

Translate this page into:
Catalyzed-like water enhanced mechanism of CO2 conversion to methanol
⁎Corresponding author at: Institute of Chemistry, University of Miskolc, 3515 Miskolc-Egyetemváros, Hungary. kemfiser@uni-miskolc.hu (Béla Fiser)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Converting carbon dioxide to fine chemicals such as methanol using electrolytic hydrogen could be an efficient way of renewable energy storage. The conversion of CO2 to methanol is a rather complicated multistep process which is usually performed catalytically in gas phase. However, the aqueous phase conversion of CO2 is also feasible in certain conditions. Thus, a catalyzed-like water enhanced mechanism of CO2 hydrogenation to methanol has been designed and studied by using the highly accurate W1U composite method. The initial reactant mixture was CO2 + 6H• + 8H2O + H3O+, where the hydrogen atoms are added one-by-one to mimic the catalytic effect of a metal surface. The presence of water and H3O+ further enhance the reaction by lowering the reaction barriers. By computing the thermodynamic properties of the reaction mechanism, it was found that the highest relative energy barrier in the most preferred pathway is 212.67 kJ/mol. By taking this into account, the energy efficiency of the pathway has been calculated and it was found to be equal to 92.5%.
Keywords
Carbon dioxide hydrogenation
Climate change
Computational study
Energy storage

1 Introduction
The continuously increasing CO2 emissions since the industrial revolution (Chandler, 2018) can be one of the major factors causing global warming and the acidification of the oceans (Gattuso and Hansson, 2011). These detrimental effects of carbon dioxide (CO2) release force more and more researchers to work on environmental protection (Vo et al., 2019). Most of the proposed solutions aiming to decrease the CO2 emissions till now are not definitive, and are based mainly on Carbon Capture and Storage (CCS) methods (Larkin et al., 2019). Instead of storing CO2 somewhere or letting it into the atmosphere, the best solution would be its total transformation into added value products (Islam et al., 2018). Even at ideal conditions, CO2 transformation reactions will consume a certain amount of energy. If this energy is coming from renewable resources, the process can be considered as an energy storage solution as well. Thus, another problem can be solved which is related to the non-stable production and consumption of renewable energy (Banerjee et al., 2019; Weitemeyer et al., 2015). By chemically converting carbon dioxide into different molecules such as methanol or methane, the applied energy is transformed and stored in chemical bonds (Castellani et al., 2017; Leonzio, 2018). Due to their high energy content, methanol and methane are the most attractive compounds to achieve from CO2. They can also be used as feedstock for other chemical processes to produce more complex compounds (Centi et al., 2013; Hu and Daasbjerg, 2019). The necessary hydrogen can be obtained by the electrolysis of water using renewable energy (Steinlechner and Junge, 2018) or other sources such as the steam reforming of natural gas (Xia et al., 2019). Carbon dioxide can be collected from where it is usually released, namely the industrial or biochemical processes (Pain et al., 2019). All in all, this can contribute to the decrease of CO2 emission (Olah et al., 2006) and it can solve the energy storage problems of renewable processes. In the last decades, CO2 hydrogenation to methanol has been a subject of interest, and a large variety of solid catalysts have been designed and tested (Sheldon, 2017). Better understanding of the reduction mechanism (Huš et al., 2017) could give us a chance to design more efficient catalysts. To achieve this, first, the uncatalyzed CO2 hydrogenation has been studied in gas and aqueous phase and the mechanisms have been compared (Hadjadj et al., 2020, 2019). It was found that the aqueous phase process is more feasible, but the hydrogenations were the rate limiting steps, and the energy barriers of these reactions are higher than the rest of the elementary steps. The challenge would be then to find alternative reactions to reduce the hydrogenation barriers. This can be done by imitating the role of catalysts which are able to adsorb and break the hydrogen molecules into atoms. The possibility of bond dissociation occurring in the adsorption process of the H2 molecule on the surface of catalysts is discussed in several works in the literature (Panczyk et al., 2005; Righi et al., 2019; Yang et al., 2010). In this way, hydrogen atoms would be ready to react at the surface of the catalyst. Thus, to mimic this process, in the current study, hydrogen addition reaction steps have been replaced with atomic hydrogenations (H•). Thereby, new insights can be achieved into the catalytic CO2 conversion which can be applicable in catalyst design and development.
2 Computational methods
The reactant mixture is (CO2 + 6H•+H2O + H3O+) which has been selected as a derivative of the ones chosen in a previously studied uncatalyzed mechanism (Hadjadj et al., 2020), where we have substituted the hydrogen molecules by hydrogen atoms, but the total number of electrons and atoms are kept the same. The thermodynamic properties of the studied species have been computed using the Gaussian 09 program package (Frisch et al., 2019). The B3LYP density functional theory (DFT) method (Kim and Jordan, 1994; Stephens et al., 1994) in combination with the 6-31G(d) basis set (Ditchfield et al., 1971) have been used to get a first approximation of the structures. Then, all of these have been recalculated by using the highly accurate W1U (Unrestricted Weizmann-1) composite method (Barnes et al., 2009; Martin and de Oliveira, 1999; Parthiban and Martin, 2001) to improve the accuracy of the results. To verify the selection of W1U, calculations of elementary reaction steps of a simple mechanism which is similar to the studied system have been carried out (Hadjadj et al., 2020). The results have been compared to experimental values available in the literature by using the heat of formations of the species. The highest absolute deviation between the computed and the experimental results is small (4.30 kJ/mol) and thus, W1U is an excellent choice for the calculations. IRC (Internal Reaction Coordinates) calculations (Deng et al., 1993) have been carried out to verify that the transition states are located between the corresponding minima. Relaxed energy scans have been carried out to verify the barrierless reactions. In one case, a rigid energy scan was performed, by freezing an inter atomic angle to avoid some undesirable interactions. Since an aqueous phase process is envisaged, solvent effects have also been mimicked by using the conductor-like polarizable continuum model (CPCM)(Barone and Cossi, 1998; Cossi et al., 2003). This model mimics a homogeneous polarizable medium where the solute is encapsulated in a cavity, which will provoke a change in the continuous dielectric field of the solvent, and this defines the solvation potential.
It has to be pointed out that all the calculations have been carried out under standard conditions. The increase in temperature will lift the system to a higher energy level which will increase the probability of reaction occurrence (Key, 2014).
3 Results and discussion
A special catalyzed-like CO2 hydrogenation mechanism to achieve methanol is envisaged and studied. The catalytic effect of a metal surface has been mimicked by considering hydrogen atoms instead of hydrogen molecules as reaction partners (Fig. 1). The presence of water (and H3O+) further enhances the reaction by lowering reaction barriers and thus, behave like additional catalyst even though its effect is modest rather than dramatic.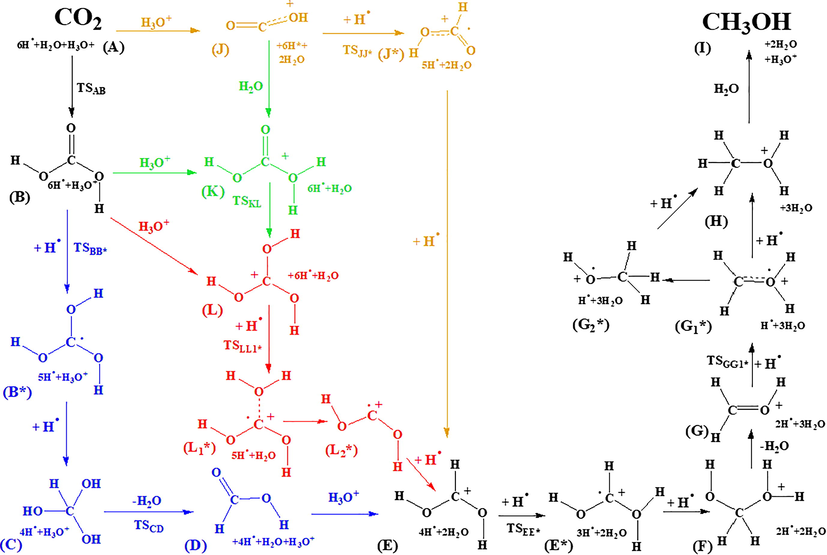
Reaction pathways of the envisaged CO2 – methanol conversion mechanism using atomic hydrogenations. Letters are assigned to every structure, and each transition state is named as TS followed with the letter referring to the reactant and then the product (e.g. TSAB), respectively. The +H• refers to a hydrogen atom addition.
As a first step CO2 (A) can either be protonated or hydrated and thus, (J) or (B) can be formed, respectively. To reach the central element of the mechanism which is the protonated formic acid (E), four pathways can be followed going through a two-step atomic hydrogenation in each case:
-
Hydration-hydrogenation route (ABB*CDE, Fig. 1, blue): by the hydration of CO2 (A), carbonic acid (B) will be formed (three conformations are possible, the one considered here is energetically higher by 3.14 kJ/mol than the most stable conformer). After that, a sequence of two atomic hydrogenations (TSBB* and B*C) have to occur to produce methanetriol (C). Then, a water elimination (TSCD), leads to formic acid (D) and via a protonation step (E) is formed.
-
Protonation-hydrogenation route (AJJ*E, Fig. 1, brown): this route consists of three elementary steps which connects CO2 with the desired protonated formic acid (E) intermediate. A protonation (AJ) followed by two atomic hydrogenations (TSJJ*, J*E) will lead to (E). It has to be noted that this route is a part of the preferred pathway of the mechanism (the reason will be discussed later).
-
Hydration-protonation route (ABLL1*L2*E, Fig. 1, red): this route is diverted from the hydration-hydrogenation route (blue) after (B) is formed. The protonation of carbonic acid (B) can lead to (L). Then, the first atomic hydrogenation occurs (TSLL1*). After that, a water subtraction (L1*L2*) followed by the second atomic hydrogenation (L2*E) leads to the protonated formic acid (E).
-
Protonation-hydrogenation/hydration-protonation route (A[B/J]KLL1*L2*E, Fig. 1, green): this route starts with either a protonation (AJ) which is followed by a hydration (JK) or with a hydration (TSAB) which is followed by a protonation (BK) to reach protonated carbonic acid (K). Then, (L) can be formed via a hydrogen shift (KL), which will put this to the track of the red (hydration-protonation) route. From here, (E) can be achieved through the reactions (TSLL1*, L1*L2*, L2*E) as in the case of the hydration-protonation route.
All the routes lead to the formation of (E), protonated formic acid. After that, another two atomic hydrogenations (TSEE* and E*F) will occur and (F) will be formed. Then, a water elimination will lead to (G), which is protonated formaldehyde. From here, there are two possible ways to reach (H), and in both cases, the first step would be the formation of (G1*). The shortest way to reach (H) is a direct hydrogen atom addition (G1*H). The other way will include the formation of (G2*) through a hydrogen shift (TSG1*G2*), and then, through a hydrogen atom addition (G2*H) the desired intermediate (H) will be reached. As a final step, a water mediated proton release (HI) will lead to the formation of methanol (I) and a hydronium ion. The relative thermodynamic properties of the individual steps have been computed as e.g. Δ
= G(X) − Gref, where G(X) and Gref are the Gibbs free energy of structure X and the reference, respectively (Fig. 2 and Table 1). The (CO2 + 6H• + H2O + H3O+) are considered as the reference throughout the reaction.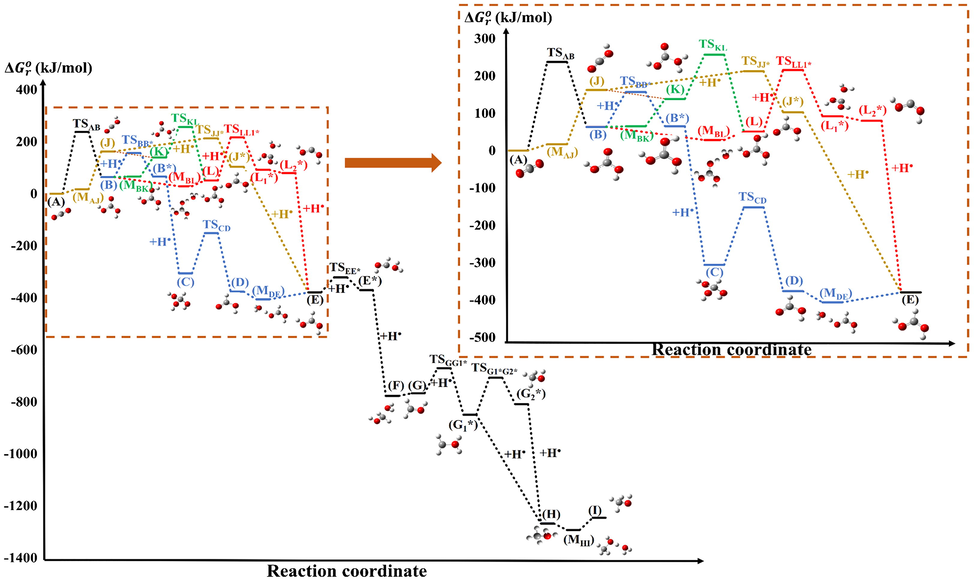
Gibbs free energy change (Δ
, kJ/mol) of the catalyzed-like conversion of CO2 to methanol calculated at the W1U level of theory. The transition states are named as TS followed by the reactant and the product (e.g. TSAB), and the atomic hydrogenation steps are highlighted with (+H•). The complexes involved in double Morse potentials are noted by M followed by the letter of the reactant and then the product (e.g. MAJ), respectively.
Species
Δ
Δ
S
kJ/mol
J/mol*K
A
CO2
0.00
0.00
0.00
B
H2CO3
23.96
63.41
−132.32
B*
H2CO3-H
−1.01
65.48
−223.01
C
HC(OH)3
−408.42
−306.14
−343.05
D
HCOOH
−434.26
−376.27
−194.51
E
HCOOH2+
−441.03
−379.69
−205.74
L1*
HCOOH+-H2O
26.50
92.01
−219.70
L2*
HOCOH+
54.20
79.77
−85.74
J
HCO2+
166.11
162.31
12.74
J*
HCO2+-H
77.18
102.99
−86.54
E*
HCOOH2+-H
−458.97
−370.40
−297.07
F
H2O-H2COH+
−901.29
−777.71
−414.51
G
H2COH+
−847.90
−767.68
−269.07
G1*
H2COH+-H
−957.84
−849.56
−363.18
G2*
H3COH+
−918.46
−809.17
−366.58
H
H3COH2+
−1411.50
−1267.97
−481.40
I
H3COH
−1386.75
−1245.43
−473.98
k
H3CO3+
98.83
138.67
−133.64
L
C(OH)3+
7.97
50.75
−143.48
TSAB
A → B
197.86
237.28
−132.23
TSBB*
B → B*
87.24
156.97
−233.89
TSCD
C → D
−255.65
−151.49
−349.37
TSLL*
L → L*
144.20
216.31
−241.86
TSJJ*
J → J*
188.90
212.67
−79.70
TSEE*
E → E*
−412.56
−321.32
−306.03
TSGG1*
G → G1*
−780.73
−670.72
−369.00
TSG1*G2*
G1* → G2*
−818.20
−707.39
−371.66
TSKL
K → L
213.66
256.78
−144.65
MDE
D-H3O+
−503.06
−406.09
−325.24
MBL
B-H3O+
−49.12
28.49
−260.30
MAJ
A-H3O+
−13.29
17.08
−101.89
MHI
H-H3O+
−1472.85
−1292.99
−603.24
MBK
B- H3O+
−9.17
65.54
−250.56
The mechanism can be divided into two sections: [A-E] and [E-I] (Fig. 2). In the section [A–E] the conversion of CO2 (A) to protonated formic acid (E) could occur through several different pathways. All the routes start (or can start, Fig. 1) with a hydration of CO2 (A) to get carbonic acid (B) except the protonation-hydrogenation (brown) pathway. This goes directly from CO2 (A) through a protonation followed by two atomic hydrogenations to the protonated formic acid (E) with one single barrier (Δ = 212.67 kJ/mol). It is the lowest relative energy barrier in the [A-E] section, and this makes it the preferred pathway. Thus, the overall preferred pathway would be then: [A-TSAJ-J-TSJJ* -J*-E-TSEE* -E*-F-G-TSGG1* -G1*-H-I].
Through the hydration-hydrogenation (Fig. 2, blue) pathway (E) could be reached within 5 reaction steps. The highest relative barrier height here is 237.28 kJ/mol which corresponds to (TSAB). There are two other transition states which are more preferred, and their relative Gibbs free energies are significantly lower (TSBB*, Δ = 156.97 kJ/mol and TSCD, Δ = −151.49 kJ/mol). This pathway also involves an immediate hydrogen atom addition (B*C) and a barrierless reaction with an intermediate (MDE) having the lowest relative energy value (Δ = −406.09 kJ/mol) in the [A-E] section.
Protonated formic acid (E) can also be reached through 5 reaction steps within the hydration-protonation (Fig. 2, red) pathway. The first step is the same as before (TSAB), which is followed by a barrierless processes which includes an intermediate (MBL). Then, two atomic hydrogenations occur, with a barrierless water removal reaction in between (L1*L2*). The first atomic hydrogenation goes through (TSLL1*) (Δ = 216.31 kJ/mol), while the second (L*E) is a barrierless step. It is possible to link the protonation-hydrogenation (brown) and hydration-protonation (red) pathways through a hydration (JK) followed by a hydrogen shift (TSKL, Δ = 256.78 kJ/mol) which has the highest relative energy among all the routes. This TS is a part of the green reaction channel as well.
The [E-I] is one single route where (E) will be converted to methanol (I) after 6 consecutive reaction steps or 7 if the side reaction between (G1*) and (G2*) is considered. The relative Gibbs free energy difference between these two molecules (ΔΔG0(G2*-G1*)) is 40 kJ/mol, but since TSGG1*>TSG1*G2*, a preferred side reaction route cannot be chosen as both processes could occur.
It has to be mentioned that in some cases several conformers can be formed, and several transition states leading to these conformers are possible. In each case, the most appropriate conformer has been chosen and included into the discussion. Among all the consecutive hydrogen atom additions, the second step is always a barrierless radical recombination reaction (Morse potential). Therefore, the second hydrogen atom in each case, is attached to the rest of the molecule without any additional energy needed (Fig. 3). There are six barrierless atomic hydrogenation steps (radical recombination), (B*C), (L2*E), (J*E), (E*F), (G1*H) and (G2*H). There were also barrierless water addition/subtraction reactions such as (JK), (L1*L2*) and (FG). The association of two Morse potentials is another barrierless reaction type involved in the discussed mechanism (Fig. 2). It goes through a minimum, a molecular complex such as (MAJ), (MBK), (MBL), (MDE) and (MHI) instead of going through a transition state. These reactions are always protonations and thus, the intermediate molecular complexes are always formed by the starting structure and an oxonium ion (H3O+). To show the energetic properties of these barrierless reaction steps, (E*F) have been examined in detail (Fig. 3) and the corresponding total energy change has been computed.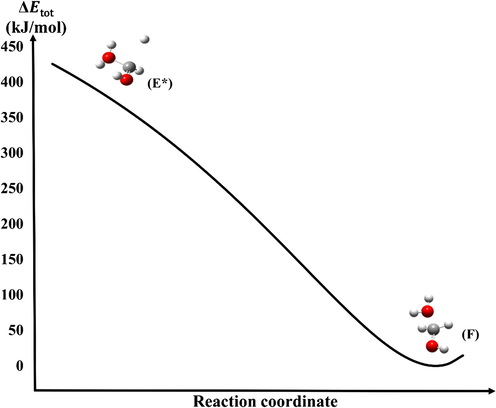
Total energy change (
) of the (E*F) barrierless reaction step (Morse potential).
The total energy decreases from the reactant’s energy level (E*) directly to the energy level of the product (F) without going through a barrier. The energy level of the product has been considered as a reference for the calculation of the total energy change.
After choosing 200 kJ/mol as an arbitrary reference for high energy structures, four transition states have been found which are above this limit, Δ = 237.28 kJ/mol, Δ = 216.31 kJ/mol, Δ = 212.67 kJ/mol and Δ = 256.78 kJ/mol (Table 1). The corresponding reaction steps are a hydration (TSAB, water molecule addition), two atomic hydrogenations (H atom addition, TSLL* and TSJJ*) and a hydrogen atom shift (TSKL).
It is possible to calculate the energy storage efficiency (η) of the preferred pathway (Protonation-hydrogenation, brown) as follows:
This corresponds to the ratio of the stored enthalpy
(
= −1386.75 kJ/mol) and the invested enthalpy (the relative enthalpy of the transition state with the highest relative activation energy of the reaction path Δ
is equal to
= 188.90 kJ/mol). However, in this way, the theoretical efficiency of methanol formation is 734.1%, which is not possible, as the efficiency would be > 100%. Nevertheless, in this case, the invested energy is not equal to the maximal barrier height only. The energy demand to break three hydrogen–hydrogen bonds (Bond Dissociation Energy of H2, BDEH2) which will provide the 6 hydrogen atoms has to be also taken in account to get the corrected efficiency (ηcorr) as follows:
Calculated BDEH2 (436.56 kJ/mol) has been used in the correction, but it was also compared to the experimentally determined value and the difference is < 1 kJ/mol (ΔBDEH2 = 0.56 kJ/mol (Darwent, 1970)), which also verifies the method selection. All in all, was found to be equal to 92.5%.
The efficiency increased a lot compare to the uncatalyzed gas phase ( = 14.4%)(Hadjadj et al., 2019) and aqueous phase mechanisms ( = 27.1%)(Hadjadj et al., 2020). Even though, the number of electrons and atoms were kept the same compared to the previous water enhanced case (Hadjadj et al., 2020), the difference in efficiency arises from the fact that hydrogen molecules were part of the reactant mixture (CO2 + H2O + H3O++3H2) previously, while in the catalyzed-like case, H atoms are considered (CO2 + H2O + H3O++6H•). It has to be mentioned that the presence of water (and H3O+) will enhance the reactions by lowering the reaction barriers. Thus, it acts like a catalyst even though its effect is modest rather than dramatic. The reactant mixture in the catalyzed-like case is less stable compared to the previous system. However, if the reaction occurs at the surface of a metal catalyst, the hydrogen atoms would be bonded to the catalyst along with the rest of the molecules. Thus, the whole system would be more stable, and the barriers could decrease even more.
3.1 Comparison between the uncatalyzed and the catalyzed-like water enhanced mechanisms
The pathway of the catalyzed-like CO2 - methanol conversion have been compared to the corresponding uncatalyzed reaction (Hadjadj et al., 2020) (Table 2).
Water enhanced (Hadjadj et al., 2020)
Catalyzed-like
Reactant mixture
CO2 + 3H2 + H2O + H3O+
CO2 + 6H• + H2O + H3O+
Barrierless reactions
Yes
Yes
Ionic reactions
Yes
Yes
Hydrogen atom addition reactions
No
Yes
Highest relative energy barrier (kJ/mol)
355.52
212.67
Efficiency (η)
27.1%
92.5% *
It has to be emphasized that the mechanisms do not have the same initial reactant mixtures as it was mentioned above. Both mechanisms involve barrierless and ionic reaction steps, but hydrogen atom additions obviously occur only in the catalyzed-like case. In the catalyzed-like pathway, there is only one transition state with a relative barrier higher than 200 kJ/mol (Δ = 212.67 kJ/mol). Unlike in the other case, where all the barriers are above > 200 kJ/mol. In the case of the uncatalyzed mechanism the efficiency is 27.1%, which is far lower than what can be achieved with catalyzed–like mechanism (92.5%).
4 Conclusion
The reduction of CO2 to methanol in aqueous phase is a complicated process and still a subject of mechanistic discussions. In this work, a newly developed catalyzed-like mechanism mimicking the role of a heterogeneous catalyst (breaking the hydrogen bond) has been studied thermodynamically using computational chemistry tools. The relative Gibbs free energy change of the mechanism have been calculated and the preferred pathway have been compared to a previously studied uncatalyzed aqueous phase mechanism.
After analyzing the catalyzed-like mechanism, further improvement and a significant decrease of the energy barriers was observed in the overall process and the corresponding energy barriers are significantly lowered. In the preferred pathway, the highest barrier is only 212.67 kJ/mol which is compared to the uncatalyzed system almost 1.7 times smaller. Furthermore, an enormous increase has been achieved in the energy storage efficiency. The catalyzed-like mechanism is 3.4 times more efficient (92.5%) than the corresponding aqueous phase uncatalyzed process (27.1%). The results are an important step further to understand the carbon dioxide hydrogenation and to design new catalyst with better performance.
Acknowledgment
This research was supported by the European Union and the Hungarian State, co-financed by the European Regional Development Fund in the framework of the GINOP-2.3.4-15-2016-00004 project, aimed to promote the cooperation between the higher education and the industry. BF thanks the support by the ÚNKP-20-4 New National Excellence Program of The Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Carbon Nanomaterials in Renewable Energy Production and Storage Applications. Cham: Springer; 2019. p. :51-104.
- Unrestricted coupled cluster and Brueckner doubles variations of W1 theory. J. Chem. Theory Comput.. 2009;5:2687-2693.
- [CrossRef] [Google Scholar]
- Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A. 1998;102:1995-2001.
- [CrossRef] [Google Scholar]
- Experimental investigation on CO2 methanation process for solar energy storage compared to CO2-based methanol synthesis. Energies. 2017;10:855.
- [CrossRef] [Google Scholar]
- Catalysis for CO2 conversion: a key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci.. 2013;6:1711.
- [CrossRef] [Google Scholar]
- Chandler, W., 2018. Energy and Environment in the Transition Economies. Routledge. https://doi.org/10.4324/9780429500817.
- Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem.. 2003;24:669-681.
- [CrossRef] [Google Scholar]
- Bond Dissociation Energies in Simple Molecules. Washington: U.S. National Bureau of Standards; 1970.
- A combined density functional and intrinsic reaction coordinate study on the ground state energy surface of H2CO. J. Chem. Phys.. 1993;99:3823-3835.
- [CrossRef] [Google Scholar]
- Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys.. 1971;54:724-728.
- [CrossRef] [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Petersson, G.A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A.,V., Bloino, J., Janesko, B.G., Gomperts, R., Mennucci, B., Hratchian, H.P., Ortiz, J.V., Izmaylov, A.F., Sonnenberg, J.L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V.G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J.A., Peralta, J.E., 6 R. Hadjadj et al. / Journal of Cleaner Production 241 (2019) 118221 Ogliaro, F., Bearpark, M.J., Heyd, J.J., Brothers, E.N., Kudin, K.N., Staroverov, V.N., Keith, T.A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A.P., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Millam, J.M., Klene, M., Adamo, C., Cammi, R., Ochterski, J.W., Martin, R.L., Morokuma, K., Farkas, O., Foresman, J.B., Fox, D.J., 2013. Gaussian 09, Revision E.01. Gaussian Inc., Wallingford CT
- Ocean Acidification. Oxford University Press; 2011.
- Water enhanced mechanism for CO2 – Methanol conversion. Chem. Phys. Lett.. 2020;746:137298
- [CrossRef] [Google Scholar]
- Renewable energy and raw materials – The thermodynamic support. J. Clean. Prod.. 2019;241:118221
- [CrossRef] [Google Scholar]
- Mechanism, kinetics and thermodynamics of carbon dioxide hydrogenation to methanol on Cu/ZnAl2O4 spinel-type heterogeneous catalysts. Appl. Catal. B Environ.. 2017;207:267-278.
- [CrossRef] [Google Scholar]
- A comprehensive review of state-of-the-art concentrating solar power (CSP) technologies: Current status and research trends. Renew. Sustain. Energy Rev.. 2018;91:987-1018.
- [CrossRef] [Google Scholar]
- Key, J.A., 2014. Activation Energy and the Arrhenius Equation.
- Comparison of density functional and MP2 calculations on the water monomer and dimer. J. Phys. Chem.. 1994;98:10089-10094.
- [CrossRef] [Google Scholar]
- Risk assessment and management frameworks for carbon capture and geological storage: A global perspective. Int. J. Risk Assess. Manage.. 2019;22:254-285.
- [CrossRef] [Google Scholar]
- State of art and perspectives about the production of methanol, dimethyl ether and syngas by carbon dioxide hydrogenation. J. CO2 Util.. 2018;27:326-354.
- [CrossRef] [Google Scholar]
- Towards standard methods for benchmark quality ab initio thermochemistry—W1 and W2 theory. J. Chem. Phys.. 1999;111:1843.
- [CrossRef] [Google Scholar]
- Olah, G.A., Goeppert, A., Prakash, G.K.S., 2006. Beyond oil and gas: the methanol economy, Focus on Catalysts. WILEY-VCH. https://doi.org/10.1016/S1351-4180(06)71901-8.
- Pain, A.J., Martin, J.B., Young, C.R., 2019. Sources and sinks of CO2 and CH4 in siliciclastic subterranean estuaries. Limnol. Oceanogr. https://doi.org/10.1002/lno.11131.
- Hydrogen adsorption on nickel (100) single-crystal face. A Monte Carlo study of the equilibrium and kinetics. J. Phys. Chem. B. 2005;109:10986-10994.
- [CrossRef] [Google Scholar]
- Assessment of W1 and W2 theories for the computation of electron affinities, ionization potentials, heats of formation, and proton affinities. J. Chem. Phys.. 2001;114:6014-6029.
- [CrossRef] [Google Scholar]
- H2 dissociation on noble metal single atom catalysts adsorbed on and doped into CeO2 (111) J. Phys. Chem. C. 2019;123:9875-9883.
- [CrossRef] [Google Scholar]
- Methanol production – a technical history. Johnson Matthey Technol. Rev.. 2017;61:172-182.
- [CrossRef] [Google Scholar]
- Renewable methane generation from carbon dioxide and sunlight. Angew. Chemie Int. Ed.. 2018;57:44-45.
- [CrossRef] [Google Scholar]
- Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem.. 1994;98:11623-11627.
- [CrossRef] [Google Scholar]
- Vo, D.H., Nguyen, H.M., Vo, A.T., McAleer, M., 2019. CO2 Emissions, Energy Consumption and Economic Growth. Econom. Inst. Res. Pap.
- Integration of Renewable Energy Sources in future power systems: The role of storage. Renew. Energy. 2015;75:14-20.
- [CrossRef] [Google Scholar]
- Hydrogen Production from Biological Sources. In: Fuel Cells and Hydrogen Production. New York, New York, NY: Springer; 2019. p. :833-863.
- [CrossRef] [Google Scholar]
- Fundamental studies of methanol synthesis from CO2 hydrogenation on Cu(111), Cu clusters, and Cu/ZnO(0001) Phys. Chem. Chem. Phys.. 2010;12:9909-9917.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
See supplementary materials for the structural parameters of all the involved molecules and transition states.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.102955.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




