

Translate this page into:
Novel uracil derivatives depicted potential anticancer agents: In Vitro, molecular docking, and ADME study
⁎Corresponding authors at: Hormones Department, Medical Research and Clinical Studies Institute, National Research Centre, Dokki, Giza, Egypt (M.A. Tantawy). s.elkalyoubi@hotmail.com (Samar El-Kalyoubi), mohamed_tantawy@daad-alumni.de (Mohamed A. Tantawy)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Using a simple technique to prepare many uracil derivatives as Schiff base of uracil 4, bis 6-amino[5-(1-(4-aryl)ethylidene)amino)]pyrimidine-2,4-dione 5–12 by heating of 5,6-diamino-1-substituteduracils 1a-e in few drops of DMF with acetophenones by light torch flame and imidazolopyrimidine-2,4-dione 13 which prepared from 5,6-diaminouracil hydrochloride 3a by heating under reflux with 4-bromoacetophenone. Moreover, 6-cyano-7-ethoxypyridopyrimidines 14–16 were prepared by refluxing of 6-amino-1-(2-chlorobenzyl)uracil 1d with ethyl benzylidene cyanoacetate in TEA through conjugate addition followed by intramolecular heterocyclization. All novel synthesized compounds were evaluated for their anticancer activity against three kinds of cancer including liver cancer (HepG2, and Huh7) cell lines, breast cancer (MCF7) cell line, and lung cancer (A549) cell line. Moreover, toxicity of the most cytotoxic compound was evaluated against normal cells (MDCK). Compounds 6, 7, 15, and 16 were the best cytotoxic compounds against most of treated cancer cell lines, and most interestingly they had no toxicity against normal cells. Furthermore, Compound 7 had high binding affinity with cyclin dependent kinase 2 (CDK2), with ability to induce apoptosis in lung cancer cells. Further investigations are needed to elaborate with these novel synthesized compounds with potential cytotoxic activity to develop new effective chemotherapy.
Keywords
N-Alkyluracils
Schiff base of 6-aminouracil
Bis(6-aminouracils)
Xanthine
Anticancer
Molecular docking
Cyclin dependent kinase 2

1 Introduction
Synthesis of fused heterocycles especially pyrimidine derivatives has remained promising area in organic synthesis due to their abundance in natural as well as synthetic molecules with diverse applications in pharmaceuticals chemistry for their therapeutic applications (Segal et al., 1962; Cumming et al., 2004), such as, analgesic, antihypertensive (Cannito et al., 1990), antipyretic (Smith and Kan, 1964), and anti-inflammatory (Vega et al., 1990) drugs. Also applications of pyrimidine compounds are extended to agriculture as a pesticides (Vega et al., 1990), and plant growth regulators (Shishoo and Jain, 1992). The functionality of the 6-position of pyrimidines showed important anti-HIV-1 activity (Botta et al., 2001; Garg et al., 1999) and antirubella virus activity (Botta et al., 1999; Zanatta et al., 2006). A variety of anticancer drugs made from pyrimidine derivatives are currently in clinical use, as for example some of these compounds applied successfully for the treatment of several neoplastic diseases such as leukemia and testicular cancer.
Chemotherapy is the mainstay of cancer treatment approaches, and it continues to be one of the most worrisome issues, affecting millions of people globally (Kartal-Yandim et al., 2016). Despite the fact that a huge number of anticancer medications have been used in clinical practice, their use is severely limited due to adverse effects (Wilson et al., 2019). As a result, research has been conducted out in order to discover novel analogues (Fan et al., 2021). On the basis of the histological type of cancer, cancer stage, and patient state, treatment and prognosis strategies are developed. Surgery, chemotherapy, and radiotherapy are among choices for treatment (Oser et al., 2015; Goldstraw et al., 2011). Chemotherapy is when chemicals or medications are used to kill cancer cells. Alkylating agents (e.g., cisplatin), mitotic inhibitors (e.g., paclitaxel), and epidermal growth factor receptor (EGFR) inhibitors are all commonly used anticancer medications (e.g., gefitinib), VEGF (vascular endothelial growth factor) or VEGF receptor inhibitors (bevacizumab, for example) (Huang et al., 2017; Wu et al., 2021).
In 1957, 5-Fluro uracil has been synthesized (Heidelberger et al., 1957), and since that time a lot of fluropyrimidine derivatives have been developed and investigated as potent anti-cancer agents. The short serum half life time for 5-fluro uracil (8–12 min) (Diasio and Harris, 1989; Baker et al., 1996) stimulated many researchers to develop different derivatives of uracil to sustain high serum concentrations, improve selectivity and reduce toxicity. As one attempt to achieve this goal, Tegafur, which is a precursor of 5-fluoro uracil, has been developed, to be activated to 5-fluoro uracil by cytochrome P450-2A6.
Several uracil derivatives have been assessed with potential activity towards breast and (MCF7) and liver cell lines by inhibiting the cycline dependent kinase, with up-regulating the expression of P21 and P27 proteins (Marchal et al., 2007; Nassar et al., 2020; Sanduja et al., 2020; El-Naggar et al., 2017). In addition, Peng et al showed that uracil derivatives have a potential cytotoxic effect against panel of cancer cell lines including Lung cancer (A549) in a dose and time dependent manner (Peng et al., 2014). For the above reasons, there is a rationality of using different uracil derivatives for treatment of different solid tumors, so, the objective of this study was explore to the potency of novel synthetic uracil derivatives against different types of tumor cell lines including lung, liver, and breast.
Based on the aforementioned literature, there is solid rational of using different uracil derivatives to treat different solid cancer, therefore, the novel synthetic uracil derivatives have been tested against three types of solid cancer including lung, liver, and breast cancer. Cell viability assay was done, followed by detection of apoptosis using flow cytometry. Drug development is a tedious, and very expensive process (Kohli, 2018). Owing to the significant improvement in the structural molecular biology, molecular docking as virtual screening procedure was emerged in the medicinal chemistry, and drug design for efficient drug design procedure or even to predict the way of interaction of small molecules (ligands) to macromolecules (proteins) (Morris and Lim-Wilby, 2008). In addition, good drug should fulfil different criteria such as good distribution, metabolism, and efficient action. Another detrimental in drug design is the toxicity, and other side effects, which are associated mainly with Absorption, Distribution, Metabolism, and Excretion behavior of the tested drug, therefore, molecular docking, and in silico ADMET prediction save a lot of time, money and effort in order to discover new drugs with potential anticipated pharmacological activity (Srivastava et al., 2020). In this aspect, in silico study has been imitated to speculate the mode of action, pharmacokinetics and dynamics for the most promising cytotoxic compounds.
2 Results and discussion
Various substituted pyridopyrimidine derivatives (Farokhian et al., 2019) have been synthesised using various methods and catalysts, including the reaction of arylaldehyde, 6-aminouracil, and malononitrile in a catalyst-free condition using glycerol (Singh et al., 2014), electrocatalytic synthesis in ethanol (Kazemi-Rad et al., 2016), and the reaction of arylaldehyde, 6-aminouracil, and malononitrile in using catalyst-free condition DAHP (diammonium hydrogen phosphate) (Shahrzad and Saeed, 2012), Pyrido[2,3-d]pyrimidines have been studied extensively as quinazoline analogues. This scaffold is associated with a wide range of biological activities including dihydrofolate reductase (DHFR) inhibition, antitumor activity (Kovacs et al., 1988; Gangjee et al., 1999; Gangjee et al., 2003), adenosine kinase inhibition (Lee et al., 2001) and tyrosine kinase inhibition (Trumpp-Kallmeyer et al., 1998; Smaill et al., 2000), among other properties (Gangjee et al., 1999; Pyrido(3,2-d)pyrimidines useful for treating viral infections, 2021).
In continuing of our research into the synthesis of medically significant compounds in ecologically friendly settings, we've expanded our research into uracil moieties (El-Kalyoubi and Agili, 2020; El-Kalyoubi and Agili, 2016; El-kalyoubi et al., 2015). Different types of pyrimidines or condensed pyrimidines can be generated depending on the reaction conditions and the nature of electronic and steric components of substituted alkyl. The diazolopyrimidine and pyrimidine derivatives were furnished from diaminouracil of different nucleophilicity while azinopyrimidine was furnished from conjugate addition of uracil of cyclic enaminone. We tried to use a new technique, to save time and solvents. Heating of different 5,6-diaminouracils 3a-e (El-Kalyoubi and Agili, 2020; El-Kalyoubi and Agili, 2016; El-kalyoubi et al., 2015) (Scheme 1) [which prepared via the nitrosation of 1a-e followed by reduction using ammonium sulfide] with appropriate acetophenones namely, acetophenone, 4-bromo-, 4-nitroacetophenone in presence of few drops of DMF using lighter torch flame till complete fusion afforded excellent yields of the desired products.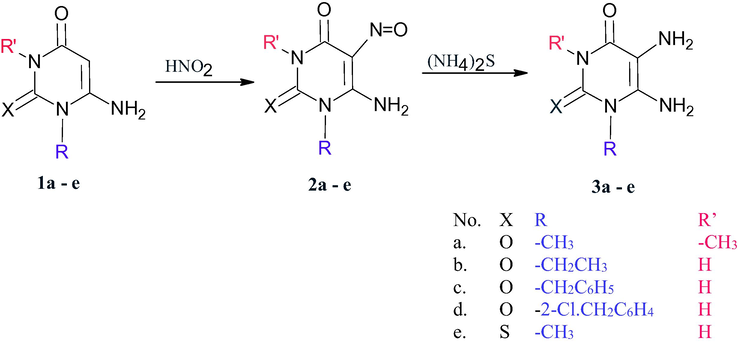
Synthesis of 5,6-diamino-1,3-disubstituted uracils.
On the other hand, the heating of 5,6-diamino-1-(2-chlorobenzyl)uracil (3d) with p-nitroacetophenone separate the intermediate Schiff base 4 which was created by the simple condensation of ketone with 5-amino- group of uracil 3d due to the involvement of exocyclic amino in hydrogen bond with acidic benzyl proton (Scheme 2). Compound 4 was confirmed by 1H NMR which exhibits a singlet signal for methyl group at δ 2.25 ppm and a singlet for 6-NH2 group at δ 6.85 ppm which exchangeable by D2O and showed a peak at υ 1575 cm−1 characterized for C⚌N group of Schiff.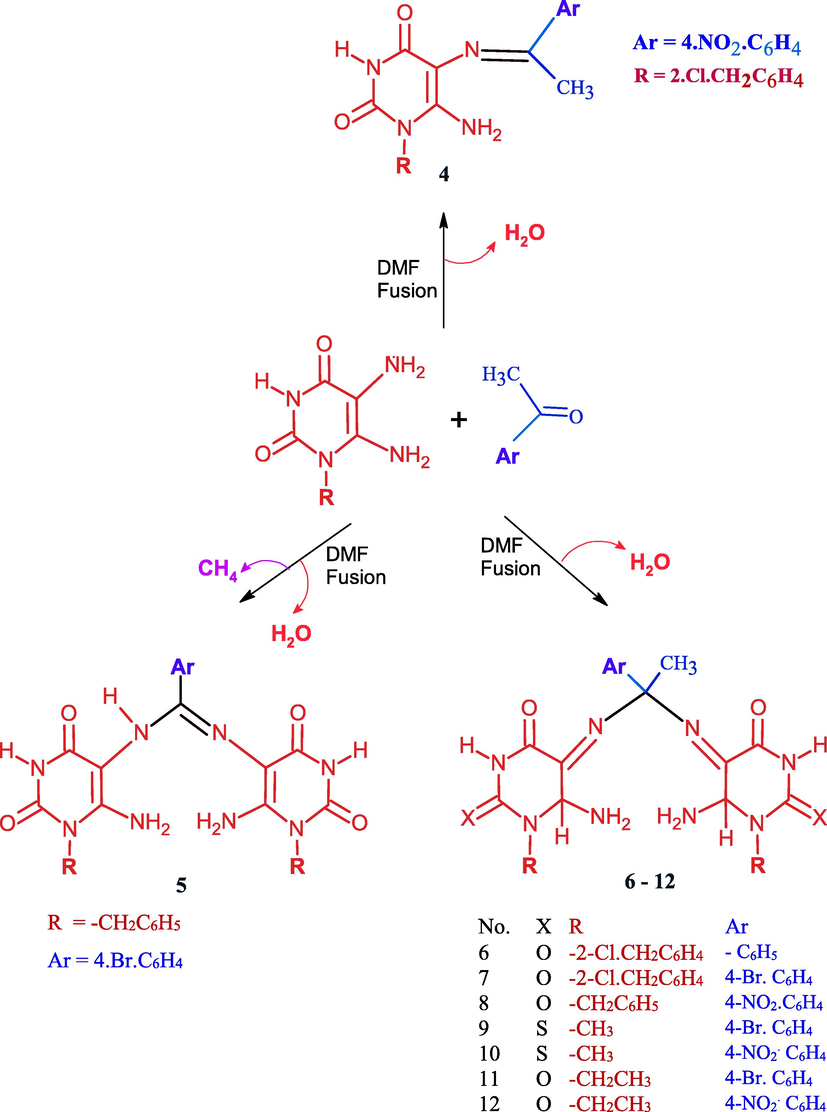
Reaction of uracils with different acetophenones.
The fusion of 3c and 4-bromoacetophenone in few drops of DMF leads to Schiff’s product through intermolecular aza Michael followed by loss of methane molecule producing N,N-Bis(6-amino-1-benzyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidn-5-yl)-4-bromobenzimidimide 5 (Scheme 2) which confirmed by proton NMR indicated the disappearance of methyl group of acetophenone and appear a signal at δ 2.9 ppm for NH(5), a singlet signal at δ 6.09 ppm for NH2 (6). The mechanistic pathway was shown in (Scheme 3).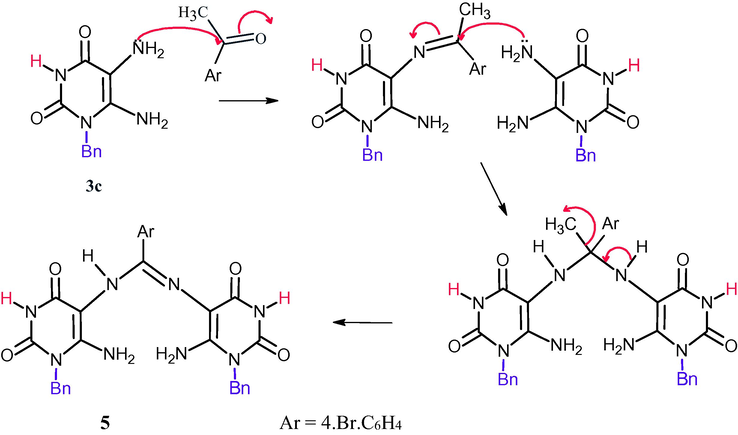
Mechanistic pathway for the formation of compound 5.
It was observed that acetophenone with 4-bromoatom undergo Schiff base formation, Michael addition of another pyrimidine followed by double 1,3-sigma tropic shift. So, The fusion of 3b-e with appropriately substituted acetophenones in few drops of DMF for 6–8 min afforded excellent yields of 5,5′-((1-(4-substituted phenyl)ethane-1,1-diyl)bis (azanediyl)bis(6-amino-1-substituted pyrimidine-2,4(1H,3H)-dione) 6–12 (Scheme 4). The confirmation of these compounds depends on the interpretation of 1H NMR, 13C NMR, and IR which exhibit signals between δ 6.09–6-19 ppm characterized for NH2 (6), signals at δ 2.8–3.4 ppm characterized for C—H at position 6, The mechanistic pathway for compounds 6–12 was suggested as shown in (Scheme 4).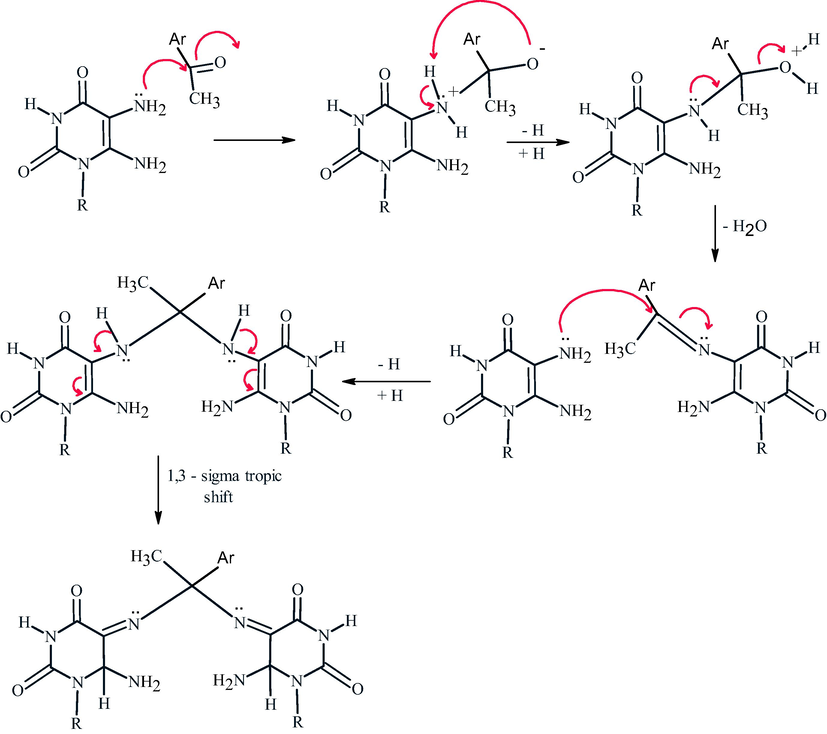
Mechanistic pathway for the formation of compounds 6–12.
The synthetic strategy for the synthesis of imidazolopyrimidine depends on the formation of Schiff bases followed by aza Michael addition, thus, the heating of 1,3-dimethyl-5,6-diaminouracil 3a with p-bromoacetophenone in DMF (1 ml) afforded 8-(4-bromophenyl)-1,3,8-trimethylxanthine (13) in high yield (Scheme 5) which confirmed by 1H NMR showed a singlet signal at δ 2.84 ppm and 6 protons for the two N-methyl groups of uracil .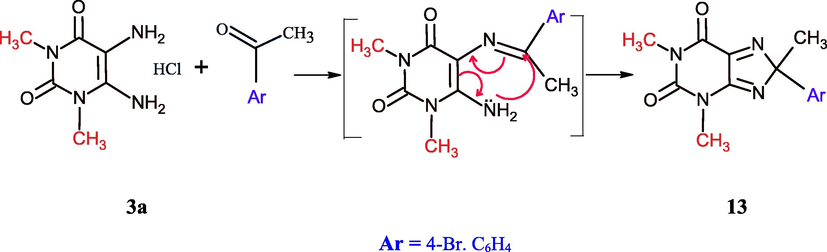
Synthesis of 8-aryl-8-methyl-1,3-dimethylxanthine.
On the other hand, the heterocyclization of 1d using α,β-unsaturated system was investigated, thus the refluxing of 6-amino-1-(2-chlorobenzyl)uracil (1d) with different arylidene ethyl cyanoacetate in DMF in the presence of triethylamine for 10 h afforded 1-(2-chlorobenzyl)-5-(4-substitutedphenyl)-7-ethoxy-2,4-dioxo-1,2,3,4,5,8-hexahydropyrido[2,3-d]pyrimidine-6-carbonitrile 14–16 and none of pyridopyrimidine I was obtained (Scheme 6). This series was proved by the appearance of CN group in IR spectra at υ 2222 cm−1, and the absence of NH2 group between υ 3442–3380 cm−1 and the significant appearance of the ethyl group of ether in 1H NMR at δ 3.03–3.29 ppm as a quartet protons for CH2 and a triplet protons for CH3 group at δ 1.03–1.20 ppm.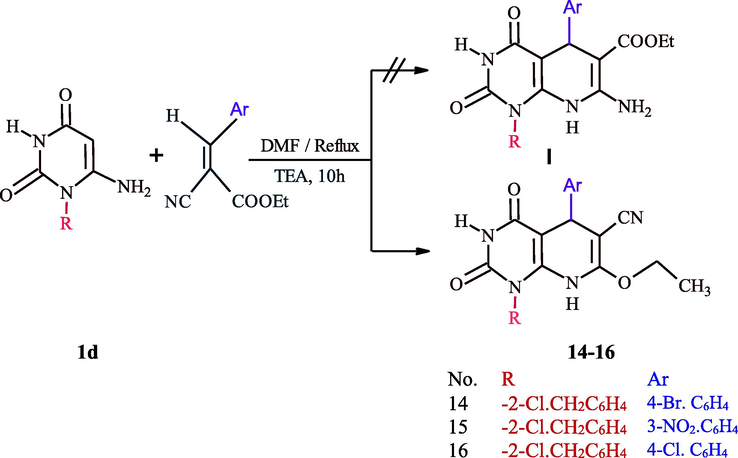
Synthesis of pyridopyrimidines 14–16.
The reaction takes place presumably via The nucleophillic attack of C-5 of uracil to β- position of benzyledine cyanoacetae leads to the formation of non-isolable acyclic Michael type adduct that undergoes cyclocondensation affording the final product 14–16. A plausible mechanism for this reaction may be as follows (see Scheme 7):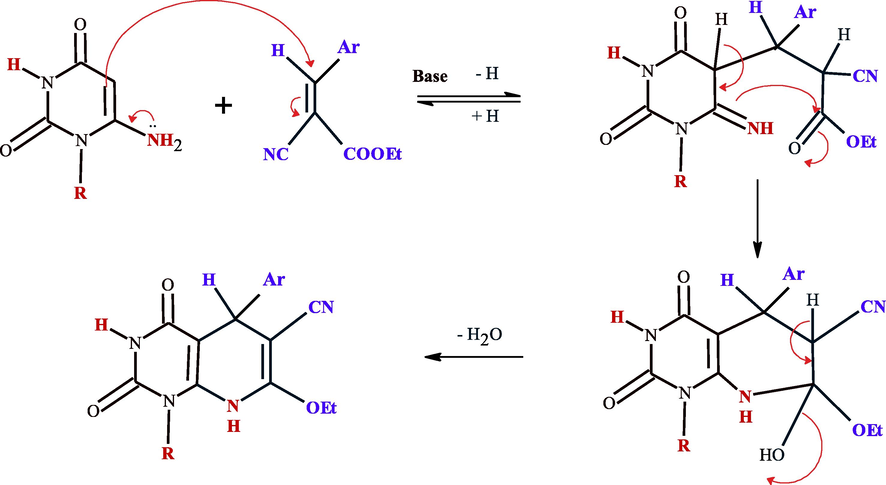
Mechanistic pathway for the synthesis of compounds 14–16.
3 Biological activity
3.1 In vitro anti-liver cancer activity
Hepatocellular carcinoma is one of the most highly aggressive cancer, which threatens the life of millions worldwide. A lot of studies have been done to discover new entities that could control HCC, and overcome the chemotherapy resistance associated with the treatment. Different uracil derivatives have been investigated as promising anti-HCC agents (Ishikawa, 2008). In this context, the cytotoxic activity for the novel prepared uracil derivatives has been investigated against liver cancer on two different cell lines including HepG2, Huh7 for 48 h. On HepG2 cells, compounds 4, 5, 8, 9, 10, 11, 12, and 16 had weak cytotoxic activity, and we could not calculate the IC50 values for these compounds in the range of concentration from 0.1 to 100 µM. While compounds 7, and 15 had moderate cytotoxicity with IC50 values (38.35, and 32.42 µM, respectively), compared to 5-Fluro uracil as a reference drug (10.32 µM). Compounds 6, and 14 were the most cytotoxic compounds with IC50 values comparable to the reference drug (13.14, and 12.45 µM, respectively) (Fig. 1, and Table 1). With the same scenario, compounds 4, 5, 8, 9, 10, 11, 12, in addition to compounds 14, and 15 had week activity against Huh7 cell line, while compounds 6, and 7 had moderate cytotoxicity with IC50 values (37.51, and 98.27 µM), while compound 16 was the most effective compound with IC50 less than the 5-Fluro uracil (14.08, and 14.89 µM, respectively) (Fig. 2, and Table 1). The different of effect of the tested compounds on liver cancer cell lines is correlated mainly due to the structure activity relationship. In addition, there are some variation between HepG2, and Huh7 cells in different drug-metabolizing enzymes and transporters (DMETs), therefore, different activity for the same drug could be observed on the two cell lines (Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes, n.d.).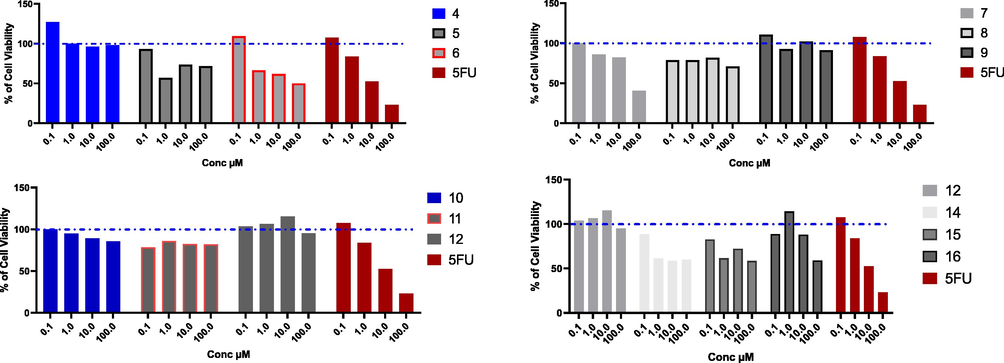
Cytotoxic effect of the tested compounds on HepG2 cell line. HepG2 % cell viability upon treatment with series of novel tested compounds using concentrations starting from 0.1 to 100 µM for 48 h, and cytotoxic effect was detected by MTT assay (n = 3). Blue dotted line indicated cell viability for control which is 100%.
Compds
IC50s µM ± SD
HepG2
Huh7
MCF7
A549
4
ND
ND
ND
ND
5
ND
ND
ND
32.63 ± 0.078
6
13.14 ± 0.062
37.51 ± 0.125
ND
13.28 ± 0.079
7
38.35 ± 0.049
98.278 ± 0.109
99.66 ± 0.089
5.46 ± 0.032
8
ND
ND
ND
43.88 ± 0.099
9
ND
ND
ND
16.27 ± 0.097
10
ND
ND
51.98 ± 0.069
9.54 ± 0.056
11
ND
ND
ND
8.51 ± 0.042
12
ND
ND
ND
ND
14
12.45 ± 0.048
ND
12.38 ± 0.063
ND
15
32.43 ± 0.036
ND
33.30 ± 0.074
94.21 ± 0.027
16
ND
14.08 ± 0.108
14.37 ± 0.075
96.31 ± 0.053
5FU
10.32 ± 0.055
14.89 ± 0.058
11.79 ± 0.062
19.66 ± 0.041
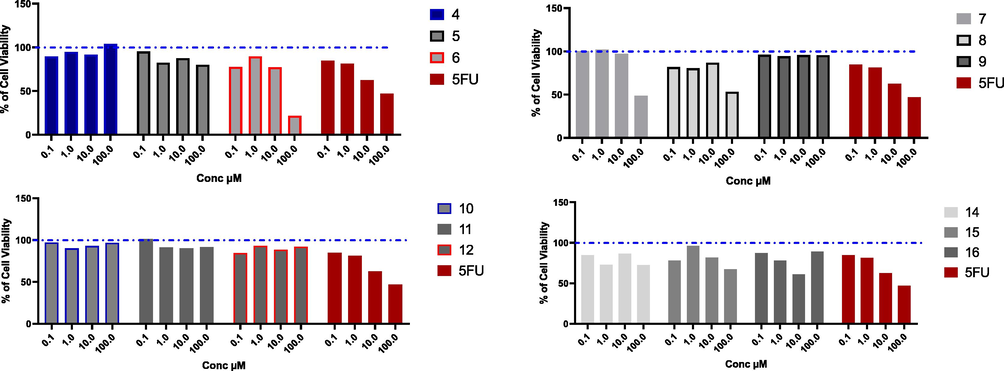
Cytotoxic effect of the tested compounds on Huh7 cell line. Huh7 % cell viability upon treatment with series of novel tested compounds using concentrations starting from 0.1 to 100 µM for 48 h, and cytotoxic effect was detected by MTT assay (n = 3). Blue dotted line indicated cell viability for control which is 100%.
3.2 In vitro anti-breast cancer activity
With the high incidence, and mortality rate, breast cancer affects and kills millions of patients annually especially women, therefore, there is a pressing need to discover new agents to eradicate this disease. Cell line provides a good tool to discover new anticancer agents, and MCF7 (Michigan Cancer Foundation-7) cell line, afford excellent platform to investigate the mechanism of action for new anti-breast cancer agents (Comşa et al., 2015; Hernández-Vargas et al., 2006). Different uracil derivatives showed a good inhibition activity on MCF7 breast cancer cell line (Marchal et al., 2007; Nassar et al., 2020). The novel uracil derivatives have been investigated using MTT assay on MCF7 cell line. The results presented in Fig. 3, and Table 1 indicate that compounds 4, 5, 6, 8, 9, 11, and 12 had weak cell inhibition activity, while compounds 7, and 10 had a moderate cytotoxic activity with IC50 values (99.66, 51.98 µM, respectively). In addition, compounds 14, 15, and 16 had potential cytotoxic activity with IC50 values (12.38, 33.30, and 14.37 µM, respectively in comparison to 5-Fluro uracil (11.79 µM).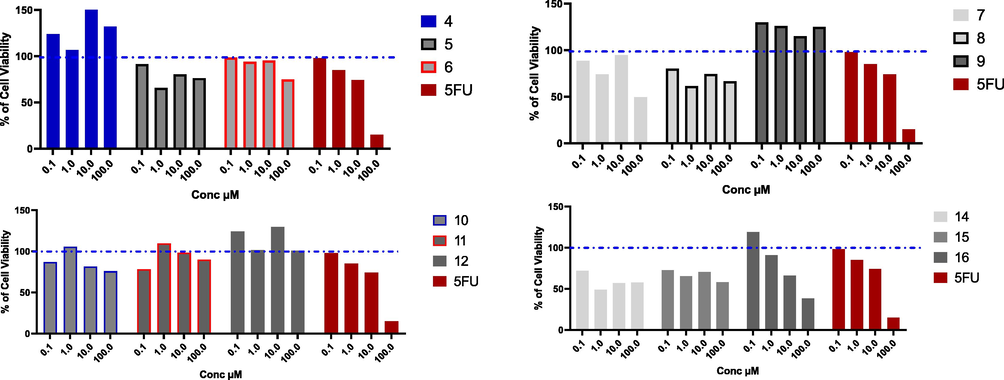
Cytotoxic effect of the tested compounds on MCF7 cell line. MCF7 % cell viability upon treatment with series of novel tested compounds using concentrations starting from 0.1 to 100 µM for 48 h, and cytotoxic effect was detected by MTT assay (n = 3). Blue dotted line indicated cell viability for control which is 100%.
3.3 In vitro anti-lung cancer activity
Novel synthesized compounds have been investigated against A549 cells, which represent a good model for non-small lung adenocarcinoma. Only compounds 4, 12, and 14 did not show good cytotoxic activity against A549 cells, while compounds 15, and 16 had moderate cytotoxicity with IC50 values (94.21, and 96.31 µM, respectively). Interestingly, compounds 6, 7, 9, 10, and 11 were very promising cytotoxic agents with IC50 values (13.28, 5.46, 16.27, 9.54, and 8.51 µM, respectively), which were even better than 5-fluro uracil with IC50 (19.66 µM) (see Fig. 4).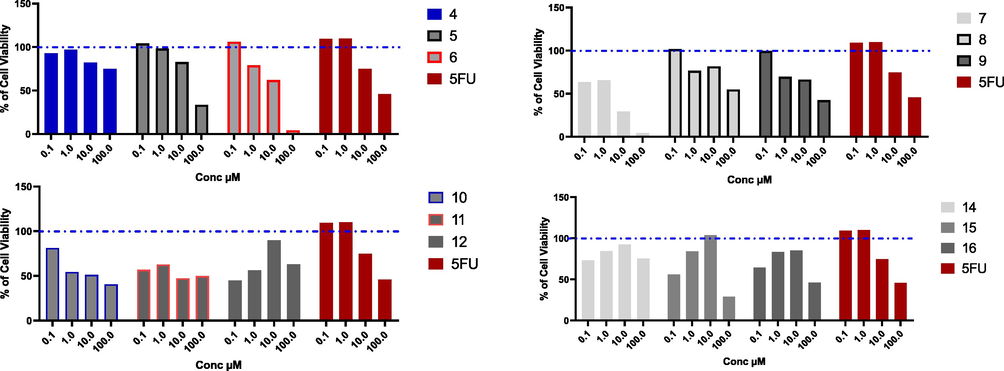
Cytotoxic effect of the tested compounds on A549 cell line. A549 % cell viability upon treatment with series of novel tested compounds, using concentrations starting from 0.1 to 100 µM for 48 h, and cytotoxic effect was detected by MTT assay (n = 3), Blue dotted line indicated cell viability for control which is 100%.
3.4 Toxicity against normal cells
Based on the initial screening for all tested compounds against three types of cancer cell lines, compounds 6, 7, 15, and 16 were the most effective cytotoxic compounds. As the main draw back for the most chemotherapeutic agents is the toxicity of these drugs on normal cells, therefore, we have chosen these most promising cytotoxic agents to elucidate their toxicity on normal Madin-Darby Canine Kidney cells (MDCK). Results showed that we could not observe any toxicity for the tested compounds comparing to 5-Fluro uracil, as IC50 could not be calculated for any of novel compounds, while the IC50 for 5-Fluro uracil was 9.36 µM (Fig. 5, and Table 2). Therefore, further assessment of the toxicity of these compounds on normal human cells should be investigated to further confirm the specificity of these compounds in targeting cancer cells while sparing normal cells.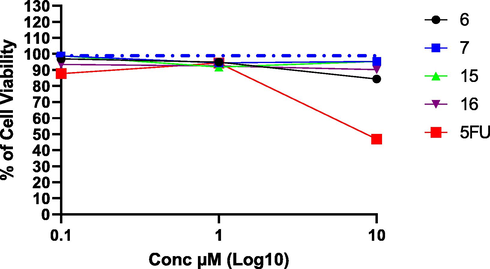
Cytotoxic effect for the most promising compounds on non-cancerous MDCK cell line. Blue dotted line indicated cell viability for control which is 100%. Concentrations used starting from 0.1 to 10 µM for 48 h, and cytotoxic effect was detected by MTT assay (n = 3).
Compounds
IC50s
6
ND
7
ND
15
ND
16
ND
5FU
9.36
3.5 Molecular docking
To take one step further to determine the mode of action of the tested compounds as potential anti-cancer agents, molecular-docking study was employed to determine the binding modes against two important proteins implicated in cancer progression including cyclin dependent kinase 2 (CDK2), and dihydrofolate reductase (DHFR). These targets were selected based on structure activity relationship with the corresponding co-crystalized ligands with our tested compounds. Moreover, different uracil derivatives have been investigated as cyclin dependent kinase inhibitor (Marchal et al., 2007; Fatahala et al., 2021), or as dihydrofolate reductase inhibitor (Sanduja et al., 2020). The cocrystal ligand for the two proteins (DTQ for CDK2 protein, MTX (methotrexate) for DHFR) were redocked to assure the validity of the docking parameters and methods to represent the position and orientation of the ligand detected in the crystal structure. The difference of RMSD value between cocrystal ligands to the original cocrystal ligand was <2 Å which approved the accuracy of the docking protocols and parameters.
To elucidate the mode of action for the most promising anticancer compounds, precisely compounds 6, 7 ([bis(6-amino-1-(2-chlorobenzyl) dihydropyrimidine-2,4(1H,3H)-dione) and 15, 16 (pyridopyrimidine-6- carbonitriles) were docked against CDK2 protein. Compounds 6, 7, and 16 showed potential binding affinity against to the active site of this protein, this was evident by low energy of binding compared to reference ligand (−9.2, −9.3, −8.7, −8.3 Kcal/mol, respectively) (Table 1), while compound 15 has a comparable value of free binding energy to the reference ligand (-8.2 Kcal/mole). Compound 7 was the best docked compound against CDK2, with two hydrogen bonds formation (ASN132: 2.228 Å, ILE10: 3.241 Å), and hydrophobic interaction (LEU83, LEU134, LEU143, LEU148, LEU2989, VAL18, VAL64, ILE10) (Table 3, Fig. 6). In addition, docking results against DHFR, depicted high binding affinity for the tested compounds as the free of binding energy were less than the reference ligand (-9.0, −9.6, −8.8, −8.8, and −7.8 Kcal/mol, respectively). Interestingly, compound 7 has the highest binding affinity with DHFR protein, with low free binding of energy, and it has two hydrogen bonds with the key amino acid residues in the pocket of the protein (ILE94: 2.117 Å, TRP22: 2.236 Å), moreover, compound 7 has hydrophobic interaction with ILE5, ILE14, ILE50, ILE94, LEU24, LEU28, LEU54, PHE51 amino acid residues (Table 3, Fig. 7).
Compounds
CDK2 protein (PDB: 1DI8)
Free binding of energy (Kcal/mole)
H-bond
No of H-Bond
Amino acid residues
Length Å
6
−9.2
3
ILE10
2.017
ILE10
3.240
ASN132
2.165
7
−9.3
3
ASN132
2.228
ILE10
3.241
15
−8.2
2
GLU12
2.397
ASP86
2.445
16
−8.7
1
LEU83
2.337
DTQ
−8.3
2
LEU83
3.162
LYS33
3.245
Compounds
DHFR protein (PDB: 4DFR)
Free binding of energy (Kcal/mole)
H-bond
No of H-Bond
Amino acid residues
Length Å
6
−9.0
2
TRP22
2.107
ILE94
2.002
7
−9.3
3
ILE94
2.117
TRP22
2.236
15
−8.8
0
–
–
16
−8.8
1
Met20
2.575
MTX
−7.8
3
SER49
2.066
SER49
2.135
THR46
2.002
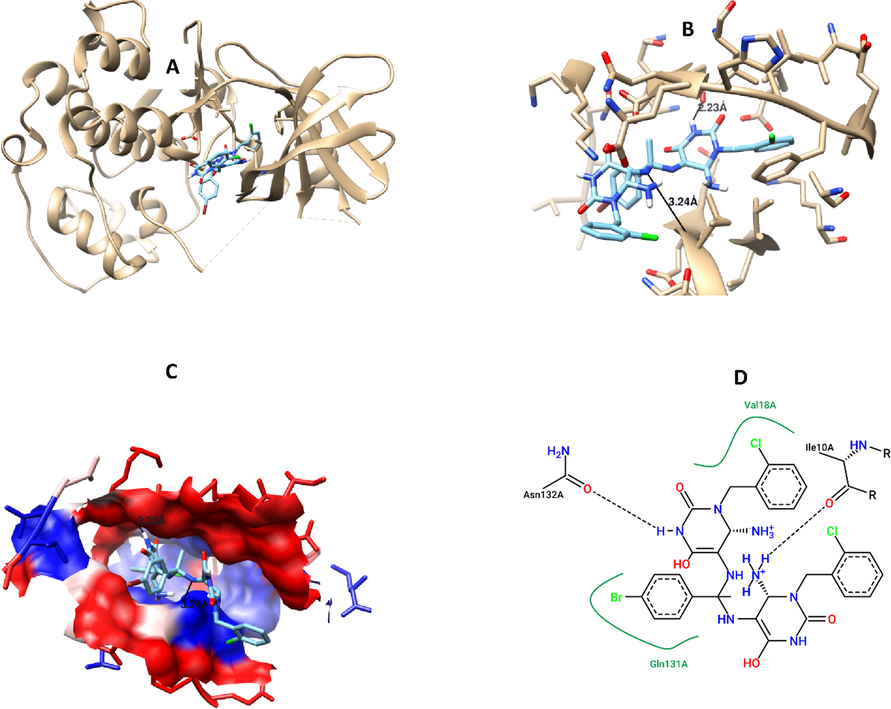
Compound 7 interaction with CDK2 protein, A) 3D interaction, B) 3D hydrogen bond formation, C) Hydrophobic interaction (represented by blue color), D) 2D interaction, B) 3D hydrogen bond formation.
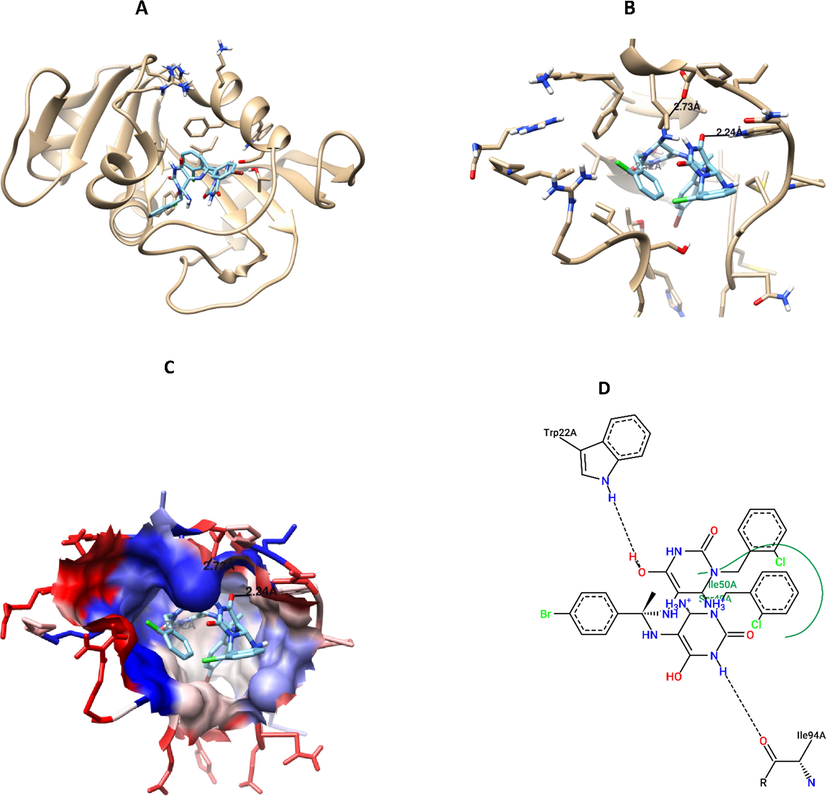
Compound 7 interaction with DHFR protein, A) 3D interaction, B) 3D hydrogen bond formation, C) Hydrophobic interaction (represented by blue color), D) 2D interaction, B) 3D hydrogen bond formation.
3.6 Flow cytometry assay
Apoptosis is one of the most important defense mechanism against cancer progression, therefore, a lot of chemotherapeutic drugs have been investigated to induce apoptosis of cancer cells, or modulate the apoptosis pathway (Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes, n.d.; Comşa et al., 2015; Hernández-Vargas et al., 2006; Fatahala et al., 2021). As the most promising compound against lung cancer, compound 7 have been tested to induce apoptosis, and cell cycle arrest in the treated lung cancer cell line, based on double staining of cells with Propidium Iodide (PI) “which can intercalate with nucleic acid in the nucleus”, and Annexin “which bind with the phosphatidylserine (PS) major component in the cell membrane that become exposed to outside upon apoptosis”. Results disclosed that compound 7 was a potential apoptosis inducer, as it can induce early apoptosis (60.38%), compared to untreated control (0.04%), which was evident by Annexin + staining, while it slightly induce late apoptosis (1.05%), compared to untreated control (0.16%), this was observed by double staining of both Annexin/PI+, while it did not induce necrosis in the treated cells (Fig. 8A). In addition, compound 7 was able to induce cell cycle arrest at S phase, compared to control (17.62%, 0.05%, respectively) (Fig. 8B).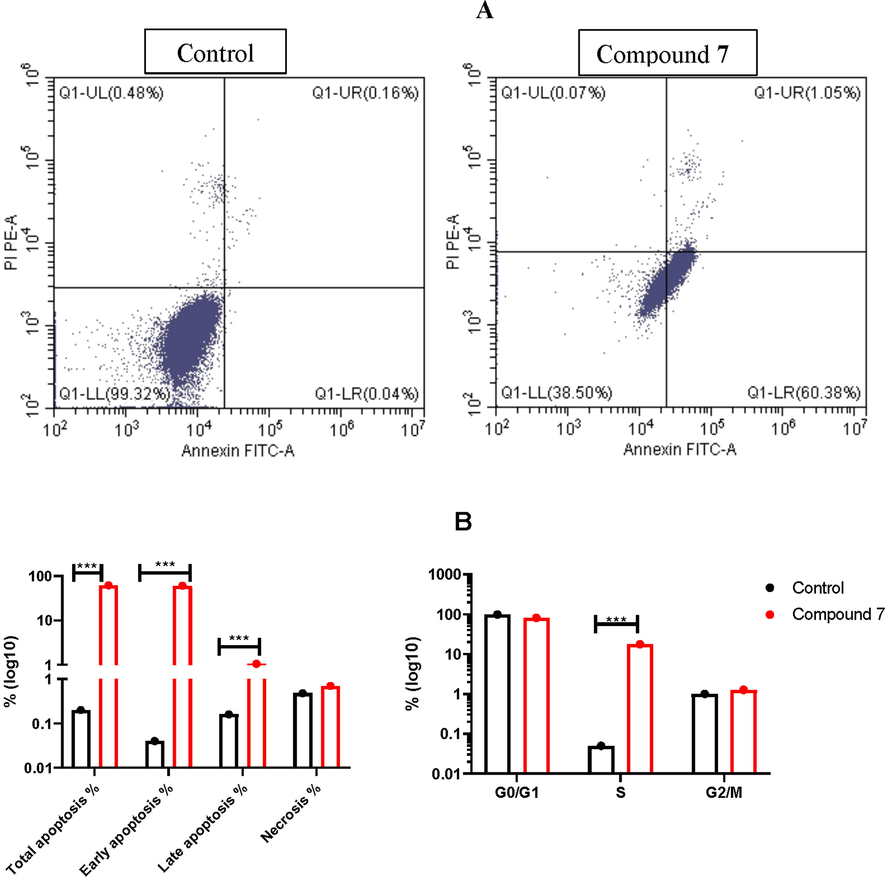
Flow cytometry assay for A549 treated with compound 7: A) Apoptosis detection using Annexin/PI staining, B) cell cycle analysis using PI staining for A549 cells treated with the IC50 value of compound 7 for 48 h.
3.7 Pharmacokinetics, and ADME activity
To predict the physicochemical, pharmacokinetics and drug-likeness properties for the most cytotoxic compounds 6, 7, 15, and 16, bioinformatics analysis based on website data base were done (Table 4-9) (Nagy et al., 2021). Bioavailability Rada has been generated to check the suitable physicochemical properties for oral bioavailability, based on 13 characters including molecular weight (MW: optimal 100–600), Number of rings (nRig: optimal 0–6), formal charge (fChar: optimal −4 to 4), Number of heteroatoms (nHet:optimal 1–15), number of atoms in the biggest ring (MaxRing: optimal 0–18), number of rigid bonds (nRing: optimal 0–30), number of rotatable bonds (nRot: optimal 0–11), Topological Polar Surface Area (TPSA: optimal 0–140), number of hydrogen bond donors (nHD: optimal 0–7), number of hydrogen bond acceptors (nHA, optimal 0–12), log of the aqueous solubility (LogS: optimal −4 to 0.5 log mol/L), Log of the octanol/water partition coefficient (LogD: optimal 0–3), logP at physiological pH 7.4 (LogP: optimal 1–3). Results indicate that all tested compounds have good physicochemical properties, and fulfil most of criteria documents (Table 4, and Fig. 9). In addition, all of tested compounds showed an acceptable medicinal chemistry, and ADMET properties (Supplementary Tables 1–6).
Parameter
Compounds
Comment
6
7
15
16
MW
634.16
712.07
479.1
468.08
Contain hydrogen atoms. Optimal:100–600
nHA
12
12
10
7
Number of hydrogen bond acceptors. Optimal:0–12
nHD
8
8
1
1
Number of hydrogen bond donors. Optimal:0–7
TPSA
185.82
185.82
143.38
100.24
Topological Polar Surface Area. Optimal:0–140
nRot
9
9
6
5
Number of rotatable bonds. Optimal:0–11
nRing
5
5
4
4
Number of rings. Optimal:0–6
MaxRing
6
6
10
10
Number of atoms in the biggest ring. Optimal:0–18
nHet
14
15
11
9
Number of heteroatoms. Optimal:1–15
fChar
0
0
0
0
Formal charge. Optimal:-4 to 4
nRig
34
34
27
26
Number of rigid bonds. Optimal:0–30
Flex
0.265
0.265
0.222
0.192
Flexibility = nRot/nRig
nStereo
0
0
2
2
Optimal: ≤ 2
LogS
−4.385
−4.457
−3.431
−3.687
Log of the aqueous solubility. Optimal: −4 to 0.5 log mol/L
LogD
3.006
3.369
3.188
3.651
Log of the octanol/water partition coefficient. Optimal: 0–3
LogP
3.542
4.312
2.812
3.816
logP at physiological pH 7.4. Optimal: 1–3
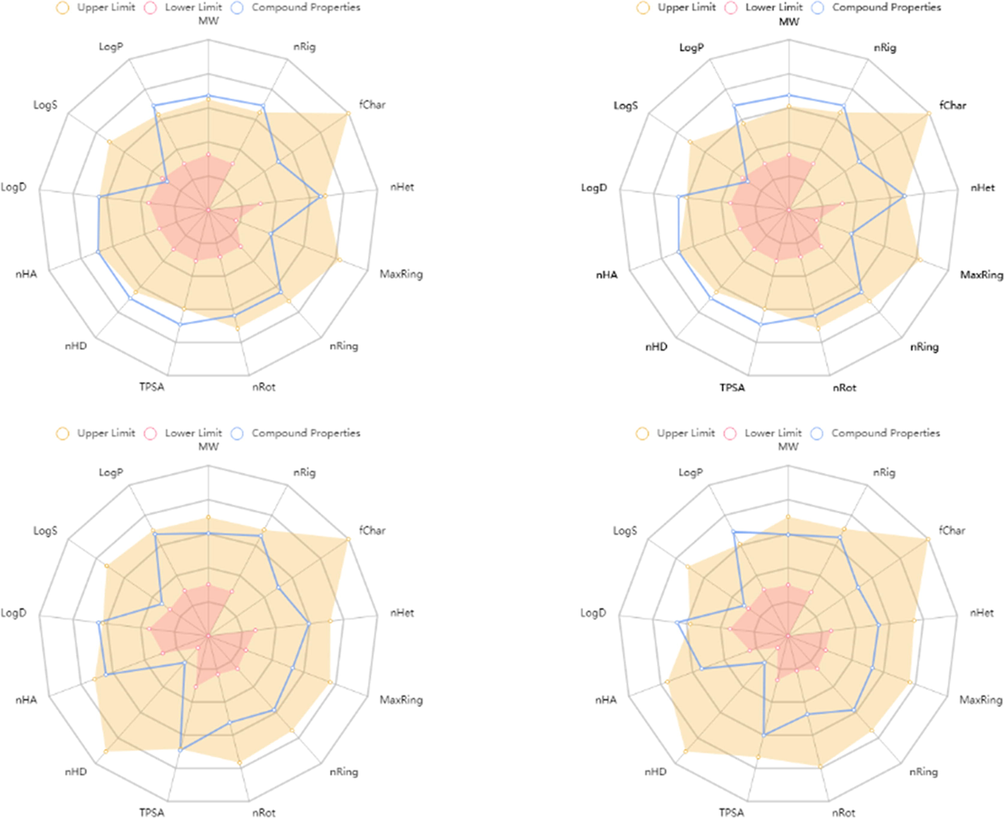
The Bioavailability Radar shows that compounds 6 (A), 7 (B), 15 (C), and 16 (D) represented by central blue lines fits in the pink area which is the optimal range for each properties.
4 Experimental part
4.1 Chemistry
All melting points were determined with an Electrothermal Mel.-Temp. II apparatus and were uncorrected. Element analyses were performed at the Regional Center for Mycology and Biotechnology at Al-Azhar University. IR spectra A Brucker FT-IR spectrophotometer was used to collect the IR spectra of the solid products for wave number from 4000 to 400 cm−1 were performed at nucleic acid center at faculty of Science, Zagazig University. The proton nuclear magnetic resonance (1H NMR) spectra were recorded on Varian Gemini 400 MHz Spectrometer using DMSO‑d6 as a solvent (Chemical shift in δ, ppm), Faculty of Science, Chemistry Department, Zagazig University. Mass spectra were recorded on DI-50 unit of Shimadzu GC/MS-QP 5050A at the Regional Center for Mycology and Biotechnology at Al-Azhar University. All reactions were monitored by TLC using precoated plastic sheets silica gel (Merck 60 F254) and spots were visualized by irradiation with UV light (254 nm). The used solvent system was chloroform: methanol (9:1) & ethyl acetate: toluene (1:1).
Synthesis of 5,6-diamino-1-substituteduracil 3a-e
These compounds were prepared according to the reported method (El-Kalyoubi and Agili, 2020; El-Kalyoubi and Agili, 2016; El-kalyoubi et al., 2015).
4.1.1 6-Amino-1-(2-chlorobenzyl)-5-((1-(4-nitrophenyl)ethylidene)amino)pyrimidine-2,4-dione (4)
A mixture of 5,6-diamino-1-(2-chlorobenzyl)uracil (3d) (1.2 mmol) with p-nitroacetophenone (1.2 mmol), and DMF (1 ml) was heated under fusion for 3 – 4 min. The residue was treated with ethanol (10 ml). The precipitate was filtered, washed with ethanol and crystallized from DMF/ethanol (2:1).
Yield:73%; M.p.: 264–266 °C; IR spectra (υmax, cm−1): 3390, 3320, 3280 (NH); 3080 (CH arom), 2950 (CH aliph.), 1705, 1631 (C⚌O), 1575 (C⚌N), 1504, 1341 (NO2), 748 (C—Cl); 1H NMR (DMSO‑d6): 10.90 (s, H, NH exchangeable by D2O), 8.31–8.28 (d, 2H, J = 8.30 Hz, H arom), 8.26–8.23 (d, 2H, J = 8.30 Hz, H arom), 7.53–7.51 (m, 1H, H arom), 7.35–7.33 (m, 2H, H arom), 6.98 (s, 1H), 6.82 (s, 2H, 2NH exchangeable by D2O), 5.17(s, 2H, CH2), 2.25 (s, 3H, CH3); 13C NMR (DMSO‑d6): 20.50 (CH3), 44.01, (CH2), 96.40, 100.97, 123.23, 123.84, 126.36, 127.53, 128.39, 136.43, 137.02, 145.19, 147.76, 150.22, 154.16, 159.60, 162.43; MS: m/z (%) = M + 2, 415 (25), M+, 413 (8), 397 (35), 335 (66), 297 (100), 239 (87), 107 (64), 43 (46); Anal. calcd for C19H16ClN5O4: C 55.27, H 3.90, N 16.92; Found: C 55.19, H 3.82, N 16.32.
4.1.2 N,N-Bis(6-amino-1-benzyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidn-5-yl)-4-bromo-benzimidimide (5)
A mixture of 5,6-diamino-1-benzyluracil (3c) (1.2 mmol) with p-bromoacetophenone (0.6 mmol), DMF (1 ml) was heated under fusion for 12 min. The residue was treated with ethanol (10 ml). The precipitate was filtered, washed with ethanol and crystallized from DMF/ethanol (3:1).
Yield: 74%; M.p.: 268–270 °C; IR spectra (υmax, cm−1): 3390, 3321, 3240 (NH), 3115 (CH arom), 2830 (CH aliph), 1686, 1657, 1627 (C⚌O), 594 (C⚌Br); 1H NMR (DMSO‑d6): 10.70 (s, 2H, 2NH), 7.34–7.19 (m, 14H, Harom), 6.09 (s, 4H, 2NH2), 5.07 (s, 4H, 2CH2), 2.90 (s, 1H, NH); 13C NMR (DMSO‑d6): 26.64, 36.22, 44.01, 48.56 (2CH2), 96.42, 126.30, 126.43, 126.60, 127.05, 128.18, 127.27, 127.57, 127.76, 127.85, 127.91, 128. 00, 128.25, 128.38, 128.65, 136.65, 136.91, 145.29, 149.64, 159.59; MS: m/z (%) = M + 2, 631 (40), M+, 629 (55), 626 (46), 599 (89), 558 (55), 443 (47), 336 (83), 277 (100), 200 (89), 66 (34); Anal. calcd for C29H25BrN8O4: C 55.33, H 4.00, N 17.80; Found: C 55.49, H 4.18, N 17.69.
5,5′-((1-arylethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-substituted-dihydropyrimidine-2,4(1H,3H)-dione) 6–8, 11,12
5,5′-((1-arylethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-methyl-2-thioxotetrahydropyrimidin-4(1H)-one) 9,10.
4.2 General method
A mixture of 5,6-diamino-1-substituteduracils (3b-e) (1.2 mmol) with appropriate acetophenones (0.6 mmol), DMF (0,5 ml) was heated under fusion for 6–8 min. The residue was treated with ethanol (10 ml). The precipitate was filtered, washed with ethanol and crystallized from DMF/ethanol (3:1).
4.2.1 5,5′-((1-phenylethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-(2-chlorobenzyl)dihydro-pyrimidine-2,4(1H,3H)-dione) (6)
Yield: 81%; M.p.: 238–240 °C; IR spectra (υmax, cm−1): 3380, 3320, 3270 (NH), 3151 (CH arom), 2981 (CH aliph), 1675, 1633 (C⚌O), 1573 (C⚌C), 744 (C—Cl); 1H NMR (DMSO‑d6): δ 10.79, 10.52 (s, 2H, 2NH), 8.06–8.04 (d, 1H, Harom), 7.56–7.45 (m, 6H, Harom), 7.31–7.23 (m, 6H, Harom), 6.84 (s, 2H, NH2), 6.17 (s, 2H, NH2), 5.25 (s, 1H, CH), 5.13 (s, 2H, CH2), 5.02 (s, 2H, CH2), 4.66 (s, 1H, CH), 1.67 (s, 3H, CH3); 13C NMR (DMSO‑d6): 26.92 (CH3), 36.10, 43.33, 42.75 (2 CH2), 75.76, 125.53, 126.42, 127.28, 127.66, 127.74, 128.21, 128.92, 129.03, 129.33, 129.47, 129.73, 130.02, 131.79, 132.31, 133.49, 133.95, 134.40, 150.25, 150.91, 151.24, 155.27, 156.30, 162.94; MS: m/z (%) = M + 4, 639 (18), M + 2, 637 (28), M+, 635 (63), 611 (48), 556 (44), 469 (86), 390 (100), 298 (36), 191 (81), 120 (40), 82 (68), 65 (15); Anal. calcd for C30H28Cl2N8O4: C 56.70, H 4.44, N 17.63; Found: C 56.88, H 4.69, N 17.51.
4.2.2 5,5′-((1-(4-bromophenyl)ethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-(2-chlorobenzyl) dihydropyrimidine-2,4(1H,3H)-dione) (7)
Yield: 82%; M.p.: 248–250 °C; IR spectra (υmax, cm−1): 3396, 3315, 3230 (NH), 3051 (CH arom), 2830 (CH aliph), 1703, 1630 (C⚌O), 1580 (C⚌C), 1503 (C—N), 747 (C—Cl), 563 (C—Br). 1H NMR (DMSO‑d6): 10.84, 10.53 (2 s. 2H, 2NH), 7.99–7.97 (d, 2H, J = 8.7 Hz, Harom), 7.62–7.60 (d, 2H, J = 8.7 Hz, Harom), 7.52–7.50 (m, 2H, Harom), 7.35–7.30 (m, 4H, Harom), 6.97–6.95 (m, 1H, H arom), 6.87–6.63 (m, 1H, H arom), 6.63 (s, 2H, 2NH), 5.16, 5.07 (2 s, 4H, 2CH2), 5.02 (s, 1H, CH(6)), 4.66 (s, 1H, CH(6), 2. 18 (s, 3H, CH3); 13C–NMR (DMSO‑d6): 19.78 (CH3), 42.47, 43.29 (2CH2), 75.53, 104.83, 123.22, 125.49, 125.56, 127.47, 128.52, 128.79, 129.24, 129.35, 130.21, 130.91, 131.04, 131.75, 134.24, 139.10, 145.20, 149.30, 155.89, 159.63, 162.34, 164.31; MS: m/z (%) = M + 4, 714 (30), M + 2, 712 (16), M+, 710 (8), 698 (35), 612 (42), 569 (38), 501 (20), 484 (37), 454 (65), 366 (38), 350 (100), 317 (65), 243 (48), 171 (35), 157 (38), 127 (87), 93 (16), 91 (18), 60 (30); Anal. calcd for C30H27BrCl2N8O4: C 50.44, H 3.81, N 15.69. Found: C 50.38, H 3.97, N 15.90.
4.2.3 5,5′-((1-(4-nitrophenyl)ethane-1,1-diyl)bis(azanediyl))bis(6-amino-1-benzyldihydro-pyrimidine-2,4(1H,3H)-dione) (8)
Yield: 79%; M.p.: 258–260 °C; IR spectra (υmax, cm−1): 3395, 3320, 3290 (NH), 3090 (CHarom), 2980 (CH aliph), 1693, 1633 (C⚌O), 1575 (C⚌C), 1505, 1340 (NO2); 1H NMR (DMSO‑d6): 10.84, 10.69 (2NH), 8.28–8.22 (dd, 4H, H arom), 7.38–7.19 (m, 12H, 10Harom, 2NH), 6.73 (s, 2H, NH2), 6.09 (s, 2H, NH2), 5.19, 5.06 (2 s, 4H, 2CH2), 2.20 (s, 3H, CH3); MS: m/z (%) = M+, 611 (27), 581 (15), 494 (43), 364 (100), 331 (43), 273 (17), 218 (17), 170 (20), 91 (79), 77 (9); Anal. calcd for C30H29N9O6: C 58.91, H 4.78, N 20.61; Found: C 58.38, H 4.94, N 20.38.
4.2.4 5,5′-((1-(4-bromophenyl)ethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-methyl-2-thioxotetrahydropyrimidin-4(1H)-one) (9)
Yield: 83%; M.p.: 260–262 °C; IR spectra υ (υmax, cm−1): 3360, 3320, 3280 (NH), 3130 (CH arom), 2810 (CH aliph), 1621 (C⚌O), 1603 (C⚌C), 1485 (C⚌N), 623 (C—Br); 1H–NMR (DMSO‑d6): 12.06 (s, 2H, 2NH), 8.0–7.97 (d, 2H, J = 8.7 Hz, H arom), 7.63–7.61 (d, 2H, J = 8.7 Hz, H arom), 6.70 (s, 2H, 2NH), 6.19 (s, 4H, 2NH2), 3.80, 3.73 (2 s, 6H, 2CH3), 3.44 (s, 2H, 2CH), 2.12 (s, 3H, CH3); 13C–NMR (DMSO‑d6): 19.76 (CH3), 36.11, 36.37 (2CH3), 48.60, 104.20, 129.43, 130.22, 13.76, 138.69, 142.52, 149.48, 152.04, 156.11, 165.77, 169.32, 172.94; MS: m/z (%) = M + 2, 527 (31), M+, 525 (19), 492 (34), 432 (100), 403 (70), 354 (49), 310 (96), 296 (38), 270 (50), 261 (83), 165 (52), 94 (39), 79 (61); Anal. calcd for C18H21BrN8O2S2: C 41.15, H 4.03, N 21.33: Found: C 41.47, H 4.19, N 21.60.
4.2.5 5,5′-((1-(4-nitrophenyl)ethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-methyl-2-thioxo-tetrahydropyrimidin-4(1H)-one) (10)
Yield: 81%; M.p.: 248–250 °C; IR spectra (υmax, cm−1): 3380, 3340, 3270 (NH), 3100, 3020 (CH arom), 2810 (CH aliph), 1652, 1621 (C⚌O), 1550, 1486 (C⚌N). 1H NMR (DMSO‑d6): 12.06 (s, 2H, 2NH), 8.31–8.29 (d, 2H, J = 9 Hz, H arom), 8.27–8.35 (d, 2H, J = 9.1, H arom), 6.88 (s, 2H, 2NH2 exchangeable by D2O), 6.19 (s, 2H, 2NH2 exchangeable by D2O), 3.81, 3.73 (2 s, 6H, 2CH3), 3.43 (s, 2H, 2CH), 2.2 (s, 3H, CH3); 13C NMR (DMSO‑d6): 19.88 (CH3), 36.54, 36.76 (2CH3), 103.95, 105.25, 112.06, 124.46, 125.16, 130.20, 140.19, 142.12, 144.57, 157.03, 160.13, 170.09; MS: m/z (%) = M+, 491 (18), 459 (25), 319 (100), 312 (55), 278 (53), 268 (68), 184 (46), 106 (43), 93 (33), 75 (29); Anal. calcd for C18H21N9O4S2: C 43.98, H 4.31, N 25.65; Found: C 44.16, H 4.58, N 25.43.
4.2.6 5,5′-((1-(4-bromophenyl)ethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-ethyldihydro-pyrimidine −2,4(1H,3H)-dione) (11)
Yield: 71%; M.p.: 288–290 °C; IR spectra (υmax, cm−1): 3376, 3312, 3280 (NH), 3135 (CH arom), 2973, 2934 (CH aliph), 1680, 1640 (C⚌O), 1600 (C⚌C), 1552 (C⚌N), 547 (C—Br); 1H–NMR (DMSO‑d6): 10.62, 10.51 (2 s, 2H, 2NH), 7.96–7.94 (d, 2H, J = 8.4 Hz, Harom), 7.60–7.58 (d, 2H, J = 8.4 Hz, Harom), 6.58 (s, 2H, NH2), 6.15 (s, 2H, NH2), 3.93–3.79 (qq, 4H, 2CH2), 2.62 (s, 2H, 2CH). 2.10 (s, 3H, CH3), 1.16–1.07 (tt, 6H, 2CH3); 13C–NMR (DMSO‑d6): 12.34 (CH3), 19.74 (CH3), 36.78 (CH2), 76.97, 96.97, 123.13, 129.69, 130.22, 131.83, 139.16, 149.57, 154.35, 160.30, 163.59, 171.37; MS: m/z (%) = M + 2, 523 (36), M+, 521 (9), 497 (54), 336 (92), 333 (100), 305 (13), 277 (38), 238 (20), 170 (14), 125 (41), 86 (34), 78 (38), 53 (35); Anal. calcd for C20H25BrN8O4: C 46.07, H 4.83, N 21.49; Found C 46.21, H 4.11, N 21.79.
4.2.7 5,5′-((1-(4-nitrophenyl)ethane-1,1-diyl)bis(azaneylylidene))bis(6-amino-1-ethyldihydro-pyrimidine-2,4(1H,3H)-dione) (12)
Yield: 78%; M.p.: 242–244 °C; IR spectra (υmax, cm−1): 3390, 3285, 3210 (NH), 3151 (CH arom), 2815 (CH aliph), 1687, 1640 (C⚌O), 1607 (C⚌C), 1553 (C—N), 1508, 1341 (NO2); 1H–NMR (DMSO‑d6): 10.63, 10.50 (2 s, 2H, 2NH), 8.28–8.26 (d, 2H, J = 9.3 Hz, Harom), 8.25–8.23 (d, 2H, J = 9.3 Hz, Harom), 6.77 (s, 2H, NH2), 6.14 (s, 2H, NH2), 3.94–3.92, 3.82–3.80 (qq, 4H, J = 7 Hz, 2CH2), 2.81 (s, 2H, 2CH), 2.17 (s, 3H, CH3), 1.17–1.15, 1.10–1.07 (tt, 6H, J = 7 Hz, 2CH3). 13C–NMR (DMSO‑d6): 13.18, 13.34 (2CH3), 20.34 (CH3), 36.84, 36.46 (2CH2), 96.06, 123.14, 128.24, 145.28, 125.83, 147.66, 149.09, 149.41, 150.13, 154.02, 159.53, 161.84; MS: m/z (%) = M+, 487 (10), 431 (37), 384 (16), 302 (100), 286 (60), 273 (40), 269 (34), 231 (37), 203 (22), 182 (50), 156 (45), 156 (45), 143 (44), 119 (54), 77 (84), 42 (29); Anal. calcd for C20H25N9O6: C 49.28, H 5.17, N 25.86; Found: C 49.56, H 5.43, N 26.13.
4.2.8 8-(4-bromophenyl)-1,3,8-trimethyl-3,8-dihydro-1H-purine-2,6-dione (13)
Yield: 67%; M.p.: 292–294 °C; IR spectra (υmax, cm−1): 3251 (NH), 3070 (CH arom), 2957 (CH aliph), 1714, 1635 (C⚌O), 1562 (C⚌C), 624 (C—Br); 1H NMR (DMSO‑d6): 10.29 (s, 1H, NH), 8.87–8.86 (d, 1H, H arom), 7.62 (m, 3H, H arom), 3.57 (s, 6H, 2CH3), 2.84 (s, 3H, CH3); 13C NMR (DMSO‑d6): 26.30 (CH3), 28.70 (CH3), 29.38 (CH3), 29.68 (C aliph), 123.41, 127.40, 138.40, 149.40, 150.68, 151.68, 158.88, 165.20; Anal. calcd for C14H13BrN4O2: C 48.16, H 3.75, N 16.05; Found: C 48.40, H 3.98, N 16.33.
4.2.9 5-(4-Substitutedphenyl)-1-(2-chlorobenzyl)-7-ethoxy-2,4-dioxo-1,2,3,4,5,6-hexahydropyrido [2,3-d]pyrimidine-6-carbonitrile 14–16
General method: To (2.3 mmol) of 6-aminouracils in DMF (5 ml) was added ethyl arylidenecyanoacetate in the presence of triethylamine (0.5 ml). The reaction mixture was refluxed for 10 hrs. The formed precipitate on cooling was filtered of, washed with ethanol, dried and recrystallized from a mixture of DMF/ethanol (2–1).
4.2.10 5-(4-bromophenyl)-1-(2-chlorobenzyl)-7-ethoxy-2,4-dioxo-1,2,3,4,5,8-hexahydropyrido[2,3-d]pyrimidine-6-carbonitrile (14)
Yield: 56%; M.p.: >300 °C; IR spectra (υmax, cm−1): 3359, 3174 (NH), 3062 (CH arom), 2919, 2854 (CH aliph), 2221 (CN), 1714, 1661 (C⚌O), 1561 (C⚌C), 1481 (C—N), 754 (C—Cl), 556 (C—Br). 1H NMR (DMSO‑d6): 10.83 (s. 1H, 1NH), 8.67 (s. 1H, 1NH), 7.94 (s, 1H, C-5), 7.60–7.57 (d, 4H, Harom), 7.22–7.19 (t, 4H, H arom), 5.31 (s, 2H, CH2), 3.15–3.03 (q, 2H, CH2), 1.20–1.17 (t, 3H, CH3); 13C NMR (DMSO‑d6): 10.89 (CH3), 34.01, (OCH2), 34.79 (NCH2), 85.34, 94.65, 116.57, 126.95, 127.62, 128.62, 129.35, 130.95, 131.62, 133.70, 135.95, 150.86, 154.93, 158.86, 160.01, 162.32, 164.84; MS: m/z (%) = M + 4, 517 (23), M + 2, 515 (29), M+, 513 (15), 489 (61), 433 (52), 405 (71), 387 (59), 351 (62), 267 (100), 238 (60), 202 (76), 182 (80), 105 (43), 92 (56), 88 (74), 45 (45); Anal. calcd for C23H18BrClN4O3: C 53.77, H 3.53, N 10.91; Found: C 53.94, H 3.70, N 11.20.
4.2.11 1-(2-chlorobenzyl)-7-ethoxy-5-(3-nitrophenyl)-2,4-dioxo-1,2,3,4,5,8-hexahydropyrido[2,3-d]pyrimidine-6-carbonitrile (15)
Yield: 58%; M.p.: >300 °C; IR spectra (υmax, cm−1): 3290, 3160 (NH), 3022 (CH arom), 2913, 2817 (CH aliph), 2222 (CN), 1720, 1648 (C⚌O), 1560 (C⚌C), 1486, 1367 (NO2), 751 (C—Cl); 1H–NMR (DMSO‑d6): 11.35 (s. 1H, 1NH), 8.38 (s. 1H, 1NH), 7.94 (s, 1H, C-5), 7.54–7.29 (m, 8H, Harom), 5.8 (s, 2H, CH2), 3.09–3.08 (q, 2H, CH2), 1.17 (t, 3H, CH3); MS: m/z (%) = M + 2, 481 (28), M+, 479 (42), 425 (81), 399 (45), 318 (31), 258 (19), 227 (48), 187 (47), 125 (54), 115 (100), 111 (58), 91 (18), 78 (19), 54 (36); Anal. calcd for C23H18ClN5O5: C 57.57, H 3.78, N 14.59; Found: C 57.78, H 3.96, N 14.63.
4.2.12 1-(2-chlorobenzyl)-5-(4-chlorophenyl)-7-ethoxy-2,4-dioxo-1,2,3,4,5,8-hexahydropyrido[2,3-d]pyrimidine-6-carbonitrile (16)
Yield: 59%; M.p.: 272–274 °C; IR spectra (υmax, cm−1): 3280, 3170 (NH), 3022 (CH arom), 2908, 2814 (CH aliph), 2222 (CN), 1705, 1655 (C⚌O), 1574 (C⚌C), 738 (C—Cl); 1H NMR (DMSO‑d6): 11.94 (s. 1H, 1NH), 8.11 (s. 1H, 1NH), 7.72 (s, 1H, C-5), 7.70–6.83 (m, 8H, Harom), 5.08 (s, 2H, CH2), 3.29–3.16 (q, 2H, CH2), 1.06–1.03 (t, 3H, CH3); 13C NMR (DMSO‑d6): 14.23 (CH3), 31.02 (OCH2), 43.62 (NCH2), 85.64, 122.87, 123.26, 125.67, 127.83, 129.13, 129.78, 130.39, 131.22, 132.00, 133.80, 134.74, 147.44, 150.31, 157.13, 163.02, 165.11; MS: m/z (%) = M + 4, 473 (54), M + 2, 471 (18), M+, 469 (16), 412 (50), 381 (37), 352 (63), 324 (77), 321 (70), 286 (36), 269 (73), 218 (48), 185 (50), 146 (50), 96 (43), 76 (56), 64 (100), 56 (29); Anal. calcd for C23H18Cl2N4O3: C 58.86, H 3.87, N 11.94. Found: C 59.12, H 4.05, N 11.85.
5 Biological activity
5.1 In-vitro cytotoxic assay
5.1.1 Cell culture and maintenance
Human hepatoma Huh7, HepG2, breast cancer cells MCF7, lung small cell adenocarcinoma (A549), and normal Madin-Darby Canine Kidney MDCK cell lines were propagated in Dulbecco’s modified Eagle’s medium (DMEM). All medium were supplemented with 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin antibiotics (Seralab, UK). The cells were incubated in 5% CO2 humidified at 37 °C for growth maintenance. All tissue culture work has been done according to our established tissue culture protocols (Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes, n.d.; Sroor et al., 2021; Tantawy et al., 2020).
5.1.2 Evaluation of cell proliferation by MTT assay
The percentages of viable HepG2, Huh7, A549 and MCF7 cells after treatment with different concentrations of the compounds were evaluated by the 3-[4,5-methylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay as reported previously (Fatahala et al., 2021; Tantawy et al., 2020; Abu Almaaty et al., 2021; Xiong et al., 2021), with slight modification. In brief, after evaluation of cell count and viability by trypan blue dye-based method, cancer cells (1 × 104 cells/well) were seeded in a 96-well plate and then kept overnight for attachment. At the next day, the complete medium was replaced with fresh one, and then various concentrations of the synthesized compounds (0, 0.1, 1, 10, and 100 μM) were investigated on each cell line. After that, cells were allowed to grow for 24 h. Four hours before completion of the incubation period, 10 μl of the MTT (5 mg/ml) was added in each well. After completing the incubation, 100 μl of Dimethyl sulfoxide (DMSO) was added to each well and left for 20 min to dissolve the formazan crystals, then the 96 well plates were shook for 5 min to ensure a homogeneous dye in the solution. After the reaction, color development was measured at 490 nm using Bio-Tek microplate reader. Based on the MTT results, we have chosen the most promising cytotoxic compound to elucidate its mode of action.
5.1.3 Molecular docking study
The mode of action for the most active compounds was investigated using molecular docking technique. Using the Chemsketch software (http://www.acdlabs.com/resources/freeware/), the chemical structure for the novel compounds, as well as the corresponding reference ligand for each protein under study, were built. The structures were prepared, optimized and energy minimized and saved as PDB format using VEGAZZ. Using AutoDockTools 1.5.6, all compounds were converted to PDBQT format (Pedretti et al., 2004; Kattan et al., 2020). The optimized compounds were used to perform molecular docking against two proteins that represents vital target for chemotherapeutic drugs, including cyclin dependent kinase-2 (CDK2), (https://www.rcsb.org/structure/1DI8), and dihydrofolate reductase (DHFR) (https://www.rcsb.org/structure/4DFR). The PDB file for each protein was downloaded from Protein Data Bank (PDB) (www.rcsb.org). Receptors were prepared by removal of heteroatoms (water and ions), the addition of polar hydrogen, and the assignment of charge. The active sites were defined using grid boxes of appropriate sizes around the bound cocrystal ligands. The docking study was performed using Autodock vina (Trott and Olson, 2010) and Chimera for visualization (Pettersen et al., 2004; Abdel-Motaal et al., 2020). All docking procedures and scoring were recorded according to established protocols (Tantawy et al., 2019; Kattan et al., 2020; Nafie et al., 2019; El-Far et al., 2020). 2D interaction diagrams were generated by uploading docking file results in PDB format to the protein plus website (Zentrum für Bioinformatik, 2021).
5.1.4 Cell cycle analysis
Cell cycle analysis and apoptotic assay for treated lung cancer cells were done according to established protocols (Balan et al., 2007; Tantawy et al., 2014; Molecular Docking Study, 2019). A549cells were seeded (1.0 × 106 cells/flask) for 24 h, then, cells were treated with the IC50 value (5.46 µM) of compound 7. After 48 h incubation, the cells were harvested using trypsin, and fixed following the instructions mentioned in Annexin V-FITC Detection Kit (Catalog #: K101-25, BioVision), and propidium iodide (PI) stain. Then, flow cytometry analysis was performed, to determine in which phase cells would be arrested and also to calculate the percentage of apoptotic cells.
5.2 Physicochemical property, pharmacokinetics, and ADME activity
As pharmacokinetics and toxicity are major determinants for the efficacy of the new developed drugs, we have initiated an in silico study, to investigate the properties of the most active compounds against tested cancer cell lines. This study was based on database function integrated on Swiss institute Bioinformatics tools (Daina et al., 2017), and ADMETlab 2.0 (Xiong et al., 2021).
6 Conclusion
In summary, we have prepared different compounds from uracil derivatives, xanthine and pyridopyrimides. The biological activity for the twelve novel synthesized compounds were evaluated as anti-cancer agents in vitro in comparison to 5-fluro uracil, against 4 cell lines, and toxicity also was included against normal non-cancerous cell line. Results disclosed that compounds 6, 7, 12, and 15 were the best cytotoxic agents against most tested cancer cell lines, with low toxicity against normal cells compared to 5-FU. Moreover, docking study revealed the potential inhibition activity for these compounds on CDK2, and DHFR proteins, which was augmented by apoptosis induction especially for A549 cells treated with compound 7. As, another step forward to elaborate with these compounds, theoretical physicochemical property, and ADME study highlighted the ability of these compounds to be used as platform to develop chemotherapeutic drugs. In vivo, and preclinical study for these compounds as anticancer agents are being an interesting focus for future studies.
Author contributions
S.E.K conceived and designed the work. S.E.K wrote the manuscript. S. E. K., and F. A. performed the experiments and analyzed the data; M.T. perform the biological analysis with I.A. and perform the ADME data. All authors have read and agreed to the published version of the manuscript.
Funding
The work has been funded by our own money.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Antimicrobial evaluation and docking study of some new substituted benzimidazole-2yl derivatives. Bioorg. Chem.. 2020;101:103972
- [CrossRef] [Google Scholar]
- 5-Aryl-1-Arylideneamino-1H-Imidazole-2(3H)-Thiones: Synthesis and In Vitro Anticancer Evaluation. Molecules. 2021;26:1706.
- [CrossRef] [Google Scholar]
- Pharmacokinetic, oral bioavailability, and safety study of fluorouracil in patients treated with 776C85, an inactivator of dihydropyrimidine dehydrogenase. J. Clin. Oncol.. 1996;14:3085-3096.
- [CrossRef] [Google Scholar]
- Antiproliferative activity and induction of apoptosis in human colon cancer cells treated in vitro with constituents of a product derived from Pistacia lentiscus L. var. chia. Phytomedicine. 2007;14:263-272.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of 2-methoxy- and 2-methylthio-6-[2’-alkylamino)ethyl]-4(3H)-pyrimidinones with anti-rubella virus activity. Bioorg. Med. Chem.. 1999;7:1925-1931.
- [CrossRef] [Google Scholar]
- Research on anti-HIV-1 agents. Part 2: Solid-phase synthesis, biological evaluation and molecular modeling studies of 2,5,6-trisubstituted-4(3H)-pyrimidinones targeting HIV-1 reverse transcriptase. Tetrahedron. 2001;57:8357-8367.
- [CrossRef] [Google Scholar]
- Synthèse et propriétés pharmacologiques de quelques thiéno[2,3-d]pyrimidin-4-one 2-thiones. Eur. J. Med. Chem.. 1990;25:635-639.
- [CrossRef] [Google Scholar]
- The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res.. 2015;35:3147-3154. https://ar.iiarjournals.org/content/35/6/3147 (accessed August 30, 2021)
- [Google Scholar]
- Novel, potent and selective anilinoquinazoline and anilinopyrimidine inhibitors of p38 MAP kinase. Bioorg. Med. Chem. Lett.. 2004;14:5389-5394.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- Clinical pharmacology of 5-fluorouracil. Clin. Pharmacokinet.. 1989;16:215-237.
- [CrossRef] [Google Scholar]
- Thymoquinone-chemotherapeutic combinations: new regimen to combat cancer and cancer stem cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020
- [CrossRef] [Google Scholar]
- Novel 2-Thioxanthine and Dipyrimidopyridine Derivatives: Synthesis and Antimicrobial Activity. Molecules. 2015;20:19263-19276.
- [CrossRef] [Google Scholar]
- A Novel Synthesis of Fused Uracils: Indenopyrimidopyridazines, Pyrimidopyridazines, and Pyrazolopyrimidines for Antimicrobial and Antitumor Evalution. Molecules. 2016;21:1714.
- [CrossRef] [Google Scholar]
- Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido[2,3-d]pyrimidine, Xanthine and Lumazine Derivatives. Molecules.. 2020;25:E5205.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and molecular docking studies of thiouracil derivatives as potent thymidylate synthase inhibitors and potential anticancer agents. Mol. Divers.. 2017;21:967-983.
- [CrossRef] [Google Scholar]
- Insight Into the Molecular Mechanism of Podophyllotoxin Derivatives as Anticancer Drugs. Front. Cell Dev. Biol.. 2021;9:2216.
- [CrossRef] [Google Scholar]
- An expeditious one-pot synthesis of pyrido[2,3-d]pyrimidines using Fe3O4–ZnO–NH2–PW12O40 nanocatalyst. J. Chem. Res.. 2019;43:135-139.
- [CrossRef] [Google Scholar]
- Synthesis of Novel 2-Thiouracil-5-Sulfonamide Derivatives as Potent Inducers of Cell Cycle Arrest and CDK2A Inhibition Supported by Molecular Docking. Int. J. Mol. Sci.. 2021;22:11957.
- [CrossRef] [Google Scholar]
- Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase inhibitors and antitumor agents: synthesis and biological activities of 2,4-diamino-5-methyl-6-[(monosubstituted anilino)methyl] pyrido[2,3-d]pyrimidines. J. Med. Chem.. 1999;42:2447-2455.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of 2,4-Diamino-6-(arylaminomethyl)pyrido[2,3-d]pyrimidines as Inhibitors of Pneumocystis carinii and Toxoplasma gondii Dihydrofolate Reductase and as Antiopportunistic Infection and Antitumor Agents. J. Med. Chem.. 2003;46:5074-5082.
- [CrossRef] [Google Scholar]
- Comparative Quantitative Structure−Activity Relationship Studies on Anti-HIV Drugs. Chem. Rev.. 1999;99:3525-3602.
- [CrossRef] [Google Scholar]
- Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179:663-666.
- [CrossRef] [Google Scholar]
- Transcriptional profiling of MCF7 breast cancer cells in response to 5-Fluorouracil: relationship with cell cycle changes and apoptosis, and identification of novel targets of p53. Int. J. Cancer. 2006;119:1164-1175.
- [CrossRef] [Google Scholar]
- A review on the effects of current chemotherapy drugs and natural agents in treating non-small cell lung cancer. Biomedicine (Taipei). 2017;7:23.
- [CrossRef] [Google Scholar]
- Chemotherapy with enteric-coated tegafur/uracil for advanced hepatocellular carcinoma. World J. Gastroenterol. WJG.. 2008;14:2797.
- [CrossRef] [Google Scholar]
- Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol.. 2016;36:716-726.
- [CrossRef] [Google Scholar]
- Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: Implication of PI3K/AKT/mTOR pathway. J. Steroid Biochem. Mol. Biol.. 2020;198:105604
- [CrossRef] [Google Scholar]
- Electrocatalytic multicomponent assembling of aminouracils, aldehydes and malononitrile: An efficient approach to 7-amino-pyrido[2,3-d]pyrimidine-6-carbonitrile derivatives. J. Serb. Chem. Soc.. 2016;81:29-34.
- [CrossRef] [Google Scholar]
- Integrated Approach to Nature as Source of New Drug Lead. IntechOpen 2018
- [CrossRef] [Google Scholar]
- Potent antipneumocystis and antitoxoplasma activities of piritrexim, a lipid-soluble antifolate. Antimicrob. Agents Chemother.. 1988;32:430-433.
- [CrossRef] [Google Scholar]
- Discovery of 4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin-3-yl)pyrido[2,3-d]pyrimidine, an orally active, non-nucleoside adenosine kinase inhibitor. J. Med. Chem.. 2001;44:2133-2138.
- [CrossRef] [Google Scholar]
- A synthetic uracil derivative with antitumor activity through decreasing cyclin D1 and Cdk1, and increasing p21 and p27 in MCF-7 cells. Breast Cancer Res. Treat.. 2007;105:237-246.
- [CrossRef] [Google Scholar]
- Molecular Docking Study, Cytotoxicity, Cell Cycle Arrest and Apoptotic Induction of Novel Chalcones Incorporating Thiadiazolyl Isoquinoline in Cervical Cancer | Bentham Science, n.d. http://www.eurekaselect.com/node/176127/article/molecular-docking-study-cytotoxicity-cell-cycle-arrest-and-apoptotic-induction-of-novel-chalcones-incorporating-thiadiazolyl-isoquinoline-in-cervical-cancer (accessed November 16, 2019).
- Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents. Steroids. 2019;152:108485
- [CrossRef] [Google Scholar]
- Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein. Pharmaceuticals (Basel).. 2021;14
- [CrossRef] [Google Scholar]
- Synthesis of new uracil derivatives and their sugar hydrazones with potent antimicrobial, antioxidant and anticancer activities. Nucleos. Nucleot. Nucl. Acids. 2020;39:991-1010.
- [CrossRef] [Google Scholar]
- Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol.. 2015;16:e165-e172.
- [CrossRef] [Google Scholar]
- VEGA–an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des.. 2004;18:167-173.
- [Google Scholar]
- 1-calcium phosphate-uracil, a synthesized pyrimidine derivative agent, has anti-proliferative, pro-apoptotic and anti-invasion effects on multiple tumor cell lines. Mol. Med. Rep.. 2014;10:2271-2278.
- [CrossRef] [Google Scholar]
- UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25:1605-1612.
- [CrossRef] [Google Scholar]
- Pyrido(3,2-d)pyrimidines useful for treating viral infections - Patent WO-2008077651-A1 - PubChem, n.d. https://pubchem.ncbi.nlm.nih.gov/patent/WO-2008077651-A1 (accessed September 8, 2021).
- Uracil-coumarin based hybrid molecules as potent anti-cancer and anti-bacterial agents. J. Saudi Chem. Soc.. 2020;24:251-266.
- [CrossRef] [Google Scholar]
- Synthesis and biological activity of some 4-(substituted) aminopyrimidines. J. Med. Pharm. Chem.. 1962;91:871-876.
- [CrossRef] [Google Scholar]
- An Efficient Synthesis of Pyrido[2,3-d]pyrimidine Derivatives via One-Pot Three-Component Reaction in Aqueous Media. Int. J. Org. Chem.. 2012;2012
- [CrossRef] [Google Scholar]
- Synthesis of some novel azido/tetrazolothienopyrimidines and their reduction to 2,4-diaminothieno[2,3-d]pyrimidines. J. Heterocycl. Chem.. 1992;29:883-893.
- [CrossRef] [Google Scholar]
- Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes, n.d. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3061558/ (accessed August 30, 2021).
- Development of five membered heterocyclic frameworks via [3+2] cycloaddition reaction in an aqueous micellar system. New J. Chem.. 2014;38:2756-2759.
- [CrossRef] [Google Scholar]
- Tyrosine Kinase Inhibitors. 17. Irreversible Inhibitors of the Epidermal Growth Factor Receptor: 4-(Phenylamino)quinazoline- and 4-(Phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides Bearing Additional Solubilizing Functions. J. Med. Chem.. 2000;43:1380-1397.
- [CrossRef] [Google Scholar]
- Cyclization of Isothiocyanates as a Route to Phthalic and Homophthalic Acid Derivatives 1,2. J. Org. Chem.. 1964;29:2261-2265.
- [CrossRef] [Google Scholar]
- Molecular docking and ADMET study of bioactive compounds of Glycyrrhiza glabra against main protease of SARS-CoV2. Mater. Today:. Proc. 2020
- [CrossRef] [Google Scholar]
- Synthesis, antimicrobial, anti-cancer and in silico studies of new urea derivatives. Bioorg. Chem.. 2021;112:104953
- [CrossRef] [Google Scholar]
- The interferon-induced gene Ifi27l2a is active in lung macrophages and lymphocytes after influenza A infection but deletion of Ifi27l2a in mice does not increase susceptibility to infection. PLoS ONE. 2014;9:e106392
- [CrossRef] [Google Scholar]
- Molecular Docking Study, Cytotoxicity, Cell Cycle Arrest and Apoptotic Induction of Novel Chalcones Incorporating Thiadiazolyl Isoquinoline in Cervical Cancer. Anticanc. Agents Med. Chem. 2019
- [CrossRef] [Google Scholar]
- Synthetic antiprotozoal thiazolide drug induced apoptosis in colorectal cancer cells: implications of IL-6/JAK2/STAT3 and p53/caspases-dependent signaling pathways based on molecular docking and in vitro study. Mol. Cell. Biochem.. 2020;469:143-157.
- [CrossRef] [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [CrossRef] [Google Scholar]
- Development of a Binding Model to Protein Tyrosine Kinases for Substituted Pyrido[2,3-d]pyrimidine Inhibitors. J. Med. Chem.. 1998;41:1752-1763.
- [CrossRef] [Google Scholar]
- Synthesis of 3-substituted-4-phenyl-2-thioxo-1,2,3,4,5,6,7,8-octahydrobenzo[4,5]thieno[2,3-á]pyrimidines. J. Heterocycl. Chem.. 1990;27:269-273.
- [CrossRef] [Google Scholar]
- Estimates of global chemotherapy demands and corresponding physician workforce requirements for 2018 and 2040: a population-based study. Lancet Oncol.. 2019;20:769-780.
- [CrossRef] [Google Scholar]
- The Anti-Cancer Effects of a Zotarolimus and 5-Fluorouracil Combination Treatment on A549 Cell-Derived Tumors in BALB/c Nude Mice. Int. J. Mol. Sci.. 2021;22:4562.
- [CrossRef] [Google Scholar]
- ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res.. 2021;49:W5-W14.
- [CrossRef] [Google Scholar]
- A convenient two-step synthesis of 6-methylenesubstituted-4-trichloromethyl-2-methylsulfanyl pyrimidines. Tetrahedron Lett.. 2006;4:573-576.
- [CrossRef] [Google Scholar]
- Zentrum für Bioinformatik: Universität Hamburg - Proteins Plus Server, n.d. https://proteins.plus/ (accessed December 16, 2021).
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103669.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




