pyrimidine 112 was produced by hydrostannylation of 111 with palladium, again with good -regioselectivity. Reductive N-heterocyclization, debenzylation, and the following steps—Kosugi-Migita-Stille cross-coupling of 112 with 113—all proceeded well and produced Hyrtinadine A(1 1 6). Hyrtinadine A (1 1 6) produced from 115 had a 12 % total yield across five stages. Last but not least, the remaining alkaloid hyrtinadine B was created using 5-ethenylpyrimidine. Hyrtinadine B was made in four synthetic stages, yielding 28 % overall, by following the same processes used to make hyrtinadine A: hydrostannylation, cross-coupling reaction, reductive cyclization, and debenzylation (Scheme 17).
Palladium catalyzed synthesis of Hyrtinadine A alkaloid.
The overall yields were 13 % (four steps), 27 % (six steps), 28 % (four steps), and 12 % (five steps), respectively (Ansari & Söderberg, 2016).
3.9 Fargesine and cimitrypazepine
The naturally occurring azepino indoles alkaloids are extracted from several sources. For instance, this group of alkaloids includes cimitrypazepine from the stems and roots of black cohosh and aurantioclavine from the fungus Penicillium aurantiovirens, clavicipic acid from the fungus Claviceps fusiformis, and fragesine from the stems and roots of Evodia fragesii. The most common method of creating the azepino[5,4,3-cd]indole ring system is to manipulate the functional groups of 3,4-disubstituted indoles in order to produce the azepine ring. In complete synthesis, the most often utilized method is the production of the azepine ring via the carbon–nitrogen bond. However, other approaches, such as the generation of carbon–carbon bonds have also been used. Sequential bond formations have been documented for the pyrrole and azepine rings but the assembly of both rings onto a functionalized benzene ring is more uncommon. When considering various functionalized analogs, the latter approaches may provide more versatile pathways than utilizing an already-assembled indole ring.(Krishnan et al., 2008; Yamada, Makita, Suzuki, & Somei, 1985). The authors described a synthesis in which the primary azepine ring-generating step was the Pictet–Spengler type cyclization of N-methylserotonin and formaldehyde, creating the C–C bond. Despite being straightforward, the process created by Somei et al(Somei, Teranishi, Yamada, & Yamada, 2001).often yields combinations of 1,2,3,4-tetrahydro-b-carbolines and azepino[5,4,3-cd]indoles using aliphatic aldehydes. Furthermore, only 5-hydroxytrytamines may produce azo[5,4,3-cd]indoles.According to Ghimire et al., the azepine ring was initially formed via an intramolecular Heck reaction, followed by the pyrrole ring by a reductive N-heterocyclization catalyzed by palladium (Cummings et al., 2011). CO2 is the only side product of the latter process, while CO serves as the final reducing agent. It is feasible to synthesize a range of cimitrypazepine analogues and azepino[5,4,3-cd]indoles naturally occurring using the flexible synthesis outlined below.
Reductive N-heterocyclization of 116 in the presence of CO (pCO = 6 atm, 120 °C), employing a catalyst system of bis(dibenzylideneacetone)palladium-1,3-bis(diphenylphosphino) propane-1,10-phenanthroline 119 and its N-protected counterpart 120 are generated in 79 % overall yield. Compound 118 was also treated using the same chemicals and reaction setup to create 120 with a lower isolated yield. The O-Boc group was taken out of 120, and the N-Boc group was changed into a CH3 group, resulting in 121, after treatment with sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al) in toluene solvent at reflux. (cimitrypazepine). Alkaloid 121 was generated in high quantities by red-Al reduction of the alkaloid 120. In all efforts, intractable compounds were created when the azepine-nitrogen of 121 was immediately oxidized with m-chloroperbenzoic acid (m-CPBA) in dichloromethane (Qu et al., 2011) or hydrogen peroxide-ammonium hydrogen carbonate in water. Based on the results of the 5-hydroxyindole oxidations, it is possible that a quinone imine was produced, followed by the breakage. Instead of simply oxidizing 121 to fargesine, we opted to stop an intermediate reported in the fargesine synthesis previously. As a result, cimitrypazepine 121 was protected by O-Boc to provide high yields of 122. This compound has recently been oxidized and deprotected using sodium hydroxide and m-CPBA to give fargesine 123 (Scheme 18) (Ghimire & Söderberg, 2016).
Palladium catalyzed synthesis of cimitrypazepine and fargesine.
3.10 (+)-spiroindimicin
Naturally occurring tryptophan containing dimeric structure is a large chemical family that has expanded considerably in recent decades. This class includes some medicinally relevant members, including staurosporine and rebeccamycin (Nakano & Ōmura, 2009; Ryan & Drennan, 2009). The spiroindimicins are a distinct subgroup of nn-planar compounds found in marine Streptomycetes within this large class. The spiroindimicins showed considerable cytotoxicity (IC50 = 9–44 M) against several cancer cell lines. It has been possible to find the action of spiroindimicins A, H, and their analogue against some parasites important to human health. It offers prospective starting points for novel therapies for the undertreated tropical illnesses and African sleeping sickness.
It is rarely unexpected that the synthetic community is interested in the spiroindimicins given their attractive frameworks and apparent bioactivities.(Nandi, Guillot, Kouklovsky, & Vincent, 2016) Sperry and Blair (15–16 steps) have described a previous racemic synthesis of spiroindimicins B and C. This synthesis focused on building the spirocenter early on using an intramolecular Heck reaction, then gradually adding the additional heterocycles.(Blair & Sperry, 2016) To the best of our knowledge, no synthetic research has been published on spiroindimicins Aor H the two C-3/C-5-linked components.Using a quick, gram-scale manufacture of a triaryl precursor and its Palladium catalyzed asymmetric spirocyclization, The first total synthesis of (+)-spiroindimicin A was reported by Zhang et al.They used the proposed approach to create lynamicins A/ D and spiroindimicin H and some structural analogs. We improved a known sequence to produce 4-iodoindole 125 from 4-nitroindole 124 in three stages on a multigram scale (Skolc, Ates, Jnoff, & Valade, 2016). After that, pyrrole stannane 127 underwent rigorous optimization through a Stille coupling in catalyst 129 with indole C3 iodination. Triaryl 130 seems to exist as two different atropisomers (dr 3:1) that progressively convert into one another at room temperature after the last iodination of the pyrrole ring and thermolytic Boc deprotection sets the way for the crucial spirocyclization. In the intended spiroindolenine 131, this 7-membered compound seems to result from direct CarboN-2 coupling rather than through the migration of C-3 to C-2 bonds. By simply applying the spirocyclization conditions to the residue left over after thermolysis, we discovered that Boc deprotection and spirocyclization could be accomplished in a single step [(+)-31: 53 %; (+)- 31: 8 %, 96 % ee]. Spiroindimicin A 132 (65–72 %) was the product of the last removal of the benzenesulfonyl group from 131 using Bu4NOH at 80 °C (Fürstner, Krause, & Thiel, 2002), concluding the first total synthesis of this target compound in 9 stages (Scheme 19) (Z. Zhang et al., 2021).
Asymmetric Palladium catalyzed Spirocyclization.
3.11 Spiro-pseudoindoxyls having N, N′-Ketals
Due to their abundant natural goods, spiro quaternary indoles are fascinating. For instance, dervaluteine exhibits by reversing many drug resistance in vincristine-resistant K-B cells, echitamine is one type of akuammiline alkaloids, Minfiensine was isolated from the African plant Strychnos minfiensis by Massiot and colleagues in 1989, and mitragynine pseudoindoxyl displays intense narcotic agonistic action in the guinea pig ileum. Wei-chen et al. described the synthesis of N, N′-ketal spiro-pseudoindoxyls from o-alkynylnitrobenzene via cycloisomerization, nucleophilic addition, and reduction reaction. They were inspired to notice the unique use of nitrostyrenes when Driver(Jana, Zhou, & Driver, 2015) described a reductive tandem reaction of nitrostyrenes with Mo(CO)6 and a Pd catalyst to make spirocyclic indoles. as a component of their continuous research into the production of bioactive compounds using domino cyclization processes.(Q. Wang, Song, Liu, Song, & Wang, 2016) With this approach, spiro-pseudoindoxyls may be easily accessed without the need to post indole derivatives. The reaction sustained various o-alkynylnitrobenzenes, and the required products were often produced in excellent quantities. The corresponding spiro-pseudoindoxyl compounds were produced in fair to excellent yields on substrates containing weak electroN-withdrawing substituents and (weak) electron-donating reagents at the C2 position. There are further transformations that may be performed using the functional group that is bromo and chloro on the benzene ring. It is important to note that the intended products were produced in reasonable quantities even when the benzene ring included a strong electroN-attracting group, such as a CF3 or a methoxy containing carbonyl. Similar results may be obtained for these reactants with substitutes at C3 position. The substituents at C1 or C4 on the benzene ring provide substantially low yields due to steric hindrance (Scheme 20).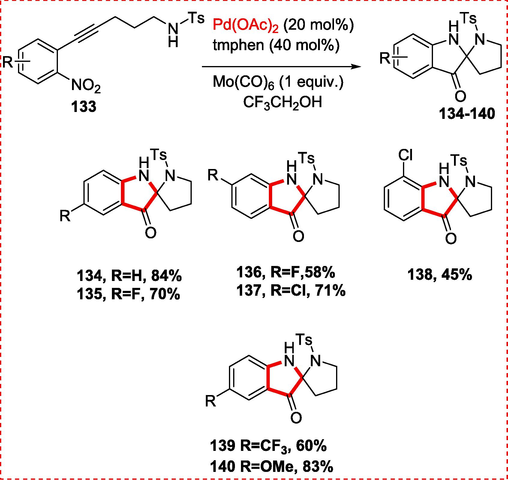
Synthesis of Spiro-pseudoindoxyls containing N, N′-ketal.
Based on the findings above, we suggest the subsequent reaction mechanism. Following an attack of nucleophiles by the oxygen of the NO2 group on the electron deficient alkyne by a 5-exo-dig cyclization to produce intermediate specie 142, internal nitroalkyne redox follows to produce intermediate metal carbene 143. The triple bond coordination of 141 with Pd(OAc)2 increases the electrophilicity of alkyne. There are two routes to get the carbene 143: either via the nitrogen of the nitroso- group (route A) or the NHTs group's nitrogen (pathway B). In process A, nitroso-group nitrogen is added to the carbene 143, which results in the isatogen 144. The isatogen 144 then experiences an intra-molecular addition of the -NHTs to produce the matching N-hydroxy indoliN-3-one 145. Using Mo(CO)6 as a reducing agent, N-O bond in 145 is subsequently broken, releasing the free amine and producing 161. Another hypothesis is that the reduction of N-O bond occurs first in the isatogen 144, followed by the cyclization that results in 161. In route B, adding -NHTs to the carbene 143 is followed by removing palladium, which creates the enol 148 and proceeds through cyclization to create 149 (Scheme 21). Unfortunately, no 149 was produced from the reaction, although the two compounds did exhibit some fungicidal properties. Both 143 and 2d = 144 draw the structure demonstrated fungicidal action against Fusarium oxysporium f. sp. cucumeris having inhibitory rate of 40.9 % at 50 mg/kg) (Armarego & Perrin, 2002; Phillips & Dumestic, 1984) and Alternaria solani having an inhibitory rate of 44.0 % at 50 mg/kg), respectively (Scheme 22) (L.-W. Chen et al., 2017).
Mechanistic study for Spiro-pseudoindoxyls containing N, N′-ketal.
3-aryl-indolinones derivatives.
3.12 3-aryl-indolinones derivatives
Children are hit mainly by malaria, which is brought on by Plasmodium parasites that are spread to individuals by stings from female Anopheles mosquitoes with the illness. The World Health Organization (WHO 2020) emphasizes how crucial it is to continue efforts to prevent, identify, and cure malaria since it affects poor people in Third World nations. Nature has been utilized to cure several ailments in Third World nations. For instance, the medicinal plant Cryptolepis sanguinolenta, widely used in West and Central Africa, contains indole alkaloids with anti-plasmodial effects (Forkuo et al., 2017). In this regard, indole alkaloids such as 6-O-(b-glucopyranosyl)hyrtiosulawesine (i) and 3,4-dihydro-hyrtiosulawesine (ii) from Aristolochia cordigera were identified, and they showed low micromolar antiplasmodial activity against P. falciparum strain (3D7 strain) and low cytotoxicity on hepatic cells (Pereira et al., 2017).
Additionally, certain sections of Brazil employ Aspidosperma olivaceum in South America to cure fever and malaria. Aspidoscarpine (iii), a potential drug against P. falciparum with minimal cytotoxicity in hepatic cells, have been isolated from A. olivaceum (Chierrito et al., 2014). Many indole-containing alkaloids with in vitro activity against P. falciparum have been produced as a result of inspiration from antimalarial alkaloids, including a 3-methylene-indolinone (v) and indole-pyrimidine hybrid (iv) (Agarwal, Srivastava, Puri, & Chauhan, 2005; Kumar et al., 2011). Additionally, indole alkaloids are a fascinating family of natural compounds with antimalarial properties that share much with our quinoline derivatives (Muscia, Bollini, Bruno, & Asís, 2006; Muscia, Bollini, Carnevale, Bruno, & Asis, 2006) (Scheme 23).
Palladium catalyzed synthesis of 3-aryl-indolinones derivatives.
Awe reported the synthesis of about thirty one derivatives of oxindole (indoliN-2-one) and aza analogues having anti-plasmodial activity.
When isatin (1 5 5) and arylamines (1 5 6) were combined in condensation reactions, 3-arylimino-indolinones (157 i–ii) were produced in low yields (30–63 %) under acid catalysis, whereas 3-arylidene-indolinones (165 i–vii) were produced in a Knoevenagel reaction using an alkaline catalyst and oxindole (1 5 9) and aryl aldehydes (1 6 0). 3-aryidene-indolinones 161 are converted to 162 by hydrogenation in the presence of palladium in methanol (Scheme 24).![Synthesis of cyclohepta[b]indole’s precursor.](/content/184/2024/17/2/img/10.1016_j.arabjc.2023.105523-fig24.png)
Synthesis of cyclohepta[b]indole’s precursor.
Some synthetic oxindole derivatives have antimalarial action. The most effective inhibitor, 8iii, had minimal cytotoxic effects on HepG2 cells (IC50 > 100 mM) and low micromolar antiplasmodial activity (IC50 5.8 mM).
3.13 Cyclohepta[b]indoles
Cyclohepta [b]indole alkaloids are a significant group of organic compounds with a many biological functions. Several pharmaceutically active molecules include the cyclohepta[b]indole motif (Bhat, Dave, MacKay, & Rawal, 2014). The actions range from selective inhibition of histone deacetylase at nanomolar doses through reduction of nitric oxide synthesis in BV2-microglial cells, Na channel blockage, anti-cholinergic and antibacterial characteristics.
To achieve the necessary enantioselective 1,2,3-substitution pattern, we used a completely new tactical strategy employing cyclopropane C–H activation. Previously Iodobenzenes were added to the cyclopropane during the C–H activation phase on analogous compounds. Iodoindoles as coupling partners in this reaction have not been studied before.(Wasa, Engle, Lin, Yoo, & Yu, 2011) The crucial process in our method is the direct introduction of a heteroaromatic molecule to chiral cyclopropane through cyclopropane C–H activation having a sp3 bond catalyzed by Pd. We searched for the ideal circumstances for the Palladium-catalyzed C–H activation using cyclopropylamides. When 163 (1 equiv) was treated with Pd(OAc)2 (10 mol%), tosylated 3-iodoindole 164 (3 equiv), and Ag2CO3 (1.2 equiv) in dry toluene (1 M) in a no N-reactive gas environment at 110 °C, we discovered that the yields were at their greatest. Typically, the reaction finished in 3–6 h, producing 165 in a 45–65 % yield. By imide synthesis (Boc protection) and reduction to the related alcohol 166 with a high amount of LiBH4, the directing group was separately removed. We showed the use of the “symmetry” that is “hidden” as an enantiomeric change using compound 166 (Scheme 25).![Synthesis of (-)-cyclohepta[b]indole.](/content/184/2024/17/2/img/10.1016_j.arabjc.2023.105523-fig25.png)
Synthesis of (-)-cyclohepta[b]indole.
Gram-scale synthesis of optically pure compound 166 followed by “amide-to-olefin” conversion to (-)-cyclohepta[b]indole 169. Wittig olefination, DVCPR, and Ley-Griffith oxidation were used to achieve this. The appropriate amount of (-)-cyclohepta[b]indoline 168 was produced quantitatively by the rearrangement, which occurred without a problem at 120 °C in toluene solvent. The required (-)-cyclohepta[b]indole 169 was produced by further TBDPS deprotection of compound 168 using Hydrogen Flouride in pyridine and following aromatization mediated by pTsOH (Scheme 26).![Various synthetic routes for cyclohepta[b]indole synthesis.](/content/184/2024/17/2/img/10.1016_j.arabjc.2023.105523-fig26.png)
Various synthetic routes for cyclohepta[b]indole synthesis.
The DVCPR precursor 166 symmetry is disrupted when the CH3– group is added to the cyclopropane. Various compounds are available through the amide-to-olefin and alcohol-to-olefin pathways, allowing for additional product diversity. The quaternary stereocenter is present in the latter group, but the cyclohepta[b]indole is only trisubstituted in the previous group (Scheme 27) (N’Goka, 1991).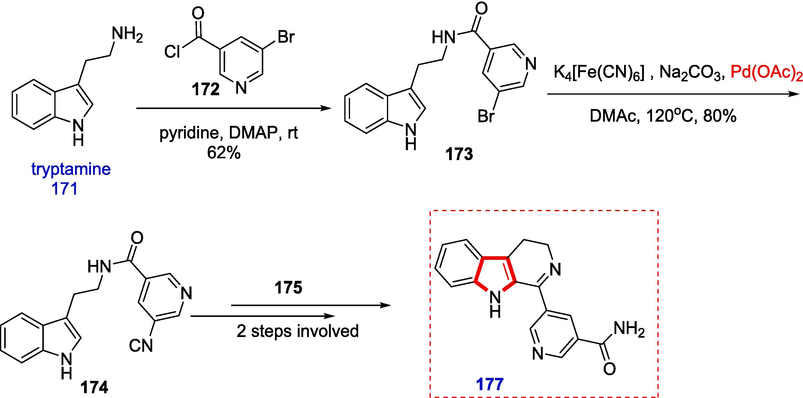
Synthesis of taberniacin B starting material.
3.14 Taberniacins A and B
Stems of the Tabernaemontana diverticata plant produce the novel indole alkaloid taberniacin A and Apocynaceae. It is a member of the Apocynaceae family and may be found in Asia and Australia. The vasorelaxant effect of these two alkaloids was noted, and their structure was reported by Hirasawa et al., who also postulated very first method for their production using palladium-catalyzed processes.
With a 62 % yield, the corresponding amide 173 was obtained via the reaction of tryptamine 171 and acid chloride 172 of 5-bromo-3-pyridinecarboxylic acid. We first looked into the Rosemund-von Braun reaction and the cyanation catalyzed by Copper. Still, both procedures only resulted in the recovery of the starting ingredients or their degradation during the creation of aromatic nitrile 174 from 173. Cyclization of amide 174 by Bischler-Napieralski in the presence of POCl3 yielded the required 3,4-dihydro—carboline, whereas the Weissman technique (K4[Fe(CN)6]/ Pd(OAc)2/Na2CO3) in N,N-dimethylacetamide (DMAc) produced 80 % yield of 175 (Weissman, Zewge, & Chen, 2005). The urea-hydrogen peroxide adduct (UHP) was used to convert nitriles into primary amides under moderate circumstances, which, in turn, afforded 176 in high yield (Scheme 28) (Balicki & Kaczmarek, 1993).
Synthesis of taberniacin B.
With the recovery of 1, the direct transformation of 173 into 174 by dehydrogenation with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) produced a poor yield of 177. Then, using 3,4-dihydro—carboline 175 as a starting point, we decided to synthesize-carboline 175, which could be quickly transformed into an amide. This effort produced the expected results as an excellent yield of 6-carboline. The primary amide was produced in 69 % yield using the same hydrolysis process to give taberniacin B (Scheme 29).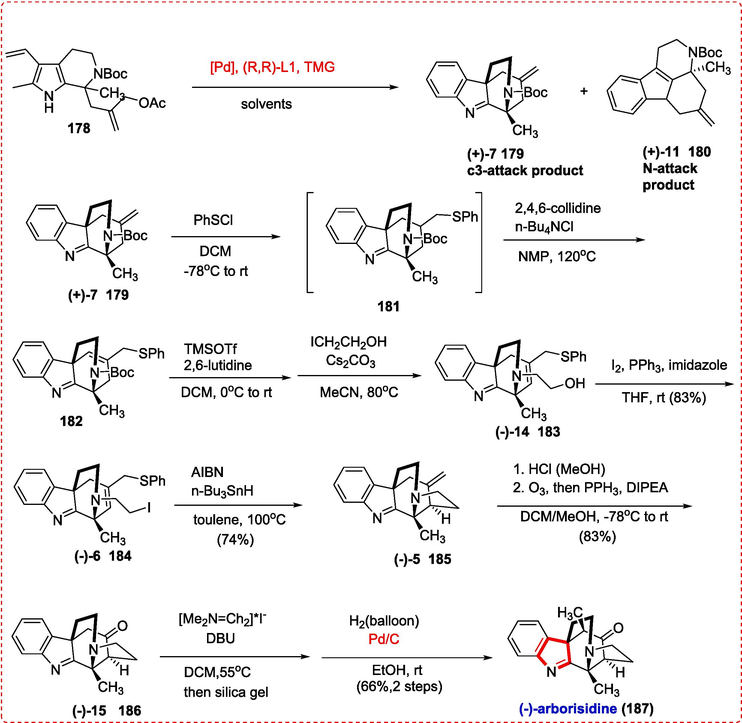
Total synthesis of (-)-arborisidine.
Taberniacins A and B had vasorelaxant activities giving IC50 values of 2.86 μM and 580 nM, respectively (Hirasawa et al., 2019).
3.15 (-)-arborisidine
Arborsidine alkaloid has two quaternary stereocenters and a unique bridging pentacyclic structure. Arborisidine has been a focus of several research projects, but it wasn't until very recently that the research groups of Snyder and Zhu published two effective total syntheses, respectively(Andres, Wang, & Zhu, 2020). Constructing the five-membered ring with a bridging nitrogen content is one of the synthetic problems. To create the C14-C15 bond, a simple method is to use an intramolecular SN2 reaction between an alkyl electrophile and ketone enolate. But as Song, Qin, and colleagues as well as Snyder have shown, a retro-Michael reaction overrides the expected SN2 route in this instance. This is most likely because an anion with the lone pair of electrons roughly coplanar to the * orbital of the C-N bond is generated.(Z. Chen, Xiao, Song, & Qin, 2018) Then Yuan et al. using a novel synthetic method to synthesize (-)-Arborisidine. The process comprises catalytic parallel kinetic resolution of the indole ring to create an intermediate for arborsidine with a bridging nitrogen containing a five-membered ring. By using a palladium-catalyzed reaction of compound 178 with various ligands, bases, and palladium complexes, in the presence of various solvents, Yuan et al. created intermediate 179 from compound 178.
The C-3 and N attack are the results of this reaction, for only using intermediate 7 to continue the synthesis of arborsidine. When (+)-7 (1 7 9) was treated with benzenesulfenyl chloride, the result was a diastereomeric mixture of crude chloride (1 8 1) (dr = 1.2:1) (Giese & Mazumdar, 1981). The base utilized in the subsequent elimination process was crucial, and the N-Bu4NCl/2,4,6-collidine system was the best option to produce allylic phenyl sulfide 182 (Snider & Burbaum, 1986). The unbreakable regioisomeric combination of (1 8 2) was immediately carried over into subsequent transformations. After the N-Boc group deprotection by using TMSOTf/2,6-lutidine and further N-alkylation with 2-iodoethanol, alcohol (-)-14 (1 8 3) could be separated from (+)-7 (1 7 9) (4 stages) to form iodide (-)-6 (1 8 4) under the influence of I2/PPh3/imidazole in 34 % of the total yield. The substrate was ready to test the predicted radical cyclization process. We were pleased to find that the traditional radical cyclization procedure (AIBN/N-Bu3SnH) was capable of encouraging the predicted 5-exo-trig radical cyclization of (-)-6 (1 8 4) to create (-)-5 (1 8 5) in a good yield, demonstrating the viability of the planned synthetic approach. Demethylarborisidine (+)-15 (1 8 6) was produced via the ozonolysis reaction of (-)-5 (1 8 5) and further reductive work-up. A first Mannich reaction with Eschenmosers salt was carried out under an immediate environment to install the 19-methyl group. The product of the Mannich reaction was eliminated on a silica gel to create an intermediate amethylenated ketone. In the end, (-)-arborisidine (1 8 7) was obtained in a high yield and with outstanding diastereoselectivity by hydrogenating the exocyclic C = C bond by Palladium/C- catalyzed reaction. This reaction added hydrogen to the side of the molecule that was less hindered (Scheme 30) (Z. Zhou et al., 2019).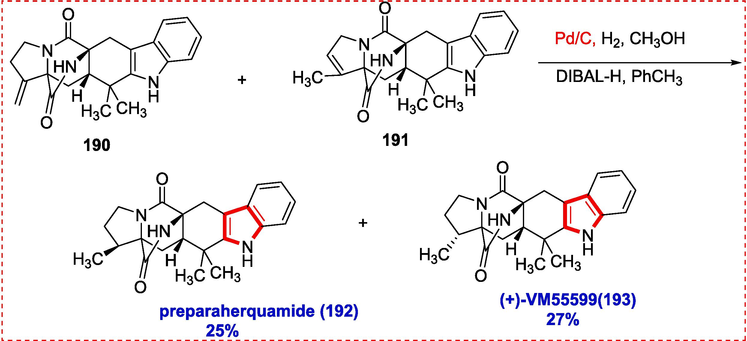
Total syntheses of preparaherquamide and (+)-VM55599.
3.16 Premalbrancheamide, (+)-VM-55599 and preparaherquamide indole alkaloids
The chemical synthesis community continues to be intrigued by secondary metabolites identified from fungi belonging to the genera Penicillium and Aspergillus because of their intricate structural makeup and varied biological functions. A group of reverse-prenylated indole alkaloids, among these natural compounds, demonstrate a variety of bioactivities, including but not limited to cytotoxic, insecticidal, anti-bacterial, and anti-bacterial and parasiticidal characteristics. The paraherquamides, stephacidins, and marcfortines all have a bicyclo[2.2.2]diazaoctane core ring structure, representing this category (Roque et al., 2020).
(+)-VM55599 193 and Preparaherquamide 192 were synthesized in a four-step process, giving a five-membered ring necessary functionality. The first attempts to olefinate ketone 188 failed because Wittig olefinations produced starting material. We investigated nucleophilic addition and alcohol elimination. For instance, the CH3MgBr addition produced considerable recovered starting material and the tertiary alcohol 189 in low yields. Low conversion is presumably caused by unfavorable events such as alpha deprotonation (enolization). When the crude mixture was put through another cycle, 48 % of the necessary tertiary alcohol was produced (1 8 9). The Ce(III) chloride addition did not increase conversion, making attempts to reduce the basic nature of the Grignard reagent ineffective. Instead, it was discovered that the LiCl addition resulted in a 51 % yield (85 % BRSM) of the desired tertiary alcohol. Exocyclic (1 9 0) and endocyclic (1 9 1) alkenes were produced in a mixture after 189 was treated with the Burgess reagent in a 71 % yield (1:2 mixture). Preparaherquamide (1 9 2) and (+)-VM-55599 (1 9 3), two epimeric natural products, were produced via hydrogenation of alkenes mixture (i.e., 190 & 191) by Pd/C, along with reduction tertiary amide via DIBAL-H, with yields of 25 % and 27 %, respectively (Scheme 31).
Total synthesis of premalbrancheamide.
Notably, successive reduction methods can reach indole alkaloids from 189 that lack substituents on ring A. For instance, the pyrrolidone ketone group in 189 was reduced using the Wolff-Kishner method to produce ketopremalbrancheamide (1 9 4). Premalbrancheamide (1 9 5) was produced from ketopremalbrancheamide using the Williams and associates' method as a guide. As a result, the tertiary amide was reduced chemoselectively by treating 194 with an excess of DIBAL-H (Scheme 32).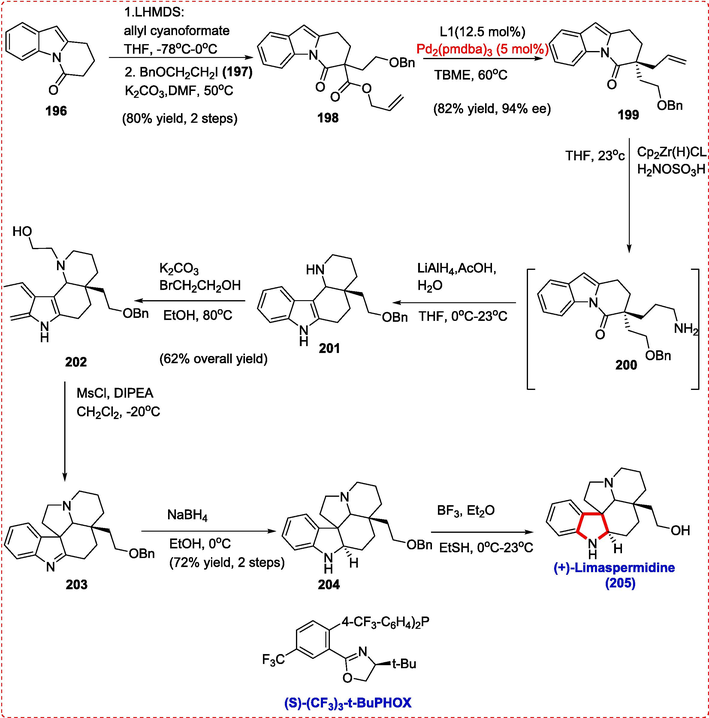
Total synthesis of (+)-limaspermidine.
3.17 (+)-limaspermidine and (+)-kopsihainanine A
Kopsia and Aspidosperma alkaloids like Kopsihainanine A and Limaspermidine are examples of the monoterpene indole alkaloid family. Aspidosperma and Kopsia alkaloids have an azadecaline motif, however, the one in Kopsia is trans-fused, while the one in Aspidosperma is cis-fused. The synthesize of kopsia and aspidosperma alkaloids, was described for the first time an enantioselective allylic alkylation of dihydropyrido[1,2-a]indolone (DHPI) substrates catalyzed by palladium, which was then used in indole-iminium cyclization processes by Pritchett et al.. C-acylation with allyl cyanoformate was the first step in the Limaspermidine synthesis, which ultimately led to C-alkylation of DHPI 196 in the presence of (2-benzylozy)ethyl iodide (1 9 7) and an 80 % yield of d bamidoester 198. To get DHPI 199 in 82 % yield and 94 % ee, we subjected 198 to the solution of Pd2(pmdba)3 (5 mol%) and (S)-(CF3)3-tBuPHOX (12.5 mol%) in TBME at 60 °C. Hartwig and coworkers' hydrozirconation/amination approach allowed for completing a formal anti-Markovnikov hydroamination. After synthesizing the primary amine (200, not isolated), lithium aluminium hydride was added. The mixture was carefully quenched with acetic acid and water to facilitate indole-iminium cyclization. The yield of cis-fused tetracycle 201 from this one-pot sequence was 60 %. The primary alcohol 202, with a yield of 83 % (50 % over two phases), was obtained by a chemoselective alkylation of piperidine. After obtaining Obenzyl limaspermidine (2 0 4) by pyrrolidine annulation and hydride reduction, we carried out a debenzylation using BF3•Et2O in ethanethiol to get (+)-limaspermidine (2 0 5) in a 60 % yield over three steps (Scheme 33) (Strom & Hartwig, 2013).
Enantioselective formal synthesis of (+)-kopsihainanine A.
-amidoester 206 was produced in two phases by the C-acylation of compound DHPI 196 and Michael addition with methyl acrylate, yielding 92 % of the product. A-quaternary DHPI 210 was successfully produced in 90 % yield and 92 % efficiency by exposing 206 to our enantioselective Palladium catalyzed decarboxylative allylic alkylation conditions. Much to our dismay, we found that the methyl ester in 207 was reduced after being treated with Schwartz's reagent, making the aforementioned hydrozirconation/amination procedure ineffective. Despite this setback, we used hydroboration catalyzed by Rhudenium to produce primary alcohol 208 in an 87 % yield (Evans, Fu, & Hoveyda, 1988). In two phases, easy conversion of alcohol 208 into azide 209 happened with an 88 % yield. With the aid of polymer-bound triphenylphosphine, a Staudinger reduction was carried out with a simultaneous translactamization to produce d-lactam 210 in an 81 % yield. When trifluoroacetic acid and 2-chloropyridine were combined, the Bischler-Napieralski cyclization of 210 went without a hitch, yielding the trans-fused tetracycle 211 in 84 % of the cases (White, Mewald, & Movassaghi, 2015). The nitrogen of piperidine and the pendant methyl ester was then attempted to be lactamized. After examining many Lewis acids and Bronsted bases, it was found that the guanidine base 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) could effectively promote the required cyclization to obtain the isolated pentacycle 212 in 65 % efficiency. When (+)-218 is exposed to lithium dimethylamide (LDMA), bis(trimethylsilyl) peroxide, and hexamethylphosphoramide (HMPA) act as an additive, (+)- kopsihainanine A (2 1 3) may be produced in 91 % of the time. As a consequence, starting with N-acyl indole 196, we have finished a very effective enantioselective synthesis of (+)-kopsihainanine A (2 1 3) (Scheme 34).
Synthesis of (+)-Minifiensine via palladium-catalyzed cyclization.
3.18 (+)-minfiensine
Monoterpene indole alkaloids comprise a broad class of natural compounds. Because of their distinct cage-like structure and intriguing biological activity, a variety of akuammiline and strychnos alkaloids are particularly appealing among them (Lewis, 2006; Saraswathi, Mathuram, Subramanian, & Govindasamy, 1999). The heterocycle-fused tetrahydrocarbazole skeleton, which contains two neighboring quaternary stereocenters, is the fundamental structural component of these natural compounds. The creation of techniques for effective asymmetric assembly of the core structure is required to overcome the synthetic difficulties given by these complex alkaloids.
Monoterpene indole alkaloids, akuammiline, and strychnos alkaloids are among a vast collection of natural compounds. This appeal due to their wide-ranging biological activity and distinctive cage-like structure. In the Strychnos family, Menefiensine is a tetracyclic compound with a broad range of biological activity. Jiao and colleagues have created a method for synthesizing minfensine using palladium for the first time. In this case, the indole moiety is alkylated using palladium before the iminium ion intermediate is trapped intramolecularly with a secondary amine. Exo-olefin produced from this process was then utilized to synthesize natural goods.
Six steps were required to convert the 2-methyltryptamine derivative 214 into the allylic alkylation precursor 215. Tetracycle 220 was produced with great enantioselectivity and good yield when 215 was treated with Pd2(dba)3 and the diphosphine ligand 216. TFA was then used to reclose the tetrahydropyrrole ring after triphosgene and methanol were used to protect the indole amine. Exo-olefin epoxidation produced a minimal mixture of diastereomers. A very regioselective epoxide opening process that followed produced 220 in a good yield. The synthesis of Minifiensine(2 2 4) was eventually finished by palladium-catalyzed cyclization of compound 223, which was initiated by N-alkylation (Scheme 35) (Z. X. Zhang et al., 2016).
Synthesis of TMC-205 alkaloid starting material.
3.19 TMC-205 and its analogues
Masaaki and Sakurai discovered the alkaloid TMC-205, a derivative of indole-3-carboxylic acid with an isoprene side-like chain at position 6, from the fungus strain TC 1630. TMC-205 inhibits the proliferation of human cancer cell lines. Only one group was able to complete the synthesis of TMC-205 in the beginning of 2014 the product was obtained in 5 steps using the longest linear sequence ever reported (6 total steps) and in 64 % total yield by using Suzuki-Miyaura coupling, Friedel-Crafts acylation, and esterification (using TMSCHN2) as the critical steps. A clear convergent strategy was employed in the first total synthesis to produce the C6 − C9 bond by the Suzuki-Miyaura cross-coupling reaction.(Yang Gao, Osman, & Koide, 2014) Nevertheless, the reported methods had inherent drawbacks like the need for expensive and toxic reagents and starting materials, as well as the requirement for independent isoprene portion preparation and multiple steps before building the 3-carboxy group could be achieved. Because of this, the earlier synthesis techniques are complicated. Li et al. revealed the first two stages of the complete synthesis of TMC-205 and its analogues from affordable and easily accessible starting materials under gentle circumstances due to their biological activity.
The first stage in diene production is the Heck-dehydration reaction, which occurs in the presence of Palladium complex by reacting molecule 225 with BHT as an additive and Acetonitrile as a solvent (Scheme 36).
Synthesis of TMC-205 alkaloid.
Compound 227, which has an aldehyde group, is oxidized to the target TMC-205 (2 3 1) in mixed solvents (THF-t-BuOH-H2O) in the second stage with a 78 % yield (Scheme 37).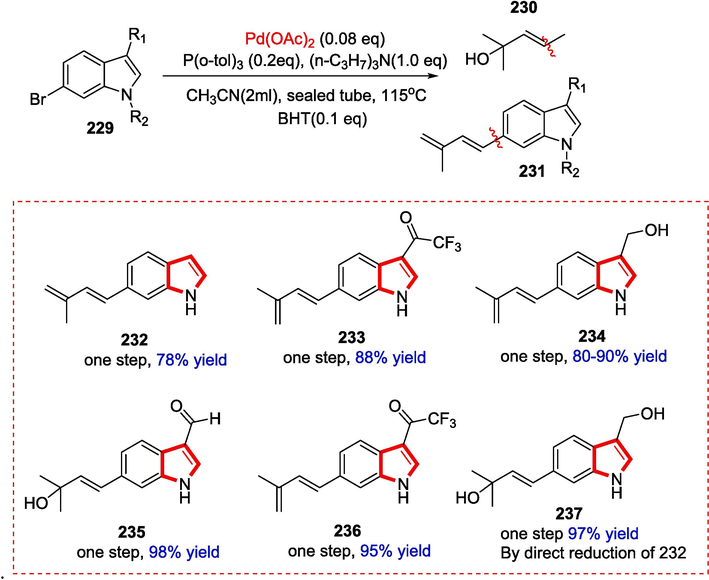
TMC-205 analogues and their yields.
Compounds 235, 236, and 237 are possible active compounds of TMC-205, whereas compounds 232, 233, and 234 have anti-proliferative activity against HCT-116 colon cancer cells (Scheme 38) (T. Li et al., 2021).
Palladium-catalyzed synthesis of (-)-Rhazinoline alkaloid.
3.20 Akuammiline alkaloids (–)- strictamine and (–)-rhazinoline
A significant class of monoterpenoid indole alkaloids, the akuammiline alkaloids were mainly isolated from the Apocynaceae family's plants. Representative methanoquinolizidine-containing akuammiline alkaloids with different stereochemistry at the C16 position include rhazinoline and strychnine. The enantioselective total synthesis of these two compounds is reported using a unified method. The main procedures in this synthesis include a late-stage intramolecular N-alkylation reaction, palladium- or nickel-mediated cyclization, a Tsuji-Trost allylation, and an intramolecular type II radical cascade reaction.
After positioning the necessary side chain at the 4th nitrogen position, the tricyclic intermediate 238 contains functions such as the C3 amine group, the C16 substituent, and the C7 quaternary stereogenic centre, served as an ideal compound for the synthesis of the E ring. According to scheme given below, the oxidative cleavage of the last alkene in 16 using OsO4/NaIO4 was followed by the selenation of the aldehyde and elimination, which effectively produced enal 239. With two steps—a typical Pinnick oxidation and esterification with TMSCHN2 in methanol—the aldehyde in 239 was converted into the appropriate ester, resulting in 240 with an 84 % yield (Bal, Childers Jr, & Pinnick, 1981). The PMB protective group had to be removed, and we had to add an unsaturated side chain at position N4. But under other circumstances, we discovered that the TBS group at the C5 location was labile. The development of a -lactone between the C16 ester and the C5 free hydroxyl group due to removing the TBS group occurred when the PMB in 240 was deprotected using various techniques. Consequently, the TBS group was converted to a pivaloyl (Piv) group in two phases by desilylation with HCl selectively and suppressing the hydroxyl group that resulted using PivCl/DMAP, yielding 241 in 95 % of the cases. Compound 242 (92 % yield) was produced when the PMB group was deprotected with FeCl3. To remove the Ns group, the latter was treated with HSCH2CO2H/K2CO3, and the amine that resulted was then treated with bromide 21 in the presence of LiOH·H2O, producing vinyl iodide 313 in 68 % over two stages. E ring formation was then ready to begin. Fortunately, Pd2(dba)3/PPh3/HCO2Na treatment of 243 produced the azabicyclo[3.3.1]nonane ring system. With the help of Boc2O and Na2CO3, the unstable secondary amine that resulted was disguised as a carbamate, yielding 244 in two stages with a 48 % yield. The reductive Heck cyclization process catalyzed by palladium revealed a single diastereomer, and the -ester at C16 was identified using NOE tests and further validated by the changing of 245 to the 16-epi-strictramine (2 4 6) and (-)-rhazinoline [(-)-2] (2 4 7). To be more precise, compound 23′s pivalate and indoline N-Bz groups were selectively reduced using LiAlH4 at −60 °C to give amino alcohol 245 (78 % yield). The two-step process of converting 245 to 16-epi-strictramine (2 4 6) went well owing to the mesylation of the free hydroxyl group's one-pot oxidation of the indoline N atom to an imine by PCC and the concomitant intramolecular N-alkylation and deprotection of the N-Boc group using TFA. As a consequence, ester 246 was converted into (-)-rhazinoline [(-)-2] (2 4 7) in a 68 % all-over yield by DIBAL-H reduction followed by Swern oxidation of the resulting alcohol (Scheme 39).
Palladium-catalyzed reductive synthesis of (+)-geissoschizine and (-)-nor-excelsinidine.
3.21 (-)-17-norexcelsinidine
A structurally complex family of bioactive natural compounds known as monoterpene indole alkaloids may be found all over the globe. Related biological studies discovered that alkaloids have an extensive range of medical benefits and biological functions, including cancer treatment, lung diseases, some fungal and bacterial infections, and headaches.
The efficient complete synthesis of (-)-17-norexcelsinidine 253 via NBS-promoted oxidative cyclization and palladium-catalyzed heck cross-couplingwas was reported by Yuan et.al, which is reductive. From 248, the benzyl ester was eliminated conventionally by debenzylation into acid 249, followed by the synthesis of phenylselenoester and decarboxylation under the influence of reductive radicals to produce enantiopure 250. The most extended linear sequence was used to manufacture (+)-geissoschizine 251 after the formylation of 250 under LDA/HCO2Me. Using geissoschizine 251 as a starting point, one may acquire 17-nor-Excelsinidine by adding carbon selectively to iN-situ produced ammonia salt and other kinds. Smooth generation of the 17-nor-excelsinidine precursor (2 5 2) using the NBS/pyridine method resulted in a 91 % yield which is reacted with NaOH to form (-)-17-nor-excelsinidine (2 5 3) (Scheme 40)(Yuan, Chen, Yan, Gao, & Wang, 2021).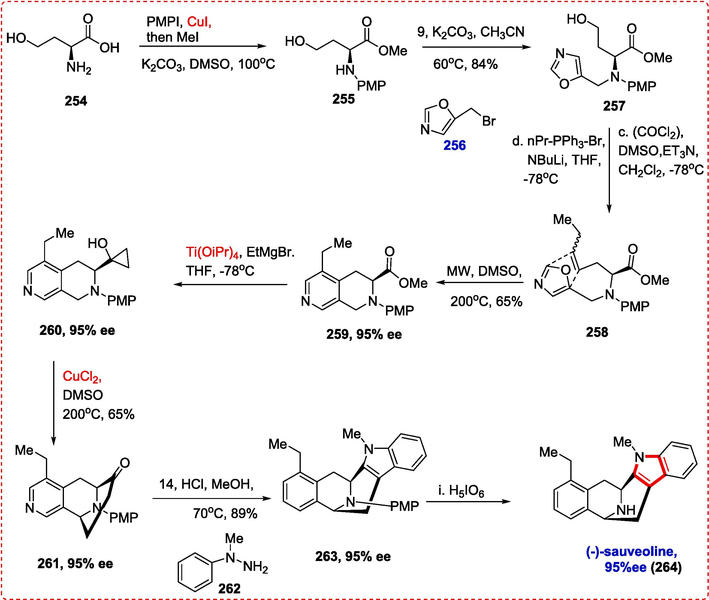
Copper-catalyzed total synthesis of (-)-Suaveoline.
4 Role of copper in total synthesis of indole alkaloids
4.1 (−)-suaveoline
Suaveoline is an indole alkaloid discovered in plants belonging to the Apocynaceae family and has significant biological significance. It contains pyridinyl and indolyl rings attached to its azabicyclo[3.3.1]nonane core substructure on both sides. For decades, scientists have been actively pursuing synthetic targets that possess the azabicyclo[3.3.1]nonane core structure due to its biological relevance and architectural complexity. The synthesis many indole alkaloids with this structural characteristic was reported by Cook et al. by the use of the Dieckmann condensation technique to create the bridging framework.(Rahman, Deschamps, Imler, & Cook, 2018) Despite these developments, the majority of current techniques need several functional group pre-installations and transformations, whereas greener approaches—such as C–H functionalization—that profit from few functional group manipulations are still little researched. Suaveoline alkaloid synthesis using a copper catalyst was first described by Tan et al. Starting with the readily accessible (S)-homoserine 254, a series of N-arylation/esterification and N-alkylation reactions led to product 257 in excellent yield. Wittig olefination and Swern oxidation of 257 produced the insignificant mixture of E/Z isomers 258, which prepared the groundwork for the crucial oxazole-olefin Diels-Alder reaction to create the pyridine ring (Ohba, Natsutani, & Sakuma, 2004, 2007; Q. Tan et al., 2019). The Diels-Alder reaction, aided by the microwave, produced 259 in 65 % yield and 95 % ee. Cyclopropanol 260 was created using the typical Kulinkovich cyclopropanation process (Ti(OiPr)4, EtMgBr) in an 85 % yield. When 260 was subjected to the usual oxidative cyclization conditions, it produced 261 in 55 % yield and 95 % ee, proving that enantiopurity decreased during oxidative cyclization steps and the Kulinkovich cyclopropanation. The nitrogen atom of pyridine, a potential copper-coordination site, did not cause any problems. Last but not least, using the ketone functionality in Fisher indole synthesis and afterward removing the PMP group produced (-)-suaveoline (2 6 4) in excellent yield with great purity (95 % ee) (Scheme 41) (Q. Tan et al., 2019).
Synthesis of HPI precursor.
4.2 Dimeric HPI alkaloids
The therapy for glaucoma, myasthenia gravis, delayed stomach emptying, Alzheimer's disease, and orthostatic hypotension uses physostigimine, a natural substance derived from the seeds of the clabar bean plant that has the hexahydropyrroindole (HPI) structural unit. The first enantioselective synthesis of the dimeric alkaloids (-)-chimonanthine and (-)-calecanthine was accomplished in 1999 by Overman and his group(Link & Overman, 1996).A reductive radical dimerization technique for the synthesis of dimeric HPI alkaloids was presented by Mavassaghi in 2007.(Movassaghi & Schmidt, 2007).After carefully reviewing the literature on the entire synthetics of dimeric HPI alkaloids, they discovered that tryptophan derivatives or tryptamine itself was often utilized as a starting material.(J. Kim & Movassaghi, 2010; Movassaghi, Ahmad, & Lathrop, 2011; Sun, Xing, Wang, Su, & Li, 2014; Tayu, Higuchi, Ishizaki, & Kawasaki, 2014).
Tryptamine and tryptophan derivatives may have restricted the availability of structurally different dimeric counterparts to some degree. For the sake of medicinal chemistry starting material, it is crucial to discover alternative methods for synthesizing the target dimeric HPI natural molecules as well as its analogues.(Y. Zhou et al., 2011).
Shen et al. focused on using metal catalyzed reactions to create key structures units for the first arylation N-oxidative dimerization of o-haloanilides having an intramolecular sulfinyl amide unit using copper as the catalyst. Based on this technique, a generic synthetic approach for the total synthesis of folicanthine, chimonanthine, ditryptophenaline and calycanthine had been devised.
Compound 266 was produced in a 78 % yield by the key arylation-oxidative dimerization of 265 (5.42 g, 10 mmol). This diastereomeric mixture was treated with 4 N HCl in methanol, which produced diamine 267 in a 95 % yield. HPLC examination showed that the isomeric purity of di-amine 267 was better than 99 % after recrystallization in CH3OH with 2 N HCl aqueous solution (2.0 eq (Scheme 10. Diamide 268 (96 %) was produced via the reductive amination of 267 with NaBH(OAc)3 and formaldehyde (Movassaghi & Schmidt, 2007; Overman, Paone, & Stearns, 1999). By using a-chloroethyl chloroformate (ACE-Cl) to selectively debenzylate the amine benzyl group in compound 268, proceeded by reflux in CH3OH, diamine 269 (95 % purity) was produced (Albert Mocholí, 2010). Desired HPI precursor (2 7 0) was produced in 54 % yield by reductive aminocyclization of diamine 269 in THF in the presence of diisobutylaluminum hydride (DIBAL-H). Finally, a Birch reduction that removed the benzyl-protecting groups produced (+)-chimonanthine (2 7 1) in 95 % of the cases. The reaction of formaldehyde and chimonanthine in the presence of sodium triacetoxyborohydride produced (+)-folicanthine(2 7 2). Chimonanthine is converted to (-)-calycanthine after being exposed to acid at reflux for 8 h (Scheme 42).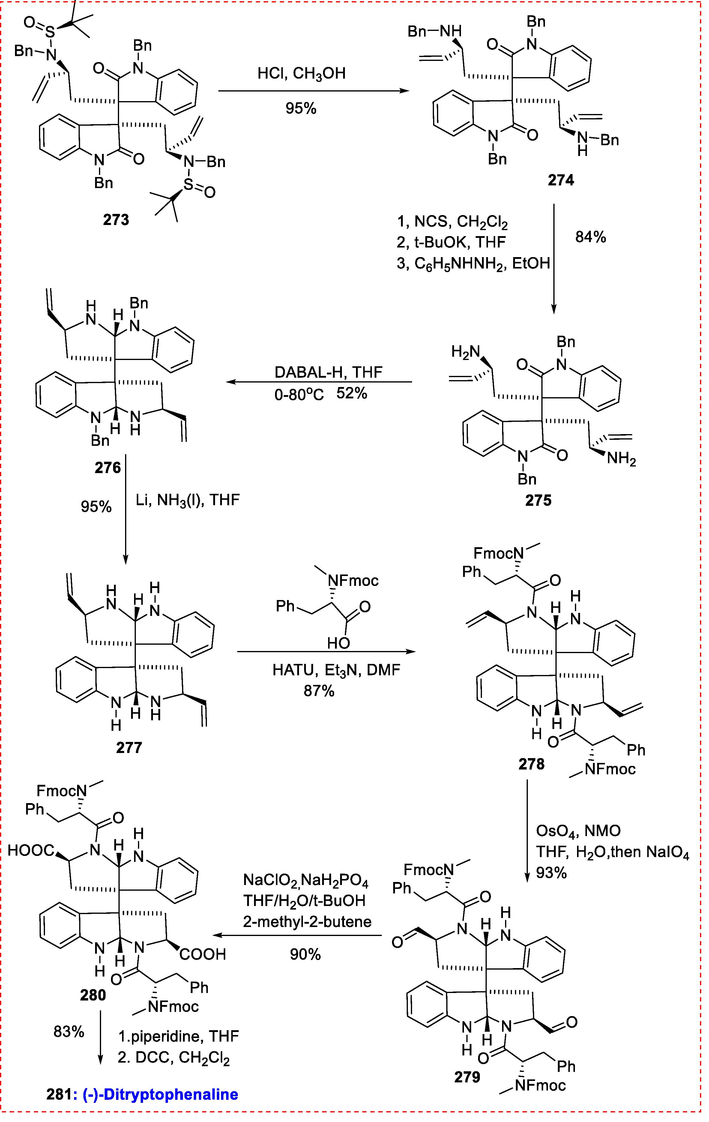
Total synthesis of (-)-Ditryptophenaline.
Investigation on the synthesis of (-)-ditryptophenaline starting from intermediate 273. Diamine 274 was produced when 273 was treated with 4 N HCl in CH3OH to remove the tert-butanesulfinyl group. Due to the existence of double bonds, it was challenging but eventually possible to deprotect the benzyl groups binding to amines selectively. Diamine 274 was treated with N-chlorosuccinamide and deprotonated with t-BuOK to produce imine (Olofson, Martz, Senet, Piteau, & Malfroot, 1984). This imine was then exposed to phenylhydrazine in ethanol, producing the main diamine 275 in an overall yield of 84 % (Vidal, Magnier, & Langlois, 1998). Intermediate 277 was produced in two phases with a 49 % yield after reductive aminocyclization with DABAL-H and debenzylation with metal lithium in liquid ammonia. Compound 278 (FMOC-(S)-MePh-OH, HATU, and Et3N in DMF) was produced by condensation of intermediate 277 with Fmoc-methylphenaline in an 87 % yield (Pérez‐Balado, Rodríguez‐Graña, & de Lera, 2009). The broken double bond produced aldehyde 279, and diacid 280 was produced by oxidizing it with buffered NaClO2 (Overman & Paone, 2001). The diacid (2 8 0) was ultimately changed into 281(-)-ditryptophenaline (Scheme 43) (Shen, Zhou, Xi, Zhao, & Zhang, 2016).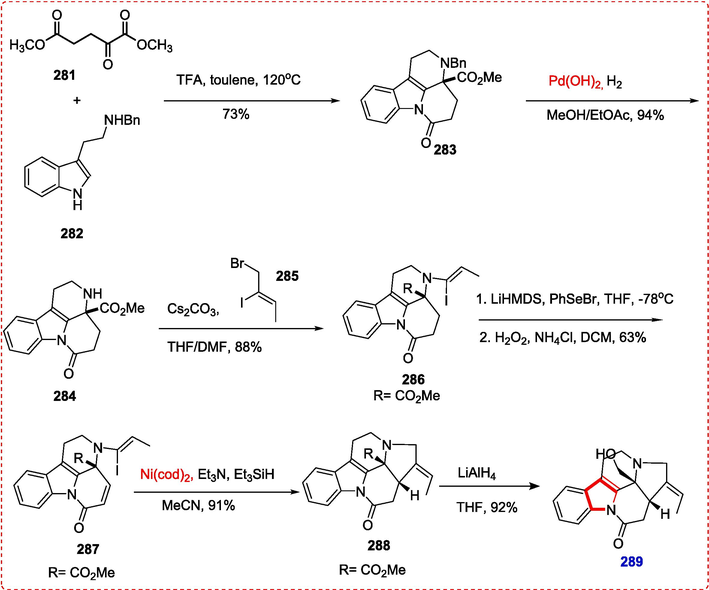
Nickel-catalyzed synthesis of monoterpene alkaloid.
5 Role of nickel in total synthesis of indole alkaloids
5.1 (±)-arbornamine
In 2016, Kam and coworkers discovered the monoterpene indole alkaloid arbornamine from Malayan Kopsia arborea. Dimethyl ester 281, 2 equiv of TFA, and benzyl tryptamine 282 are heated in refluxing toluene The synthesis of arbornamine was reported by Zheng et al. by crucial cascade cyclization of tetracyclic δ-lactam 283 in 73 % isolated yield. Using Pearlman's catalyst, -δlactam 283 was the first hydrogenolysis under atmospheric pressure to produce vinyl iodide under the conditions necessary to eliminate the protecting group, i.e., benzyl from Nb-nitrogen atom, and transform it into 284. After that, readily accessible (Z)-1-bromo-2-iodobut-2-ene 285 was used to alkylate the product to produce vinyl iodide 286. Using a typical selenenylatioN-elimination procedure, vinyl iodide 286 was transformed into a conjugated tetracyclic δ-lactam 287. The scene was prepared for the last ring's completion with δ-lactam 287 in hand. The required pentacyclic compound 288 was produced in 91 % yield using a reductive cyclization called Heck that was moderated by Ni(cod)2. Finally, arbornamine (2 8 9) was produced by globally reducing pentacyclic -δlactam 288 with lithium aluminum hydride (Scheme 44) (Zheng, Yue, Wei, & Yang, 2018).
Total synthesis of (-)-Strictamine (-)-1.
5.2 Akuammiline alkaloids (–)- strictamine
The total synthesis of compounds containing methanoquinolizidine core have not yet discovered.The whole synthesis of (-)-strictamine [(-)-1], starting with the advanced intermediate 290. Diol 291 was produced by converting the aldehyde functional group in 292 to an alcohol group under Luche reduction reaction conditions and removing the PMB and TBS protecting groups with FeCl3. Vinyl iodide 293 was produced by treating 291 with 292/Cs2CO3, which created a side chain at N4. The secondary amine 294 (with a 90 % yield) that was produced after the Ns group at N4 in 293 was deprotected was prepared for a transition-metal cyclization to create an E ring. To our delight, we discovered that Ni(cod)2/Et3N treatment of a solution of 294 in DMSO/MeCN (6:1) produced the required cyclization compound 295 with 61 % yield, enabling us to make the C ring under the conditions of 2-fluoropyridine/Tf2O to give the pentacyclic 296 with outstanding efficiency (95 % yield). As anticipated, the exocyclic alkene 296 was treated with 9- BBN in PhCH3 at 110 °C, following oxidation with NaBO3·4H2O, resulting in alcohol 297 as a diastereomer in 54 % result product. This process was successful in producing the necessary C16 stereochemistry. Finally, (-)-strictamine [(-)-1] (2 9 8) was produced via a three-step oxidation, oxidation, and esterification process from 297 (Scheme 45) (W. Li et al., 2019).
Total synthesis of (-)-Aspidophyline A(-)-2.
6 Role of iridium in synthesis and total synthesis of indole alkaloids
6.1 Aspidophylline A
Different research groups have made efforts in the synthesis of Alkuammiline alkaloids like Garg group did this by Fischer indolization(Zu, Boal, & Garg, 2011) and Ma group synthesized this alkaloid by Oxidative coupling occurring intramolecularly. The synthesis of Alkuammiline alkaloids was reported by Shi et.al by using metal catalyzed asymmetric cyclization.(Dounay et al., 2005) One of the well-known alkaloids is (-)-aspidophylline A an akuammiline alkaloid, which was discovered to stop drug resistance in drug-resistant KB cells by Kam and coworkers in 2007 after being isolated from Malayan Kopsia singapurensis (Subramaniam et al., 2007). This pentacyclic alkaloid is structurally composed of a bridging [3,3,1] bicycle, a high substituent containing a cyclohexyl ring carrying five continuous stereogenic centers, and a tricyclic furoindoline motif.
It started with the generation of necessary 2,3-disubstituted indole derivative 304 for the crucial cyclization catalyzed by iridium, which was the first step in the total synthesis of the akuammiline alkaloid. We were happy to discover that the required substrate 304 could be made from the known 3-(2-(tert-butyldimethylsilyloxy)ethyl)-1H-indole) in five stages. Using the Jiao and Bach technique, direct C2 alkylation of 299 proceeded without incident, and the compound 300 produced was advanced to substrate 304 using a conventional four-step methodology (Jiao & Bach, 2011; Jiao, Herdtweck, & Bach, 2012). Given that 307 was recovered in 70 % yield using flash column chromatography and the complete stereochemistry of the main tetracyclic furoindoline 16′h had been determined by X-ray crystallographic analytic process, we chose to employ (R)-L. Accordingly, the RheeneN-developed conditions for copper catalysis allowed for easily converting the acquired aldehyde into 308 in 75 % yield and 99 % efficiency (Van Rheenen, 1969). The tetracyclic a,b-unsaturated ester 308 was produced in good yield by vinyl triflating and then methoxycarbonylating it with palladium. According to Zhu's technique, putting the nitrogen atom at position C3 allowed for separating the required (-)-configured azide 308 in a respectable 42 % yield, with 40 % of its a-configured epimer. The primary amine 312 was produced by reducing 310 under Staudinger conditions, and it was then alkylated with (Z)-1-bromo-2-iodobut-2-ene, producing the vinyl iodide 312 (Sirasani & Andrade, 2011; Staudinger & Meyer, 1919). Under conventional reaction conditions, 312 was N-formylated to produce N-formamide 313. Following the method of Ma and colleagues, a cyclization catalyzed by nickel was used to close the last ring, and the required pentacyclic compound 314 was subsequently produced with 45 % yield. In the end, the carbamate was cleaved at N1, releasing (aspidophyline-A-)-2 (3 1 5)(Scheme 46) (Jiang et al., 2016).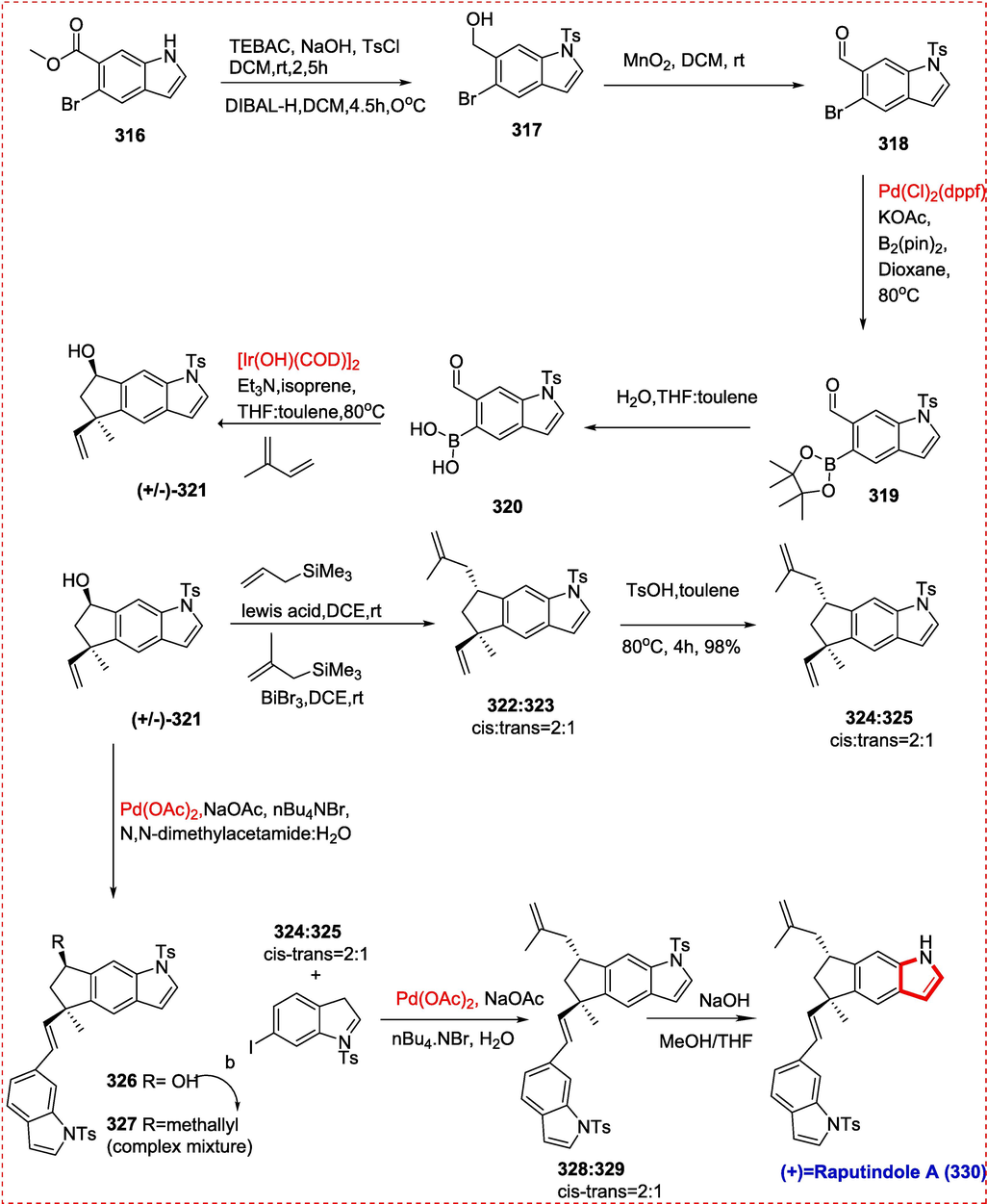
Total synthesis of (+)-Raputindole A.
6.2 (+)-raputindole A
Raputindole A (3 3 0) and its three isomers 2–4 were shown to have modest efficacy in inhibiting GSK-3B, DYRK1, and CDK2 kinases (IC50 > 10 uM). They were isolated from the Raputia simullans kalunki tree in the Peruvian Amazon jungle in 2010. The first total synthesis of raputindole A was done by Lindel by using Au(I) catalyzed cyclization and for isobutyle side chain Pd-catalyzed reaction was used.(Kock, Jones, & Lindel, 2017) But it has drawback of low diastereoselectivity. To construct the raputindole A (3 3 0) scaffold, our method uses N-tosyl indoles in the structure's southern and northern regions and an iridium-catalyzed diastereoselective cyclization, a Heck cross-coupling reaction. (Nishimura, Yasuhara, & Hayashi, 2007). Through stereoselective iridium-catalyzed cyclization and enzymatic resolution, we were able to complete the diastereoselective total synthesis of (+)-raputindole A (3 3 0) and obtain the northern portion of the compound as a 2:1 mixture of cis/trans 322/323 following the addition of the isobutenyl side chain at C-6. After preparative chiral HPLC separation in 10 stages (LLS) and 10 % overall yield, (+)-raputindole A (3 3 0) was isolated. It was combined with 6-iodo-indole (southern portion) by Heck reaction, and both tosyl groups were eliminated.
It was still necessary to add the isobutenyl side chain and include the southern indole moiety to finish our synthetic route to raputindole A (3 3 0). The former was intended to be created by allylating the (R,R)-321-derived benzylic carbocation with methallyltrimethylsilane, which needed evaluating several Lewis and Bronsted acids [93–95]. The best option was bismuth tribromide, which produced the necessary methallyl substituted indole in 69 % yield despite having a 2:1 M ratio (cis: trans isomers). The installation of the southern indole moiety prior to the reaction with methallyltrimethylsilane was investigated to increase the ratio of the trans isomer. The Heck reaction of 321 with tosylindole 316, made following the published technique, produced bisindole 327 in a yield of 48 %, however, its further reaction with methallyltrimethylsilane assisted by bismuth tribromide produced a complicated product mixture. We continued with the 2:1 combination of cis and trans-322:323. They isomerized the double bond to change the exo double bond into the necessary isobutenyl side chain despite the low stereoselectivity seen during the installation of the isobutenyl side chain. A 2:1 combination of 324:325 resulted in a nearly measurable yield after treatment with p-TsOH in toluene at 80 °C. (Mal & Roy, 2015). The cis and trans mixture of indoles 324:325 was subjected to the Heck cross-coupling reaction under the same conditions used for 321 and produced a 2:1 cis/trans mixture of 328:329 in 71 % product yield. It wasn't easy to eliminate the two tosyl groups that helped assemble the precursor 324:325. They only saw product deterioration the first time we tried using thioglycolic acid, LiOH in THF, and TBAF in THF. The deprotection was made feasible by using KOH and CTAB in THF-H2O under transfer phase catalysis, although an insoluble mixture of raputindole A (3 3 0) and its monotosyl derivative was produced (Scheme 47) (Pilli, Regueira, & Silva Jr, 2020).
Enantioselective Total Synthesis of (−)-Alstoscholarisine A.
6.3 Enantioselective (−)-alstoscholarisine
Alstoscholarisines are derived from Alstonia scholaris, which Luo and colleagues have identified as monoterpenoid indole alkaloid categories. Only the carboxylic acid and ester moiety presence or absence on C16 and the arrangement of C19 distinguish the compounds. They all have potential biological activities in stimulating adult NSC growth, with alstoscholarisine A (2 9 2) being the most active at small doses (0.1 g/mL) and considering the complicated molecular architecture of alstoscholarisine A (2 9 2), which may be useful in the treatment of neurological diseases.The first time enantioselective synthsis of (-)-Alstoscholarisine A was reported by Xiao et.al.
Trimethylaluminum facilitated the acylation of 3-methylindole (3 3 1) by 4-vinylbutyrolactone (3 3 2) to produce secondary allylic alcohol (3 3 3) in a 75 % yield. Asymmetric catalysis Using [Ir(cod)Cl2, (R)-Carreira ligand, and Sc(OTf)3 a Lewis acid, Asymmetric catalytic Friedel-Crafts alkylation of 333 effectively produced tricycle 334 with 99 % ee and 75 % yield. After being dihydroxylated, the resultant terminal olefin of 334 was protected as the equivalent acetonide 335. The dihydroxylation catalyzed by osmium generated a 5:1 diastereomeric combination that could be separated following acetonide production without exogenous stereodirecting agents. A conjugated tricyclic amide 336 was produced when the main acetonide isomer was subjected to the common selenenylation N-elimination procedure (Pineschi, 2004). The crucial addition of the adjacent stereogenic centres in cascade fashion was established with amide 336. Fortunately, the stereospecific delivery of amide β-hydroxyl 337 in 75 % yield was achieved by CuI2-promoted vinyl Grignard reagent 1,4-addition to the unsaturated amide 336 in addition to an aldol reaction with acetaldehyde. For our needs, an 8:1 diastereomeric ratio was found to be enough. A mixture of 1:1 hemiacetals 338 was produced when the acetonide-protecting group in 337 was removed, the diol was cleaved with NaIO4, and simultaneous cyclization occurred. TriethylsilaneBF3•Et2O was used to reduce the mixture, yielding 339 (85 %). Aldehyde 340 was produced in 81 % yield by hydroboration and oxidation of the terminal olefin 339 and subsequent oxidation of the resultant Primary-alcohol utilizing the Dess-Martin periodinane. Aldehyde 340 was finally transformed into (-)-alstoscholarisine A (3 4 1) using a reductive amination and cyclization method (Scheme 48) (Liang, Jiang, Wei, & Yang, 2016).
Total synthesis of Herbindole A.
7 Role of ruthenium in synthesis and total synthesis of indole alkaloids
7.1 (±)-herbindole
One of three naturally occurring 6,7-benzannulated indoles that were discovered from the Axinella species of sponge present in Western Australia is herbindole A. The first asymmetric synthesis of (±)-Herbindole had been done in 1992.(Muratake et al., 1992) We have shown quick and effective (+)-epi-herbindole and (+)-herbindole A total syntheses A, which were accomplished using an allylation N-cycloisomerization catalyzed by ruthenium and cascade reaction. Herbindole A was previously reported to act as a fish antifeedant and have cytotoxic activity against KB cells (5 g/mL).
Racemic diol 343 was synthesized in excellent product yield and with strong diastereoselectivity by dihydroxylation of dihydromesitylene 342 with minimal catalyst load. Diol 343 undergoes diastereoselective hydrogenation to give large amounts of saturated diol 344. The stereoselectivity considerably decreased when the reaction was not immediately discontinued when it was finished. Using oxidative diol fragmentation, ketoaldehyde 345 was quantitatively created from 344 without any loss of stereoselectivity. On a gram scale, the synthesis of 345 was carried out, and the syn-building block 345 was made from dihydromesitylene (3 4 2) in three stages with an overall yield of 57 %. Following the Baati et al. technique, ketoaldehyde 345 was subjected to 5-enolexo aldolization to produce cyclopentane 346 (Ghobril, Sabot, Mioskowski, & Baati, 2008; Hammar et al., 2010). 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was found as an optimum catalyst to accelerate this reaction with the appropriate regioselectivity, following their findings into the cyclization of 6-oxoheptanal. Low yields, regioisomeric compounds, or complex mixes were produced when some other bases used, such as pyrrolidine, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), proline, or acidic catalysts were used. Aldol product 346 was produced as four monomers with a molar ratio of 10:4:2:1. While ethynylation of the cis-diastereomer 349 produced the cyclization precursor 351 in a separate 2:1 ratio and yielded about 73 %, dehydration in the presence of acid of the aldol product 346 produced the required cyclopentene in excellent product rate and moderate diastereoselectivity of 4:1 preferring the cis-isomer 349. The combination was subsequently processed to produce ()-herbindole A (3 5 2) in 76 % yield since both diastereomers of 351 provide suitable starting materials for the final allylation and cycloisomerization cascade reaction that is catalyzed by ruthenium. 8 Dihydromesitylene was converted to (±)-herbindole A (3 5 2) in seven stages with a 22 % yield (Scheme 49).
Rhuthenium catalyzed synthesis of (+)-epi-herbindole A.
A good precursor for (±)-epi-herbindole A (3 5 0) is the trans-diastereomer of the enone, which was produced from ketoaldehyde in yield of 40 % Propargyl alcohols 353 were produced by ethynylating in yield of 71 % and with a dr of 2:1. The final conversion of the diastereomeric mixture 353 to pyrrole under the catalytic action of ruthenium produced (±)-epi-herbindole A (3 5 4) with a yield of 71 % and a dihydromesitylene yield of 11 % (Scheme 50) (Thies, Stürminger, & Haak, 2017).
Synthesis of (+)-Eburnamonine.
8 Role of rhodium in total synthesis of indole alkaloids
8.1 Aspidosperma, iboga-type and corynanthe- alkaloids
Based on the skeletons of carbon of the monoterpenoid units, three primary categories of monoterpenoids containing indole alkaloids have been identified: aspidosperma-, iboga-type, and corynanthe- alkaloids. We provide a photocatalytic radical cascade to create libraries of chiral tetrahydrocarbolinones under benign circumstances, with high regio-, -stereo, and chemoselectivity. This radical cascade may produce two of the three monoterpenoid indole-alkaloid, including the aspidosperma-type and corynanthe-type alkaloids by switching up the readily available substrates.
8.2 Eburnamine-vincamine alkaloids synthesis
The total synthesis of indole alkaloids belonging to eburnamine-vincamine family was the first instance in which the established radical cascade was used. Wang et al, separated the diastereomers 356 and 357 in a radical two-step cascade of 355 on a 14-gram scale, as shown in Scheme 1A, with an 81 % total yield and a 1:1.5 ratio. The tosyl (Ts)-protecting group in the resulting 358 was eliminated with sodium naphthalenide to produce amine 359 in good yield after masking aldehyde in 357 as dioxane. After amide reduction, trifluoroacetic acid treatment, and a straightforward indoline moiety oxidation to indole (359 to 360), (+)-eburnamenine (3 6 1) was produced in three stages with a 73 % yield. 359 and 360 show what happened when 361 was hydrated with HCl/H2O, producing (-)-eburnamine (3 6 2) and (+)-isoeburnamine (3 6 3). The alkaloid (+)-eburnamonine (3 6 4) was produced by further oxidation of the combination of 362 and 363 (Scheme 51).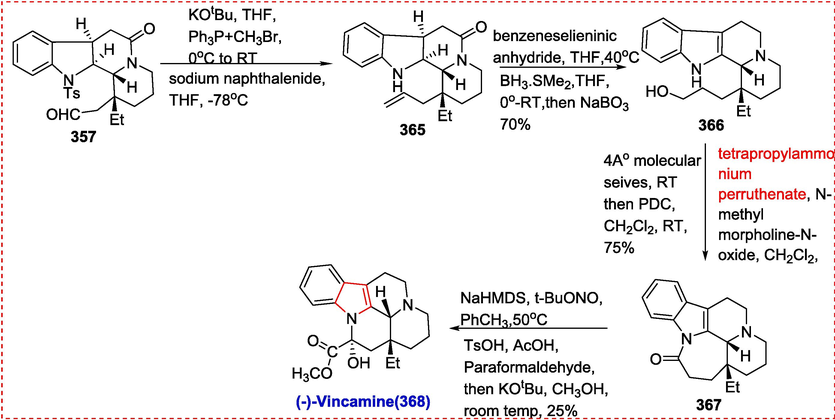
Synthesis of (-)-Vincamine.
In seven stages, intermediate 357 was employed to create (-)-vincamine (3 6 8). Compound 365 was produced with a high yield by Wittig methylenation of 357 and Ts group removal. After the indoline moiety was oxidized, the double bond in 365 underwent hydroboration oxidation, which led to the simultaneous amide reduction to produce alcohol 366. The hydroxy group in 366 was oxidized, creating lactam 367. Finally, using a published approach, 368 was synthesized from 367 (Scheme 52).
Synthesis of (-)-Vallesamidine.
With 360 as the starting material, (-)-vallesamidine (3 7 3) was synthesized (Scheme 1C). Aldehyde 369 was reacted with SmI2 to produce a ketyl radical after being methylated and deprotected twice. This ketyl radical was attached to the double bond of indole to produce 370 as diastereomers pair. After the hydroxy group in 370 was removed, the resultant 371 was reduced with rhodium(I) hydride, producing 372 in a good yield (Scheme 52) (X. Wang et al., 2017).
9 Conclusion
Transition metal-catalyzed reactions are intriguing for synthesizing indole alkaloids and indole-containing subunits. These reactions exhibit high stereoselectivity and are effective strategies for synthesizing natural products. Although promising, these methods need additional studies to enhance their efficacy in producing specific products. The review emphasizes the significance of transition metal-catalyzed transformations in the total synthesis of natural products. Transition metal-catalyzed transformations show the potential in synthesizing natural products, specifically indole alkaloids.
Further research is required to comprehensively investigate the mechanisms of these transformations to enhance our comprehension of their potential and constraints. Effective synthetic methods are essential for meeting the increasing demand for natural products. This will lead to more effective and feasible approaches to producing natural products. Moreover, investigating the application of transition metal-catalyzed reactions for producing intricate natural compounds apart from indole alkaloids presents a promising research avenue. Applying transition metal-catalyzed reactions in natural product synthesis exhibits significant potential and warrants additional investigation and advancement. Please provide me with the user's text so I can rewrite it concisely and academically.
Acknowledgement
The authors express their appreciation to the Deanship of Scientific Research at King Khalid University, Saudi Arabia, for funding this work through research group program under grant number RGP. 2/428/44. The authors would also like to acknowledge Universiti Teknologi MARA Cawangan Selangor Kampus Puncak Alam, Selangor, Malaysia for the high impact journal fund.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis of substituted indole derivatives as a new class of antimalarial agents. Bioorg. Med. Chem. Lett.. 2005;15(12):3133-3136.
- [Google Scholar]
- Ortho substituents direct regioselective addition of tributyltin hydride to unsymmetrical diaryl (or heteroaryl) alkynes: an efficient route to stannylated stilbene derivatives. Angew. Chem.. 2002;114(9):1648-1650.
- [Google Scholar]
- Albert Mocholí, L. (2010). Síntesis asimétrica de aminoalcoholes y aminoácidos fluorados.
- Andres, R. m., Wang, Q., & Zhu, J. (2020). Asymmetric total synthesis of (−)-arborisidine and (−)-19-epi-arborisidine enabled by a catalytic enantioselective Pictet–Spengler reaction. Journal of the American Chemical Society, 142(33), 14276-14285.
- Short syntheses of the indole alkaloids alocasin A, scalaridine A, and hyrtinadine AB. Tetrahedron. 2016;72(29):4214-4221.
- [Google Scholar]
- Armarego, W., & Perrin, D. (2002). Purification of laboratory chemicals butterworth heinemann: Oxford.
- An introduction to alkaloids and their applications in pharmaceutical chemistry. Pharma Innov. J.. 2015;4(10):74-75.
- [Google Scholar]
- Mild and efficient conversion of nitriles to amides with basic urea-hydrogen peroxide adduct. Synth. Commun.. 1993;23(22):3149-3155.
- [Google Scholar]
- Flexible and convergent total synthesis of cyclotheonamide B. J. Org. Chem.. 1997;62(12):3880-3889.
- [Google Scholar]
- The chemistry of hapalindoles, fischerindoles, ambiguines, and welwitindolinones. Alkaloids Chem. Biol.. 2014;73:65-160.
- [Google Scholar]
- Total syntheses of (±)-spiroindimicins B and C enabled by a late-stage Schöllkopf–Magnus–Barton–Zard (SMBZ) reaction. Chem. Commun.. 2016;52(4):800-802.
- [Google Scholar]
- Hetero diels-alder methodology in organic synthesis. Org. Chem.: A Ser. Monogr.. 1987;47:300-310.
- [Google Scholar]
- Biochemistry and molecular biology of plants. John wiley & sons; 2015.
- 2-Substituted 3-aryl-and 3-heteroarylindoles by the palladium-catalyzed reaction of o-trifluoroacetanilides with aryl bromides and triflates. Synthesis. 2003;2003(05):0728-0734.
- [Google Scholar]
- Synthetic development and mechanistic study on Pd (II)-catalyzed cyclization of enediynes to benzo [a] carbazoles. Org. Lett.. 2010;12(24):5652-5655.
- [Google Scholar]
- Asymmetric synthesis of the tetracyclic skeleton of natural product arborisidine. Chin. J. Org. Chem.. 2018;38(9):2427.
- [Google Scholar]
- Pd-Catalyzed cycloisomerization/nucleophilic addition/reduction: an efficient method for the synthesis of spiro-pseudoindoxyls containing N, N′-ketal. Org. Chem. Front.. 2017;4(9):1731-1735.
- [Google Scholar]
- Anti-malarial activity of indole alkaloids isolated from Aspidosperma olivaceum. Malar. J.. 2014;13(1):1-10.
- [Google Scholar]
- Effect of green tea (Camellia sinensis) extract on the rat diaphragm. J. Ethnopharmacol.. 1997;57(3):197-201.
- [Google Scholar]
- Ergot alkaloids: current status and review of clinical pharmacology and therapeutic use compared with other oxytocics in obstetrics and gynaecology. Drugs. 1998;56:523-535.
- [Google Scholar]
- Benzylic substitution of gramines with boronic acids and rhodium or iridium catalysts. Org. Lett.. 2007;9(6):961-964.
- [Google Scholar]
- Recent advances in the synthesis of N-containing heteroaromatics via heterogeneously transition metal catalysed cross-coupling reactions. Molecules. 2011;16(6):5241-5267.
- [Google Scholar]
- Sequential catalytic asymmetric Heck− iminium ion cyclization: Enantioselective total synthesis of the Strychnos alkaloid minfiensine. J. Am. Chem. Soc.. 2005;127(29):10186-10187.
- [Google Scholar]
- Rhodium (I)-catalyzed hydroboration of olefins. The documentation of regio-and stereochemical control in cyclic and acyclic systems. J. Am. Chem. Soc.. 1988;110(20):6917-6918.
- [Google Scholar]
- In vitro anti-malarial interaction and gametocytocidal activity of cryptolepine. Malar. J.. 2017;16(1):1-9.
- [Google Scholar]
- Efficient relay syntheses and assessment of the DNA-cleaving properties of the pyrrole alkaloid derivatives permethyl storniamide A, lycogalic acid A dimethyl ester, and the halitulin core. Tetrahedron. 2002;58(32):6373-6380.
- [Google Scholar]
- Total Synthesis and Biological Studies of TMC-205 and Analogues as Anticancer Agents and Activators of SV40 Promoter. ACS Med. Chem. Lett.. 2014;5(8):863-867.
- [Google Scholar]
- Intramolecular Larock indole synthesis for the preparation of tricyclic indoles and its application in the synthesis of tetrahydropyrroloquinoline and fargesine. Tetrahedron. 2014;70(34):5136-5141.
- [Google Scholar]
- Short syntheses of the tricyclic indole alkaloids cimitrypazepine and fargesine. Tetrahedron Lett.. 2016;57(34):3873-3876.
- [Google Scholar]
- Ghobril, C., Sabot, C., Mioskowski, C., & Baati, R. (2008). TBD‐Catalyzed Direct 5‐and 6‐enolexo Aldolization of Ketoaldehydes: Wiley Online Library.
- Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci.. 2005;78(5):442-453.
- [Google Scholar]
- Rh (III)-Catalyzed Cascade Annulation/C–H Activation of o-Ethynylanilines with Diazo Compounds: One-Pot Synthesis of Benzo [a] carbazoles via 1, 4-Rhodium Migration. Org. Lett.. 2016;18(20):5236-5239.
- [Google Scholar]
- An approach to unsymmetrical 3, 3′-diindolylmethanes through Pd-catalyzed cascade Heck cyclization of allenamides and o-ethynylanilines. Org. Chem. Front.. 2021;8(8):1783-1788.
- [Google Scholar]
- Theoretical mechanistic study of the TBD-catalyzed intramolecular aldol reaction of ketoaldehydes. J. Org. Chem.. 2010;75(14):4728-4736.
- [Google Scholar]
- Palladium-catalyzed Markovnikov terminal arylalkynes hydrostannation: application to the synthesis of 1, 1-diarylethylenes. J. Org. Chem.. 2009;74(3):1337-1340.
- [Google Scholar]
- New vasorelaxant indole alkaloids, taberniacins A and B, from Tabernaemontana divaricata. J. Nat. Med.. 2019;73(3):627-632.
- [Google Scholar]
- Enantioselective total synthesis of (+)-lysergic acid,(+)-lysergol, and (+)-isolysergol by palladium-catalyzed domino cyclization of allenes bearing amino and bromoindolyl groups. J. Org. Chem.. 2011;76(7):2072-2083.
- [Google Scholar]
- Promoting reductive tandem reactions of nitrostyrenes with Mo (CO) 6 and a palladium catalyst to produce 3 H-indoles. J. Am. Chem. Soc.. 2015;137(21):6738-6741.
- [Google Scholar]
- Iridium-Catalyzed Enantioselective Indole Cyclization: Application to the Total Synthesis and Absolute Stereochemical Assignment of (−)-Aspidophylline A. Angew. Chem.. 2016;128(12):4112-4116.
- [Google Scholar]
- Palladium-catalyzed direct 2-alkylation of indoles by norbornene-mediated regioselective cascade C-H activation. J. Am. Chem. Soc.. 2011;133(33):12990-12993.
- [Google Scholar]
- Pd (II)-catalyzed regioselective 2-alkylation of indoles via a norbornene-mediated C-H activation: mechanism and applications. J. Am. Chem. Soc.. 2012;134(35):14563-14572.
- [Google Scholar]
- Nine-step enantioselective total synthesis of (+)-minfiensine. J. Am. Chem. Soc.. 2009;131(38):13606-13607.
- [Google Scholar]
- New synthetic protocols for the preparation of unsymmetrical bisindoles. Org. Lett.. 2006;8(25):5761-5764.
- [Google Scholar]
- Kasahara, A. (1981). A new synthesis of 2-aryl-4-quinolones.
- Alkaloids-important therapeutic secondary metabolites of plant origin. J. Crit. Rev.. 2015;2(3):1-8.
- [Google Scholar]
- General approach to epipolythiodiketopiperazine alkaloids: total synthesis of (+)-chaetocins A and C and (+)-12, 12′-dideoxychetracin A. J. Am. Chem. Soc.. 2010;132(41):14376-14378.
- [Google Scholar]
- Tyrosinase inhibitory components from Aloe vera and their antiviral activity. J. Enzyme Inhib. Med. Chem.. 2017;32(1):78-83.
- [Google Scholar]
- Total synthesis and absolute configuration of raputindole A. Org. Lett.. 2017;19(23):6296-6299.
- [Google Scholar]
- Pd-catalyzed enantioselective aerobic oxidation of secondary alcohols: applications to the total synthesis of alkaloids. J. Am. Chem. Soc.. 2008;130(41):13745-13754.
- [Google Scholar]
- Design, synthesis and evaluation of 3-methylene-substituted indolinones as antimalarials. Eur. J. Med. Chem.. 2011;46(3):927-933.
- [Google Scholar]
- Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc.. 1991;113(17):6689-6690.
- [Google Scholar]
- Application of a Palladium-Catalyzed C− H Functionalization/Indolization Method to Syntheses of cis-Trikentrin A and Herbindole B. Angew. Chem. Int. Ed.. 2016;55(39):11824-11828.
- [Google Scholar]
- Total synthesis of dihydrolysergic acid and dihydrolysergol: development of a divergent synthetic strategy applicable to rapid assembly of D-ring analogs. Tetrahedron. 2015;71(35):5897-5905.
- [Google Scholar]
- Recent advances in the chemistry of macroline, sarpagine and ajmaline-related indole alkaloids. Tetrahedron. 2006;62(37):8655-8681.
- [Google Scholar]
- Asymmetric total syntheses of the akuammiline alkaloids (−)-strictamine and (−)-rhazinoline. Angew. Chem.. 2019;131(18):6120-6124.
- [Google Scholar]
- A simple and efficient total synthesis of anticancer indole alkaloids TMC-205 and its analogues. Tetrahedron Lett.. 2021;74:153137
- [Google Scholar]
- Catalytic asymmetric total synthesis of (−)-galanthamine and (−)-lycoramine. Angew. Chem. Int. Ed.. 2015;54(21):6255-6259.
- [Google Scholar]
- Enantioselective total synthesis of (−)-alstoscholarisine A. J. Am. Chem. Soc.. 2016;138(8):2560-2562.
- [Google Scholar]
- Gene-inspired mycosynthesis of skeletally new indole alkaloids. Org. Lett.. 2015;17(11):2610-2613.
- [Google Scholar]
- Stereocontrolled total syntheses of meso-chimonanthine and meso-calycanthine via a novel samarium mediated reductive dialkylation. J. Am. Chem. Soc.. 1996;118(34):8166-8167.
- [Google Scholar]
- Ir-catalyzed regio-and enantioselective Friedel–Crafts-type allylic alkylation of indoles. Org. Lett.. 2008;10(9):1815-1818.
- [Google Scholar]
- Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat. Prod. Rep.. 2000;17(2):175-191.
- [Google Scholar]
- A regioselective facile synthesis of furo [3, 4-b] carbazolones: application to the total synthesis of mafaicheenamine E and claulansine D. Org. Biomol. Chem.. 2015;13(22):6344-6352.
- [Google Scholar]
- In vitro conversion of chanoclavine-I aldehyde to the stereoisomers festuclavine and pyroclavine controlled by the second reduction step. RSC Adv.. 2012;2(9):3662-3669.
- [Google Scholar]
- Gold (I)-catalyzed N-desulfonylative amination versus N-to-O 1, 5-sulfonyl migration: A versatile approach to 1-azabicycloalkanes. Angew. Chem. Int. Ed.. 2016;55(31):9088-9092.
- [Google Scholar]
- Enantioselective total syntheses of akuammiline alkaloids (+)-strictamine,(−)-2 (s)-cathafoline, and (−)-aspidophylline a. J. Am. Chem. Soc.. 2016;138(4):1162-1165.
- [Google Scholar]
- Directed heterodimerization: stereocontrolled assembly via solvent-caged unsymmetrical diazene fragmentation. J. Am. Chem. Soc.. 2011;133(33):13002-13005.
- [Google Scholar]
- Concise total synthesis of (−)-calycanthine,(+)-chimonanthine, and (+)-folicanthine. Angew. Chem.. 2007;119(20):3799-3802.
- [Google Scholar]
- Total synthesis of (+)-Herbindole A,(+)-herbindole B, and (+)-herbindole C. Determination of the absolute configuration of the natural herbindoles. Tetrahedron Lett.. 1992;33(32):4595-4598.
- [Google Scholar]
- Evaluation of antiparasitic, antituberculosis and antiangiogenic activities of 3-aminoquinolin-2-one derivatives. J. Chil. Chem. Soc.. 2006;51(2):859-863.
- [Google Scholar]
- Microwave-assisted Friedländer synthesis of quinolines derivatives as potential antiparasitic agents. Tetrahedron Lett.. 2006;47(50):8811-8815.
- [Google Scholar]
- N’Goka, W. P. R. P. (1991). V. Hosseinzadeh F. Tamm C. Helv. Chim. Acta, 74, 1941.
- Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J. Antibiot.. 2009;62(1):17-26.
- [Google Scholar]
- Synthesis of 3, 3-spiroindolines via FeCl3-mediated cyclization of aryl-or alkene-containing 3-substituted N-Ac indoles. Org. Lett.. 2016;18(8):1716-1719.
- [Google Scholar]
- Nishimura, T., Yasuhara, Y., & Hayashi, T. (2007). Iridium-catalyzed [3+ 2] annulation of 1, 3-dienes with ortho-carbonylated phenylboronic acids. A catalytic process involving regioselective 1, 2-addition. Journal of the American Chemical Society, 129(24), 7506-7507.
- Total synthesis of suaveoline and norsuaveoline via intramolecular oxazole–olefin Diels-Alder reaction. Tetrahedron Lett.. 2004;45(34):6471-6474.
- [Google Scholar]
- Intramolecular Diels-Alder reactions of oxazole–olefins: synthesis of the Rauwolfia alkaloids suaveoline and norsuaveoline. Tetrahedron. 2007;63(41):10337-10344.
- [Google Scholar]
- A new reagent for the selective, high-yield N-dealkylation of tertiary amines: improved syntheses of naltrexone and nalbuphine. J. Org. Chem.. 1984;49(11):2081-2082.
- [Google Scholar]
- Enantioselective Total Syntheses of Ditryptophenaline and e nt-WIN 64821. J. Am. Chem. Soc.. 2001;123(38):9465-9467.
- [Google Scholar]
- Direct stereo-and enantiocontrolled synthesis of vicinal stereogenic quaternary carbon centers. Total syntheses of meso-and (−)-chimonanthine and (+)-calycanthine. J. Am. Chem. Soc.. 1999;121(33):7702-7703.
- [Google Scholar]
- Chemical composition, antiprotozoal and cytotoxic activities of indole alkaloids and benzofuran neolignan of Aristolochia cordigera. Planta Med.. 2017;83(11):912-920.
- [Google Scholar]
- Stereocontrolled and versatile total synthesis of bispyrrolidinoindoline diketopiperazine alkaloids: Structural revision of the fungal isolate (+)-asperdimin. Chem.–Eur. J.. 2009;15(38):9928-9937.
- [Google Scholar]
- Production of supported metal catalysts by the decomposition of metal carbonyls. Appl. Catal.. 1984;9(1):1-30.
- [Google Scholar]
- Pilli, R., Regueira, J. L. L. F., & Silva Jr, L. F. (2020). Diastereoselective Total Synthesis of (+)-Raputindole A: An Iridium-Catalyzed Cyclization Approach.
- Pineschi, M. (2004). Del Moro F. Gini F. Minnaard AJ. Feringa BL. Chem. Commun, 1244.
- Plemenkov, V. (2001). Vvedenie v khimiyu prirodnykh soedinenii (Introduction to the Chemistry of Natural Compounds), Kazan: Kazan. Gos. Univ.
- Provot, O., Hamze, A., Peyrat, J.-F., Brion, J.-D., & Alami, M. (2013). Discovery and hit to lead optimization of novel combretastatin A-4 analogues: dependence of C-linker length and hybridization. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents), 13(10), 1614-1635.
- Tryptamine derivatives as novel non-nucleosidic inhibitors against hepatitis B virus. Bioorg. Med. Chem.. 2011;19(10):3120-3127.
- [Google Scholar]
- Total synthesis of sarpagine-related bioactive indole alkaloids. Chem.–Eur. J.. 2018;24(10):2354-2359.
- [Google Scholar]
- Ergot alkaloid biosynthesis in Aspergillus fumigatus: Overproduction and biochemical characterization of a 4-dimethylallyltryptophan N-methyltransferase. J. Biol. Chem.. 2008;283(40):26859-26868.
- [Google Scholar]
- A unified strategy to reverse-prenylated indole alkaloids: total syntheses of preparaherquamide, premalbrancheamide, and (+)-VM-55599. Chem. Sci.. 2020;11(23):5929.
- [Google Scholar]
- Divergent pathways in the biosynthesis of bisindole natural products. Chem. Biol.. 2009;16(4):351-364.
- [Google Scholar]
- Modulation of the impaired drug metabolism in sarcoma-180-bearing mice by echitamine chloride. Cancer Biochem. Biophys.. 1999;17(1–2):79-88.
- [Google Scholar]
- Intramolecular larock indole synthesis: Preparation of 3, 4-fused tricyclic indoles and total synthesis of fargesine. Angew. Chem.. 2013;125(18):5002-5005.
- [Google Scholar]
- Intramolecular larock indole synthesis: Preparation of 3, 4-fused tricyclic indoles and total synthesis of fargesine. Angew. Chem. Int. Ed.. 2013;52(18):4902-4905.
- [Google Scholar]
- Total synthesis of dimeric HPI alkaloids. Nat. Prod. Bioprospect.. 2016;6(2):117-139.
- [Google Scholar]
- Total syntheses of trikentrins and of herbindoles. Tetrahedron. 2010;66(22):3875-3895.
- [Google Scholar]
- Recent progress in biological activities of indole and indole alkaloids. Mini Rev. Med. Chem.. 2018;18(1):9-25.
- [Google Scholar]
- Skolc, D., Ates, A., Jnoff, E., & Valade, A. (2016). Preparation of isoindoline derivatives D1 pos. allosteric modulators. PCT Int. Appl. WO, 2016055482, A1.
- Traceless solid phase synthesis of 2, 3-disubstituted indoles. Tetrahedron Lett.. 1998;39(45):8317-8320.
- [Google Scholar]
- Lewis acid induced asymmetric prins reactions of chiral acetals with alkenes. Synth. Commun.. 1986;16(12):1451-1460.
- [Google Scholar]
- Somei, M., Teranishi, S., Yamada, K., & Yamada, F. (2001). The chemistry of indoles. CVII. A novel synthesis of 3, 4, 5, 6-tetrahydro-7-hydroxy-1H-azepino [5, 4, 3-cd] indoles and a new finding on Pictet-Spengler reaction. Chemical and pharmaceutical bulletin, 49(9), 1159-1165.
- Recent progress in organocatalytic asymmetric total syntheses of complex indole alkaloids. Natl. Sci. Rev.. 2017;4(3):381-396.
- [Google Scholar]
- Staudinger, H., & Meyer, J. (1919). Über neue organische phosphorverbindungen III. Phosphinmethylenderivate und phosphinimine. Helvetica Chimica Acta, 2(1), 635-646.
- One-pot anti-Markovnikov hydroamination of unactivated alkenes by hydrozirconation and amination. J. Org. Chem.. 2013;78(17):8909-8914.
- [Google Scholar]
- Biologically active aspidofractinine, rhazinilam, akuammiline, and vincorine alkaloids from Kopsia. J. Nat. Prod.. 2007;70(11):1783-1789.
- [Google Scholar]
- Highly efficient and stereocontrolled oxidative coupling of tetrahydropyrroloindoles: synthesis of chimonanthines,(+)-WIN 64821 and (+)-WIN 64745. Org. Chem. Front.. 2014;1(8):956-960.
- [Google Scholar]
- ‘The chemistry of indoles’, Academic Press, New York, 1970. Angew. Chem. Int. Ed. Engl.. 1988;27:1113.
- [Google Scholar]
- Copper-catalyzed aerobic oxidative cyclization cascade to construct bridged skeletons: Total synthesis of (−)-suaveoline. Angew. Chem.. 2019;131(19):6486-6490.
- [Google Scholar]
- An updated asymmetric total synthesis of (+)-tronocarpine. Tetrahedron. 2020;76(49):131641
- [Google Scholar]
- Tasch, B. O., Merkul, E., & Müller, T. J. (2011). One‐Pot Synthesis of Diazine‐Bridged Bisindoles and Concise Synthesis of the Marine Alkaloid Hyrtinadine A: Wiley Online Library.
- Thionium-based one-pot construction of homo-/heterodimeric pyrroloindoline from tryptamine. Org. Lett.. 2014;16(14):3613-3615.
- [Google Scholar]
- Application of a ruthenium-catalyzed allylation-cycloisomerization cascade to the synthesis of (±)-herbindole A. Synlett. 2017;28(06):701-704.
- [Google Scholar]
- Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev.. 2003;103(8):2921-2944.
- [Google Scholar]
- Copper-catalyzed oxygenation of branched aldehydes–an efficient ketone synthesis. Tetrahedron Lett.. 1969;10(12):985-988.
- [Google Scholar]
- Formal synthesis of Manzamine C via a sila-Cope elimination. Tetrahedron. 1998;54(22):5959-5966.
- [Google Scholar]
- Copper-catalyzed trifluoromethylation and bicyclizations of 1, 7-enynes leading to fused polycycles. Adv. Synth. Catal.. 2016;358(21):3435-3442.
- [Google Scholar]
- A radical cascade enabling collective syntheses of natural products. Chem. 2017;2(6):803-816.
- [Google Scholar]
- Pd (II)-catalyzed enantioselective C-H activation of cyclopropanes. J. Am. Chem. Soc.. 2011;133(49):19598-19601.
- [Google Scholar]
- Ligand-free palladium-catalyzed cyanation of aryl halides. J. Org. Chem.. 2005;70(4):1508-1510.
- [Google Scholar]
- Direct observation of intermediates involved in the interruption of the Bischler-Napieralski reaction. J. Org. Chem.. 2015;80(15):7403-7411.
- [Google Scholar]
- A total and practical synthesis of ergot alkaloid,(±)-aurantioclavine. Chem. Pharm. Bull.. 1985;33(5):2162-2163.
- [Google Scholar]
- Synthesis of pyrroles, indoles, and carbazoles through transition-metal-catalyzed C H functionalization. Asian J. Org. Chem.. 2013;2(6):466-478.
- [Google Scholar]
- Palladium catalyzed reductive Heck coupling and its application in total synthesis of (−)-17-nor-excelsinidine. RSC Adv.. 2021;11(13):7570-7574.
- [Google Scholar]
- Synthesis of heterocycles via palladium-catalyzed oxidative addition. Chem. Rev.. 2006;106(11):4644-4680.
- [Google Scholar]
- Total synthesis of (+)-minfiensine: Construction of the tetracyclic core structure by an asymmetric cascade cyclization. Angew. Chem. Int. Ed.. 2016;55(28):8090-8094.
- [Google Scholar]
- Zhang, Y., Hubbard, J. W., Akhmedov, N. G., Petersen, J. L., & Söderberg, B. r. C. (2015). Total synthesis of the tetracyclic indole alkaloid ht-13-B. The Journal of Organic Chemistry, 80(9), 4783-4790.
- Zhang, Z., Ray, S., Imlay, L., Callaghan, L., Niederstrasser, H., Mallipeddi, P. L., . . . Smith, M. (2021). Total Synthesis of (+)-Spiroindimicin A via Asymmetric Palladium-Catalyzed Spirocyclization.
- Total synthesis of the tetracyclic indole alkaloid ht-13-A. Tetrahedron Lett.. 2016;57(26):2865-2867.
- [Google Scholar]
- Short synthesis of the monoterpene indole alkaloid (±)-arbornamine. J. Org. Chem.. 2018;83(8):4867-4870.
- [Google Scholar]
- Efficient synthesis of esermethole and its analogues. Org. Biomol. Chem.. 2011;9(11):4091-4097.
- [Google Scholar]
- Alkaloid biosynthesis: metabolism and trafficking. Annu. Rev. Plant Biol.. 2008;59:735-769.
- [Google Scholar]
- Total synthesis of (±)-aspidophylline A. J. Am. Chem. Soc.. 2011;133(23):8877-8879.
- [Google Scholar]




