

Translate this page into:
Synthesis and pharmacological evaluation of 3-[5-(aryl-[1,3,4]oxadiazole-2-yl]-piperidine derivatives as anticonvulsant and antidepressant agents
⁎Corresponding author at: Department of Pharmaceutical Sciences, Dibrugarh University, Dibrugarh 786004, Assam, India. kamaruzzaman.du@gmail.com (Kamaruz Zaman)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In the present study, we have synthesized a series of fifteen nipecotic acid 1,3,4-oxadiazole based hybrids with significant (60–78%) yields. All the compounds were characterized by using different spectroanalytical techniques such as FT-IR, 1H NMR, 13C NMR, and elemental analysis. This design strategy was validated by using in vivo anti-epileptic and anti-depressant bioassay models. Anti-convulsant activity was evaluated using subcutaneous pentylenetetrazol (scPTZ) in mice and MES induced seizure. Among a spectrum of activities, three compounds (4i, 4m, and 4n) displayed significant activity against pentylenetetrazole (scPTZ) induced seizures. No disruptions in motor co-ordination were observed in mice pretreated with the test compounds in the rotarod test. Their influence on the safety profile of elevated serum levels of biochemical markers such as hepatic and renal toxicity has been found to be safe. The derivatives also show marked anti-depressant activity, devoid of serotonergic augmentation as assessed using the despair swim test, 5-hydroxytryptophan (5-HTP)-induced head twitch test and learned helplessness test. In silico docking studies targeted on homology modelled GABA transporter 1 (GAT1) protein shows the critical enzyme-ligand interactions leading to the inhibition of the GAT1 transporter. The compound 4m was found to be the most active compound among all the synthesized compounds.
Keywords
Nipecotic acid
Piperidine
1,3,4-Oxadiazole
Anti-convulsant
Anti-depressant

1 Introduction
Epilepsy is a chronic noncommunicable brain disorder that affects more than 50 million people globally. After cerebrovascular diseases and dementia, epilepsy is one of the most common neurological condition faced by human being (Krämer, 2001). The evidence of epidemiological factsheet suggests that approximately 80% of epileptic patients are from developing regions (World Health Organization, 2018) (http://www.who.int/en/news-room/fact-sheets/detail/epilepsy). Despite the availability of a plethora of antiepileptic drugs (AEDs), their associated adverse effects lead to the treatment failure of about 25% of patients (de Kinderen et al., 2014; Perucca and Gilliam, 2012). One of the primary class of AEDs elicit their activity via enhancement of γ-aminobutyric acid (GABA) mediated inhibition of synaptic neurotransmission in the cerebral cortex. These drugs either stimulate the influx of chloride ions by acting at GABAA receptors or increase the synaptic GABA levels by inhibiting the catabolism or reuptake (Treiman, 2001). The potent in vitro neuronal and glial GABA reuptake inhibitory activity of nipecotic acid, a piperidine-3-carboxylic acid and corresponding systemic inactiveness led to the discovery of the N-substituted lipophilic congener tiagabine (Das et al., 2012). The latter demonstrated an improved ability to cross the blood-brain barrier, and to date, the only clinically approved reuptake inhibitor. Nevertheless, over the decades, the GABA reuptake inhibitors have been an area of continued interest as potential AEDs (Schousboe et al., 2014). Similarly, various ester-linked prodrugs enhanced the brain delivery of nipecotic acid and its neurotrophic activity (Manfredini et al., 2002; Wang et al., 2005).
In recent years, apart from nipecotic acid, derivatives of the former (Petrera et al., 2016; Quandt et al., 2013) together with piperidine-based natural and synthetic compounds were reported to possess potent antiepileptic activity (Mishra et al., 2015; Siddiqui et al., 2012). Our previous study on nipecotic acid derivatives reported the substitution of the nitrogen atom of the piperidine ring with aryl-substituted 1,3,4-oxadiazole (Singh et al., 2018). As part of the ongoing research, the present work investigates the level of anticonvulsant activity concerning the variation of 1,3,4-oxadiazole substitution at the third position of the piperidine ring. A series of 3-substituted piperidine were synthesized bearing 5-aryl substituted 1,3,4-oxadiazole nucleus. Herein, we esterified the carboxylic acid portion of nipecotic acid into ethyl nipecotate followed by refluxing with hydrazine hydrate. Thereafter, cyclization of piperidine-3-carbohydrazide with substituted aryl acid yielded the 1,3,4-oxadiazole based piperidine derivatives. The linkage of oxadiazole to the piperidine skeleton was contemplated to anchor a lipophilic group that will provide adequate interaction of the molecule with the target site. In addition to imparting the lipophilicity to the piperidine moiety, the covalent attachment of the 1,3,4 oxadiazole motif was carried out to achieve a hybrid fusion of two active pharmacophores. Several research groups reported the versatile nature of 1,3,4 oxadiazole nucleus that possesses a wide range of biological activities viz. antimicrobial (Bala et al., 2014), anticancer (Yadav et al., 2017), anti-inflammatory (Banerjee et al., 2015)among other activities. Furthermore, various 1,3,4 oxadiazole derivatives demonstrated potential anticonvulsant (Zarghi et al., 2008)and antidepressant activities (Jubie et al., 2015; Singh et al., 2012). On this basis, the designed compounds were synthesized, characterized, and activity assessed using in vivo antiepileptic and antidepressant bioassay models. Additionally, the study also evaluated their relative gastric, hepatic and renal safety profiles in comparison to the standard.
2 Results and discussion
2.1 Chemistry
The target compounds (4a-4o) were synthesized as per the sequence outlined in Scheme 1. Intermediate (2) was synthesized by refluxing the piperidine-3-carboxylic acid (1) with absolute ethanol in the presence of 2–3 drops of sulphuric acid for six hour. The FT-IR spectrum of (2) exhibited distinct stretching bands for the secondary amine at 3230 cm-1and an ester stretching at1712 cm−1 respectively. The intermediate piperidine-3-carbohydrazide (3) was obtained in appreciable yield by gently refluxing the ester (2) with hydrazine hydrate in ethanol. Key evidence for the formation of (3) as observed in FT-IR spectrum was the disappearance of the ester (C⚌O) stretching band and the subsequent appearance of –NH stretching bands at 3422, 3274 and 3202 cm−1 respectively. The reaction of the intermediate (3) with different substituted benzoic acid derivatives in phosphorous oxychloride afforded the target compounds (4a-4o) via dehydration followed by cyclization.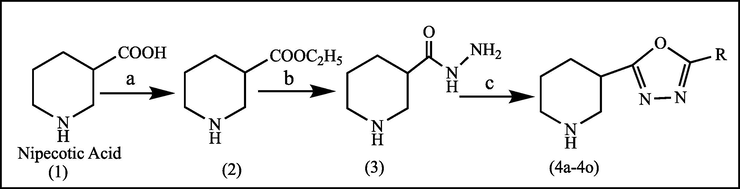
Synthesis of target compounds (4a-4o): (a) Absolute ethanol, sulphuric acid, Reflux 6 h, (b) NH2NH2·H2O, ethanol, reflux 6 h and (c) POCl3, substituted aryl acid, Reflux 6–8 h.
Analysis of 1H NMR spectra for compounds (4a-4o) exhibited the segregation of peaks in four distinct regions. The protons of the piperidine ring system were the most shielded and observed in between δ 3.73–1.37. The chemical shifts of aromatic protons were observed in between δ 9.19–6.36. Lastly, the proton of the secondary nitrogen present in piperidine ring was the most shielded and was observed at around δ 1.23–1.06. The 13C NMR peaks were as per the expected chemical shift. Results of the elemental analysis were within ±0.4% of the theoretical values. The partition coefficient was determined using the octanol/water “shake-flask” method. The Rf values, percentage yield, melting point and log P values of the compounds (4a-4o) are presented in Table 1. The experimental log P values and a mean value of 3.34 suggest that log P of the compounds are within the prescribed limit (log P ≤ 5) for CNS drugs and indicate good CNS penetration (Perucca and Meador, 2005; Pajouhesh and Lenz, 2005).
Compound
R
Rf
% yield
M.P. (°C)
Log P
4a
Phenyl
0.55
62
155–157
2.82
4b
4-Chlorophenyl
0.56
74
164–166
3.57
4c
3-Chlorophenyl
0.58
76
161–163
3.57
4d
3-Cyanophenyl
0.61
68
171–173
3.05
4e
2,4-Dihydroxyphenyl
0.57
62
185–187
3.24
4f
4-Methoxyphenyl
0.62
61
192–200
3.89
4g
3-Methoxyphenyl
0.57
62
188–190
2.89
4h
2,4-Dichlorophenyl
0.59
74
166–168
3.63
4i
4-Nitrophenyl
0.55
76
172–174
3.55
4j
2-Hydroxyphenyl
0.56
67
155–157
3.93
4k
3-Nitrophenyl
0.52
78
158–160
3.20
4l
2,4,5-Trifluorophenyl
0.53
78
162–164
3.49
4m
3,5-Dinitrophenyl
0.58
77
166–168
3.11
4n
4-Aminophenyl
0.59
78
177–179
3.21
4o
2-Amino-4,5-dimethoxyphenyl
0.54
71
170–172
2.96
2.2 Pharmacology
2.2.1 Anti-epileptic activity
2.2.1.1 Pentylenetetrazole (PTZ) induced convulsions in mice
PTZ-induced convulsions and maximal electroshock seizure (MES) test, are most frequently used validated rodents model to screen anti-epileptic activity of drugs. PTZ antagonizes the inhibitory function of GABA by binding to the picrotoxin-binding site of the postsynaptic GABAA receptor (Macdonald and Barker, 1978).
This action causes frequent depolarization and convulsions (Kondziella et al., 2003; Macdonald and Barker, 1978).
As depicted in Table 2, the test compounds 4i, 4m, and 4n protected the mice against PTZ-induced convulsions as the latency of tonic seizures increased significantly in comparison to control group. Considering the percentage of protection of 4i (83.3%), 4m (100%) and 4n (66.66%), the 3,5-dinitrophenylderivative (4m) exhibited the activity on a par with tiagabine. Standard drug tiagabine protected all the treated animals from seizures and clonic-tonic convulsions. Thus the ability of the derivatives (4i, 4m, and 4n) to prevent seizures and clonic-tonic convulsions in the experimental animals was promising and subjected to further pharmacological evaluations. Apart from compounds discussed above, the other derivatives failed to elicit a minimum level of protection compared to the “standard,” and they were excluded from further evaluation. Values are expressed as the Mean ± SEM (n = 6). *p < 0.05 vs. Control; Control: 30% w/v PEG 400 in distilled water; Tiagabine: (10 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Test compounds (4a-4o) were administered at an equimolar intraperitoneal dose relative to 10 mg/kg tiagabine in 30% w/v PEG 400 in distilled water.
Sr. No
Group
% Protection
Latency of tonic convulsion (Minutes)
1
Control
0
8.91 ± 0.97
2
4a
0.0
10.41 ± 0.97
3
4b
0.0
9.83 ± 1.0.83
4
4c
0.0
10.13 ± 1.55
5
4d
0.0
10.16 ± 1.94
6
4e
0.0
9.16 ± 1.60
7
4f
0.0
10.50 ± 1.22
8
4g
0.0
10.75 ± 1.21*
9
4h
0.0
10.66 ± 2.16*
10
4i
83.3
15.51 ± 1.06*
11
4j
0.0
9.66 ± 1.63
12
4k
0.0
10.41 ± 1.11
13
4l
0.0
9.83 ± 1.29
14
4m
100
–
15
4n
66.66
14.16 ± 1.75*
16
4o
0.0
9.25 ± 1.29
17
Tiagabine
100
–
2.2.1.2 Maximal electroshock (MES) test in mice
In this test, only those compounds that showed activity in the sc-PTZ model were selected. As observed in the present study none of the piperidine derivative (4i, 4m, and 4n) including standard drug tiagabine protected the animals against MES induced seizures (Table 3). It is well proven fact that tiagabine and structurally similar drugs are only active against PTZ-induced convulsions and fail to elicit a protective response in MES (Swinyard et al., 1952a; Shiah and Yatham, 1998). Values are expressed as the Mean ± SEM (n = 6); Control: 30% w/v PEG 400 in distilled water; Tiagabine: (10 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Test compounds (4a-4o) were administered at an equimolar intraperitoneal dose relative to 10 mg/kg tiagabine in 30% w/v PEG 400 in distilled water.
Compounds
% protection
Latency of hind limb extension (seconds)
Control
0.0
6.83 ± 030
Tiagabine
0.0
7.83 ± 030
4i
0.0
7.86 ± 0.16
4n
0.0
7.50 ± 0.34
4m
0.0
7.16 ± 0.46
Thus, it may be concluded that the inhibitory action on GAT leads to increase in the synaptic GABA levels and resultant anti-epileptic activity of the synthesized compounds.
2.2.1.3 Rota-rod test in mice
The rota-rod test is used to check the motor impairment, ataxia, loss of skeletal muscle strength, and acute neurotoxicity if any caused by the synthesized compounds. The compounds that were effective in anticonvulsant activity in a PTZ-induced convulsions model were evaluated using this validated rodent model. Significant reduction in “fall-off” time post-treatment was considered as an indication of impairment of motor performance in the test. From the results summarized in Table 4, it is clear that compounds 4i, 4m, 4n and tiagabine are devoid of any impairment in muscle co-ordination amongst the treated animals. However standard drug diazepam significantly reduced the “fall-off” time post-treatment. Values are expressed as the Mean ± SEM (n = 6). *p < 0.05 vs. Control; Diazepam: (10 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Tiagabine: (10 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Test compounds were administered at an equimolar intraperitoneal dose relative to 10 mg/kg tiagabine in 30% w/v PEG 400 in distilled water
Compounds
Before (Sec)
After (Sec)
Control
187.28 ± 2.23
188.42 ± 2.29
Diazepam
193.66 ± 4.41
96.66 ± 6.40*
Tiagabine
192.15 ± 2.19
188.24 ± 2.11
4i
178.83 ± 7.70
181.5 ± 6.28
4n
182.5 ± 2.88
186.83 ± 1.94
4m
175.66 ± 6.86
190.5 ± 1.96
2.2.2 Anti-depressant activity
Recent animal experiments and clinical observations have indicated a close link between epilepsy and depression. Although monoamine theory has been the central theme to explain the etiology of depression, a depleted level of GABA has also been identified with existing depression in both experimental animal models and clinical studies (Shiah and Yatham, 1998).
At the same time low level of GABA is hallmark of epileptic and associated conditions. Thus, considering the connecting role of GABA between depression and epilepsy it was postulated to evaluate the anti-depressant activity of the potential compounds using validated rodents model such as despair swim test, the learned helplessness test, and 5-HTP induced head twitches test. Compounds exhibiting significant anti-epileptic activity (4i, 4m, and 4n) were selected for the screening of anti-depressant activity.
2.2.2.1 Forced swim test in rats
This test uses immobility period of rats characterized by passive swimming preceded by vigorous effort to come out of bucket water, an indicator of depression. In this test reduction in immobility period during the test session subsequently after a training session; is a marker of anti-depressant activity. Compounds 4i, 4m, and 4n along with standard drug imipramine and tiagabine showed significant anti-depressant activity as indicated by decreased immobility period. Results are summarised in Table 5. Values are expressed as the Mean ± SEM (n = 6). *p < 0.05 vs. Control; Imipramine: (50 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Tiagabine: (10 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Test compounds were administered at an equimolar intraperitoneal dose relative to 10 mg/kg tiagabine in 30% w/v PEG 400 in distilled water.
Compound
Immobility period (Second)
Head twitches
Control
180.66 ± 5.35
9.33 ± 0.89
Imipramine
94.16 ± 7.88*
18.83 ± 1.64*
Tiagabine
107.50 ± 3.13*
9.78 ± 1.09
4i
114.33 ± 6.25*
10.66 ± 0.83
4n
107.83 ± 6.67*
9.83 ± 1.14
4m
103.33 ± 8.43*
10.66 ± 2.77
2.2.2.1.1 5-HTP-induced head twitches in mice
5-HTP is a precursor of 5-HT that induces a typical head twitch response (HTR) in mice. Any drug that augments serotonergic activity also potentiates the HTR caused by 5-HTP. In this test only imipramine exhibited the potentiation of 5-HTP-induced head twitches while test compounds and tiagabine were devoid of any such response indicating that the selected compounds do not elicit anti-depressant activity via interaction with serotonergic receptors. Results are summarised in Table 5.
2.2.2.2 Learned helplessness test
In this test, the ability of rats to elicit avoidance to a shock treatment by jumping to another chamber in response to a pre-shock stimulus is an indicator of anti-depressant activity. More the number of the avoidance response in 30 trials session of the test, the better is the anti-depressant activity of the administered drug. On the contrary escape failure in response to a stimulus is a sign of depression. In this test, the drug and imipramine-treated rats performed more avoidance consecutive test days in anticipation to similar to standard drug imipramine. The result is summarized in Table 6. Values are expressed as the Mean ± SEM (n = 6). *p < 0.05 vs. Control; EF: Escape failure; AR: Avoidance response; Imipramine: (50 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Tiagabine: (10 mg/kg, i.p) in 30% w/v PEG 400 in distilled water; Test compounds were administered at an equimolar intraperitoneal dose relative to 10 mg/kg tiagabine in 30% w/v PEG 400 in distilled water.
Group
Day 1
Day 2
Day 3
EF
AR
EF
AR
EF
AR
Control
19.16 ± 1.72
10.83 ± 1.72
17.66 ± 1.21
12.33 ± 1.21
15.16 ± 0.75
14.83 ± 0.75
Imipramine
12.83 ± 1.47*
17.46 ± 1.47*
8.5 ± 0.83*
21.50 ± 0.83
4.66 ± 1.21*
25.33 ± 1.21*
Tiagabine
13.16 ± 0.47*
16.83 ± 0.47*
11.16 ± 0.47*
18.83 ± 0.47*
8.50 ± 0.56*
21.50 ± 0.56*
4i
16.5 ± 1.51*
13.5 ± 1.51*
12.33 ± 0.51*
17.66 ± 0.51*
8.66 ± 0.51*
21.33 ± 0.51
4n
14.66 ± 2062*
15.33 ± 2.06*
9.83 ± 0.98*
20.16 ± 0.98*
7.50 ± 0.83*
22.50 ± 0.83*
4m
16.83 ± 2.13*
13.16 ± 2.13*
11.66 ± 0.81*
17.00 ± 3.03*
8.83 ± 0.40*
21.16 ± 0.40
Many hypotheses have been put forward to explain the anti-depressant activity of drugs out of which monoamine theory is most validated. In this theory, monoamine viz. serotonin and noradrenaline are essential neurotransmitters that play a vital role in the pathophysiology of the depression. Augmentation of these neurotransmitters at the synapse is critical in alleviating depression. The synthesized compounds showed significant anti-depressant activity in forced swim test and learned helplessness test without altering 5-HTP-induced head twitches. The absence of HTR indicates that anti-depressant activity of the compounds as seen in other two models is devoid of serotonergic augmentation (Lattmann et al., 2009).
2.2.3 Hepatic and renal functions of rats in sub-chronic toxicity study
Therapeutic intervention of epilepsy necessitates long term usage of antiepileptic drug thereby increasing the chances of adverse drug reaction. Approximately, 40% of the patients get affected leading to treatment failure (Perucca and Meador, 2005). Hepatotoxicity is the most common adverse effect associated with antiepileptic drugs (Björnsson, 2008).
Chronic usage of antiepileptic drugs can cause weakened bone, and few can affect the mood such as levetiracetam has a higher risk of causing mood disturbance whereas lamotrigine and valproic acid may have mood stabilizing effects (Shin et al., 2014).
Further, the teratogenic effect of valproic acid is also well-documented (Fathe et al., 2014).
First generation antiepileptic drugs in general and carbamazepine, phenytoin, primidone, and benzodiazepines in particular, exhibit significant risk of motor impairment (Banerjee et al., 2015). A generalized hypersensitivity reaction along with drug-induced liver injury is involved with lamotrigine, carbamazepine, phenobarbital, and phenytoin have a considerable potential to induce hypersensitivity reaction along with liver injury (Ahmed and Siddiqi, 2006).
The possible toxic effect of the synthesized compounds was assessed by a subchronic toxicity study according to OECD guideline 407. The rats were treated with the standard tiagabine and potential derivatives (4i, 4m, and 4n) for 30 consecutive days. Thereafter, blood samples were withdrawn and evaluated for hepatic and renal function parameters. Elevated levels of serum aspartate aminotransferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase (ALP) indicate hepatotoxicity whereas, increased total protein (TP) and albumin (TA) are markers of impaired renal function (Table 7). The results demonstrate that the selected derivatives are relatively safe and devoid of any significant hepatic and renal toxicity. Values are expressed as the Mean ± SEM (n = 6); AST: serum aspartate aminotransferase; ALT: Serum Alanine aminotransferase; ALP: Alkaline phosphatase; TP: Total Protein; TA: Total Albumin
Comp.
AST
(IU/L)ALT
(IU/L)ALP
(IU/L)TP
(gm/dL)TA
(gm/dL)Ulcer Index
Control
43.33 ± 0.83
39.83 ± 1.16
138.16 ± 4.11
4.48 ± 0.31
2.75 ± 0.28
0
Tiagabine
41.65 ± 0.47
43.62 ± 1.28
142.16 ± 2.04
4.18 ± 0.23
2.89 ± 0.13
20.0 ± 0.54
4i
41.50 ± 2.81
40.96 ± 0.98
140.33 ± 2.94
4.35 ± 0.29
2.73 ± 0.19
0
4m
40.50 ± 1.87
41.16 ± 1.16
139.16 ± 3.48
4.60 ± 0.26
2.75 ± 0.23
0
4n
42.50 ± 2.07
40.83 ± 1.16
142.83 ± 2.56
4.57 ± 0.28
2.75 ± 0.19
0
2.2.4 Ulcerogenic studies
Compounds (4i, 4m, and 4n) exhibiting promising anti-epileptic and anti-depressant activity profiles were further evaluated for their ulcerogenic potential expressed in terms of ulcer index (UI). The stomach of the sacrificed animals revealed no histological changes or ulcers. However, tiagabine treated rats were having mild ulcers. The lack of ulcerogenic potential might be due to absence of carboxylic group as compared to previously reported derivatives (Singh et al., 2018) that possess free carboxylic acid group and exhibited mild ulcerogenic potential. Results are depicted in Table 8. *All the interactions of protein-ligand were observed within the distance of 3 Å.
Comp.
Glide Score
Hydrogen bonding
Salt bridge
Active site residues
4i
−4.11
Asp451
Na611
Tyr 60, Ala61, Gly63, Leu64, Gly65, Asn66, Leu136, Tyr139, Tyr140, Phe294, Ser295, Tyr296, Gly297, Leu300, Ser396, Tyr452, Ser456, Gly457, Ser460
4m
−4.34
Asp451
Na611
4n
−5.72
Tyr 60, Asp451
—
Tiagabine
−4.4
Gly65, Tyr140, Phe294
Na611
2.3 Computational studies
2.3.1 In silico homology modeling and docking simulations
The homology model was built by using Drosophila melanogaster dopamine transporter (dDAT) as a template, [PDB Code: 4XP4_A] (Supplementary Fig. 1) due to unavailability of GAT1 X-ray crystal structure (Penmatsa et al., 2015). Ramachandran plot revealed that all the amino acids except Phe 174 and Ser 178 were present in the favoured/allowed regions (Supplementary Fig. 2). The quality of the model allowed us to use the best model for the docking study as the two amino acid residue as mentioned earlier are not part of the ligand-binding site (Zafar & Jabeen, 2018).
Docking simulation studies were performed using the Maestro Schrödinger program to get an insight into the possible protein-ligand interactions using the homology modeled GAT1 GABA transporter. The minimum energy conformer of tiagabine was docked using the prepared grid towards validation of the generated grid and docking protocols (Kontoyianni et al., 2004). The protein-ligand interactions confirmed the similar binding mode of tiagabine within the active site as previously reported (Jurik et al., 2015; Petrera et al., 2016).
Therefore, the parameters set for Glide-XP docking mode were competent in generating reproducible in silico binding mode for GAT1 GABA transporter. The LigPrep® module produced the low-energy conformers of the highest active compounds (4i, 4m, and 4n). The binding pose of 4i and 4m showed hydrogen bonding (H-bond) exclusively with ASP 451 with a G score of −4.11 and −4.34, respectively (Figs. 1 and 2). Alternatively, compound 4n displayed H-bond interaction with TYR 60 and ASP 451 with a G score of −5.72 (Fig. 3). However, the presence of a p-amino phenyl ring in 4n was unable to favour the non-covalent interaction with sodium via salt bridge formation. The hydrogen bond interaction reveals that the protonated hydrogen of piperidine served as hydrogen bond donor that interacts with the carbonyl group of aspartate residue.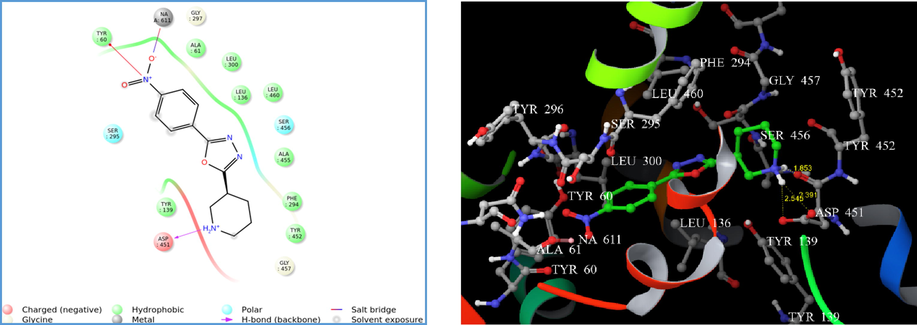
(A) 2D interaction diagram of compound 4i. (B) Docking of compound 4i on homology modelled GAT1 GABA transporter and hydrogen bonded with ASP 451 shown as yellow dash.
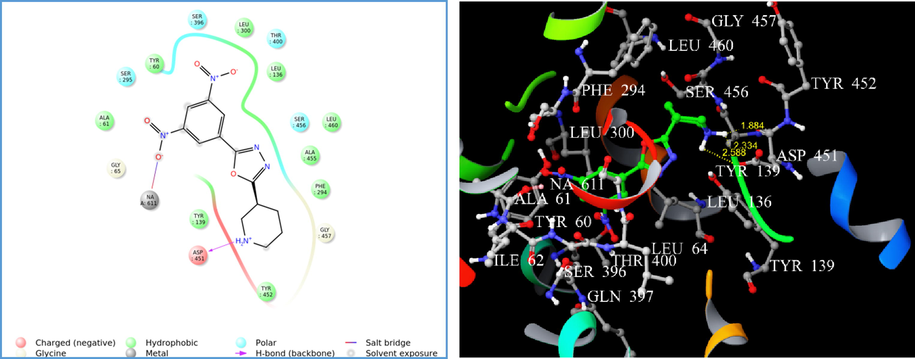
(A) 2D interaction diagram of compound 4m. (B) Docking of compound 4m on homology modelled GAT1 GABA transporter and hydrogen bonded with ASP 451 shown as yellow dash.
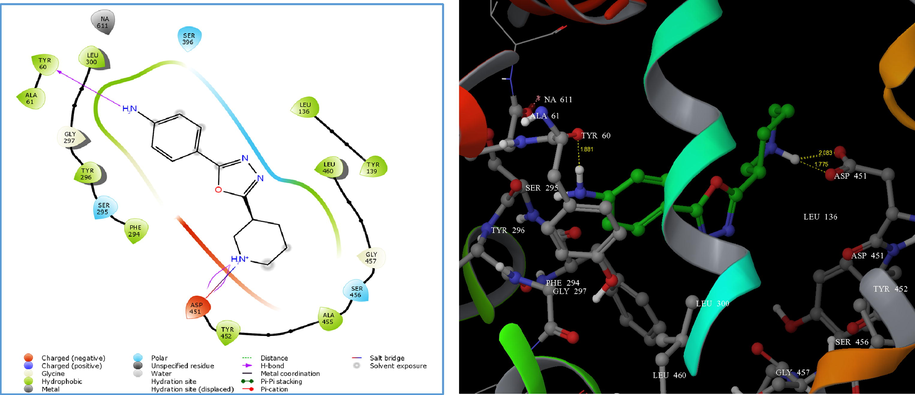
(A) 2D interaction diagram of compound 4n. (B) Docking of compound 4n on homology modelled GAT1 GABA transporter and hydrogen bonded with TYR 60 and ASP 451 shown as yellow dash.
2.3.2 In silico prediction of “drug-like” properties
Prediction of “drug-likeliness” for the active compounds (4i, 4m, and 4n) was carried out using QikProp® module of Maestro Schrödinger (Table 9). #rtvFG: Reactive function groups; PSA: polar surface area; donorHB: No. of hydrogen bonds donor; accptHB: No. of hydrogen bonds acceptor; QPlogPo/w: Predicted octanol/water partition coefficient; QplogBB: Predicted brain/blood partition coefficient; Percent human-oral absorption: human oral absorption on 0 to 100% scale; Rule of five: No. of violations of Lipinski’s rule of five.
Comp
#rtvFG (0–2)
DonorHB (0–6)
AccptHB (2–20)
QPlogBB (−3.0 to 1.2)
% Human oral absorption
(>80% high, <25% poor)Rule of five (Max 4)
4i
0
1
5.00
−0.787
63.73
0
4m
0
1
6.00
−1.847
68.63
0
4n
0
2.5
5.00
−0.433
68.57
0
Computation approaches to predicts the important properties viz. absorption, distribution, metabolism, excretion and toxicity of drugs assure high probability to develop drugs enriched with features that would translate to a safe and orally efficacious drug (Lipinski et al., 1997). Amongst the predicted parameters, the active compounds (4i, 4m, and 4n) are devoid of any reactive functional groups and they have moderate percentage oral absorption. The outcome of Lipinski’s rule of five(mol_MW < 500, QPlog Po/w < 5, donorHB < 5, accptHB < 10), along with the other predicted parameters, indicated that the potential compounds under consideration do possess considerable “drug-like” characteristics (Veber et al., 2002).
3 Conclusion
In our continuous effort of synthesising potential compounds having anti-convulsant and anti-depressant activities, we have reported fifteen new compounds consisting of 3-substituted piperidine ring bearing 5-aryl substituted 1,3,4-oxadiazole nucleus. Compounds having electron withdrawing substituent were found to be more potent than the electron releasing group. Among all the synthesised compounds, 4i, 4m, and 4n displayed promising antiepileptic activity, and, 4m (2,4-dinitro derivative) was found to be the most active compound. These compounds were also found to possess promising antidepressant activity. An in vivo and computational study suggests that the anti-epileptic effects of the synthesised compounds may be due to the inhibition of the GAT1 GABA transporter protein. None of the compounds showed neurotoxicity with good renal and hepatic safety profile.
4 Experimental
4.1 Chemistry
The chemicals and solvents used were of analytical grade or dry distilled. The progress of the reactions was monitored by thin layer chromatography on a pre–coated Merck silica gel 60 F254 aluminum sheets (Merck, Germany). Melting points were determined using open capillary tubes on Stuart melting point apparatus (SMP10) and were uncorrected. IR spectra were recorded on Shimadzu 8400S FT–IR spectrophotometer. 1H NMR (500 MHz) and 13C NMR (125 MHz) were recorded on a Bruker FT–NMR in DMSO–d6 using TMS as an internal standard. C, H, N analyses were performed on an Exeter CE–440 elemental analyzer.
4.1.1 General procedure for the synthesis of piperidine-3-carboxylic acid ethyl ester/ethylnipecotate (2)
Nipecotic acid (0.246 mol), ethanol (2.5 mol) and few drops of concentrated sulphuric acid were heated under reflux for 4 h using a water bath. The progress of the reaction was monitored by TLC using n-hexane/ethyl acetate (1:1) as the mobile phase. Excess of alcohol was evaporated using a rotary evaporator. The residue was poured into a separatory funnel having distilled water and a few ml of carbon tetrachloride was added to separate the lower ester layer. Then evaporate the liquid mixture on rota evaporator to yield the title compound (2).
4.1.2 General procedure for the synthesis of piperidine-3-carboxylic acid hydrazide (3)
Ethyl nipecotate (2) (1 Mol. Eq.) was dissolved in 10 ml methanol by gentle heating at around 60 °C. To this solution, hydrazine hydrate (4 Mol. Eq.) was added dropwise. The reaction mixture was refluxed for 6 h and the progress being monitored by TLC using DCM/methanol (2:8) as the mobile phase. Post completion, the reaction mixture was allowed to cool to room temperature and poured onto crushed ice. The solid that separated was filtered dried and recrystallized from ethanol to yield the title compound (3).
4.1.3 General procedure for the synthesis of compounds (4a–4o)
Equimolar quantities of compound 3 (1 mmol) and different substituted aryl acid (1 mmol) were placed in a round–bottom flask and cooled in an ice bath. 8–10 ml of POCl3 was slowly poured dropwise into the above reaction mixture. After complete addition, the contents were refluxed for 6–8 h. After completion of the reaction (monitored by TLC), the mixture was allowed to cool and poured onto crushed ice and kept overnight. Contents of the beaker were filtered to yield a crude product, which was recrystallized from ethanol to yield the desired compounds (4a–4o).
4.1.4 3-(5-Phenyl-[1,3,4]oxadiazol-2-yl)-piperidine (4a)
White solid; mp 155–157 °C; IR (KBr, cm−1) 3230, 3099, 2885, 1608, 1541, 1498, 1348, 1280, 1180, 1080; 1H NMR (500 MHz,DMSO–d6) δ 7.58–7.57 (2H, d, J = 5.0 Hz, phenyl-H), 7.45–7.42 (2H, d, J = 10.5 Hz, phenyl-H), 7.38–7.36 (1H, m, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10.5, J2 = 5.0 Hz, piperidine-H), 3.04–3.01 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 2.92–2.88 (1H, p, piperidine-H), 2.83–2.79 (2H, dd, J1 = 15.5, J2 = 2.5 Hz, piperidine-H), 2.23–2.19 (2H, m, piperidine-H), 1.64–1.54 (1H, m, piperidine-H), 1.22 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.69, 154.95, 131.57, 129.06, 129.06, 127.08, 127.08, 126.66, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 229.58 (100), 230.58 (M + 1). Anal. C13H15N3O: C, 68.10; H, 6.59; N, 18.33; Found: C, 67.99; H, 6.50; N, 18.01.
4.1.5 3-[5-(4-Chloro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4b)
White solid; mp 164–166 °C; IR (KBr, cm−1) 3280, 3090, 2885, 1608, 1541, 1489, 1358, 1285, 1180, 1080; 1H NMR (500 MHz, DMSO–d6) δ 7.37–7.32 (4H, dd, J1 = 10, J2 = 5.5 Hz, phenyl-H), 3.17–3.13 (1H, dd, J1 = 15.0, J2 = 5.0 Hz, piperidine-H), 2.92–2.88 (1H, dd, J1 = 15.0, J2 = 5.0 Hz, piperidine-H), 2.82–2.77 (2H, m, piperidine-H), 2.67–2.64 (1H, p, piperidine-H), 2.15–2.11 (1H, m, piperidine-H), 1.62–1.51 (2H, m, piperidine-H), 1.42–1.37 (1H, m, piperidine-H), 1.06 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.69, 154.95, 137.31, 129.46, 129.46, 128.39, 128.09, 128.09, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 263.87 (1 0 0), 264.87, 265.10 (M + 1, M + 2). Anal. C13H14ClN3O: C, 59.21; H, 5.35, N, 15.93; Found: C, 59.18; H, 5.32; N, 15.90.
4.1.6 3-[5-(3-Chloro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4c)
Pale yellow solid; mp 161–163 °C; IR (KBr, cm−1) 3282, 3090, 2933, 1604, 1545, 1479, 1398, 1269, 1166, 1091; 1H NMR (500 MHz, DMSO–d6) δ 7.66 (1H, s, phenyl-H), 7.47–7.45 (1H, m, phenyl-H), 7.38–7.37 (2H, m, phenyl-H), 3.73–3.69 (1H, p, piperidine-H), 3.03–3.00 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 15.5, J2 = 5.0 Hz, piperidine-H), 2.82–2.78 (2H, m, piperidine-H), 2.22–2.19 (1H, m, piperidine-H), 1.63–1.55 (3H, m, piperidine-H), 1.22 (1H, s, piperidine-NH).13C NMR (125 MHz, DMSO–d6) δ 163.89, 154.95, 133.48, 130.21, 130.18, 128.91, 127.21, 126.21, 48.01, 46.43, 31.55, 24.33, 24.27. MS (ESI): m/z found 263.57 (1 0 0), 264.57, 265.58 (M + 1, M + 2). Anal. C13H14ClN3O: C, 59.21; H, 5.35, N, 15.93; Found: C, 58.35; H, 5.37; N, 15.86.
4.1.7 3-(5-Piperidin-3-yl-[1,3,4]oxadiazol-2-yl)-benzonitrile (4d)
Pale yellow solid; mp 171–173 °C; IR (KBr, cm−1) 3288, 3090, 2933, 2262, 1644, 1545, 1499, 1398, 1267, 1166, 1091; 1H NMR (500 MHz, DMSO–d6) δ 8.01 (1H, s, phenyl-H), 7.90–7.88 (1H, dd, J1 = 5.0, J2 = 2.5 Hz, phenyl-H), 7.75–7.73 (1H, dd, J1 = 4.0, J2 = 2.5 Hz, phenyl-H), 7.63–7.60 (1H, t, J = 10.5, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10.5, J2 = 5.0 Hz, piperidine-H), 3.03–2.98 (1H, p, piperidine-H), 2.92–2.88 (2H, m, piperidine-H), 2.84–2.79 (1H, dd, J1 = 15.0, J2 = 5.5 Hz, piperidine-H), 2.22–2.19 (1H, m, piperidine-H), 1.64–1.55 (3H, m, piperidine-H), 1.19 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.89, 154.95, 132.10, 129.81, 129.40, 127.09, 126.27, 119.29, 115.45, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 254.25 (1 0 0), 255.25 (M + 1). Anal. C14H14N4O: C, 65.48; H, 5.49; N, 22.00; Found: C, 65.23; H, 5.55; N, 22.03.
4.1.8 4-(5-Piperidin-3-yl-[1,3,4]oxadiazol-2-yl)-benzene-1,3-diol (4e)
Pale yellow solid; mp 185–187 °C; IR (KBr, cm−1) 3423.76, 3268.35, 3064.99, 2762, 1614, 1494, 1384, 1253, 1130, 1047; 1H NMR (500 MHz, DMSO–d6) δ 7.28–7.27 (1H, d, J = 5.0 Hz, phenyl-H), 6.52–6.50 (1H,d, J = 5.5 Hz, phenyl-H), 6.45 (1H, s, phenyl-H), 4.24 (1H, s, OH), 4.03 (1H, s, OH), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 3.03–3.00 (1H, p, piperidine-H), 2.92–2.88 (2H, m, piperidine-H), 2.82–2.78 (1H, dd, J1 = 10.5, J2 = 5.0 Hz, piperidine-H), 2.40–233 (1H, m, piperidine-H), 1.64–1.56 (3H, m, piperidine-H), 1.18 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 159.33, 159.07, 156.52, 154.95, 129.77, 109.09, 105.16, 104.06, 48.00, 46.43, 31.54, 24.32, 24.26. MS (ESI): m/z found 261.58 (1 0 0), 262.58 (M + 1). Anal. C13H15N3O3: C, 58.99; H, 5.70; N, 16.00; Found: C, 59.76; H, 5.79; N, 16.08.
4.1.9 3-[5-(4-Methoxy-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4f)
Pale yellow solid; mp 192–194 °C; IR (KBr, cm−1) 3200, 3109, 2924, 2854, 2802, 1610, 1533, 1350, 1109, 1072; 1H NMR (500 MHz, DMSO–d6) δ 7.56 (2H, d, J = 5.0, phenyl-H), 7.05 (2H, d, J = 5.0, phenyl-H), 3.81 (3H, s, methoxy-H), 3.73–3.69 (1H, dd, J1 = 10.0, J2 = 5.5 Hz, piperidine-H), 3.42–3.35 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 2.83–2.76 (2H, m, piperidine-H), 2.23–2.17 (1H, m, piperidine-H), 1.72–1.56 (3H, m, piperidine-H), 1.23 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.69, 162.19, 154.95, 127.07, 127.07, 121.55, 113.88, 113.88, 56.04, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 259.48 (1 0 0), 260.48 (M + 1). Anal. C14H17N3O2: C, 64.48; H, 6.64; N, 16.21; Found: C, 64.75; H, 6.61; N, 16.20.
4.1.10 3-[5-(3-Methoxy-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4g)
Pale yellow solid; mp 188–190 °C; IR (KBr, cm−1) 3260, 3116, 2924, 2854, 2802, 1610, 1533, 1350, 1109, 1072; 1H NMR (500 MHz, DMSO–d6) δ 7.40–7.37 (1H, t, J = 10.5 Hz, phenyl-H), 7.20–7.19 (2H, d, J = 5.0, phenyl-H), 7.03–7.02 (1H, d, J = 5.0, phenyl-H), 3.81 (3H, s, methoxy-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 3.04–2.98 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 2.84–2.75 (2H, m, piperidine-H), 2.24–2.20 (1H, m, piperidine-H), 1.64–1.54 (3H, m, piperidine-H), 1.19 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.89, 161.44, 154.95, 128.96, 125.25, 120.52, 117.36, 114.11, 56.04, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 259.58 (1 0 0), 260.58 (M + 1). Anal. C14H17N3O2: C, 64.48; H, 6.64; N, 16.21; Found: C, 64.47; H, 6.71; N, 16.57.
4.1.11 3-[5-(2,4-Dichloro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4h)
White solid; mp 166–168 °C; IR (KBr, cm−1) 3269, 3029, 2885,1608, 1581, 1489, 1346, 1298, 1188, 1080; 1H NMR (500 MHz, DMSO–d6) δ 7.53(1H, s, phenyl-H), 7.50–7.48 (1H, d, J1 = 5.0, J2 = 5.0 Hz, phenyl-H), 7.35–7.33 (2H, d, J = 6 Hz, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 3.08–3.02 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 2.83–2.76 (2H, m, piperidine-H), 2.24–2.20 (1H, m, piperidine-H), 1.64–1.55 (3H, m, piperidine-H), 1.21 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 156.67, 154.95, 135.87, 134.93, 131.72, 128.52, 128.47, 127.88, 48.00, 46.43, 31.54, 24.32, 24.26. EIMS (m/z): 297.07 (1 0 0), 298.07, 299.08 (M + 1, M + 2). Anal. C14H17Cl2N3O: C, 53.52; H, 5.45; N, 13.37; Found: C, 53.72; H, 5.47; N, 13.34.
4.1.12 3-[5-(4-Nitro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4i)
Pale yellow solid; mp 172–174 °C; IR (KBr, cm−1) 3263, 3080, 2926, 2852, 1647, 1591, 1473, 1377, 1290, 1244, 1093; 1H NMR (500 MHz, DMSO–d6) δ 8.32–8.31 (2H, d, J = 5.0 Hz, phenyl-H), 7.84–7.83 (2H, d, J = 5.0, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 3.06–2.99 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 2.83–2.76 (2H, m, piperidine-H), 2.24–2.19 (1H, m, piperidine-H), 1.64–1.55 (3H, m, piperidine-H), 1.23 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.69, 154.95, 147.39, 132.08, 127.45, 127.45, 124.68, 124.68, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 274.40 (1 0 0), 275.41 (M + 1). Anal. C13H14N4O3: C, 56.93; H, 5.15; N, 20.43; Found: C, 57.19; H, 5.45; N, 20.47.
4.1.13 2-(5-Piperidin-3-yl-[1,3,4]oxadiazol-2-yl)-phenol (4j)
Pale yellow solid; mp 155–157 °C; IR (KBr, cm−1) 3423, 3262, 3064, 2882, 2762, 1614, 1494, 1384, 1338, 1253, 1130, 1047; 1H NMR (500 MHz, DMSO–d6) δ 8.32–8.31 (2H, d, J = 5.0 Hz, phenyl-H), 7.84–7.83 (2H, d, J = 5.0 Hz, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 3.06–2.99 (1H, p, piperidine-H), 2.92–2.88 (1H, p, piperidine-H), 2.82–2.78 (2H, m, piperidine-H), 2.23–2.19 (1H, m, piperidine-H), 1.64–1.54 (3H, m, piperidine-H), 1.22 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.69, 154.95, 147.39, 132.08, 127.45, 127.45, 124.68, 124.68, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 245.15 (1 0 0), 246.15 [M + 1]. Anal. C13H15N3O2: C, 63.66; H, 6.16; N, 17.13; Found: C, 64.01; H, 6.14; N, 17.17.
4.1.14 3-[5-(3-Nitro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4k)
Pale yellow solid; mp 158–160 °C; IR (KBr, cm−1) 3260, 3080, 2926, 2852, 1647, 1567, 1473, 1337, 1290, 1244, 1093; 1H NMR (500 MHz, DMSO–d6) δ 8.43 (1H, s, phenyl-H), 8.31–8.29 (1H, d, J = 5.5 Hz, phenyl-H), 7.91–7.89 (1H, d, J = 6 Hz, phenyl-H), 7.69–7.68 (1H, d, J = 2.5 Hz, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 3.05–2.99 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 2.84–2.75 (2H, m, piperidine-H), 2.26–2.19 (1H, m, piperidine-H), 1.64–1.55 (3H, m, piperidine-H), 1.19 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 163.89, 154.95, 149.03, 133.70, 130.33, 129.79, 122.67, 121.29, 48.00, 46.43, 31.54, 24.32, 24.26. EIMS (m/z): 274.14 (1 0 0), 275.14 (M + 1). Anal. C13H14N4O3: C, 56.93; H, 5.15; N, 20.43; Found: C, 56.90; H, 5.17; N, 20.44.
4.1.15 3-[5-(2,4,5-Trifluoro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4l)
Pale brown solid; mp 162–164 °C; IR (KBr, cm−1) 3229, 3059, 2885, 1608, 1541, 1498, 1348, 1280, 1180, 1080; 1H NMR (500 MHz, DMSO–d6) δ 7.37–7.33 (1H, d, J = 15.5 Hz, phenyl-H), 6.98–6.94 (1H, dt, J1 = 5.5, J2 = 5.0 Hz, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 3.07–3.01 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 2.84–2.75 (2H, m, piperidine-H), 2.24–2.18 (1H, m, piperidine-H), 1.66–1.54 (1H, m, piperidine-H), 1.23 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 158.85, 155.38, 154.95, 151.96, 147.64, 116.14, 113.78, 106.09, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 283.00 (1 0 0), 284.00 (M + 1). Anal. C13H12F3N3O: C, 55.12; H, 4.27; N, 14.84; Found: C, 55.02; H, 4.24; N, 14.81.
4.1.16 3-[5-(3,5-Dinitro-phenyl)-[1,3,4]oxadiazol-2-yl]-piperidine (4m)
Pale yellow solid; mp 166–168 °C; IR (KBr, cm−1) 3263, 3082, 2922, 2852, 1627, 1591, 1473, 1413, 1377, 1290, 1244, 1093; 1H NMR (500 MHz, DMSO–d6) δ 9.19 (1H, s, phenyl-H), 8.89 (1H, s, phenyl-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 3.06–3.03(1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 2.83–2.76 (2H, m, piperidine-H), 2.36–2.33 (1H, m, piperidine-H), 1.65–1.56 (1H, m, piperidine-H), 1.21 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 162.50, 154.95, 147.47, 147.47, 127.90, 127.33, 127.33, 119.52, 48.01, 46.43, 31.55, 24.33, 24.27. MS (ESI): m/z found 319.00 (1 0 0), 320.00 (M + 1). Anal. C13H13N5O5: C, 48.01; H, 4.17; N, 21.94; Found: C, 48.21; H, 4.10; N, 21.94.
4.1.17 4-(5-Piperidin-3-yl-[1,3,4]oxadiazol-2-yl)-phenylamine (4n)
Pale yellow solid; mp 177–179 °C; IR (KBr, cm−1) 3341, 3242, 3070, 2922, 2856, 1610, 1545, 1472, 1386, 1284, 1078; 1H NMR (500 MHz, DMSO–d6) δ 7.39–7.37 (2H, d, J = 6, phenyl-H), 6.75–6.73 (2H, d, J = 5.0 Hz, phenyl-H), 4.16 (2H, s, phenyl-NH2), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 3.06–3.00 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 2.84–2.75 (2H, m, piperidine-H), 2.25–2.20 (1H, m, piperidine-H), 1.64–1.54 (1H, m, piperidine-H), 1.22 (1H, s, piperidine-NH). 1H NMR (500 MHz, DMSO–d6) δ 7.38, 7.38, 6.74, 6.74, 4.16, 4.16, 3.71, 3.03, 2.90, 2.82, 2.78, 2.22, 1.63, 1.58, 1.56, 1.22. EIMS (m/z): 244.24 (1 0 0), 245.25 (M + 1). Anal. C13H16N4O: C, 63.91; H, 6.60; N, 22.93; Found: C, 64.01; H, 6.67; N, 22.91.
4.1.18 4,5-Dimethoxy-2-(5-piperidin-3-yl-[1,3,4]oxadiazol-2-yl)-phenylamine (4o)
Pale yellow solid; mp 170–172 °C; IR (KBr, cm−1) 3341.66, 3242, 3070, 2994, 2924, 2856, 2804, 1610, 1545, 1479, 1404, 1286, 1184, 1078; 1H NMR (500 MHz, DMSO–d6) δ 7.02 (1H, s, phenyl-H), 6.36 (1H, s, phenyl-H), 4.64 (2H, s, NH2), 3.82 (6H, s, methoxy-H), 3.73–3.69 (1H, dd, J1 = 10, J2 = 5.5 Hz, piperidine-H), 3.09–3.02 (1H, p, piperidine-H), 2.92–2.88 (1H, dd, J1 = 10, J2 = 5.0 Hz, piperidine-H), 2.84–2.75 (2H, m, piperidine-H), 2.25–2.19 (1H, m, piperidine-H), 1.66–1.54 (1H, m, piperidine-H), 1.23 (1H, s, piperidine-NH). 13C NMR (125 MHz, DMSO–d6) δ 154.95, 152.32, 151.21, 144.23, 140.98, 109.71, 109.65, 98.97, 56.79, 56.79, 48.01, 46.43, 31.55, 24.33, 24.27. EIMS (m/z): 304.15 (1 0 0), 305.11 (M + 1). Anal. C15H20N4O3: C, 59.74; H, 6.48; N, 18.71; Found: C, 59.50; H, 6.62; N, 18.41.
4.2 Determination of partition coefficient
The lipophilic constant of all the compounds (4a–4o) was determined in n–octanol and buffer (pH 7.4) by shake flask method (Takacs-Novak and Avdeel, 1996). The LogPo/w was calculated by correlating the absorbance with the concentration in the standard plot.
4.3 Pharmacology
4.3.1 Animals
Adult Charles Foster rats (150 ± 10 g) and Wistar mice (20 ± 5 g) of either sex were used in the study. Animals were obtained from the central animal house of Department of Pharmaceutical Sciences, Dibrugarh University, Dibrugarh, Assam. The animals were housed in groups of six in polypropylene cages at an ambient temperature of 25 ± 1 °C and 45–55% relative humidity, with a 12:12 h light/dark cycle. Animals were provided with commercial food pellets and water ad libitum unless stated otherwise. Animals were acclimatized to laboratory conditions for at least one week before experiments. Principles of laboratory animal care guidelines (NIH publication number 85–23, revised 1985) were followed. Protocols of the in vivo study were approved by Institutional Animal Ethical Committee of Dibrugarh University (Regd. No. 1576/GO/Ere/S/11/CPCSEA) Dated: 30-03-2015 and Approval no. (IAEC/DU/125 Dated: 29-12-2016). All standard drugs and test compounds were dissolved in 30% w/v aqueous solution of polyethylene glycol (PEG) 400 before administration to the animals. Tiagabine was administered as (10 mg/kg, i.p). The synthesized derivatives were administered at an equimolar dose relative to 10 mg/kg of tiagabine. Control group received vehicle comprising of 30% w/v aqueous solution of polyethylene glycol (PEG) 400.
4.3.2 Anti-epileptic activity
4.3.2.1 Subcutaneous pentylenetetrazole (s.c.-PTZ) induced convulsions in mice
Tiagabine (10 mg/kg) and nipecotic acid derivatives (4a-4o) were administered intraperitoneally to the standard and the test groups respectively 1 h prior to PTZ challenge. Post 1 h, the mice were challenged with PTZ (80 mg/kg, s.c.). The number of mice, which exhibited seizures, the latency to first convulsion and percent lethality was recorded (Rudzik et al., 1973).
4.3.2.2 Maximal electroshock (MES) test in mice
MES test is most common test along with s.c.-PTZ test to evaluate the antiepileptic potential of the test drugs. In this test only those compounds that were active in s.c.-PTZ test were selected for the screening. The supramaximal electroshock (50 mA at 60 Hz) was administered through a pair of corneal electrodes for 0.2 sec duration using a convulsiometer. The hind limb extensor response was taken as the positive end point (Yogeeswari et al., 2003).
Mice were pre-screened and only those showing positive hind limb tonic extensor response were used after an interval of at least 48 h. Tiagabine (10 mg/kg), phenobarbitone (60 mg/kg) and nipecotic acid analogues (4i, 4m, and 4n) were administered intraperitoneally 1 h before MES challenge.
4.3.2.3 Rota-rod performance test in mice
This test is used to assess the motor impairment or neurotoxicity caused by the drugs. For this purpose rota-rod apparatus was used. Each animal was placed on a rotating rod (6 rpm) in a pre-test session and only those animals, which stayed on the rod for not less than 3 min, were selected for the test session. The test session was performed on the same day as the pre-test session. “Fall-off” time in seconds (when the mouse falls from the rotating rod) for each animal was noted before and after drug administration (Reddy and Kulkarni, 1998).
Tiagabine (10 mg/kg), diazepam (10 mg/kg) and nipecotic acid derivatives (4i, 4m, and 4n) were administered intraperitoneally 1 h before test session.
4.3.3 Anti-depressant activity
4.3.3.1 Forced swim test in rats
The forced swim test is most commonly used validated rodent model to assess the antidepressant effect of compounds. Tiagabine (10 mg/kg), imipramine (50 mg/kg) and nipecotic acid derivatives (4i, 4m, and 4n) were administered intraperitoneally 1 h before test session. Post 1 h, rats were forced to swim individually for 10 min in a polypropylene vessel (45 × 40 × 30 cm), and a water level of 20 cm ensured that the rat's feet do not touch the floor of the vessel and that it could not climb out of it. After that, during the next 5 min, the total period of immobility, characterized by complete cessation of swimming with the head floating above water level, was noted. After the initial frenzied attempts to escape, the immobility period is postulated to represent ‘behavioral-despair’ as an experimental model of endogenous depression (Porsolt et al., 1977).
4.3.3.2 5-Hydroxytryptophan (5-HTP)-induced head twitches in mice
Tiagabine (10 mg/kg), imipramine (50 mg/kg) and nipecotic acid derivatives (4i, 4m and 4n) were administered intraperitoneally 30 min before 5-HTP administration. The number of head twitches displayed by each mouse treated with 5-HTP (100 mg/kg, i.p.) was counted by the staggering method within three 2 min intervals (19–21 min), (23–25 min) and (27–29 min) post 5-HTP administration.
4.3.3.3 Learned helplessness test in rats
The rationale behind this particular model is that the animals encountered a repeated failure to escape on exposure to a situation beyond their control. The resulting stress due to the helpless state affects their behavioral and physiological performance in the subsequent learning of tasks. (Estrin et al., 1999).
Tiagabine (10 mg/kg), imipramine (50 mg/kg) and nipecotic acid derivatives (4i, 4m, and 4n) were administered intraperitoneally 1 h prior proceeding for inescapable shock pre-treatment and conditioned avoidance training. A typical experiment involves two parts.
-
Inescapable shock pre-treatment
A chamber (20 × 10 × 10 cm) with plexiglass walls and a steel grid floor delivered the electric foot shock to the housed test animals. A constant current was utilized to produce 60 scrambled, randomized inescapable shocks (15 sec, 0.8 mA, per min) to the steel grids. Alternatively, the control rats were placed in an identical chamber with no electrical source for 1 h. Inescapable shock pre-treatment was delivered the following morning.
Conditioned avoidance training
Avoidance training was initiated 48 h after inescapable shock pre-treatment in the jumping box to evaluate the escape and avoidance performance. The jumping box was partitioned into two equal sized chambers (27 × 29 × 25 cm) by plexiglass. A space of 14 × 17 cm served as a gate that provides access from one compartment to enter the other.
The animals were subjected to test environment habituation for 5 min (for the first session only) by placing them singly in one of the partitioned chambers of the jumping box. A total of 30 avoidance trials with inter-trial intervals being 30 sec were carried out on the animals. During the initial 3 sec of each test, a conditioned stimulus in the form of a light signal was presented, allowing the animals to avoid shocks. Any absence of response within the test period initiates the application of unconditioned stimulus of 0.8 mA shock (3 sec duration) via the grid floor. The shock and light conditioned stimulus were aborted when no escape response occurs within this period. The avoidance sessions were conducted for three consecutive days in the morning. The number of escape failures (no crossing response), during shock delivery, were recorded.
4.3.3.4 Assessment of hepatic and renal functions of rats in sub-chronic toxicity study
Adult Charles Foster albino rats (150 ± 10 g) of either sex were divided into control; standard and test groups comprising of six animals in each group. OECD guideline (Organization for Economic Co-operation and Development, Guideline-407, adopted on 3rd October 2008) was followed during the study with slight modifications. Test compounds were administered orally for thirty consecutive days. Body weights of animals, as well as their food and water consumptions, were monitored daily throughout the study period. They were fasted overnight before blood collection by retro-orbital technique on the day 31st of the study. The blood samples were analyzed for biochemical parameters reflecting hepatic and renal functions.
4.3.4 Ulcerogenic studies
Ulcerogenic liability was evaluated on the same animals used in the assessment of sub-chronic toxicity. Rats under anaesthesia were sacrificed, and the stomach was removed, opened along the curvature, washed with distilled water and cleaned gently in normal saline. After washing, the stomach mucosa was examined for ulcers using a handheld lens. The lesions were counted, and an ulcer index (UI) for each animal was calculated (Szelenyi and Thiemer, 1978). where
n = number of ulcers
I = ulcer area covering less than 1 mm2
II = ulcer area covering area from 1 to 3 mm2 and
III = ulcer area covering more than 3 mm2
4.3.5 Statistical analysis
Values are expressed as mean ± SEM. Statistical analysis was performed using One-way ANOVA followed by Tukey’s multiple comparison test, p-value<0.05 was considered as significant difference.
4.3.6 In silico studies
4.3.6.1 Homology modeling
The human GAT1 (UniProtKB: P30531) from the uniprot protein knowledgebase (https://www.uniprot.org/uniprot/) and the Drosophila melanogaster dopamine transporter (dDAT) (PDB ID: 4XP4_A) served query and template sequence respectively (Penmatsa et al., 2015). The sequence alignment program Clustal W aligned the GAT1 and the template sequence, and the 4XP4 showed 46% sequence identity and 67% similarity with GAT1 (Supplementary Fig. 1). A disulfide bridge between located in the extracellular-loop 2 of GAT1 was included and Na+ and Cl- ions were retained during model generation (Jurik et al., 2015). A total of ten models were prepared using the Schrödinger suite 2016 and the Prime module refined the loop regions of generated models. Ramachandran plot (Supplementary Fig. 2) assessed the quality of the models and docking with tiagabine validated the best model.
4.3.6.2 Molecular docking
The docking study was carried out using the Schrödinger Glide module in Schrödinger Suite 10.5.014 MM Share version 3.3.014 release 2016–1 with workstation 4x Intel® Xeon® CPU E5-1607 v3 @ 3.10 GHz on Linux operating environment. The LigPrep generated the energy-minimized conformers of the drawn ligands using the OPLS 2005 force field. The protein preparation wizard module prepared and optimized the energy of the GAT1 GABA transporter protein model. A grid was generated by supplying the x, y and z coordinates (x = 29 Å, y = 27 Å and z = 22 Å) within the contour of the tiagabine binding active pocket. The protein was kept rigid while the ligands were kept flexible during the entire docking simulation using Glide extra precision (XP). The Glide score and interacting amino acid residues of the ligands under study were compared with tiagabine.
Declaration of Competing Interest
The authors declare no conflict of interests.
References
- 1,3,4-Oxadiazole derivatives: synthesis, characterization, antimicrobial potential, and computational studies. Biomed Res. Int.. 2014;2014
- [Google Scholar]
- Synthesis, characterization, evaluation and molecular dynamics studies of 5, 6–diphenyl–1, 2, 4–triazin–3 (2H)–one derivatives bearing 5–substituted 1, 3, 4–oxadiazole as potential anti–inflammatory and analgesic agents. Eur. J. Med. Chem.. 2015;101:81-95.
- [Google Scholar]
- Hepatotoxicity associated with antiepileptic drugs. Acta Neurol. Scand.. 2008;118(5):281-290.
- [Google Scholar]
- Side-effects of antiepileptic drugs: the economic burden. Seizure. 2014;23(3):184-190.
- [Google Scholar]
- Next century challenges: Scalable coordination in sensor networks. In: Paper Presented at the Proceedings of the 5th Annual ACM/IEEE International Conference on Mobile Computing and Networking. 1999.
- [Google Scholar]
- Brief report novel mechanism for valproate-induced teratogenicity. Birth Defects Res. Part A: Clin. Mol. Teratol.. 2014;100(8):592-597.
- [Google Scholar]
- Design, synthesis and antidepressant activities of some novel fatty acid analogues. Med. Chem. Res.. 2015;24(4):1605-1616.
- [Google Scholar]
- A binding mode hypothesis of tiagabine confirms liothyronine effect on γ-aminobutyric acid transporter 1 (GAT1) J. Med. Chem.. 2015;58(5):2149-2158.
- [Google Scholar]
- The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: glial–neuronal metabolic interactions. Neurochem. Int.. 2003;43(7):629-637.
- [Google Scholar]
- Evaluation of docking performance: comparative data on docking algorithms. J. Med. Chem.. 2004;47(3):558-565.
- [Google Scholar]
- Epilepsy in the elderly: some clinical and pharmacotherapeutic aspects. Epilepsia. 2001;42:55-59.
- [Google Scholar]
- In vivo evaluation of substituted 3-amino-1, 4-benzodiazepines as anti-depressant, anxiolytic and anti-nociceptive agents. Arzneimittelforschung. 2009;59(02):61-71.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23(1–3):3-25.
- [Google Scholar]
- Different actions of anticonvulsant and anesthetic barbiturates revealed by use of cultured mammalian neurons. Science. 1978;200(4343):775-777.
- [Google Scholar]
- Design, synthesis and activity of ascorbic acid prodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropic activity. J. Med. Chem.. 2002;45(3):559-562.
- [Google Scholar]
- Anticonvulsant mechanisms of piperine, a piperidine alkaloid. Channels. 2015;9(5):317-323.
- [Google Scholar]
- Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541-553.
- [Google Scholar]
- X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat. Struct. Mol. Biol.. 2015;22(6):506.
- [Google Scholar]
- Development of highly potent GAT1 inhibitors: synthesis of nipecotic acid derivatives by Suzuki-Miyaura cross-coupling reactions. ChemMedChem. 2016;11(5):519-538.
- [Google Scholar]
- Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266(5604):730.
- [Google Scholar]
- Synthesis and evaluation of N-substituted nipecotic acid derivatives with an unsymmetrical bis-aromatic residue attached to a vinyl ether spacer as potential GABA uptake inhibitors. Bioorg. Med. Chem.. 2013;21(11):3363-3378.
- [Google Scholar]
- Possible role of nitric oxide in the nootropic and antiamnesic effects of neurosteroids on aging-and dizocilpine-induced learning impairment. Brain Res.. 1998;799(2):215-229.
- [Google Scholar]
- Triazolobenzodiazepines, a new class of central nervous system-depressant compounds. The Benzodiazepines 1973:285-297.
- [Google Scholar]
- The GABA synapse as a target for antiepileptic drugs: a historical overview focused on GABA transporters. Neurochem. Res.. 2014;39(10):1980-1987.
- [Google Scholar]
- GABA function in mood disorders: an update and critical review. Life Sci.. 1998;63(15):1289-1303.
- [Google Scholar]
- Review of epilepsy-etiology, diagnostic evaluation and treatment. Int. J. Neurorehabil.. 2014;1(130) 2376-0281.1000130
- [Google Scholar]
- Synthesis of new piperidyl indanone derivatives as anticonvulsant agents. Med. Chem. Res.. 2012;21(6):726-733.
- [Google Scholar]
- Synthesis and evaluation of substituted diphenyl-1,3,4-oxadiazole derivatives for central nervous system depressant activity. Org. Med. Chem. Lett.. 2012;2(1):8.
- [Google Scholar]
- Design, synthesis, characterization, and molecular modeling studies of novel oxadiazole derivatives of nipecotic acid as potential anticonvulsant and antidepressant agents. Med. Chem. Res.. 2018;27(1):137-152.
- [Google Scholar]
- Comparative assays of antiepileptic drugs in mice and rats. J. Pharmacol. Exp. Ther.. 1952;106(3):319-330.
- [Google Scholar]
- Distention ulcer as a model for testing of drugs for ulcerogenic side effects. Arch. Toxicol.. 1978;41(1):99-105.
- [Google Scholar]
- Interlaboratory study of log P determination by shake-flask and potentiometric methods. J. Pharm. Biomed. Anal.. 1996;14(11):1405-1413.
- [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem.. 2002;45(12):2615-2623.
- [Google Scholar]
- Nipecotic acid: systemic availability and brain delivery after nasal administration of nipecotic acid and n-butyl nipecotate to rats. Pharm. Res.. 2005;22(4):556-562.
- [Google Scholar]
- Development of 1, 3, 4-oxadiazole thione based novel anticancer agents: Design, synthesis and in-vitro studies. Biomed. Pharmacother.. 2017;95:721-730.
- [Google Scholar]
- Synthesis and anticonvulsant and neurotoxicity evaluation of N 4-phthalimido phenyl (thio) semicarbazides. Eur. J. Pharm. Sci.. 2003;20(3):341-346.
- [Google Scholar]
- Structure, function, and modulation of γ-aminobutyric acid transporter 1 (GAT1) in neurological disorders: a pharmacoinformatic prospective. Front. Chem.. 2018;6:397.
- [Google Scholar]
- Design and synthesis of new 2-substituted-5-[2-(2-halobenzyloxy) phenyl]-1, 3, 4-oxadiazoles as anticonvulsant agents. Chem. Pharm. Bull.. 2008;56(4):509-512.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.03.009.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




