

Translate this page into:
Multifunctionalization of cyanuric chloride for the stepwise synthesis of potential multimodal imaging chemical entities
⁎Corresponding authors at: Department of Chemistry, Faculty of Science and Technology, University of Coimbra, Coimbra, Portugal. smpinto@qui.uc.pt (Sara M.A. Pinto), mmpereira@qui.uc.pt (Mariette M. Pereira)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
We report a synthetic strategy to combine different moieties in a single structure using cyanuric chloride (2,4,6-trichlorotriazine) as a starting platform for preparing potential bioimaging agents. This reacted with macrocycles of the porphyrin family and DOTA type metal chelators through mono-, di- and tri- substitution of its chlorine atoms by appropriate nucleophiles, controlling the stepwise by temperature, to produce a system that opens the potential for biomedicinal applications. Porphyrins were chosen as one of the sensing arms, based on their rich structural chemistry, and excellent photophysical properties, while DO3A was used since it can form a versatile aminopropionate functionalized metal ion chelator. All new compounds were fully characterized, both spectroscopically and photophysically.
Keywords
Porphyrins
Aza-chelates
Sulfonation
Fluorescence
Biomedicinal applications
Cyanuric chloride

1 Introduction
Cancer is one of the leading causes of death worldwide and it is estimated that by 2030, the number of new cancer cases may increase by about 70%. It is accepted by both medical and scientific communities that new drugs for cancer therapy and more efficient imaging diagnostic methods to detect tumors at an early stage (Singh et al., 2015; Ethirajan et al., 2011; Calvete et al., 2014) will play a major role in solving the problem of cancer. Porphyrins have potential in both these areas. We can highlight, for example, the use of tetrapyrrolic macrocycles in photodynamic therapy (Singh et al., 2015; Ethirajan et al., 2011; Arnaut, 2011; Pereira et al., 2006; Dabrowski et al., 2012), and their valuable properties for imaging. In this regard, the development of new contrast agents for medical imaging becomes imperative; currently the most widely used medical imaging processes at the clinical level are MRI (magnetic resonance imaging) (Calvete et al., 2017c; Calvete et al., 2014; Kueny-Stotz et al., 2012), SPECT (single photon emission tomography) (Calvete et al., 2017b; Srivatsan et al., 2015) and PET (positron emission tomography) (Rangacharyulu and Roh, 2015; Mewis and Archibald, 2010; Gambhir, 2002; Simoes et al., 2015).
Recently, promising techniques, such as FI (fluorescence imaging) (Josefsen and Boyle, 2012; Wagnieres et al., 1998; Lobo et al., 2016) and PAI (photoacoustic imaging) (Wu et al., 2014; Pan et al., 2013), have also gained considerable attention. Each of these techniques possesses unique strengths and weaknesses in terms of characteristics, such as spatial resolution, radiation penetration depth, contrast, imaging acquisition time, and equipment/running costs. The design/development of new chemical entities that can potentially act as multimodal contrast agents combining the advantages of each technique has gained much attention over last decade, and has the exciting prospect of overcoming the specific limitations of each technique, improving diagnosis and allowing, in best cases, the detection and characterization of small tumors (Dong et al., 2017; Luo et al., 2014).
As a strategy towards stepwise multi-functionalization to produce a multimodal chemical entity, we have turned our attention to cyanuric chloride (2,4,6-trichloro-[1,3,5]-triazine) as a useful platform that can be used to link imaging units through mono-, di- and tri- substitution of its chlorine atoms by appropriate nucleophiles. The stepwise substitution involved can be controlled by temperature, since the reactivity decreases with increasing number of substituents linked to the platform (Blotny, 2006; Puthiaraj et al., 2016; Luechai et al., 2012; Xiao et al., 2010). Given the excellent properties of porphyrins, including their straightforward structural modification, either by introduction of different functional groups or complexation with several metal ions (Ethirajan et al., 2011; Calvete et al., 2017c; Srivatsan et al., 2015; Josefsen and Boyle, 2012; Pinto et al., 2016; Henriques et al., 2016; Henriques et al., 2012; Simoes et al., 2012; Pinto et al., 2012), their photophysical properties (for example, high fluorescence quantum yields) (Arnaut, 2011; Marques et al., 2012), preferential uptake in tumors and low in vivo toxicity (Dabrowski et al., 2011), these have been chosen as one of the arms of this multimodal sensor. Another moiety is based on the versatility of chelates of the DOTA type (DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid), DO3A-N-alpha-aminopropionate (Ferreira et al., 2009), a versatile bifunctional chelator that may form low toxicity, highly thermodynamically and kinetically stable Gd3+ complexes with optimized relaxivity to generate MRI contrast (Ferreira et al., 2013), or complexes with different metal ions for other imaging applications. Herein, we envisage a synthetic strategy to combine, in a single structure, multiple chemical entities which are potentially able as reporters for different imaging techniques. The new compounds were fully characterized by spectroscopic and photophysical methods.
2 Materials and methods
2.1 General
Commercially available reagents were from Aldrich and Fluorochem. All solvents were pre-dried according to standard laboratory techniques.
UV–visible absorption spectra were recorded on a Hitachi U-2010 spectrometer using quartz cells. The molar absorption coefficients were determined using DMSO and THF as solvents. Steady-state fluorescence spectra were obtained using a Horiba-Jobin-Yvon SPEX Fluorolog 3-22 instrument (0.5 nm slits). The fluorescence quantum yields (ΦF) of the systems were measured under conditions of matched absorbance (0.01) at the excitation wavelength, and were obtained from the ratio of the integrated fluorescence bands of the sample and reference, expressed as a function of energy units, and multiplied by the fluorescence quantum yield of the reference, after correction for the difference in the refractive indexes between the sample and the reference solutions. A solution of 5,10,15,20-tetraphenylporphyrin (TPP) in toluene was used as standard (fluorescence quantum yield = 0.11) (Murov et al., 1993). 1H NMR spectra were recorded on a 400 MHz Bruker Avance III NMR spectrometer. Proton chemical shifts are given in parts per million (ppm) relative to tetramethylsilane at δ 0.00 ppm. Mass spectra (ESI-FIA-TOF) were acquired using a Bruker model Micro-TOF (University Santiago de Compostela, Spain). Elemental analysis was obtained in a FISONS model EA 1108 analyzer (University Santiago de Compostela, Spain).
2.2 Synthesis
5-(4-Hydroxyphenyl)-10,15,20-triphenylporphyrin 1 was prepared following our previously described methodologies (Calvete et al., 2017a; Silva et al., 2014) and its properties are in agreement with literature data (Calvete et al., 2017a; Tome et al., 2005; Neves et al., 2012). Protected DO3A-N-α-aminopropionate 4 was prepared according to the literature, and its characterization is in agreement with previous reports (Ferreira et al., 2009).
2.2.1 Synthesis of compound 5
Using optimized conditions, a mixture of cyanuric chloride (0.05 g, 0.27 mmol) and diisopropylethylamine (DIPEA) (0.3 mL, 3.6 mmol) was dissolved in 7 mL of tetrahydrofuran (THF). L-leucine methyl ester (0.05 g, 0.27 mmol) was added at -10 °C and the reaction was left for 30 min with stirring. Then, 5-(4-hydroxyphenyl)-10,15,20-triphenylporphyrin (0.170 g, 0.27 mmol) and DIPEA (0.3 mL, 3.6 mmol) were added, and the reaction was left to react 24 h at 30 °C. Finally, the protected DO3A-N-α-aminopropionate 4 (0.132 g, 0.27 mmol) and more DIPEA (0.3 mL, 3.6 mmol) were added and the reaction was left for 72 h at 60 °C. The control of each stage of the sequential reaction was performed using TLC. After solvent evaporation the reaction crude was purified using silica gel column chromatography, starting with CH2Cl2 as eluent to remove residual byproducts and CH2Cl2:ethanol (1:1) to obtain the pure compound. Compound 5 (Rf in CH2Cl2:ethanol (1:1) = 0.25) was obtained in 56% yield.
MS (ESI-FIA-TOF) m/z calcd for [M + Li]+: C78H87LiN13O11 1388.6808; found 1388.6732. 1H NMR (400 MHz) (CDCl3), δ, ppm: 8.83–8.81 (broad signal, 8H, β-H), 8.16 (broad signal, 8H, Ar-H), 7.71–7.52 (multiplet, 11H, Ar-H), 4.30–4.12 (2 broad singlets, 6H, –OCH3), 3.70–2.17 (multiplets, 32H, –CH2CH3, –NCH2CO2–, –NCH2CHNH–, –HNCH-aminoacid, –N(CH2)2N–), 1.50–1.46 (multiplets, 12H, CH3CHCH3-aminoacid, –CH-CH2-CH-aminoacid, –OCH2CH3), 1.41–1.40 (broad signal, CH3CHCH3-aminoacid). UV–vis (THF): λmax, nm (log ε) 419 (5.43), 515 (4.26), 550 (4.15), 589 (3.66), 646 (3.62). Elemental Anal. calcd. for C78H87N13O11·2H2O: C, 66.04; H, 6.47; N, 12.84; Found C, 65.86; H, 6.58; N, 12.73.
2.2.1.1 Synthesis of compound 5a
Compound (5) (0.070 g, 0.051 mmol) was dissolved in a mixture of ethanol (3 mL) and hydrochloric acid (6 M, 3 mL) and stirred overnight at room temperature. After the reaction was complete, the solvent was evaporated, the obtained compound was redissolved in water and the solvent evaporated several times. Then, the obtained solid was dissolved in water (10 mL), the solution was adjusted to pH 10–11 by addition of small portions of Dowex-1X2-100 OH– resin and was left with stirring for 4 h at room temperature. The resin was transferred into a column, washed with water and eluted with hydrochloric acid (0.1 M). The reddish-brown fraction was collected and the solvent was removed under reduced pressure (temperature < 40 °C) to give compound 5a (0.031 g, 0.024 mmol, 47%).
MS (ESI- TOF-INFUSION) m/z calcd for [M + Na + H]+: C70H72N13NaO11 1293.5372; found 1293.6942.
2.2.2 Synthesis of 5-(4-hydroxy-3-sulfonylphenyl)-10,15,20-(4-sulfonyltriphenyl)porphyrin 6
5-(4-Hydroxyphenyl)-10,15,20-triphenylporphyrin (0.200 g, 0.32 mmol) and chlorosulfonic acid (12 mL, 150 mmol) were stirred at 100 °C for 2 h. After this period, chloroform (400 mL) was added to the solution and a continuous water extraction was carried out, under constant stirring, until neutralization of the solution. The organic phase was washed with NaHCO3 and dried with Na2SO3. After solvent evaporation, deionized water was added (150 mL) and the mixture was left to hydrolyze under stirring, at 100 °C during 12 h. After water evaporation, the compound 5-(4-hydroxy-3-sulfonylphenyl)-10,15,20-(4-sulfonyltriphenyl)porphyrin (6) was obtained in 90% yield.
HRMS (ESI-FIA-TOF) m/z calcd for [M + Na]+: C44H30NaN4O13S4 973.0584; found 973.0575. UV–vis (DMSO): λmax, nm (log ε) 422 (4.36), 517 (3.41), 555 (3.14), 584 (2.94), 635 (1.94). 1H NMR (400 MHz) (DMSO), δ, ppm: 8.92–8.84 (m, 8H, β-H), 8.21–8.18 (m, 8H, ortho-Ph-H), 8.06–804 (m, 7H, meta-Ph-H). Elemental Anal. calcd. for C44H30NaN4O13S4·3H2O: C, 52.58; H, 3.61; N, 5.57; S, 12.76; Found C, 52.00; H, 3.98; N, 5.22; S, 12.91.
2.2.3 Synthesis of compound 8
Using optimized conditions, cyanuric chloride (0.014 g, 0.074 mmol) and DIPEA (0.2 mL, 2.4 mmol) were dissolved in DMF. A solution of 5-(4-hydroxy-3-sulfonylphenyl)-10,15,20-(4-sulfonyltriphenyl)porphyrin 6 (0.145 g, 0.150 mmol) in DMF was then added and the reaction mixture was left for 12 h at 25 °C with stirring. After confirming the disappearance of the starting material, protected DO3A-N-α-aminopropionate 4 (0.078 g, 0.16 mmol) and DIPEA (0.1 mL, 1.22 mmol) were added and the reaction was left for 72 h at 60 °C. Compound 8 was obtained after precipitation and washing with acetone, in 42% yield.
MS (ESI-FIA-TOF) m/z calcd for [M]+: C115H102N16O34S8 2507.4544; found 2507.4445. 1H NMR (400 MHz) (DMSO‑d6), δ, ppm: 8.90–8.85 (broad signal, 16H, β-H), 8.21–8.01 (broad signal, 30H, Ar-H), 4.19–4.06 (multiplets, 10H, –OCH2CH3, –OCH3, –NCH2CHNH–), 3.64–2.61 (multiplets, 24H, –NCH2CO2-, -N(CH2)2N–, –NCH2CHNH–), 1.17 (broad triplet, 9H, –OCH2CH3). UV–vis (DMSO): λmax, nm (log ε) 416 (5.06), 514 (3.91), 547 (3.79), 592 (3.29), 646 (3.07). Elemental Anal. calcd. for C115H102N16O34S8·4H2O: C, 53.52; H, 4.30; N, 8.68; S, 9.94; Found C, 53.86; H, 4.28; N, 8.63; S, 9.92.
3 Results and discussion
3.1 Synthesis
The synthetic pathways to obtain the new multimodal chelator 5 are presented in Scheme 1. First we optimized the synthesis of non-symmetric 5-(4-hydroxyphenyl)-10,15,20-triphenylporphyrin (1), using our recently developed NaY/nitrobenzene synthetic methodology (Silva et al., 2014; Calvete et al., 2017a; Henriques et al., 2015; Henriques et al., 2014); the product was obtained in 16% yield. It should be noted that this method gave a twofold increase in yield compared with other standard one-pot procedures (Adler et al., 1964; Gonsalves et al., 1991). Then, the synthesis of the protected DO3A-N-α-aminopropionate 4 (Scheme 1b), was carried out using a methodology previously described by some of us (Ferreira et al., 2009). A Michael addition of Boc2-Ser-OMe to cyclen was performed and, after isolation and purification, alkylation with ethyl bromoacetate was carried out to give the fully protected DO3A-N-α-aminopropionate derivative, in accordance with literature (Ferreira et al., 2009) with 75% yield. To obtain 4, a selective deprotection of the amine group present at the aminopropionate arm was performed, using a solution of 10% trifluoroacetic acid in dichloromethane, giving 4 in 90% yield. Both compounds 1 (Calvete et al., 2017a) and 4 (Ferreira et al., 2009) were confirmed by 1H NMR and mass spectroscopy, and the obtained data are in agreement with the literature.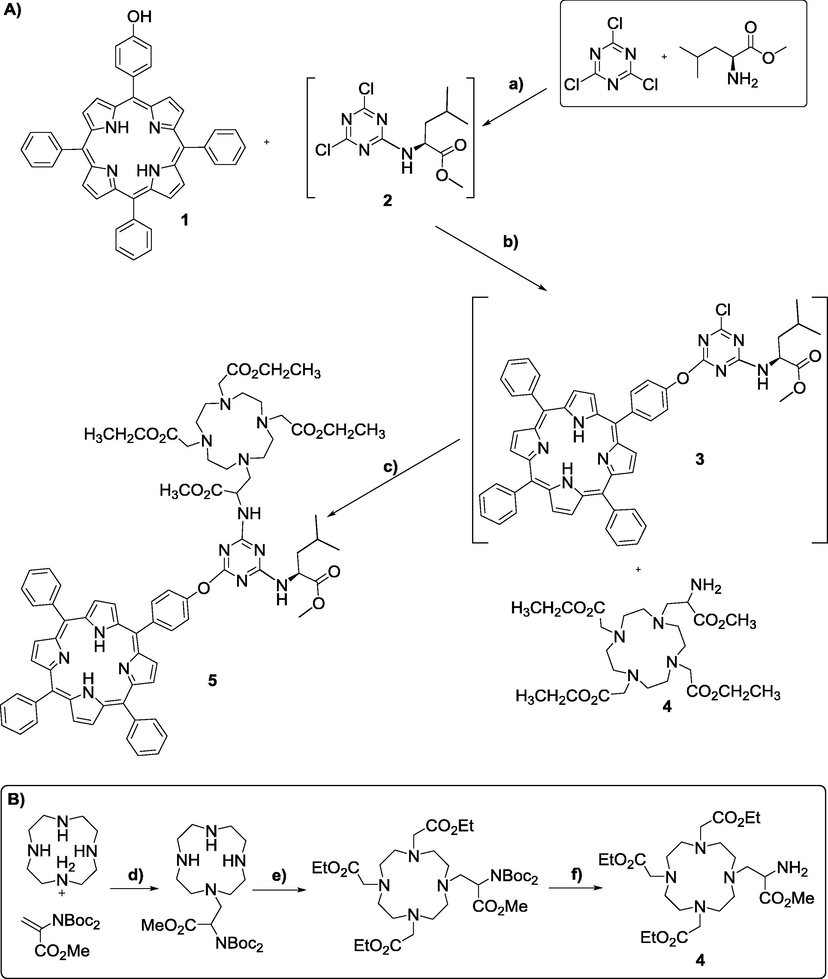
(A) Reaction pathway to obtain compound 5. Reagents and conditions: (a) −10 °C to −5 °C, 30 min, DIPEA, THF; (b) 30 °C, 24 h, DIPEA, THF; (c) 60 °C, 72 h, DIPEA, THF. (B) Reaction pathway to obtain compound 4. Reagents and conditions: (a) RT, 4 h, K2CO3, CH3CN; (b) RT, 4 h, ethylbromoacetate, K2CO3, CH3CN; (c) RT, overnight, TFA (10%), CH2Cl2.
Next, we proceeded to the preparation of compound 5. We started by reacting 1 equiv of commercially available L-leucine methyl ester (to induce more biocompatibility) (Dong et al., 1998; Mikhalenko et al., 2004; Haywood-Small et al., 2006; Drechsler et al., 1999) with 1 equiv of cyanuric chloride in THF at −10 °C in the presence of diisopropylethylamine (DIPEA). The reaction was monitored by TLC and, after 30 min, complete disappearance of cyanuric chloride was observed, concomitantly with the formation of the monoadduct derivative 2. Then, without purification of 2, 1 equiv of the previously synthesized porphyrin 1 was added, together with more DIPEA and left to react at 30 °C. Evolution of the reaction was monitored by TLC and compound 2 disappeared, being converted into 3 after 24 h. Finally, upon addition of 1 equiv of 4 and more DIPEA, the reaction was left at 60 °C for 72 h. Isolation and purification by silica gel column chromatography using CH2Cl2 to remove byproducts, followed by CH2Cl2:ethanol (1:1) as eluents, gave product 5, in 56% yield (Scheme 1).
In addition, to obtain water soluble structures, compound 5 was hydrolyzed using acid hydrolysis, with HCl 6 M in ethanol, in order to deprotect the carboxylic groups. However, to our dismay, the solubility of the supposed structure 5a (Fig. 1) was also highly pH dependent, being only soluble in aqueous solution at pH > 9 (10−3 M), which is in agreement with the formation of anionic carboxylate salts. Due to strong aggregation, it was not possible to obtain 1H NMR spectra of the final compound 5a, and the only characterization data obtained was its mass spectrum, which presented a peak at 1293.6942 for [M + Na]+ (see Fig. S7 in SI).
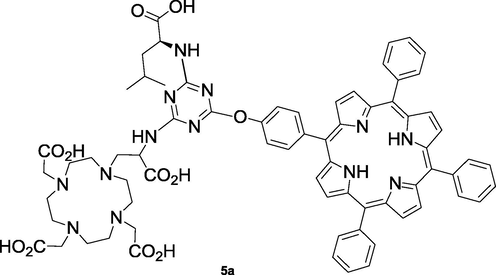
Structure of the deprotected compound 5a.
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.arabjc.2018.06.005.
In addition, to obtain water soluble structures, compound 5 was hydrolyzed using acid hydrolysis, with HCl 6 M in ethanol, in order to deprotect the carboxylic groups. However, to our dismay, the solubility of the supposed structure 5a (Fig. 1) was also highly pH dependent, being only soluble in aqueous solution at pH > 9 (10−3 M), which is in agreement with the formation of anionic carboxylate salts. Due to strong aggregation, it was not possible to obtain 1H NMR spectra of the final compound 5a, and the only characterization data obtained was its mass spectrum, which presented a peak at 1293.6942 for [M + Na]+ (see Fig. S7 in SI).
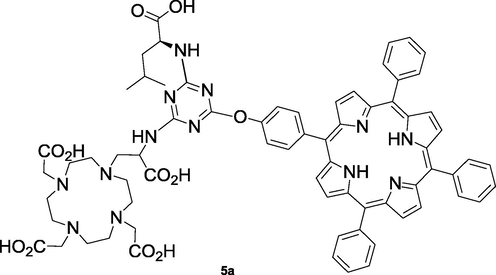
Structure of the deprotected compound 5a.
Supplementary data 1
Supplementary data 1 Mass spectra, electronic spectra and 1H NMR spectra are given as supplementary material.Given the low water solubility presented by compound 5a, we turned our attention to the synthesis of a more water-soluble system. To increase water solubility of the starting porphyrin, we have synthesized a new sulfonated porphyrin derivative, by chlorosulfonation of porphyrin 1, following our previously described methodology (Gonsalves et al., 1996; Monteiro et al., 2008), using an excess of chlorosulfonic acid, for 2 h at 100 °C (Scheme 2). After work-up, hydrolysis was performed by adding water and heating the suspension at 100 °C during 12 h. Upon full solubilization of the mixture in water and isolation/purification, 5-(4-hydroxy-3-sulfonylphenyl)-10,15,20-(4-sulfonyltriphenyl)porphyrin 6 was obtained in 90% yield. The degree of sulfonation was determined by analysis of the 1H NMR and mass spectra, which corroborates the preferred reactivity pattern of substituted benzenes (substitution in para position) and phenols (substitution in ortho position) (Cremlyn, 2002).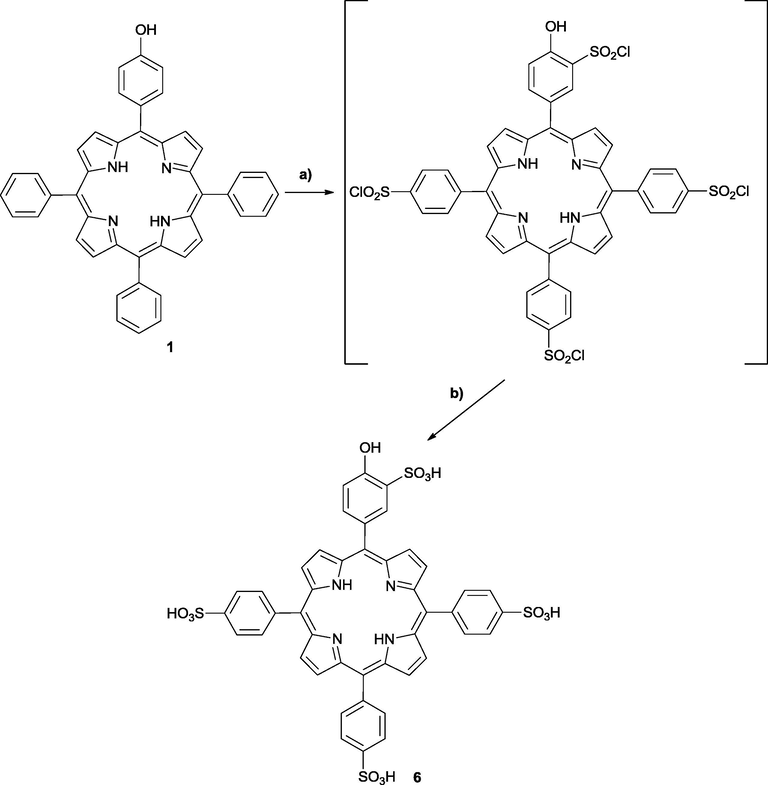
Synthesis of water soluble sulfonated porphyrin 6. (a) 100 °C, 2 h, HSO3Cl; (b) 100 °C, 12 h, water.
In our attempts to prepare a water soluble system, we experienced difficulties when attempting to modulate the cyanuric chloride first with protected aminoacid, followed by sulfonated porphyrin and finally DO3A, in a stepwise one-pot reaction. We never managed to obtain the desired compound bearing one “aminoester”, a sulfonated porphyrin and DO3A. Since attempts to isolate intermediates were unsuccessful, we hypothesized that the intermediate bearing an aminoacid was not stable in presence of sulfonated porphyrin in the second step. We focused then on skipping the first step, and promoted the multifunctionalization directly with sulfonated porphyrin, followed by DO3A.
The synthetic pathway to prepare the target compound, involving the sequential nucleophilic substitution pattern, using cyanuric chloride was similar to that previously described for compound 5, with some modifications. In this case we started by reacting 1 equiv of cyanuric chloride with 2 equiv of 6 in DMF, at 25 °C in the presence of 2.4 equiv of DIPEA (Scheme 3). After disappearance of the starting materials (12 h, checked by TLC), 2.1 equiv of 4 and 1.2 equiv of DIPEA were added, the reaction was left at 60 °C for 72 h. Evolution of the reaction was monitored by TLC and the disappearance of compound 7. Isolation and purification of the product was achieved by precipitation with acetone and a product was obtained in 42% yield.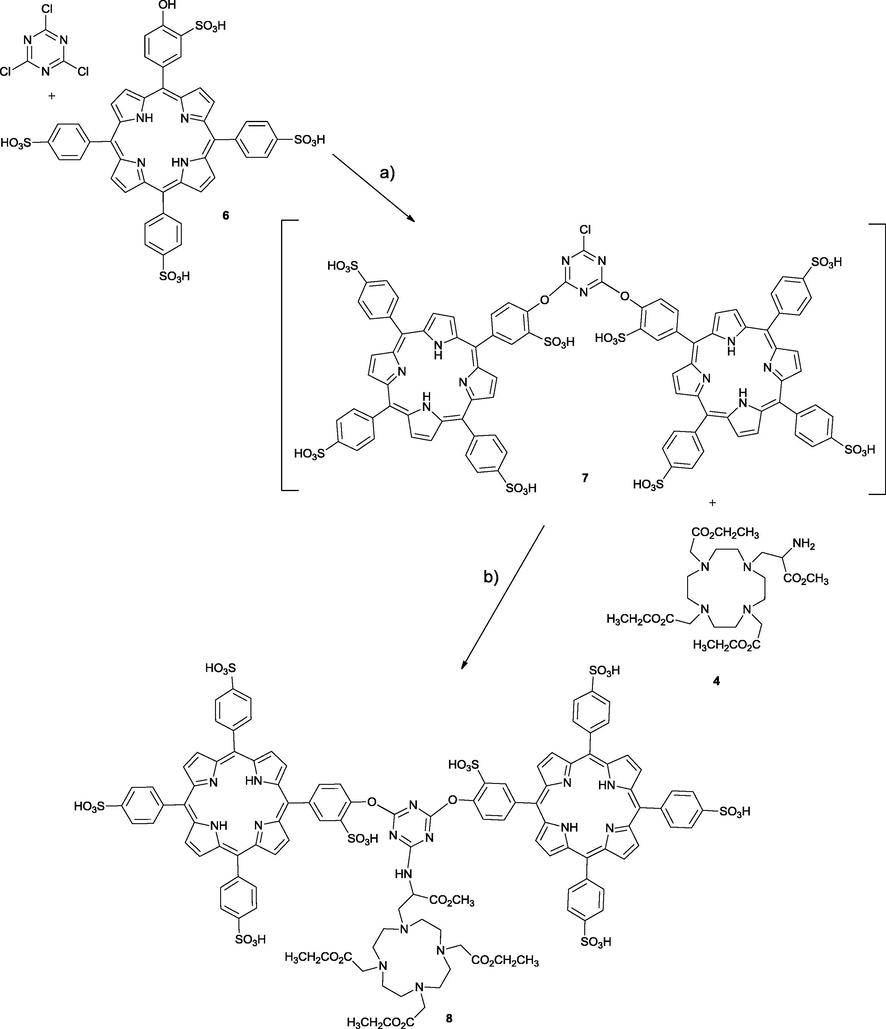
Reaction pathway to obtain compound 8. Reagents and conditions: (a) 25 °C, 12 h, DIPEA, DMF; (b) 60 °C, 72 h, DIPEA, DMF.
Next, tests were carried out in order to evaluate the solubility, and we observed that, even without deprotection of the ethyl groups of the DO3A counterpart, the compound 8 presents satisfactory solubility (10−3 M) in polar solvents, such as DMSO, DMF, ethanol, methanol and water.
3.2 UV/visible absorption and fluorescence spectral properties
The absorption spectra of structures 5 and 8 were recorded in THF and DMSO, respectively, and the typical five bands, B and Soret, Qy(1-0), Qy(0-0), Qx(1-0) and Qx(0-0) of porphyrins can be seen in Fig. 2a–b (solid line). Molar absorption coefficients (ε) were determined using the Beer-Lambert law for both compounds (Table 1) and are in the typical range of ε for porphyrins reported in literature (Pinto et al., 2012; Martinez-Diaz et al., 2010).
UV–Visible and fluorescence spectra of (a) compound 5 in THF; (b) compound 8 in DMSO.
λmáx (nm), ε (L mol−1 cm−1)
ΦF
1
(in THF)B(0-0)
417 nm
1.19 × 105
Qy(1-0)
513 nm
7.21 × 103
Qy(1-0)
548 nm
3.39 × 103
Qx(1-0)
589 nm
2.07 × 103
Qx(0-0)
650 nm
1.78 × 103
0.06
(Pinto et al., 2012)
5
(in THF)B(0-0)
419 nm
2.71 × 105
Qy(1-0)
515 nm
1.82 × 104
Qy(1-0)
550 nm
6.04 × 103
Qx(1-0)
589 nm
4.56 × 103
Qx(0-0)
646 nm
4.21 × 103
0.05
6
(in DMSO)B(0-0)
419 nm
1.04 × 105
Qy(1-0)
517 nm
6.43 × 103
Qy(1-0)
551 nm
2.56 × 103
Qx(1-0)
589 nm
1.67 × 103
Qx(0-0)
643 nm
1.10 × 103
0.05
8
(in DMSO)B(0-0)
416 nm
1.14 × 105
Qy(1-0)
514 nm
8.17 × 103
Qy(0-0)
547 nm
6.23 × 103
Qx(1-0)
592 nm
1.96 × 103
Qx(0-0)
646 nm
1.18 × 103
0.03
In order to evaluate the potential of these structures for fluorescence imaging, their fluorescence spectra and quantum yields were also determined. As can be seen in Fig. 2a–b (dashed lines), both fluorescence spectra show two bands: at 650 nm and 719 nm for 5 and 652 nm and 717 nm for 8. These are within the 650 – 1450 nm spectral window required for imaging, where tissue components have their lowest absorption (Pansare et al., 2012). Although fluorescence quantum yields in THF (for 5) and DMSO (for 8) are modest, they are acceptable for imaging since they fall within the important near infrared window in biological tissues, and are comparable to values found in the literature for free base porphyrins (Pinto et al., 2012; Pinto et al., 2011) (Table 1). However, we were expecting that compound 8, possessing two porphyrin units in its structure, could have a higher fluorescence quantum yield, as we know from our previous studies (Pinto et al., 2012). Future time-resolved fluorescence measurements are proposed to obtain a deeper understanding of the photophysics of these multimodal systems with the objective of improving emission quantum yields.
4 Conclusions
In summary, we have reported the synthesis, structural characterization and spectral evaluation of two new multifunctional chemical entities, based on the triazine molecule as starting platform, for the potential development of multimodal imaging agents. These have modulated lipophilicity, and have three arms bearing macrocycles of the porphyrin family, chelates of the DOTA type (DO3A) and, in the case of the triad molecule 5, an aminoacid to increase biocompatibility. We have managed to overcome the limited solubility of triad 5, by synthesizing compound 8, which presents reasonable water solubility, even without deprotection of DO3A ethyl groups. The absorption and fluorescence spectra and quantum yields were also determined, and the preliminary findings suggest that the optical properties presented by these systems indicate that they have potential as fluorescence imaging agents and, following complexation of the DO3A ligand with Gd3+, also as MRI agents. These studies are undergoing, and future developments will be published elsewhere.
Authors contributions
Mário J.F. Calvete and Sara M.A. Pinto synthesized and characterized the compounds. Sara M.A. Pinto performed the photophysical assessment. Mariette M. Pereira was in article conception, result discussion and writing. Hugh D. Burrows, M. Margarida C.A. Castro and Carlos F. G. C. Geraldes assisted them in the discussion of the results and writing of the manuscript.
Acknowledgements
The authors thank FCT-Portugal (Portuguese Foundation for Science and Technology) and FEDER – European Regional Development Fund through the COMPETE Programme (Operational Programme for Competitiveness) for funding with PEst-OE/QUI/UI0313/2014, UID/QUI/00313/2013 and PTDC/QEQ-MED/3521/2014. S.M.A.P. and M.J.F.C. are grateful for SFRH/BPD/84619/2012 and SFRH/BPD/99698/2014, respectively.
References
- Mechanistic Investigations of porphyrin syntheses. I. Preliminary studies on Ms-tetraphenylporphin. J. Am. Chem. Soc.. 1964;86:3145.
- [CrossRef] [Google Scholar]
- Design of porphyrin-based photosensitizers for photodynamic therapy. Adv. Inorg. Chem.. 2011;63:187-233.
- [CrossRef] [Google Scholar]
- Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis. Tetrahedron. 2006;62:9507-9522.
- [CrossRef] [Google Scholar]
- A cost-efficient method for unsymmetrical meso-aryl porphyrin synthesis using NaY zeolite as an inorganic acid catalyst. Molecules. 2017;22:741.
- [CrossRef] [Google Scholar]
- Tetrapyrrolic Macrocycles as Molecular Probes for Radionuclide-Based Imaging Techniques. In: Berhardt L.V., ed. Advances in Medicine and Biology. New York: Nova Science Publishers; 2017.
- [Google Scholar]
- Metal coordinated pyrrole-based macrocycles as contrast agents for magnetic resonance imaging technologies: Synthesis and applications. Coordin. Chem. Rev.. 2017;333:82-107.
- [CrossRef] [Google Scholar]
- Tetrapyrrolic macrocycles: potentialities in medical imaging technologies. Curr. Org. Synth.. 2014;11:127-140.
- [CrossRef] [Google Scholar]
- Sulfonation and chlorosulfonation of aromatic compounds using chlorosulfonic acid. In: Chlorosulfonic Acid: A Versatile Reagent Ch. 4. Cambridge: The Royal Society of Chemistry; 2002. p. :35-145.
- [Google Scholar]
- Combined effects of singlet oxygen and hydroxyl radical in photodynamic therapy with photostable bacteriochlorins: Evidence from intracellular fluorescence and increased photodynamic efficacy in vitro. Free Radical. Bio Med.. 2012;52:1188-1200.
- [CrossRef] [Google Scholar]
- Tissue uptake study and photodynamic therapy of melanoma-bearing mice with a nontoxic, effective chlorin. Chemmedchem. 2011;6:1715-1726.
- [CrossRef] [Google Scholar]
- PEGylated GdF3: Fe nanoparticles as multimodal T-1/T-2-weighted MRI and X-ray CT imaging contrast agents. Acs Appl. Mater. Inter.. 2017;9:20426-20434.
- [CrossRef] [Google Scholar]
- Oxidative biomacromolecular damage from novel phthalocyanine. Sci. China B. 1998;41:45-49.
- [CrossRef] [Google Scholar]
- Synthesis of novel functionalised zinc phthalocyanines applicable in photodynamic therapy. Eur. J. Org. Chem. 1999:3441-3453.
- [CrossRef] [Google Scholar]
- The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev.. 2011;40:340-362.
- [CrossRef] [Google Scholar]
- Amide conjugates of the DO3A-N-(alpha-amino)propionate ligand: leads for stable, high relaxivity contrast agents for MRI? Contrast Media Mol. I. 2013;8:40-49.
- [CrossRef] [Google Scholar]
- Gd(DO3A-N-alpha-aminopropionate): a versatile and easily available synthon with optimized water exchange for the synthesis of high relaxivity, targeted MRI contrast agents. Chem. Commun. 2009:6475-6477.
- [CrossRef] [Google Scholar]
- Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer. 2002;2:683-693.
- [CrossRef] [Google Scholar]
- Some new aspects related to the synthesis of mesosubstituted porphyrins. J. Heterocyclic Chem.. 1991;28:635-640.
- [CrossRef] [Google Scholar]
- New procedures for the synthesis and analysis of 5,10,15,20-tetrakis(sulphophenyl)porphyrins and derivatives through chlorosulphonation. Heterocycles. 1996;43:829-838.
- [CrossRef] [Google Scholar]
- Phthalocyanine-mediated photodynamic therapy induces cell death and a G(0)/G(1) cell cycle arrest in cervical cancer cells. Biochem. Bioph. Res. Commun.. 2006;339:569-576.
- [CrossRef] [Google Scholar]
- Unsymmetrical porphyrins: the role of meso-substituents on their physical properties. J. Porphyr. Phthalocya.. 2012;16:290-296.
- [CrossRef] [Google Scholar]
- Ecofriendly porphyrin synthesis by using water under microwave irradiation. Chemsuschem. 2014;7:2821-2824.
- [CrossRef] [Google Scholar]
- Synthesis of low melting point porphyrins: A quest for new materials. J. Porphyr. Phthalocya.. 2016;20:843-854.
- [CrossRef] [Google Scholar]
- Solventless metallation of low melting porphyrins synthesized by the water/microwave method. Rsc Adv.. 2015;5:64916-64924.
- [CrossRef] [Google Scholar]
- Unique diagnostic and therapeutic roles of porphyrins and phthalocyanines in photodynamic therapy, imaging and theranostics. Theranostics. 2012;2:916-966.
- [CrossRef] [Google Scholar]
- Manganese-enhanced MRI contrast agents: from small chelates to nanosized hybrids. Eur. J. Inorg. Chem. 2012:1987-2005.
- [CrossRef] [Google Scholar]
- Phthalocyanine labels for near-infrared fluorescence imaging of solid tumors. J. Med. Chem.. 2016;59:4688-4696.
- [CrossRef] [Google Scholar]
- Photosensitizing porphyrin-triazine compound for bulk heterojunction solar cells. J. Mater. Chem.. 2012;22:23030-23037.
- [CrossRef] [Google Scholar]
- Tetranuclear gadolinium(III) porphyrin complex as a theranostic agent for multimodal imaging and photodynamic therapy. Inorg. Chem.. 2014;53:4184-4191.
- [CrossRef] [Google Scholar]
- Energy transfer from fluorene-based conjugated polyelectrolytes to on-chain and self-assembled porphyrin units. J. Polym. Sci. Pol. Chem.. 2012;50:1408-1417.
- [CrossRef] [Google Scholar]
- Lighting porphyrins and phthalocyanines for molecular photovoltaics. Chem. Commun.. 2010;46:7090-7108.
- [CrossRef] [Google Scholar]
- Biomedical applications of macrocyclic ligand complexes. Coordin. Chem. Rev.. 2010;254:1686-1712.
- [CrossRef] [Google Scholar]
- Phthalocyanines and related compounds: XXXVII. Synthesis of covalent conjugates of carboxy-substituted phthalocyanines with alpha-amino acids. Russ. J. Gen. Chem.+. 2004;74:451-459.
- [CrossRef] [Google Scholar]
- Synthesis of amphiphilic sulfonamide halogenated porphyrins: MALDI-TOFMS characterization and evaluation of 1-octanol/water partition coefficients. Tetrahedron. 2008;64:5132-5138.
- [CrossRef] [Google Scholar]
- Handbook of Photochemistry. New York: Marcel Dekker; 1993.
- Routes to synthesis of porphyrins covalently bound to poly(carbazole)s and poly(fluorene)s: Structural and computational studies on oligomers. J. Mol. Struct.. 2012;1029:199-208.
- [CrossRef] [Google Scholar]
- A brief account of nanoparticle contrast agents for photoacoustic imaging. Wires Nanomed. Nanobi.. 2013;5:517-543.
- [CrossRef] [Google Scholar]
- Review of long-wavelength optical and NIR imaging materials: contrast agents, fluorophores, and multifunctional nano carriers. Chem. Mater.. 2012;24:812-827.
- [CrossRef] [Google Scholar]
- Pereira, M.M., Arnaut, L.G., Simoes, S.J.F., Monteiro, C., 2006. Novel Derivatives of Porphyrin, Particularly Chlorins and/or Bacteriochlorins, and Uses Thereof in Photodynamic Therapy. US Patent WO/2006/053707, 26 June 2006.
- Synthesis of meso-substituted porphyrins using sustainable chemical processes. J. Porphyr. Phthalocya.. 2016;20:45-60.
- [CrossRef] [Google Scholar]
- Synthesis of new metalloporphyrin triads: efficient and versatile tripod optical sensor for the detection of amines. Inorg. Chem.. 2011;50:7916-7918.
- [CrossRef] [Google Scholar]
- Metalloporphyrin triads: Synthesis and photochemical characterization. J. Photoch. Photobio. A. 2012;242:59-66.
- [CrossRef] [Google Scholar]
- Triazine-based covalent organic polymers: design, synthesis and applications in heterogeneous catalysis. J. Mater. Chem. A. 2016;4:16288-16311.
- [CrossRef] [Google Scholar]
- Isotopes for combined PET/SPECT imaging. J. Radioanal. Nucl. Ch.. 2015;305:87-92.
- [CrossRef] [Google Scholar]
- Size and ability do matter! Influence of acidity and pore size on the synthesis of hindered halogenated meso-phenyl porphyrins catalysed by porous solid oxides. Chem. Commun.. 2014;50:6571-6573.
- [CrossRef] [Google Scholar]
- Amphiphilic meso(sulfonate ester fluoroaryl)porphyrins: refining the substituents of porphyrin derivatives for phototherapy and diagnostics. Tetrahedron. 2012;68:8767-8772.
- [CrossRef] [Google Scholar]
- Synthesis of a new F-18 labeled porphyrin for potential application in positron emission tomography. In vivo imaging and cellular uptake. Rsc Adv.. 2015;5:99540-99546.
- [CrossRef] [Google Scholar]
- Glycosylated porphyrins, phthalocyanines, and other porphyrinoids for diagnostics and therapeutics. Chem. Rev.. 2015;115:10261-10306.
- [CrossRef] [Google Scholar]
- Porphyrin-based photosensitizers and the corresponding multifunctional nanoplatforms for cancer-imaging and phototherapy. J. Porphyr. Phthalocya.. 2015;19:109-134.
- [CrossRef] [Google Scholar]
- Synthesis of glycoporphyrin derivatives and their antiviral activity against herpes simplex virus types 1 and 2. Bioorgan. Med. Chem.. 2005;13:3878-3888.
- [CrossRef] [Google Scholar]
- In vivo fluorescence spectroscopy and imaging for oncological applications. Photochem. Photobiol.. 1998;68:603-632.
- [CrossRef] [Google Scholar]
- Contrast agents for photoacoustic and thermoacoustic imaging: a review. Int. J. Mol. Sci.. 2014;15:23616-23639.
- [CrossRef] [Google Scholar]
- Selective synthesis and biological activity of triazine-porphyrins as potential anti-cancer agents. J. Porphyr. Phthalocya.. 2010;14:123-127.
- [CrossRef] [Google Scholar]




