

Translate this page into:
Design, synthesis, and biological evaluation of novel bromo-pyrimidine analogues as tyrosine kinase inhibitors
This work is dedicated to Professor Yejella Rajendra Prasad for taking the charge as Principal of Andhra University College of Pharmaceutical Sciences, Andhra University, Visakhapatnam, Andhra Pradesh, India.
⁎Corresponding authors at: East West College of Pharmacy, #63, I Phase, BEL Layout, Bharathnagar, Vishwaneedam PO, Bangalore 560091, Karnataka, India (Suresh Kumar G.V.). Department of Pharmaceutical Sciences, College of Pharmacy & Health Sciences, Ajman University, Ajman PO Box 346, United Arab Emirates (R.R. Bhandare). Department of Pharmaceutical Chemistry, Vignan Pharmacy College, Vadlamudi-522213, Guntur, Andhra Pradesh, India (Afzal B. Shaik). sureshkumargv@gmail.com (G.V. Suresh Kumar), r.bhandareh@ajman.ac.ae (Richie R. Bhandare), bashafoye@gmail.com (Afzal B. Shaik)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
In the present investigation we designed, synthesized and evaluated a novel series of bromo-pyrimidine analogues (6a-j, 7a-e, 9a-f, and 10a-f) as anticancer agents. The compounds were characterized using spectroscopic studies and elemental analysis and screened for their in vitro cytotoxic activity by MTT assay against four cancer cell lines including HCT116 (human colon cancer cell line), A549 (human lung cancer cell line), K562 (human chronic myeloid leukemia cell line), U937 (human acute monocytic myeloid leukemia cell line) as well as the normal human liver cell line, L02. Most of the compounds showed potent activity on K562 cells. Considering this, the compounds were evaluated for Bcr/Abl tyrosine kinase inhibitory activity by ADP-Glo assay. Dasatinib was used as standard drug for both cytotoxicity and tyrosine kinase inhibition studies. The compounds, 6g, 7d, 9c, and 10e emerged as potent Bcr/Abl kinase inhibitors. Hence, the potent compounds that arose out of this investigation are potential lead molecules to develop as an alternative to existing dasatinib therapy.
Keywords
Anticancer
Bromo-pyrimidine
Cytotoxicity
Dasatinib derivatives
MTT assay
Bcr/Abl tyrosine kinase

1 Introduction
Chronic myelogenous leukemia (CML), also called as chronic myeloid leukemia is an important type of cancer of bone marrow where there is an uncontrolled production of myeloid cells that form platelets, most kinds of white blood cells (except lymphocytes) and red blood cells. The myeloid cells will undergo cell division in bone marrow and accumulate in blood. This leukemia is as a result of mutual translocation among chromosomes 9 and 22 (Philadelphia chromosome or t(9;22)). Another distinguishing feature of CML is the coding of an abnormal fusion protein Bcr/Abl with dysregulated tyrosine kinase activity (Rossari et al., 2018; Quintás-Cardama and Cortes, 2009). Imatinib (ST1571) is the first orally active and potent Bcr/Abl kinase inhibitor developed. Despite of good activity, its clinical use was deprived due to the advent of drug resistance (Shah, 2004; Quintás-Cardama and Cortes, 2008; Lee et al., 2008). The point mutations in the kinase domain of Bcr/Abl was the major mechanism for its resistance. Additionally, patients under imatinib therapy land into molecular residual disease.
To overcome imatinib resistance, second generation tyrosine kinase inhibitors (TKIs)-(Dasatinib and Nilotinib) and third generation TKI (bosutinib) were developed and these agents possess substantial clinical outcome. Dasatinib (BMS-354825) is the choice of drug for treatment of CML and Philadelphia chromosome-positive acute lymphoblastic leukemia and is active against 14 of the 15 clinically proved resistant mutants (Tokarski, 2006). Hence, it is a multi-targeted tyrosine kinase inhibitor. Dasatinib interrupts the major cellular functions that are essential for metastasis of cancer cells and angiogenesis1. The three-dimensional structure of Abl kinase and SRC kinase complexed with Dasatinib demonstrated that a couple of hydrogen bonds were formed in the hinge region of the ATP-binding site. The two hydrogen bonds were formed with amino acid residue Met318 wherein the nitrogen of the thiazole ring of dasatinib bonds with amide nitrogen of Met318 and 2-amino hydrogen of dasatinib with the carbonyl oxygen of Met318. Additionally, a hydrogen bond was formed between amide nitrogen of dasatinib and the hydroxyl oxygen of amino acid Thr315. Nonetheless, no hydrogen bonds were formed between the pyrimidine of dasatinib and the protein regardless of its importance in increased binding affinity (Tokarski, 2006).
We previously reported novel bromopyrimide analogs (1a & 1b) as tyrosine kinase inhibitors via modification of dasatinib (Fig. 1) (Munikrishnappa, 2016). In continuation of our search for new generation Bcr/Abl kinase inhibitors and to continue investigation on novel bromo-pyrimidine as another class of dasatinib analogs (Fig. 2), we hereby report further structure activity relationship (SAR), design, synthesis, and evaluation of novel bromo-pyrimidine as potent Bcr/Abl inhibitors by well-established ADP-Glo and MTT assay method (Tokarski, 2006; Cohen, 2003; Cohen, 2005; Kris, 2003; Wu, 2010; Das, 2006; Wityak, 2003). Our aim was to investigate novel bromo-pyrimidine derivatives as potent kinase inhibitors. The strategy to obtain newer bromo-pyrimidine derivatives was based on the following approaches: (1) Replacement of hydroxyethyl piperazine fragment with hydrogen based on the literature. This fragment was shown to be interacting with the solvent side of the tyrosine kinase (Tokarski, 2006; Păunescu et al., 2015). (2) Substitution of H-atom and CH3 group with bromine and morpholine as suggested by our previously published work (compounds 1a and 1b; Fig. 1). (3) Based on the SAR and docking studies of 2-aminothiazole template containing kinase inhibitors, it is known that the aminothiazole containing 2-chloro-6-methyl benzamide interacts with the kinase active site. Hence, an attempt was made to substitute 2-chloro-6-methyl-benzamide fragment with substituted acetohydrazide and phenyl substituted triazole. We retained aminothiazole in our present series as a privilege fragment.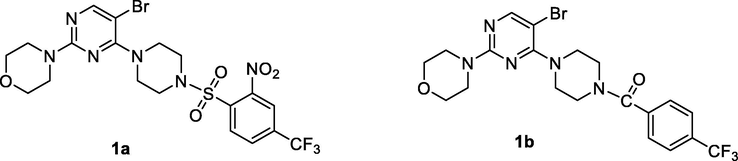
Previously reported novel bromo pyrimidine analogs as tyrosine kinase inhibitors.
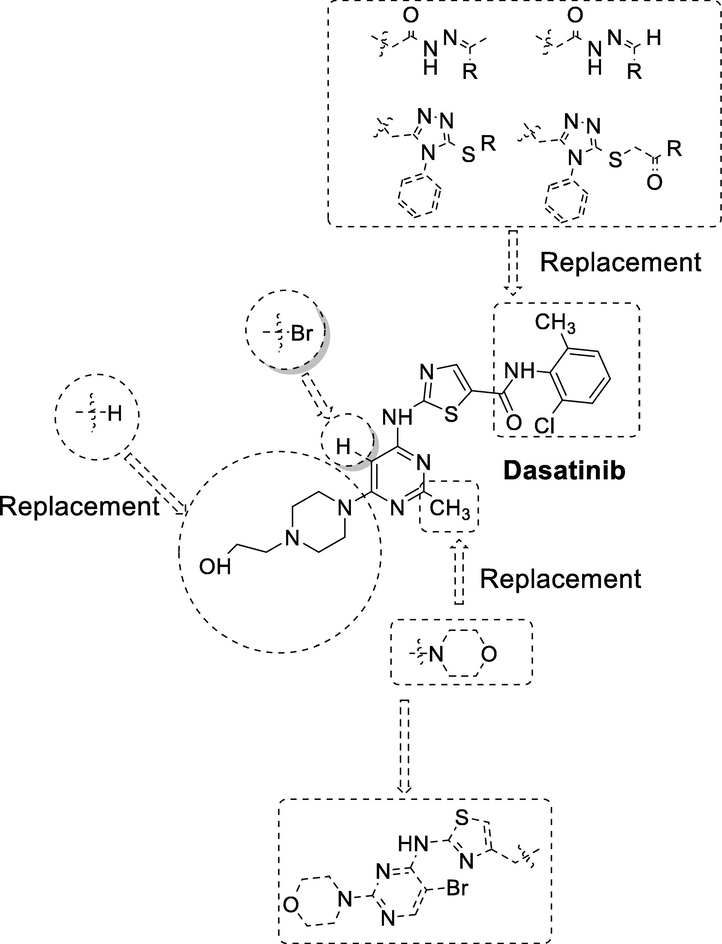
A design for synthesis of Dasatinib derivatives (6a-j, 7a-e, 9a-f, and 10a-f).
Additionally, considering that synthetic accessibility is an important factor that needs to be considered during the design phase, the starting materials required for incorporation of phenyl substituted triazole and substituted acetohydrazide were readily available. The designed derivatives were tailored to localize to the hinge region of kinase active site via aminothiazole privilege fragment. The R groups (Fig. 2) were modified to evaluate the effect on the potency of kinase inhibition.
2 Results and discussion
2.1 Chemistry
The reaction sequences employed for synthesis of titled bromo-pyrimidine derivatives 6a-j, 7a-e, 9a-f, and 10a-f were illustrated in Scheme 1 and the analytical and physicochemical properties of these compounds are displayed in Table 1. NA- Not applicable.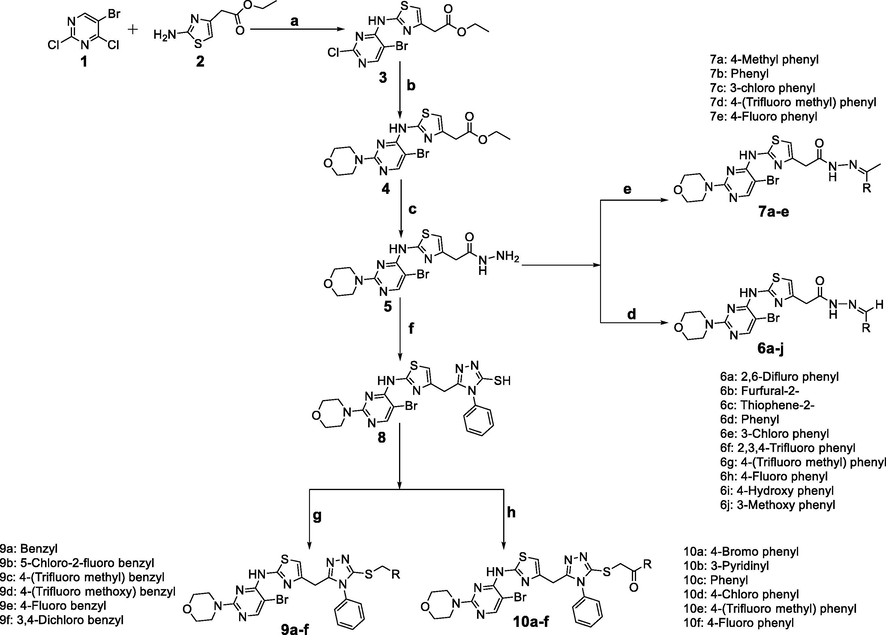
Synthetic pathway for the preparation of 5-bromo-pyrimidine derivatives. a) Potassium carbonate, 1,4 Dioxane, refluxed for 24 h. b) Ethanol, morpholine, and triethylamine, refluxed for 12 h. c) Hydrazine hydrate, ethanol, refluxed for 8 h. d) Substituted aldehyde, glacial acetic acid, ethanol, refluxed for 3 h. e) Substituted acetophenone, glacial acetic acid, ethanol, refluxed for 3 h. f) Phenyl isothiocyanate, ethanol, sodium hydroxide, refluxed for 4 h. g) Substituted benzylbromides, ethanol, potassium hydroxide, 25–30 °C for 5 h. h) Substituted phenacylbromides, ethanol, potassium hydroxide, 25–30 °C for 5 h.
Compound
R
Molecular Formula
M.Wa
M.p. (°C)b/Crystallization solvent
Yield (%)
%Analysis of C, H, N found (calc.)c
C
H
N
2
NA
C7H10N2O2S
186.23
90–95/n-heptane
80
NA
NA
NA
3
NA
C11H10BrClN4O2S
377.64
122–126/ethanol
84
NA
NA
NA
4
NA
C15H18BrN5O3S
428.3
96–98/ethanol
88
NA
NA
NA
5
NA
C13H16BrN7O2S
414.28
206–210/ethanol
89
NA
NA
NA
6a
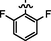
C20H18BrF2N7O2S
538.37
224–230/ethanol
83
44.62(44.65)
3.37(3.38)
18.21(18.22)
6b
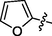
C18H18BrN7O3S
492.35
198–203/ethanol
80
43.91(43.95)
3.68(3.68)
19.91(19.94)
6c
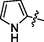
C18H19BrN8O2S
491.36
200–204/ethanol
78
44.00(44.10)
3.90(3.92)
22.80(22.82)
6d
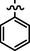
C20H20BrN7O2S
502.39
215–218/ethanol
84
47.81(47.84)
4.01(4.02)
19.52(19.54)
6e
C20H19BrClN7O2S
536.83
214–218/ethanol
81
44.74(44.75)
3.58(3.57)
18.27(18.26)
6f
C20H17BrF3N7O2S
556.36
234–238 /ethanol
83
43.18(43.20)
3.08(3.09)
17.62(17.64)
6 g
C21H19BrF3N7O2S
570.39
220–223/ethanol
80
44.23(44.22)
3.35(3.36)
17.18(17.19)
6 h
C20H19BrFN7O2S
520.38
215–218/ethanol
79
46.16(46.18)
3.66(3.65)
18.83(18.84)
6i
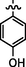
C20H20BrN7O3S
518.39
217–220/ethanol
75
46.33(46.34)
3.89(3.89)
18.90(18.91)
6j
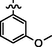
C21H22BrN7O3S
532.42
214–217/ethanol
81
47.36(47.37)
4.18(4.17)
18.42(18.42)
7a
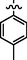
C22H24BrN7O2S
530.44
104–107/ethanol
77
49.81(49.85)
4.56(4.58)
18.48(18.50)
7b
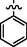
C21H22BrN7O2S
516.41
110–114/ethanol
86
48.84(48.85)
4.29(4.28)
18.99(18.98)
7c
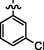
C21H21BrClN7O2S
550.86
112–116/ethanol
82
45.79(45.80)
3.84(3.89)
17.80(17.84)
7d
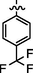
C22H21BrF3N7O2S
584.42
117–120/ethanol
70
45.22(45.21)
3.62(3.62)
16.79(16.78)
7e
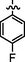
C21H21BrFN7O2S
534.43
115–117/ethanol
81
47.21(47.20)
3.95(3.96)
18.34(18.35)
8
NA
C20H19BrN8OS2
531.45
224–226/ethanol
75
NA
NA
NA
9a
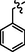
C27H25BrN8OS2
621.57
108–112/ethanol
81
52.17(52.18)
4.05(4.06)
18.03(18.05)
9b
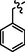
C27H23BrClFN8OS2
674.01
106–110/ethanol
80
48.11(48.11)
3.44(3.45)
16.62(16.68)
9c
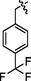
C28H24BrF3N8OS2
689.57
112–116/ethanol
80
48.76(48.77)
3.50(3.51)
16.25(16.25)
9d
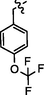
C28H24BrF3N8O2S2
705.57
84–87/ethanol
78
47.66(47.68)
3.43(3.45)
15.88(15.85)
9e
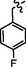
C27H24BrFN8OS2
639.57
103–106/ethanol
82
50.72(50.71)
3.77(3.78)
17.53(17.52)
9f
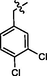
C27H23BrCl2N8OS2
690.46
115–118/ethanol
84
46.97(46.92)
3.36(3.38)
16.23(16.25)
10a
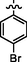
C28H24Br2N8O2S2
728.48
202–206/ethanol
79
46.16(46.18)
3.32(3.34)
15.38(15.39)
10b
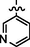
C27H24BrN9O2S2
650.57
195–198/ethanol
81
49.85(50.0)
3.72(3.74)
19.38(19.39)
10c
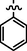
C28H25BrN8O2S2
649.58
204–208/ethanol
80
51.77(51.75)
3.88(3.90)
17.25(17.28)
10d
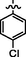
C28H24BrClN8O2S2
684.03
208–211/ethanol
82
49.16(49.20)
3.54(3.54)
16.38(16.39)
10e
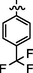
C29H24BrF3N8O2S2
717.58
212–214/ethanol
83
48.55(48.54)
3.38(3.37)
15.61(15.62)
10f
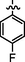
C28H24BrFlN8O2S2
667.58
204–208/ethanol
79
50.37(50.38)
3.63(3.62)
16.78(16.79)
The key starting material 2 was prepared as per the literature (Campaigne and Selby, 1980), which further on Buchwald amination reaction with 5-bromo-2,4-dichloropyrimidine produced intermediate 3 with superior yield (Connors et al., 2008). The intermediate 4 and 5 were obtained by steps as described in literature (Aquila et al., 2005; Prasad et al., 2013). A series of acetohydrazide derivatives (6a-j) and (7a-e) were concurrently prepared by treating intermediate 5 with commercially available substituted aldehydes and acetophenones in the presence of few drops of glacial acetic acid and ethanol (Mallikarjuna, 2009). Finally, the target compounds (9a-f) and (10a-f) were concomitantly synthesized by treating scaffold 8 with substituted benzyl bromide/phenacyl bromide in the presence of potassium hydroxide and ethanol (Karthikeyan et al., 2008; Bayraka et al., 2009) (Scheme 1).
The sharp singlet at δ 13.23 accountable for NH in 1H NMR spectrum and stretch band at 1728 cm−1 corresponding to functional group C⚌O in IR spectrum. Further, molecular ion peak (m/z, %): 377 (M + 1,) in LC-MS spectrum, confirmed the formation of crucial intermediate 3. 1H NMR spectrum of intermediate 4 displayed multiplets at δ 3.67–3.68 and 3.75–3.76 due to morpholine. Further, disappearance of ethyl group signals and appearances of NH-NH2 signals at δ 5.74–5.95, which were absent on D2O exchange, and molecular ion peak (m/z, %): 414.0 (M + 1, 99% purity) in LC-MS spectrum authenticated the formation of intermediate 5.
The formation of acetohydrazide derivatives (6a-j) and (7a-e) were established by the lack of characteristic peak at δ 5.95 of NH2 in 1H NMR spectrum. Further, 13C NMR spectrum depict signals at around δ 141.98 (HC⚌N-N), 144.73 (C⚌N-N), molecular ion peak data in LC-MS spectrum and stretching band at 2351 cm-1 of C⚌N of IR spectrum established the formation of target compounds (6a-j) and (7a-e).
1H NMR spectrum of the synthesized scaffold 8 showed the characteristic peaks δ 13.80 due to SH proton. Lack of characteristic peak at δ 13.80 (SH) in the 1H NMR spectrum of alkylated derivatives (9a-f & 10a-f). Further, elemental analysis, exact number of hydrogen and carbon atoms at appropriate chemical shift values in 13C NMR spectrum authenticated the formation of designed compounds.
2.2 Pharmacological activity and structure activity relationship (SAR)
2.2.1 Anticancer activity
All the titled compounds (6a-j, 7a-e, 9a-f, and 10a-f) were evaluated for their in vitro cytotoxic activity and Bcr/Abl tyrosine kinase inhibitory activity. The cytotoxic activity was performed by MTT assay Mosmann’s method against tumor cell line panel consisting of HCT116 (human colon cancer cell line), A549 (human lung cancer cell line), K562 (human chronic myeloid leukemia cell line), U937 (human acute monocytic myeloid leukemia cell line), and L02 (human normal liver cell line) whereas the Bcr/Abl tyrosine kinase inhibitory activity was done using the well-established ADP-Glo assay. For both cell based and enzymatic assays, Dasatinib was used as positive control and all the values are represented in μM.
The SAR observed with four set of titled compounds was based on the results outlined in Table 2. The in vitro cytotoxic activity and antiproliferative studies showed that the biological activity of these compounds depends on (i) the nature and site of substituents on aromatic ring (ii) the length and type of substitution on thiazole. (iii) effect of either substituted phenyl/phenyl-triazole all the compounds demonstrate anti-proliferation effects with IC50 values comparable to the control standard dasatinib.
Compounds
Human tumor cells
Human normal liver cells
Bcr-Abl
HCT116
A549
U937
K562
L02
6a
7.22 ± 0.58
6.38 ± 0.26
6.02 ± 1.21
6.08 ± 0.95
>40
0.385
6b
8.11 ± 0.65
7.34 ± 0.11
9.12 ± 0.56
8.82 ± 0.56
>40
0.018
6c
8.21 ± 0.45
8.58 ± 0.23
8.03 ± 0.21
7.43 ± 0.23
>40
16.2
6d
8.29 ± 0.28
12.22 ± 0.87
6.08 ± 0.58
9.11 ± 0.14
>40
11.8
6e
9.81 ± 0.18
9.38 ± 0.18
8.25 ± 0.23
8.67 ± 0.57
>40
12.1
6f
9.51 ± 0.49
13.22 ± 0.28
9.23 ± 0.41
8.53 ± 0.43
>40
16.2
6g
3.22 ± 0.44
5.62 ± 0.71
5.36 ± 0.53
6.15 ± 0.65
>40
0.008
6h
4.34 ± 0.34
6.67 ± 0.09
7.45 ± 0.23
6.47 ± 0.09
>40
0.024
6i
7.34 ± 0.23
12.11 ± 0.23
14.34 ± 0.98
8.21 ± 0.23
>40
2.58
6j
8.21 ± 0.41
9.19 ± 0.45
9.22 ± 0.43
7.49 ± 0.55
>40
12.25
7a
5.01 ± 0.11
7.45 ± 0.87
7.04 ± 0.09
6.09 ± 1.71
>40
0.052
7b
6.52 ± 0.91
8.81 ± 0.48
9.31 ± 0.51
8.82 ± 1.01
>40
0.025
7c
6.54 ± 0.66
12.61 ± 0.76
12.39 ± 0.23
9.98 ± 1.76
>40
0.255
7d
3.43 ± 0.34
5.92 ± 0.34
6.22 ± 0.11
6.51 ± 1.23
>40
0.010
7e
5.07 ± 0.41
7.25 ± 0.24
7.14 ± 0.22
7.41 ± 1.89
>40
0.22
9a
8.11 ± 0.45
9.34 ± 0.11
11.11 ± 0.65
9.32 ± 0.43
>40
4.52
9b
8.11 ± 0.43
9.71 ± 0.43
11.61 ± 0.23
8.99 ± 0.11
>40
ND
9c
3.56 ± 0.67
4.23 ± 0.54
6.12 ± 0.54
6.32 ± 0.56
>40
0.008
9d
6.23 ± 0.45
6.56 ± 0.23
7.55 ± 0.23
9.11 ± 0.67
>40
>500
9e
4.02 ± 0.22
5.65 ± 0.28
6.14 ± 0.74
6.65 ± 0.45
>40
0.022
9f
7.32 ± 0.22
6.25 ± 0.28
7.54 ± 0.74
9.95 ± 0.45
>40
0.019
10a
5.56 ± 0.71
7.17 ± 0.34
13.09 ± 0.86
9.23 ± 0.14
>40
0.89
10b
6.43 ± 0.22
9.76 ± 0.91
13.09 ± 0.40
9.23 ± 0.19
>40
>500
10c
6.25 ± 0.43
7.87 ± 0.38
10.18 ± 0.65
8.22 ± 0.68
>40
0.72
10d
5.56 ± 0.71
10.67 ± 0.44
9.89 ± 0.76
9.45 ± 0.54
>40
0.024
10e
3.45 ± 0.67
4.44 ± 0.67
5.88 ± 0.11
5.17 ± 0.86
>40
0.006
10f
5.11 ± 0.48
7.22 ± 0.45
9.11 ± 0.40
7.55 ± 0.54
>40
0.052
Dasatinib (control)
5.14 ± 0.22
8.65 ± 0.13
9.63 ± 0.25
11.95 ± 0.31
>40
0.019
The SAR studies indicated that the derivatives displayed moderate to high inhibitory activity against the Bcr/Abl protein. In the series (6a-j), various analogs containing methoxy, halo (fluorine, bromine, chlorine), methyl, and hydroxyl groups on various positions of phenyl ring were synthesized to study the cytotoxic potential of designed compounds. In vitro cytotoxicity and antiproliferative inhibition results specify that among the derivatives, compound 6 g with CF3 at 4th position exhibits best activity in these series with IC50 0.008 μM. To further assess the significance of 4-CF3, we replaced these groups in compound 6 h with 4-F and with 2,3,4-F in compound 6f. Both these experiments resulted in partial or total loss of activity. The pharmacological evaluation of Schiff bases (7a-e) prepared with substituted ketones occasioned in additional terminal methyl group in comparison to series (6a-j). The IC50 values against tested cell lines and Bcr/Abl kinase of compound 7d confirmed that the terminal methyl does not influence the biological activity.
The pharmacological activity of triazole substituted derivatives (9a-f and 10a-f), synthesized to determine the significance of terminal bulk group, confirm that there is considerable influence on the cytotoxicity results. The 4-CF3 substituted derivatives 9c and 10e exhibit much higher potency towards tumor cells in comparison to 6g, which exhibited best cytotoxic inhibition among the series (6a-j and 7a-e). Cytotoxicity analyses illustrated that compounds with CF3 at fourth position exhibit best activity in the series. To further study the importance of number and position of fluorine on the phenyl ring, various fluorine derivatives (9b, 9e and 10f) were synthesized, and their biological activity accomplished that the replacement or alteration in positions of fluorine substituents results in forfeiture of activity compared to compounds 9c and 10e.
From the biological data, we could arrive to a conclusion that the synthesized compounds are potent Bcr/Abl kinase inhibitors. They may be potential lead compounds to be developed as an alternative to current dasatinib therapy.
3 Conclusions
In the present investigation, we designed and synthesized four novel series of compounds (6a-j, 7a-e, 9a-f, and 10a-f) utilizing dasatinib as the lead compound. All the target compounds were evaluated for in vitro cytotoxic activity against a panel of cancer cell lines comprising of HCT116 (human colon cancer cell line), A549 (human lung cancer cell line), K562 (human chronic myeloid leukemia cell line), U937 (human acute monocytic myeloid leukemia cell line), and L02 (human normal liver cell line) employing MTT assay. Majority of the compounds showed excellent activity against K562. Hence, all of the prepared compounds were screened for Bcr/Abl tyrosine kinase inhibitory activity using ADP-Glo assay. As most of the compounds are highly potent against K562 cells, Bcr/Abl tyrosine kinase inhibitory activity of all the synthesized compounds was evaluated by using the well-established ADP-Glo assay. Among others, compounds 6g, 7d, 9c, and 10e were the most active Bcr/Abl kinase inhibitors. Therefore, these compounds are promising lead molecules to be developed as an alternative to current dasatinib therapy.
4 Experimental
4.1 Materials and methods
All starting materials, solvents and reagents were purchased from commercial vendors and used without purification. Melting points were recorded using an open capillary tube in a Thomas Hoover melting point apparatus and are uncorrected. Infrared spectra were performed on Shimadzu FT-IR 157. Proton and carbon magnetic resonance (1H NMR, and 13C NMR) were recorded in CDCl3 or DMSO‑d6 using Bruker spectrometer at 300/400 MHz having TMS as an internal standard and reported in ppm. LCMS was recorded on Agilent LC-MS spectrometer. Elemental analysis was performed on Thermo Finnigan Flash (EA 1112 CHNS Analyzer). Thin layer chromatography (TLC) was performed on Merck silica gel GF254 aluminium sheets using mixture of different polar and nonpolar solvents in varying proportions throughout the reaction and spots were observed using iodine as visualizing agent (Munikrishnappa, 2016).
4.2 Synthesis
4.2.1 General procedure for the synthesis of ethyl-2-(2-aminothiazol-4-yl)acetate (2)
The reaction mixture of thiourea (50 g, 0.6578 mol), ethyl-4-chloroactoacetate (119.19 g, 0.7236 mol) and ethanol (1.0 L) was stirred and refluxed for 2 h. After the reaction mixture was cooled, ethanol was removed and the mixture was slowly added into water (500 mL). The pH of reaction mixture was adjusted to 8–8.5 with sodium bicarbonate and the product was extracted using ethyl acetate. Further, organic layer was concentrated and triturated using n-heptane. The product was filtered and dried at 45–50 °C for 5 h and obtained as a yellowish to brown solid (196 g, yield 80%); mp 90–95 °C; IR (KBr) νmax/cm−1 3401 (N—H), 1712 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 1.15–1.18 (t, J = 7.2 Hz, 3H, methyl), 3.43 (s, 2H, CH2, thiazole-CH2-CO), 4.02–4.07 (q, J = 6.8 Hz, 2H, CH2, —O—CH2—), 6.30 (s, 1H, CH, thiazole), 6.89 (s, 2H, NH2, D2O exchangeable); LC-MS (m/z, %): 187.3 (M + 1, 99.16).
4.2.2 General procedure for the synthesis of ethyl-2-(2-(5-bromo-2-chloropyrimidin-4-ylamino)thiazol-4-yl)acetate (3)
The reaction mixture of 5-Bromo-2,4-dichloropyrimidin 1 (50.0 g, 0.2194 mol), potassium carbonate (75.81 g, 0.5485 mol), 1,4 dioxane (500 mL) and ethyl-2-(2-aminothiazol-4-yl)acetate 2 (44.94 g, 0.2413 mol) was refluxed for 24 h. Upon completion of reaction, reaction mixture was quenched into ice cold water and stirred at 25–30 °C for 1 h. Further, the product was filtered and recrystallized using ethanol and dried at 45–50 °C. The compound was obtained as a brown colored solid (69.6 g, yield 84%); mp 122–126 °C; IR (KBr) νmax/cm−1 2983 (N—H), 1728 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 1.18–1.22 (t, J 7.2, 3H, methyl), 3.74 (s, 2H, CH2, thiazole-CH2-CO), 4.08–4.14 (q, J = 6.8 Hz, 2H, CH2, —O—CH2—), 6.96 (s, 1H, CH, thiazole), 8.48 (s, 1H, CH, Pyrimidine), 13.23 (s, 1H, NH, pyrimidine-NH-thiazole, D2O exchangeable); LC-MS (m/z, %): 377 (M + 1, 99).
4.2.3 General procedure for the synthesis of ethyl-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetate (4)
The reaction mixture of ethyl-2-(2-(5-bromo-2-chloropyrimidin-4-ylamino)thiazol-4-yl)acetate 3 (60 g, 0.1588 mol), triethylamine (32.12 g, 0.3177 mol), morpholine (27.70 g, 0.3177 mol) and ethanol (600 mL) was refluxed for 12 h. Upon completion of reaction, ethanol was removed and quenched into cold water. Further, reaction mixture was stirred at 25–30 °C for 1 h and the solids formed were filtered. Product was recrystallized using ethanol and dried at 45–50 °C. The compound was obtained as a light Yellowish solid (59.88 g, yield 88%); mp 96–98 °C; IR (KBr) νmax/cm−1 3342 (N—H), 1727 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 1.17–1.21 (t, J = 7.08 Hz, 3H, methyl), 3.67–3.68 (m, 4H, 2CH2, morpholine), 3.70 (s, 2H, CH2, thiazole-CH2-CO), 3.75–3.76 (m, 4H, 2CH2, morpholine), 4.07–4.12 (q, J = 7.08 Hz, 2H, CH2, —O—CH2—), 6.99 (s, 1H, CH, thiazole), 8.24 (s,1H, CH, Pyrimidine), 10.39 (s, 1H, NH, pyrimidine-NH-thiazole, D2O exchangeable); LC-MS (m/z, %): 428 (M + 1, 99.38).
4.2.4 General procedure for the synthesis of 2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (5)
The reaction mixture of ethyl 2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetate 4 (55 g, 0.1284 mol), ethanol (500 mL) and 98% hydrazine hydrate (50 mL) was refluxed for 8 h. Upon completion of reaction, product was filtered and recrystallized using ethanol and dried at 45–50 °C. The compound was obtained as a pale brown solid (47.34 g, yield 89%); mp 206–210 °C; IR (KBr) νmax/cm−1 3242 (N—H), 1671 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.38 (s, 2H, CH2, thiazole-CH2-CO), 3.65–3.67 (m, 4H, 2CH2, morpholine), 3.73–3.74 (m, 4H, 2CH2, morpholine), 5.74–5.95 (m, 3H, NH—NH2, D2O exchangeable), 6.78 (s, 1H, CH, thiazole), 8.13 (s, 1H, CH, Pyrimidine), 9.12 (s, 1H, NH, pyrimidine-NH-thiazole, D2O exchangeable); LC-MS (m/z, %): 414.0 (M + 1, 99).
4.2.5 General procedure for the synthesis of (Z)-N'-(substituted benzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6a-j)
The reaction mixture of 2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide 5 (0.01 mol), substituted aldehydes (0.01 mol) and ethanol (5 V) was refluxed for 3 h in the presence of few drops of glacial acetic acid. Upon completion of reaction, solvent was evaporated and residue was quenched using cold water (5 V). Further, product was filtered and crude solid was recrystallized using appropriate solvent systems to obtain target products (6a-j).
4.2.5.1 Synthesis of (Z)-N'-(2,6-difluorobenzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6a)
Recrystallized using ethanol, off-white to pale brown solid (yield 83%); mp 224–230 °C; IR (KBr) νmax/cm−1 3206 (N—H), 2351 (C⚌N), 1670 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.68 (m, 4H, 2CH2, morpholine), 3.76 (m, 4H, 2CH2, morpholine), 3.99 (s, 2H, CH2, thiazole-CH2-CO), 6.94 (s, 1H, CH, thiazole), 7.17 (m, 2H, 2CH, 2,6-difluorobenzyl), 7.48 (m, J 7.2 , 1H, CH, 2,6-difluorobenzyl), 8.15 (s, 1H, CH, Pyrimidine), 8.25 (s, 1H, CH, N⚌CH, benzylidenimin), 10.34 (t, 1H, NH, sec amine, D2O exchangeable), 11.62 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.6 (thiazole-CH2-), 45.16, 66.33 (morpholine), 95.5 (pyrimidine-C5), 104.4 (thiazole-C3), 105.6, 111.87, 112.71, 131.86 (2,6-diflurobenzyl-C1, C3, C5, C4), 141.98 (HC⚌N-N), 145.53 (pyrimidine-C2), 150.8, 158.18 (thiazole-C1, C2), 160.14 (pyrimidine-C6), 161.94 (2,6-difluro benzyl-C2, C6), 165.75 (CO, sec amide), 171.41 (pyrimidine-C4); LC-MS (m/z, %): 538.0 (M + 1, 99).
4.2.5.2 Synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(furan-2-ylmethylene)acetohydrazide (6b)
Recrystallized using ethanol, pale brown solid (yield 80%); mp 198–203 °C; IR (KBr) νmax/cm−1 3245 (N—H), 2354 (C⚌N), 1672 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.67 (m, 4H, 2CH2, morpholine), 3.76 (m, 4H, 2CH2, morpholine), 3.98 (s, 2H, CH2, thiazole-CH2-CO), 6.92 (s, 1H, CH, thiazole), 7.09 (m, 1H, CH, furan), 7.41 (m, 1H, CH, furan), 8.03 (m, 1H, CH, furan), 8.10 (s,1H, CH, Pyrimidine), 8.40 (s, 1H, CH, N⚌CH, benzylidenimin), 10.30 (t, 1H, NH, sec amine, D2O exchangeable), 11.60 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.8 (thiazole-CH2-), 45.20, 66.35 (morpholine), 95.4 (pyrimidine-C5), 104.5 (thiazole-C3), 112.6, 118.90 (furan-C3, C2), 141.4 (HC⚌N—N), 144.2 (furan-C4), 145.55 (pyrimidine-C2), 148.34 (furan-C1), 150.82, 158.20 (thiazole-C1, C2), 160.12 (pyrimidine-C6), 165.77 (CO, sec amide), 171.40 (pyrimidine-C4); LC-MS (m/z, %): 492 (M + 1, 99.10).
4.2.5.3 Synthesis of (Z)-N'-((1H-pyrrol-2-yl)methylene)-2-(2-(5-bromo-2-morpholino pyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6c)
Recrystallized using ethanol, pale brown solid (yield 78%); mp 200–204 °C; IR (KBr) νmax/cm−1 3235 (N—H), 2349 (C⚌N), 1675 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.65 (m, 4H, 2CH2, morpholine), 3.77 (m, 4H, 2CH2, morpholine), 3.97 (s, 2H, CH2, thiazole-CH2-CO), 6.35 (m, 1H,CH, pyrrole), 6.93 (s, 1H, CH, thiazole), 7.05 (m, 1H, CH, pyrrole), 7.54 (m, 1H, CH, pyrrole), 8.20 (s, 1H, CH, Pyrimidine), 8.58 (s, 1H, CH, N⚌CH, benzylidenimin), 10.32 (t, 1H, NH, sec amine, D2O exchangeable), 11.62 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.6(thiazole-CH2-), 45.16, 66.33 (morpholine), 95.5 (pyrimidine-C5), 104.41 (thiazole-C3), 110.9, 119.2, 124.8, 132.71 (pyrrole-C4, C3, C2, C1), 142.5 (HC⚌N-N), 146.51 (pyrimidine-C2), 150.80, 158.21 (thiazole-C1, C2), 160.14 (pyrimidine-C6), 165.75 (CO, sec amide), 171.41 (pyrimidine-C4); LC-MS (m/z, %): 491 (M + 1, 99.12).
4.2.5.4 Synthesis of (Z)-N'-benzylidene-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6d)
Recrystallized using ethanol, off-white to pale brown solid (yield 84%); mp 215–218 °C; IR (KBr) νmax/cm−1 3214 (N—H), 2350 (C⚌N), 1674 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.68 (m, 4H, 2CH2, morpholine), 3.75 (m, 4H, 2CH2, morpholine), 4.02 (s, 2H, CH2, thiazole-CH2-CO), 6.90 (s, 1H, CH, thiazole), 7.55–7.76 (m, 5H, 5CH, benzyl), 7.96 (s, 1H, CH, Pyrimidine), 8.24 (s, 1H, CH, N⚌CH, benzylidenimin), 10.32 (t, 1H, NH, sec amine, D2O exchangeable), 11.54 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.62 (thiazole-CH2-), 45.13, 66.32 (morpholine), 95.6 (pyrimidine- C5), 104.3 (thiazole-C3), 128.8, 129.21, 131.03, 133.6 (benzyl-C3, C5, C2, C6, C4, C1), 142.81 (HC⚌N-N), 145.55 (pyrimidine-C2), 150.85, 158.20 (thiazole-C1, C2), 160.18 (pyrimidine-C6), 165.77 (CO, sec amide), 171.42 (pyrimidine-C4); LC-MS (m/z, %): 502 (M + 1, 99).
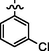
4.2.5.5 Synthesis of (Z)-N'-(3-chlorobenzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6e)
Recrystallized using ethanol, off-white to pale brown solid (yield 81%); mp 214–218 °C; IR (KBr) νmax/cm−1 3194 (N—H), 2276 (C⚌N), 1672 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.69 (m, 4H, 2CH2, morpholine), 3.76 (m, 4H, 2CH2, morpholine), 4.05 (s, 2H, CH2, thiazole-CH2-CO), 6.93 (s, 1H, CH, thiazole), 7.22 (t, J = 7.96 Hz, 1H, CH, 3-chlorobenzyl), 7.44–7.55 (m, 3H, 3CH, 3-chlorobenzyl), 7.99 (s, 1H, CH, Pyrimidine), 8.22 (s, 1H, CH, N⚌CH, benzylidenimin), 10.33 (t, 1H, NH, sec amine, D2O exchangeable), 11.59 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.60 (thiazole-CH2-), 45.13, 66.32 (morpholine), 95.5 (pyrimidine-C5), 104.3 (thiazole-C3), 113.59, 117.29, 123.76, 131.29, 137.20 (3-chlorobenzyl-C1, C5, C3, C4, C2), 141.98 (HC⚌N-N), 145.53 (pyrimidine-C2), 150.8, 158.18 (thiazole-C1, C2), 160.14 (pyrimidine-C6), 161.65 (3-chlorobenzyl-C6), 165.75 (CO, sec amide), 171.41 (pyrimidine-C4); LC-MS (m/z, %): 536 (M + 1, 99.03).
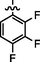
4.2.5.6 Synthesis of (Z)-N'-(2,3,4-trifluorobenzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6f)
Recrystallized from ethanol, off-white to pale brown solid (yield 83%); mp 234–238 °C; IR (KBr) νmax/cm−1 3201 (N—H), 2298 (C⚌N), 1679 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.69 (m, 4H, 2CH2, morpholine), 3.76 (m, 4H, 2CH2, morpholine), 4.04 (s, 2H, CH2, thiazole-CH2-CO), 6.92 (s, 1H, CH, thiazole), 7.11–7.54 (m, 2H, 2CH, 2,3,4-trifluorobenzyl), 7.98 (s, 1H, CH, Pyrimidine), 8.24 (s, 1H, CH, N⚌CH, benzylidenimin), 10.35 (t, 1H, NH, sec amine, D2O exchangeable), 11.57 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.60 (thiazole-CH2-), 45.15, 66.35 (morpholine), 95.8 (pyrimidine-C5), 104.3 (thiazole-C3), 115.5, 120.89, 128.0, 140.20 (2,3,4-trifluorobenzyl-C1, C5, C6, C3), 141.98 (HC⚌N—N), 145.54 (pyrimidine-C2), 145.63 (2,3,4-trifluorobenzyl-C2), 150.81 (thiazole-C1), 153.42 (2,3,4-trifluorobenzyl-C4), 158.23 (thiazole-C2), 160.14 (pyrimidine-C6), 165.76 (CO, sec amide), 171.43 (pyrimidine-C4); LC-MS (m/z, %): 556 (M + 1, 98.6).
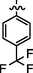
4.2.5.7 Synthesis of (Z)-N'-(4-(trifluoromethyl)benzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino) thiazol-4-yl)acetohydrazide (6 g)
Recrystallized using ethanol, off-white to pale brown solid (yield 80%); mp 220–223 °C; IR (KBr) νmax/cm−1 3193 (N—H), 2277 (C⚌N), 1674 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.68 (m, 4H, 2CH2, morpholine), 3.75 (m, 4H, 2CH2, morpholine), 4.04 (s, 2H, CH2, thiazole-CH2-CO), 6.92 (s, 1H, CH, thiazole), 7.19 (s, 2H, 2CH, 4-(trifluoromethyl)phenyl), 7.33 (s, 2H, 2CH, 4-(trifluoromethyl)phenyl), 7.97 (s, 1H, CH, Pyrimidine), 8.22 (s, 1H, CH, N⚌CH, benzylidenimin), 10.32 (t, 1H, NH, sec amine, D2O exchangeable), 11.58 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.61 (thiazole-CH2-), 45.14, 66.33 (morpholine), 95.51 (pyrimidine-C5), 104.32 (thiazole-C3), 122.85 (trifluoro carbon), 127.33, 128.64 (4-(trifluoromethyl)phenyl-C3, C5, C2, C6), 134.74 (4-(trifluoromethyl)phenyl-C4), 141.99 (HC⚌N—N), 145.54 (pyrimidine-C2), 149.60 (4-(trifluoromethyl)phenyl-C1), 150.80, 158.18 (thiazole-C1, C2), 160.15 (pyrimidine-C6), 165.75 (CO, sec amide), 171.41 (pyrimidine-C4); LC-MS (m/z, %): 570 (M + 1, 99.0).
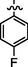
4.2.5.8 Synthesis of (Z)-N'-(4-fluorobenzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6h)
Recrystallized using ethanol, off-white to pale brown solid (yield 79%); mp 215–218 °C; IR (KBr) νmax/cm−1 3194 (N—H), 2276 (C⚌N), 1676 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.67 (m, 4H, 2CH2, morpholine), 3.74 (m, 4H, 2CH2, morpholine), 4.02 (s, 2H, CH2, thiazole-CH2-CO), 6.93 (s, 1H, CH, thiazole), 7.12 (s, 2H, 2CH, 4-fluorophenyl), 7.30 (s, 2H, 2CH, 4-fluorophenyl), 7.98 (s, 1H, CH, Pyrimidine), 8.23 (s, 1H, CH, N⚌CH, benzylidenimin), 10.33 (t, 1H, NH, sec amine, D2O exchangeable), 11.59 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.60 (thiazole-CH2-), 45.13, 66.32 (morpholine), 95.50 (pyrimidine-C5), 104.31 (thiazole-C3), 128.8, 130.25 (4-fluorophenyl- C3, C5, C2, C6), 132.83 (4-fluorophenyl-C1), 141.99 (HC⚌N—N), 145.54 (pyrimidine-C2), 159.20 (4-fluorophenyl-C4), 150.80, 158.18 (thiazole-C1, C2), 160.15 (pyrimidine-C6), 165.75 (CO, sec amide), 171.41 (pyrimidine-C4); LC-MS (m/z, %): 520 (M + 1, 98.8).
4.2.5.9 Synthesis of (Z)-N'-(4-hydroxybenzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6i)
Recrystallized using ethanol, off-white to pale brown solid (yield 75%); mp 217–220 °C; IR (KBr) νmax/cm−1 3195 (N—H), 2278 (C⚌N), 1675 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.66 (m, 4H, 2CH2, morpholine), 3.73 (m, 4H, 2CH2, morpholine), 4.04 (s, 2H, CH2, thiazole-CH2-CO), 6.92 (s, 1H, CH, thiazole), 7.16 (s, 2H, 2CH, 4-hydroxyphenyl), 7.38 (s, 2H, 2CH, 4-hydroxyphenyl), 7.99 (s, 1H, CH, Pyrimidine), 8.22 (s, 1H, CH, N⚌CH, benzylidenimin), 9.10 (s, 1H, benzene-OH), 10.34 (t, 1H, NH, sec amine, D2O exchangeable), 11.58 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.61 (thiazole-CH2-), 45.12, 66.30 (morpholine), 95.51 (pyrimidine-C5), 104.30 (thiazole-C3), 116.0, 126.4 (4-hydroxyphenyl-C3,5, C1), 130.6 (4-hydroxyphenyl-C2,6), 141.99 (HC⚌N—N), 145.54 (pyrimidine-C2), 150.80, 158.18 (thiazole-C1, C2), 160.16 (pyrimidine-C6), 160.80 (4-hydroxyphenyl-C4), 165.76 (CO, sec amide), 171.42 (pyrimidine-C4); LC-MS (m/z, %): 518 (M + 1, 98.6).
4.2.5.10 Synthesis of (Z)-N'-(3-methoxybenzylidene)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide (6j)
Recrystallized using ethanol, off-white to pale brown solid (yield 81%); mp 214–217 °C; IR (KBr) νmax/cm−1 3195 (N—H), 2278 (C⚌N), 1675 (C⚌O); 1H NMR (400 MHz, DMSO‑d6): δ 3.68 (m, 4H, 2CH2, morpholine), 3.72 (m, 4H, 2CH2, morpholine), 3.82 (s, 3H, phenyl-O-CH3), 4.05 (s, 2H, CH2, thiazole-CH2-CO), 6.93 (s, 1H, CH, thiazole), 7.30 (t, 1H, CH, 3-methoxy phenyl), 7.47–7.59 (m, 3H, 3CH, 3-methoxy phenyl), 7.98 (s, 1H, CH, Pyrimidine), 8.22 (s, 1H, CH, N⚌CH, benzylidenimin), 10.35 (t, 1H, NH, sec amine, D2O exchangeable), 11.57 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 38.60 (thiazole-CH2-), 45.13, 66.30 (morpholine), 55.9 (CH3, aliphatic methyl), 95.51 (pyrimidine-C5), 104.30 (thiazole-C3), 111.2, 116.6, 121.5 (3-methoxy phenyl-C2, C4, C6), 129.8 (3-methoxy phenyl-C5), 138.2 (3-methoxyphenyl-C1), 141.99 (HC⚌N-N), 145.54 (pyrimidine-C2), 150.80, 158.18 (thiazole-C1, C2), 160.16 (pyrimidine-C6), 160.6 (3-methoxy phenyl-C3), 165.76 (CO, sec amide), 171.40 (pyrimidine-C4); LC-MS (m/z, %): 532 (M + 1, 98.9).
4.2.6 General procedure for the synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(1-(substitutedphenyl)ethylidene)acetohydrazide (7a-e).
The reaction mixture of 2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide 5 (0.01 mol), substituted acetophenones (0.01 mol) and ethanol (5 V) was refluxed for 3 h in presence of few drops of glacial acetic acid. Upon completion of reaction, solvent was evaporated and the product was quenched using cold water (5 V). The product was filtered, dried, and recrystallized using appropriate solvent systems to obtain target products (7a-e).
4.2.6.1 Synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(1-p-tolylethylidene)acetohydrazide (7a)
Recrystallized using ethanol, off-white to pale brown solid (yield 77%); mp 104–107 °C; IR (KBr) νmax/cm−1 3309 (N—H), 2326 (C⚌N), 1674 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 2.15 (s, 3H, CH3), 2.22 (m, 3H, CH3, methyl benzene), 3.81 (m, 4H, 2CH2, morpholine), 3.85 (m, 4H, 2CH2, morpholine), 4.26 (s, 2H, CH2, thiazole-CH2-CO), 6.82 (s, 1H, CH, thiazole), 7.28–7.70 (m, 4H, 4CH, methyl benzene), 7.72 (s, 1H, CH, Pyrimidine), 8.89 (t, 1H, NH, sec amine, D2O exchangeable), 10.44 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, CDCl3): δ 12.54 (methyl, methyl benzene), 13.78 (methyl), 38.34 (thiazole-CH2-), 44.88, 66.69 (morpholine), 91.39 (pyrimidine-C5), 110.4 (thiazole-C3), 127.01, 129.13, 134.48, 139.90 (methyl benzene-C2, C6, C3, C5, C1, C4), 144.73 (C⚌N—N), 146.01 (pyrimidine-C2), 152.94, 157.38 (thiazole-C1, C2), 157.49 (pyrimidine-C6), 159.78 (CO, sec amide), 172.75 (pyrimidine-C4); LC-MS (m/z, %): 530.40 (M + 1, 99.2).
4.2.6.2 Synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(1-phenylethylidene)acetohydrazide (7b)
Recrystallized using ethanol, off-white to pale brown solid (yield 86%); mp 110–114 °C; IR (KBr) νmax/cm−1 3323 (N—H), 2324 (C⚌N), 1673 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 2.16 (s, 3H, CH3), 3.80 (m, 4H, 2CH2, morpholine), 3.86 (m, 4H, 2CH2, morpholine), 4.25 (s, 2H, CH2, thiazole-CH2-CO), 6.83 (s, 1H, CH, thiazole), 7.53 (m, 3H, 3CH, phenyl), 7.70 (s,1H, CH, Pyrimidine), 7.94 (m, 2H, 2CH, phenyl), 8.89 (t, 1H, NH, sec amine, D2O exchangeable), 10.43 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, CDCl3): δ 13.77 (methyl), 38.35 (thiazole-CH2-), 44.87, 66.68 (morpholine), 91.40 (pyrimidine- C5), 110.2 (thiazole- C3), 128.20, 128.89, 131.01, 137.50 (methyl benzene-C2, C6, C3, C5, C4, C1), 144.72 (C⚌N—N), 146.0 (pyrimidine-C2), 152.93, 157.39 (thiazole-C1, C2), 157.48 (pyrimidine-C6), 159.77 (CO, sec amide), 172.77 (pyrimidine-C4); LC-MS (m/z, %): 516 (M + 1, 98.9).
4.2.6.3 Synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(1-(3-chlorophenyl)ethylidene)acetohydrazide (7c)
Recrystallized from ethanol, off-white to pale brown solid (yield 82%); mp 112–116 °C; IR (KBr) νmax/cm−1 3311 (N—H), 2328 (C⚌N), 1670 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 2.16 (s, 3H, CH3), 3.80 (m, 4H, 2CH2, morpholine), 3.86 (m, 4H, 2CH2, morpholine), 4.25 (s, 2H, CH2, thiazole-CH2-CO), 6.83 (s, 1H, CH, thiazole), 7.28 (m, 2H, 2CH, 3-chlorophenyl), 7.59 (d, J 7.24, 1H, CH, 3-chlorophenyl), 7.70 (s,1H, CH, Pyrimidine), 8.11 (s, 1H, CH, 3-chlorophenyl), 8.88 (t, 1H, NH, sec amine, D2O exchangeable), 10.42 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, CDCl3): δ 13.77 (methyl), 38.34 (thiazole-CH2-), 44.88, 66.69 (morpholine), 91.38 (pyrimidine-C5), 110.4 (thiazole-C3), 126.17, 126.76, 129.23, 129.55, 134.44, 139.39 (3-chlorophenyl-C6, C2, C5, C4, C3, C1), 144.73 (C⚌N—N), 146.01 (pyrimidine-C2), 152.94, 157.38 (thiazole-C1, C2), 157.49 (pyrimidine-C6), 159.78 (CO, sec amide), 172.75 (pyrimidine-C4); LC-MS (m/z, %): 550 (M + 1, 98.25).
4.2.6.4 Synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(1-(4-(trifluoromethyl) phenyl)ethylidene)acetohydrazide (7d)
Recrystallized using ethanol, off white to pale brown solid (yield 70%); mp 117–120 °C; IR (KBr) νmax/cm−1 3308 (N—H), 2326 (C⚌N), 1675 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 2.14 (s, 3H, CH3), 3.80 (m, 4H, 2CH2, morpholine), 3.84 (m, 4H, 2CH2, morpholine), 4.25 (s, 2H, CH2, thiazole-CH2-CO), 6.83 (s, 1H, CH, thiazole), 7.16 (s, 2H, 2CH, 4-(trifluoromethyl)phenyl), 7.30 (s, 2H, 2CH, 4-(trifluoromethyl)phenyl), 7.74 (s, 1H, CH, Pyrimidine), 8.90 (t, 1H, NH, sec amine, D2O exchangeable), 10.44 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, CDCl3): δ 13.78 (methyl), 38.35 (thiazole-CH2-), 44.88, 66.69 (morpholine), 91.39 (pyrimidine-C5), 110.4 (thiazole-C3), 122.83 (trifluoro carbon), 127.34, 128.63 (4-(trifluoromethyl)phenyl-C3, C5, C2, C6), 134.64 (4-(trifluoromethyl)phenyl-C4), 144.73 (C⚌N—N), 146.01 (pyrimidine-C2), 147.60 (4-(trifluoromethyl)phenyl-C1), 152.94, 157.38 (thiazole-C1, C2), 157.50 (pyrimidine-C6), 159.78 (CO, sec amide), 172.76 (pyrimidine-C4); LC-MS (m/z, %): 584 (M + 1, 99.0).
4.2.6.5 Synthesis of (Z)-2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)-N'-(1-(4-fluorophenyl)ethylidene)acetohydrazide (7e)
Recrystallized using ethanol, off-white to pale brown solid (yield 81%); mp 115–117 °C; IR (KBr) νmax/cm−1 3306 (N—H), 2326 (C⚌N), 1674 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 2.15 (s, 3H, CH3), 3.81 (m, 4H, 2CH2, morpholine), 3.83 (m, 4H, 2CH2, morpholine), 4.26 (s, 2H, CH2, thiazole-CH2-CO), 6.82 (s, 1H, CH, thiazole), 7.11 (s, 2H, 2CH, 4-fluorophenyl), 7.30 (s, 2H, 2CH, 4-fluorophenyl), 7.73 (s, 1H, CH, Pyrimidine), 8.89 (t, 1H, NH, sec amine, D2O exchangeable), 10.43 (s, 1H, NH, sec amide, D2O exchangeable); 13C NMR (100 MHz, CDCl3): δ 13.77 (methyl), 38.34 (thiazole-CH2-), 44.87, 66.68 (morpholine), 91.40 (pyrimidine-C5), 110.4 (thiazole-C3), 128.81, 130.26 (4-fluorophenyl- C3, C5, C2, C6), 132.82 (4-fluorophenyl-C1), 144.73 (C⚌N—N), 146.01 (pyrimidine-C2), 152.94, 157.38 (thiazole-C1, C2), 157.50 (pyrimidine-C6), 159.20 (4-fluorophenyl-C4), 159.78 (CO, sec amide), 172.76 (pyrimidine-C4); LC-MS (m/z, %): 534 (M + 1, 98.8).
4.2.7 General procedure for the synthesis of 5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazole-3-thiol (8)
2-(2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)acetohydrazide 5 (25 g, 0.0603 mol), phenylisothiocyanate (12.23 g, 0.0905 mol) and ethanol (250 mL) mixture was refluxed for 4 h to give a white solids. The resulting solids were dissolved in 2 N sodium hydroxide (100 mL) and refluxed for 4 h. The resulting solution was cooled to 25–30 °C and acidified to pH 3–4 with conc. Hydrochloric acid to give off-white solid. The solid formed was filtered and recrystallized using ethanol to obtain as an off-white solid (24 g, yield 75%); mp 224–226 °C; IR (KBr) νmax/cm−1 3212 (N—H), 2845 (S—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.67 (m, 4H, 2CH2, morpholine), 3.74 (m, 4H, 2CH2, morpholine), 3.91 (s, 2H, CH2, thiazole-CH2-triazole), 6.59 (s, 1H, CH, thiazole), 7.31 (s, 2H, 2CH, phenyl triazole), 7.46 (s, 3H, 3CH, phenyl triazole), 8.24 (s, 1H, CH, Pyrimidine), 10.35 (s, 1H, NH, sec amine, D2O exchangeable), 13.80 (s, 1H, SH, thiol); LC-MS (m/z, %): 530.8 (M + 1, 98.15).
4.2.8 General procedure for the synthesis of N-(4-((5-(substituted benzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9a-f)
The reaction mixture of 5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazole-3-thiol 8 (0.10 mol), ethanol (5 V) and potassium hydroxide (0.25 mol) was stirred at 25–30 °C for 30 min. Further, substituted benzyl bromide (0.11 mol) was added and stirred at 25–30 °C for 5 h. Upon completion of reaction ethanol was evaporated and quenched into the water (5 V). The obtained product was filtered and recrystallized using suitable solvents to obtain target compounds (9a-f).
4.2.8.1 Synthesis of N-(4-((5-(benzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9a)
Recrystallized using ethanol, pale brown solid (yield 81%); mp 108–112 °C; IR (KBr) νmax/cm−1 3216 (N—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.66 (m, 4H, 2CH2, morpholine), 3.71 (m, 4H, 2CH2, morpholine), 3.97 (s, 2H, CH2, thiazole-CH2-triazole), 4.36 (s, 2H, CH2, S-CH2-phenyl), 6.50 (s, 1H, CH, thiazole), 7.25–7.27 (m, 3H, 3CH, phenyl), 7.38–7.48 (m, 6H, 6CH, phenyl, phenyl triazole), 7.65 (m, 1H, CH, phenyl triazole), 8.24 (s, 1H, CH, Pyrimidine), 10.29 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 22.7 (thiazole-CH2-triazole), 34.70 (-S-CH2-phenyl), 45.1, 66.28 (morpholine), 94.50 (pyrimidine-C5), 110.10 (thiazole-C3), 127.1, 127.7, 128.8, 130.25 (phenyl, phenyl triazole), 137.10 (phenyl-C1), 145.5 (phenyl triazole-C1), 147.97 (triazole-C1), 150.20 (thiazole-C2, pyrimidine-C2), 153.62 (triazole-C2), 158.45 (thiazole-C1, pyrimidine-C6),159.96 (pyrimidine-C4); LC-MS (m/z, %): 621 (M + 1, 99.2).
4.2.8.2 Synthesis of N-(4-((5-(5-chloro-2-fluorobenzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9b)
Recrystallized using ethanol, pale brown solid (yield 80%); mp 106–110 °C; IR (KBr) νmax/cm−1 3221 (N—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.66 (m, 4H, 2CH2, morpholine), 3.70 (m, 4H, 2CH2, morpholine), 3.99 (s, 2H, CH2, thiazole-CH2-triazole), 4.32 (s, 2H, CH2, S-CH2-phenyl), 6.51 (s, 1H, CH, thiazole), 7.09–7.32 (m, 3H, 3CH, 5-chloro-2-fluorobenzyl), 7.38 (s, 2H, 2CH, phenyl triazole), 7.62 (s, 3H, 3CH, phenyl triazole), 8.21 (s, 1H, CH, Pyrimidine), 10.28 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 22.70 (thiazole-CH2-triazole), 34.71 (-S-CH2-phenyl), 45.10, 66.27 (morpholine), 95.6 (pyrimidine-C5), 110.10 (thiazole-C3), 116.21, 128.40 (5-chloro-2-fluorobenzyl-C5, C1), 128.80 (phenyl triazole-C3, C4, C5), 128.9, 129.10 (5-chloro-2-fluorobenzyl-C4, C2), 130.25 (phenyl triazole-C2, C6), 131.10 (5-chloro-2-fluorobenzyl-C3), 145.5 (phenyl triazole-C1), 147.91(triazole- C1), 150.12 (thiazole-C2, pyrimidine- C2), 153.61 (triazole-C2), 158.45 (thiazole-C1, pyrimidine-C6), 160.03 (5-chloro-2-fluorobenzyl-C6), 160.30 (pyrimidine-C4); LC-MS (m/z, %): 674 (M + 2, 98.89).
4.2.8.3 Synthesis of N-(4-((5-(4-(trifluoromethyl)benzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9c)
Recrystallized using ethanol, pale brown solid (yield 80%); mp 112–116 °C; IR (KBr) νmax/cm−1 3214 (N—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.66 (m, 4H, 2CH2, morpholine), 3.71 (m, 4H, 2CH2, morpholine), 3.99 (s, 2H, CH2, thiazole-CH2-triazole), 4.34 (s, 2H, CH2, S-CH2-phenyl), 6.51 (s, 1H, CH, thiazole), 7.16 (s, 2H, 2CH, 4-(trifluoromethyl)benzyl), 7.28 (s, 2H, 2CH, 4-(trifluoromethyl)benzyl), 7.40–7.49 (m, 5H, 5CH, phenyl triazole), 8.23 (s, 1H, CH, Pyrimidine), 10.28 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 22.70 (thiazole-CH2-triazole), 35.90 (—S—CH2-phenyl), 45.10, 66.28 (morpholine), 95.60 (pyrimidine-C5), 110.13 (thiazole-C3), 121.75 (trifluoro carbon), 127.60, 128.63 (4-(trifluoromethyl)benzyl-C3, C5, C2, C6), 129.97, 130.25, 131.29 (phenyl triazole-C3, C4, C5, C2, C6), 133.30 (4-(trifluoromethyl)benzyl- C4), 145.5 (phenyl triazole-C1), 147.30 (triazole-C1), 147.97 (4-(trifluoromethyl)benzyl-C1), 150.16 (thiazole-C2, pyrimidine-C2), 153.63 (triazole-C2), 158.53 (thiazole-C1, pyrimidine-C6), 159.95 (pyrimidine-C4); LC-MS (m/z, %): 689 (M + 1, 98.67).
4.2.8.4 Synthesis of N-(4-((5-(4-(trifluoromethoxy)benzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9d)
Recrystallized using ethanol, pale brown solid (yield 78%); mp 84–87 °C; IR (KBr) νmax/cm−1 3216 (N—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.66 (m, 4H, 2CH2, morpholine), 3.71 (m, 4H, 2CH2, morpholine), 3.99 (s, 2H, CH2, thiazole-CH2-triazole), 4.34 (s,2H, CH2, S—CH2-phenyl), 6.51 (s, 1H, CH, thiazole), 7.14 (s, 2H, 2CH, 4-(trifluoromethoxy)benzyl), 7.26 (s, 2H, 2CH, 4-(trifluoromethoxy)benzyl), 7.40–7.49 (m, 5H, 5CH, phenyl triazole), 8.23 (s, 1H, CH, Pyrimidine), 10.28 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 22.7 (thiazole-CH2-triazole), 35.90 (-S-CH2-phenyl), 45.10, 66.28 (morpholine), 95.50 (pyrimidine-C5), 110.13 (thiazole-C3), 119.21 (4-(trifluoromethoxy)benzyl-C3, C5), 127.60 (4-(trifluoromethoxy)benzyl-C2, C6), 128.80 (phenyl triazole-C3, C4, C5), 129.97 (4-(trifluoromethoxy)benzyl-C1), 130.25 (phenyl triazole-C2, C6), 131.29 (trifluoro carbon), 145.51 (phenyl triazole-C1), 146.0 (4-(trifluoromethoxy)benzyl-C4), 147.97 (triazole-C1), 150.16 (thiazole- C2, pyrimidine-C2), 153.63 (triazole-C2), 158.53 (thiazole-C1, pyrimidine- C6), 159.95 (pyrimidine-C4); LC-MS (m/z, %): 704.4 (M + 1, 98.81).
4.2.8.5 Synthesis of N-(4-((5-(4-fluorobenzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9e)
Recrystallized using ethanol, pale brown solid (yield 82%); mp 103–106 °C; IR (KBr) νmax/cm−1 3221 (N—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.66 (m, 4H, 2CH2, morpholine), 3.70 (m, 4H, 2CH2, morpholine), 3.97 (s, 2H, CH2, thiazole-CH2-triazole), 4.36 (s,2H, CH2, S—CH2-phenyl), 6.50 (s, 1H, CH, thiazole), 7.11 (s, 2H, 2CH, 4-fluorobenzyl), 7.32 (s, 2H, 2CH, 4-fluorobenzyl), 7.38–7.48 (m, 4H, 4CH, phenyl triazole), 7.62 (m, 1H, CH, phenyl triazole), 8.22 (s,1H, CH, Pyrimidine), 10.29 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 22.8 (thiazole-CH2-triazole), 34.70 (-S-CH2-phenyl), 45.14, 66.29 (morpholine), 94.50 (pyrimidine-C5), 110.10 (thiazole-C3), 115.5, 128.6, 128.8, 130.25 (4-fluorobenzyl, phenyl triazole), 132.7 (4-fluorobenzyl-C1), 145.50 (phenyl triazole-C1), 147.96 (triazole-C1), 150.20 (thiazole-C2, pyrimidine-C2), 153.62 (triazole-C2), 158.54 (thiazole-C1, pyrimidine-C6), 159.1 (4-fluorobenzyl-C4), 159.97 (pyrimidine-C4); LC-MS (m/z, %): 639 (M + 1, 98.75).
4.2.8.6 Synthesis of N-(4-((5-(3,4-dichlorobenzylthio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)thiazol-2-yl)-5-bromo-2-morpholinopyrimidin-4-amine (9f)
Recrystallized using ethanol, pale brown solid (yield 84%); mp 115–118 °C; IR (KBr) νmax/cm−1 3214 (N—H); 1H NMR (400 MHz, DMSO‑d6): δ 3.67 (m, 4H, 2CH2, morpholine), 3.71 (m, 4H, 2CH2, morpholine), 3.97 (s, 2H, CH2, thiazole-CH2-triazole), 4.36 (s, 2H, CH2, S—CH2-phenyl), 6.50 (s, 1H, CH, thiazole), 7.22 (m, 1H, CH, 3,4-dichlorobenzyl), 7.38–7.48 (m, 4H, 4CH, phenyl triazole), 7.58 (m, 1H, CH, phenyl triazole), 7.62 (m, 1H, CH, 3,4-dichlorobenzyl), 7.73 (m, 1H, CH, 3,4-dichlorobenzyl), 8.23 (s, 1H, CH, Pyrimidine), 10.28 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6): δ 22.7 (thiazole-CH2-triazole), 34.40 (-S-CH2-phenyl), 45.10, 66.28 (morpholine), 94.55 (pyrimidine-C5), 110.15 (thiazole-C3), 128.8, 129.1, 130.25, 130.60, 131.8, 132.0 (phenyl triazole, 3,4-dichlorobenzyl), 135.91 (3,4-dichlorobenzyl-C1), 145.51 (phenyl triazole-C1), 147.98 (triazole-C1), 150.21 (thiazole- C2, pyrimidine-C2), 153.64 (triazole-C2), 158.55 (thiazole-C1, pyrimidine-C6), 160.0 (pyrimidine-C4); LC-MS (m/z, %): 690 (M + 2, 99.1).
4.2.9 General procedure for the synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-(substituted phenyl) ethanone (10a-f)
The reaction mixture of 5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazole-3-thiol 8 (0.10 mol), ethanol (5 V) and potassium hydroxide (0.25 mol) was stirred at 25–30 °C for 30 min. Further, substituted phenacyl bromide (0.11 mol) was added and stirred at 25–30 °C for 5 h. Upon completion of reaction, ethanol was evaporated and quenched into the water (5 V). Product was filtered and recrystallized using suitable solvents to obtain target compounds (10a-f).
4.2.9.1 Synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-(4-bromophenyl)ethanone (10a)
Recrystallized using ethanol, off-white to pale brown solid (yield 79%); mp 202–206 °C; IR (KBr) νmax/cm−1 3216 (N—H), 1694 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 3.79 (m, 4H, 2CH2, morpholine), 3.86 (m, 4H, 2CH2, morpholine), 4.05 (s, 2H, CH2, thiazole-CH2-triazole), 4.88 (s, 2H, CH2, S-CH2-CO-phenyl), 6.50 (s, 1H, CH, thiazole), 7.22 (s, 2H, 2CH, phenyl triazole),7.28 (s, 2H, 2CH, phenyl triazole), 7.50 (s, 1H, CH, phenyl triazole), 7.64 (s, 2H, 2CH, 4-bromophenyl), 7.90 (m, 2H, 2CH, 4-bromophenyl), 8.16 (s, 1H, CH, Pyrimidine), 8.56 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (75 MHz, CDCl3): δ 27.8 (thiazole-CH2-triazole), 40.67 (-S-CH2-CO phenyl), 44.87, 66.60 (morpholine), 91.32 (pyrimidine-C5), 110.04 (thiazole-C3), 127.05 (4-bromophenyl-C4), 129.19, 129.87 (phenyl triazole- C3, C5, C4), 129.98 (phenyl triazole-C2, C6), 130.15 (4-bromophenyl-C2, C6), 132.09, 132.74 (4-bromophenyl-C3, C5), 133.91 (4-bromophenyl-C1), 145.11 (phenyl triazole-C1), 150.98 (triazole-C1), 152.85 (thiazole-C2, pyrimidine-C2), 153.49 (triazole-C2), 157.15, 157.48 (thiazole-C1, pyrimidine-C6), 159.75 (pyrimidine-C4), 192.27 (CO); LC-MS (m/z, %): 728.48 (M + 2, 98.44).
4.2.9.2 Synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-(pyridin-3-yl)ethanone (10b)
Recrystallized using ethanol, off-white to pale brown solid (yield 81%); mp 195–198 °C; IR (KBr) νmax/cm−1 3218 (N—H), 1693 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 3.78 (m, 4H, 2CH2, morpholine), 3.85 (m, 4H, 2CH2, morpholine), 4.05 (s, 2H, CH2, thiazole-CH2-triazole), 4.77 (s, 2H, CH2, S-CH2-CO-phenyl), 6.52 (s, 1H, CH, thiazole), 7.24 (s, 2H, 2CH, phenyl triazole), 7.29 (s, 2H, 2CH, phenyl triazole), 7.52 (s, 1H, CH, phenyl triazole), 7.64 (s, 1H, CH, pyridine), 8.17 (s, 1H, CH, Pyrimidine), 8.34 (s, 1H, CH, pyridine), 8.56 (s, 1H, NH, sec amine, D2O exchangeable), 8.88 (s, 1H, CH, pyridine), 9.18 (s, 1H, CH, pyridine); 13C NMR (75 MHz, CDCl3): δ 27.6 (thiazole-CH2-triazole), 40.65 (—S—CH2—CO phenyl), 44.89, 66.60 (morpholine), 91.32 (pyrimidine-C5), 110.10 (thiazole-C3), 123.60 (pyridine-C2), 129.18, 129.88 (phenyl triazole-C3, C5, C4), 129.97 (phenyl triazole-C2, C6), 132.30 (pyridine-C6), 136.20 (pyridine-C1), 145.14 (phenyl triazole-C1), 150.99 (triazole-C1), 151.86 (thiazole-C2, pyrimidine-C2), 152.01, 152.99 (pyridine-C3, C5), 153.48 (triazole-C2), 157.16, 157.45 (thiazole- C1, pyrimidine-C6), 159.48 (pyrimidine-C4), 192.32 (CO); LC-MS (m/z, %): 650 (M + 1, 98.80).
4.2.9.3 Synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-phenylethanone (10c)
Recrystallized using ethanol, off-white to pale brown solid (yield 80%); mp 204–208 °C; IR (KBr) νmax/cm−1 3215 (N—H), 1695 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 3.78 (m, 4H, 2CH2, morpholine), 3.87 (m, 4H, 2CH2, morpholine), 4.03 (s, 2H, CH2, thiazole-CH2-triazole), 4.86 (s, 2H, CH2, S-CH2-CO-phenyl), 6.51 (s, 1H, CH, thiazole), 7.23 (s, 2H, 2CH, phenyl triazole), 7.29 (s, 2H, 2CH, phenyl triazole), 7.51 (s, 1H, CH, phenyl triazole), 7.59 (s, 2H, 2CH, phenyl), 7.62 (s, 1H, CH, phenyl), 7.92 (m, 2H, 2CH, phenyl), 8.14 (s, 1H, CH, Pyrimidine), 8.58 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (75 MHz, CDCl3): δ 27.6 (thiazole-CH2-triazole), 40.65 (—S—CH2—CO phenyl), 44.89, 66.60 (morpholine), 91.35 (pyrimidine- C5), 110.08 (thiazole- C3), 128.6 (phenyl- C3, C5), 128.8 (phenyl- C2, C6), 129.20, 129.85 (phenyl triazole- C3, C5, C4), 129.96 (phenyl triazole- C2, C6), 133.10 (phenyl- C4), 135.40 (phenyl- C1), 145.10 (phenyl triazole- C1), 150.95 (triazole- C1), 152.86 (thiazole- C2, pyrimidine- C2), 153.44 (triazole- C2), 157.20, 157.47 (thiazole- C1, pyrimidine- C6), 159.79 (pyrimidine- C4), 192.26 (CO); LC-MS (m/z, %): 649 (M + 1, 98.45).
4.2.9.4 Synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-(4-chlorophenyl)ethanone (10d)
Recrystallized using ethanol, off-white to pale brown solid (yield 82%); mp 208–211 °C; IR (KBr) νmax/cm−1 3218 (N—H), 1697 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 3.78 (m, 4H, 2CH2, morpholine), 3.86 (m, 4H, 2CH2, morpholine), 4.03 (s, 2H, CH2, thiazole-CH2-triazole), 4.86 (s, 2H, CH2, S-CH2-CO-phenyl), 6.50 (s, 1H, CH, thiazole), 7.21 (s, 2H, 2CH, phenyl triazole), 7.29 (s, 2H, 2CH, phenyl triazole), 7.50 (s, 1H, CH, phenyl triazole), 7.65 (s, 2H, 2CH, 4-chlorophenyl), 7.91 (m, 2H, 2CH, 4-chlorophenyl), 8.18 (s, 1H, CH, Pyrimidine), 8.57 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (75 MHz, CDCl3): δ 27.6 (thiazole-CH2-triazole), 40.66 (—S—CH2—COphenyl), 44.89, 66.61 (morpholine), 91.33 (pyrimidine-C5), 110.04 (thiazole-C3), 127.01 (4-chlorophenyl-C4), 129.18, 129.86 (phenyl triazole-C3, C5, C4), 129.97 (phenyl triazole-C2, C6), 130.14 (4-chlorophenyl-C2, C6), 132.08, 132.73 (4-chlorophenyl-C3, C5), 133.90 (4-chlorophenyl-C1), 145.11 (phenyl triazole-C1), 150.97 (triazole-C1), 152.86 (thiazole-C2, pyrimidine-C2), 153.50 (triazole-C2), 157.16, 157.48 (thiazole-C1, pyrimidine-C6), 159.79 (pyrimidine-C4), 192.26 (CO); LC-MS (m/z, %): 684.0 (M + 2, 99.24).
4.2.9.5 Synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-(4-(trifluoromethyl)phenyl)ethanone (10e)
Recrystallized using ethanol, off-white to pale brown solid (yield 83%); mp 212–214 °C; IR (KBr) νmax/cm−1 3216 (N—H), 1694 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 3.80 (m, 4H, 2CH2, morpholine), 3.87 (m, 4H, 2CH2, morpholine), 4.08 (s, 2H, CH2, thiazole-CH2-triazole), 4.89 (s, 2H, CH2, S-CH2-CO-phenyl), 6.50 (s, 1H, CH, thiazole), 7.22 (s, 2H, 2CH, phenyl triazole), 7.28 (s, 2H, 2CH, phenyl triazole), 7.52 (s, 1H, CH, phenyl triazole), 7.66 (m, J = 6.80 Hz, 2H, 2CH, 4-(trifluoromethyl)phenyl), 7.93 (m, J 6.77, 2H, 2CH, 4-(trifluoromethyl)phenyl), 8.16 (s, 1H, CH, Pyrimidine), 8.56 (s, 1H, NH, sec amine, D2O exchangeable); 13C NMR (75 MHz, CDCl3): δ 27.80 (thiazole-CH2-triazole), 40.68 (—S—CH2-CO-phenyl), 44.87, 66.60 (morpholine), 91.32 (pyrimidine-C5), 110.05 (thiazole-C3), 124.11 (trifluoro carbon), 127.05, 129.19 (4-(trifluoromethyl) phenyl-C3, C5, C2, C6), 129.87, 129.98 (phenyl triazole- C3, C5, C4), 130.15 (phenyl triazole-C2, C6), 133.91 (4-(trifluoromethyl) phenyl- C4), 145.11 (phenyl triazole-C1), 150.50 (4-(trifluoromethyl) phenyl-C1), 150.98 (triazole-C1), 152.85 (thiazole-C2, pyrimidine-C2), 153.49 (triazole-C2), 157.15, 157.48 (thiazole-C1, pyrimidine-C6), 159.75 (pyrimidine-C4), 192.27 (CO); LC-MS (m/z, %): 717.2 (M + 1, 98.41).
4.2.9.6 Synthesis of 2-(5-((2-(5-bromo-2-morpholinopyrimidin-4-ylamino)thiazol-4-yl)methyl)-4-phenyl-4H-1,2,4-triazol-3-ylthio)-1-(4-fluorophenyl)ethanone (10f)
Recrystallized using ethanol, off-white to pale brown solid (yield 79%); mp 204–208 °C; IR (KBr) νmax/cm−1 3216 (N—H), 1692 (C⚌O); 1H NMR (400 MHz, CDCl3): δ 3.76 (m, 4H, 2CH2, morpholine), 3.84 (m, 4H, 2CH2, morpholine), 4.02 (s, 2H, CH2, thiazole-CH2-triazole), 4.88 (s, 2H, CH2, S-CH2-CO-phenyl), 6.50 (s, 1H, CH, thiazole), 7.21 (s, 2H, 2CH, phenyl triazole), 7.28 (s, 2H, 2CH, phenyl triazole), 7.51 (s, 1H, CH, phenyl triazole), 7.37 (m, 2H, 2CH, 4-fluorophenyl), 8.43 (s, 1H, CH, Pyrimidine), 8.55 (s, 1H, NH, sec amine, D2O exchangeable); 8.67 (m, 2H, 2CH, 4-fluorophenyl); 13C NMR (75 MHz, CDCl3): δ 27.8 (thiazole-CH2-triazole), 40.66 (—S—CH2-COphenyl), 44.85, 66.60 (morpholine), 91.31 (pyrimidine-C5), 110.04 (thiazole-C3), 115.30, 128.83 (4-fluorophenyl-C3, C5, C2), 129.19, 129.87 (phenyl triazole- C3, C5, C4), 129.98 (phenyl triazole-C2, C6), 130.32, 131.28 (4-fluorophenyl-C6, C1), 145.11 (phenyl triazole-C1), 150.98 (triazole-C1), 152.85 (thiazole-C2, pyrimidine-C2), 153.49 (triazole-C2), 157.15, 157.47 (thiazole-C1, pyrimidine-C6), 159.76 (pyrimidine-C4), 163.31 (4-fluorophenyl-C4), 192.28 (CO); LC-MS (m/z, %): 667 (M + 1, 98.24).
4.3 Biological protocols
4.3.1 MTT assay
The synthesized bromo-pyrimidine derivatives (6a-j, 7a-e, 9a-f, and 10a-f) were tested in vitro for their cytotoxic properties against tumor cell lines panel consisting of HCT116 (human colon cancer cell line), A549 (human lung cancer cell line), K562 (human chronic myeloid leukemia cell line), U937 (human acute monocytic myeloid leukemia cell line), and L02 (human normal liver cell line) by using MTT assay Mosmann’s method. The MTT assay is based on the reduction of the soluble MTT
(0.5 mg mL−1, 100 µL), into a blue-purple formazan product, mainly by mitochondrial reductase activity inside living cells (Mosmann, 1983).
The cells used in cytotoxicity assay were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, penicillin, and streptomycin at 37 °C and humidified at 5% CO2. Briefly, cells were placed on 96-well plates at 100 µL total volume with density of 1–2.5 × 104 cells per mL and were allowed to adhere for 24 h before treatment with tested drugs in DMSO solution (10-5, 10-6, 10-7 mol L-1 final concentration). Triplicate wells were treated with media and agents. Cell viability was assayed after 96 h of continuous drug exposure with a tetrazolium compound. The supernatant medium was removed, and 150 µL of DMSO solution was added to each well. The plates were gently agitated using mechanical plate mixer until the color reaction was uniform and the OD570 was determined using microplate reader. The 50% inhibitory concentration (IC50) was defined as the concentration that reduced the absorbance of the untreated wells by 50% of vehicle in the MTT assay. Assays were performed in triplicate on three independent experiments. The results had good reproducibility between replicate wells with standard errors below 10%.
4.3.2 Bcr/Abl inhibitory activity assay
The Bcr/Abl inhibitory activity assay was performed using ADP-GloTM Kinase assay kit (Promega, Catalog: V9101) according to the manufacturer’s instructions. The Abl1 reaction utilizes ATP and generates ADP. Then the ADP-GloTM reagent is added to simultaneously terminate the kinase reaction and deplete the remaining ATP. Finally, the Kinase detection reagent is added to convert ADP to ATP and the newly synthesized ATP is converted to light using the luciferase reaction (Wang, 2014; Zhang, 2011).
Abl was incubated with substrates, inhibitors, and ATP in a final buffer of 25 mM HEPES (pH 7.4), 10 mM MgCl2, 0.01% Triton X-100, 100 µ mL−1 BSA, 2.5 mM DTT in 384-well plate with the total volume of 10 µL. Then the ADP-GloTM kinase assay was performed in two steps once the kinase reaction is complete. Subsequently, 5 µL ADP-Glo Reagent was added to stop the kinase reaction and deplete the unconsumed ATP. Only ADP and very low background of ATP were left. Then the mixture was incubated at room temperature for 40 min and added 10 µL of Kinase detection reagent to convert ADP to ATP and introduced luciferase and luciferin to detect ATP. At last, the mixture was incubated at room temperature for 30–60 min and the luminescence was measured with a plate-reading luminometer. The signal was correlated with the amount of ATP present in the reaction and was inversely correlated with the kinase activity.
Author contributions
All the authors (CSM, GVSK, RRB and ABS) contributed equally for preparing the manuscript and all the authors reviewed the manuscript.
Acknowledgement
CSM and SKGV Figure 1 would like to thank management of Rallis India Ltd and East West College of Pharmacy, Bangalore, for providing necessary facilities. We sincerely thank Dr. Senthil Duraisamy, Director, and E.G. Rama Kishore, Manager of G7 Synergon Private Limited, Bangalore for the anticancer activity. We also wish to thank IISC, Bangalore, India for providing IR, NMR, mass spectra, and elemental analysis data. R.R.B and A.B.S would like to thank the Dean’s office of College of Pharmacy and Health Sciences, Ajman University, UAE & Vignan Pharmacy College, Vadlamudi, Andhra Pradesh, India for their support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Aquila, B., Ioannidis, S., Lyne, P., Pontz, T., 2005. Azine-carboxamides as anti-cancer agents. WO Patent 2006-003378, filled Jun 29, 2005, issued Jan 12, 2006.
- Synthesis of some new 1,2,4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur. J. Med. Chem.. 2009;44:1057-1066.
- [CrossRef] [Google Scholar]
- Reactions of 4-chloroacetoacetic esters with thioureas. J. Het. Chem.. 1980;17:1255-1257.
- [CrossRef] [Google Scholar]
- FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist.. 2003;8:303-306.
- [CrossRef] [Google Scholar]
- FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist.. 2005;10:461-466.
- [CrossRef] [Google Scholar]
- Connors, R.V., et al., 2008. Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors. WO Patent 2009-085185, filled December 18, 2008, issued Jul 9, 2009.
- 2-Aminothiazole as a Novel Kinase Inhibitor Template. Structure-Activity Relationship Studies toward the Discovery of N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a Potent pan-Src Kinase Inhibitor. J. Med. Chem.. 2006;49:6819-6832.
- [CrossRef] [Google Scholar]
- Synthesis and antimicrobial studies of novel dichlorofluorophenyl containing aminotriazolothiadiazines. Eur. J. Med. Chem.. 2008;43:309-3014.
- [CrossRef] [Google Scholar]
- Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA.. 2003;290:2149-2158.
- [CrossRef] [Google Scholar]
- Overcoming kinase resistance in chronic myeloid leukemia. Int. J. Biochem. Cell. Biol.. 2008;40:334-343.
- [CrossRef] [Google Scholar]
- Synthesis of new 4-isopropylthiazole hydrazide analogs and some derived clubbed triazole, oxadiazole ring systems – A novel class of potential antibacterial, antifungal and antitubercular agents. Eur. J. Med. Chem.. 2009;44:4739-4746.
- [CrossRef] [Google Scholar]
- Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods.. 1983;65:55-63.
- [CrossRef] [Google Scholar]
- Part-1: Design, synthesis and biological evaluation of novel bromopyrimidine analogs as tyrosine kinase inhibitors. Eur. J. Med. Chem.. 2016;119:70-82.
- [CrossRef] [Google Scholar]
- Păunescu, E., et al., 2015. Improved angiostatic activity of dasatinib by modulation with hydrophobic chains. ACS Med. Chem. Lett. 6, 313−317. doi: 10.1021/ml500496u.
- Synthesis and biological evaluation of novel 4,5-dihydropyrazole derivatives as potent anticancer and antimicrobial agents. Med. Chem. Res.. 2013;22:2061-2078.
- [Google Scholar]
- Therapeutic options against BCR-ABL1 T315I-positive chronic Myelogenous Leukemia. Clin. Cancer. Res.. 2008;14:4392-4399.
- [CrossRef] [Google Scholar]
- Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood.. 2009;113:1619-1630.
- [CrossRef] [Google Scholar]
- Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J. Hematol. Oncol.. 2018;11:1-14.
- [CrossRef] [Google Scholar]
- Overriding imatinib resistance with a novel ABL kinase inhibitor. Science.. 2004;305:399-401.
- [CrossRef] [Google Scholar]
- The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer. Res.. 2006;66:5790-5797.
- [CrossRef] [Google Scholar]
- Biphenyl derivatives incorporating urea unit as novel VEGFR-2 inhibitors: Design, synthesis and biological evaluation. Bioorg. Med. Chem.. 2014;22:277-284.
- [CrossRef] [Google Scholar]
- Discovery and Initial SAR of 2-Amino-5-carboxamidothiazoles as Inhibitors of the Src-family Kinase p56Lck. Bioorg. Med. Chem. Lett.. 2003;13:4007-4010.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel Gefitinib analogues with improved anti-tumor activity. Bioorg. Med. Chem.. 2010;18:3812-3822.
- [CrossRef] [Google Scholar]
- Exploration of (S)-3-amino pyrrolidine as a potentially interesting scaffold for discovery of novel Abl and PI3K dual inhibitors. Eur. J. Med. Chem.. 2011;46:1404-1414.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103054.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




