

Translate this page into:
Ruthenium tetroxide oxidation of N-methyl-isoxazolidine: Computational mechanistic study
⁎Corresponding author. laura.legnani@unimib.it (Laura Legnani)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
In this paper, we report a mechanistic study of RuO4-catalyzed oxidation on the 2-methylisoxazolidine through computational methods. The investigation was performed taken into consideration that the oxidation could take place on different sites of the substrate. This reaction occurs in two steps, involving a double H-transfer. In particular, the rate-determining one implies a [3 + 2] one-step, but asynchronous mechanism. In the first step, when methyl propanoate is used as solvent, the formation of an ion pair, which affords to the product, is involved. Furthermore, the study highlights that all carbon atoms of the isoxazolidine system, near to the heteroatoms, can undergo the oxidation process. The detected selectivity is correlated to the stability of the corresponding carbocations, leading to the N-methylisoxazolidin-3-one as preferred product.
Keywords
Oxidation
DFT calculations
Dipolar cycloaddition
Oxazolidinone
Transition metal
Selectivity

1 Introduction
The first example of organic substrates oxidation with ruthenium tetroxide (RuO4) dates back to 1953 (Djerassi, et al., 1953). Since then, despite its high reactivity, reactions employing RuO4 have not found general applicability, due to their lack of selectivity. Only in the 1990 s, the first successful applications of this oxidation method in organic chemistry appeared in literature (Piccialli, et al., 1993; Shing, et al., 1994; Shing, et al., 1996). RuO4 demonstrated to be a powerful and very selective oxidant for the dihydroxylation of olefins, resulting fast and efficient, and proceeding better than the analogous reaction using OsO4.
More interestingly, several research groups have discovered new and highly selective oxidative reactions, using RuO4 for the oxyfunctionalization of saturated hydrocarbons. The high reactivity and selectivity of this oxidant avoids the obtainment of side products (Piccialli, et al., 1993; Shing, et al., 1994; Shing, et al., 1996).
The commonly used protocol involves a biphasic system of an organic solvent and water, using RuO4 in a convenient catalytic route. RuO4 could be obtained in situ from hydrated ruthenium trichloride or ruthenium dioxide. Once reduced, it can be regenerated by various reoxidizing agents such as sodium periodate.
A very useful application of RuO4 is the oxidation of cycloalkanes; these oxidations have been thoroughly investigated by different research groups with particular attention to the possible reaction mechanisms. Based on either kinetic isotope or solvent and substituent effects Bakke et al. (Bakke, et al., 1986) suggested in 1986, the formation of cations during the oxidation reaction of some cyclic hydrocarbons. Carbocations have been assumed to be formed by the extraction of hydride ions from a reactant or by the abstraction of a hydrogen atom from an initially formed radical cation.
In 1989, Waegell et al. proposed a cyclic (2 + 2) addition mechanism and in the next year, the same research group discarded the ionic mechanism, and proposed a concerted but asynchronous (3 + 2) addition reaction involving a five-membered ring as rate-determining transition state (Tenaglia, et al., 1990).
Finally, in 1992, Bakke et al. considered both the via ion-pair and (3 + 2) addition mechanisms (Bakke, et al., 1992). In this work, the assumed oxidation reaction model involves the concomitant formation of the carbocation nearby the nucleophilic species HRuO4- leading to very fast collapse of the ion-pair, to give the reaction product. Nevertheless, also a mechanism with a cyclic transition state with a well-developed positive charge on the C atom was not excluded.
More recently, papers concerning theoretical studies on these oxidation reactions appeared in literature. Deng et al. (Deng, et al., 1997) used a DFT method to investigate the oxidation of methanol to formaldehyde by oxometal complexes. Frunzke et al. (Frunzke, et al., 2004) reported a systematic quantum chemical investigation of ethylene oxidation with RuO4, leading to the scission of the C-C bond. Taking up the mechanistic controversy in 1990 s, the RuO4-mediated catalytic oxidations of various cycloalkanes (adamantane, cis- and trans-decalin, methane) were theoretically studied by Strassner though DFT calculations at the B3LYP/6-31G(d) level, in order to investigate the corresponding reaction mechanisms. The analysis of the transition states allowed a comparison with the experimentally determined activation enthalpies and free energies and the obtained results supported a (3 + 2) mechanism, involving the interaction of the σ-CH with the O-Ru-O moiety (Drees, et al., 2006).
The first example of a direct oxidation of the isoxazolidine nucleus to the 3-isoxazolidone was previously reported (Piperno, et al., 2007). In this study, the reaction was carried out using RuO2/NaIO4, under ethyl acetate/water biphasic conditions. High regioselectivity was observed, since the oxidation reaction occurred exclusively at position 3 of the isoxazolidine ring. More recently, we performed a preliminary study of ruthenium tetroxide-mediated oxidation of cyclopentane, tetrahydrofuran, tetrahydrothiophene and N-substituted pyrrolidines through DFT and topological methods, confirming that, on these substrates, the rate-limiting step of the reaction takes place through a highly asynchronous (3 + 2) concerted cycloaddition reaction (Pedrón, et al., 2019). Starting from these bases, and considering our expertise concerning cycloaddition reactions and synthesis of heterocyclic compounds (Legnani, et al, 2007; Legnani, et al, 2016; Legnani, et al, 2015; Chiacchio, et al., 2017; Merino, et al., 2017; Merino, et al., 2017; Giofrè, et al., 2016; Chiacchio, et al., 2019), in this paper, we report a complete computational mechanistic study on the oxidation reaction of 2-methylisoxazolidine with RuO4. The study concerns the trend of the regioselectivity, focusing attention on the possible oxidation of all the carbon atoms of 2-methylisoxazolidine 1.
The study carefully accounts for the origin of the selective oxidation of the C3 of 2-methylisoxazolidine 1 with the aim of trying to clarify not only the mechanism of the oxidation reaction, but also the selectivity.
2 Results and discussion
2.1 Theoretical calculations
All calculations were performed using the Gaussian16 program package (Frisch, et al., 2016), through optimizations with the Thrular’s functional M062x (Zhao, et al., 2008; Zhao, et al., 2008) and 6-31G(d) basis set for all atoms, while the Stuttgart/Dresden ECP was used for Ru, as reported in literature (Drees, et al., 2006). Optimizations were also performed in solvent (methyl propanoate) at the same level in the classical polarizable continuum model (PCM) (Cances, et al., 1997.; Cossi, et al., 1996; Barone, et al., 1998). Moreover, to check the validity of the obtained results, calculations were repeated using both the larger basis set 6–31 + G(d,p) with the m062x functional, and a different DFT method (b3lyp/def2svp/emp = gd3bj/int = ultrafine/solvent = water).
The supposed oxidation mechanism of isoxazolidine ring, reported in literature, (Piperno, et al., 2007) presents two separate steps: the first one is the activation of the R-CH bond, giving product P1, coordinated to the Ru(VI); the second one regards the second hydrogen transfer with the final obtainment of the oxygenated compound and Ru(IV). Accordingly, the oxidation reaction of 2-methylisoxazolidine 1, using RuO4, can lead to 2-methylisoxazolidin-3-one 2 or isoxazolidine-2-carbaldehyde 3 (Scheme 1), because of the possible attack of the oxidizing species on C3 or C1′, respectively.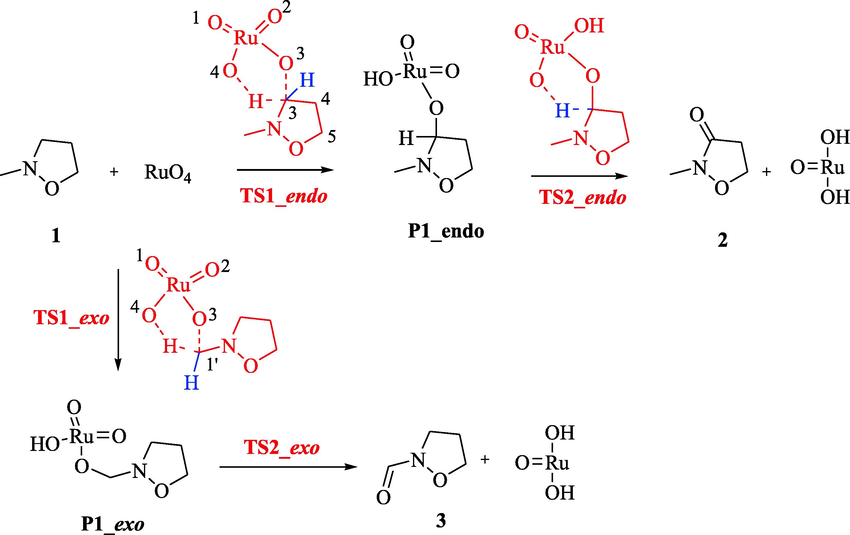
Mechanistic pathways of the oxidation reaction of 2-methylisoxazolidine 1 with RuO4, leading to 2-methylisoxazolidin-3-one 2 and isoxazolidine-2-carbaldehyde 3, respectively.
Reagents, intermediate products, and transition states (TSs) along the reaction pathways have been taken into consideration and located.
The geometries of isolated reacting species, i.e., N-methylisoxazolidine 1 and RuO4, were optimized.
We considered, as first event in the oxidation reaction, the formation of a bimolecular pre-reaction complex between N-methylisoxazolidine and RuO4, as recently reported in literature (Mitka, et al., 2021; Dresler, et al., 2018; Jasiński, et al., 2018).
Actually, also in our case, the two addends pass through a bimolecular complex with nonbonding interactions between the oxygen atoms of RuO4 and hydrogen atoms of the isoxazolidine system. Nevertheless, considering the different level of calculations, this specie is not a minimum in the free energy profiles, but only in the energy ones (see Figures S5-S7 in the Supporting Information).
Subsequently, the modeling study considered the first step, giving the endo and exo products P1.
All the possible minimum energy conformers of P1 were taken under consideration and in Fig. 1 the three-dimensional plots of the preferred ones are reported. In the case of the endo pathway, considering the free energy relative values, the preferred geometry showed the N-methyl in a pseudo-equatorial position with formation of a N···HO hydrogen bond with the O(3) atom (P1_endo_A). The other favorite conformations had the N-methyl in a pseudo-axial position and differ each other for the orientation of the O(3)-Ru bond (see Scheme 1). In both these cases, there is the formation of a hydrogen bond with the nitrogen of the isoxazolidine ring (P1_endo_B) or the O(3) atom (P1_endo_C).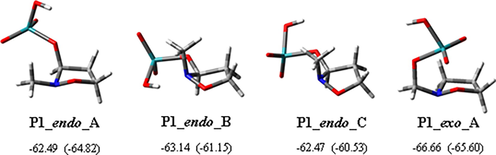
Three-dimensional plots of the most stable conformers of intermediates P1 in the endo and exo pathways of the oxidation reaction of 2-methylisoxazolidine with RuO4. The corresponding relative Gibbs energies in methyl propanoate (kcal/mol) and Gibbs energies in vacuo (in parentheses), with respect to the isolated reacting species, are reported. All imaginary frequencies are positive.
For the second hydrogen transfer, conformation P1_endo_C represents the starting point. The transition from a conformation to the other one does not necessarily occur through a conformational change overcoming a very low energy barrier. Also the exit or entrance of a hydrogen atom in another position can give the starting structure for the second reaction step. All the other located conformations of P1_endo resulted to be less stable by about 10 kcal/mol.
Concerning the exo pathway, conformation P1_exo_A is the preferred one (Fig. 1).
It was reported (Piperno, et al., 2007) that the product P1 is obtained by the activation of the R − CH bond, in a one-step five-membered transition state, with the simultaneous transfer of hydrogen and the formation of the new C(3)-O(3) bond.
Moreover, for the oxidation of a saturated hydrocarbon (Coudret and Waegell, 1994), a competitive mechanism was reported with a higher energy linear transition state to form an ion pair. This hypothesis can be also applied to our case, as shown in Scheme 2.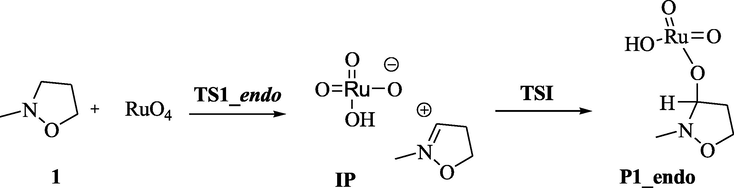
Alternative proposed mechanistic pathways for the first step of the oxidation reaction of 2-methylisoxazolidine 1 with RuO4, leading to 2-methylisoxazolidin-3-one 2.
The transition state (TS1), leading to P1 for both the endo and exo pathways, was located and the corresponding IRC analysis was performed in both the forward and the backward directions (Figure S1). In the forward direction for TS1_endo pathway, we obtained a species very similar to a ion pair with a partial character of double bond between C(3) and N and the O(3)-oxygen negative charged, with a distance C(3)-O(3) of 2.74 Å. However, when optimized in the gas phase, the ion pair it straight fell into the energy hole corresponding to the P1_endo compound. Contrary to the unsuccessful attempts in vacuo, the ion pair was located through optimization in methyl propanoate and in water, while in carbonium tetrachloride, as expected, it was not a minimum. We selected methyl propanoate as solvent, and in this media, starting from the ion pair IP, through a scan analysis, performed progressively approaching C(3) and O(3), also the transition state, corresponding to the formation of this bond (TSI) and connecting the ion pair IP to P1_endo, was located (Fig. 2).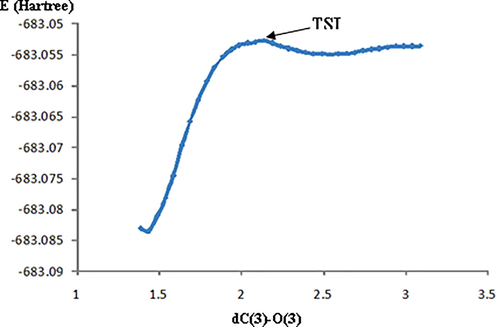
Relaxed scan from IP to P1_endo_A showing a very low barrier of PES corresponding to TSI.
The same IRC analysis was repeated starting from P1_exo_A (Figure S2). Also in this case, the forward direction involved a species presenting a partial character of double bond between the methylene carbon atom (C1′) and the N atom, and with the O(3) atom negative charged, even if with a shorter distance between C1′-O(3) (2.50 Å) with respect to the endo C3-O(3) detected distance. In this case, the ion pair was not located neither in vacuo nor in solvent, and the optimization directly gave P1_exo_A. This result might be justified by the lower stability of the less substituted iminium ion of the exo pathway with respect to the more substituted endo one.
In Fig. 3 are reported the 3D-plots of the preferred conformations of the first step transition states (TS1) for both the endo and exo pathways and the TSI, optimized in methyl propanoate.
Three-dimensional plots of the transition states TS1 of both the endo and exo pathways and TSI in methyl propanoate. Displacement vectors for TS imaginary frequencies (-760.15, −762.31, −58.82 for TS_endo_A, TS_exo_A, and TSI, respectively) are shown as dotted lines and distances are reported in angstroms.
The nature of ion pair IP and the transition state TSI was established by natural bond order (NBO) analysis (Fig. 4). When the secondary hydrogen atom in alpha to the nitrogen one of the isoxazolidine ring is transferred to the oxygen of the ruthenium, the values of the NBO analysis show that it presents a negative charge (-0.83), while the corresponding carbon and nitrogen atoms positive charges (0.105 and 0.52, respectively). These charge differences resulted from the involvement of the intimate ion pair along the reaction pathway, with the formation of the iminium ion. The transition state (TSI) leading to P1 shows very similar characteristics. In both ion pair (IP) and transition state (TSI), large dipole values were observed (IP: 14.4 D; TSI:14.5 D).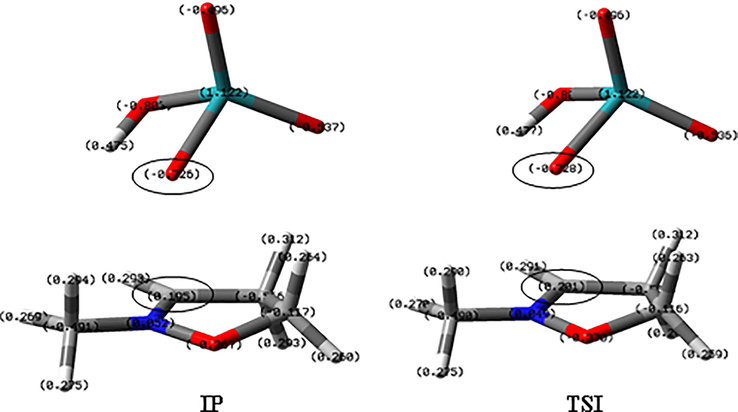
NBO analysis of the Ion Pair IP and the following transition state TSI, leading to P1_endo_A.
Moreover, the careful examination of the timing of bond formation and breaking (Fig. 5), for both the endo (A) and exo (B) pathways, showed for C(3)-O(3) (red one in graph) a gentler slop of the trend line with respect to C(1′)-O(3), thus justifying in the latter case, a one-step, though asynchronous mechanism (Domingo, et al, 2018).![Length (Å) of the forming H − O(4), O(3)-C(3,1′) and breaking C(3,1′) − H bonds [d(H − O(4)), d(O − C(3,1′)) and d(C(3,1′) − H), respectively] at the forward (positive) and backward (negative) points in the IRC analysis, starting from (A) TS1_endo (point 0) and (B) TS1_exo (point 0).](/content/184/2022/15/9/img/10.1016_j.arabjc.2022.104063-fig7.png)
Length (Å) of the forming H − O(4), O(3)-C(3,1′) and breaking C(3,1′) − H bonds [d(H − O(4)), d(O − C(3,1′)) and d(C(3,1′) − H), respectively] at the forward (positive) and backward (negative) points in the IRC analysis, starting from (A) TS1_endo (point 0) and (B) TS1_exo (point 0).
As clearly shown by the profiles reported in Fig. 6, a competition between oxidation on position 3 of the ring, and on the alpha carbon of the side chain C(1′), both promoted by the presence of the activating nitrogen atom, might be considered. The regioselectivity-determining step for the oxidation reaction corresponds to TS1. The transition state TS1_endo (black), that gives P1_endo, and consequently the oxidation on the ring, in solvent is lower by about 2 kcal/mol than TS1_exo (red), related to the oxidation on the methyl group. Therefore, by computational data, the oxidation reaction is faster on the ring than on the N-methyl substituent, confirming the reactivity decrease of primary C-H bond, with respect to the secondary one (Plietker, 2005.).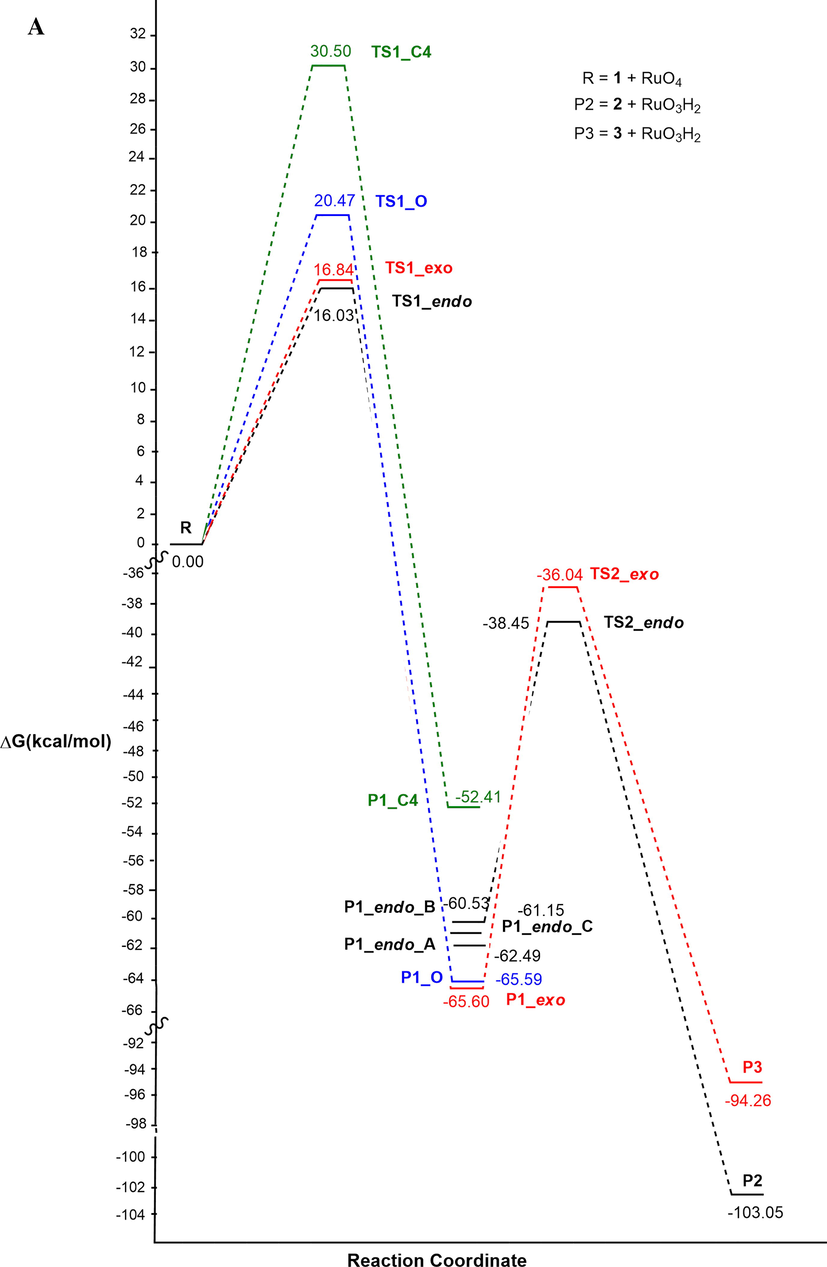
Energy profile in vacuo (A) and methyl propanoate (B) for the reaction pathway of the oxidation reaction of 2-methylisoxazolidine with Ruthenium Tetroxide at C3 (endo, black) or at C1′ methyl (exo, red).
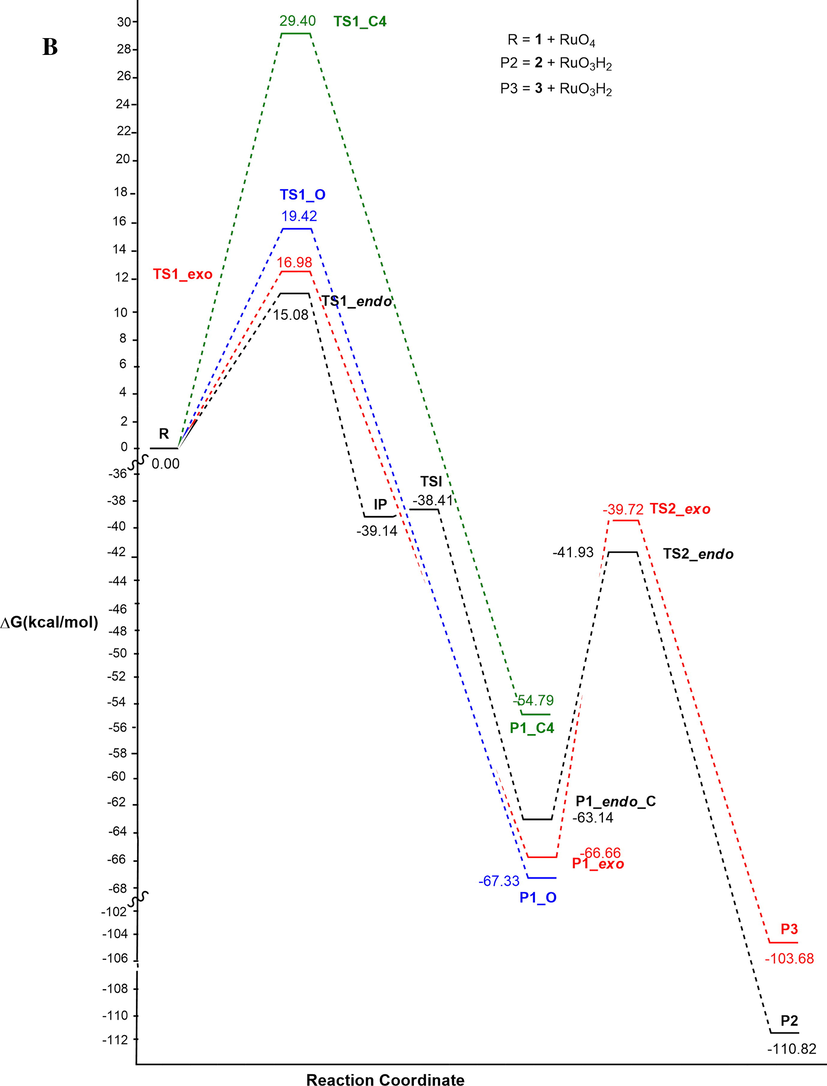
Energy profile in vacuo (A) and methyl propanoate (B) for the reaction pathway of the oxidation reaction of 2-methylisoxazolidine with Ruthenium Tetroxide at C3 (endo, black) or at C1′ methyl (exo, red).
Moreover, in literature (Plietker, 2005; Florea, et al., 2017) is reported that various heteroatom-containing compounds undergo oxidation of methylene groups at the α-position. Not only amines and amides are oxidized to give amides and imides, respectively, but also ethers can be converted into esters and/or lactones. Because of the presence into the isoxazolidine ring of both nitrogen and oxygen atoms, the possible competition with oxidation on the ring at position 5 was also investigated.
The transition state TS1_O, leading to isoxazolidin-5-one, was located and resulted to be less stable than the preferred TS1_endo by about 4.34 kcal/mol, even if the energy barriers do not exclude a possible competition between the two oxidation reactions.
The IRC analysis (Figure S3), performed for TS1_O, in both the forward and backward directions clearly indicates the formation of a product with the new C(5)-O(3) bond almost completely formed (d = 1.95 Å), as evidenced by the trend for the forming bond, reported in Fig. 7. This different behavior, with respect to that of the forming bond C(3)/C1′-O(3) for TS1_endo and TS1_exo (Fig. 5), might be due to the lower electronegativity of the N atom compared to the O one and therefore, to its greater availability to give the electron pair for the formation of the double bond and the stabilization of the isoxazolidinium ion. Therefore, for the N atom the pathway through the ionic pair seems to be preferred.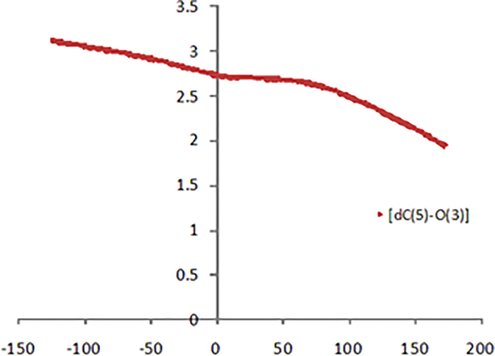
Length (Å) of the forming O(3)-C(5) bond at the forward (positive) and backward (negative) points in the IRC analysis, starting from TS1_O (point 0).
Moreover, to complete the study, the oxidation reaction on C4, which is not adjacent to any heteroatom, was also considered. As expected, it presents very high barriers (about 30 kcal/mol).
With regard to reaction progression, the second step easily occurred with a new H transfer, with higher barriers for the exo pathway with respect to the endo one, both in vacuo and in solvent (ΔΔGvacuo(TS2_endo) = 22.70 kcal/mol, ΔΔGvacuo(TS2_exo) = 29.56 kcal/mol; ΔΔGsolvent(TS2_endo) = 21.21 kcal/mol, ΔΔG vacuo (TS2_exo) = 26.94 kcal/mol). The corresponding three-dimensional plots of transition states TS2_endo and TS2_exo are reported in Fig. 8. During the second reaction step, a ring conformational change occurred at the same time as the H transfer. The second transition state (TS2) is formally like the TSs located for thermal beta-elimination of carboxylic acids from esters (Kącka, A, et al., 2018; Kącka, B. A., et al. 2017; Kącka-Zych, A., et al., 2016).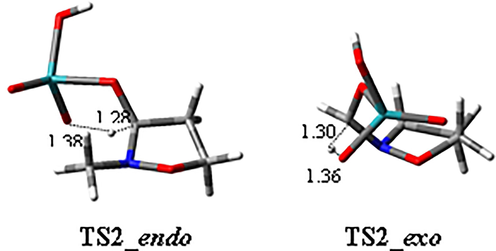
Three-dimensional plots of the transition states TS2 of both the endo and exo pathways in methyl propanoate. Displacement vectors for TS imaginary frequencies (-1523.94, −1606.04 for TS2_endo, TS2_exo, respectively) are shown as dotted lines and distances are reported in angstroms.
Because of the higher barriers, the second step can be defined as the rate-determining one. Conversely, as above discussed, the first step determines the regioselectivity of the reaction. Indeed, from a kinetic point of view, the energy profile is relative to a non-steady state reaction with two irreversible steps. Although the highest transition state is the first, the corresponding step shows smaller activation energy, when compared to the second one. Therefore, reagents will be swiftly converted to P1 that will give slowly the final products.
Considering the exo route, once the P1_exo has been obtained, another possibility is that the second hydrogen atom is extracted not yet at C(1′), but at position C(3), with a competition between TS2_exo and TS2_sec (Scheme 3).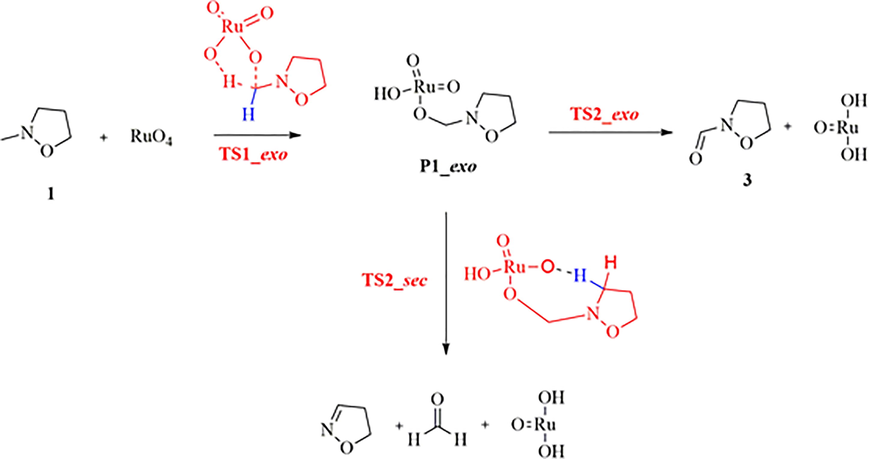
Alternative proposed mechanism for the second step of the oxidation reaction of 2-methylisoxazolidine 1 with RuO4 on C(1′) carbon atom.
With respect to the reagents, the new located transition state TS2_sec presents relative Gibbs energies of −43.67 kcal/mol and −36.72 kcal/mol in methyl propanoate, and in vacuo, respectively (vs −39.72 kcal/mol and −36.04 kcal/mol of TS2-exo), confirming the possible competition of the two reactions.
The IRC analysis in the forward direction (Figure S4) gives a complex (Complex_TS2_sec) that evolves to formaldehyde, 4,5-dihydroisoxazole (see Scheme 3) and to the precursor, for the regeneration of the oxidizing agent.
Finally, in order to check the validity of the obtained results, calculations were repeated with the same functional, but taking under consideration diffuse and polarization functions, using the larger basis set 6–31 + G(d,p), and also evaluating a different DFT method (b3lyp/def2svp/emp = gd3bj/int = ultrafine/solvent = water). The corresponding energy profiles (Figures S5-S7) are in all cases very similar to those reported in Fig. 6, confirming the reliability of the presented data.
3 Conclusions
In this paper, we report a computational mechanistic study of the oxidation reaction of 2-methyl isoxazolidine using RuO4, taking into consideration the different sites where the oxidation could take place. Firstly, we considered the oxidation at position 3 (endo) and 1′ (exo), nearby the nitrogen atom. In vacuo, we confirmed a highly asynchronous [3 + 2] one-step mechanism. In particular, the first step of the reaction consists in a H-transfer and a O-C bond formation that occur separately, within the same reaction coordinate, through a five-membered transition state. When using methyl propanoate as solvent, in the first step, exclusively for the endo route, when the H-transfer occurs, an ion pair that evolves to the P1-endo product is located.
However, since all the hydrogen atoms of the isoxazolidine system, near to the heteroatoms, could be transferred by oxidation with RuO4, the reaction at position 5 was also taken under consideration. In general, the selectivity, tuned by the stability of the corresponding transient carbocation, was detected. The energy barriers for the oxidation of the various carbon atoms of the isoxazolidine system (C3, C4, C5, C1′) are correlated to the stability of the carbocation, leading to N-methylisoxazolidin-3-one as preferred product.
The IRC analyses showed that, during the first step, the C-H distance of the breaking bond is stable until the transition state and then, starts to grow up. Calculations allowed to suppose an electron transfer, in which the electron jumps from isoxazolidine system to Ru. The negatively charged RuO- moiety carries away the proton and a subsequent rearrangement takes place with the formation of the ion pair.
The energy barrier of the second step is higher than that of the first one. Thus, the second step can be defined as rate-determining, taking into account all the catalytic cycle. Conversely, the first step, that is irreversible, determines the regioselectivity of the reaction, and can be considered as the regioselectivity-determining step.
CRediT authorship contribution statement
Maria Assunta Chiacchio: Conceptualization, Writing – original draft, Data curation, Investigation. Daniela Iannazzo: Validation, Visualization, Funding acquisition, Resources. Salvatore V. Giofrè: Data curation, Investigation, Validation, Visualization. Roberto Romeo: Validation, Visualization. Laura Legnani: Conceptualization, Writing – original draft, Data curation, Investigation.
Acknowledgements
Authors are very grateful to Professor Pedro Merino and Professor Ugo Chiacchio for helpful discussions and Dr. Stefano Piacentini for his contribution to preliminary data.
Authors thank Universities of Catania, Messina and Milano-Bicocca for partial financial support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The mechanism of RuO4-mediated oxidations of saturated hydrocarbons. reactivity, kinetic isotope effect and activation. Acta Chem. Scand.. 1992;46:644-649.
- [Google Scholar]
- The RuO4 oxidation of cyclic saturated hydrocarbons. formation of alcohols. Acta Chem. Scand. B. 1986;B40:430-433.
- [Google Scholar]
- Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem.. 1998;19:404-417.
- [Google Scholar]
- A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys.. 1997;107:3032-3042.
- [Google Scholar]
- Pivotal neighbouring group participation in substitution vs elimination reactions: computational evidence for ion pairs in the thionation of alcohols with Lawesson's reagent. Eur. J. Org. Chem. 2017:1952-1960.
- [Google Scholar]
- 1,2,4-Oxadiazole-5-ones as analogues of tamoxifen: synthesis and biological evaluation. Org. Biomol. Chem.. 2019;17:4892-4905.
- [Google Scholar]
- Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. J. Chem. Phys. Lett.. 1996;255:327-335.
- [Google Scholar]
- Oxidation of cis- and trans-pinane with RuO4 generated in situ. Inorg. Chim. Acta. 1994;222:115-122.
- [Google Scholar]
- Theoretical study of the oxidation of alcohol to aldehyde by d0 transition-metal−oxo complexes: combined approach based on density functional theory and the intrinsic reaction coordinate method. Organometallics. 1997;16:716-724.
- [Google Scholar]
- The mysticism of pericyclic reactions: a contemporary rationalisation of organic reactivity based on electron density analysis. Eur. J. Org. Chem. 2018:1107-1120.
- [Google Scholar]
- Ruthenium tetraoxide oxidations of alkanes: DFT calculations of barrier heights and kinetic isotope effects. J. Org. Chem.. 2006;71:1755-1760.
- [Google Scholar]
- Regioselectivity, stereoselectivity, and molecular mechanism of [3 + 2] cycloaddition reactions between 2-methyl-1-nitroprop-1-ene and (Z)-C-aryl-N-phenylnitrones: a DFT computational study. J. Mol. Model. 2018;24:329.
- [Google Scholar]
- RuO4-mediated oxidation of secondary amines. 2. Imines as main reaction intermediates. J. Serb. Chem. Soc.. 2017;82:627-640.
- [Google Scholar]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B. et al., 2016. Gaussian 16, Revision C.01, Inc., Wallingford, CT.
- Why are olefins oxidized by RuO4 under cleavage of the carbon−carbon bond whereas oxidation by OsO4 yields cis-diols? J. Am. Chem. Soc.. 2004;126:3642-3652.
- [Google Scholar]
- Synthesis of spiro[isoindole-1,5’-isoxazolidin]3(2H)-ones as potential inhibitors of the MDM2-p53 interaction. Beilstein J. Org. Chem.. 2016;12:2793-2807.
- [Google Scholar]
- Competition between one-step and two-step mechanism in polar [3 + 2] cycloadditions of (Z)-C-(3,4,5-trimethoxyphenyl)-N-methyl-nitrone with (Z)-2-EWG-1-bromo-1-nitroethenes. Comput. Theor. Chem.. 2018;1125:77-85.
- [Google Scholar]
- Does a fluorinated Lewis acid catalyst change the molecular mechanism of the decomposition process of nitroethyl carboxylates? Res. Chem. Intermed.. 2018;44:325-337.
- [Google Scholar]
- Alternative mechanistic paths in the hetero-diels-alder reaction of α-oxothiones: a theoretical study. Eur. J. Org. Chem. 2007:3547-3554.
- [Google Scholar]
- Computational mechanistic study of the Julia-Kocieński reaction. J. Org. Chem.. 2015;80:3092-3100.
- [Google Scholar]
- Computational mechanistic study of thionation of carbonyl compounds with Lawesson's reagent. J. Org. Chem.. 2016;81:7733-7740.
- [Google Scholar]
- Introducing topology to assess synchronicity of organic reactions. dual reactivity of oximes with alkenes as a case study. Org. Chem. Front.. 2017;4:1541-1554.
- [Google Scholar]
- BET & ELF quantum topological analysis of neutral 2-aza-cope rearrangement of γ-alkenyl nitrones. Molecules. 2017;22:1371-1384.
- [Google Scholar]
- On the question of zwitterionic intermediates in the [3 + 2] cycloaddition reactions between C-arylnitrones and perfluoro 2-methylpent-2-ene. Molecules. 2021;26:7147.
- [Google Scholar]
- Transient and intermediate carbocations in ruthenium tetroxide oxidation of saturated rings. Beilstein J. Org. Chem.. 2019;15:1552-1562.
- [Google Scholar]
- Studies towards the synthesis of polyoxygenated steroids. reaction of some tri- and tetra-substituted monoene steroids with RuO4. Tetrahedron. 1993;49:4211.
- [Google Scholar]
- First example of direct RuO4-catalyzed oxidation of isoxazolidines to 3-isoxazolidones. J. Org. Chem.. 2007;72:3958-3960.
- [Google Scholar]
- Selectivity versus reactivity - recent advances in RuO4-catalyzed oxidations. Synthesis. 2005;15:2453.
- [Google Scholar]
- Practical and rapid vicinal hydroxylation of alkenes by catalytic ruthenium tetraoxide. Angew. Chem. Int. Ed. Engl.. 1994;33:2312-2313.
- [Google Scholar]
- Shing, T. K. M, Tam, E. K. W., Tai, V. W. F.et al, 1996. Ruthenium-Catalyzed cis-Dihydroxylation of Alkenes: Scope and Limitations. Chem. Eur. J., 2, 50−57.
- Tenaglia, A., Terranova, E., Waegell, 1990. Ruthenium-catalysed C–H bond activation. Evidence for a concerted mechanism in oxyfunctionalization of cyclic saturated hydrocarbons. J. Chem Soc. Chem. Commun., 1344−1345.
- The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor.Chem. Acc. 2008:215-241.
- [Google Scholar]
- Density functionals with broad applicability in chemistry. Acc. Chem. Res.. 2008;41:157-167.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104063.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




