

Translate this page into:
Theoretical design and prediction of novel fluorene-based non-fullerene acceptors for environmentally friendly organic solar cell
⁎Corresponding authors. crlu@dhu.edu.cn (Changrui Lu), Safwan.akram@tees.ac.uk (Muhammad Safwan Akram)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Organic solar cells (OSCs) with electron-withdrawing cyano-group (−C≡N) have created massive interest by exhibiting higher efficiencies. Nevertheless, introducing the − C≡N group through malononitrile during their synthesis is highly toxic and harmful to the environment. Therefore, the development of environmentally friendly OSCs (EFOSCs) is of utmost importance. Taking inspiration from the recent reports, this manuscript looks to suggest more efficient novel fluorene-based EFOSCs based on DTC-T-F type molecule containing fluorene as central core and thiophene as the linker. The − C≡N of 2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene) malononitrile (TC) present as an end-capped acceptor in DTC-T-F is replaced with non-toxic electron-pulling units -CF3, -SO3H, –NO2 and a novel series developed through quantum chemical calculation of fluorene-based photovoltaic materials (C1- C9). Frontier molecular orbital, density of state, heat maps of TDM, and finally, the charge shift process is observed by blending the C3 acceptor with donor polymer PTB7-Th (HOMOPTB7-Th-LUMOC3). The designed molecules gave comparable and better results from reference C. The minimum energy gap is observed in C3, C6 and C9 molecules with 2.29 eV, 2.28 eV and 2.27 eV energy gap values respectively. The proposed C9 compound exhibits a prominent redshift in solvent (681.82 nm) and gas phase (618.88 nm). Open circuit voltage (Voc) is the key parameter while assessing the OSC efficiency. The highest Voc is possessed by C5 (2.05 V). Exciton binding energy (Eb) is computed, and C7 has the lowest value of Eb = 0.38 eV. Finally, the reorganization energy values shown by proposed molecules indicate that fluorene-based series are effective candidates for manufacturing EFOSCs. All of the data showed that the designed molecules not only have superior optoelectronic properties to those of the synthetic compound, but also possess environmentally benign properties. This theoretical understanding will provide a C—N—free strategy for developing next-generation EFOSCs and novel fluorene-based environmentally friendly materials for OSCs applications.
Keywords
DFT
Fluorene
Non-fullerene acceptors
Environmental friendly organic solar cells
Photovoltaic properties

1 Introduction
Organic polymer solar cells (OSCs) are getting significant consideration due to apparent advantages of flexible deployment, transparency, facile manufacturing process; material structure diversity; ultra-thin and lightweight structure leading to convenient transportation. These characteristics are thought to be a viable option for next-generation solar cells (Cheng et al., 2018, Hou et al., 2018). This area has seen sustained advancement during the last decades, particularly innovative novel materials with optimized device architecture (Li et al., 2012, Lu et al., 2015, Collins et al., 2017). The continual appearance of novel materials offers a foundation for OSCs performance enhancement (Yan et al., 2018). The recent discovery of organic semiconductors of n-type has given this field a new engine, propelling OSCs into a new era of development and attaining several milestones (Cheng et al., 2018, Hou et al., 2018). Single junction devices' power conversion efficiencies (PCEs) that are founded on non-fullerene acceptor (NFAs) and polymer donor materials are approaching 16 % to 19 % (Fan et al., 2019, Cui et al., 2020, Cui et al., 2021).
Apart from the high PCE, another major problem to be solved to attain the full commercialization potential of OSCs is to reduce the cost of goods. Most of the promising fullerene free acceptor molecules have ladder-type A–D–A conformation with fused-ring central core that includes ITIC (Lin et al., 2015), IDIC (Lin et al., 2016), IHIC (Wang et al., 2017), IEICO (Yao et al., 2016), and COi8DFIC (Xiao et al., 2017), etc. This ensures BHJ (bulk heterojunction) for utilizing solar photons with increased carrier mobility in the near-infrared (NIR) spectra. Creating ladder architecture with fused-multi-ring has challenges regarding synthesis, particularly multiple purification steps leading to an increase in the cost of the material. Therefore, there is a great need to investigate novel designs and new compounds to get efficient and low-cost OSCs. In recent times, few significant efforts have been made in this area with hopeful outcomes (Li et al., 2018, Huang et al., 2019). However, there are still plethora of compounds and architectures to explore.
Carbazole and fluorene are commonly employed as electron-donating units in organic photoelectric materials because of their low cost and excellent hole mobility (Li and Grimsdale 2010, Jeon et al., 2018). They are also employed to make NFAs comprising deep HOMO and large bandgaps (Holliday et al., 2015). On the other hand, it has been demonstrated that TC (2-(6-oxo-5, 6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene) malononitrile) has the potential to be an excellent end-cap acceptor because of its high light-harvesting ability, which is achieved through electron delocalization, as well as its good charge transport, which is achieved through strong intermolecular S..S contacts (Zhang et al., 2017). Keeping these in mind, Liao et al. synthesized fluorene-based efficient and simple NFAs namely DTC-T-F, which consisted of central core 9, 9-dioctylfluorene, thiophene as bridging units, and 2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile (TC) as an end-capped acceptor (Liao et al., 2021). The DTC-T-F is recognized as an excellent OSCs candidate due to its high extinction coefficients, significant absorption in NIR spectra, and reduced HOMO-LUMO gap. These fluorene-based acceptors exhibited superior optoelectronic properties for OSCs.
End-capped amendments is recognized as a credible approach for finding novel OSC materials (Khalid et al., 2021, Khalid et al., 2021, Khan et al., 2021, Rafiq et al., 2022). Yao et al. have proposed that swapping the − C≡N unit with environmentally friendly and non-toxic groups such as − CF3, −SO3H, or − NO2 can render molecules environmentally friendly without having a considerable trade-off on efficiency (Yao et al., 2020). These groups bring the energy gap down while simultaneously increasing the affinity of the vertical electrons and giving the backbone a positive charge. From these reports, TC of DTC-T-F is replaced with an end-capped acceptor made of 1,1-dicyanomethylene-3-indanone (IC), and the double − C≡N groups on both end-capped IC units are replaced with non-toxic NO2, SO3H, and CF3 groups. This way, nine new environment-friendly, inexpensive and efficient molecules (C1-C9) are designed for their potential use in OSCs applications. Computations based on DFT and TDDFT are used to investigate the photovoltaic potential of proposed compounds. This theoretical understanding will open up new avenues for computational scientists, and give them access to a productive method for creating OSC materials that are environmentally friendly. It will also make it possible for experimental scientists to make use of highly efficient predicted molecules in the production of photovoltaics for use in solar cell applications.
2 Computational procedure
Gaussian 09 W (Frisch et al., 2009) program was used to perform quantum calculations. GaussView 5.0 program was used to make all input files (Dennington et al., 2008). DFT calculations at five different functionals B3LYP (Civalleri et al., 2008), ωB97XD (Chai and Head-Gordon 2008), CAM-B3LYP (Yanai et al., 2004), M06-2X (Zhao and Truhlar 2008), and MPW1PW91 (Adamo and Barone 1998) with conjunction of 6-31G(d,p) basis set were performed for optimization of reference molecule C. The absorption maximum (λmax) values were estimated from TDDFT computations at above mentioned five functionals. In order to choose the most applicable functional, we compared the λmax value of the C that was computed theoretically with the experimentally determined λmax value that was reported (Liao et al., 2021). Findings from B3LYP/6-31G(d,p) exhibited the closest match with the experimentally published values (Liao et al., 2021), providing the basis to undertake further computations of designed molecules utilizing this functional and basis set combination. Reference C and designed molecules (C1-C9) UV–Vis spectral analysis was estimated using the CPCM model (Barone and Cossi 1998) and chloroform solvent at TDDFT/B3LYP/6-31G(d,p) functional. Frontier molecular orbital analysis (FMOs), the density of state (DOS) calculations, reorganizational energies, transition density matrixes (TDM) analysis, open circuit voltage (Voc), analysis of charge transfer of C and C1-C9 compounds were performed at DFT/B3LYP/6-31G(d,p) functional. Reorganization energies can be explained according to Marcus theory (Marcus 1993), which further divides it into two branches. The first one is λint known as internal reorganization energy, while the second one is λext recognized as external reorganization energy. Internal structure and their changes depend upon the internal reorganization energy of a molecule and external energy concerns with external environment. In this report, effect of external energy was ignored. For the calculations of reorganization energies of λe (electron) and λh (hole) following equations are helpful (Khan et al., 2019, Khan et al., 2020).
In these equations, Eo− and Eo+ indicate single point energy values of anions and cations, respectively, obtained through simple optimization of neutral molecules. The and E+ represent cationic and anionic energies via optimized geometries of the cations and anions. and represent neutral molecule energies through anion and cation optimization. The energy of a neutral molecule in its ground state is denoted by the symbol Eo (Tang and Zhang 2012). All results obtained from Gaussian calculations were executed using Origin, PyMOlyze 2.0 (O'boyle et al., 2008), Multiwfn 3.7 program (Lu and Chen 2012), Chemcraft (Andrienko 2010), and Avogadro (Hanwell et al., 2012) programs.
3 Results and discussion
In present study, DTC-T-F-based novel NFAs are designed that exhibit superior photo-electronic properties for OSCs. The structure of DTC-T-F consists of 9, 9-dioctylfluorene as the central core, bridging units thiophene, and TC as a terminal acceptor (Liao et al., 2021). The − C≡N group of TC end-capped unit is replaced with − CF3, −SO3H, and − NO2 groups containing end-capped acceptors A1 = 3-chloro-6-(perfluoropropan-2-ylidene)-5,6-dihydrocyclopenta[c]thiophen-4-one, A2=(3-chloro-6-oxo-5,6-dihydrocyclopenta[c]thiophen-4-ylidene)methanedisulfonic acid, A3 = 3-chloro-6-(dinitromethylene)-5,6-dihydrocyclopenta[c]thiophen-4-one and C1-C3 molecules are designed respectively. Further, TC end-capped unit of DTC-T-F is substituted with 1,1-dicyanomethylene-3-indanone (IC) end-capped acceptors, and double C≡N groups on both end-capped units of IC are replaced with non-toxic − CF3, −SO3H, and − NO2 groups. In this way, six new environment friendly, inexpensive and efficient molecules (C4-C9) are proposed with the end-capped units A4 = 5,6-difluoro-3-(perfluoropropan-2-ylidene)-2,3-dihydroinden-1-one, A5 = 5,6-difluoro-3-oxo-2,3-dihydroinden-1-ylidene)methane- disulfonic acid, A6 = 5,6-dichloro-3-(dinitromethylene)-2,3-dihydroinden-1-one, A7 = 5,6-dichloro-3-(perfluoropropan-2-ylidene)-2,3-dihydroinden-1-one, A8 =(5,6-dichloro-3-oxo-2,3-dihydroinden-1-ylidene)methanedisulfonic acid and A9 = 5,6-dichloro-3-(dinitromethylene)-2,3-dihydroinden-1-one respectively (Scheme 1).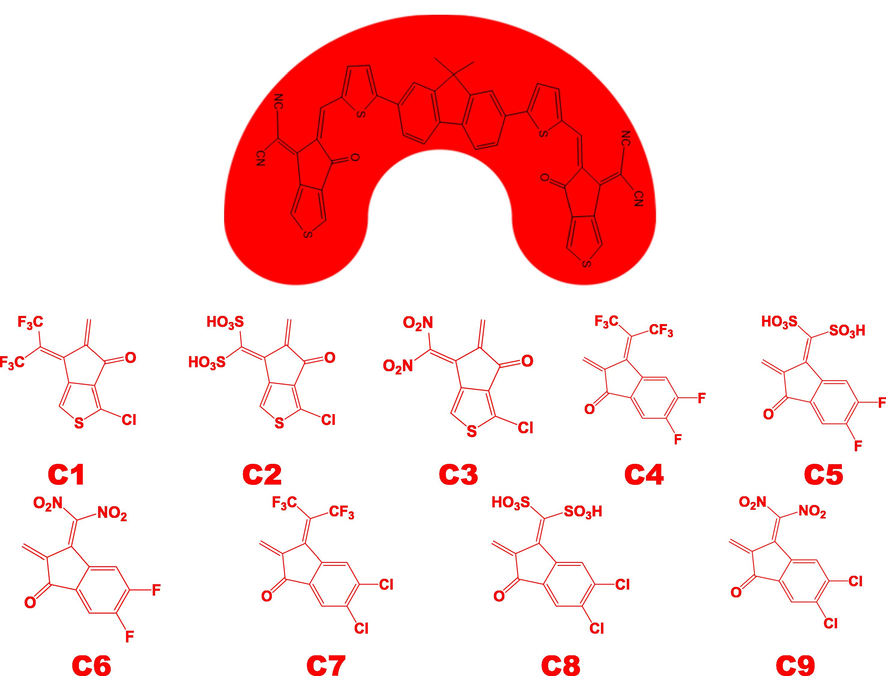
Sketch map of DTC-T-F-based non-fullerene acceptor with different end-capped structures.
Considering the computational cost, long alky chains of designed molecules (C1-C9) are replaced with methyl groups for simplification. According to earlier reports, these simplifications do not disturb molecules' results, properties, and characteristics (Ma et al., 2013).
Primarily, λmax values of reference molecule (C) was calculated at B3LYP, CAM-B3LYP, M062X, MPW1PW91, and ωB97XD at 6-31G(d,p)/DFT functionals in chloroform as solvent using CPCM model and the best method was selected. The values of λmax for C molecule are computed as 600 nm, 455 nm, 459 nm, 561 nm, and 440 nm at B3LYP, CAM-B3LYP, M062X, MPW1PW91, and ωB97XD functionals respectively. The experimental λmax value of C is 591 nm (Liao et al., 2021). A bar chart at above-mentioned basis set and levels of theory combinations representing the comparison amongst experimental and DFT computed λmax of reference molecule (C) is presented in Fig. 1. It is evident from the bar chart that B3LYP/6-31G (d,p) functional computed value exhibited good harmony with the experimental value and can be used for further optoelectronic calculations of C and designed (C1-C9) molecules.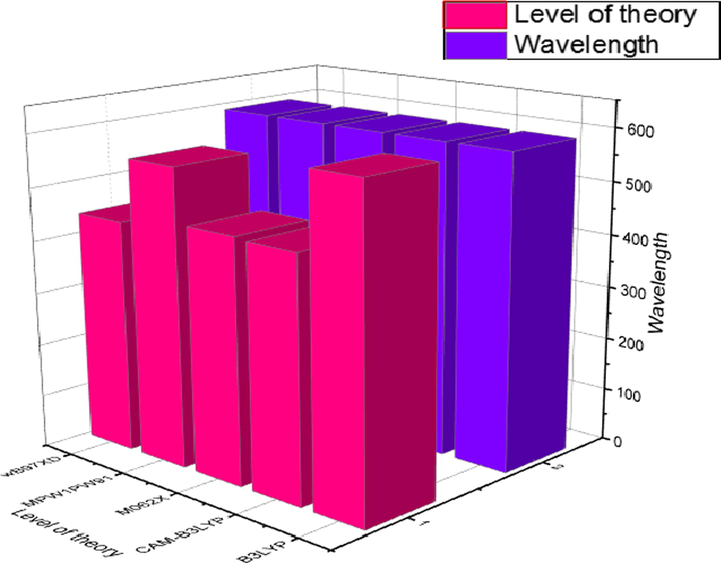
Selection of level of theory “Graphical representation of comparison between experimentally and calculated UV–vis results of reference (C) at five DFT based functionals in solvent (CHCl3) by utilizing origin 8.5 version (
https://www.originlab.com/). All out put files of entitled compounds were accomplished by Gaussian 09 version D.01 (
https://gaussian.com/g09citation/)”.
Using B3LYP/6-31G(d,p) functional, optimized molecular structures of reference (C) and designed (C1-C9) compounds are illustrated in Fig. 2.
Optimized geometries of reference C and designed C1-C9 molecules.
4 Frontier molecular orbitals (FMOs) analysis
FMO analysis can be performed to explain the optoelectronic properties of molecules in this case, C and C1-C9. It allowed insight into the charge transfer characteristics of investigated molecules and their photovoltaic features (Khalid et al., 2021, Khalid et al., 2021). A low energy gap promotes efficient charge transfer in OSCs (Khan et al., 2020). Energy calculations were made at DFT/ B3LYP/6-31G(d,p) functional to find out the HOMO and LUMO energy levels and the energy gap of C and C1-C9 molecules.
The reference molecule, as shown in Table 1, has HOMO energy −5.68 eV, LUMO energy − 3.23 eV, and an energy gap of 2.45 eV, indicating the reference molecule as a promising molecule to fabricate the small molecule OSCs. A small energy gap promotes the faster charge transfer. Results indicate that the energy gap of designed molecules fabricated using environmentally friendly end-capped moieties are comparable to reference C. At the same time, C3, C6, and C9 are superior to the reference, with reduced energy gap values of 2.29 eV, 2.28 eV, and 2.27 eV, respectively. In C1-C3, the energy gap of SO3H and NO2-containing molecules (C2, C3) are marked lower, implying the strong pulling nature of these functional groups compared to CN and CF3 used in C and C1 molecules, respectively. Similarly, a higher energy gap is seen in C4, and C5 molecules comprising IC-2F end-capped acceptors with CF3 and SO3H groups in place of CN. However, IC-2F end-capped acceptor with NO2 group in C6 abridged the energy gap to the second lowest value (2.29 eV) among C and C1-C9 molecules. The results of C7-C8 with IC-2Cl end-capped acceptors containing CF3, and SO3H, groups are found to be slightly large but comparable to the reference C molecule. The C9 molecule with NO2 containing IC-2Cl end-capped acceptor is marked with the lowest energy gap among all studied compounds. HOMO energy values for C1-C9 are computed as −5.65, −5.82, −5.93, −5.68, −5.49, −5.95, −5.74, −5.55 and −6.03 respectively (Table 1). Similarly, LUMO energy values for C1-C9 are measured as −3.10, −3.40, −3.64, −3.07, −2.85, −3.67, −3.18, −2.96, −3.76 respectively. The decrease in energy gap is observed in molecules with NO2 groups, i.e., C3, C6, and C9 as NO2 has a more potent electron-withdrawing group than other molecule that efficiently pulls the electron density. As a result, LUMO energy level decreases, lowering the energy gap. Compared to NO2, the molecules containing SO3H and CF3 do not pull the electron density strongly. Therefore, a subtle alteration of the energy gap is observed in such compounds. But the results of SO3H and CF3 containing groups are still comparable to the energy gap results of reference C. These results indicate that using cyanide-free end caps is an efficient strategy for architect simple, low-cost, and environmentally friendly organic solar cells.
Molecules
HOMO (EHOMO)
LUMO (ELUMO)
Egap = ELUMO-EHOMO
C (Ref)
−5.68
−3.23
2.45
C1
−5.65
−3.10
2.55
C2
−5.82
−3.40
2.42
C3
−5.93
−3.64
2.29
C4
−5.68
−3.07
2.61
C5
−5.49
−2.85
2.64
C6
−5.95
−3.67
2.28
C7
−5.74
−3.18
2.56
C8
−5.55
−2.96
2.59
C9
−6.03
−3.76
2.27
The distribution pattern of HOMO, LUMO charge density is portrayed in Fig. 3. The cyan color in Fig. 3 indicates the positive end, while the red shows the negative end. About molecule C, the charge resides on the donor core in the case of HOMO orbitals. On the other hand, in the case of the LUMO orbitals of the C molecule, the charge is distributed on the acceptor moieties. If the designed molecule is compared with the reference C, we come across that proposed molecules C1-C9 exhibit similar patterns indicating that our designed molecules are correctly fabricated. Furthermore, the proposed molecules with non-toxic groups are as effective as toxic CN-based reference C. Therefore, environmentally friendly proposed molecules C1-C9 can be targeted for solar cell applications instead of C.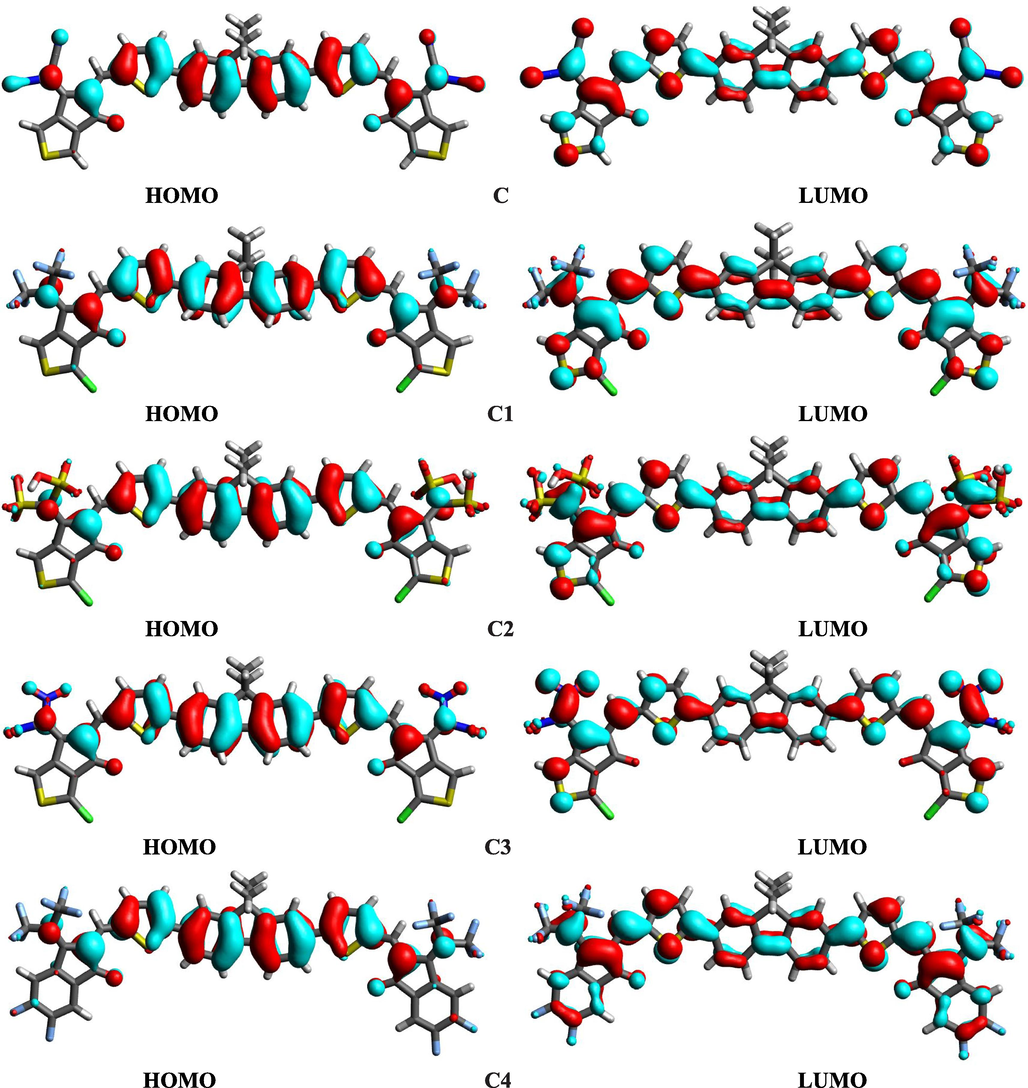
HOMOs and LUMOs of investigated molecules.
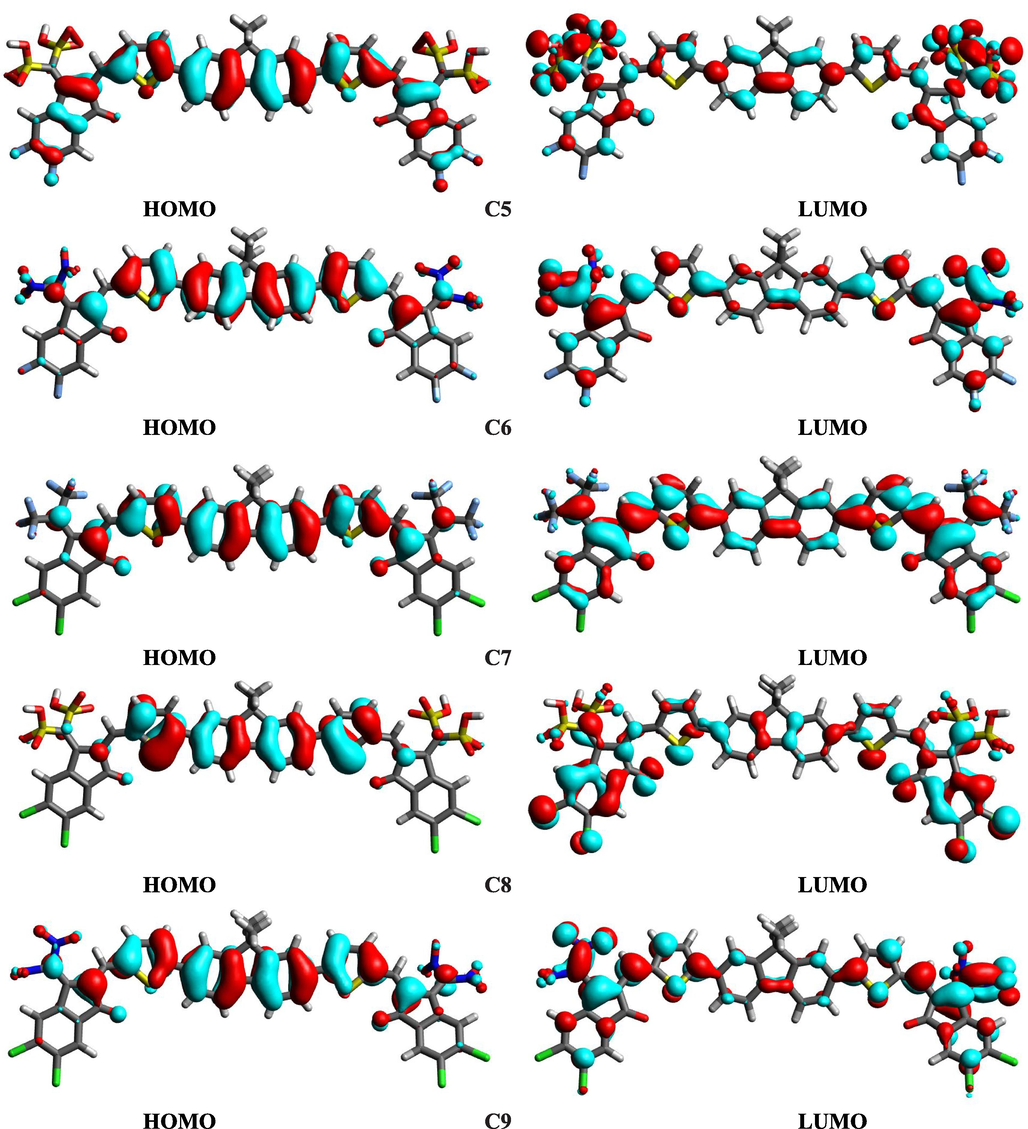
HOMOs and LUMOs of investigated molecules.
5 Density of state and overlap density of state analysis
Density of states (DOS) and overlap density of state (ODOS) analysis are performed at the same theory level and the basis set as supporting analysis to FMOs that indicate the contribution of each fragment of molecule in charge transfer (Mehboob et al., 2021). DOS and ODOS graphs are shown in Fig. 4. For this purpose, each molecule is divided into two parts i.e. donor core and acceptor attached on both ends of the core. LUMO percentage contribution for reference C and designed molecules C1-C9 donor core are 11.6 %, 13.2 %, 11.2 %, 8.4 %, 18 %, 18.4 %, 7.9 %,17.1 %, 17.4 % and 7.9 % respectively. LUMO percentage contribution for reference and designed molecules C1-C9 acceptors is 63.7 %, 60.3 %, 63.9 %, 71.5 %, 51.3 %, 50.8 %, 67.5 %, 52.6 %, 52.2 % and 67.6 % respectively. HOMO percentage contribution for reference and designed molecules C1-C9 donor core is 41 %, 42.8 %, 44.3 %, 44.6 %, 55.8 %, 52.8 %, 56.5 %, 55.9 %, 52.9 % and 56.6 % respectively. On the other hand, HOMO percentage contribution for reference and designed molecules C1-C9 acceptors is 27.5 %, 24.9 %, 25 %, 25.1 %, 17.7 %, 20.5 %, 18.4 %, 18 %, 20.8 % and 18.8 % respectively. For efficient charge transfer, HOMO value of the donor should be higher than HOMO value of the acceptor. Contrarily, LUMO energy value of the donor core should be lower, and the acceptor should be high. These facts are well proven in our investigated compounds. The successful completion of an intramolecular charge transfer can be identified by the movement of charge density from the donor to the end-capped acceptors. This exemplifies the efficacy of our end-capped alterations strategy for creating new and efficient NFAs based on fluorene that are friendly to the environment and exhibit promising photovoltaic characteristics for use in OSCs applications.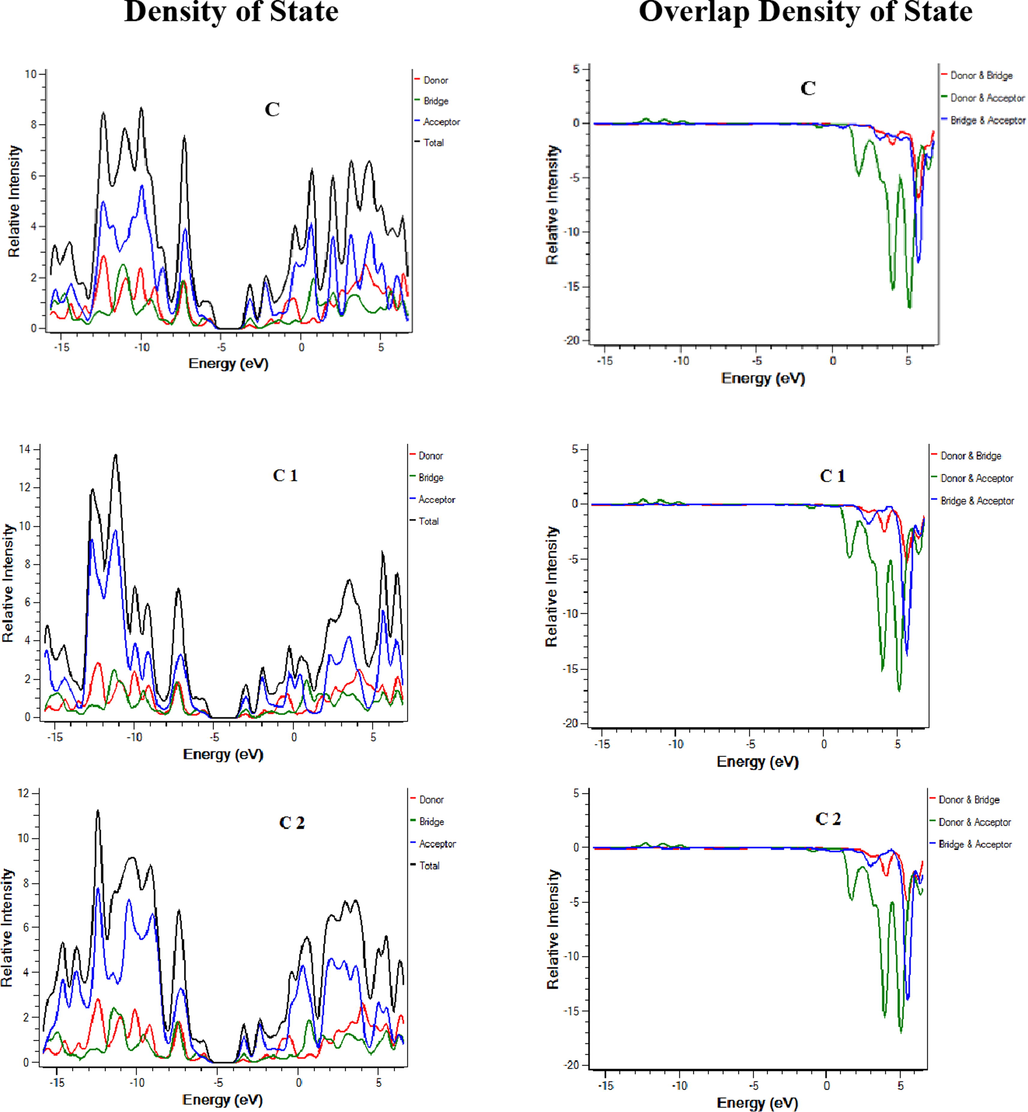
DOS and ODOS for C and C1-C9.
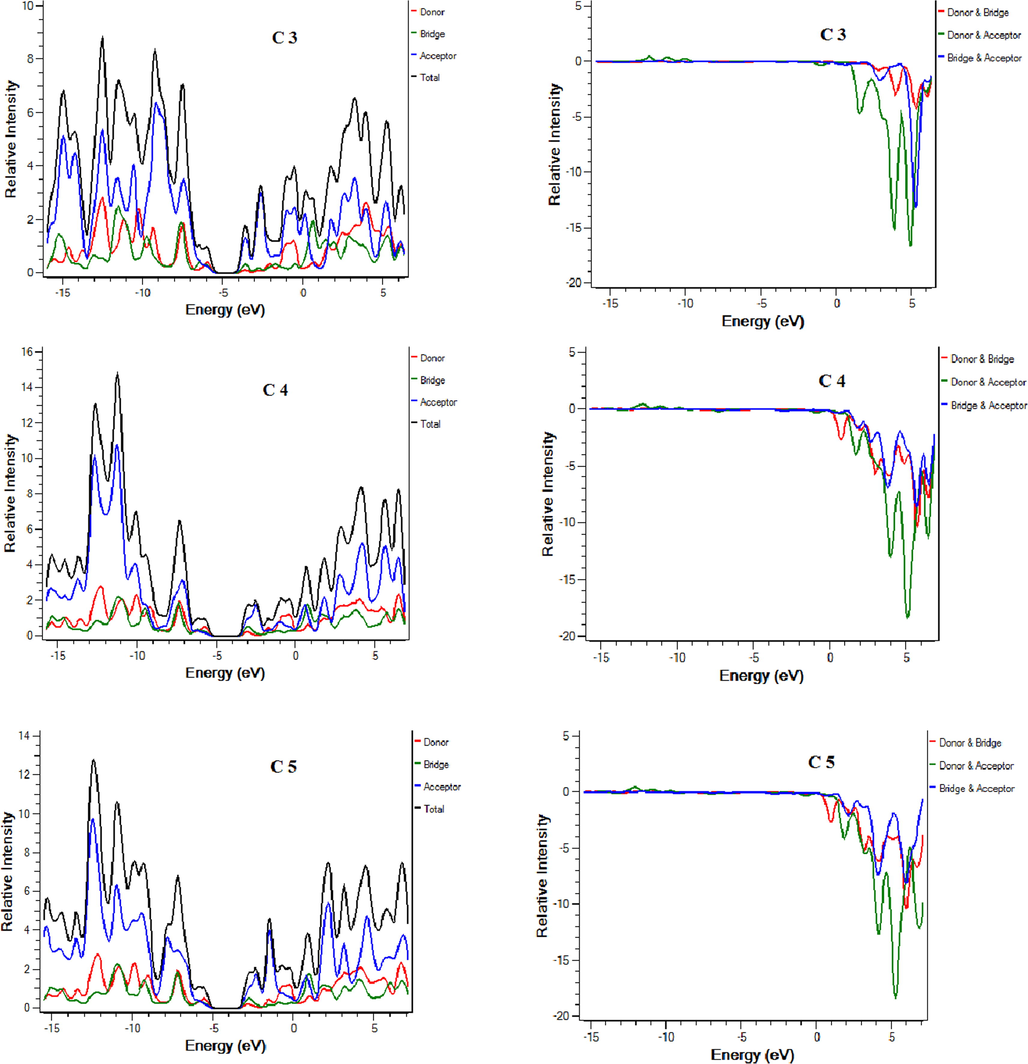
DOS and ODOS for C and C1-C9.
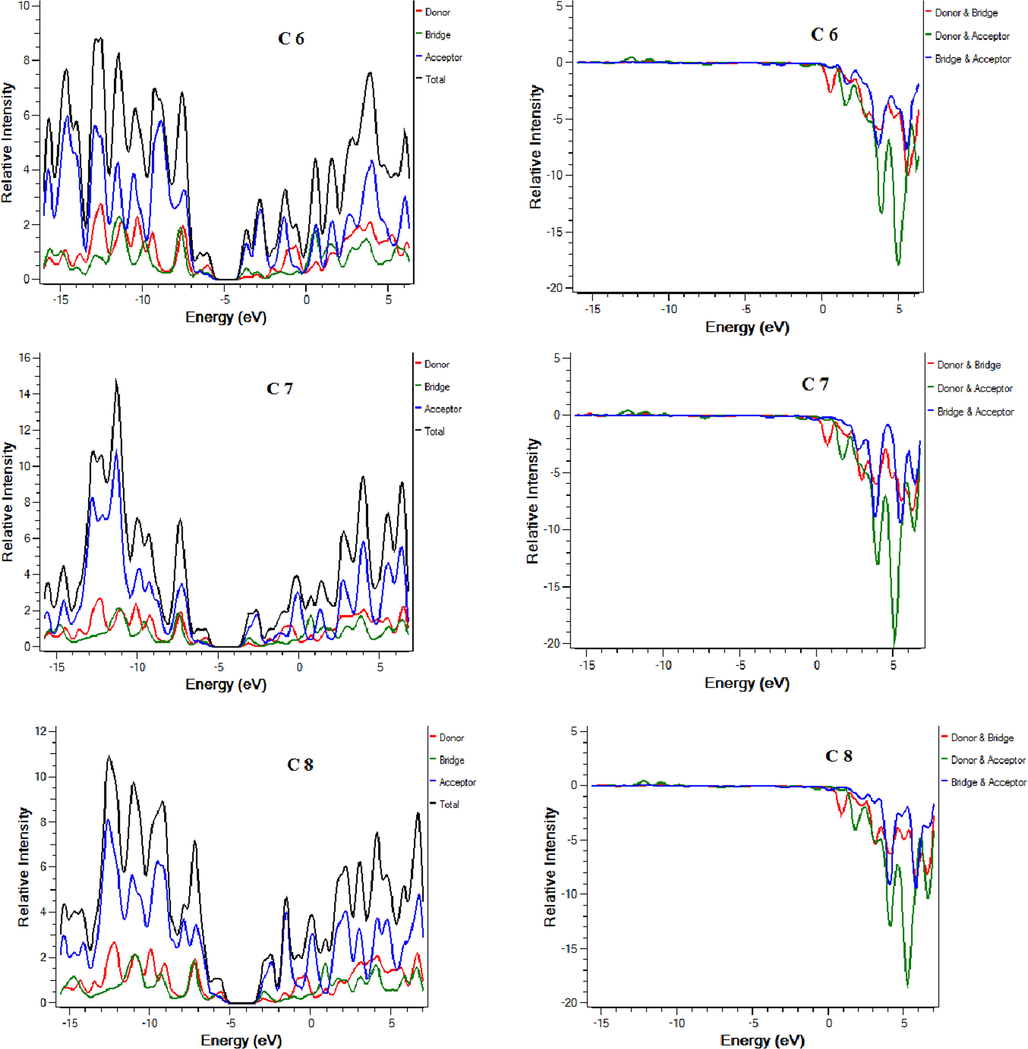
DOS and ODOS for C and C1-C9.
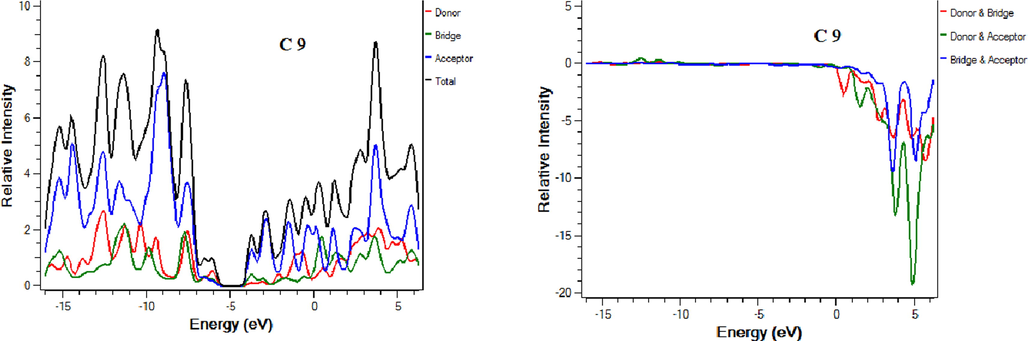
DOS and ODOS for C and C1-C9.
6 Optical properties
To explore the optoelectronic features in both gas and solvent phases, TDDFT/B3LYP/6-31G(d,p) and TDDFT/B3LYP/6-31G(d,p)/chloroform/CPCM model are used for UV–vis calculations respectively. The data from solvent and gas phase calculations is compiled in Tables 2 and 3.
Molecules
Calculated λmax(nm)
Experimental λmax(nm)
Ex(eV)
ƒos
Major MO Assignments
C
600.17
591
2.06
2.47
HOMO → LUMO (97 %)
C1
573.41
2.16
2.30
HOMO → LUMO (97 %)
C2
617.26
2.00
2.45
HOMO → LUMO (97 %)
C3
657.45
1.88
2.10
HOMO → LUMO (97 %)
C4
557.88
2.22
2.35
HOMO → LUMO (97 %)
C5
555.65
2.23
2.44
HOMO → LUMO (97 %)
C6
673.27
1.84
1.73
HOMO → LUMO (97 %)
C7
568.54
2.18
2.39
HOMO → LUMO (97 %)
C8
565.61
2.19
2.47
HOMO → LUMO (97 %)
C9
681.82
1.81
1.81
HOMO → LUMO (97 %)
Molecules
Calculated λmax(nm)
Experimental λmax(nm)
Ex(eV)
ƒos
Major MO Assignments
C
570.11
653
2.17
2.20
HOMO → LUMO (99 %)
C1
547.36
2.26
2.08
HOMO → LUMO (98 %)
C2
583.75
2.12
2.20
HOMO → LUMO (98 %)
C3
605.89
2.04
1.93
HOMO → LUMO (99 %)
C4
533.49
2.32
2.14
HOMO → LUMO (98 %)
C5
533.67
2.32
2.21
HOMO → LUMO (98 %)
C6
613.83
2.01
1.63
HOMO → LUMO (97 %)
C7
542.14
2.28
2.20
HOMO → LUMO (98 %)
C8
541.76
2.28
2.2651
HOMO → LUMO (98 %)
C9
619.88
2.00
1.7217
HOMO → LUMO (98 %)
The experimental λmax value was reported to be 591 nm in the solvent phase (Liao et al., 2021). All proposed compounds exhibit λmax in the UV–Visible region. CF3-containing molecules possess a lower value of λmax, while the molecules with NO2 groups exhibit a higher value of λmax. The effect of SO3H lies in between CF3 and NO2-containing molecules. Therefore, the increasing order is seen as: C9 > C6 > C3 > C2 > C > C1 > C7 > C8 > C4 > C5 in solvent phase and C9 > C6 > C3 > C2 > C > C1 > C7 > C8 > C4 = C5 in gas phase. When comparing the designed molecule C1-C3 to the C molecule, the λmax values of C2, C3 rose significantly to 617 nm, 657 nm (in solvent), and 583 nm, 605 nm (in gas), showing a greater effect of the end-capped acceptor having SO3H and NO2 groups on each arm in C2, C3 as compared to CF3 in C1 and CN in C results. The C6 molecule has a higher λmax value in the solvent and gas phase with 673 nm and 613 nm, respectively. Possible reasons for the shift to higher λmax values include the extended conjugation present in IC-2F end-capped acceptors with the NO2 group of the C6 molecule, and the presence of electron-withdrawing 2F groups. The proposed C9 molecule exhibits the most significant λmax values of 619 nm and 681 nm amongst C, C1-C9, which are correspondingly 49, and 81 nm more prominent in contrast to reference in the gas and the solvent. The joint action of IC-2Cl (end-capped) of C9 containing NO2 groups and prolonged conjugation successfully shifts the λmax to a higher value. Compared to end-capped acceptors of C, C1-C8 molecules, C9 results verified the greater tendency of its end-capped acceptor units due to wide conjugation and durable pulling character. 97 % transition originated in HOMO → LUMO is observed in the solvent phase and > 97 % transition from HOMO → LUMO in the gas phase is noted in all the studied molecules. The optical results make it abundantly clear that designed C1-C9 molecules exhibited comparable and better results than the reference C. The non-toxic functional groups usage in designed compounds give them an edge over toxic CN based reference C molecule, hence, have a strong proclivity to excel as photovoltaic aspirants for building up highly efficient environmentally friendly, low cost solar cells devices.
7 Open circuit voltage
Voc is a critical parameter in determining the maximum operating capability of OSCs. Voc values can be utilized to estimate the functional ability of OSCs. The Voc is a term that refers to the maximum amount of electric current that can be drawn from an optical device (Tang and Zhang 2012). Different factors also affect the values of Voc, such as light intensity, the temperature of solar cell device, types of materials. energy levels, and working functions of the electrodes. Equation (3) (Scharber et al., 2006) calculates Voc values of C and C1-C9.
The LUMO and HOMO Voc's value is contingent on the acceptor and donor concentrations, so they play a pivotal role in the calculation. The probability of electron transfer from the donor's HOMO level to the acceptor's LUMO level increases, leading to better Voc when the acceptor's LUMO is located lower in the energy level spectrum. A narrow-bandgap donor polymer PTB7-Th is used to manufacture inverted BHJSCs because of the absorption spectrum's energy level matching and significant complementarity. The equation (3) results calculated regarding HOMOPTB7-Th –LUMOacceptor Eg is visible in Fig. 5.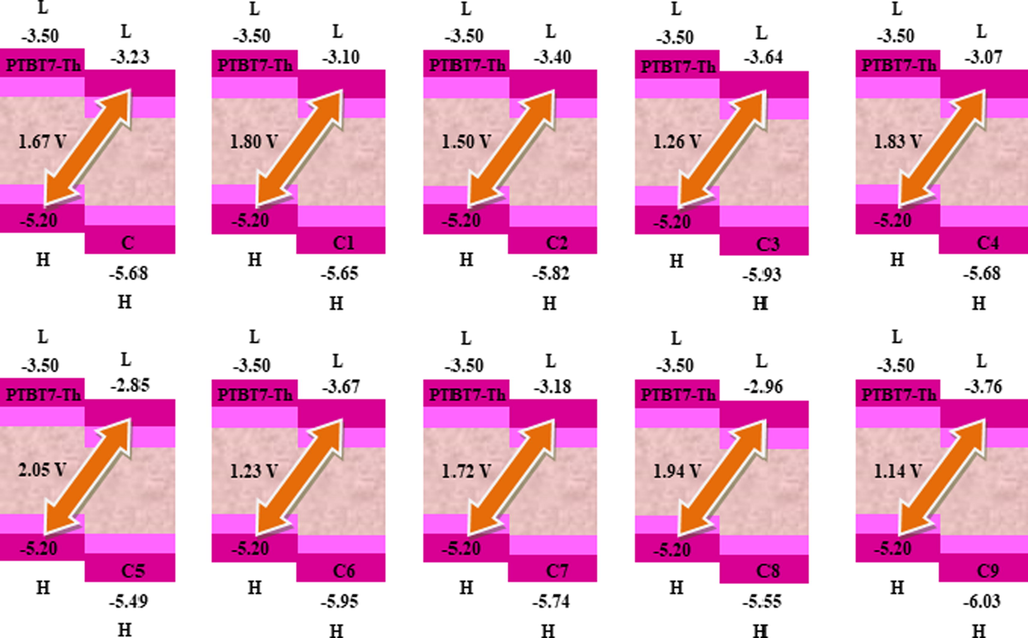
The Voc results of the reference C and the designed C1-C9 compounds. The output files of the designed compounds were computed by Gaussian 09 version D.01.
The values of Voc for molecules C, C1-C9 are computed as 1.67, 1.80, 1.50, 1.26, 1.83, 2.05, 1.23, 1.72, 1.94, 1.14 V correspondingly. The declining Voc order for C1-C9 is C5 ˃ C8 ˃ C4 ˃ C1 ˃ C7 ˃ C ˃ C2 ˃ C3 ˃ C6 ˃ C9. Designed molecules C2 and C3 exhibit slightly smaller Voc results than the reference C molecule. C5 molecules depict a higher Voc value than reference C molecule, while C9 has the lowest value of Voc than other molecules. On the other hand, C1, C4, and C8 molecules have high open circuit voltage than C. Results exposed that Voc in C8 and C5 molecules is higher than the reference molecule. Overall, C5 depicts the highest Voc value 2.05 V, which is 0.38 V greater than C. These findings suggest that C1-C9 molecules with non-toxic groups have comparable and better Voc results than the C molecule with toxic functional group, hence, can be potentially used for the construction of environmental friendly solar cells.
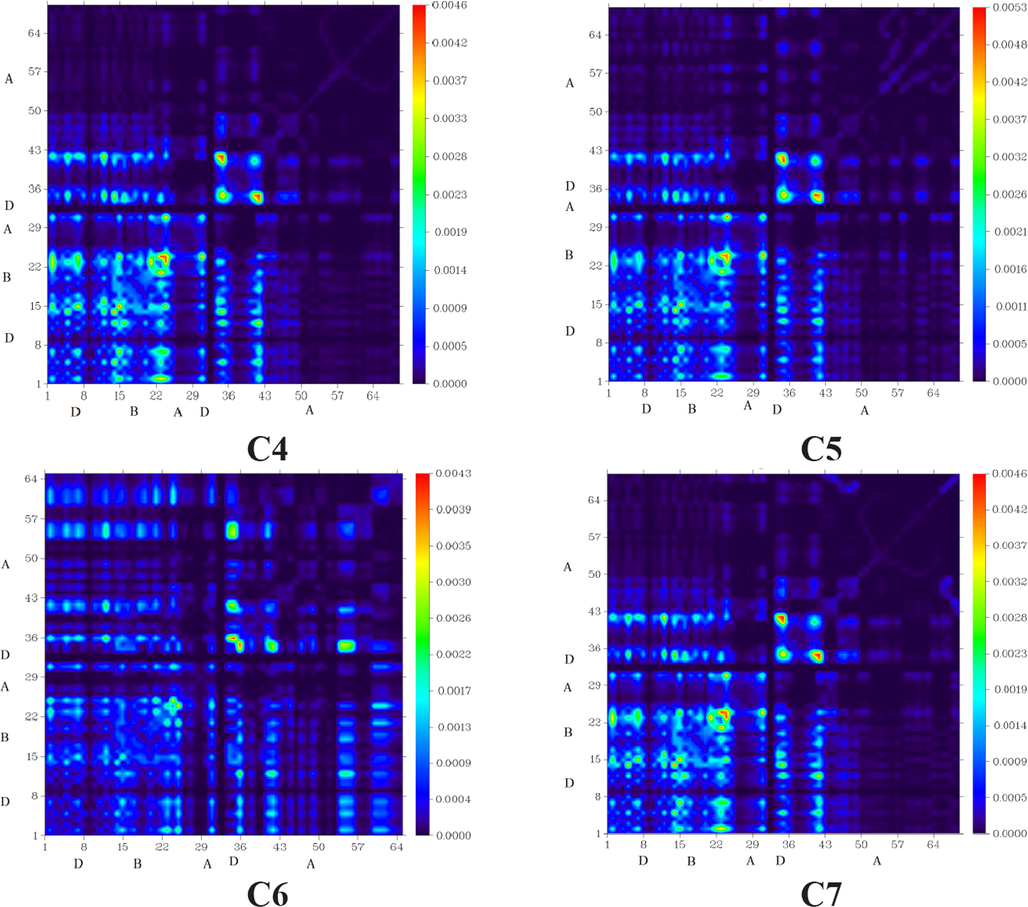
8 Transition density matrix (TDM) heat map analysis
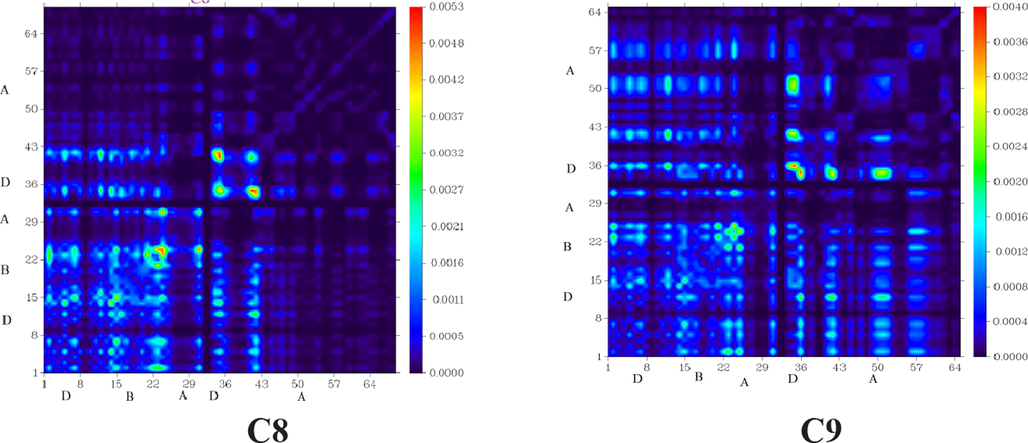
TDM analysis explains solar cell performance by calculating the density of electron transfer from donor to acceptor moieties via-conjugation, as well as donor and acceptor functional group interaction in the excited state and electron hole localization. As hydrogen atoms play such a minor role in such transitions, they are often neglected (Tahir et al., 2020). The C and C1-C9 molecules are classified into three types: core (D), acceptor (A), and bridge (B). Fig. 6 illustrates the TDM results for C and C1-C9. The number of atoms that can be found in each of these molecules can be seen in the diagram's left side and bottom right corner.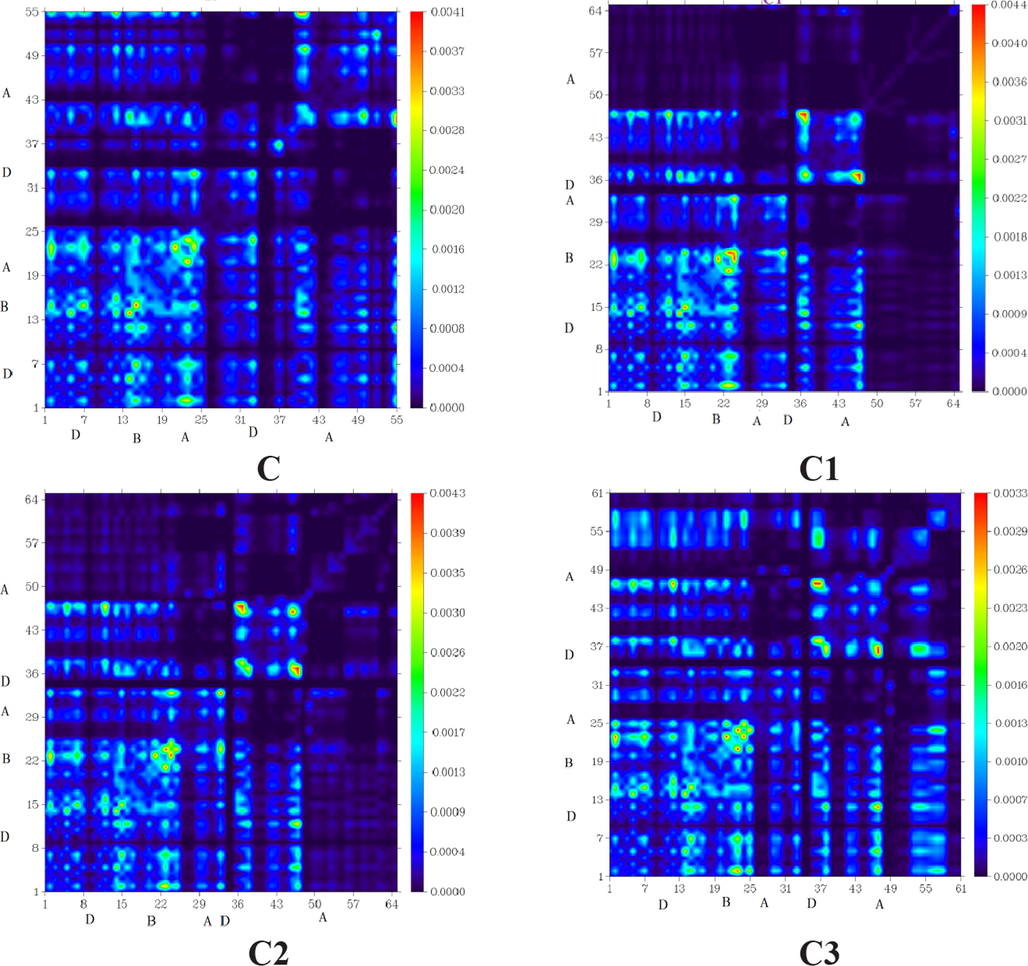
“Transition Density Matrix (TDM) heats maps of C and C1-C9 at S1 state. They were prepared with the help of Multiwfn 3.7 software (http:// sober eva. com/ multi wfn/). All the output files of designed compounds were completed by Gaussian 09 version D.01 (https:// gauss ian. com/ g09ci tation/)”.
“Transition Density Matrix (TDM) heats maps of C and C1-C9 at S1 state. They were prepared with the help of Multiwfn 3.7 software (http:// sober eva. com/ multi wfn/). All the output files of designed compounds were completed by Gaussian 09 version D.01 (https:// gauss ian. com/ g09ci tation/)”.
“Transition Density Matrix (TDM) heats maps of C and C1-C9 at S1 state. They were prepared with the help of Multiwfn 3.7 software (http:// sober eva. com/ multi wfn/). All the output files of designed compounds were completed by Gaussian 09 version D.01 (https:// gauss ian. com/ g09ci tation/)”.
All molecules exhibit charge coherence as depicted by TDM diagrams. Electron coherence in the diagonal transfer is perceived in all the investigated molecules. It substantiates the fact that charge coherence was effectively transferred from the donor to the bridge, allowing electron density to change without being trapped in the end group acceptors. Fig. 6 represents the distribution of charges in TDM maps, which is essential for developing OSCs.
The hole-electron overlap maps are presented in Fig. 7.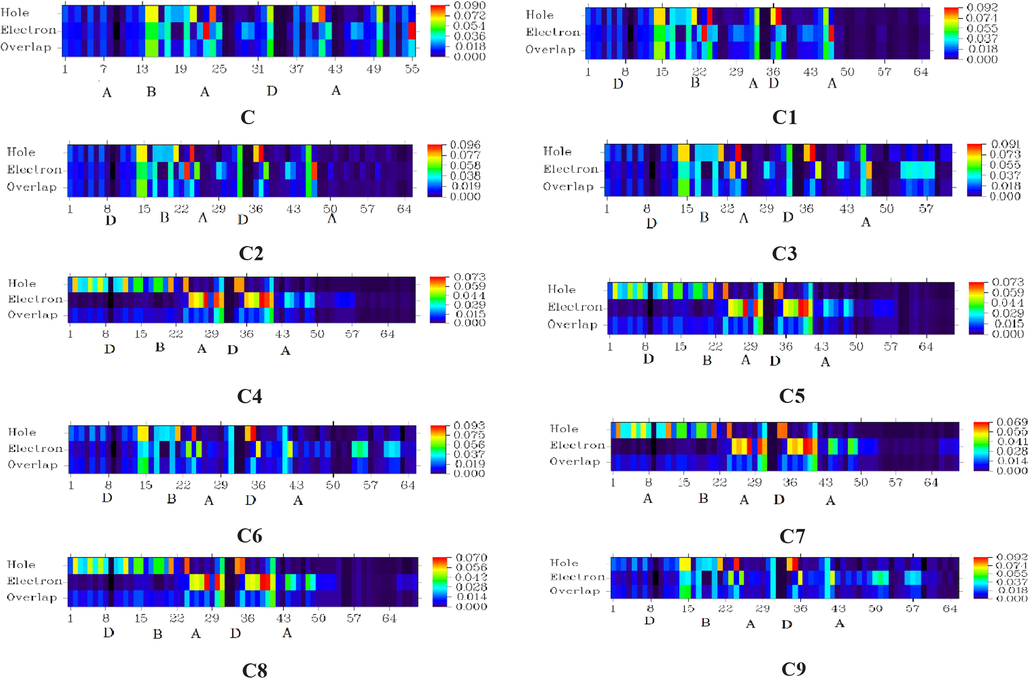
Electron hole overlap surfaces of C, C1-C9.
9 Exciton binding energy (Eb)
Another critical thing to consider when evaluating optoelectronic properties is binding energy. By lowering the columbic force amongst electrons and holes, decreased binding energy enhances the exciton dissociation. The binding energy (Eb) is calculated by equation (4), and the findings are shown in Table 4:
Molecules
EH-L (eV)
Eopt (eV)
Eb (eV)
C
2.45
2.07
0.38
C1
2.55
2.16
0.39
C2
2.42
2.01
0.41
C3
2.29
1.89
0.40
C4
2.61
2.22
0.39
C5
2.64
2.23
0.41
C6
2.28
1.84
0.44
C7
2.56
2.18
0.38
C8
2.59
2.19
0.40
C9
2.27
1.81
0.46
Calculations show that C has a binding energy of 0.38 eV. The Eb values of C1-C9 are noted as 0.39, 0.41, 0.40, 0.39, 0.41, 0.44, 0.38, 0.40 and 0.46 eV correspondingly. Hence proven that all designed moieties have a comparable value of binding energy. Out of all the suggested molecules, C7 has the lowest Eb. Since the lower the binding energy, the higher the charge transfer rate because lower Eb assists exciton dissociation by breaking the columbic forces between the hole and the electron. That’s why C7 molecule can act as the best acceptor molecule among proposed molecules for solar cell applications. Overall, proposed compounds hold comparable to reference C molecule Eb values. These results also support our end-capped modification strategy for building simple and low-cost non-fullerene environmentally friendly photovoltaic materials by improving device current density and PCE.
10 Reorganization energy
The overall charge transfer between a molecule's donor and acceptor segments is estimated using overall reorganization energy. The overall reorganisation energy is comprised of both the internal reorganisation energy (λint) and the external reorganization energy (λext). Internal structural changes are explained by λint, while the impact of polarization on the external environment is demonstrated by λext. Variations in the solvent of the surrounding media have been ignored in this study, which exclusively considers the internal reorganization energy of conjugated complexes.
It is possible to make use of the Marcus equation, which is utilised to quantify the amount of energy consumed whenever there is a change in the molecular geometry. When the molecular geometry shifts from neutral to charged species or from charge to natural, energy is expended, and this can be quantified using the Marcus equation.
In equation (5),
is the transfer rate of electrons. The electronic coupling present among various states is denoted with symbol T, the electron λe and hole λh reorganization energy values of C and C1- C9 are computed and the results are mentioned in Table 5. [c] reorganization energy of electron [d] reorganization energy of hole.
Molecules
λe (eV) [c]
λh [d]
C
0.0063
0.0072
C1
0.0097
0.0087
C2
0.0176
0.0109
C3
0.1189
0.1152
C4
0.0113
0.0090
C5
0.0122
0.0085
C6
0.0099
0.0087
C7
0.0144
0.0044
C8
0.0117
0.0083
C9
0.0098
0.0086
It can be seen from Table 5 that λe and λh values are extremely promising. The λe value of C is computed as 0.0063 eV, while designed molecules: C1, C2, C3, C4, C5, C6, C7, C8 and C9 are found to have 0.0097, 0.0176, 0.1189, 0.0113, 0.0122, 0.0099, 0.0144, 0.0117and 0.0098 eV λe values respectively. The increasing order for λe values of C and C1-C9 is found to be C < C1 < C9 < C6 < C4 < C8 < C5 < C7 < C2 < C3. In terms of electron reorganisation, the C1 molecule and the reference molecule are comparable. Whereas λe values of other molecules are higher than reference molecule. The λh value of C is calculated as 0.007262 eV. Whereas, 0.0087, 0.0109, 0.1152, 0.0090, 0.0085, 0.0087, 0.0044. 0.0083,0.0086 eV are the λh values that are detected in proposed compounds C1-C9 correspondingly. The λh values order is found to be C7 < C < C8 < C5 < C9 < C1 < C6 < C4 < C2 < C3. In this ranking, it can be seen that all of the designed molecules, with the exception of C7, have greater hole mobility than the C reference molecule. The λh value of C7 is low than the reference and all other designed molecules. This low hole mobility of C7 might be due to the CF3 group. The preceding discussion has established that some compounds are excellent candidates for hole and electron mobilities. These calculations revealed that these acceptors could be better hole transporters.
11 Charge transfer analysis
Charge transfer analysis was used to support the discovery of improved electron transport mobilities in the examined compounds. Charge transfer is investigated using the NO2-containing C3 molecule, and complex formation with the polymer PTB7-Th is then performed because of the smaller HOMO-LUMO energy gap values between these two species. The optimized shape of the C3: PTB7-Th complex suggests that the orientation of C3 and the polymer PTB7-Th in terms of electron transmission is particularly interesting. The C3 molecule and the center section of the PTB7-Th polymer are positioned parallel to each other, aiding in the efficient transfer of charge between the donor and the recipient units. The FMO diagram (Fig. 8) indicates that the charge density for HOMO is concentrated on the PTB7-Th donor polymer, while that for LUMO is concentrated on the C3 acceptor molecule. Donor and acceptor components undergo charge transfer, as evidenced by the HOMO and LUMO distribution patterns, which is a substantial proof of charge shifting between two distinct moieties.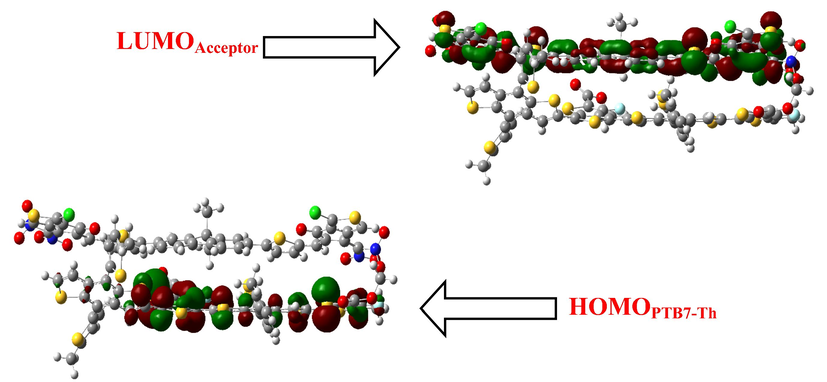
Charge transfer HOMOPTB7-Th-LUMOAcceptor C3 blend. The images are made with the help of GaussView 5.0 and Gaussian 09 version D.01.
12 Conclusion
Herein, we devised a fluorene-based series (C1-C9) of environmental friendly organic solar cells (EFOSCs) by replacing toxic − C≡N of 2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile (TC) present as an end-capped acceptor in DTC-T-F with non-toxic electron-withdrawing groups -CF3, -SO3H, –NO2. The results of the proposed molecules are found to be comparable and even better than the reference C molecule. Reduction in the energy band gap of up to 0.15–0.17 eV than C (2.44 eV) is observed in designed molecules C3, C6, and C9 with band gap values of 2.29 eV, 2.28 eV, and 2.27 eV respectively. Molecules with lower energy gaps have NO2 electron-withdrawing groups. C9 molecule containing IC-2Cl with NO2 groups exhibited a robust red shift in both the solvent (681 nm) and the gas phase (619 nm) than reference 600 nm (solvent) and 570 nm (gas phase) values. DOS analysis confirms that donor moiety in C1-C9 contributes a more significant percentage to HOMO and a smaller portion to LUMO and vice versa. The open circuit voltage is the most critical component to consider while assessing OSC efficiency. C5 exhibited the highest Voc 2.05 V with respect to HOMOPTB7-Th–LUMOacceptor. The lowest value of binding energy (Eb) was found to be in C7 = 0.38 eV. Finally, the reorganization energy values revealed that the proposed C-series is an excellent option for producing EFOSCs. This research shows that the proposed fluorene-based cyano (−C≡N) free NFAs materials are better and should be used to construct simple and low-cost non-fullerene environmentally friendly organic solar cells. This report also outlines an effective strategy for designing environmentally friendly organic solar cells by substituting non-toxic electron-withdrawing groups (-CF3, -SO3H, –NO2 groups) for toxic CN groups. It could pave the way for future research to speed up the development of environmentally friendly solar cells.
Acknowledgment
Funding in the Lu lab was provided by the Fundamental Research Funds for the Central Universities (2232021G-04), Shanghai Science and Technology Committee (19ZR1471100). M.A.A. appreciates the support of the Research Center for Advanced Materials Science (RCAMS) at King Khalid University Abha, Saudi Arabia, through grant KKU/RCAMS/22.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: the m PW and m PW1PW models. J. Chem. Phys.. 1998;108:664-675.
- [Google Scholar]
- Andrienko, G. A., 2010. Chemcraft. Graphical Software for Visualization of Quantum Chemistry Computations
- Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A. 1998;102:1995-2001.
- [Google Scholar]
- Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys.. 2008;10:6615-6620.
- [Google Scholar]
- Next-generation organic photovoltaics based on non-fullerene acceptors. Nat. Photonics. 2018;12:131-142.
- [Google Scholar]
- B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm.. 2008;10:405-410.
- [Google Scholar]
- Small is powerful: recent progress in solution-processed small molecule solar cells. Adv. Energy Mater.. 2017;7:1602242.
- [Google Scholar]
- Single-junction organic photovoltaic cells with approaching 18% efficiency. Adv. Mater.. 2020;32:1908205.
- [Google Scholar]
- Single-junction organic photovoltaic cell with 19% efficiency. Adv. Mater.. 2021;33:2102420.
- [Google Scholar]
- Dennington, R. D., T. A. Keith and J. M. Millam, 2008. GaussView 5.0. 8. Gaussian Inc.
- Achieving over 16% efficiency for single-junction organic solar cells. Sci. China Chem.. 2019;62:746-752.
- [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, et al., 2009. D. 0109, Revision D. 01, Gaussian. Inc., Wallingford, CT.
- Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform.. 2012;4:17.
- [Google Scholar]
- A rhodanine flanked nonfullerene acceptor for solution-processed organic photovoltaics. J. Am. Chem. Soc.. 2015;137:898-904.
- [Google Scholar]
- Organic solar cells based on non-fullerene acceptors. Nat. Mater.. 2018;17:119-128.
- [Google Scholar]
- Noncovalently fused-ring electron acceptors with near-infrared absorption for high-performance organic solar cells. Nat. Commun.. 2019;10:1-10.
- [Google Scholar]
- A fluorene-terminated hole-transporting material for highly efficient and stable perovskite solar cells. Nat. Energy. 2018;3:682-689.
- [Google Scholar]
- Exploration of promising optical and electronic properties of (non-polymer) small donor molecules for organic solar cells. Sci. Rep.. 2021;11:1-15.
- [Google Scholar]
- Exploration of efficient electron acceptors for organic solar cells: rational design of indacenodithiophene based non-fullerene compounds. Sci. Rep.. 2021;11:1-15.
- [Google Scholar]
- Structural modulation of π-conjugated linkers in D–π–A dyes based on triphenylamine dicyanovinylene framework to explore the NLO properties. Royal Soc. Open Sci.. 2021;8:210570
- [Google Scholar]
- Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc.. 2021;25:101305
- [Google Scholar]
- Designing triazatruxene-based donor materials with promising photovoltaic parameters for organic solar cells. RSC Adv.. 2019;9:26402-26418.
- [Google Scholar]
- In silico modeling of new “Y-Series”-based near-infrared sensitive non-fullerene acceptors for efficient organic solar cells. ACS Omega. 2020;5:24125-24137.
- [Google Scholar]
- Designing star-shaped subphthalocyanine-based acceptor materials with promising photovoltaic parameters for non-fullerene solar cells. ACS Omega. 2020;5:23039-23052.
- [Google Scholar]
- Novel W-shaped oxygen heterocycle-fused fluorene-based non-fullerene acceptors: first theoretical framework for designing environment-friendly organic solar cells. Energy Fuels. 2021;35:12436-12450.
- [Google Scholar]
- Carbazole-based polymers for organic photovoltaic devices. Chem. Soc. Rev.. 2010;39:2399-2410.
- [Google Scholar]
- An unfused-core-based nonfullerene acceptor enables high-efficiency organic solar cells with excellent morphological stability at high temperatures. Adv. Mater.. 2018;30:1705208.
- [Google Scholar]
- Construction of simple and low-cost acceptors for efficient non-fullerene organic solar cells. Org. Electron.. 2021;89:106026
- [Google Scholar]
- An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv. Mater.. 2015;27:1170-1174.
- [Google Scholar]
- A facile planar fused-ring electron acceptor for as-cast polymer solar cells with 8.71% efficiency. J. Am. Chem. Soc.. 2016;138:2973-2976.
- [Google Scholar]
- Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33:580-592.
- [Google Scholar]
- Recent advances in bulk heterojunction polymer solar cells. Chem. Rev.. 2015;115:12666-12731.
- [Google Scholar]
- Modeling charge recombination in dye-sensitized solar cells using first-principles electron dynamics: effects of structural modification. Phys. Chem. Chem. Phys.. 2013;15:17187-17194.
- [Google Scholar]
- Electron transfer reactions in chemistry. theory and experiment. Rev/ Modern Phys.. 1993;65:599.
- [Google Scholar]
- Designing of benzodithiophene core-based small molecular acceptors for efficient non-fullerene organic solar cells. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc.. 2021;244:118873
- [Google Scholar]
- O'boyle, N. M., A. L. Tenderholt and K. M. Langner, 2008. Cclib: a library for package‐independent computational chemistry algorithms. J. Comput. Chem. 29, 839-845.
- Novel star-shaped benzotriindole-based nonfullerene donor materials: toward the development of promising photovoltaic compounds for high-performance organic solar cells. Energy Technol. 20222100751
- [Google Scholar]
- Design rules for donors in bulk-heterojunction solar cells—towards 10% energy-conversion efficiency. Adv. Mater.. 2006;18:789-794.
- [Google Scholar]
- Well-designed 3D/2D/2D WO3/Bt/g-C3N4 Z-scheme heterojunction for tailoring photocatalytic CO2 Methanation with 2D-layered bentonite-clay as the electron moderator under visible light. Energy Fuels. 2020;34:14400-14418.
- [Google Scholar]
- Design of donors with broad absorption regions and suitable frontier molecular orbitals to match typical acceptors via substitution on oligo (thienylenevinylene) toward solar cells. J. Comput. Chem.. 2012;33:1353-1363.
- [Google Scholar]
- Fused hexacyclic nonfullerene acceptor with strong near-infrared absorption for semitransparent organic solar cells with 9.77% efficiency. Adv. Mater.. 2017;29:1701308.
- [Google Scholar]
- Ternary organic solar cells offer 14% power conversion efficiency. Sci. Bull.. 2017;62:1562-1564.
- [Google Scholar]
- A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem. Phys. Lett.. 2004;393:51-57.
- [Google Scholar]
- Design and synthesis of a low bandgap small molecule acceptor for efficient polymer solar cells. Adv. Mater.. 2016;28:8283-8287.
- [Google Scholar]
- Elucidating the key role of the Cyano (− C≡ N) Group to Construct Environmentally friendly fused-ring electron acceptors. J. Phys. Chem. C. 2020;124:23059-23068.
- [Google Scholar]
- Achieving over 10% efficiency in a new acceptor ITTC and its blends with hexafluoroquinoxaline based polymers. J. Mater. Chem. A. 2017;5:11286-11293.
- [Google Scholar]
- The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc.. 2008;120:215-241.
- [Google Scholar]




