

Translate this page into:
Design, synthesis and biological evaluation of novel quinazoline derivatives as potential NF-κb inhibitors
⁎Corresponding authors. kevinzhlj@163.com (Long-Jiang Zhang), rggaofeng@hotmail.com (Feng Gao), cjr.luguangming@vip.163.com (Guang-Ming Lu)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A series of novel 4-aminoquinazoline derivatives were designed, synthesized and biological properties on nuclear factor-kappaB (NF-κb) pathway inhibitory and potential in vitro anti-proliferation against breast cancer lines were also evaluated. Among them, LU1501 exhibited potent inhibition with IC50 values in SK-BR-3 (10.16 ± 0.86 µM) and HCC1806 (10.66 ± 1.01 µM) cell lines. In vivo studies in breast cancer tumor model proved the correlation between anticancer activity of LU1501 and proliferation inhibition through the NF-κb signal pathway. The molecular docking studies also portrayed the potential binding mechanism between LU1501 and the key proteins of p65 and IkBα in NF-κb pathway. Accordingly, compound LU1501 could serve as a potent agent against breast cancer for further investigation.
Keywords
4-aminoquinazoline
Molecular docking
Anticancer activity
NF-κb
Breast cancer

1 Introduction
Tyrosine Kinase (TK) is a tyrosine-specific protein kinase and its receptor like epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), insulin-like growth factor receptor-1(IGFR-1) and hepatic growth factor receptor (HGFR) (Yin et al., 2019; Sun et al., 2020) could bind to a ligand and phosphorylates tyrosine residues. The EGFR TK family includes four receptor tyrosine kinases, namely, EGFR (HER1), ERBB-2 (HER2), ERBB-3 (HER3), and ERBB-4 (HER4). EGFR, a transmembrane protein, is the first tumor-associated cell surface receptor (Sassen et al., 2008; Jiang et al., 2021). Tyrosine kinase plays an important role in the occurrence and development of cancer and also represents an important target for cancer treatment (Fang et al., 2020; Cheng et al., 2021; Jiang et al., 2019). Dozens of drugs targeting TK have been designed and approved for clinical treatment of cancer. For example, erlotinib, which could selectively target EGFR, has been applied to treat non-small-cell lung carcinoma (non-small cell lung cancer) in clinics; and lapatinib, the HER2 targeted drug, is highly effective against breast cancer (Roskoski, 2019; Wu et al., 2021; Ding et al., 2018).
Small molecular tyrosine-kinase inhibitors (TKI) can be divided into quinazoline, quinoline, pyrimidine, pyrimidine, and indole (Han et al., 2021; Sayed et al., 2018; Faisal and Saeed, 2021). Quinazolines are a class of compounds with a wide range of biological activities, such as anti-cancer, anti-bacterial, anti-inflammatory, anti-malaria, anti-hypertension and other effects (Abuelizz et al., 2017; Yang et al., 2014; Krapf and Wiese, 2016). Quinazoline derivatives could inhibit EGFR or EGF receptor tyrosine kinase in Pro-epidermal growth factor cells, resulting in anti-cancer activity, and therefore they could be used to fight prostate cancer, lung cancer, gastric cancer and bile cancer. At present, over 40 quinazoline inhibitors, such as gefitinib, erlotinib, afatinib, lapatinib, icotinib, vandetanib and so on, have been applied in clinics (Alagarsamy et al., 2018; Wdowiak et al., 2021; Ashmawy et al., 2020).
Quinazoline inhibitors have been shown to possess several attractive pharmacological activities. Para-aryl modification of the 4-aminoquinazoline ring account for the vast majority of anticancer activities. Based on the above analysis and the structures of newly developed clinical drugs (Fig. 1), we thought modification of para-aryl of the 4-aminoquinazoline ring was another clue for designing new derivatives.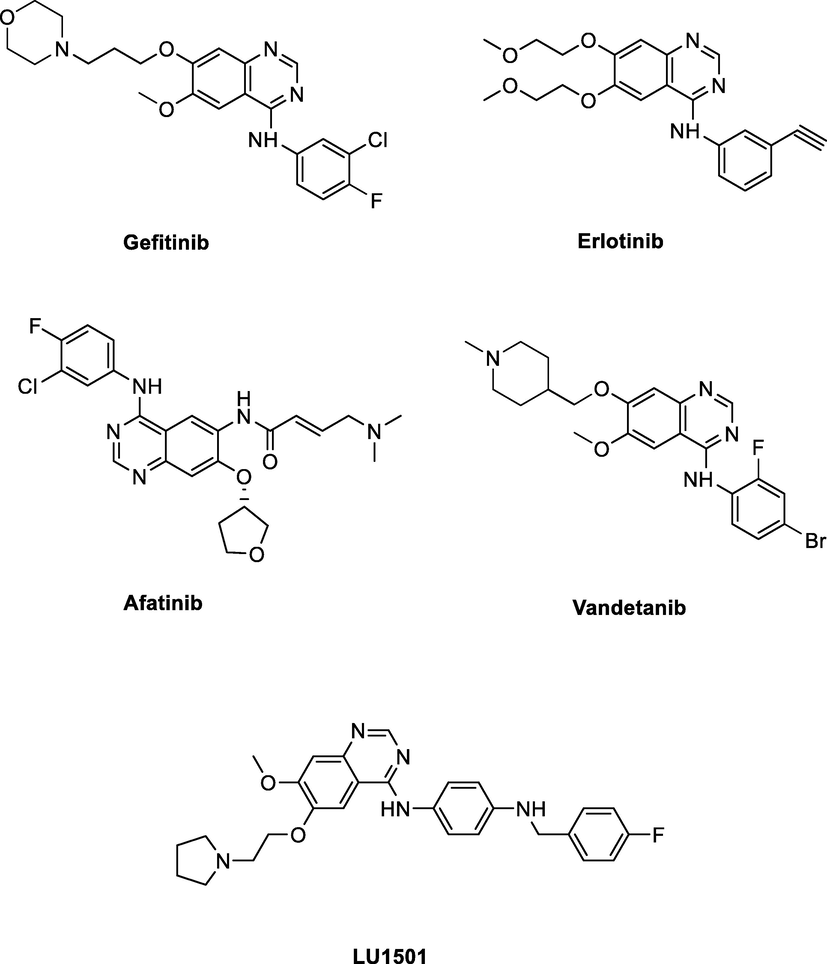
Chemical structures of representative quinazoline inhibitors and the target compound LU1501.
Here, we reported a series of novel quinazoline derivatives, which can effectively inhibit the growth and proliferation of breast cancer cells by suppressing the activation of NF-κb signaling pathway, especially HER-2 and EGFR-positive cell lines such as SK-BR-3.
2 Results
2.1 Chemistry
The preparation routes of the target compounds were shown in Scheme 1 and Scheme 2. The preparation yield and purity of the target compounds were shown in Table 1. Compound 1 was obtained successfully by adding 7-methoxy-4-oxo-3,4-dihydroquinazolin-6-yl acetate, sulfurooyl dichloride into N, N-dimethylformamide dropwise in an atmosphere of nitrogen, Then compound 1 reacted with NH3 in methanol, leading to compound 2. Later, conjugating compound 2 with 2-(pyrrolidin-1-yl)ethan-1-ol in tetrahydrofuran under DTAD and PPh3 gave compound 3. Finally, target compounds 5a–5e were obtained by the reaction of compounds 3 and 4a–4e in the presence of 4-methylbenzene-1-sulfonic acid, and propan-2-ol (Liang et al., 2021; Gao et al., 2019).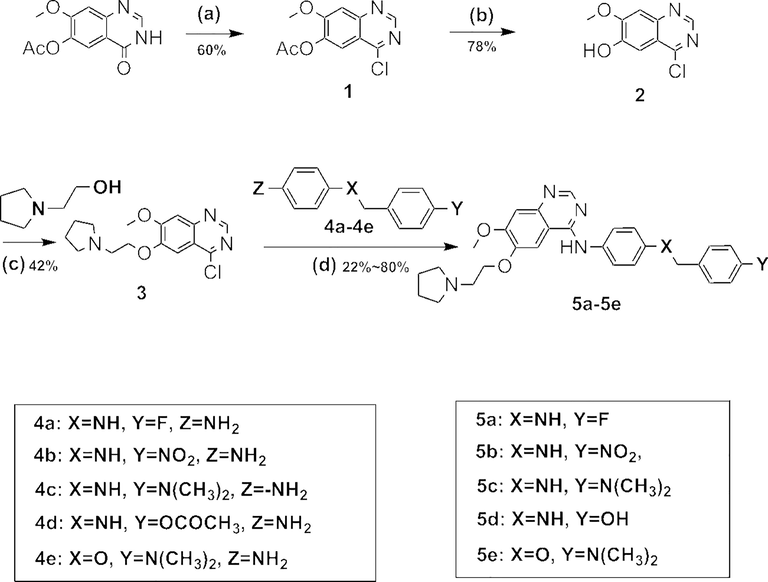
Synthetic procedure for compounds 5a–5e.
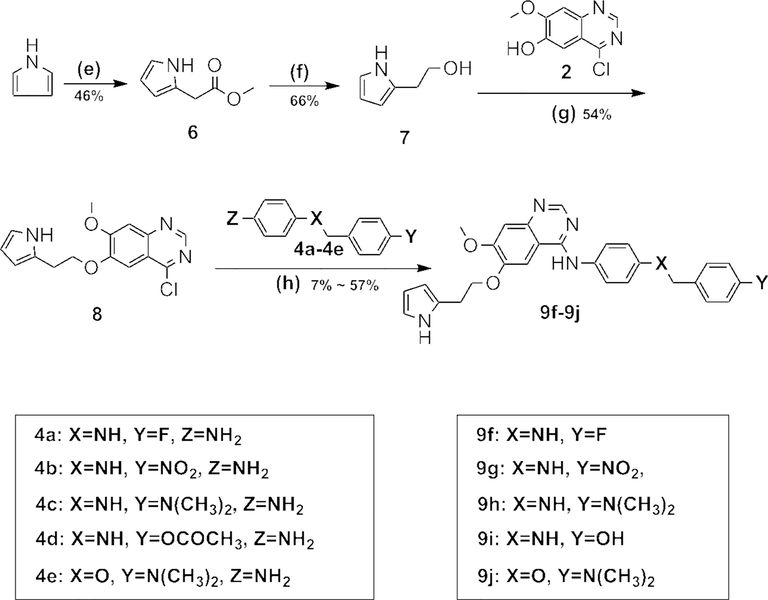
Synthetic procedure for compounds 9f–9j.
Compound
Yield (%)
Purity (%)
LU1501
41
98.0
5b
26
99.3
5c
54
98.1
5d
80
98.0
5e
22
95.2
9f
33
96.6
9g
26
99.9
9h
36
91.9
9i
57
99.2
9j
7
92.1
In order to get the target compounds 9f–9j, compound 6 was firstly synthesized from 1H-pyrrole and methyl 2-bromoacetate. Afterwards, compound 6 was reduced by LiAlH4 to afford compound 7. Compound 8 was achieved via conjugation of compound 2 and 7, and finally compounds 8 reacted with materials 4a–4e under trifluoroacetic acid, yielding the target compounds 9f–9j.
Reagents and conditions: (a) SOCl2,DMF, 3h; (b) NH3/MeOH, 10 °C; (c) PPh3, DTAD, THF; (d) n-BuOH, TFA, 75 °C; (e) EtMgBr, THF,methyl 2-bromoacetate, HCl, 0 °C, 1 h; (f) THF, LiAlH4, 0 °C; (g) PPh3, DTAD, THF, r.t, overnight; (h) n-BuOH, TFA, 75 °C,1.5 h.
2.2 Biological evaluation
2.2.1 In vitro cytotoxic activity of novel quinazoline derivatives against MDA-MB-468, SK-BR-3, HCC1806, MCF-7 breast cancer cell lines and HEK 293T cells
The inhibitory activity of target compounds (IC50 ± SD μM) towards breast cancer cells was listed in Table 2. Erlotinib, a first-generation EGFR inhibitor, was used as a positive control. As shown in Table 2, the IC50 of compound 5a (LU1501), 5c, 9f, 9g, 9j was lower than 20 µM on MDA-MB-468. LU1501 and 5b showed strong inhibition on MCF-7 with IC50<20 µM. Additionally, LU1501, 5b, 5c, 9f displayed high cytotoxic activity on SK-BR-3 (IC50 < 20 µM) and LU1501 together with 5b could obviously suppress the proliferation of HCC1806 cells with IC50 < 20 µM. Generally, most of the tested target compounds seemed to be more effective against SK-BR-3 than against other cell lines. LU1501, the most active compound among the tested derivatives, exhibited significantly lower IC50 values than the positive control on the MCF-7, SK-BR-3, and HCC1806 cells. Meanwile, compounds on HEK 293T cells displayed minimal toxicity at the dosage of 100 µM for 48hrs as shown in Table 2.
Compound
IC50 (µM)
MDA-MB-468
MCF-7
SK-BR-3
HCC1806
HEK 293 T
LU1501
19.45 ± 1.33
16.13 ± 3.47
10.16 ± 0.86
10.66 ± 1.01
˃100
5b
26.91 ± 1.74
4.06 ± 1.08
10.41 ± 1.13
14.35 ± 1.31
˃100
5c
4.68 ± 0.43
33.33 ± 3.15
3.95 ± 0.46
25.60 ± 2.18
˃100
5d
34.32 ± 1.89
>100
51.29 ± 3.87
47.32 ± 1.54
˃100
5e
52.33 ± 7.56
49.66 ± 6.87
60.31 ± 9.31
>100
˃100
9f
5.22 ± 1.68
32.54 ± 3.81
12.30 ± 1.02
21.78 ± 3.11
˃100
9g
17.48 ± 1.29
41.03 ± 6.50
31.27 ± 2.36
37.58 ± 1.22
˃100
9h
21.02 ± 3.34
89.22 ± 6.17
25.26 ± 4.49
˃100
˃100
9i
24.49 ± 2.89
40.68 ± 1.81
27.26 ± 4.16
34.57 ± 1.29
˃100
9j
10.26 ± 1.08
54.69 ± 3.79
23.73 ± 4.03
33.25 ± 1.18
˃100
Elotinib
16.1 ± 1.46
23.81 ± 1.25
51.65 ± 0.38
˃100
˃100
The cell viability and inhibition of cell proliferation, as well as dose–response curves of LU1501, Elotinib and Afatinib on SK-BR-3 and HCC1806 breast cancer cell lines were illustrated in Fig. 2. The results showed that cell viability decreased nearly 100% with significant inhibition on HCC1806 and SK-BR-3 proliferation with LU1501 treatment for 72 h, which was similar with the inhibition of Afatinib. Moreover, the inhibitory activity of LU1501 on HCC1806 and SK-BR-3 cells was higher than that of Elotinib. All these indicated that LU1501 possessed a potential anti-tumor effect on breast cancer, especially on HCC1806 and SK-BR-3 cell line.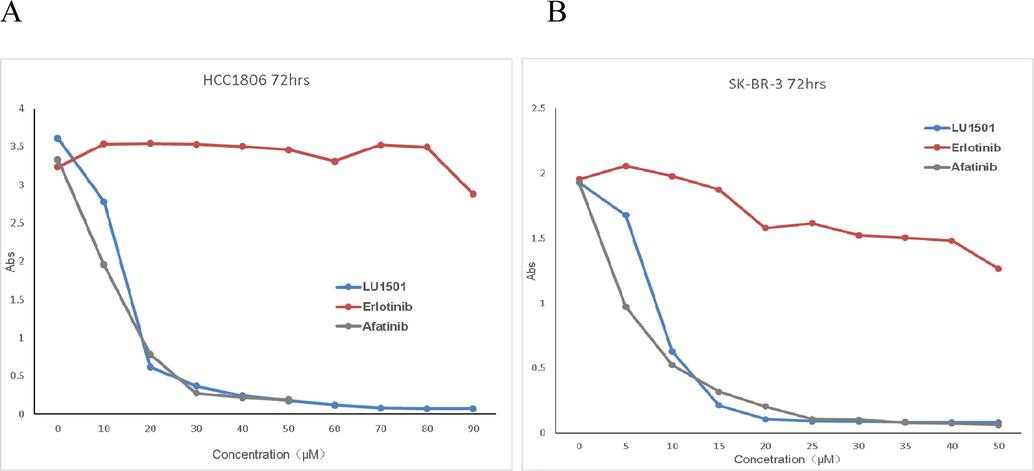
Compound LU1501, Elotinib and Afatinib inhibit proliferation in breast cancer cell lines SK-BR-3 and HCC1806 after treated for 72hrs. (A) HCC1806 cell lines (B) SK-BR-3 cell lines.
A B.
2.2.2 LU1501 inhibits EGFR/NF-κB signal pathway activity in SK-BR-3
Because LU1501 demonstrated the highest anticancer activity among all the tested derivatives, we investigated the regulatory mechanism of LU1501 in breast cancer cells by Agilent microarray analysis. From the IC50 value and cell viability curve results listed above, SK-BR-3 cell line was quite sensitive to LU1501, therefore SK-BR-3 was used to explore the differentially expressed genes (DEGs) responding to LU1501 treatment. As showed in Fig. 3A–C, 534 genes were up-regulated and 1872 genes were down-regulated. NF-κb signaling pathway was enriched by the KEGG analysis. The protein expression was further examined by western blot analysis. The level of NF-κb phospho-p65 and IkBα protein expression were significantly up-regulated in LU1501 treatment group(Fig. 3D–F). These results revealed that LU1501 inhibited the cell growth and proliferation of breast cancer by suppressing the EGFR/NF-κb pathway.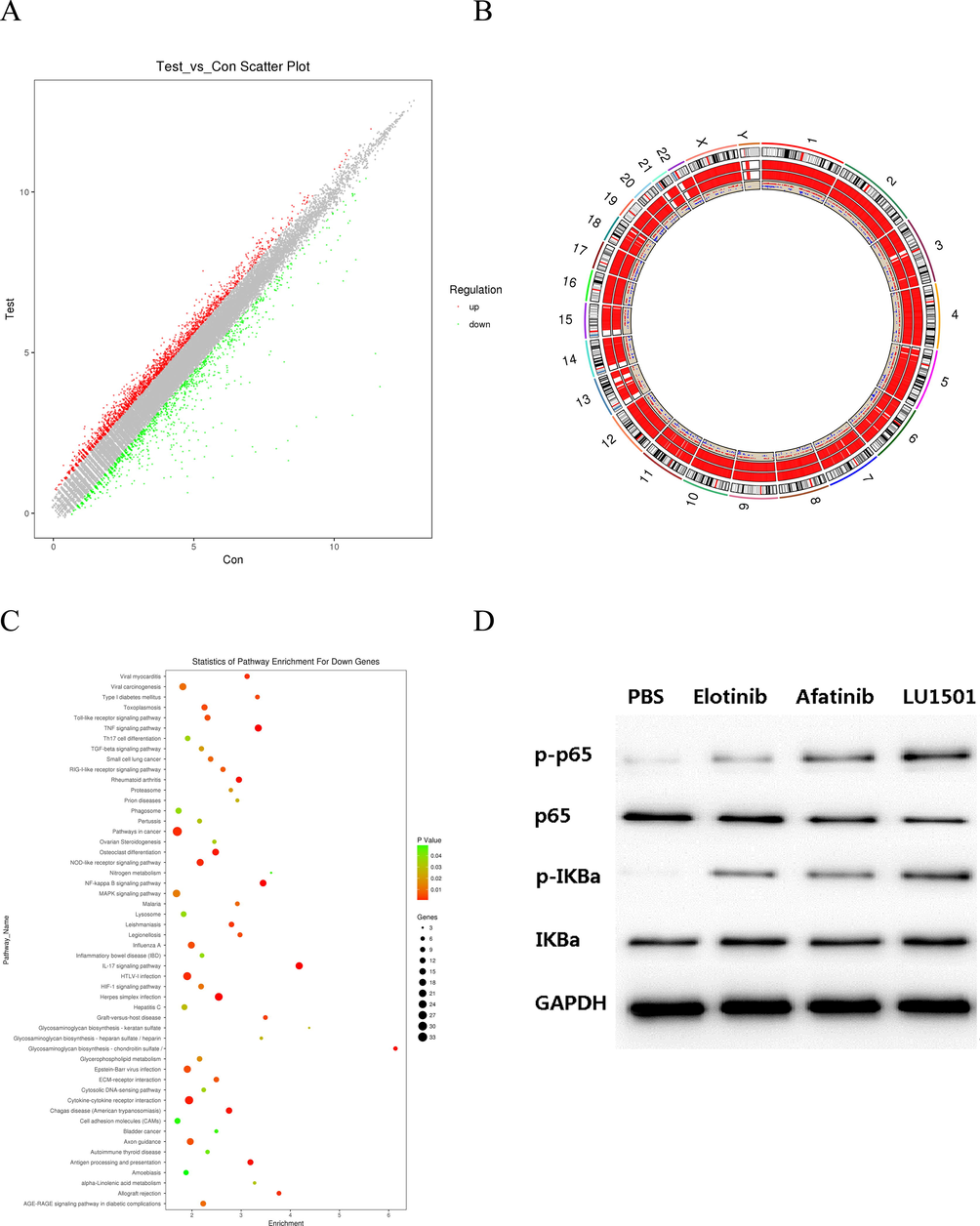
LU1501 inhibits EGFR/NF-κb signal pathway activity in SK-BR-3 cell line. (A) Scatter plot of differential genes showed genes up-regulated and down-regulated. (B) Differential genes were distributed specific positions on chromosomes. (C) NF-κb signaling pathway was enriched by the KEGG analysis. (D) The expression levels of EGFR/NF-κb pathway related proteins were evaluated using Western blot analysis. (E) Corresponding grayscale values analysis of EGFR/NF-κb p65 and phospho-p65. (F) Corresponding grayscale values analysis of EGFR/NF-κb IkBα and phospho-IkBα.
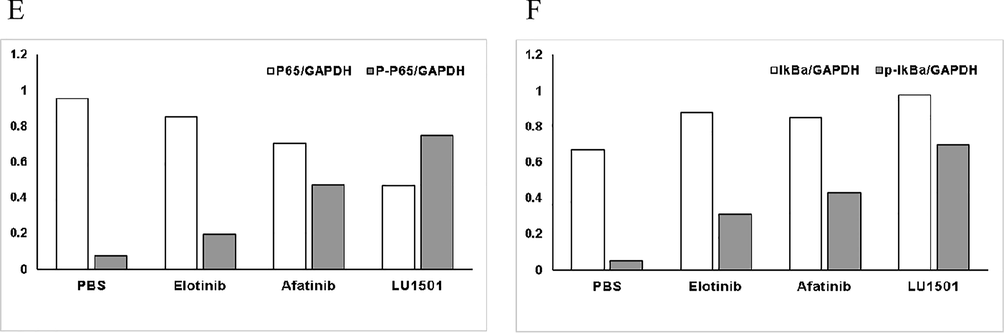
LU1501 inhibits EGFR/NF-κb signal pathway activity in SK-BR-3 cell line. (A) Scatter plot of differential genes showed genes up-regulated and down-regulated. (B) Differential genes were distributed specific positions on chromosomes. (C) NF-κb signaling pathway was enriched by the KEGG analysis. (D) The expression levels of EGFR/NF-κb pathway related proteins were evaluated using Western blot analysis. (E) Corresponding grayscale values analysis of EGFR/NF-κb p65 and phospho-p65. (F) Corresponding grayscale values analysis of EGFR/NF-κb IkBα and phospho-IkBα.
2.2.3 LU1501 inhibits breast cancer growth in vivo
To further study the anti-tumor effect of LU1501 in vivo, tumor bearing mice model was established. SK-BR-3 tumor models were subsequently injected with saline, Erlotinib, Afatinib or LU1501 (in the dosage of 1.2 mg/kg/day) within two weeks of continuous intraperitoneal injected (group = 4, n = 6). The mice were sacrificed after 14 days of administration of saline, Erlotinib, Afatinib or LU1501. Tumor volume and weight were analyzed to determine tumor growth rate. Tumor volume of LU1501 group was notably smaller than that of Erlotinib and Afatinib group (Fig. 4A), while the ratio of tumor and body weight of LU1501 group was significantly less than that of Erlotinib and Afatinib groups (Fig. 4B).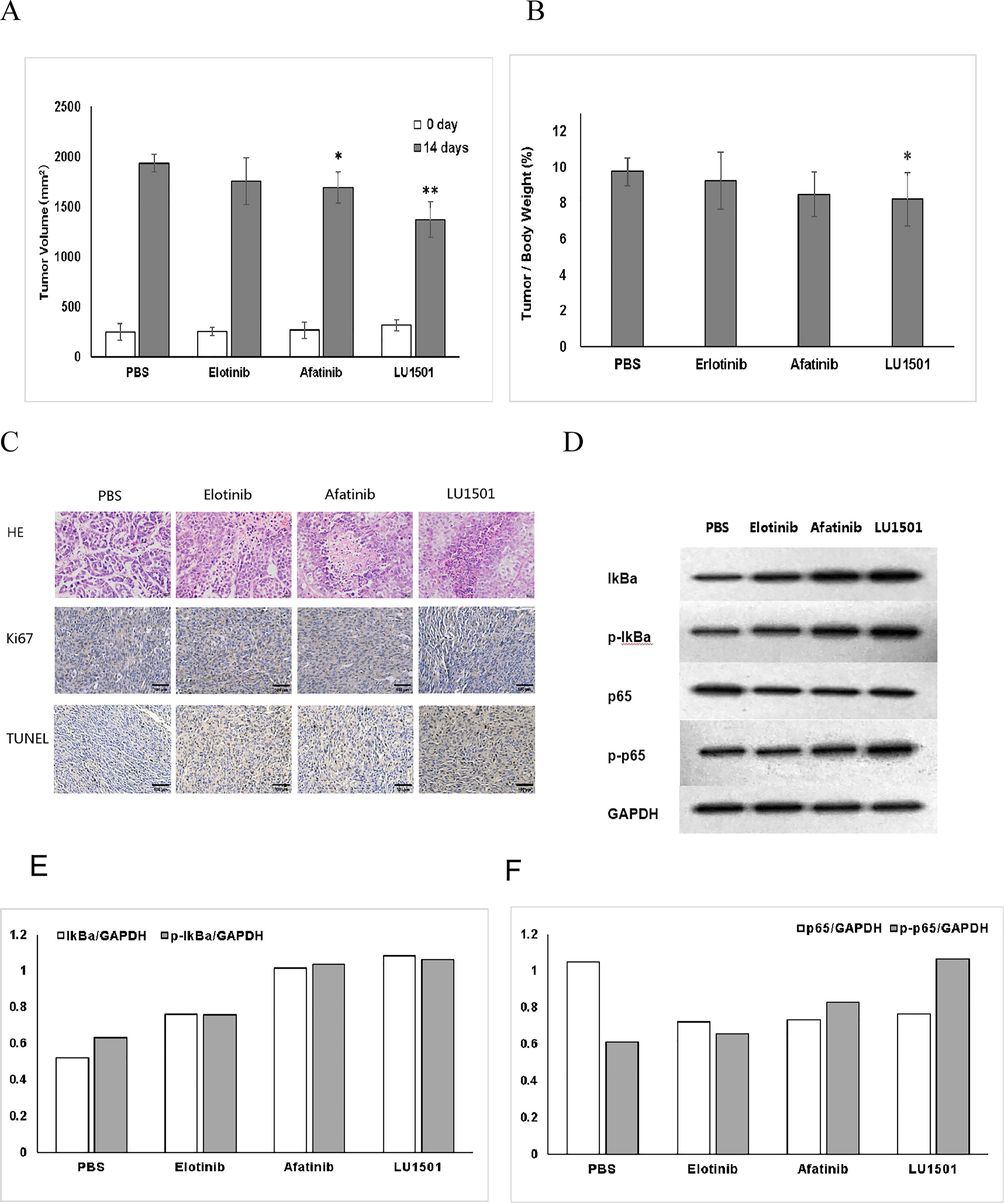
(A) Tumor-bearing mice of different groups. (B) Tumor volumes after 14 days treatment. (C) HE, Tunel analysis and protein Ki-67 was analyzed by IHC staining. (D) The expression levels of EGFR/ NF-κb pathway related proteins in vivo were evaluated using Western blot analysis. *p < 0.05, **p < 0.01 when compared with control group. (E) Corresponding grayscale values analysis of EGFR/NF-κb p65 and p-p65. (F) Corresponding grayscale values analysis of EGFR/NF-κb IkBα and p-IkBα.
Moreover, HE analysis, Tunel analysis and cell proliferation associated protein Ki-67 were analyzed by IHC staining (Fig. 4C). Graded scoring results were shown as Table 3. After administration in each group,. The necrotic areas of tumor cells in Elotinib, Afatinib and LU1501 groups were higher than those in PBS group. The infiltration degree of inflammatory cells in Elotinib group was significantly higher than that in other groups. Fibrosis in the necrotic area of the tumor can be seen in Afatinib group and LU1501 group.
Group
Score
PBS
1
Elotinib
3
Afatinib
2.5
LU1501
2.5
The finding was quite in agreement with in vitro results. The protein level of NF-κb phospho-p65, and IkBα similarly increased in LU1501 group in vivo (Fig. 4D–4F). In summary, LU1501 could inhibit the breast cancer cell proliferation and invasion both in vitro and in vivo.
2.3 Molecular docking study
In order to test the interaction and the binding modes of 5a (LU1501) with the functional protein, the docking study of the compound with the p65 (PDB: 1NFI) and IkBα (PDB: 1IKN) protein were scored by Discover Studio 2020. The results showed that LU1501 interacted with p65 protein mainly via hydrogen bond interaction (ARG30, ASP80, ARG164), hydrophobic interaction (PRO81, PRO82), strong π - π stacking hydrophobic interaction(PHE184) and halogen interaction between F atom and ASN190(Fig. 5). For the interaction between LU1501 and IkBα protein, hydrogen bond interaction (GLN107, LYS323, PHE106, ASN105, CYS135, ALA133 and HIS171), hydrophobic interaction (PHE106, LEU104, CYS135 and PRO137) and halogen interaction between F atom and ASP136 were the main effects (Fig. 6). According to the binding pattern and chemical structure of LU1501, we speculated this compound was a potential NF-κb inhibitor.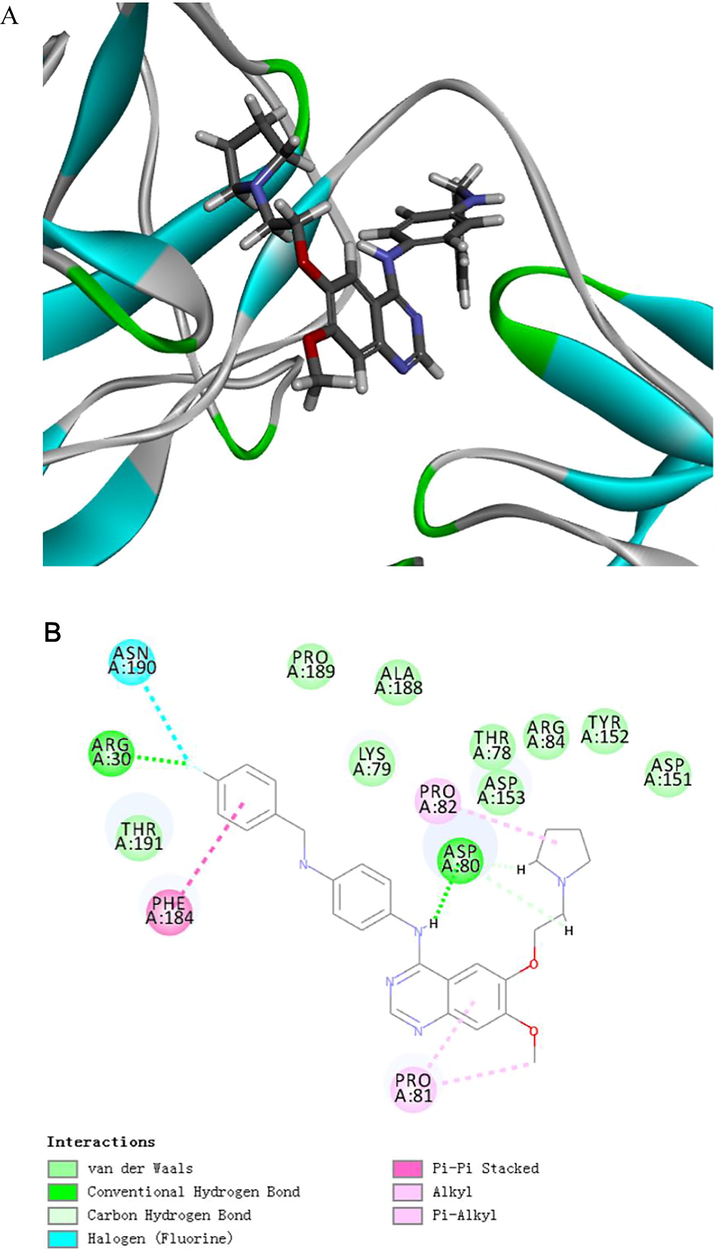
Representations of lowest energy docking poses of LU1501 bound to p65 proteins. (A) 2D interactions (B) 3D interactions.
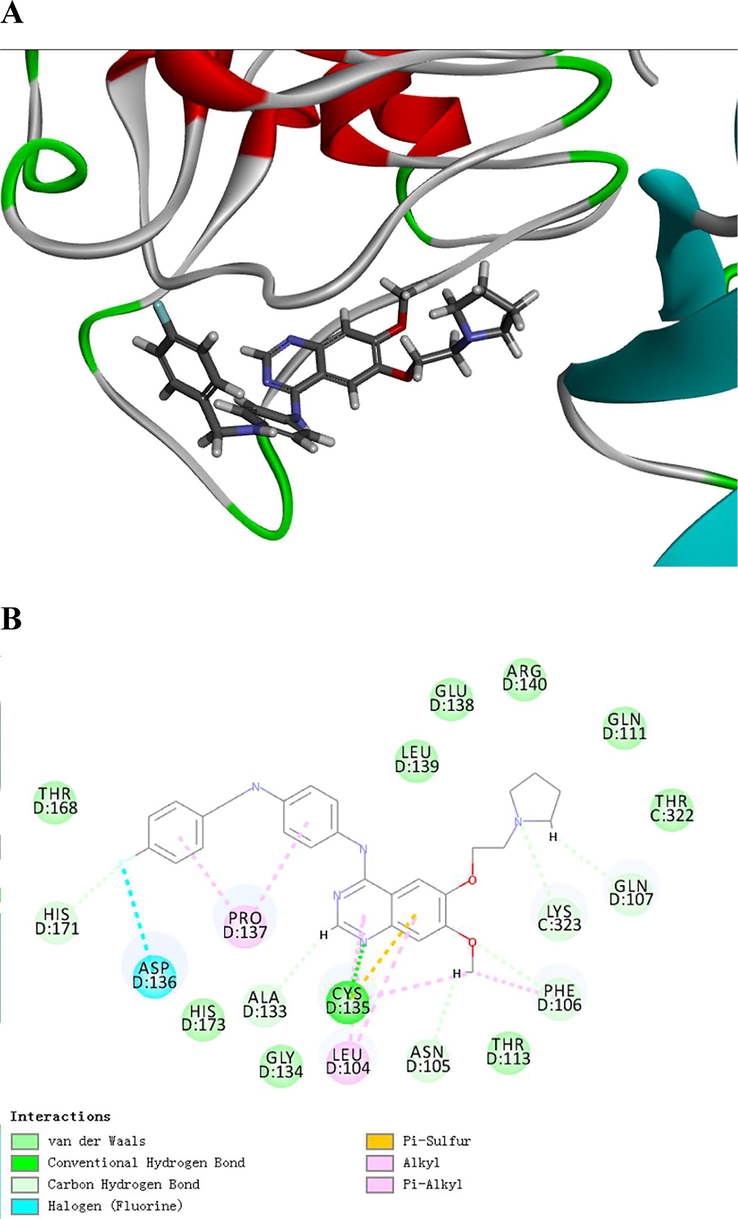
Representations of lowest energy docking poses of LU1501 bound to IkBα proteins. (A) 2D interactions (B) 3D interactions.
3 Discussion
The pathological morphology of breast cancer is complex. Usually, there are various types, and even more than two types may exist in the same cancer tissue or the same section (Calderon et al., 2020). In recent years, with the application of microarray technology and multi-gene RT-PCR quantitative detection method, breast cancer was divided into four types: triple negative breast cancer (ER-/PR-, HER2-), luminal A (ER+/PR+, HER2-), Luminal B (ER+/PR+, HER2+) and HER2+ (ER-/PR-/HER2+) (Weinberg et al., 2020; Akhtar et al., 2017; Chow et al., 2020; Stanley et al., 2015; Hamed et al., 2017). With the development of signaling pathway, apoptosis and other molecular biological methods in tumor research, molecular targets and targeted therapy of breast cancer have gradually become the trend and hotspot of anti-breast cancer research.
EGFR-Tyrosine Kinase Inhibitor (EGFR-TKI) blocks the EGFR signaling pathway by competitively binding to adenosine triphosphate (ATP) in the EGFR tyrosine kinase region (Ji et al., 2016). According to binding characteristics and action sites of different drugs, EGFR-TKI can be divided into three generations. The first generation of EGFR-TKI reversibly inhibits the tyrosine kinase activity of EGFR, and the representative drugs are Gefitinib and Erlotinib (Li et al., 2020; Liu et al., 2021). The second generation of EGFR-TKI such as Afatinib could irreversibly inhibit the tyrosine kinase activity of EGFR and other members of ERBB family as well (Takeda et al., 2021). The third generation of EGFR-TKI is characterized by effective mutation of EGFR 20 exon T790M, a common drug resistance target of the first and second generation of drugs, and it is easier to penetrate the blood–brain barrier (Tamiya et al., 2021). The representative drugs are oxitinib, amitinib and vometinib. However, the core structure of both amitinib and vometinib is similar to that of oxitinib, and the main purpose of the optimizing the structure is to further improve the binding of the drug to the mutant EGFR (Jiang et al., 2019; Roskoski, 2019; Wu et al., 2021). Alternatively, drugs can be developed with higher selectivity, that is, they can inhibit only mutant EGFR and have little inhibition on wild-type EGFR without mutation, thus reducing adverse reactions and improving safety (Wang et al., 2015; Pan et al., 2016).
From the structure and the protein binding prediction, LU1501 is more selective in binding to EGFR receptors than erlotinib and afatinib. In vitro and in vivo breast cancer cytotoxicity tests also showed stronger inhibitory effects than these two positive drugs, especially in SK-BR-3 cells which were HER-2 and EGFR positive.
The intracellular signal transduction of EGFR is mainly through cytoplasmic MAPK, PI3K, c-SRC and nuclear NFκB signal pathway. In this study, it was found that LU1501 can competitively bind to the ATP site of EGFR, blocking the transmission of its downstream signal pathway, especially inhibiting the activation of NF-κB/p65 and IκBα proteins. It is worth noting that molecular mRNA profiling results and signal pathway experiments showed that LU1501 inhibited SK-BR-3 as well as MAPK signal pathway. Therefore, the regulation mechanism of LU1501 on NF-κb, together with the pathway crosstalk between NF-κb and MAPK responding to LU1501 may need further study in the future.
4 Conclusions
In this study, we designed, synthesized a series of novel 4-aminoquinazoline derivatives and also evaluated the biological activity against breast cancer by NF-κb pathway. From the MTT assay, most of the tested target compounds were found more effective towards SK-BR-3 than towards other three human breast cancer cell lines. LU1501, the most active compound among the tested derivatives exhibited comparable or lower IC50 values than control drugs on all the tested cells, suggesting LU1501 had high inhibitory activity. In vivo studies were carried out to determine the activity of LU1501 against breast cancers. After LU1501 were injected into SK-BR-3 tumor bearing mice for 14 days, tumors were obviously inhibited. HE analysis, Tunel analysis and cell proliferation associated protein Ki-67 were analyzed by IHC staining, and the results further verified that LU1501 appeared inhibitory effect through the NF-κb signal pathway by adjusting p65 and IkBα proteins. Later, the molecular docking studies were performed and the compound LU1501 was found to bind well towards the proteins of p65 and IkBα on the NF-κb pathway, which were consistent with the biological data. Accordingly, LU1501 was considered as a preferable and promising anti-tumor drug candidate, which could be used for further investigation in clinics.
Acknowledgments
The authors thank Department of Medical Imaging, Jinling Hospital, Medical School of Nanjing University for providing the necessary infrastructural facilities to carry out this work. We gratefully acknowledge the financial support provided by the National Key Basic Research Program of the PRC (2014CB744504) and the National Natural Science Funds of China (No. 81601555, 81530054 and 81971681).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular modeling, enzyme activity, anti-inflammatory and antiarthritic activities of newly synthesized quinazoline derivatives. Fut. Med. Chem.. 2017;9(17):1995-2009.
- [CrossRef] [Google Scholar]
- Structure-activity relationship (SAR) study and design strategies of nitrogencontaining heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem.. 2017;125:143-189.
- [Google Scholar]
- An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem.. 2018;151:628-685.
- [CrossRef] [Google Scholar]
- Discovery and SAR of novel disubstituted quinazolines as dual PI3Kalpha/mTOR inhibitors targeting breast cancer. ACS Med. Chem. Lett.. 2020;11:2156-2164.
- [CrossRef] [Google Scholar]
- Calderon, O.H., Pérez, A.F.Y., Saumeth, J.Q., Armas, J.P.R., Pacheco, M.P., Sánchez, J.M.O., et al. (2020). Carvacrol: An In Silico Approach of a Candidate Drug on HER2, PI3Kalpha, mTOR, hER-alpha, PR, and EGFR Receptors in the Breast Cancer. Evid Based Complem. Alternat. Med. 1-12. https://doi.org/10.1155/2020/8830665.
- Osimertinib versus comparator EGFR TKI as first-line treatment for EGFR-mutated advanced NSCLC: FLAURA China, A Randomized Study. Target Oncol.. 2021;16:165-176.
- [CrossRef] [Google Scholar]
- Advances in EGFR/HER2-directed clinical research on breast cancer. Adv. Cancer. Res.. 2020;147:375-428.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of novel 4-aminoquinazolines as dual target inhibitors of EGFR-PI3Kα. Eur. J. Med. Chem.. 2018;146:460-470.
- [CrossRef] [Google Scholar]
- Chemical insights into the synthetic chemistry of quinazolines: recent advances. Front. Chem.. 2021;8:1204-1227.
- [CrossRef] [Google Scholar]
- PI3K-AKT-mTOR pathway alterations in advanced NSCLC patients after progression on EGFR-TKI and clinical response to EGFR-TKI plus everolimus combination therapy. Transl. Lung Cancer Res.. 2020;9:1258-1267.
- [CrossRef] [Google Scholar]
- Design, synthesis and antibacterial activity evaluation of moxifloxacin-amide-1,2,3-triazole-isatin hybrids. Bioorg. Chem.. 2019;91:103162
- [CrossRef] [Google Scholar]
- First bispecific inhibitors of the Epidermal Growth factor receptor kinase and the NF-#B activity as novel anti-cancer agents. J. Med. Chem.. 2017;60(7):2853-2868.
- [CrossRef] [Google Scholar]
- SH-1028, an irreversible third-generation EGFR TKI, overcomes T790M-mediated resistance in non-small cell lung cancer. Front. Pharmacol.. 2021;12:983-994.
- [CrossRef] [Google Scholar]
- Ivermectin reverses the drug resistance in cancer cells through EGFR/ERK/Akt/NF-κB pathway. J. Exp. Clin. Canc. Res.. 2019;38:265-282.
- [CrossRef] [Google Scholar]
- Toripalimab plus chemotherapy as second-line treatment in previously EGFR-TKI treated patients with EGFR-mutant-advanced NSCLC: a multicenter phase-II trial. Signal Transduct. Target Ther.. 2021;6:355-363.
- [CrossRef] [Google Scholar]
- Pharmacokinetic-pharmacodynamic modeling of the antitumor effect of TM208 and EGFR-TKI resistance in human breast cancer xenograft mice. Acta. Pharmacol. Sin.. 2016;37:825-833.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of 4-Anilino-quinazolines and -quinolines as Inhibitors of Breast Cancer Resistance Protein (ABCG2) J. Med. Chem.. 2016;59:5449-5461.
- [CrossRef] [Google Scholar]
- 1,2,3-triazole-containing compounds as anti-lung cancer agents: current developments, mechanisms of action, and structure-activity relationship. Front. Pharmacol.. 2021;12:661173
- [CrossRef] [Google Scholar]
- Restricting glutamine uptake enhances NSCLC sensitivity to third-generation EGFR-TKI almonertinib. Front. Pharmacol.. 2021;12:1202-1215.
- [CrossRef] [Google Scholar]
- Efficacy of osimertinib for the treatment of previously EGFR TKI treated NSCLC patients: a meta-analysis. Clin. Transl. Oncol.. 2020;22:892-899.
- [CrossRef] [Google Scholar]
- The CBM complex underwrites NF-κB activation to promote HER2-associated tumor malignancy. Mol. Cancer. Res.. 2016;14(1):93-102.
- [CrossRef] [Google Scholar]
- Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res.. 2019;139:395-411.
- [CrossRef] [Google Scholar]
- Cytogenetic analysis of HER1/EGFR, HER2, HER3 and HER4 in 278 breast cancer patients. Breast Cancer Res.. 2008;10(1):R2.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: molecular docking study. J. Enzym. Inhib. Med. Chem.. 2018;32:199-209.
- [CrossRef] [Google Scholar]
- PARP1 and phospho-p65 protein expression is increased in human HER2-positive breast cancers. Breast Cancer Res. Treat.. 2015;150(3):569-579.
- [CrossRef] [Google Scholar]
- Neoadjuvant EGFR-TKI Therapy for EGFR-Mutant NSCLC: A Systematic Review and Pooled Analysis of Five Prospective Clinical Trials. Front. Oncol.. 2020;10:586-596.
- [CrossRef] [Google Scholar]
- A phase II study to assess the efficacy of Osimertinib in patients with EGFR mutation-positive NSCLC who developed isolated CNS progression (T790M-negative or unknown) during first- or second-generation EGFR-TKI or systemic disease progression (T790M-negative) after treatment with first- or second-generation EGFR-TKI and platinum-based chemotherapy (WJOG12819L) Clin. Lung Cancer. 2021;22:376-380.
- [CrossRef] [Google Scholar]
- The ratio of T790M to EGFR-activating mutation predicts response of osimertinib in 1st or 2nd generation EGFR-TKI-refractory NSCLC. Sci. Rep.. 2021;11:9629-9635.
- [CrossRef] [Google Scholar]
- Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem.. 2015;22(2):264-289.
- [CrossRef] [Google Scholar]
- Quinazoline derivatives as potential therapeutic agents in urinary bladder cancer therapy. Front. Chem.. 2021;9:955-968.
- [CrossRef] [Google Scholar]
- EGFR expression in HER2-driven breast cancer cells. Int. J. Mol. Sci.. 2020;21:9008-9026.
- [CrossRef] [Google Scholar]
- First-generation EGFR-TKI plus chemotherapy versus EGFR-TKI alone as first-line treatment in advanced NSCLC With EGFR activating mutation: a systematic review and meta-analysis of randomized controlled trials. Front. Oncol.. 2021;11:883-894.
- [CrossRef] [Google Scholar]
- Synthesis, Molecular docking and biological evaluation of glycyrrhizin analogs as anticancer agents targeting EGFR. Molecules. 2014;19:6368-6381.
- [CrossRef] [Google Scholar]
- Natural products as important tyrosine kinase inhibitors. Eur. J. Med. Chem.. 2019;182:111664
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103908.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




