

Translate this page into:
1,3,4-Thiadiazoline−coumarin hybrid compounds containing D-glucose/D-galactose moieties: Synthesis and evaluation of their antiproliferative activity
⁎Corresponding author. nguyendinhthanh@hus.edu.vn (Nguyen Dinh Thanh)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We synthesized a new series of 1,3,4-thiadiazoline−coumarin hybrid compounds that contain d-glucose and d-galactose moieties (9a-g) and evaluated their cytotoxic activity against breast adenocarcinoma (MCF-7), human liver cancer (HepG2), human cervical cancer (HeLa), human melanoma cancer (SK-Mel-2), and human lung cancer (LU-1) cells. To reveal their selectivity toward cancer cells, the compounds were also tested against human fibroblast cell line, MRC-5. Synthesized compounds exhibited potent cytotoxic activity against the tested cell lines with IC50 values of 1.18–11.81, 1.72–9.43, 1.98–13.16, 1.82–11.25, and 2.25–14.62 μM (against MCF-7, HepG2, HeLa, SK-Mel-2, and LU-1 cells, respectively) compared to Sorafenib, 5-FU, and DOX. The long and branched-terminal carbon chains often increased activity. Interestingly, compounds 9a-g displayed selectivity toward cancer cell lines over MRC-5 (IC50 3.93–25.55 μM). These compounds also displayed potent inhibitory activity against EGFR and HER2 kinases (IC50 0.22–0.47 and 0.13–0.35 μM, respectively) compared to the standard drug, Sorafenib (IC50 = 0.11 and 0.13 μM, respectively). Molecular docking study also employed to identify the structural features required for the EGFR/HER2 inhibitory activity of the new series.
Keywords
4-Formylcoumarins
1,3,4-Thiadiazolines
EGFR
HER2
Antiproliferative
Molecular docking
Sugar

1 Introduction
WHO reports that in 2020 cancer still is a serious health problem in all populations, regardless of wealth or social status, and is one of the major causes of death in the world. The predicted global burden will double to about 29–37 million new cancer cases by 2040, with the greatest increases in low- and middle-income countries (WHO, 2020). Lung, prostate, colorectal, stomach and liver cancer are the most common types of cancer in men, while breast, colorectal, lung, cervical and thyroid cancer are the most common among women. In 2018, there were an estimated 18 million new cases of cancer and 10 million deaths from cancer worldwide. According to reports by WHO, breast cancer is the most frequently diagnosed cancer among women, , while cervical cancer is the fourth most common cancer in women. Colorectal cancer is the third most commonly occurring cancer in men and the second most commonly occurring cancer in women. Lung cancer (both small cell and non-small cell) is the second most common cancer in both men and women (not counting skin cancer). Melanoma is the 19th most common cancer in men and women, and viral hepatitis B and C infections are key causes of primary liver cancer, the second most common cancer in the world, with 788.000 people die from primary liver cancer every year (WHO, 2020).
Anticancer therapy has progressed significantly, but the management of malignancies in patients still constitutes a major concern for contemporary medicine. In addition to surgical treatment and radiotherapy, cancers are also treated with chemicals. This treatment minimizes the pain caused by surgery or the danger caused by nuclear radiation to the patients.
Thus, many anticancer agents were synthesized (Spaczyńska et al., 2019, Bernat et al., 2020) or isolated from plants (Lichota and Gwozdzinski, 2018, Lee et al., 2020) and their cytotoxicity have been studied to develop novel and efficient drugs for treating cancer. These studies showed that many anticancer agents exert their effects through destruction of rapidly dividing cells, and these cytotoxic agents remain the primary resource in cancer chemotherapy, despite advances in the understanding of the cell cycle that could facilitate development of more selective chemotherapeutic agents (Akhdar et al., 2012). For example, compounds A exhibited antiproliferative activity against human colon carcinoma cell lines HCT-116 (Spaczyńska et al., 2019)(Fig. 1); compounds B inhibited protein kinases Bcr-Abl (Bernat et al., 2020). Ellipticine [5,11-dimethyl-6H-pyrido-(4,3-b)-carbazole], was an alkaloid isolated from the leaves of Ochrosia elliptica Labill, has been shown to be effective against human breast cancer cells (MCF-7), leukemia (HL-60 and CCRF-CEM) cells, neuroblastoma (IMR-32, UKF-NB-3 and UKF-NB-4) cells, and glioblastoma cells (U87MG) (Poljaková et al., 2007).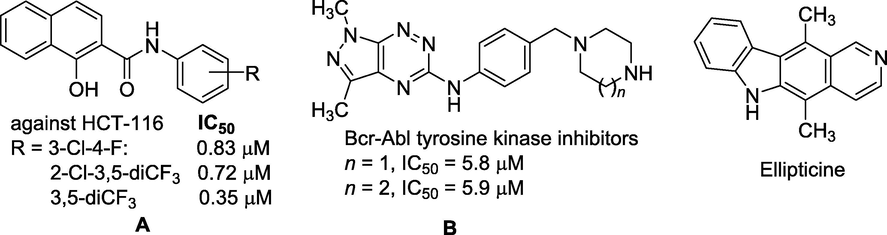
Some typical potential compounds against cancer cell lines.
Amongst the synthetic compounds with the above-mentioned biological activity, thiadiazole rings (including 1,2,3-, 1,2,4-, 1,2,5-, and 1,3,4-thiadiazoles) as well as their derivatives have gained great interest due to their remarkable broad spectra of activity, such as anticancer (Yusuf and Jain, 2014, Chandra Sekhar et al., 2019), antitubercular (Chandra Sekhar et al., 2019), antibacterial (Noolvi et al., 2016), antifungal (Karaburun et al., 2018), antiviral (Brai et al., 2019), antioxidant (Mathew et al., 2016) activity, etc. 1,3,4-Thiadiazole scaffold and its derivatives also contained well-known anticancer agents that can target different types of cancer cells (Alireza, 2016, Janowska et al., 2020, Szeliga, 2020), and many compounds have been proven under in vivo conditions (Swathi et al., 2020). Fig. 2 displayed some bioactive 2,3-dihydro-1,3,4-thiadiazolidine derivatives. For examples, K858 [N-(4-acetyl-4,5-dihydro-5-methyl-5-phenyl-1,3,4-thiadiazol-2-yl)acetamide] is a significant antitumor agent against human prostate cancer PC3 and melanoma SK-MEL-5 and SK-MEL-28 cells (De Iuliis et al., 2016). Filanesib, [ARRY-520, (2S)-2-(3-aminopropyl)-5-(2,5-difluorophenyl)-N-methoxy-N-methyl-2-phenyl-1,3,4-thiadiazole-3(2H)-carboxamide trifluoroacetate], is a kinesin spindle protein (KIF11) inhibitor which has recently been proposed as a cancer treatment, specifically for multiple myeloma (Khoury et al., 2012). Litronesib, [LY2523355, N-[(5R)-4-(2,2-dimethylpropanoyl)-5-[[2-(ethylamino)ethylsulfonylamino]methyl]-5-phenyl-1,3,4-thiadiazol-2-yl]-2,2-dimethylpropanamide], showed marked antitumor activity in most of the xenograft tumor models and resulted in a dose-dependent mitotic arrest of HCT-116 cells and subsequent cell death (Ye et al., 2015). Two compounds, ARRY-520 and LY2523355, are two analogues of K858 (Fig. 2).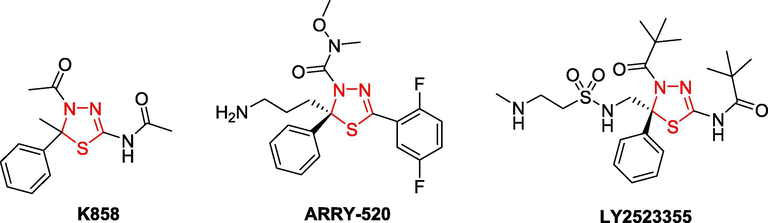
Some bioactive compounds having 2,3-dihydro-1,3,4-thiadiazole moiety.
The presence of sugar components can also elicit significant anticancer activity from 1,3,4-thiadiazoles (Flefel et al., 2017, Alminderej et al., 2019, Khalaf et al., 2020). Therefore, some 1,3,4-thiadiazole and 1,2,3-triazole hybrid glycosides were synthesized and evaluated as cytotoxic agents against HepG-2 (human liver cancer), HCT-116 (human colorectal carcinoma), MCF-7 (human breast adenocarcinoma), and RPE-1 (human normal retinal pigmented epithelial cell line) (Alminderej et al., 2019, Kassem et al., 2019, Khalaf et al., 2020). Due to their mesoionic nature, 1,3,4-thiadiazoles are able to cross the cellular membranes. Their relatively good liposolubility is most likely attributed to the presence of the sulfur atom (Haider et al., 2015). 1,3,4-Thiadiazoline showed broad-spectrum anticancer activities against human cancers and target proteins involved in proliferation, survival, and metastasis, including carbonic anhydrase (CA), matrix metalloproteinases (MMPs), histone deacetylases (HDALs), B-cell lymphoma 2 (Bcl-2), Bcl-XL, Bcl-2-associated X protein (Bax), Akt/PKB, tubulin, focal adhesion kinase (FAK), protein tyrosine kinases, microtubule (MT)-stimulated ATPase activity of Eg5, dihydrofolate reductase (DHFR) (Khathi et al., 2018, Singh et al., 2018).
Coumarin (2H-chromen-2-one) was a compound of cinnamon and was a family of benzopyrone (1,2-benzopyrone or 2H-1-benzopyran-2-ones) that widely distributed in the nature in many plants (Venugopala et al., 2013, Stefanachi et al., 2018). The compound having coumarin ring had been reported to possess pharmacological activities (Stefanachi et al., 2018, Zhu and Jiang, 2018). Numerous compounds containing coumarin ring were studied for their anticancer activity through multiple different mechanisms and targets (Liang et al., 2019, Al-Warhi et al., 2020, Wang et al., 2020, Xavier et al., 2020). These such compounds exhibited antitumor activity against different cancer cell lines, such as human liver cancer HepG2 (Ding et al., 2020, Kasmi et al., 2020), breast cancer MCF-7 (Ding et al., 2020, Kasmi et al., 2020), cervical carcinoma HeLa (Ding et al., 2020, Maleki et al., 2020), melanoma SK-MEL-5 (Goel et al., 2015), etc.
Owing to the pharmacological significance of 1,3,4-thiadiazoline derivatives, we synthesized 1,3,4-thiadizole scaffold containing coumarin and d-glucose/d-galactose moieties, and examined in vitro their anticancer activities, such as MCF7 (human breast adenocarcinoma cells), HepG2 (human liver cancer cells), HeLa (human cervical cancer cells), SK-Mel-2 (human melanoma cancer cells), and LU-1 (human lung cancer cells). Molecular docking study was performed to better understand the molecular basis for the inhibition of target enzymes in these cancer cells by the newly synthesized derivatives.
Molecular docking study was performed to better understand the molecular basis for the inhibition of target enzymes in these cancer cells by the newly synthesized derivatives.
2 Experimental
2.1 General methods
Melting points were determined by open capillary method on STUART SMP3 instrument (BIBBY STERILIN, UK) and are uncorrected. IR spectra (KBr disc) were recorded on an Impact 410 FT-IR Spectrometer (Nicolet, USA). 1H and 13C NMR spectra were recorded on Bruker Avance Spectrometer AV500 (Bruker, Germany) at 500 MHz and 125 MHz, respectively, using DMSO‑d6 as solvent and TMS as an internal standard. ESI-EI-HRMS and ESI/HR-mass spectra were recorded on LC-EI-HRMS LTQ Orbitrap XL or Thermo Scientific Exactive Plus Orbitrap spectrometers (ThermoScientific, USA) in methanol using ESI method. The analytical thin-layer chromatography (TLC) was performed on silica gel 60 WF254S aluminum sheets (Merck, Germany) and was visualized with UV light. Chemical reagents in high purity were purchased from the Merck Chemical Company (in Viet Nam). Starting materials were prepared according to the procedures described in cited literatures, including 6- and 7-alkoxy-4-formylcoumarins (6a-g) (Ngoc Toan and Dinh Thanh, 2020) and N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)thiosemicarbazide (7a) and N-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)thiosemicarbazide (7b) (Thanh et al., 2016).
2.2 General methods for synthesis of N-(2,3,4,6-tetra-O-acetyl-β-d-glycopyranosyl)thiosemicarbazones of 6- and 7-alkoxy-2-oxo-2H-chromene-4-carbaldehydes (8a-g)
A suspension mixture consisted of appropriate 6- and 7-alkoxy-2-oxo-2H-chromene-4-carbaldehydes 6a-g (1 mmol) and thiosemicarbazides 7a/7b (1 mmol) in methanol (5 mL). Glacial acetic acid (8.26 mol%, 0.01 mL) was added to this mixture. The reaction mixture was irradiated at microwave power of 600 W for 9–13 min. After irradiating for about 5 min, the suspension mixture became clear solution. The irradiation was continued in given time. In the end of reaction, the precipitate was appeared. After reaction completion, the mixture was cooled to room temperature; the formed precipitate was filtered, washed with cold methanol and crystallized from 96% ethanol or a mixture of toluene and ethanol (1:1 by volume) to afford the titled compounds 8a-g.
2.2.1 6-Ethoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-galactopyranosyl)thiosemicarbazone (8a)
From 6a (2 mmol, 218 mg) and 7b (2.2 mmol, 421 mg). Reaction time: 9 min. Yield: 397 mg (64%) of 8a as yellow crystals. M.p.: 189–191 °C (from 96% ethanol) (Thanh and Toan, 2013), + 76.1 (c = 0.21, CHCl3). IR (KBr), ν (cm−1): 3330 and 3250 (νNH), 1748 (νC=O ester), 1730 (νC=O lactone), 1518 (νC=C arene), 1235 & 1042 (νCOC ester), 1090 (νC=S); 1H NMR (500 M Hz, DMSO‑d6), δ (ppm): 12.06 (s, 1H, NHa), 9.24 (d, J = 8.5 Hz, 1H, NHb), 8.53 (s, 1H, CH = N), 7.40 (d, J = 9.0 Hz, 1H, H-8), 7.37 (s br, 1H, H-3), 7.30 (s, 1H, H-5), 7.26 (dd, J = 9.25, 2.75 Hz, 1H, H-7), 5.98 (t, J = 9.0 Hz, 1H, H-1′), 5.38 (dd, J = 10.0, 3.5 Hz, 1H, H-3′), 5.35 (d, J = 9.0 Hz, 1H, H-2′), 5.33 (d, J = 3.5 Hz, 1H, H-4′), 4.35 (t, J = 6.5 Hz, 1H, H-5′), 4.11 (q, J = 7.0 Hz, 2H, 6-OCH2CH3), 4.05 (d, J = 6.5 Hz, 2H, H-6′a & H-6′b), 2.14–1.94 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.37 (t, J = 7.0 Hz, 3H, 6-OCH2CH3); 13C NMR (125 M Hz, DMSO‑d6), δ (ppm): 178.9 (C = S), 170.0, 169.6, 169.4 (4 × COCH3), 161.2 (C-6), 160.0 (coumarin C = O), 154.8 (CH = N), 147.8, 144.0, 136.9, 120.0, 118.1, 117.5, 112.4, 107.5 (coumarin-C), 82.1, 71.8, 70.7, 68.7, 67.5, 61.3 (d-glucopyranose-C), 20.5, 20.4, 20.3, 20.2 (4 × COCH3), 63.8, 14.5 (6-ethoxy-C); ESI-HRMS(+): C27H31N3O12S, calc. for. M + H = 622.1701 Da, M + Na = 644.1526 Da, found: m/z 622.1713 ([M + H]+), 644.1531 ([M + Na]+).
2.2.2 6-Butoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazone (8b)
From 6b (2 mmol, 246 mg) and 7a (2.2 mmol, 421 mg). Reaction time: 13 min. Yield: 402 mg (62%) of 8b as yellow crystals. M.p.: 119–121 °C (from 96% ethanol), + 81.7 (c = 0.24, CHCl3). IR (KBr), ν (cm−1): 3286 and 3251 (νNH), 1753 (νC=O ester), 1718 (νC=O lactone), 1530, 1500 (νC=C arene), 1241 & 1042 (νCOC ester), 1089 (νC=S); 1H NMR (500 M Hz, DMSO‑d6), δ (ppm): 12.04 (s, 1H, NHa), 9.27 (d, J = 8.0 Hz, 1H, NHb), 8.52 (s, 1H, CH = N), 7.36 (d, J = 9.5 Hz, 1H, H-8), 7.34 (s, 1H, H-3), 7.23 (d, J = 7.0 Hz, 1H, H-7), 7.22 (s, 1H, H-5), 5.98 (t, J = 9.25 Hz, 1H, H-1′), 5.39 (dd, J = 10.0, 3.5 Hz, 1H, H-3′), 5.35 (d, J = 10.0 Hz, 1H, H-2′), 5.33 (d, J = 2.3 Hz, 1H, H-4′), 4.35 (t, J = 6.25 Hz, 1H, H-5′), 4.06 (d, J = 6.5 Hz, 2H, H-6′a & H-6′b), 4.01 (t, J = 7.5 Hz, 2H, 6-OCH2CH2CH2CH3), 2.15–1.94 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–COCH3 ester), 1.72 (quintet, J = 7.5 Hz, 2H, 6-OCH2CH2CH2CH3), 1.44 (sextet, J = 7.5 Hz, 2H, 6-OCH2CH2CH2CH3), 0.94 (t, J = 7.5 Hz, 3H, 6-OCH2CH2CH2CH3); 13C NMR (125 M Hz, DMSO‑d6), δ (ppm): 178.9 (C = S), 170.0, 169.6, 169.4, 169.3 (COCH3), 161.3 (C-6), 160.0 (coumarin C = O), 155.0 (CH = N), 147.8, 143.9, 136.7, 119.9, 118.0, 117.5, 111.9, 107.4 (coumarin-C), 82.1, 71.8, 70.7, 68.7, 67.5, 61.3 (d-glucopyranose-C), 20.5, 20.4, 20.3 (4 × COCH3), 67.8, 30.7, 18.7, 13.6 (6-butoxy-C); ESI-HRMS(+): C29H35N3O12S, calc. for. M + H = 650.2020 Da, M + Na = 672.1839 Da, found: m/z 650.2025 ([M + H]+), 672.1834 ([M + Na]+).
2.2.3 6-Pentoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazone (8c)
From 6c (2 mmol, 260 mg) and 7a (2.2 mmol, 421 mg). Reaction time: 13 min. Yield: 497 mg (75%) of 8c as yellow crystals. M.p.: 166–168 °C (from 96% ethanol), + 80.3 (c = 0.26, CHCl3). IR (KBr), ν (cm−1): 3203 and 3263 (νNH), 1756 (νC=O ester), 1715 (νC=O lactone), 1571, 1527 (νC=C arene), 1224 & 1038 (νCOC ester), 1089 (νC=S); 1H NMR (500 M Hz, DMSO‑d6), δ (ppm): 12.04 (s, 1H, NHa), 9.28 (d, J = 8.0 Hz, 1H, NHb), 8.53 (s, 1H, CH = N), 7.37 (d, J = 9.0 Hz, 1H, H-8), 7.36 (s, 1H, H-5), 7.24 (d, J = 7.5 Hz, 1H, H-7), 5.98 (t, J = 8.75 Hz, 1H, H-1′), 5.39 (dd, J = 10.0, 4.5 Hz, 1H, H-3′), 5.35 (d, J = 9.0 Hz, 1H, H-2′), 5.33 (d, J = 4.5 Hz, H-4′), 4.35 (t, J = 6.25 Hz, 1H, H-5′), 4.05 (d, J = 6.5 Hz, 2H, H-6′a & H-6′b), 4.01 [t, J = 7.0 Hz, 2H, 6-OCH2CH2(CH2)2CH3], 2.15–1.94 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–COCH3 ester), 1.74 (quintet, J = 7.0 Hz, J = 7.0 Hz, 6-OCH2CH2CH2CH2CH3), 1.42–1.32 (m, 4H, 6-OCH2CH2CH2CH2CH3), 0.87 (t, J = 7.0 Hz, 3H, 6-OCH2CH2CH2CH2CH3); 13C NMR (125 M Hz, DMSO‑d6), δ (ppm): 178.9 (C = S), 170.0, 169.5, 169.3 (COCH3), 161.7 (C-6), 160.1 (coumarin C = O), 155.0 (CH = N), 147.8, 144.0, 136.6, 119.9, 118.0, 117.6, 112.0, 107.4 (coumarin-C), 82.2, 71.8, 70.7, 68.7, 68.0, 61.3 (d-glucopyranose-C), 20.5, 20.4, 20.3, 20.2 (COCH3), 67.5, 28.3, 27.6, 21.8, 13.8 (6-pentoxy-C); ESI-HRMS(+): C30H37N3O12S, calc. for. M + H = 664.2171 Da, M + Na = 686.1990 Da, found: m/z 664.2181 ([M + H]+), 686.1991 ([M + Na]+).
2.2.4 6-Isopentoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazone (8d)
From 6d (2 mmol, 260 mg) and 7a (2.2 mmol, 421 mg). Reaction time: 13 min. Yield: 464 mg (70%) of 8d as yellow crystals. M.p.: 179–181 °C (from 96% ethanol), + 80.5 (c = 0.26, CHCl3). IR (KBr), ν (cm−1): 3570 and 3270 (νNH), 1753 (νC=O ester), 1719 (νC=O lactone), 1600, 1530 (νC=C arene), 1241 & 1043 (νC−O ester), 1090 (νC=S); 1H NMR (DMSO‑d6) δ (ppm): 12.09 (s, 1H, NHa), 9.17 (d, 1H, J = 9.0 Hz, NHb), 8.54 (s, 1H, CH = N), 8.45 (s, 1H, H-3), 7.83–7.45 (m, 3H, H-5, H-7 & H-8), 6.03 (t, J = 9.25 Hz, 1H, H-1ʹ), 5.43 (t, J = 9.25 Hz, 1H, H-3ʹ), 5.29 (t, J = 9.25 Hz, 1H, H-2ʹ), 4.97 (t, J = 9.75 Hz, 1H, H-4ʹ), 4.22 (dd, J = 12.25, 4.75 Hz, 1H, H-6ʹa), 4.12 (ddd, J = 10.5, 4.75, 2.0 Hz, H-5ʹ), 4.08 [t, J = 6.5 Hz, 2H, 6-OCH2CH2CH(CH3)2], 3.99 (dd, J = 10.5, 2.0 Hz, 1H, H-6ʹb), 2.00–1.92 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester); 1.81 [septet, J = 6.5 Hz, 1H, 6-OCH2CH2CH(CH3)2], 1.65 [q, J = 6.5 Hz, 2H, 6-OCH2CH2CH(CH3)2], 0.95 [d, J = 6.5 Hz, 6H, 6-OCH2CH2CH(CH3)2]; 13C NMR (DMSO‑d6) δ (ppm): 179.0 (C = S), 170.0, 169.6, 169.4, 169.3 (COCH3), 161.6 (C-6), 160.1 (coumarin C = O), 155.0 (CH = N), 147.0, 144.0, 137.4, 120.0, 118.1, 117.6, 107.6 (coumarin-C), 81.7, 72.7, 72.4, 71.0, 67.8, 61.8 (d-glucopyranose-C), 20.6, 20.4, 20.3, 20.2 (COCH3), 66.6, 37.4, 24.6, 14.2 (6-isopentoxy-C); ESI-HRMS(+): C30H37N3O12S, calc. for. M + H = 664.2176 Da, M + Na = 686.1996 Da, found: m/z 664.2182 ([M + H]+), 686.1990 ([M + Na]+).
2.2.5 7-Ethoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-galactopyranosyl)thiosemicarbazone (8e)
From 6e (2 mmol, 218 mg) and 7b (2.2 mmol, 421 mg). Reaction time: 9 min. Yield: 416 mg (67%) of 8e as yellow crystals. M.p.: 137–139 °C (from 96% ethanol) (Thanh and Toan, 2013), + 76.7 (c = 0.21, CHCl3). IR (KBr) ν cm−1: 3506 and 3224 (νNH), 1751 (νC=O ester), 1702 (νC=O lactone), 1614, 1529 (νC=C arene), 1222 & 1051 (νC−O ester), 1090 (νC=S); 1H NMR (DMSO‑d6), δ (ppm): 12.22 (s, 1H, NHa), 9.11 (d, J = 9.5 Hz, 1H, NHb), 8.45 (s, 1H, CH = N), 7.83 (d, J = 8.5 Hz, 1H, H-5), 7.07 (s, 1H, H-3), 7.03 (dd, J = 7.25, 2.25 Hz, 1H, H-6), 7.02 (d, J = 2.0 Hz, 1H, H-8), 5.99 (t, J = 9.0 Hz, H-1′), 5.43 (t, J = 9.25 Hz, 1H, H-3′), 5.34 (t, J = 9.25 Hz, 1H, H-2′), 4.97 (t, J = 9.75 Hz, H-4′), 4.23 (dd, J = 12.5, 4.75 Hz, 1H, H-6′a), 4.11 (ddd, J = 9.75, 4.75, 2.25 Hz, 1H, H-5′), 4.15 (q, 2H, J = 7.0 Hz, 7-OCH2CH3), 3.99 (dd, J = 12.25, 1.75 Hz, 1H, H-6′b), 2.00–1.94 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.36 (t, 3H, J = 7.0 Hz, 7-OCH2CH3); 13C NMR (125 M Hz, DMSO‑d6), δ (ppm): 178.9 (C = S), 169.9, 169.5, 169.3, 169.2 (COCH3), 161.7 (C-7), 160.2 (coumarin C = O), 155.5 (CH = N), 144.3, 137.3, 125.8, 112.9, 110.2, 109.2, 101.6 (coumarin-C), 81.6, 72.7, 72.3, 70.8, 67.8, 61.7 (d-glucopyranose-C), 20.5, 20.3, 20.3, 20.1 (COCH3), 64.0, 14.3 (7-ethoxy-C); ESI-HRMS(+): C27H31N3O12S, calc. for. M + H = 622.1701 Da, M + Na = 644.1526 Da, found: m/z 622.1717 ([M + H]+), 644.1532 ([M + Na]+).
2.2.6 7-Isobutoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazone (8f)
From 6f (2 mmol, 246 mg) and 7a (2.2 mmol, 421 mg). Reaction time: 13 min. Yield: 519 mg (80%) of 8f as yellow crystals. M.p.: 191–193 °C (from 96% ethanol), + 82.3 (c = 0.24, CHCl3). IR (KBr) ν cm−1: 3362 and 3312 (νNH), 1732 (νC=O ester), 1706 (νC=O lactone), 1611, 1507 (νC=C arene), 1235 & 1047 (νC−O ester), 1090 (νC=S); 1H NMR (DMSO‑d6), δ (ppm): 12.22 (s, 1H, NHa), 9.11 (d, J = 9.0 Hz, 1H, NHb), 8.45 (s, 1H, CH = N), 7.84 (d, J = 8.5 Hz, 1H, H-5), 7.07–7.04 (m, 2H, H-6 & H-8), 7.04 (s, 1H, H-3), 5.99 (t, J = 9.25 Hz, 1H, H-1′), 5.43 (t, J = 9.5 Hz, 1H, H-3′), 5.35 (t, J = 9.0 Hz, 1H, H-2′), 4.97 (t, J = 9.75 Hz, 1H, H-4′), 4.22 (dd, J = 12.25, 4.75 Hz, 1H, H-6′a), 4.11 (ddd, J = 9.75, 4.75, 2.25 Hz, 1H, H-5′), 3.99 (dd, J = 12.25, 1.75 Hz, 1H, H-6′b), 3.88 [d, J = 6.5 Hz, 2H, 7-OCH2CH(CH3)2]; 2.05 [septet, J = 6.5 Hz, 1H, 7-OCH2CH(CH3)2], 2.00–1.93 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester); 0.99 [d, J = 6.5 Hz, 6H, 7-OCH2CH(CH3)2]; 13C NMR (DMSO‑d6), δ (ppm): 178.9 (C = S), 170.1, 169.6, 169.4, 169.3 (4 × COCH3), 161.9 (C-7), 160.3 (coumarin C = O), 155.5 (CH = N), 144.4, 137.4, 125.8, 113.0, 110.3, 109.2, 101.6 (coumarin-C), 81.6, 72.7, 72.3, 70.8, 67.8, 61.7 (d-glucopyranose-C), 20.5, 20.4, 20.3, 20.2 (4 × COCH3), 67.8, 27.6, 18.9 (7-isobutoxy-C); ESI-HRMS(+): C29H35N3O12S, calc. for. M + H = 650.2020 Da, M + Na = 672.1839 Da, found: m/z 650.2026 ([M + H]+), 672.1844 ([M + Na]+).
2.2.7 7-Isopentoxy-2-oxo-2H-chromene-4-carbaldehyde N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazone (8g)
From 6g (2 mmol, 260 mg) and 7a (2.2 mmol, 421 mg). Reaction time: 13 min. Yield: 490 mg (74%) of 8g as yellow crystals. M.p.: 201–203 °C (from 96% ethanol), + 81.5 (c = 0.26, CHCl3). IR (KBr) ν cm−1: 3289 and 3170 (νNH), 1744 (νC=O ester), 1614, 1522 (νC=C arene), 1224 & 1045 (νC−O ester), 1090 (νC=S); 1H NMR (DMSO‑d6), δ (ppm): 12.21 (s, 1H, NHa), 9.08 (d, J = 8.0 Hz, 1H, NHb), 8.45 (s, 1H, CH = N), 7.84 (d, J = 8.0 Hz, 1H, H-5), 7.04 (s, 1H, H-3), 7.04–7.03 (m, 2H, H-6 & H-8), 5.99 (t, J = 9.25 Hz, H-1′), 5.42 (t, J = 9.5 Hz, 1H, H-3′), 5.35 (t, J = 9.25 Hz, 1H, H-2′), 4.98 (t, J = 9.75 Hz, H-4′), 4.23 (dd, J = 12.25, 4.75 Hz, 1H, H-6′a), 4.13–4.12 (m, 1H, H-5′); 4.12–4.11 [m, 2H, 7-OCH2CH2CH(CH3)2], 3.99 [d, J = 12.25 Hz, 1H, H-6′b), 2.00–1.94 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.78 [septet, J = 6.5 Hz, 1H, 7-OCH2CH2CH(CH3)2], 1.64 [t, J = 6.5 Hz, 2H, 7-OCH2CH2CH(CH3)2], 0.94 [d, J = 6.5 Hz, 6H, 7-OCH2CH2CH(CH3)2]; 13C NMR (DMSO‑d6), δ (ppm): 179.0 (C = S), 170.0, 169.5, 169.4, 169.3 (4 × COCH3), 161.9 (C-7), 160.3 (coumarin C = O), 155.5 (CH = N), 144.4, 137.4, 125.8, 113.0, 110.3, 109.2, 101.6 (coumarin-C), 81.6, 72.7, 72.3, 70.8, 67.8, 61.7 (d-glucopyranose-C), 20.5, 20.4, 20.3, 20.2 (4 × COCH3), 66.9, 37.1, 24.6, 14.3 (7-isopentoxy-C); ESI-MS(−): C30H37N3O12S, calc. for M − H = 662 Da, found: m/z 662 ([M − H]−).
ESI-HRMS(+): C30H37N3O12S, calc. for. M + H = 664.2171 Da, M + Na = 686.1990 Da, found: m/z 664.2181 ([M + H]+), 686.1991 ([M + Na]+).
2.3 General methods for synthesis of substituted 2,3-dihydro-1,3,4-thiadiazoles (9a-g)
To a solution of appropriate thiosemicarbazones 8a-g (1 mmol) in dry dichloromethane (40 mL) acetic anhydride (10 mL, 10.6 mmol) was added. The reaction mixture was heated under reflux for 45–47 h (see Scheme 3). After reaction completion, the mixture was cooled to room temperature, the product was separated as yellow oil and the organic liquids (acetic acid, excess acetic anhydride and dichloromethane) were removed under reduced pressure. The residue was grinded with brine (2 × 10 mL) and then filtered to obtain the solid product. Purification of the obtained product was performed by column chromatography (silica gel, gradient solvent systems of n-hexane and ethyl acetate, from 2:1 to 1:1 to 1:2 to 0:1) to afford compounds 9a-g as white solids.
2.3.1 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-galactopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-6-ethoxycoumarin (9a)
From 8a (1 mmol, 621 mg). Reaction time: 45 h. Yield: 558 mg (82%) of 9a as white solids. M.p.: 112–114 °C (from 96% ethanol), + 83.2 (c 0.25, CHCl3). IR (KBr) ν cm−1: 1745 (νC=O ester), 1620 (νC=N), 1237 & 1048 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.54 (d, J = 14.5 Hz, 1H, H-5), 7.41 (dd, J = 9.0, 3.5 Hz, 1H, H-7), 7.35 (s, 1H, H-3), 7.28–7.27 (m, 1H, H-8), 6.03 (s, 1H, thiadiazoline H-2′), 5.29–5.27 (m, 2H, H-1′′ & H-3′′), 5.16–5.08 (m, 2H, H-2′′ & H-4′′), 4.30 (t, J = 5.75 Hz, 1H, H-6′′a), 4.11–4.07 (m, 1H, H-6′′b), 4.15–4.12 (m, 1H, H-5′′), 4.01–4.00 (m, 2H, 6-OCH2CH3), 2.86 (s, 3H, >N − COCH3, 3′-acetamido), 2.21 (s, 3H, thiadiazoline 2′–CH3), 1.99–1.90 (s, 12H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.35 (t, J = 6.5 Hz, 3H, 6-OCH2CH3); 13C NMR (DMSO‑d6), δ (ppm): 169.9, 169.8, 169.7, 169.4 (COCH3), 168.8, 168.6 (N − COCH3), 159.8 (C-6), 159.7 (coumarin C = O), 152.0 (>C = N), 154.9, 151.3, 147.9, 120.3, 118.2, 116.0, 110.1, 108.3 (coumarin-C), 82.3, 71.2, 70.8, 68.2, 67.9, 61.5 (d-glucopyranose-C), 61.4 (thiadiazoline C-2), 26.3, 21.5 (N − COCH3), 20.5, 20.4, 20.3, 20.2 (COCH3), 64.0, 14.4 (6-ethoxy-C); ESI-MS(−): C31H35N3O14S, M = 705.2 Da, calc. for M − H = 704.2 Da, found: m/z 704.5 [M − H]−; HRMS(+): C31H35N3O14S, calc. for M + H = 706.1912 Da, M + Na = 728.1732 Da, found: m/z 706.1921 ([M + H]+), 728.1739 ([M + Na]+).
2.3.2 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-glucopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-6-butoxycoumarin (9b)
From 8b (1 mmol, 649 mg). Reaction time: 45 h. Yield: 553 mg (80%) of 9b as white solids. M.p.: 84–86 °C (from 96% ethanol), + 82.6 (c 0.24, CHCl3). IR (KBr) ν cm−1: 1753 (νC=O ester), 1610 (νC=N), 1237 & 1037 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.61 (s, 1H, H-3), 7.41 (d, J = 9.0 Hz, 1H, H-5), 7.32 (d, J = 2.0 Hz, 1H, H-7), 7.28 (dd, J = 9.0 Hz, 1H, H-8), 6.18 (s, 1H, thiadiazoline H-2′), 6.00 (d, J = 9.0 Hz, 1H, H-1′′), 5.42 (t, J = 9.5 Hz, 1H, H-3′′), 5.34–5.33 (m, 1H, H-2′′), 4.88 (t, J = 9.5 Hz, 1H, H-4′′), 4.19 (d, J = 10.0 Hz, 1H, H-6′′a), 4.09–4.08 (m, 1H, H-5′′), 4.08–4.05 (m, 1H, H-6′′b), 4.01 (t, J = 6.5 Hz, 2H, 6-OCH2CH2CH2CH3), 2.37 (s, 3H, >N − COCH3, 3′-acetamido), 1,97–1,78 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.71 (quintet, J = 6.5 Hz, 2H, 6-OCH2CH2CH2CH3), 1.45 (sextet, J = 6,5 Hz, 2H, 6-OCH2CH2CH2CH3), 0.94 (t, J = 7.25 Hz, 3H, 6-OCH2CH2CH2CH3); 13C NMR (DMSO‑d6), δ (ppm): 170.6, 169.6, 169.5, 169.1 (COCH3), 168.8, 168.6 (N − COCH3), 159.5 (C-6), 155.1 (>C = N), 154.5, 151.1, 147.8, 120.2, 118.2, 115.7, 110.4, 108.2 (coumarin-C), 82.0, 72.6, 72.3, 68.5, 68.1, 60.9 (d-glucopyranose-C), 60.8 (thiadiazoline C-2), 22.5, 21.7 (N − COCH3), 20.3, 20.2, 20.1 (COCH3), 67.0, 30.6, 18,6, 13.7 (6-butoxy-C); ESI-HRMS(+): C33H39N3O14S, calc. for M + H = 734.2231 Da, M + Na = 756.2050 Da, found: m/z 734.2234 ([M + H]+), 756.2054 ([M + Na]+).
2.3.3 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-glucopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-6-pentoxycoumarin (9c)
From 8c (1 mmol, 663 mg). Reaction time: 47 h. Yield: 515 mg (83%) of 9c as white solids. M.p.: 94–96 °C (from 96% ethanol), + 83.5 (c 0.24, CHCl3). IR (KBr) ν cm−1: 3284 (νNH), 1752 (νC=O ester), 1615 (νC=N), 1233 & 1038 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.60 (d, J = 7.5 Hz, 1H, H-8), 7.41 (s, 1H, H-3), 7.36 (s, 1H, H-5), 7.29–7.28 (m, 1H, H-7), 6.02 (s, 1H, thiadiazoline H-2′), 5.39–5.32 (m, 1H, H-3′′), 5.25 (t, J = 9.0 Hz, 1H, H-1′′), 4.93–4.84 (m, 2H, H-2′′ & H-4″′), 4.17–4.12 (m, 1H, H-6′′a), 4.10–4.07 (m, 1H, H-5′′), 4.06–4.04 (m, 1H, H-6′′b), 4.02–3.97 [m, 2H, 6-OCH2(CH2)3CH3], 2.29 (s, 3H, >N − COCH3), 2.01–1.92 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.80–1.70 [m, 2H, 6-OCH2CH2(CH2)2CH3], 1.42–1.35 [m, 4H, 6-OCH2CH2(CH2)2CH3], 0.90 (t, J = 7,5 Hz, 3H, 6-OCH2(CH2)3CH3]; 13C NMR (DMSO‑d6), δ (ppm): 170.0, 169.9, 169.6, 168.9 (COCH3), 167.4 (N − COCH3), 159.7 (C-6), 152.2 (>C = N), 155.1, 151.4, 147.9, 129.2, 120.4, 118.2, 116.0, 110.0, 108.3 (coumarin-C), 82.2, 72.7, 72.1, 70.6, 68.4, 61.7 (d-glucopyranose-C), 63.7 (thiadiazoline C-2), 21.9, 21.7 (N − COCH3), 20.5, 20.4, 20.3 (COCH3), 63.8, 28.2, 27.7, 13.9 (6-pentoxy-C); ESI-HRMS(+): C34H41N3O14S, calc. for M + H = 748.2387 Da, M + Na = 770.2207 Da, found: m/z 748.2383 ([M + H]+), 770.2210 ([M + Na]+).
2.3.4 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-glucopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-6-isopentoxycoumarin (9d)
From 8d (1 mmol, 663 mg). Reaction time: 46 h. Yield: 599 mg (85%) of 9d as white solids. M.p.: 100–102 °C (from 96% ethanol), + 82.9 (c 0.23, CHCl3). IR (KBr) ν cm−1: 1750 (νC=O ester), 1614 (νC=N), 1238 & 1040 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.60 (t, J = 9.0 Hz, 2H, H-8), 7.42–7.36 (m, 2H, H-3 & H-5), 7.30–7.28 (m, 1H, H-7), 6.02 (s, 1H, thiadiazoline H-2′), 5.38–5.32 (m, 1H, H-1′′), 5.25–5.24 (m, 1H, H-3′′), 4.93–4.84 (m, 2H, H-2′′ & H-4′′), 4.17–4.14 (m, 1H, H-6′′), 4.12–4.08 (m, 1H, H-5′′), 4.07–4.04 (m, 1H, H-6′′b), 4.04–3.97 (m, 2H, 6-OCH2CH2CH(CH3)2], 2.29 (s, 3H, >N − COCH3, 3′-acetamido), 1.96–1.90 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester); 1.83–1.78 [m, 2H, 6-OCH2CH2CH(CH3)2], 1.66–1.62 [m, 1H, 6-OCH2CH2CH(CH3)2], 0.95–0.94 [dd, J = 4.0, 2.5 Hz, 6H, OCH2CH2CH(CH3)2]; 13C NMR (DMSO‑d6), δ (ppm): 169.9, 169.8, 169.5, 168.9 (COCH3), 167.4 (N − COCH3), 159.7 (C-6), 152.2 (>C = N), 154.9, 151.3, 147.9, 120.4, 118.2, 115.9, 109.9, 108.2 (coumarin-C), 82.1, 72.7, 72.0, 70.5, 66.8, 61.7 (d-glucopyranose-C), 63.6 (thiadiazoline C-2), 22.4, 21.5 (N − COCH3), 20.4, 20.3, 20.2, 20.1 (COCH3), 63.7, 37.3, 24.5, 14.7 (6-isopentoxy) ; ESI-HRMS(+): C34H41N3O14S, calc. for M + H = 748.2387 Da, M + Na = 770.2207 Da, found: m/z 748.2390 ([M + H]+), 770.2204 ([M + Na]+).
2.3.5 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-galactopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-7-ethoxycoumarin (9e)
From 8e (1 mmol, 621 mg). Reaction time: 46 h. Yield: 557 mg (84%) of 9e as white solids. M.p.: 120–122 °C (from 96% ethanol), + 83.7 (c 0.24, CHCl3). IR (KBr) ν cm−1: 1746 (νC=O ester), 1612 (νC=N), 1231 & 1041 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.83 (d, J = 2.0 Hz, 1H, H-5), 7.43 (s, 1H, H-3), 7.04 (t, J = 8.0 Hz, 1H,H-8), 6.99–6.98 (m, 1H, H-6), 5.85 (s, 1H, thiadiazoline H-2′), 5.37–5.36 (m, 1H, H-3′′), 5.26 (t, J = 9.0 Hz, 1H, H-1′′), 4.94–4.93 (m, H-2′′), 4.89–4.86 (m, 1H, H-4′′), 4.18–4.17 (m, 1H, H-6′′a), 4.14–4.10 (m, 1H, H-5′′), 4.06–4.04 (m, 1H, H-6′′b), 3.99–3.98 (m, 2H, 7-OCH2CH3), 2.31 (s, 3H, >N − COCH3, 3′-acetamido), 2.01–1.88 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.35 (t, J = 6.75 Hz, 3H, 7-OCH2CH3); 13C NMR (DMSO‑d6), δ (ppm): 169.9, 169.8, 169.5, 168.9, (COCH3), 167.4, 162.0 (N − COCH3), 151.9 (>C = N), 155.6 (C-7), 147.9, 125.9, 118.2, 114.0, 108.7, 106.3, 101.8 (coumarin-C), 82.0, 72.8, 72.0, 71.2, 67.9, 64.2 (d-glucopyranose-C), 61.8 (thiadiazoline C-2), 21.5, 20.5, 20.4, 20.3, 20.2 (COCH3), 64.1, 14.5 (7-ethoxy-C); ESI-MS(−): C31H35N3O14S, M = 705.2 Da, calc. for M − H = 704.2 Da, found: m/z 703.7 [M − H]−. HRMS(+): C31H35N3O14S, calc. for M + H = 706.1912 Da, M + Na = 728.1732 Da, found: m/z 706.1922 ([M + H]+), 728.1737 ([M + Na]+).
2.3.6 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-glucopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-7-isobutoxycoumarin (9f)
From 8f (1 mmol, 649 mg). Reaction time: 47 h. Yield: 622 mg (90%) of 9f as white solids. M.p.: 110–112 °C (from 96% ethanol), + 82.9 (c 0.23, CHCl3). IR (KBr) ν cm−1: 3323 (νNH), 1749 (νC=O ester), 1615 (νC=N), 1231 & 1039 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.82 (d, J = 9.0 Hz, 1H, H-5), 7.44 (s, 1H, H-3), 7.05 (t, J = 2.5 Hz, 1H, H-8), 7.00 (dd, J = 9.0, 2.5 Hz, 1H, H-6), 5.85 (s, 1H, thiadiazoline H-2′), 5.38 (t, J = 9.5 Hz, 1H, H-3′′), 5.26 (t, J = 9.0 Hz, 1H, H-1′′), 4.91–4.89 (m, 1H, H-2′′), 4.87–4.85 (m, 1H, H-4′′), 4.16–4.13 (m, 1H, H-6′′a), 4.10–4.08 (m, 1H, H-5′′), 3.99 (t, J = 10.0 Hz, 1H, H-6′′b), 3.89 [d, J = 6.5 Hz, 2H, 7-OCH2CH(CH3)2], 2.28 (s, 3H, >N − COCH3), 2.07–1.99 [m, 1H, 7-OCH2CH(CH3)2], 1.98–1.92 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 0.96 [d, 6H, J = 6.75, 7-OCH2CH(CH3)2]; 13C NMR (DMSO‑d6), δ (ppm): 170.0, 169.9, 169.5, 168.9 (COCH3), 167.4, 162.3 (N − COCH3), 155.6 (C-7), 152.0 (>C = N), 154.9, 151.9, 125.9, 112.8, 108.7, 106.3, 101.9 (coumarin-C), 82.1, 74.4, 72.7, 71.8, 70.5, 61.7 (d-glucopyranose-C), 63.6 (thiadiazoline C-2), 21.5, 20.5, 20.4, 20.3, 20.2 (COCH3), 67.9, 27.5, 18.9 (7-isobutoxy-C); ESI-HRMS(+): C33H39N3O14S, calc. for M + H = 734.2231 Da, M + Na = 756.2050 Da, found: m/z 734.2234 ([M + H]+), 756.2054 ([M + Na]+).
2.3.7 4-(3′-Acetyl-5′-(N-(2′′,3′′,4′′,6′′-tetra-O-acetyl-β-d-glucopyranosyl)acetamido-2′-methyl-2′,3′-dihydro-1′,3′,4′-thiadiazol-2′-yl)-7-isopentoxycoumarin (9g)
From 8g (1 mmol, 663 mg). Reaction time: 47 h. Yield: 622 mg (88%) of 9g as white solids. M.p.: 90–92 °C (from 96% ethanol), + 85.1 (c 0.25, CHCl3). IR (KBr) ν cm−1: 3309 (νNH), 1748 (νC=O ester), 1614 (νC=N), 1227 & 1041 (νC−O ester); 1H NMR (DMSO‑d6), δ (ppm): 7.82 (d, J = 8.75 Hz, 1H, H-5), 7.44 (s, J = 8.75 Hz, 1H, H-8), 7.07 (s, 1H, H-3), 6.99–6.97 (m, 1H, H-6), 5.86 (s, 1H, thiadiazoline H-2′), 5.38 (t, 1H, J = 9.0 Hz, H-3′′), 5.26 (t, J = 9.0 Hz, 1H,H-1′′), 4.93–4.91 (m, 1H, H-2′′), 4.89–4.86 (m, 1H, H-4′′), 4.17–4.16 (m, 1H, H-6′′a), 4.13–4.12 (m, 1H, H-5′′), 3.99 (t, J = 11.75 Hz, 1H, H-6′′b), 4.10–4.09 [m, 2H, 7-OCH2CH2CH(CH3)2], 2.28 (s, 3H, >N − COCH3, 3′-acetamido), 2.01–1.92 (s, 4 × 3H, 2′′-, 3′′-, 4′′- & 6′′–CH3CO ester), 1.79–1.77 [m, 1H, 7-OCH2CH2CH(CH3)2], 1.64 [dd, 2H, J = 6.25 Hz, 7-OCH2CH2CH(CH3)2], 0.94 [d, J = 6.5 Hz, 6H, 7-OCH2CH2CH(CH3)2]; 13C NMR (DMSO‑d6), δ (ppm): 170.0, 169.9, 169.5, 169.3, 168.9 (COCH3), 167.3, 162.2 (N − COCH3), 155.6 (C-7), 152.0 (>C = N), 151.9, 125.9, 112.8, 108.7, 106.3, 106.2, 101.8 (coumarin-C), 82.1, 72.7, 72.0, 70.5, 67.9, 61.7 (d-glucopyranose-C), 66.9 (thiadiazoline C-2), 20.5, 20.4, 20.3, 20.2 (4 × COCH3), 63.5, 37.1, 24.5, 14.7 (7-isopentoxy-C); ESI-HRMS(+): C34H41N3O14S, calc. for M + H = 748.2387 Da, M + Na = 770.2207 Da, found: m/z 748.2391 ([M + H]+), 770.2211 ([M + Na]+).
2.4 Cytotoxicity assay
Cell viability following exposure to synthetic compounds was estimated by using the MTT reduction assay. Dilution series (128, 32, 8, 2, and 0.5 µg/mL of each compound 9a-g) were prepared and used for 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Scudiero et al., 1988). Tested cancer cell lines were seeded at a density of 3 × 104 cells/well and treated with a range of concentrations in triplicate in 96-well cell culture plates, whereupon cell proliferation was assessed using a standard MTT assay. Specifically, the growth inhibitory activity of pyrimidines was determined using MTT, which correlates the cell number with the mitochondrial reduction of MTT to a blue formazan precipitate. In brief, the cells were plated in 96-well plates and allowed to attach overnight. The medium was then replaced with serum-free medium containing the test compounds and cells were incubated at 37 °C for 72 h. The medium was then replaced with fresh medium containing 1 mg/mL MTT. Following incubation at 37 °C for 2–4 h, the wells were aspirated, the dye was solubilized in DMSO and the absorbance was measured at 540 nm using a Tecan™ GENios® Microplate Reader (Conquer Scientific, USA). The viability of cells was compared with that of the control cells. The slope of the absorbance change was used for calculating the reaction rate. Negative controls were performed in the absence of enzyme and compound, and positive controls in the presence of enzyme and 100% DMSO. The percentage of residual activity was calculated as the difference in absorbance between the time 6 and 2 min, obtained by the average of two experiments carried out in triplicate. The obtained rate was related to the rate when the inhibitor was absent. IC50 values were calculated from linear extrapolations of reaction rate (as a function of the logarithm of the concentration). The IC50 values were determined with increasing concentrations of inhibitor (128, 32, 8, 2, and 0.5 µg/mL) versus % of inhibition, in triplicate in two independent experiments. The percent inhibition of viability for each concentration of the compound was calculated with reference to the control and IC50 values were calculated by using a program Graph-Pad PRISM version 5 (GraphPad Software, San Diego, CA, USA).
2.5 Molecular docking
The two-dimensional structures (.mae) of selected compounds (ligands) 9a,9c.9d,9f, and 9g and the standard drug (Sorafenib), were drawn and the structure was analyzed by using 2D sketcher and 3D builder of Maestro 12.5 according to previous method (Toan et al., 2020). The three-dimensional structures of these compounds (ligands) were generated from three-dimensional structures prepared by conformational search tool using OPLS-2005 force field for geometrically minimizing with MacroModel 12.9 followed by conformational analysis using MMFFs force field. (Toan et al., 2020). Monte Carlo Multiple Minimum (MCMM) conformational search was used with 2500 iterations and convergence threshold of 0.05 kJ/mol. Water was chosen as solvent. Truncated Newton Conjugate Gradient minimization was used with 2500 iterations and convergence threshold of 0.05 kJ/mol. Other parameters were used as default.
The X-ray crystal structures of EGFR kinase domain when complexed with tak-285 (PDB ID: 3POZ) and HER2 kinase domain complexed with tak-285 (PDB ID: 3CRD) were retrieved from the PDB Data Bank (http://www.rcsb.org) and considered as the targets for docking simulations. Coordinates of the protein–ligand complex were fixed for errors in atomic representations and optimized using Protein Preparation Wizard in Epik v. 5.3. The bond orders were assigned to residues, hydrogen atoms were added at pH 7.0 ± 2.0. The restrained minimizations were carried out using the OPLS 2005 force field with an RMSD cut-off value of 0.3 Å for heavy atom convergences. The molecular docking was accomplished and analyzed via the Glide v. 8.8 docking tool (Halgren et al., 2004). Protein was prepared by Protein Preparation Wizard used OPLS-2005 force field for structural optimization and minimization and for Receptor Grid Generation tool of Glide v.8.8. The Glide HTVS 8.8 algorithm (High-Throughput Virtual Screening Mode) was employed using a grid box volume of 10 × 10 × 10 Å. Briefly, Glide approximates a systematic search of positions, orientations and conformations of the ligand in the receptor binding site using a series of hierarchical filters. The bond orders were assigned to residues, hydrogen atoms were added at pH 7.0 ± 2.0. The restrained minimizations were carried out using the OPLS 2005 force field with an RMSD cut-off value of 0.3 Å for heavy atom convergences.
3 Results and discussion
3.1 Chemistry
Based on the significant antitumor activity of compounds K858 against human prostate and melanoma cancer cells and our interest in developing new agents for cancer therapy (Thanh et al., 2016, Toan et al., 2020), and the various literature data about the antitumor effect of 1,3,4-thiadiazolines (De Monte et al., 2015, Gomha et al., 2018, Sobhi et al., 2018, Chandra Sekhar et al., 2019, Sobhi et al., 2019, Bondock et al., 2020, Gomha et al., 2020, Rashdan et al., 2020, Swathi et al., 2020), we designed and synthesized several compounds that consists of 1,3,4-thiadiazolines (9a-g) provided with 4-coumarinyl substituents at C-5 of this ring, asymmetrical amidic chains at C-2 with sugar moiety, and at position 4 (N-4) with acetamido group (Fig. 3). The choice of different substituents on coumarin ring, which was replaced for benzene ring in above-mentioned K858, was to explore the chemical space at C-5 and highlight the biological differences after the proposed chemical modifications.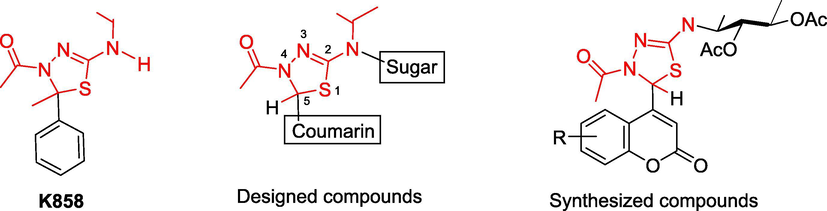
Chemical structures of the K858-based 2,3-dihydro-1,3,4-thiadiazoles for development of new anticancer agents having coumarin and sugar moieties.
3.1.1 Synthesis of N-(tetra-O-acetyl-β-d-glycopyranosyl)thiosemicarbazones of 6- and 7-alkoxy-2-oxo-2H-chromene-4-carbaldehydes
The required initial materials were 6- and 7-alkoxy-2-oxo-2H-chromene-4-carbaldehydes (i.e. 6-/7-alkoxy-4-formylcoumarins (6a-g, Scheme 1). These aldehydes of coumarin were prepared from substituted phenols, including hydroquinone (1) and resorcinol (2), through reaction with ethyl acetoacetate in the presence of 85% sulfuric acid as catalyst to obtain two hydroxycoumarins, namely, 6-hydroxy-4-methylcoumarin (3) and 7-hydroxy-4-methylcoumarin (4). These 6- and 7-hydroxyl-4-methylcoumarins were converted into corresponding 6- and 7-alkoxy-4-methylcoumarins (5a-g) by reaction with alkyl bromide or alkyl iodide, followed by subsequent selenium dioxide oxidation of 4-methyl group to yield the corresponding 6- and 7-alkoxy-oxo-2H-chromene-4-carbaldehydes (6a-g). The synthesis of these aldehydes of coumarin were performed according to our previous procedures (Ngoc Toan and Dinh Thanh, 2020).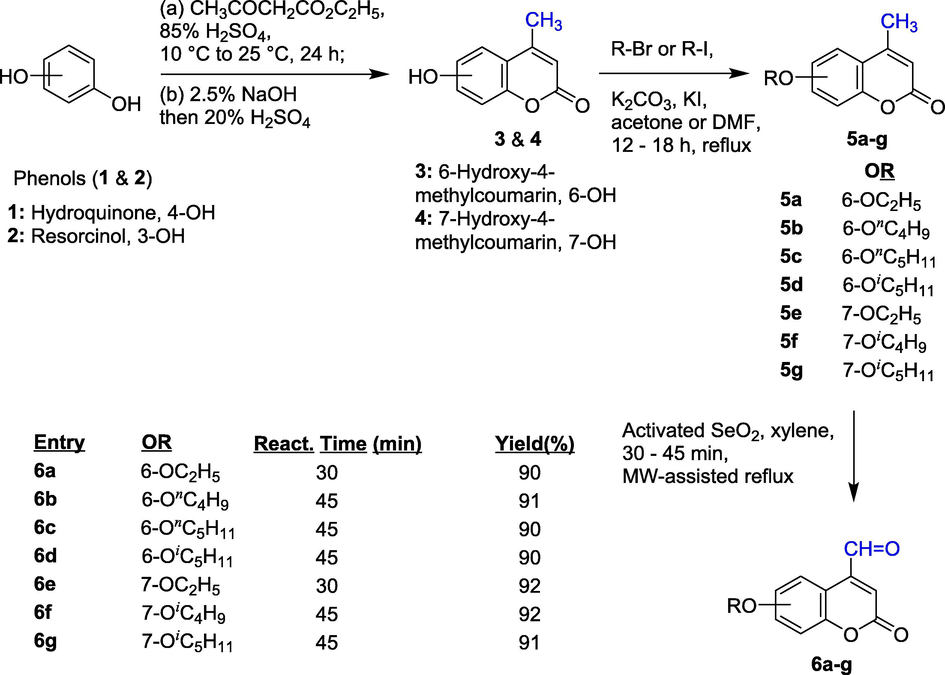
Synthetic path to substituted 6- and 7-alkoxy--oxo-2H-chromene-4-carbaldehydes (6a-g).
Reactions of thiosemicarbazides (7a/7b) of d-glucose or d-galactose with corresponding synthesized 6- and 7-alkoxy-4-oxo-2H-chromene-4-carbaldehydes (6a-g) were carried out in methanol in the presence of glacial acetic acid as catalyst. Reaction was performed using microwave-assisted heating method (Scheme 2) at power of 600 W for 9–13 min. Synthesized thiosemicarbazones 8a-g were obtained with yields of 62–80%.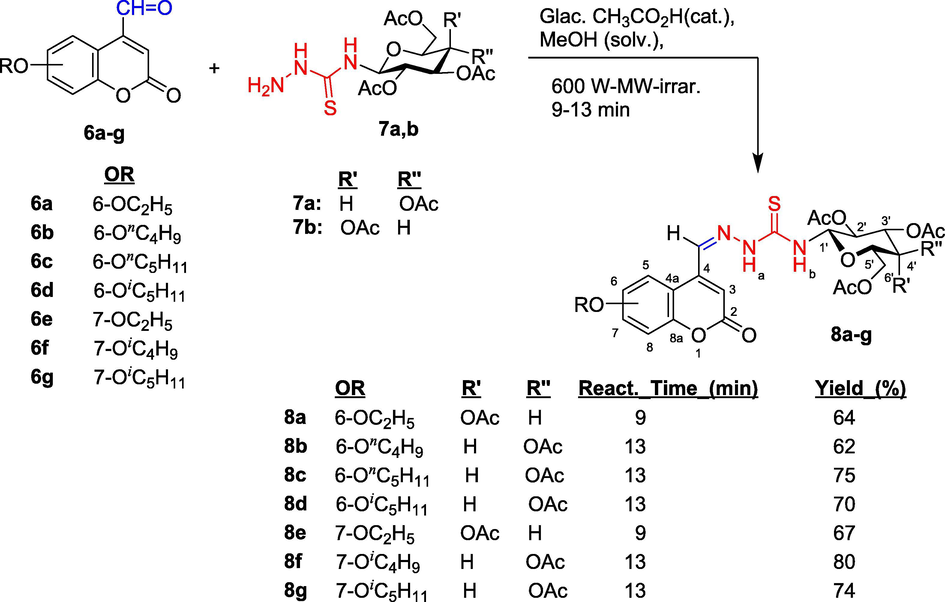
Synthetic path to N-(tetra-O-acetyl-β-d-glycopyranosyl)thiosemicarbazones (8a-g) having coumarin ring. Molecular skeletons were numbered for assigning 1H and 13C NMR spectral data.
Chemical shifts that appeared at δ = 11.15–9.28 (singlet) and 9.05–8.66 ppm (doublet) in 1H NMR spectra confirmed the presence of N−H groups in molecular structure of thiosemicarbazone 8a-g. The former chemical shift was assigned to proton Hb and the latter specified to proton Ha; this proton had coupling interaction to proton H-1 on pyranose ring with coupling constant J = 9.25–8.50 Hz. These values agreed with trans-axial H−H disposition and a β-anomeric configuration. Proton resonance signal at δ = 8.53–8.45 ppm in singlet belonged to proton in azomethine group (CH = N); carbon atom in this group had chemical shift at δ = 137.4–136.6 ppm. The appearance of six proton resonance signals in region at δ = 6.05–4.05 ppm and six carbon-13 resonance signals at δ = 82.0–61.1 ppm belonged protons and carbon atoms on pyranose rings. Proton H-3 of coumarin ring had chemical shift at δ = 8.45–7.04 ppm in singlet. Resonance of carbon atom C-1 on pyranose ring was easily recognized, at δ = 81.5 ppm, lying in the weakest field in resonance region of pyranose′s carbon. Resonance signals at δ = 181.6–180.0 ppm belonged to carbon atom in thiosemicarbazone C=S group. Carbon atoms in coumarin ring had chemical shifts in region at δ = 155.4–110.1 ppm. Acetyl groups had characteristic chemical shifts in region at δ = 21.5–21.1 ppm for methyl groups and at δ = 170.5–168.8 ppm for carbon atoms in C=O bond of acetyl groups. Carbon atom in lactone carbonyl group on coumarin ring had chemical shift at δ = 155.1–158.7 ppm.
3.1.2 Synthesis of 2,3-dihydro-1,3,4-thiadiazole−coumarin hybrid compounds
1,3,4-Thiadiazole and 4,5-dihydro-1,3,4-thiadiazole rings may be formed by 1,3-dipolar cycloaddition of nitrilimines to functionalized sulfur dipolarophiles, followed by β-elimination of simple molecule from the initially formed cycloadducts (Jain et al., 2013, Shawali, 2014, Serban et al., 2018), such as condensation reaction of hydrazones of oxamic acid thiohydrazides with an acid chlorides (Yarovenko et al., 2003), of thiosemicarbazones with acetic anhydride (Szczepankiewicz et al., 2001, Shih and Wu, 2005), by treating of acid derivatives with thiosemicarbazide and POCl3 or PPA (Noolvi et al., 2016), via regioselective [3 + 2]-cycloaddition of nitrile imines to polyfluoroalkanethioamides (Mykhaylychenko et al., 2017), etc. Solvent used in these reactions were ethanol (Abdelhamid et al., 2000; Yarovenko et al., 2003; Anhar, 2014), dichloromethane (Szczepankiewicz et al., 2001, Shih and Wu, 2005), chloroform (Mykhaylychenko et al., 2017).
In this study, the thiosemicarbazones 8a-g were converted into corresponding 2,3-dihydro-1,3,4-thiadiazoles 9a-g by reacting with excess amount of acetic anhydride (Scheme 3) in the presence of anhydrous sodium acetate as base. Based on prior literatures, we used dichloromethane as solvent for this conversion reaction. The reaction mixture was heated under reflux for 45–47 h (Scheme 3). Under these reaction conditions, NH bond on position 3ʹ in 2,3-dihydro-1,3,4-thiadiazole ring and the secondary amino NH group on position 5ʹ (Thanh et al., 2016) (9ʹa-g, not obtained) were acetylated to afford the corresponding N-acetyl derivatives. 2,3-Dihydro-1,3,4-thiadiazole − coumarin hybrid compounds were obtained with yields of 80–90%.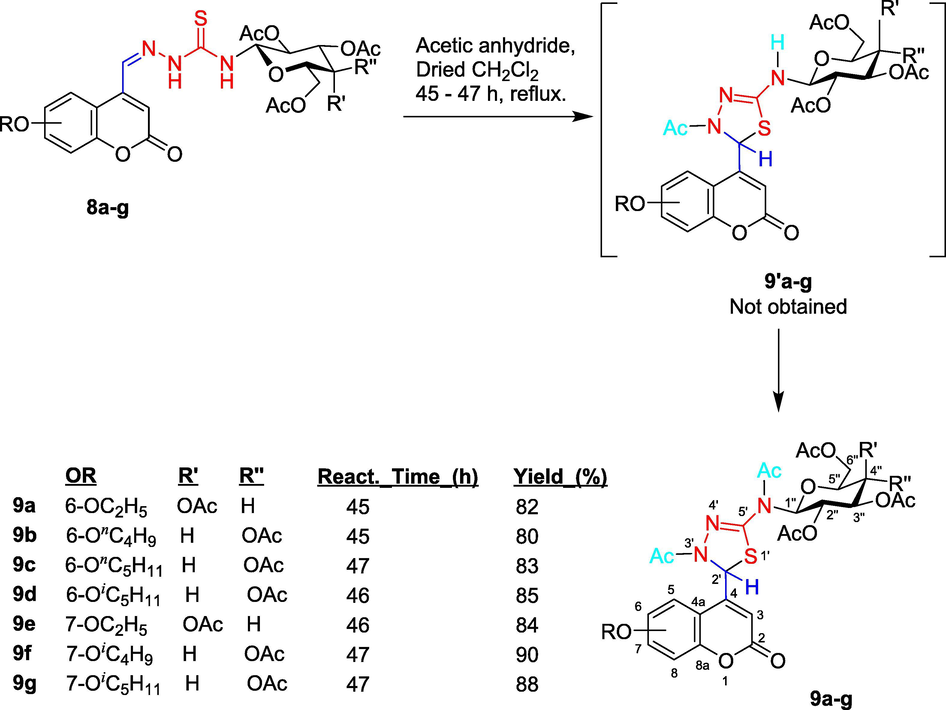
Synthetic pathway for 1,3,4-thiadiazoline − coumarin hybrid compounds 9a-g having d-glucose/d-galactose moiety: Reaction of thiosemicarbazones 8a-g with acetic anhydride. Molecular skeletons were numbered for assigning 1H and 13C NMR spectral data.
There were some changes in the magnetic resonance signals of typical chemical shifts in the 1H NMR spectra that help to confirm the structure of 1,3,4-thiadiazolines derived from thiosemicarbazones. The formation of 1,3,4-thiadiazoline ring could be recognized by the disappeared of proton resonance signal of CH=N azomethine (at δ = 8.53–8.45 ppm in singlet), simultaneously, new signal appeared at δ = 6.18–6.02 ppm that assigned to proton H-2ʹ of 1,3,4-thiazoline ring. The 1H NMR spectra of compounds 9a-g had two new N-acetyl signals (at δ = 2.89–2.28 ppm) beside other O-acetyl signals on pyranose ring when compared with the spectra of thiosemicarbazones 8a-g. Chemical shifts at δ = 181.6–180.0 ppm (belonged to C = S carbon in thiosemicarbazones 8a-g) was moved downfield in 1,3,4-thiadiazolines ring, at δ = 108.3–106.3 ppm, due to this carbon atom became imine carbon in this ring.
A proposed plausible mechanism for the formation of 2,3-dihydro-1,3-thiadiazole ring is depicted in Fig. 4, which includes two steps. Initially, acyl nucleophilic substitution SN(CO) to acetyl group of acetic anhydride by the nitrogen atom of the nucleophilic azomethine bond (due to the presence of unsplit electron pair on nitrogen atom and due to the polarity of the azomethine link towards the this atom) formed the intermediate A. The thione-thiol tautomerism of thiourea linkage in A formed two tautomers B1 and B2 having a strong nucleophilic thiol group (Uda and Kubota, 1979, Gastaca et al., 2019). Thiol tautomer B1 was stable thermodynamically and was preferred due to its conjugation stabilization by hydrazono moiety, whereas tautomer B2 was less stable. Therefore, the B1 pathway was favored over the B2 pathway, which subsequently led to the formation of product C via intramolecular nucleophilic attack of thiol group to carbon atom of azomethine bond. Deprotonation of C produced D, followed by acetylation to yield compound 9.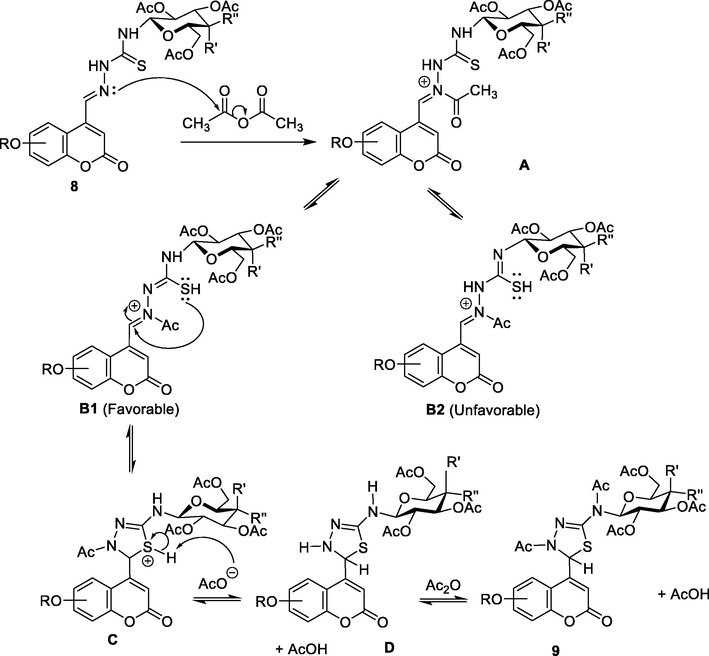
Plausible mechanism for the reaction of thiosemicarbazones 9 with acetic anhydride to form 2,3-dihydro-1,3,4-thiadiazole derivatives.
3.2 Biological activity
3.2.1 In vitro cytotoxic activity
All synthesized compounds 9a-g were screened against five human cancer cell lines, including MCF7, HepG2, HeLa, SK-Mel-2, and LU-1 by using the standard 3-(4,5-dimethylthiadiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Scudiero et al., 1988). Sorafenib, 5-fluorouracil (5-FU), and doxorubicin (DOX) were used as the reference drugs; their IC50 values were displayed in Table 1.
Entry (R group)
IC50 (μM)
MCF-7
HepG2
HeLa
SK-Mel-2
LU-1
MRC-5
9a (6-OC2H5)
11.81 ± 0.31
3.27 ± 0.38
6.52 ± 0.31
6.86 ± 0.31
10.81 ± 0.12
13.46 ± 2.19
9b (6-OnC4H9)
5.65 ± 0.32
9.43 ± 0.21
1.98 ± 0.19
8.24 ± 0.42
14.62 ± 0.18
25.55 ± 3.03
9c (6-OnC5H11)
3.28 ± 0.42
5.92 ± 0.32
3.79 ± 0.32
5.31 ± 0.32
8.26 ± 0.32
4.50 ± 0.63
9d (6-OiC5H11)
6.28 ± 0.12
5.23 ± 0.25
4.67 ± 0.32
1.82 ± 0.43
5.73 ± 0.21
3.93 ± 0.43
9e (7-OC2H5)
5.18 ± 0.23
7.25 ± 0.22
2.15 ± 0.24
5.52 ± 0.21
6.21 ± 0.11
7.07 ± 0.41
9f (7-OiC4H9)
1.92 ± 0.11
1.97 ± 0.21
13.16 ± 0.31
9.21 ± 0.32
9.52 ± 0.28
4.19 ± 0.74
9g (7-OiC5H11)
1.18 ± 0.23
1.32 ± 0.12
4.16 ± 0.42
11.25 ± 0.41
2.25 ± 0.22
7.27 ± 0.41
Sorafenib
2.74 ± 0.14
1.81 ± 0.14
4.32 ± 0.14
3.34 ± 0.21
2.84 ± 0.24
24.12 ± 1.34
5-Fluorouracil
1.81 ± 0.07
1.72 ± 0.08
1.83 ± 0.09
1.93 ± 0.08
1.98 ± 0.08
−
Doxorubicin
1.28 ± 0.08
1.21 ± 0.06
1.27 ± 0.07
1.23 ± 0.07
1.34 ± 0.07
−
With the exception of negligible inhibitory effect (IC50 > 6 μM) on tested cell lines, such as MCF-7 (9a), HepG2 (9b,9e), HeLa (9a,9f), SK-Mel-2 (9a,9b,9f,9g), and LU-1 (9a,9b,9c,9e,9f), the remaining compounds had a good (with IC50 values of 1.82–1.92 μM) to medium inhibitory effect (IC50 = 3.27–5.92 μM).
Compound 9d, which incorporates the isopentoxy group at position 6 on coumarin ring, possessed strong anticancer activity against some of the investigated cell lines, with IC50 values of 1.97 and 1.82 μM against HepG2 and SK-Mel-2 cell lines, respectively, compared to Sorafenib, 5-FU and DOX. This compound had medium activity against MCF-7 and LU-1 cancer cell lines. Compound 9f with isobutoxy group at position 7 exhibited strong cytotoxicity for MCF-7 and HeLa cell lines. Compounds 9f and 9g had the best activity against MCF-7 and HepG2 cell lines; 9d had the good inhibitory activity for SK-Mel-2. Two compounds 9b and 9e had better anticancer activity against HeLa cell lines, and 9g expressed the highest cytotoxicity against three cell lines (MCF-7, HepG2 and LU-1).
All synthesized compounds were tested against the normal human embryonic lung fibroblast cell line, MRC-5. All tested compounds showed high selectivity toward some cancer cells against MRC-5 cell with IC50 = 3.93–25.55 μM (Table 1).
3.2.1.1 Structure-activity relationship (SAR)
Compounds 9a-d with alkoxy groups on position 6 exhibited the better antiproliferative activity than compounds 9e-g with alkoxy groups on position 7 against HepG2, HeLa, and SK-Mel-2 than MCF-7 and LU-1 cells. Alkoxy groups with longer carbon chains gave the better antiproliferative activity, for examples, compound 9f (with isobutoxy group) for MCF-7 and HepG2 cells, 9d (with isopentoxy group) for HeLa and SK-Mel-2 cells. The length of carbon chains and the branching of carbon chains had certain effects on cytotoxicity for tested cell lines. Isopentoxy group at position 7 on coumarin ring (compound 9g) bring on medium-to-weak cytotoxicity for tested cancer cell lines, except for HepG2 and LU-1 cell lines (with IC50 value of 1.32 and 2.25 μM, respectively). The increase in the chain length of the alkoxy groups lead to increase cytotoxicity, with the branching at chain end, such as in compounds 9a and 9f, resulting in a sharp increase in anticancer activity. For HepG2 and SK-Mel-2 cell lines, the cytotoxicity depended on the substituted position and the length of carbon chains, with the greatest values seen in 9d and the weakest values in 9g.
3.2.2 Kinase inhibitory activity
EGFR is an EGF receptor that is involved in cell proliferation and signal transduction, and belongs to the epidermal growth factor receptor (HER) family. This family includes HER1 (erbB1, EGFR), HER2 (erbB2, NEU), HER3 (erbB3) and HER4 (erbB4). EGFR is overexpressed on the membranes of MDR tumor cells (Araújo et al., 2012, Barbuti et al., 2019). It is known that this protein is overexpressed in approximately 30% of breast cancer cases (Howe and Brown, 2011; Mitri et al., 2012), therefore, this may explain the result of the most potent cytotoxic activity being displayed against the breast cancer cell line, MCF-7, compared to the other cell lines. Some selected compounds, including 9b,9c,9e,9f, and 9g (i.e. the compounds had stronger antiproliferative activity), were tested against EGFR and HER2 tyrosine kinases (Table 2).
Entry (R group)
Kinase IC50 (μM)
MCF-7 IC50 (μM)
EGFR
HER2
9b (6-OnC4H9)
0.31
0.32
5.65 ± 0.32
9c (6-OnC5H11)
0.21
0.18
3.28 ± 0.42
9e (7-OC2H5)
0.15
0.25
5.18 ± 0.23
9f (7-OiC4H9)
> 0.50
0.15
1.92 ± 0.11
9g (7-OiC5H11)
0.43
0.27
1.18 ± 0.23
Sorafenib
0.11
0.13
2.11 ± 0.14
The compounds showed potent activities against these two kinases with HER2 displaying the most sensitivity followed by EGFR. IC50 values of two respective kinases EGFR and HER2 were 0.15–0.43 μM and 0.15–0.32 μM, respectively. Compound 9e (R = 7-ethoxy) exhibited the most potent inhibitory activity against EGFR kinase. Compound 9g (R = 7-isopentoxy) showed moderate activity compared to 9b and 9c (R = 6-butoxy and 6-pentoxy, respectively). The long carbon chains on position 7 had a significant reduction in their EGFR inhibitory activity; compounds 9b and 9c which had butoxy and pentoxy groups at the position 6 of coumarin ring displayed the better potent EGFR inhibitory activity. The substitution of isobutoxy and isopentoxy groups at position 7 of coumarin ring produced compounds 9f and 9g which had a decrease in activity against EGFR (IC50 > 50 and 0.43 μM, respectively), but shorter carbon chain in this position (ethoxy group, compound 9e) increased activity against EGFR (IC50 = 0.15 μM).
All above-selected compounds were also tested for HER2, and displayed potent inhibitory activity against HER2. Compounds 9c (R = 6-butoxy) and 9f (R = isobutoxy) had an inhibitory activity comparable to Sorafenib against HER2, with IC50 values of 0.18, 0.13 and 0.13 μM, respectively (Table 2). However, by introducing the shorter carbon chain on position 6 (9b vs. 9c) kinase inhibitory activity was reduced two folds). The shorter carbon chain on position 7 of coumarin ring (compound 9f, R = 7-isobutoxy) increased the inhibitory activity.
3.2.3 Molecular docking study
In order to understand the anticancer mechanisms via EGFR and HER2 inhibition, we performed the molecular docking study based on the interactions of selected synthesized compounds 9b,9c,9e,9f, and 9g (ligands) and the standard drug (Sorafenib) with the active sites of EGFR kinase domain complexed with tak-285 and HER2 kinase domain complexed with TAK-285 (Abdellatif et al., 2020). Selected compounds were docked at active site of enzymes EGFR and HER2 using our previously reported modelling tools and procedures (Toan et al., 2020). The standard drug, Sorafenib was also docked at these enzymes in order to compare the active interactions of these ligands together. Docking scoring, rescoring, and evaluation were performed with Glide HTVS 8.8 (Schrödinger et al., 1988). Overall Glide score (Table 3) often correlates with the measured cytotoxic activity (Tables 1 and 2). In addition, the significance of individual substituents, protein residues, and interactions among a collection of compounds can be verified via quantification.
Ligand
R
Binding free energy (kcal/mol)
EGFR (3POZ)
HER2 (3RCD)
9b
6-OnC4H9
−5.572
−4.534
9c
6-OnC5H11
−5.701
−5.346
9e
7-OC2H5
−6.687
−4.805
9f
7-OiC4H9
−3.978
−6.332
9g
7-OiC5H11
−5.110
−4.772
Sorafenib
−
−7.713
−7.120
Results of molecular docking study showed that at the binding cavity of EGFR and HER2, compounds 9b,9c,9e,9f, and 9g displayed different binding modes (Figs. 5 and 6). These differences were also reflected in the binding energy of these compounds to the active sites of these two enzymes. The modelled 9b,9c,9e,9f,9g, and Sorafenib complexes with EGFR are shown in Fig. 5 while those modelled with HER2 are shown in Fig. 6. The 2D ligand-enzyme interactions and 3D alignment positions of 9b,9c,9e,9f,9g, and Sorafenib on enzymes EGFR and HER2 were displayed in Sections 1 (Supplementary Data of online version of this article).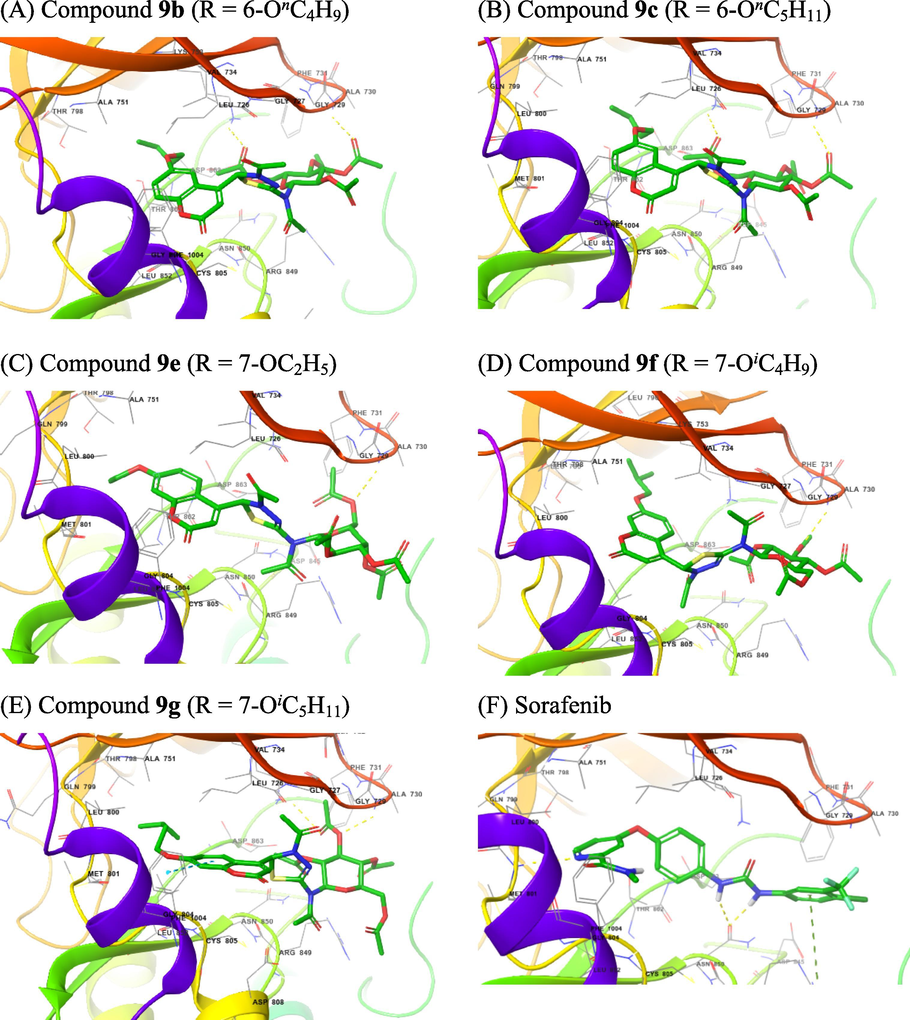
The 3D interactions with EGFR of selected compounds 9a,9c.9d,9f, and 9g (A − E) and of Sorafenib (F).
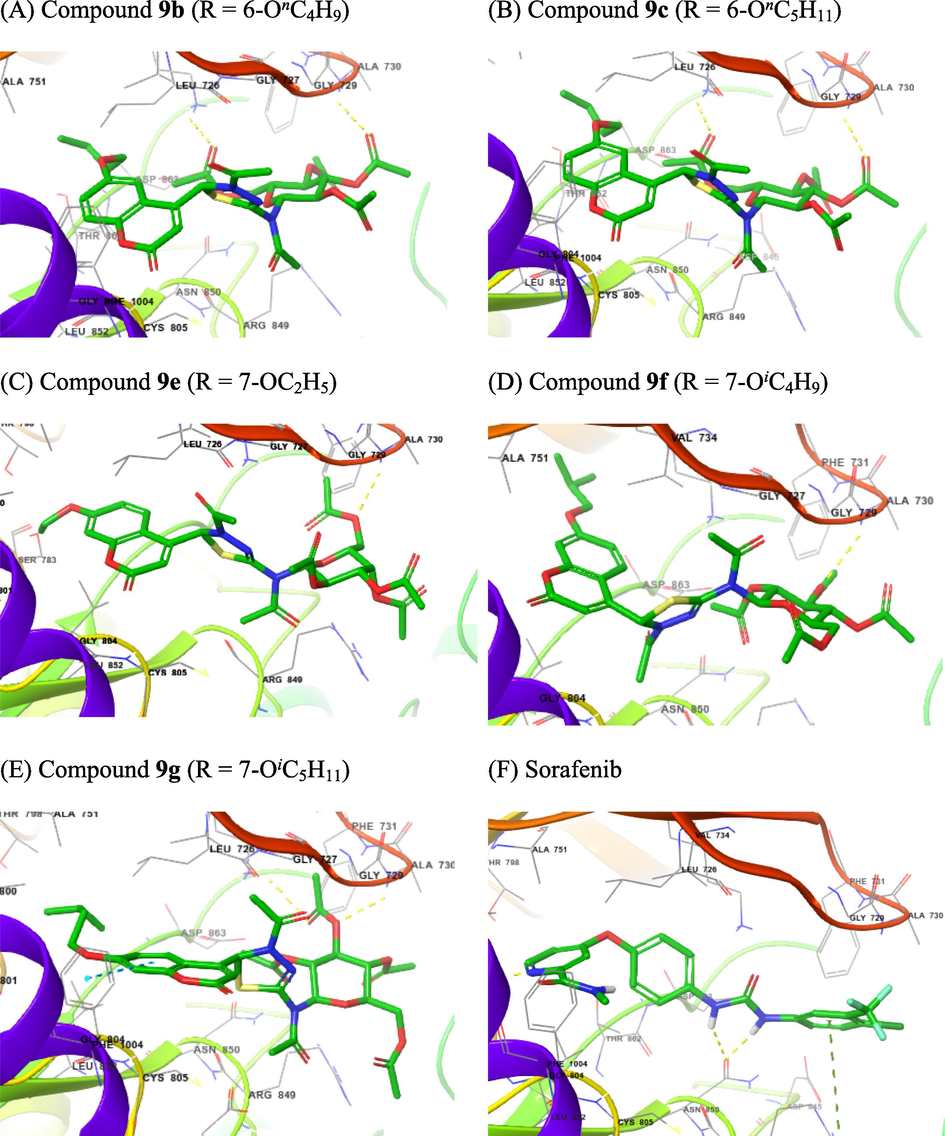
The 3D interactions with HER2 of selected compounds 9a,9c.9d,9f, and 9g (A − E) and of Sorafenib (F).
Compounds 9 consisted of two different parts in terms of polarity: two heterocyclic rings, coumarin and 1,3,4-thiadiazoline, were the non-polar part, while the sugar moiety was the polar. At the EGFR binding cavity, compound 9b formed hydrogen bonds with Lys745 and Ser720, and hydrophobic interactions at the hydrophobic cavity containing Leu792, Gly796, Cys797 and Asp800. Other hydrophobic interactions were also found to occur at the hydrophobic cavity containing Leu718 and Gly719 (Fig. 5A). Compound 9c formed hydrogen bond with Ala722 and hydrophobic interactions at the hydrophobic cavity containing Leu792, Met793, Gly796, Cys797 and Asp800. A hydrophobic interaction also occurred at the hydrophobic cavity containing Ala743 and Lys745 (Fig. 5B). Compound 9e formed hydrogen bond with Lys745, and hydrophobic interactions at the hydrophobic cavity containing Leu777, Cys775, Leu788, Ile789, Thr790and Gln791. A hydrophobic interaction also occurred at the hydrophobic cavity containing Thr854, Asp855, Phe856, and Leu858 (Fig. 5C). Compound 9f formed hydrogen bonds with Ser720 and Arg803, and hydrophobic interactions at the hydrophobic cavity containing Arg841, Asn842, Leu844, Thr790, and Thr 854. A hydrophobic interaction also occurred at the hydrophobic cavity containing Thr725, Val726, Lys745, and Ala743 (Fig. 5D). Compound 9g formed hydrogen bonds with Lys745 and Lys875, and hydrophobic interactions at the hydrophobic cavity containing Leu792, Met793, Gly796, Cys797, Leu799, Asp800 and Arg803, and at other one containing Leu718, Gly719, and Ser720. Two π-cation interactions occurred between Lys745 and coumarin ring. (Fig. 5E). The hydrogen binding interactions often occurred between acetate function and appropriate amino acid residues. Sorafenib had two hydrogen bonds with The854 and Asp800, while hydrophobic interactions occurred at the hydrophobic cavity containing Leu777, Arg778, Cys775, Leu788, and Thr790; the other hydrophobic interactions were also observed at the hydrophobic cavity containing Asp855, Phe856, Leu858, and Met766 (Fig. 5F). Based on the Glide score (Table 3), 7-isopentoxy group appeared to be a better substituent than 7-isobutoxy group (9 g vs. 9f); however, it confirms that compared to 6-butoxy and 6-pentoxy groups (compounds 9b and 9c, respectively), 7-isopentoxy group was not loosely inserted into the subpocket because of its lower hydrophobicity and larger size due to the terminal branching of this alkoxy group.
For HER2 binding cavity, we found that compound 9b formed hydrogen bonds with Ala730 and Lys753, while hydrophobic interactions occurred at the hydrophobic cavity containing Ala751, Thr798, Leu800, Met801, Gly804, Cys805, Leu807, and Asp808 (Fig. 6A). Compound 9c formed hydrogen bonds also with Ala730 and Lys753. At the hydrophobic cavity containing Leu796, Thr798, Leu800, Met801, Gly804, and Cys805, hydrophobic interactions were also observed (Fig. 6B). Compound 9e formed hydrogen bond with only Ala730. It had hydrophobic interactions at the hydrophobic cavity containing Ser783, Thr798, Gln799, Leu800, Met801, Gly804, Cys805, and Leu807. The hydrophobic cavity containing Leu726 and Gly727 also had a hydrophobic interaction (Fig. 6C). Compound 9f also formed hydrogen bonds with Ala730. Hydrophobic interactions were also observed at the hydrophobic cavity containing Leu785, Leu796, Val797, Thr798, Leu800, Met801, Gly804, and Cys805, and that containing Leu726 and Gly727 (Fig. 6D). Compound 9g also formed hydrogen bonds with Ala730 and Lys753. The hydrophobic cavity containing Ser783, Thr798, Gln799, Leu800, Met801, Gly804, Cys805, Leu807, and Asp808, and that containing Leu726, Gly727, Ser728, and Gly729 also experienced a hydrophobic interaction (Fig. 6E). The hydrogen binding interactions often occurred between acetate function and appropriate amino acid residues. Sorafenib had three hydrogen bonds with Arg849, Asn850, and Met801, while hydrophobic interactions occurred at the hydrophobic cavity containing Leu726, Gly729, Ala730, Phe731 and Val734; the other hydrophobic interactions were also observed at the hydrophobic cavity containing Leu800, Gln799, Thr798, Ala751 and Lys753 (Fig. 6F). Based on the Glide score (Table 3), 7-isobutoxy group appeared to be the best substituent at all; 6-butoxy, 7-ethoxy, and 7-isopentoxy groups were poorer substituents compared to 6-pentoxy and 7-isobutoxy groups. (compounds 9b and 9c, respectively, 7-isopentoxy group was not loosely inserted into the subpocket because of its lower hydrophobicity and larger size due to the terminal branching of this alkoxy group. The values of binding energies in Table 3 showed that compound 9c with 6-butoxy substituent and 9f with 7-isobutoxy can be inserted into the subpockets as the hydrophobic cavity possesses a sufficient fitting size.
Superimposed poses displayed in Fig. 7 showed that ligands Sorafenib (red), and the best kinase inhibitory active compounds 9c (magenta), 9f (green), and 9g (blue) on EGFR kinase domain complexed with tak-285 (top) and HER2 kinase domain complexed with TAK-285 (bottom). Ligands 9e gave better glide scores when compared with ligands 9b and 9c for the target protein EGFR, with binding score of − 6.687, −5.572 and − 5.701 kcal/mol, respectively. Ligands 9f and 9c gave better glide scores than 9e and 9g on the target protein HER2.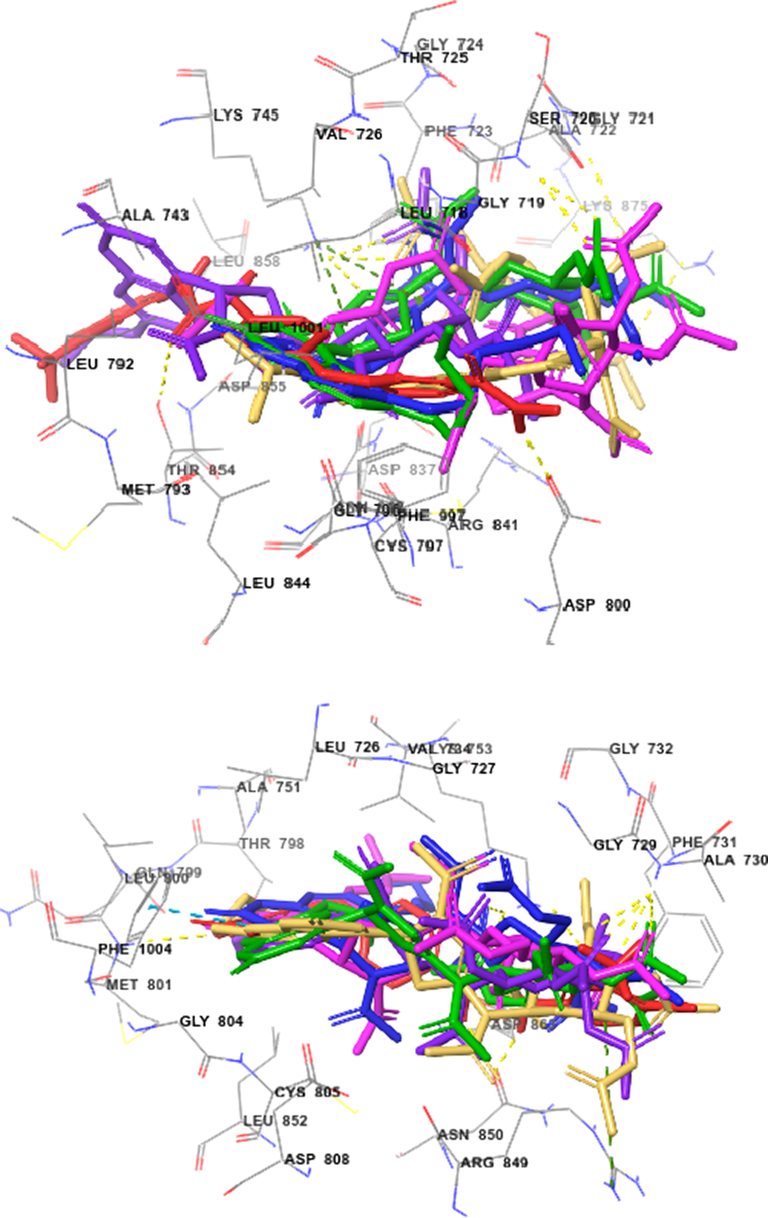
Docked (superimposed) poses showed Sorafenib (red) and the best kinase inhibitory active compounds 9b (green), 9c (blue), 9e (violet), 9f (magenta), and 9g (orange) on EGFR (top) and HER2 (bottom).
The important intermolecular protein–ligand interactions (Section 1.1 in Supplementary Data of online version of this article) of ligands 9b,9c,9e,9f,9g and Sorafenib on EGFR kinase showed that the highest active compound 9e were similar to docking position of this drug. On HER2 kinase, compounds 9c and 9f had similar docking positions when compared to Sorafenib.
4 Conclusion
Some 2,3-dihydro-1,3,4-thiadiazole − coumarin hybrid compounds were synthesized with yields of 80–90% by reaction of 6- and 7-alkoxy-4-formylcoumarin N-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)thiosemicarbazones with excess acetic anhydride amount. The target hybrid compounds were evaluated in vitro for their cytotoxic activity against MCF-7, HepG2, HeLa, SK-Mel-2, and LU-1 human cancer cell lines. Obtained results showed that almost all tested compounds displayed significant inhibitory activities in vitro against these cancer cell lines. By evaluating the compounds’ selectivity for cancer cells against the normal cell line, MRC-5, some target compounds were found to display potent cytotoxic activity with IC50 values of 2.93–25.55 μM. The tested compounds were also found to be potent inhibitors of EGFR and HER2 receptor tyrosine kinases (IC50 values of 0.22–0.50 and 0.13–0.35 μM, respectively). The structural features required for the inhibitory activity of EGFR and HER2 were identified by molecular modelling studies which demonstrated the importance of the coumarin and 1,3,4-thiadiazole rings for kinase inhibitory activity. The anticancer mechanisms were studied via EGFR, HER2 inhibition and molecular docking. Molecular docking also showed that the hydrogen binding interactions often occurred between acetate function and appropriate amino acid residues played a key role in enhancing its potency against both enzymes.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Phosphorus Sulfur Silicon Relat. Elem.. 2000;156:35-52.
- [CrossRef]
- Bioorg. Chem... 2020;101:103976
- [CrossRef]
- Akhdar, H., Legendre, C., Aninat, C., and More, F., 2012. Anticancer Drug Metabolism: Chemotherapy Resistance and New Therapeutic Approaches. In Paxton, J. (Ed.), Topics on Drug Metabolism, pp. 137-170. Doi: 10.5772/30015.
- Bioorg. Chem... 2020;103:104163
- [CrossRef]
- Anti-Cancer Agents Med. Chem... 2016;16:1301-1314.
- [CrossRef]
- Molecules.. 2019;24:3738.
- [CrossRef]
- Am. J. Clin. Oncol.. 2012;35
- Chapter 1 - EGFR and HER2 Inhibitors as Sensitizing Agents for Cancer Chemotherapy. In: Chen Z.-.S., Yang D.-.H., eds. Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy. Academic Press; 2019. p. :1-11.
- [CrossRef] [Google Scholar]
- Molecules.. 2020;25:3948.
- J. Sulfur Chem... 2020;1–39
- [CrossRef]
- Molecules.. 2019;24:3988.
- [CrossRef]
- Russ. J. Gen. Chem... 2019;89:770-779.
- [CrossRef]
- Invest. New Drugs.. 2016;34:399-406.
- [CrossRef]
- Eur. J. Med. Chem.. 2015;105:245-262.
- [CrossRef]
- Bioorg. Chem... 2020;101:104023
- [CrossRef]
- Molecules.. 2017;22:170.
- [CrossRef]
- Int. J. Anal. Mass Spectrom. Cromatogr.. 2019;7:19-34.
- [CrossRef]
- RSC Adv... 2015;5:37887-37895.
- [CrossRef]
- Drug Des. Dev. Ther.. 2018;12:1511-1523.
- [CrossRef]
- Polycycl. Aromat. Compd.. 2020;1–13
- [CrossRef]
- Eur. J. Med. Chem... 2015;92:156-177.
- [CrossRef]
- J. Med. Chem... 2004;47:1750-1759.
- [CrossRef]
- Cancer Prev. Res.. 2011;4:1149-1157.
- [CrossRef]
- Chem. Biol. Drug Des... 2013;81:557-576.
- [CrossRef]
- Molecules.. 2020;25:4309.
- [CrossRef]
- Molecules.. 2018;23:3129.
- [CrossRef]
- Heliyon.. 2020;6:e04514
- [CrossRef]
- Chem. Pharm. Bull... 2019;67:888-895.
- [CrossRef]
- Nucleosides, Nucleotides Nucleic Acids. 2020;39:1036-1056.
- [CrossRef]
- Bioorg. Med. Chem. Lett... 2018;28:2930-2938.
- [CrossRef]
- Cancer.. 2012;118:3556-3564.
- [CrossRef]
- ACS Omega.. 2020;5:14586-14596.
- [CrossRef]
- Eur. J. Med. Chem... 2019;171:129-168.
- [CrossRef]
- Int. J. Mol. Sci... 2018;19:3533.
- [CrossRef]
- Toxicol. In Vitro.. 2020;63:104745
- [CrossRef]
- J. Saudi Chem. Soc.. 2016;20:S426-S432.
- [CrossRef]
- Chem. Heterocycl. Compd... 2017;53:1268-1276.
- [CrossRef]
- Synth. Commun... 2020;1–13
- [CrossRef]
- Arab. J. Chem.. 2016;9:S1283-S1289.
- [CrossRef]
- Cancer Lett... 2007;252:270-279.
- [CrossRef]
- Molecules.. 2020;25:4997.
- [CrossRef]
- Mitri, Z., Constantine, T., and O'regan, R., 2012. Chemother. Res. Pract. 2012, 743193. https://doi.org/10.1155/2012/743193.
- Schrödinger Release 2020-3. Schrödinger, Inc., New York, NY (USA), (2020).
- Cancer Res... 1988;48:4827-4833.
- Drug Des. Dev. Ther.. 2018;12:1545-1566.
- [CrossRef]
- J. Adv. Res... 2014;5:1-17.
- [CrossRef]
- Tetrahedron.. 2005;61:10917-10925.
- [CrossRef]
- Sci. Rep... 2018;8:3190.
- [CrossRef]
- Mini-Rev Med. Chem.. 2018;18:1670-1682.
- [CrossRef]
- Mini-Rev Med. Chem.. 2019;19:437-447.
- [CrossRef]
- Sci. Rep... 2019;9:6387.
- [CrossRef]
- Molecules.. 2018;23:250.
- [CrossRef]
- Lett. Drug Des. Discovery.. 2020;17:434-444.
- [CrossRef]
- J. Med. Chem... 2001;44:4416-4430.
- [CrossRef]
- Pharmacol. Rep... 2020;72:1079-1100.
- [CrossRef]
- Eur. J. Med. Chem... 2016;123:532-543.
- [CrossRef]
- Asian J. Chem... 2013;25:6609-6611.
- [CrossRef]
- Arab. J. Chem.. 2020;13:7860-7874.
- [CrossRef]
- J. Heterocycl. Chem... 1979;16:1273-1275.
- [CrossRef]
- Biomed Res. Int... 2013;2013:963248
- [CrossRef]
- Bioorg. Chem... 2020;95:103530
- [CrossRef]
- Int. J. Pharm. Pharm. Sci.. 2014;7
- Chem. Heterocycl. Compd... 2003;39:1633-1639.
- [CrossRef]
- Bioorg. Chem... 2020;102:104081.
- [CrossRef]
- Mol. Cancer Ther... 2015;14:2463.
- [CrossRef]
- Arab. J. Chem.. 2014;7:525-552.
- [CrossRef]
- Mol. Nutr. Food Res... 2018;62:1701073.
- [CrossRef]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103053.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




