

Translate this page into:
Ultrafast 99% Polyethylene terephthalate depolymerization into value added monomers using sequential glycolysis-hydrolysis under microwave irradiation
⁎Corresponding author at: Materials Research Institute, Technological University of the Shannon Midlands Midwest, Athlone N37HD68, Ireland. oadly@ait.ie (Olivia A. Attallah)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Combined green and fast glycolysis-hydrolysis depolymerization of polyethylene terephthalate (PET) was carried out under microwave irradiation (MW) with excellent efficiencies.
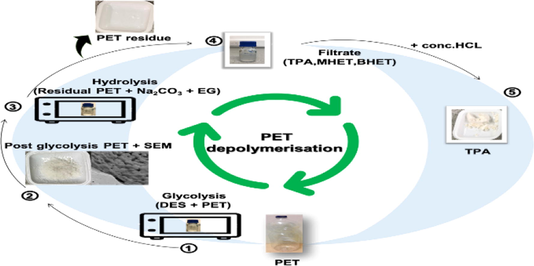
Abstract
Effective and efficient hybrid depolymerisation technologies are emerging as high potential sustainable routes with considerable benefits over conventional recycling methods for the achievement of circular economies for plastics. Herein, combined green and fast glycolysis-hydrolysis depolymerization of polyethylene terephthalate (PET) was carried out under microwave irradiation (MW) with excellent efficiencies. In MW assisted glycolysis of PET, the catalytic activity of two deep eutectic solvents (DES) based on (choline chloride-urea (DES 1)) and (choline chloride-thiourea (DES 2)) was evaluated and compared. Optimised glycolysis conditions were determined using Box Behnken Design (BBD) to attain maximum weight loss of PET, low crystallinity and increased carbonyl index of residual PET. DES volume of 4 mL, 5.5–6 mL of ethylene glycol, and 0.5 min MW irradiation time resulted in a prominent rise in PET weight loss and carbonyl index of residual PET. DES 2 showed an improved catalytic activity than that of DES 1 which is associated to its stronger interaction with EG and PET polymer chains during the course of the reaction. Residual PET obtained post glycolysis reaction was further depolymerized using MW assisted hydrolysis in the presence of weakly basic Na2CO3 and EG. Within 3-minute, the proposed sequential depolymerization technologies facilitated ≈99% conversion of PET to terephthalic acid (TPA), monohydroxyethyl terephthalate (MHET), and bis (2-hydroxyethyl) terephthalate (BHET) monomers produced at a yield of 62.79–80.66%, 17.22–34.79% and 0.54–0.59% respectively. Application on post-consumer PET sample also revealed very satisfactory results with 96.77–98.25% PET conversion and 60.98–78.10% yield of TPA.
Keywords
Depolymerization
Microwave
Deep eutectic solvent
Polyethylene terephthalate
Monomers
Recycling

1 Introduction
Polyethylene terephthalate (PET) is a thermoplastic polymer with ideal physiochemical properties for a number of high consumption applications in particular fibers, packaging and bottles. PET product manufacturing has markedly increased and as the fourth largest produced polymer accounts for 12% volume of the world’s total solid waste (Chan and Zinchenko, 2021).
Chemical recycling has been recently proposed as one of the promising techniques for PET recycling where the waste plastics are broken down into monomers or other value-added products. Among the conventional chemical recycling techniques, glycolysis and hydrolysis are being explored for sufficient efficiency and feasibility for investment as industrial scale applications. In a typical glycolysis process for PET, ester linkages are replaced by hydroxyl groups generating feed stock monomer, bis (2-hydroxyethyl) terephthalate (BHET). In the hydrolysis of PET wastes, PET is depolymerised in the presence of steam, acid or base giving rise to ethylene glycol (EG) and terephthalic acid (TPA) monomer (Padhan and Sreeram, 2019).
The simultaneous application of glycolysis-hydrolysis, glycolysis-methanolysis and methanolysis-hydrolysis directed towards more efficient PET depolymerization have also been presented (Güçlü et al., 2003). Doerr et al. stated the reduction in PET molecular weight from 30,000 to 1000 by mixing small amounts of EG in an extruder with molten PET under a reaction temperature of 280 °C. In a subsequent hydrolysis reaction, the treated PET was converted to EG and TPA, with the glycolic pre-treatment step reducing the time required for the hydrolysis reaction from 45 to 15 min (Doerr, 1986). In Simon et al. patent initial glycolysis of PET to BHET, followed by a purification stage using adsorption and filtration and subsequent hydrolysis of BHET to yield TPA in a separate, glass-lined reactor at 200 °C and predetermined reaction time was presented (West, Simon, 1993). A recent study by Güçlü et al. also introduced a simultaneous glycolysis and neutral hydrolysis of PET using constant amount of EG and increasing amounts of water, in the presence of xylene. The reactions were carried out at 170 and 190 °C to produce a highly pure monohydroxyethyl terephthalate (MHET) monomer of significant yield (Güçlü et al., 2003). Another study introduced a simultaneous hydrolysis-amino-glycolysis of PET waste by diethanolamine (DEA) in the presence of xylene as well. Depolymerised products containing carboxyl and hydroxyl end groups were obtained at reaction temperature of 170–220 °C and moderate pressure. It was postulated that the DEA during the glycolysis reaction led to the formation of piperazine and terephtalamide which upon further hydrolysis contributed to the production of carboxylic and hydroxylic groups. Thus, giving rise to carboxyl and hydroxyl end group intermediates (IsılAcar, 2011).
Notwithstanding chemical recycling progress to date, the industrial implementation of these methods remains limited, due to slow reaction rates, bulk quantities of reactants, high volumes of solvents and high energy consumption during the depolymerization processes. Practically, the development of novel energy efficient methods including supercritical technologies and catalytic systems are currently the main focus in overcoming these drawbacks (Bartolome et al., 2014; Genta et al., 2010; Kazarian and Martirosyan, 2002).
Catalysts in depolymerization reactions have shown strong potential in converting PET and other plastics into their building block monomers or into high value intermediate products under mild reaction conditions (Ellis et al., 2021). This has paved the way for the current chemical recycling approaches designed to meet the demands of sustainability. The emergence of green catalysts that can upcycle and facilitate efficient PET depolymerisation are being new development. For example, the vital role of ionic liquids as purpose specific catalysts is vastly explored. Marullo et al. was able to convert PET into BHET via glycolysis reaction in the presence of cholinium-based ionic liquids. Under optimum conditions, 85% conversion of PET was achieved at 150 °C after 6 h (Marullo et al., 2021). Another study by Sun et al. investigated the use of cholinium phosphate as an ionic liquid catalyst for PET glycolysis reaction. At relatively low temperatures (120 °C, 3 h), approximately 100 % conversion of PET and 60.6 % yield of BHET were obtained (Sun et al., 2018).
Deep eutectic solvents (DESs) unlike ionic liquids, are mixtures of different compounds with many applications in organic, analytical, and polymer chemistry fields. Recently, DESs were proposed as green catalysts in plastics chemical recycling (Gómez et al., 2019; Martins et al., 2019). Urea based DESs in particular have shown remarkable potential as catalysts in PET depolymerization reactions. Wang et al.(Wang et al., 2015) investigated the effect of a DES of urea and metal salt for catalysing the glycolysis of PET waste. The results showed fast reaction rates and high conversion (up to 100%) of PET into BHET within 30 min at 170 °C. In a study by Liu et al. 1,3-dimethylurea based DESs was evaluated for the glycolysis depolymerization of PET with the active sites of urea playing a critical role in the conversion of PET into BHET (82%) at 190 °C with a 20 min reaction time (Liu et al., 2019). Sert et al. studied PET glycolysis under catalysed reaction conditions using urea and urea derivative based DESs such as urea/choline chloride and dimethyl urea/choline chloride, increasing the conversion of PET into BHET to 55–60% at 180 °C temperature and 5 h reaction time (Sert et al., 2019b).
Microwave (MW) technology has been investigated as an efficient method for fast PET depolymerization via glycolysis and hydrolysis reactions. The rapid heating achievable via MW while utilising ionic liquids or DESs as model catalysts has allowed significant shortening of reaction times while delivering acceptable monomers conversion yields. This could be attributed to the heating behaviour and MW absorption properties of these solvents when considered for catalysing depolymerisation reactions under MW irradiation. Thus, solvents with strong MW absorbing capabilities are highly desirable (Attallah et al., 2021; González-Rivera et al., 2021, 2020). For instance, Rivera et al. investigated the MW heating response of different choline chloride based DESs and found that the MW absorption was mainly dependant on the intrinsic polarity and ionic behaviour properties of the DESs. A significant rise in MW absorption was observed in all DESs while choline chloride-glycerol based DES showed a maximum MW absorption capability due to the formation of hydrogen bond links and polarity enhancement under MW irradiations (González-Rivera et al., 2020). In our previous work for PET depolymerization using MW assisted DES/hydrolysis technique, it was also found that due to the strong MW absorption of the proposed DES catalyst of choline chloride and m-cresol, a PET conversion of 85% was achieved in 92 sec of MW irradiation (Attallah et al., 2021).
A series of studies of MW assisted glycolysis reactions comprising the use of ionic liquids as catalysts to evaluate the efficiency of PET depolymerization have also been reported. In one study, depolymerised products mainly constituting BHET monomer were obtained at a yield of 64% within 2 h at 175 °C temperature in a glycolysis reaction catalysed by 1-butyl-3-methylimidazolium bromide ionic liquid (Alnaqbi et al., 2015). In another study, PET glycolysis in MW while employing an oxalate-bridged binuclear iron(III) ionic liquid resulted in the conversion of PET into 80% BHET monomer yield within 3 h compared with 24 h under conventional heating (Cot et al., 2019). For MW assisted hydrolysis reactions, Choi et al. performed alkaline hydrolysis reaction in MW using urea/choline chloride DES as catalyst. Enhanced hydrophilicity of PET fabric and minimal hydrolysis products were observed within 1 min reaction time (Choi and Cho, 2016). Another study by Choi et al. implicated the use of glycerol/choline chloride DES to depolymerise PET in blend fabrics in the presence of sodium hydroxide (NaOH) under MW irradiation. A 38 to 61% weight loss of PET were achieved in <100 sec, under different NaOH concentrations (Choi and Choi, 2019). In our previous work, MW assisted DES techniques for depolymerization of PET was also investigated. In one study, MW assisted DES/hydrolysis of PET was performed using DES of choline chloride and m-cresol in the presence of 10% NaOH. An overall 91.55% TPA yield was achieved within 92 sec of MW irradiation (Attallah et al., 2021). In another study, a 3 min, green MW assisted DES of choline chloride, urea and glycerol pre-treatment of PET followed by enzymatic hydrolysis was carried out and a total monomer yield of 16% (w/w) without adding any depolymerising agent was obtained (Attallah et al., 2022).
Moreover, trials at large scale are now being implemented to improve the sustainability of MW assisted depolymerization of PET. Practically, the use of high concentrations of caustic reagents such as NaOH can corrode the reaction vessels which poses a limitation for the MW assisted PET hydrolysis. Thus, the proposition of green PET depolymerizing agents is recommended to decrease the carbon footprint imposed by the current PET depolymerizing techniques.
Sodium carbonate (Na2CO3) is the disodium salt of carbonic acid having strong alkalinizing properties. It is found as a white powder that usually absorbs moisture from the air. Sodium carbonate is commonly used to make glass and as a water softener (Blackshaw, 2012). Recently, Na2CO3 has been proposed as a depolymerizing agent for hydrolysis reaction of PET. In one study, Weerasooriya et al. used Na2CO3 to depolymerize PET under the reaction time of 35 min and 170 °C operation temperature. The obtained yield of TPA was found to be 77.95% (Weerasooriya et al., 2011). López-Fonseca et al. studied the effect of Na2CO3 (0.06% (w/v)) as a catalyst in glycolysis of PET waste under excess EG. Within 1 h and around 196°C temperature, 50% yield of BHET was achieved (López-Fonseca et al., 2010a). Another study by López-Fonseca et al. investigated the glycolysis of PET bottles using excess EG and Na2CO3 (0.45% (w/v)) as a depolymerising catalyst. Under optimised particle size (0.25 mm), 600 rpm stirring rate and 1 h reaction time, Na2CO3 efficiently catalysed PET conversion into BHET monomer and 80 % yield was obtained (López-Fonseca et al., 2011).
Noting the capacity of DESs as green catalysts and good MW absorbers in PET depolymerisation reactions and the proposition of Na2CO3 in EG as potential environmentally friendly PET depolymerizing agent. In this work, we present an ultrafast, green, sequential glycolysis-hydrolysis depolymerization of PET under MW irradiation to obtain value added monomers which can be reused in the production of commercial PET. Glycolysis reaction was carried out in the presence of EG and urea derivatives based DESs as catalysts and the optimization of the process was performed using Box Behnken design (BBD). Na2CO3 in EG was then used as depolymerizing agent in the hydrolysis of residual PET obtained post the optimized glycolysis processes to complete the depolymerization of the original PET sample. Characterization of the residual PET and the produced monomers was performed using DSC, FTIR and HPLC.
2 Experimental
2.1 Materials
PET granules were obtained from Alpek Polyester UK Ltd. (UK), and were converted into micron sized fine powder using centrifugal miller. Ethylene glycol (EG) (99%), sodium carbonate (98%), choline chloride (98%, ChCl), thiourea (98%) and urea (98%) were purchased from Sigma Aldrich (UK). All other chemicals were of analytical grade and readily available to use without any purification.
2.2 Preparation of DESs
Prior to the preparation of DESs, ChCl was dried overnight at 65 °C in the oven. Two DESs of composition based on ChCl:urea and ChCl:thiourea were prepared with (1:2) molar ratio by continuously mixing and heating at 80 °C until homogeneous and transparent liquids were observed.
2.3 Sequential glycolysis-hydrolysis depolymerization of PET
2.3.1 MW Assisted DESs/glycolysis of PET
The experiments were carried out by mixing one gram of powdered PET in varied volumes of ethylene glycol (EG) and the prepared DESs (ChCl:urea as DES 1 and ChCl:thiourea as DES 2) separately while stirring for 10 min. The prepared suspensions were exposed to MW irradiation of 350 W power at specified MW irradiation times in a domestic 1200 W MW oven. After MW treatments, PET residues were collected by filtration, dried in an oven at 70 °C overnight and kept in sealed containers for further analysis by FTIR and DSC. Distilled water was then added to the filtrate and resultant BHET was precipitated by cooling overnight in the fridge at 2 °C. The DES/EG mixtures were regenerated by distillation to be ready for reuse.
2.3.2 Optimization of MW assisted DESs/glycolysis using BBD
Two BBDs (Design Expert, trial version 10.0.5.0, Stat-Ease Inc., Minneapolis, MN) were performed to optimize the proposed MW assisted PET glycolysis using DES 1 and DES 2 as catalysts. Fifteen runs were arranged for the designs to study volume of DES (mL) (X1), volume of EG (mL) (X2), and MW irradiation time (min) (X3) as independent factors. Experiments were run in triplicates and the responses were determined as the carbonyl and crystallinity indices of residual PET and PET weight loss (%) (Table 1). Results were reported as the mean for triplicate measurements. BBD optimization was then used to optimize the studied responses.
level
Independent variables
−1
0
1
Constrains
X1: Volume of DES (mL)
4
5
6
In the range
X2: Volume of EG (mL)
4
5
6
In the range
X3: MW time (min)
0.50
1.25
2.00
In the range
2.3.3 MW Assisted hydrolysis of residual PET obtained post glycolysis
The efficiency of MW assisted hydrolytic depolymerization of PET was evaluated using different PET samples including virgin PET (1.00 gm), residual PET of optimized MW assisted DES 1/glycolysis (0.85 gm) and residual PET of optimized MW assisted DES 2/glycolysis (0.82 gm). The PET samples were separately mixed in 10%(w/v) Na2CO3 in 20 mL EG and stirred for 10 min. The samples suspensions were then MW irradiated at 350 W in the MW oven for 3 min. Dissolved PET was precipitated by addition of distilled water. Finally, the obtained mixture was filtered, and the filtrate containing soluble monomers was analyzed by high performance liquid chromatography (HPLC). The residual PET samples were dried overnight at 70 °C. The depolymerization of PET was calculated using the following equation (Du et al., 2020):
The selectivity of TPA, MHET and BHET was quantified by peak area normalization method from the HPLC chromatograms and the yield of TPA was calculated using the following equation (Du et al., 2020):
Hydrochloric acid (HCl) (34%) of 2 mL was added to the cooled filtrate to precipitate the TPA monomer found as sodium terephthalate. The obtained TPA from each sample was then washed with water, dried overnight at 70 °C and kept in sealed bags for FTIR analysis.
*All the instrumentation and calculations details are provided in supplementary materials as S1.
3 Results and discussion
3.1 Properties of prepared DESs
The selected DESs; ChCl:urea as DES 1 and ChCl:thiourea as DES 2 are emerging as green and sustainable solvents that play a significant role in improving PET degradation reactions as elaborated in previous reports (Liu et al., 2019; Sert et al., 2019b; Wang et al., 2015). It is postulated that the amino and carbonyl groups in urea derivatives can efficiently enhance the catalytic activity of depolymerization reactions by interacting with both the depolymerizing agent and the carbon chain of the polymer to be degraded (IsılAcar, 2011). This is in addition to the known catalytic activity posed by ChCl as quaternary ammonium compound in mild glycolysis reactions of PET (Choi and Choi, 2019; Liu et al., 2019; Sert et al., 2019b; Wang et al., 2015). Physical properties of the prepared DESs are illustrated in Table 2. The pH values of the DESs varied greatly where DES 1 had basic nature of pH 10.55 while DES 2 was acidic with a pH value of 3.02. Based on previous literature it is evident that acidic solvents enhance the dissolution of polymers making them amenable to substantial depolymerisation reactions (Tan et al., 2018). Additionally, by considering the optimum density and viscosity values of the DES that were used in literature for PET depolymerization (Sert et al., 2019a). The proposed DESs showed acceptable ranges of these properties for the catalysis of PET glycolysis reaction. *values were measured at room temperature (25 °C).
DES 1
DES 2
pH
10.55
3.02
Density (Kg/m3)
1.176
1.198
Viscosity (mPa.s)
1571
1862
Furthermore, the FTIR analysis of the prepared DESs (Fig. 1) confirmed the presence of the expected functional groups together with some shifting in the peaks’ frequency which indicated the new characteristics of the formed DESs. As illustrated in Fig. 1 [a] the spectrum of DES 1 showed the characteristic peaks of C—N, C⚌O and N—H functional groups of urea at 1446 cm−1, 1670.44 cm−1 and 3432 cm−1 respectively. The shift of C—N stretching peak in urea from 1084 cm−1 to 1064 cm−1 and the appearance of the finger print region of ChCl in the DES 1 spectra also indicated the successful interaction between urea and ChCl to form the DES. In the FTIR spectrum of DES 2 (Fig. 1[b]), characteristic peaks for asymmetric and symmetric stretching of N—H groups in thiourea were observed at 3364.5 cm-1 and 3254 cm−1. Similarly peaks at 1465 cm−1 and 1097 cm−1 corresponded to C—N asymmetric and stretching vibrations while C⚌S rocking vibrations were observed at 729 nm−1. Formation of new bonds at 3107 cm−1 and 2060 cm−1 and shifting of N—H bending peak from 1610 cm−1 to 1593.3 cm−1 was also observed which depicted the new linkages between N-C-S and ChCl active sites.![FTIR spectra of [a] DES 1 (Choline chloride:urea (1:2)) and [b] DES 2 (Choline chloride:thiourea (1:2)).](/content/184/2022/15/7/img/10.1016_j.arabjc.2022.103903-fig2.png)
FTIR spectra of [a] DES 1 (Choline chloride:urea (1:2)) and [b] DES 2 (Choline chloride:thiourea (1:2)).
3.2 MW Assisted DESs/glycolysis of PET experimental design
Response surface methodology was used to study the models of MW assisted glycolysis of PET using DES 1 and DES 2 as catalysts. Based on the designed plan by BBD for the studied parameters (DES volume, EG volume and MW irradiation time) the experimental runs were carried out. As recorded in Table 3, the studied responses comprising crystallinity and carbonyl indices of residual PET and PET weight loss (%) were determined after each run. X1: volume of DES, X2: volume of EG, X3: MW irradiation time, Y1:,Carbonyl index, Y2: Crystallinity index and Y3: Weight loss (%).
Independent variable
Dependent variable
DES 1
DES 2
Run
X1 (mL)
X2 (mL)
X3 (min)
Y1
Y2
Y3(%)
Y1
Y2
Y3(%)
1
6
5
0.5
3.63
26.83
13.97
3.87
26.00
11.86
2
6
4
1.25
3.43
28.02
11.20
3.84
38.32
9.76
3
5
4
2
3.46
34.96
22.38
3.16
29.22
26.18
4
5
6
0.5
4.59
27.09
19.36
3.85
24.65
15.35
5
6
6
1.25
3.38
33.71
27.50
3.90
42.02
1.22
6
4
4
1.25
3.19
31.09
15.00
3.91
36.23
1.00
7
4
5
0.5
4.15
26.41
16.87
4.65
27.68
20.76
8
4
5
2
3.15
35.27
13.51
3.69
35.38
20.07
9
5
4
0.5
3.48
25.93
18.40
3.72
26.47
16.37
10
5
5
1.25
3.81
28.69
7.41
3.70
34.01
7.41
11
5
5
1.25
3.78
28.77
6.85
3.69
34.20
6.68
12
5
6
2
3.06
36.99
31.10
3.50
40.65
28.71
13
6
5
2
3.23
37.27
32.38
3.90
39.13
31.35
14
5
5
1.25
3.75
29.12
5.45
3.70
34.80
6.62
15
4
6
1.25
3.92
27.62
9.51
4.32
42.02
7.63
Different regression models were tested and it was found that for both MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis techniques, the quadratic model was the best fitting model. The relationship between the carbonyl index (Y1), crystallinity index (Y2) and percentage weight loss of PET (Y3) and the studied parameters; volume of DES (X1), volume of EG (X2), and MW irradiation time (X3) are demonstrated in Table S1 and Table S2 for MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis, respectively.
For carbonyl index (Y1) of MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis treatments, all treated PET residues had higher carbonyl index than that of the untreated PET (2.80). Both volume of DES and MW irradiation time showed a significant negative effect on the carbonyl index values while the volume of EG showed a positive effect. Nevertheless, the interactions of EG volume with both MW irradiation time and volume of DES showed significant negative effects on the carbonyl index of PET residue. Thus, it can be indicated that the increase in both MW irradiation time and DES volume led to some form of non-oxidative PET degradation which was observed through the low values of carbonyl index of residual PET. On the other hand, high levels of EG volume with low levels of MW irradiation time and DES volume caused a significant increase in the carbonyl groups on the surface of PET due to the hydrophilic nature of EG.
For crystallinity index (Y2), the coefficients of the quadratic model equation in both treatments indicated that the increase in almost all the studied factors together with their interactions led to a significant increase in the crystallinity index of residual PET. Such results elaborate that the increase in the studied factors led to an increase in the degradation of PET which initially occurs in PET’s amorphous phase (Beltrán-Sanahuja et al., 2020). Such behavior resulted in an increase in the overall crystallinity of treated samples and an elevation in PET’s crystallinity index than that of virgin PET (31.4%) at high levels of studied factors.
For PET weight loss (Y3), the coefficients of the model equations in both MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis showed that MW irradiation time and its interactions with DES volume and EG volume had positive effects on PET weight loss. Such results indicate that high levels of MW irradiation time allow more interaction between the PET chains and DES and EG leading to a greater degree of PET depolymerization and increase in its weight loss percentage.
As demonstrated in Tables S1 and S2, the adequacy of the proposed model to describe the studied responses for MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis was evaluated and the obtained F-values of the quadratic model were large compared to other models’ values. Hence, the proposed treatments can be modeled effectively. The statistics tests also showed high coefficients of determination for both MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis where the adjusted R2 values for both treatments were higher than 0.99.
Moreover, based on the analysis of variance (ANOVA), the obtained p-values for both MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis indicated that all the studied responses fitted the model well with a probability value (p-value) < 0.05. Additionally, the lack-of-fit test performed on the studied responses showed a highly desirable non-significant lack-of-fit (p˃0.1) with p-values ranging between 0.107 and 0.352 for crystallinity index, 0.497 and 0.518 for carbonyl index and 0.134 and 0.889 for percentage weight loss of PET.
3.3 Response surface analysis
Analysis of surface response was performed to determine the combinatorial effects of the studied variables on the responses resulting from MW assisted glycolysis of PET using DES 1 and DES 2. Contour plots of carbonyl index, crystallinity index and PET weight loss for MW assisted glycolysis using DES 1 and DES 2 are demonstrated in Figs. 2, 3 and 4 respectively. The 3-D surface plots are provided as supplementary material in as Fig. S1, Fig. S2 and Fig. S3.![Contour plots of the effect of the interaction of [a] DES volume (X1) and EG volume (X2), [b] DES volume (X1) and MW time (X3) and [c] EG volume (X2) and MW time (X3) on carbonyl index for MW assisted glycolysis of PET using DES 1 and 2.](/content/184/2022/15/7/img/10.1016_j.arabjc.2022.103903-fig3.png)
Contour plots of the effect of the interaction of [a] DES volume (X1) and EG volume (X2), [b] DES volume (X1) and MW time (X3) and [c] EG volume (X2) and MW time (X3) on carbonyl index for MW assisted glycolysis of PET using DES 1 and 2.
![Contour plots of the effect of the interaction of [a] DES volume (X1) and EG volume (X2), [b] DES volume (X1) and MW time (X3) and [c] EG volume (X2) and MW time (X3) on crystallinity index for MW assisted glycolysis of PET using DES 1 and 2.](/content/184/2022/15/7/img/10.1016_j.arabjc.2022.103903-fig4.png)
Contour plots of the effect of the interaction of [a] DES volume (X1) and EG volume (X2), [b] DES volume (X1) and MW time (X3) and [c] EG volume (X2) and MW time (X3) on crystallinity index for MW assisted glycolysis of PET using DES 1 and 2.
![Contour plots of the effect of the interaction of [a] DES volume (X1) and EG volume (X2), [b] DES volume (X1) and MW time (X3) and [c] EG volume (X2) and MW time (X3) on weight loss of PET (%) for MW assisted glycolysis of PET using DES 1 and 2.](/content/184/2022/15/7/img/10.1016_j.arabjc.2022.103903-fig5.png)
Contour plots of the effect of the interaction of [a] DES volume (X1) and EG volume (X2), [b] DES volume (X1) and MW time (X3) and [c] EG volume (X2) and MW time (X3) on weight loss of PET (%) for MW assisted glycolysis of PET using DES 1 and 2.
Fig. 2 shows the carbonyl index (Y1) dependence on the studied variables; volume of DES, volume of EG and MW irradiation time. Generally, carbonyl index is one of the important responses used to evaluate the extent of surface oxidation of the treated polymers. It can be observed that the increase in both volume of DES and MW irradiation time did not cause a significant increase in the carbonyl index of treated PET samples. Thus, it can be assumed that high levels of DES volume and MW irradiation time result in PET degradation rather than surface oxidation giving rise to slight increase in carbonyl index values. On the other hand, the interactions of high levels of EG volume with low levels of both DES volume and MW irradiation time showed a significant increase of carbonyl index confirming PET surface oxidation at these levels of studied factors.
Contour plots for crystallinity index (Y2) are demonstrated in Fig. 3. Noticeably, high level of MW irradiation time in combination with different levels of DES and EG volumes caused an increase in the crystallinity index of PET residue. Such result indicates that MW irradiation time has the upper hand in affecting the crystallinity of PET residue where increasing the reaction time led to the initial degradation of the amorphous phase of PET causing an increase in the overall crystallinity of the polymer (Cho et al., 2016). A significant increase in the crystallinity index was also observed at high levels of DES and EG volumes combinations especially in MW assisted glycolysis using DES 2. Hence, performing the MW assisted glycolysis of PET using the proposed DESs at high levels of all studied factors results in more effective initial degradation of PET and increased crystallinity of PET residues reaching 40%.
PET weight loss is also considered one of the critical responses to assess initial degradation of PET. The higher the value of weight lost the greater the degree of initial degradation of the treated PET samples. As elaborated in Table S1 and S2, MW irradiation time and its interactions influenced PET weight loss significantly (term P-value < 0.05) which indicates that the reaction time in the MW has the upper hand in PET initial degradation. Such conclusion was also supported by the surface response results of MW irradiation time interactions with both DES and EG volumes. As shown in Fig. 4, in the case of MW assisted glycolysis using DES 1, an increased PET weight loss was observed at the interactions of high levels of MW irradiation time and DES and EG volumes. Alternatively, in MW assisted glycolysis using DES 2, significant PET weight loss was observed at the interactions of high levels of MW irradiation times and all the levels of DES and EG volumes indicating the significance of MW irradiation time in the degradation process. These results come in accordance with those of crystallinity index indicating that the weight loss and crystallinity of PET are directly proportionally influenced by the studied independent variables.
3.4 Optimization of MW assisted DES/glycolysis of PET
BBD optimization was performed on the studied responses and optimum conditions were chosen to obtain maximum initial PET degradation and residual PET characteristics that allow enhanced depolymerization in the MW assisted hydrolysis step. As previously discussed in the results, maximum initial PET degradation was observed with increased PET weight loss. Additionally, enhanced PET hydrolytic depolymerization can be achieved through low crystallinity and high carbonyl index of residual PET. Thus, the conditions of the MW assisted DES/glycolysis were adjusted to attain minimum crystallinity index and maximum carbonyl index of residual PET and maximum PET percentage weight loss. Two batch experiments were carried out at the optimized conditions for the MW assisted DES/glycolysis using DES 1 and DES 2. Typically, one gram of powdered PET was mixed with 5.5–6 mL EG and 4 mL of the prepared DES while stirring for 10 min. The prepared suspensions were exposed to MW irradiation of 350 W power for 0.5 min. After MW treatments, PET residues were collected by filtration and dried. Excess distilled water was added to the filtrate and resultant BHET was precipitated by cooling overnight. As illustrated in Table 4, the studied responses values recorded a fine agreement between the observed and predicted results. Hence, the validity of the model was confirmed and BBD proved its success in optimizing the proposed MW assisted DES/glycolysis technique for PET initial degradation. Precipitated BHET showed a percentage yield of 9.33 to 11.6% which confirmed the presence of some initial degradation of PET during the 0.5 min MW irradiation. Noticeably, MW assisted PET glycolysis using DES 2 as catalyst showed better results with respect to the studied responses than that of DES 1. This could be attributed to the enhanced catalytic activity of DES 2 where thiourea provided better interaction with EG and PET polymer. Such efficient catalytic activity gave rise to improved PET depolymerization which was manifested in the high PET weight loss percentage and increased crystallinity and carbonyl index of residual PET.
Independent Variable
Optimized level
DES 1
DES 2
X1: Volume of DES (mL)
4
4
X2: Volume of EG (mL)
6
5.5
X3: MW time (min)
0.5
0.5
Over all desirability
0.788
0.817
Dependent variables
Desirability
DES 1
DES 2
Expected
Observed
Expected
Observed
Y1: PET carbonyl index
Maximize
4.8
4.2
4.65
4.52
Y2: PET crystallinity index
Minimize
25.07
28.65
27.95
29.32
Y3: PET weight loss (%)
Maximize
18.61
16.75
21.45
18.66
Yield of BHET (%)
–
–
9.33
–
11.65
3.5 High PET conversion via MW assisted hydrolysis technique
Following the successful optimization of MW assisted DES/glycolysis conditions, virgin PET and residual PET obtained post glycolysis treatments underwent depolymerization via MW assisted hydrolysis using Na2CO3 in EG. The residual PET obtained was weighed and conversion of PET (%) was calculated. The reaction products dissolved in the cooled filtrate were analyzed by HPLC as shown in Fig. S4. The effects of different PET treatments on the conversion of PET, the selectivity of BHET, MHET and the yield of TPA are illustrated in Fig. 5. It was observed that the conversion of PET was only 66.80% when applying hydrolysis reaction on untreated virgin PET, and then rapidly increased to 98.62 and 99.31% when hydrolysing the treated residues of MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis respectively. Such significant increase in depolymerization efficiency with the MW assisted DES/glycolysis treated PET could be attributed to the new characteristics that the residual PET gained with respect to crystallinity, hydrophilicity and weight loss upon treatment with MW assisted DES/glycolysis. These modifications in the PET residues increased the efficiency of their hydrolysis using Na2CO3 at only 3.0 min MW irradiation time.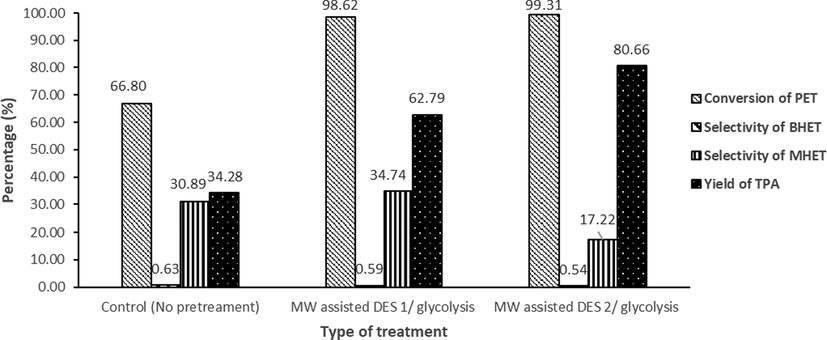
The effect of type of treatment on the conversion of PET and the selectivity of BHET, MHET and the yield of TPA.
Moreover, the selectivity of MHET was much higher than that of BHET for all the treated samples as shown in Fig. 5. It was also observed that the selectivity of BHET and MHET dramatically decreased while hydrolysing the PET residues obtained post glycolysis. Simultaneously, the yield of TPA increased from 34.28% for virgin PET sample to 63.67% and 81.23% for PET residues of MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis, respectively. Noticeably, the obtained results indicated that the formation of MHET was preferential to BHET and that the increased MW irradiation time and initial glycolysis step was beneficial to the yield of TPA.
It is also worth mentioning that the extent of hydrolytic depolymerization of PET residue obtained post MW assisted glycolysis using DES 2 into TPA was slightly higher than that using DES 1. Such finding confirmed that DES 2 as a catalyst resulted in a PET residue more amenable for efficient hydrolytic depolymerization using the weakly alkaline Na2CO3 in EG.
The identity of the TPA white powder monomer precipitated post the MW assisted hydrolysis process was confirmed using FTIR analysis. Fig. 6 demonstrates the FTIR spectra of precipitated TPA from all treated PET samples which showed great similarity to that of TPA standard. Carboxylic acid—OH was indicated by the peaks at 3064 cm−1, acidic carbonyl group was indicated by 1673 cm−1 peak while the peak at 1280 cm−1 indicated the presence of ether C—O stretching (Musale and Shukla, 2016).![FTIR spectra of: [a] TPA standard, [b] TPA obtained from PET depolymerization via hydrolysis only, [c] TPA obtained from PET depolymerization via hydrolysis post MW assisted DES 1/glycolysis and [d] TPA obtained from PET depolymerization via hydrolysis post MW assisted DES 2/glycolysis.](/content/184/2022/15/7/img/10.1016_j.arabjc.2022.103903-fig7.png)
FTIR spectra of: [a] TPA standard, [b] TPA obtained from PET depolymerization via hydrolysis only, [c] TPA obtained from PET depolymerization via hydrolysis post MW assisted DES 1/glycolysis and [d] TPA obtained from PET depolymerization via hydrolysis post MW assisted DES 2/glycolysis.
3.6 Mechanism of the sequential glycolysis-hydrolysis of PET under MW irradiation
The sequential glycolysis-hydrolysis reactions and catalytic activity of DESs for PET depolymerization is explored in detail. A possible catalytic mechanism of the glycolysis of PET using DES 1 and 2 is presented in Fig. 7. The H-bond actions between EG and the chloride of choline chloride and between EG and the urea (C⚌O) and thiourea (C⚌S) of DES 1 and 2 catalysts is expected to change the charge density of the hydroxyl (OH) group in EG and increase the electronegativity of the oxygen atom in OH group of EG. Hence, the nucleophilicity of the oxygen becomes stronger supporting preferential attack to the carbon of the ester group in PET (Wang et al., 2012). Under MW irradiation, PET treated with DES/EG initially changes to a more amorphous material, but with longer MW irradiation time, cleavage of the ester C—O bond and the formation of a new C—O bond with EG occur (Attallah et al., 2021; Wang et al., 2012). It is also worth mentioning that the enhanced catalytic activity of DES 2 as compared to DES 1 could be related to unique characteristic of lower C—S bond energy in thiourea which allowed the DES 2 to be more interactive with EG, thus, progressing enhanced PET chain cleavages (Ohno A., 1977). Furthermore, difference in the electrochemical properties of DES 1 and DES 2 could also have a major impact on their catalytic performances. The higher conductivity offered by thiourea in DES 2 when compared to that of urea in DES 1 could have affected the heating rate of materials under MW irradiation and facilitated faster PET depolymerization (Boghosian and Howson, 1990; Hoppe et al., 2019).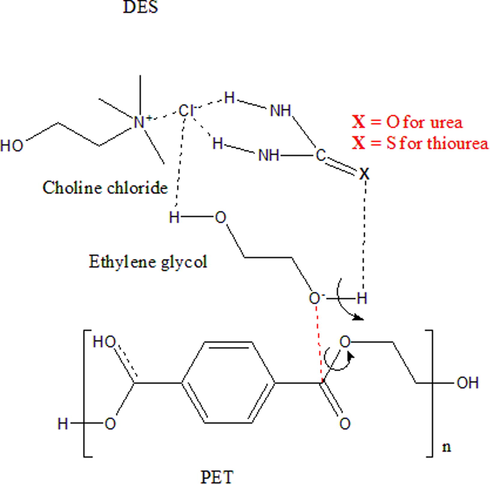
Schematic diagram for the interactions of DESs with EG and PET.
Based on previous reports for hybrid glycolysis/hydrolysis reactions (Güçlü et al., 2003; López-Fonseca et al., 2011, 2010b; Zanela et al., 2018), it is postulated that PET is converted to oligomers, and the oligomers are then converted to BHET and MHET and, finally, to TPA and EG. Fig. 8 shows the proposed scheme for PET conversion to oligomers and ester hydrolysis of BHET producing TPA (a two-carboxylated compound) or MHET (a one-carboxylated compound). EG is formed in both cases. Technically, the water content causing BHET hydrolysis can be either produced during the reaction of PET with Na2CO3 in EG as previously reported by (Yoon et al., 1993) and (Güçlü et al., 2003) or could be due to moisture content from air.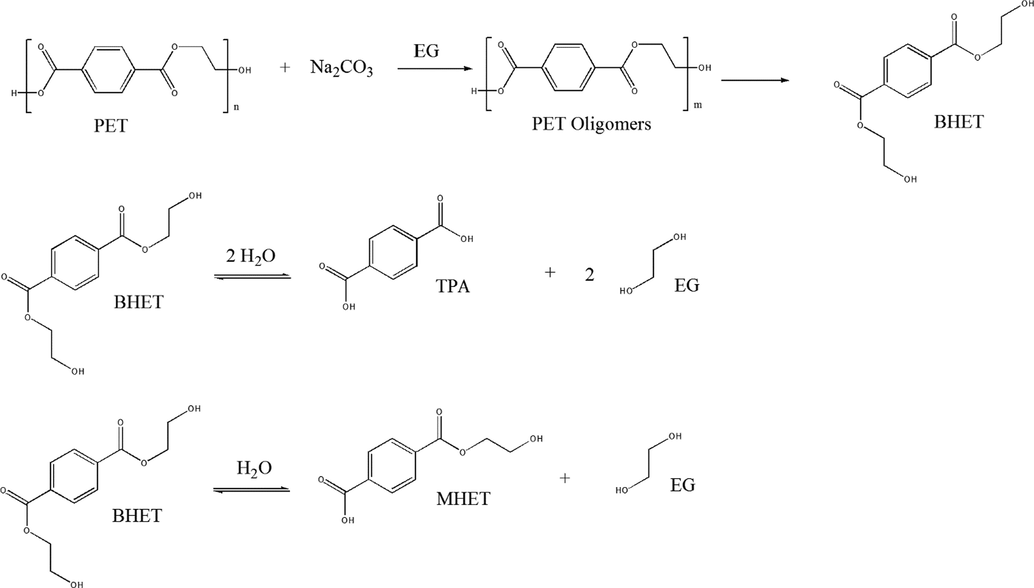
Suggestion mechanism for production of compounds during depolymerization of PET.
3.7 Application on postconsumer PET bottles
The efficiency of the proposed hybrid PET depolymerization technique involving MW assisted glycolysis followed by MW assisted hydrolysis was evaluated on post-consumer PET sample provided by SHABRA Plastics and Recycling ltd. (Ireland). The MW assisted DES/glycolysis was performed using the optimized conditions obtained from BBD (1 gm post-consumer PET, 4 mL DES, 6 mL EG and 0.5 min MW irradiation time). Residual PET samples obtained post glycolysis reaction were then mixed in 20 mL EG/10% (w/v) Na2CO3 and treated with MW irradiation for 3.0 min. Insoluble matter contents were separated, washed, dried and weighed. Percentage of PET conversion was calculated to be 96.77% and 98.25% for MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis, respectively. The yield of TPA obtained after precipitation with HCl was determined by HPLC and estimated to be 60.98% and 78.10% for PET residues of MW assisted DES 1/glycolysis and MW assisted DES 2/glycolysis, respectively.
Based on the obtained results, it is postulated that the proposed hybrid depolymerization technique can achieve acceptable PET conversion percentages despite the presence of other additives or polymers in the post-consumer PET sample.
3.8 Evaluation of performance
Practically speaking, for chemical recycling of plastics, it is noted that high ratios of solvents, harsh chemicals and longer reaction times significantly restrict its sustainable large-scale applications. Based on previous reports, it was found that a wide range of solvents to PET ratios of 4:1 to 50:1 are being used. Caustic chemicals such as NaOH, KOH, HCl and H2SO4 have also been selected as depolymerizing agents for hydrolytic depolymerization of PET. Reaction times ranging between 2 min and 8 h were set to complete the depolymerization reactions under conventional heating or MW irradiation while obtaining a monomer yield of 64% to 88%. Table 5 illustrates the reaction conditions and monomer yields of already established PET depolymerisation techniques using DESs as catalysts. Noticeably, the DESs proposed in the current study have proved themselves as efficient MW absorbers and PET depolymerization reaction catalysts owing to their unique properties. In the current work, the reaction time required for complete depolymerization of PET was only 3.5 min which is considerably short when compared to other studies presenting green approaches for PET degradation. Furthermore, the significant PET conversion (99%) and monomer yield reaching up to 80.66% can render the proposed technique ideal for PET depolymerisation and probably other polyesters from an industrial prospect. * Hydrogen bond donor (HBD); hydrogen bond acceptor (HBA); (2-hydroxyethyl)-terephthalamide (THETA); Terephthalic acid: (TPA); bis (2-hydroxy ethylene) terephthalamide: (BHETA); bis(hydroxyethyl) terephthalate (BHET); sodium hydroxide (NaOH).
DES composition
Reaction conditions
PET conversion(%)
Monomer yield (%)
Reference
HBA
HBD
Ratio (HBA:HBD)
Reaction time
Depolymerising agent
Temperature
1,3-dimethylurea
Zn(OAc)2
4:1
20 min
ethylene glycol
190 °C
100%
BHET (82)
(Liu et al., 2019)
Choline chloride
Glycerol
1:2
1.3–1.6 min
Glycerol, NaOH
–
38–61%
(Choi and Choi, 2019)
Choline chloride
Zinc chloride
1:1–1:3
30 min
Diethanolamine, ethanolamine
–
–
THETA (82), TPA (83), BHETA (95)
(Musale and Shukla, 2016)
Choline chloride
m-cresol
1:2
1.5 min
NaOH
–
84%
TPA (91.55)
(Attallah et al., 2021)
Potassium carbonate
Ethylene glycol,
1:6
2 h
ethylene glycol
180 °C
-
BHET (88)
(Güçlü et al., 2003)
Choline chloride
Urea, Glycerol
1:1:1
3 min
–
–
1.8% weight loss
TPA, MHET, BHET 16 (w/w)
(Attallah et al., 2022)
Zinc chloride, zinc acetate, manganese acetate
Urea
1:12 to 1:6
30 min
ethylene glycol
170 °C
100%
bis(hydroxyalkyl) terephthalate (83)
(Wang et al., 2015)
1-methyl-3-butylimidazolium chloride
Zinc chloride, Manganese chloride
1:1
5.0 h
ethylene glycol
190 °C
–
BHET (83.8)
(Yue et al., 2013)
Choline chloride
Ethylene glycol
1:2
2 min
ethylene glycol,NaOH
144 °C
45.8% weight loss of PET
–
(Cho et al., 2016)
Choline chloride
Urea,
1:2
3.5 min
sodium carbonate, ethylene glycol
–
98.62%
TPA (62.79), MHET (34.79), BHET (0.59)
This work
Choline chloride
Thiourea
1:2
3.5 min
sodium carbonate, ethylene glycol
–
99.31%
TPA (80.66), MHET (17.22), BHET (0.54)
This work
4 Conclusion
A hybrid depolymerization process for PET comprising a fast, low energy MW assisted glycolysis-hydrolysis was presented. In comparison to reported MW assisted PET depolymerization approaches, the proposed hybrid technique achieved improved PET depolymerization with a total of ≈99% PET conversion under environmentally friendly operating conditions in only 3.5 min. The catalytic activities of urea derivatives based DESs for MW assisted glycolysis of PET were compared. DES 2 showed better results as catalyst with respect to the studied responses than that of DES 1 which was related to the enhanced interaction of thiourea of DES 2 with EG and PET polymer chains. The MW assisted hydrolysis of PET residue obtained post glycolysis reaction was then performed and a total of 99% weight loss of glycolytically treated PET in comparison to 67% weight loss of virgin PET undergoing only hydrolysis reaction was demonstrated. HPLC analysis of the reaction mixture filtrates obtained post hydrolytic depolymerization showed the presence of TPA, MHET and traces of BHET as the produced monomers. Application on Post-consumer PET sample also revealed very satisfactory results with 96.77–98.25% PET conversion rates and 60.98–78.10% yield of TPA.
Furthermore, taking into consideration the industrial scale depolymerization of PET with factors like plant installation and maintenance costs, skilled labour requirements and safety, the route demonstrated by the proposed technique can present an attractive economic and environmentally sustainable technology for PET and other polyester recycling and circularity.
Acknowledgements
This project received funding from the European Union’s Horizon 2020 research, the Irish Research Council (GOIPG/2021/1739) and innovation programme under grant agreement No. 870292 (BIOICEP) and was supported by the National Natural Science Foundation of China (grant numbers: Institute of Microbiology, Chinese Academy of Sciences: 31961133016; Beijing Institute of Technology: 31961133015; Shandong University: 31961133014)
Data Availability Statement
All data generated or analysed during this study are included in the article (and its Supplementary Information Files).
Author contribution
M.A. practical experimenting, writing original draft, Investigation, Data curation, funding acquisition, visualization; M.B.F. Supervision, Validation, writing original draft, resources, funding acquisition, visualization, project administration; O.A. Conceptualization, Data curation, Investigation, Writing and reviewing original draft, formal analysis, visualization;
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Microwave assisted glycolysis of poly(ethylene terephthalate) catalyzed by 1-butyl-3-methylimidazolium bromide ionic liquid. J. Appl. Polym. Sci.. 2015;132:1-7.
- [CrossRef] [Google Scholar]
- Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment. Polym. 2022
- [CrossRef] [Google Scholar]
- Fast, High Monomer Yield from Post-consumer Polyethylene Terephthalate via Combined Microwave and Deep Eutectic Solvent Hydrolytic Depolymerization. ACS Sustain. Chem. Eng.. 2021;9:17174-17185.
- [CrossRef] [Google Scholar]
- Superparamagnetic γ-Fe2O3 nanoparticles as an easily recoverable catalyst for the chemical recycling of PET. Green Chem.. 2014;16:279-286.
- [CrossRef] [Google Scholar]
- Monitoring polymer degradation under different conditions in the marine environment. Environ. Pollut.. 2020;259
- [CrossRef] [Google Scholar]
- Temperature dependence of the electrical conductivity of amorphous VxSi1-x. Phys. Rev. B. 1990;41:7397-7401.
- [CrossRef] [Google Scholar]
- Conversion of waste bottles’ PET to a hydrogel adsorbent via PET aminolysis. J. Environ. Chem. Eng.. 2021;9
- [CrossRef] [Google Scholar]
- Rapid hydrophilic modification of poly(ethylene terephthalate) surface by using deep eutectic solvent and microwave irradiation. Text. Res. J.. 2016;86:1318-1327.
- [CrossRef] [Google Scholar]
- Microwave-mediated rapid tailoring of PET fabric surface by using environmentally-benign, biodegradable Urea-Choline chloride Deep eutectic solvent. Fibers Polym.. 2016;17:847-856.
- [CrossRef] [Google Scholar]
- Eco-friendly, Expeditious Depolymerization of PET in the Blend Fabrics by Using a Bio-based Deep Eutectic Solvent under Microwave Irradiation for Composition Identification. Fibers Polym.. 2019;20:752-759.
- [CrossRef] [Google Scholar]
- An Oxalate-Bridged Binuclear Iron(III) Ionic Liquid for the Highly Efficient Glycolysis of Polyethylene Terephthalate under Microwave Irradiation. Chempluschem. 2019;84:786-793.
- [CrossRef] [Google Scholar]
- Doerr, M.L., 1986. Depolymerization of Condensation Polymers Involving a Pre-Molecular Weight Reduction Step.
- ZnO nanodispersion as pseudohomogeneous catalyst for alcoholysis of polyethylene terephthalate. Chem. Eng. Sci.. 2020;220:115642.
- [CrossRef] [Google Scholar]
- Chemical and biological catalysis for plastics recycling and upcycling. Nat. Catal.. 2021;4:539-556.
- [CrossRef] [Google Scholar]
- Heterogeneous continuous kinetics modeling of PET depolymerization in supercritical methanol. J. Supercrit. Fluids. 2010;52:266-275.
- [CrossRef] [Google Scholar]
- Use of Natural Deep Eutectic Solvents for Polymerization and Polymer Reactions. J. Braz. Chem. Soc.. 2019;30:717-726.
- [CrossRef] [Google Scholar]
- Insights into microwave heating response and thermal decomposition behavior of deep eutectic solvents. J. Mol. Liq.. 2020;300
- [CrossRef] [Google Scholar]
- Combining acid-based deep eutectic solvents and microwave irradiation for improved chestnut shell waste valorization. Green Chem.. 2021;23:10101-10115.
- [CrossRef] [Google Scholar]
- Simultaneous glycolysis and hydrolysis of polyethylene terephthalate and characterization of products by differential scanning calorimetry. Polymer (Guildf).. 2003;44:7609-7616.
- [CrossRef] [Google Scholar]
- Deep eutectic solvents based on choline cation - Physicochemical properties and influence on enzymatic reaction with β-galactosidase. Int. J. Biol. Macromol.. 2019;136:296-304.
- [CrossRef] [Google Scholar]
- Aminoglycolysis of Waste Poly(ethylene terephthalate) With Diethanolamine and Evaluation of the Products as Polyurethane Surface Coating Materials. Polym. Eng. Sci. 2011:746-754.
- [CrossRef] [Google Scholar]
- ATR-IR spectroscopy of superheated water and in situ study of the hydrothermal decomposition of poly(ethylene terephthalate) Phys. Chem. Chem. Phys.. 2002;4:3759-3763.
- [CrossRef] [Google Scholar]
- Lewis Acid-Base Synergistic Catalysis for Polyethylene Terephthalate Degradation by 1,3-Dimethylurea/Zn(OAc)2 Deep Eutectic Solvent. ACS Sustain. Chem. Eng.. 2019;7:3292-3300.
- [CrossRef] [Google Scholar]
- Chemical recycling of post-consumer PET wastes by glycolysis in the presence of metal salts. Polym. Degrad. Stab.. 2010;95:1022-1028.
- [CrossRef] [Google Scholar]
- Kinetics of catalytic glycolysis of PET wastes with sodium carbonate. Chem. Eng. J.. 2011;168:312-320.
- [CrossRef] [Google Scholar]
- Insights into the Nature of Eutectic and Deep Eutectic Mixtures. J. Solution Chem.. 2019;48:962-982.
- [CrossRef] [Google Scholar]
- Amino Acid-Based Cholinium Ionic Liquids as Sustainable Catalysts for PET Depolymerization. ACS Sustain. Chem. Eng.. 2021;9:15157-15165.
- [CrossRef] [Google Scholar]
- Deep eutectic solvent as effective catalyst for aminolysis of polyethylene terephthalate (PET) waste. Int. J. Plast. Technol.. 2016;20:106-120.
- [CrossRef] [Google Scholar]
- Ohno, A., 1977. Thiones. In: Oae S. (Eds.), Organic Chemistry of Sulfur. Springer, Boston, MA. https://doi.org/10.1007/978-1-4684-2049-4_5, n.d..
- Padhan, R.K., Sreeram, A., 2019. Chemical Depolymerization of PET Bottles via Combined Chemolysis Methods, Recycling of Polyethylene Terephthalate Bottles. Elsevier Inc. https://doi.org/10.1016/b978-0-12-811361-5.00007-9.
- Chemical Recycling of Polyethlylene Terephthalate by Glycolysis Using Deep Eutectic Solvents. J. Polym. Environ.. 2019;27:1-7.
- [CrossRef] [Google Scholar]
- Chemical Recycling of Polyethlylene Terephthalate by Glycolysis Using Deep Eutectic Solvents. J. Polym. Environ.. 2019;27:2956-2962.
- [CrossRef] [Google Scholar]
- High loading solubilization and upgrading of poly (ethylene terephthalate) in low cost bifunctional ionic liquid. ChemSusChem. 2018;11:781-792.
- [Google Scholar]
- Evaluation of fractionation and delignification efficiencies of deep eutectic solvents on oil palm empty fruit bunch. Ind. Crops Prod.. 2018;123:271-277.
- [CrossRef] [Google Scholar]
- Deep eutectic solvents as highly active catalysts for the fast and mild glycolysis of poly(ethylene terephthalate)(PET) Green Chem.. 2015;17:2473-2479.
- [CrossRef] [Google Scholar]
- Urea as an efficient and reusable catalyst for the glycolysis of poly(ethylene terephthalate) wastes and the role of hydrogen bond in this process. Green Chem.. 2012;14:2559-2566.
- [CrossRef] [Google Scholar]
- Weerasooriya, P.R.D., Karunanayake, U.P.A.B.P., Udayakumara, S. V, Gunapala, O., 2011. Use of soda ash as a recycling agent to reduce Non-degradable PET Waste in Sri Lanka.
- West, Simon, M., 1993. Improved Polyethlene terephthalate Decontamination. world Intellect. Prop. Organ. https://doi.org/93/23465.
- Diffusion of ethylene glycol in solid state poly(ethylene terephthalate) Polym. J.. 1993;25:227-236.
- [CrossRef] [Google Scholar]
- The glycolysis of poly(ethylene terephthalate) waste: Lewis acidic ionic liquids as high efficient catalysts. Polymers (Basel).. 2013;5:1258-1271.
- [CrossRef] [Google Scholar]
- Chemical recycling of poly(Ethylene terephthalate) (PET) by alkaline hydrolysis and catalyzed glycolysis. Orbital. 2018;10:226-233.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary Information (Instrumentation, HPLC chromatograms and 3D surface plots of response surface analysis of BBD). Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103903.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




