

Translate this page into:
Electronic structure theory investigation on the electrochemical properties of cyclohexanone derivatives as organic carbonyl-based cathode material for lithium-ion batteries
⁎Corresponding authors at: Computational and Bio-Simulation Research Group, University of Calabar, Calabar, Nigeria (H. Louis). louismuzong@gmail.com (Hitler Louis), aadeyinka@UJ.ac.za (Adedapo S. Adeyinka)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Organic carbonyl-based compounds with redox-active site have recently gained full attention as organic cathode material in lithium-ion batteries (LIBs) owing to its high cyclability, low cost, high abundance, tunability of their chemical structure compared to traditionally used inorganic material. However, the utilization of organic carbonyl-based compounds in LIBs is limited to its poor charge capacity and dissolution of lower molecular weight species in electrolytes. In this study, we theoretically investigated five set of cyclohexanone derivatives (denoted as: H1, H2, H3, H4, and H5) and influence of functional groups (-F and -NH2) on their electrochemical properties using advanced level density functional theory (DFT) with the Perdew-Burke-Ernzenhof hybrid functional (PBE0) at 6-31+G(d,p) basis set. In line with the result gotten, the HOMO-LUMO results revealed that compound H5 is the most reactive among the studied cyclohexanone derivatives exhibiting energy gap values of 0.552, 0.532, 0.772 eV for free optimized structures and structurally engineered structures with electron withdrawing group (EWG) and electron donating group (EDG) respectively. Also, results from electrochemical properties of the studied compounds lithiated with only one lithium atom displayed that compound H2 exhibited interesting redox potential and energy density for all the studied structures in free optimized state (1108.28 W h kg−1, 4.92 V vs Li/Li+), with EWG (648.22 W h kg−1, 3.313 V Li/Li+), and with EDG (1002.4 W h kg−1, 5.011 V vs Li/Li+). From our result, we can infer that compound H2 and H3 with corresponding redox potential, energy density and theoretical charge capacity value of 4.92 V vs Li/Li+, 1108.28 W h kg−1, 225.26 mA h g−1 and 5.168 V, 1041.61 W h kg−1, 201.55 mA h g−1 lithiated with only one lithium atom in free optimized state are the most suitable compounds to be employed as organic cathode material in lithium-ion batteries among all the investigated cyclohexanone derivatives.
Keywords
Cyclohexanone
Organic carbonyl
Cathode material
Lithium-ion battery
DFT
Electrochemical properties

1 Introduction
Organic carbonyl – based compounds are one of the promising organic electrode materials employed as cathode material in rechargeable lithium – ion batteries (LIBs) that is commonly used in high performance energy storage devices such as electric vehicles (EVs) and portable rechargeable electronic devices (laptops, mobile phones) which is contributed from its high energy density and fast charge transfer (Shi et al., 2021; Lyu et al., 2021; Ye and Li, 2021; Amin et al., 2018). Presently, redox-active organic compounds with carbonyl moiety have been found to be of great advantage as cathode materials in LIBs due to the ease of production, good cyclability, reversible multi-electron reaction, sustainability, high abundance, benign and tune-ability of their chemical structures in order to modify their electrochemical properties (Onori et al., 2016; Esser et al., 2021; Wu et al., 2016; Kerman et al., 2017; Horstmann et al., 2021; Trahey et al., 2020) compared to conventionally used transition metal-based inorganic cathode material such as LiCoO2, LiFePO4, LiNiO2, LiMn2O4 which suffers major problems of poor diffusion of lithium ions through them (Kim et al., 2021; Amin and Chiang, 2016; Yang et al., 2021; Chung et al., 2002; Chen et al., 2017; Li et al., 2016). In as much as organic carbonyl-based compounds aids in enhancing the electrochemical performance of lithium – ion batteries, the main bottle neck of organic compounds as electrode material is that the lower molecular weight species tends to attain dissolution in the electrolyte used and their low theoretical charge capacity (<600 mA h g−1) (Gunnarsdóttir et al., 2020; Amanchukwu et al., 2020; Wang et al., 2020; Yu et al., 2020; Le et al., 2017).
In recent years, several researchers have reported the use of organic carbonyl compounds as cathode material in LIBs and also suggested several methods of increasing their electrochemical properties for proper utilization in LIBs. For instance, Kim and coworkers conducted DFT studies on the stability and redox potential of seven selected quinone derivatives namely; 1,4-Benzoquinone, 1,4-Naphthoquinone, 9,10-Anthraquinone, 2-Aminoanthraquinone, Anthraquinone-2-carboxylic acid, 2,6-Diaminoanthraquinone, Anthraquinone-2,6-dicarboxylic acid using two functionals (PBE0 and PWB6K) with 6-31+G(d,p) basis set for possible application as organic cathode material in Lithium-ion batteries (Kim et al., 2016). Their result revealed that modifying the studied compounds with functional groups EWG (COOH) and EDG (NH2), presence of EWG tends to increase their redox potential than those designed with EDG due to increase in electron affinity of the compounds with EWG. Park and coworkers successfully performed DFT calculation using PBE0/6-31+G(d,p) level of theory on two ketone derivatives (phenalenyl and anthracene) by incorporating on them different number of carbonyl group (Park et al., 2017). They pointed out that redox potential of the investigated compounds increases as the number of attached redox-active carbonyl group increases. Luo and coworkers also carried out theoretical study on tetra-(phthalimido)-benzoquinone (TPB) with rigid ring as insoluble cathode material for LIBs by utilizing DFT/B3LYP/6-311G(d,p) theory level (Luo et al., 2017). They reported that, on insertion of 2Li atom and 4Li atom, TPB demonstrated redox potential of 3.63 V and 2.28 V respectively. Recently, Jung et al., systematically performed DFT study on carbonitrile and quinone derivatives for possible use as organic positive electrode material in rechargeable Li-ion batteries by employing DFT/PBE0/6-31+G(d,p) method (Jung et al., 2020). They inferred that introducing EWG functional group is an effective method of enhancing redox potential of the abovementioned compounds. Lu and coworkers investigated the electrochemical characteristics of cyclohexanehexone experimentally and computationally using DFT method for prospective application as cathode material for LIBs (Lu et al., 2019). They highlighted that C6O6 exhibited ground breaking result with theoretical and experimental charge capacity of 957 mA h g−1 and 902 mA h g−1 respectively which is attributed to ultima electron transfer on all the six redox active sites present on the compound.
Herein, we report the use of density functional theory (DFT) method to theoretically examine the electrochemical properties of five set of cyclohexanone derivatives with and without functional groups (EWG: fluorine and EDG: NH2) for possible utilization as organic cathode material in lithium-ion batteries (LIBs) by employing DFT/PBE0/6-31+G(d,p) level of theory. Frontier molecular orbital (FMO) of the studied cyclohexanone derivatives was also estimated to verify their stability and reactivity as organic cathode material in LIBs. Natural bond orbital (NBO) analysis was also considered in order to visualize the form of interaction taking place within the orbitals of the investigated compounds. The electrochemical properties of the studied compounds were investigated with respect to the redox potential, energy density, and charge capacity.
2 Methodology
2.1 Computational details
In this study, all density functional theory (DFT) computations were executed using Gaussian 09 (Trucks et al., 2013) and GaussView 6.0.16 software packages. We employed the Perdew-Burke-Ernzenhof hybrid functional (PBE0) level of theory and 6-31+G(d,p) basis set as utilized by several authors (Kim et al., 2016; Park et al., 2017; Jung et al., 2020) for geometry optimization of all the structures implemented in this research work. Frequency calculations were also performed on the optimized structures using the aforementioned DFT level of theory and basis set to validate that the resultant geometries of the investigated structures attain minima potential energy on the surface (Park et al., 2017). Due to the atomic size of the considered structures, the chosen function sufficiently captures all possible electron–electron interaction by considering all dispersion corrections. Throughout this study, redox potential of the cyclohexanone derivatives was computed both in neutral and in anionic phase by utilizing Born-Haber’s thermodynamics cycle proposed by Truhler and coworkers (Kim et al., 2016; Park et al., 2017; Jung et al., 2020; Kushwaha et al., 2017) which is an application of Hess law. In addition, to determine solvation effect, we deploy the conductor – like polarizable continuum model (CPCM) (Itkis et al., 2021) which is an implicit solvation model by employing water as solvent in order to aid in calculation of redox potentials and assess all possible electronic effects imposed by ions in solution. Also, Natural Bond Orbital (NBO) (Undiandeye et al., 2022; Patrick-Inezi et al., 2022; Nemykin et al., 2021; Louis et al., 2021) calculations were implemented on the in-built NBO 3.1 module (Glendening et al., 2003) in the Gaussian 09 software in order to investigate intra and inter molecular interactions and charge transfer taking place within the molecules (Ji et al., 2021).
Redox potential of the investigated compounds without and with Li atom were evaluated respectively using equation (1) and (2);
From equation (5), designate theoretical charge capacity, reflects amount of electron stored, specifies Faraday’s constant (96500C/mol) and denotes molecular weight of the selected compounds under study.
Binding energy (B.E) involved during the lithiation of cyclohexanone derivatives was computed with the aid of equation (6);
Here, , , signifies the total energies of cyclohexanone derivatives with lithium atom, bare cyclohexanone derivatives and the Li atom.
2.2 Screening of cyclohexanone derivatives
We conducted DFT calculations for the screening of 11 models of the cyclohexanone derivatives using M06-2X meta-hybrid functional with 6-311 + G(d,p) basis set in order determine the best stable derivatives and the result is shown in Fig S1 of the supporting information. Selected stable cyclohexanone derivatives for this study can be seen in Fig. 1.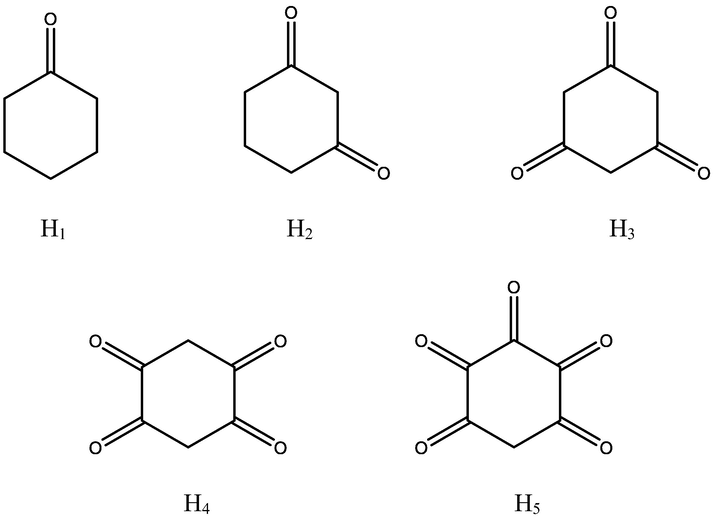
Selected stable cyclohexanone derivatives employed in this study.
3 Results and discussion
3.1 Cyclohexanone
3.1.1 Frontier molecular orbital (FMO)
To understand the nature of stability and reactivity of cyclohexanone derivatives, the highest occupied molecular orbital (HOMO) (Udoikono et al., 2022), the lowest unoccupied molecular orbital (LUMO) (Unimuke et al., 2022) and energy gap (Eg) (Benjamin et al., 2022) of the cyclohexanone derivatives were computed at BPE0/6-31+G(d,p) (Kim et al., 2016; Park et al., 2017; Jung et al., 2020) level of theory. In accordance with FMO theory, the HOMO behaves as an electron donor and the LUMO accepts electron while energy gap (Eg) signifies the difference between the HOMO and the LUMO energy levels (Benjamin et al., 2022). Energy gaps obtained from the HOMO and LUMO values of the compounds critically depicts the stability and reactivity of the redox active organic compound under study. Also, from the aforementioned FMO theory, a higher energy gap indicates a more stable and less reactive compound while lower energy gap implies that the compound is less stable and very reactive (see Figs. 2–9).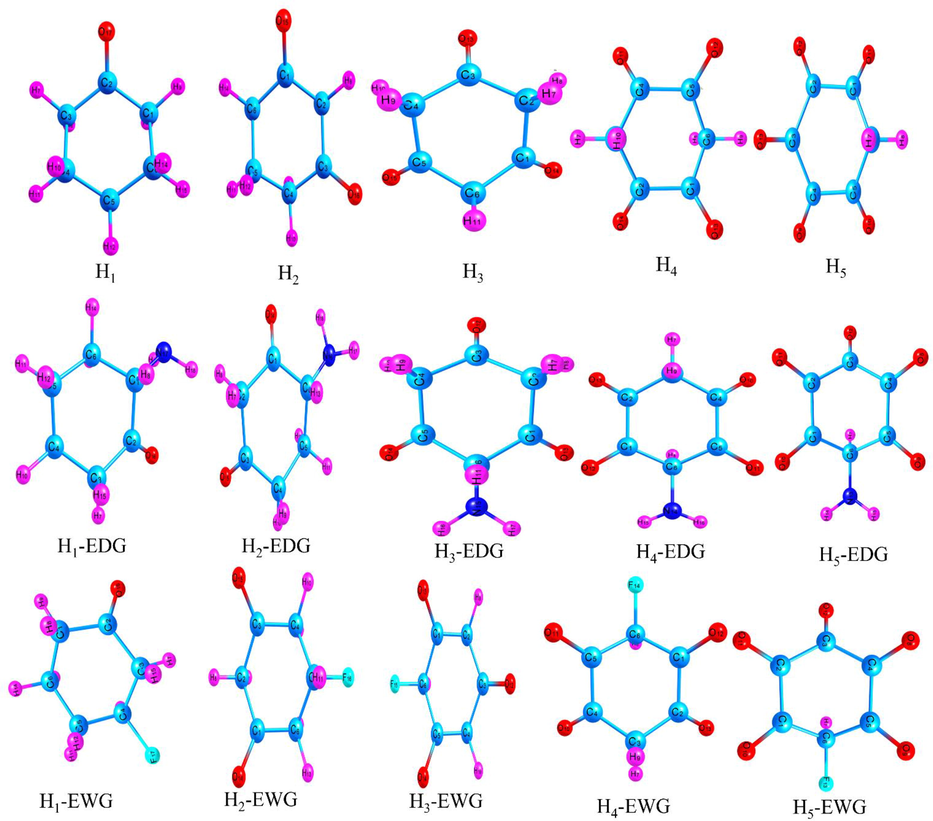
Optimized structures of the free and functionalized cyclohexanone derivatives.
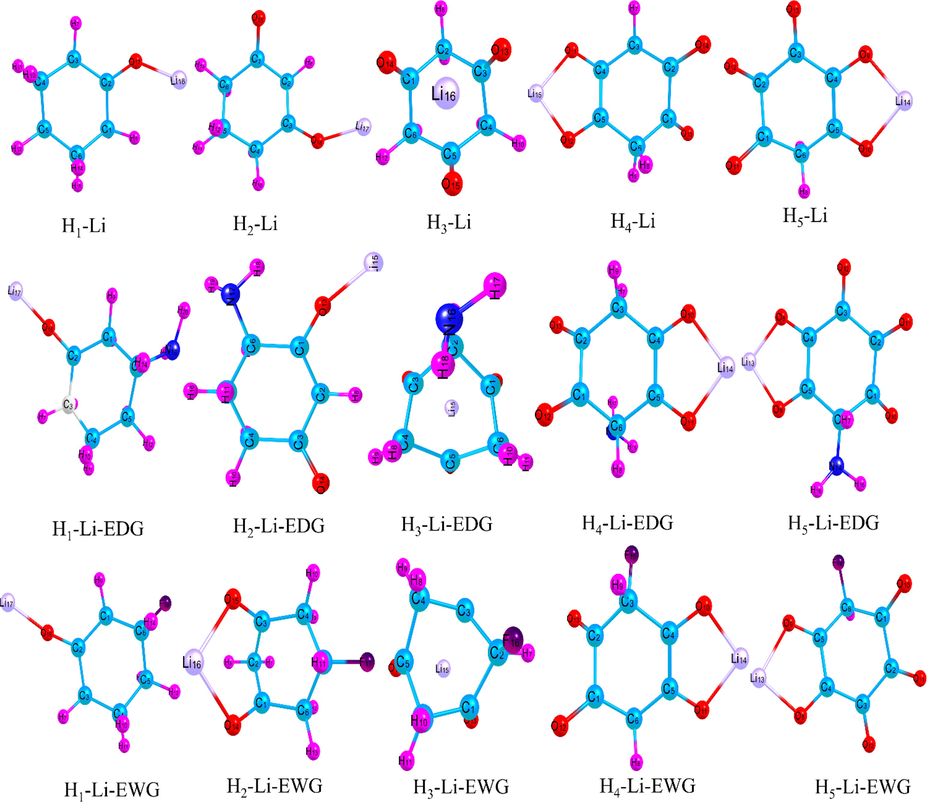
Optimized structures of the free and functionalized cyclohexanone derivatives with one Li atom.
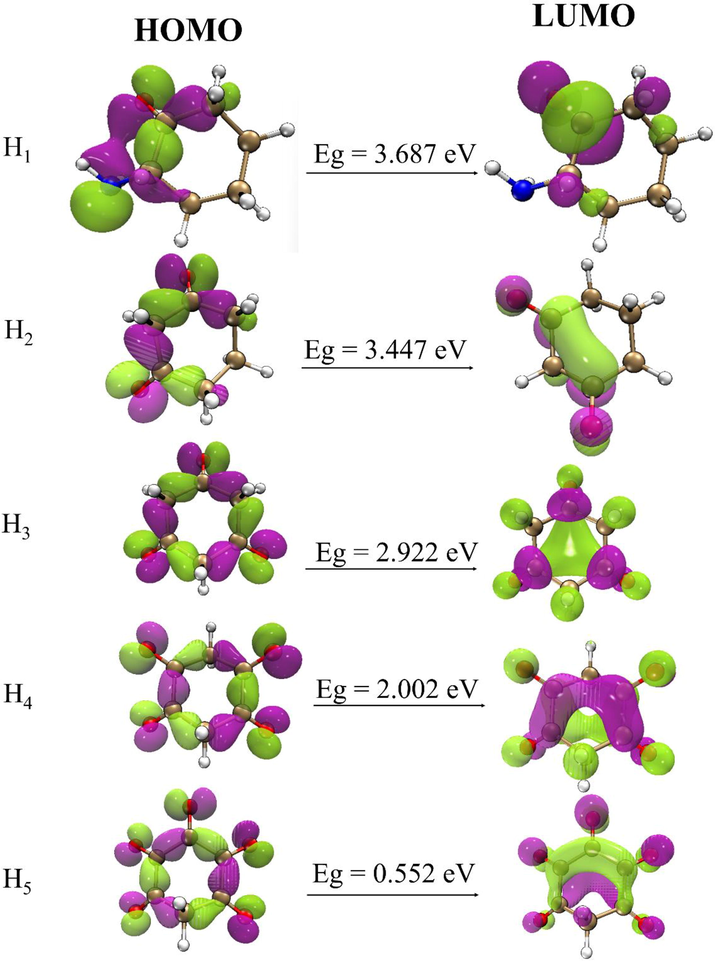
HOMO-LUMO plot of the free optimized structures.
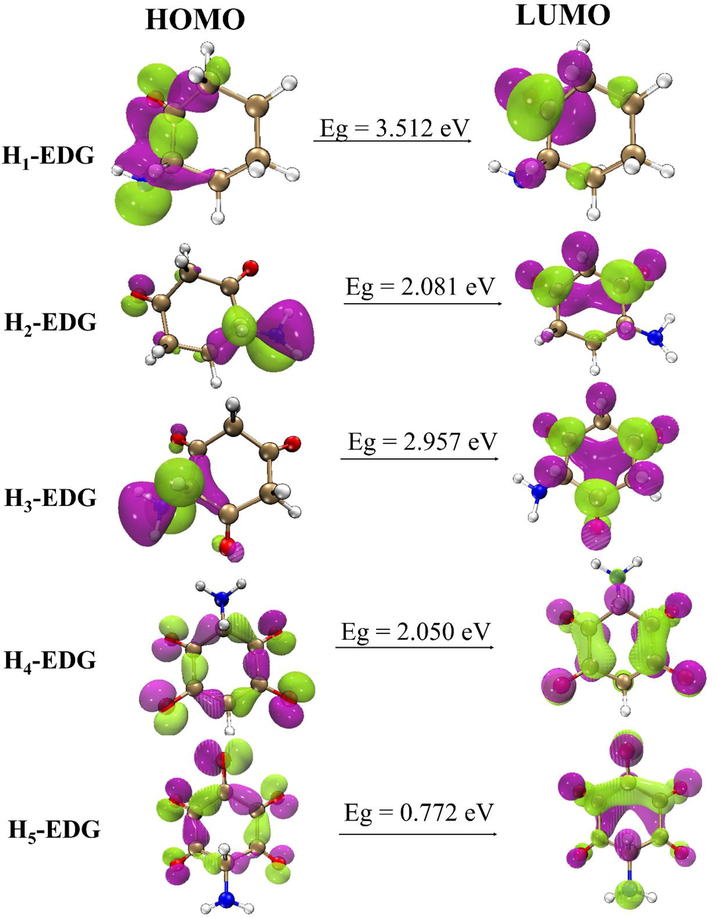
HOMO – LUMO plot of the optimized structures structurally designed with EDG.
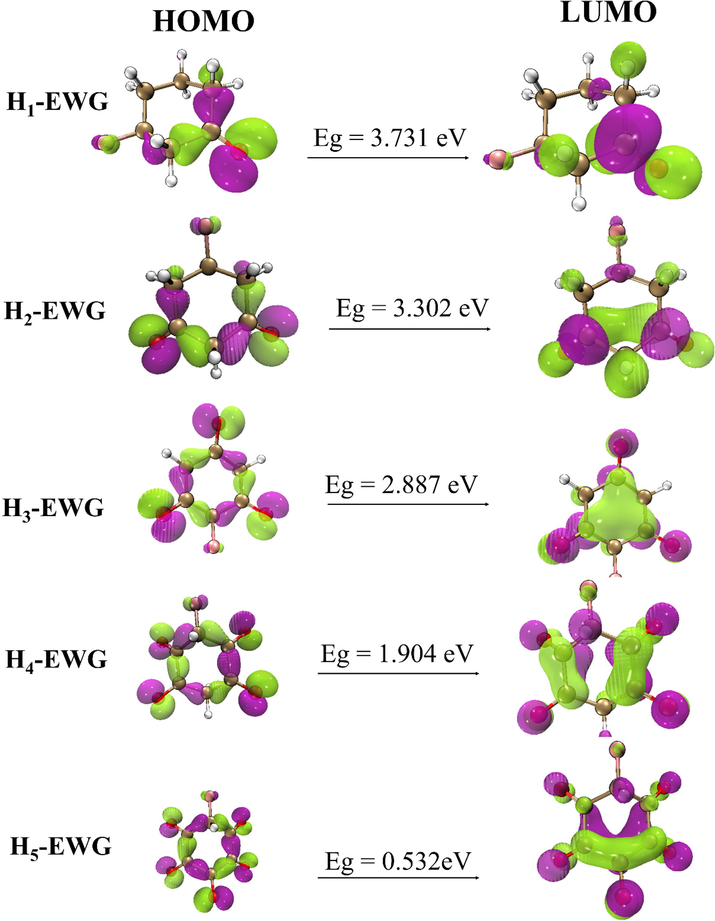
HOMO – LUMO plot of the optimization structures fine-tuned with EWG.
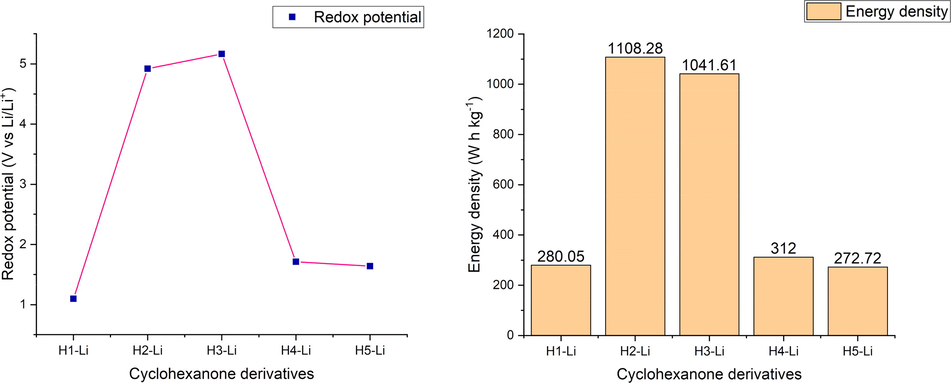
Redox potential of the lithiated cyclohexanone derivatives in free optimized state (b) Energy density of the lithiated cyclohexanone derivatives in free optimized state.
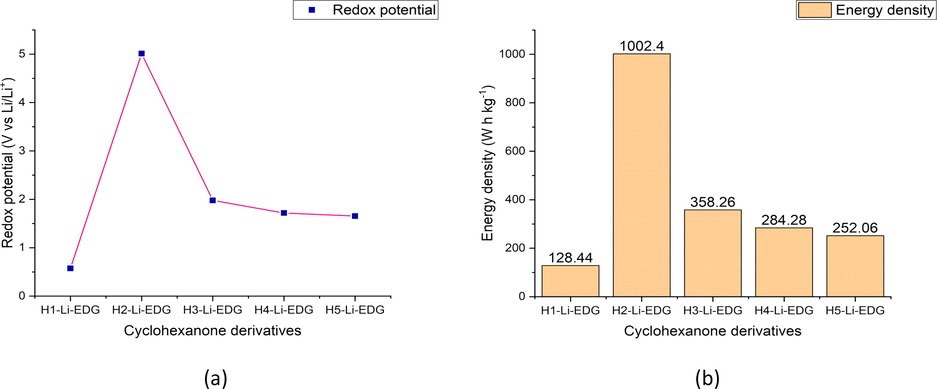
Redox potential of the lithiated cyclohexanone derivatives fine-tuned with EDG. (b) Energy density of the lithiated cyclohexanone derivatives fine-tuned with EDG.
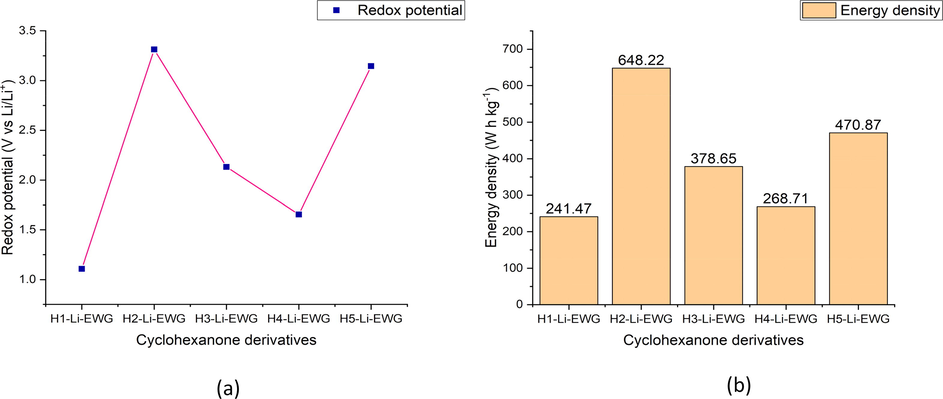
Redox potential of the lithiated cyclohexanone derivatives fine-tuned with EWG (b) Energy density of the lithiated cyclohexanone derivatives fine-tuned with EWG.
HOMO – LUMO energy levels and energy gaps (Eg) of the free optimized structures of the compounds under investigation is presented in Table S1. As evident from the table, results from the energy gap of the studied compounds reflects a progressive trend of H5 < H4 < H3 < H2 < H1 with specific values of 0.552, 2.002, 2.922, 3.447, 3.687 eV respectively. From the trend of the results, there is a clear indication that H5 possesses the least Eg value while compound H1 possesses the highest energy gap. The high Eg of 3.687 eV for H1 is as a result of attachment of one oxygen atom to one carbon atom of the cyclo ring while the low Eg of 0.552 eV for H5 is of the fact that five oxygen atoms are bonded to five carbon atoms of the cyclo ring which in turn increases the electron density of the cyclo ring as a result of lone pair of electrons on the five oxygen atoms that is been delocalized into the cyclo ring. Thus, it can be stressed that the five oxygen atoms with lone pair of electrons conferred to H5 makes it the least stable and most reactive with least Eg among the studied compounds while H1 with only one atom with lone pair of electrons on it makes it the most stable and least reactive with highest Eg among the studied compounds.
In addition, molecular orbital composition of the HOMO and LUMO orbitals of all the cyclohexanone derivatives was also analyzed. For compound H1, the HOMO orbital is majorly distributed on O17, C2, C4 atoms with orbital contribution of 66.96%, 23.08%, 3.67% respectively while its LUMO orbital is mainly localized on C4, O17, C3 having individual orbital composition of 111.05%, 39.88%, 14.61%. In the same line, HOMO orbital of compound H2 is largely distributed on O15, C2, C3 atoms of which their orbital compositions are 37.99%, 17.72%, 10.03% respectively while the LUMO orbital is dominantly concentrated on C5, C1, O15 atoms possessing specific orbital composition of 28.27%, 25.42%, 19.12%. In the case of compound H3, the HOMO orbital is chiefly spread on O13, O14, C5, C4 atoms of which their respective orbital composition was 24.32%, 23.52%, 8.35%, 6.19% while that of LUMO orbital is principally dispersed on C4, C6, O14, O13 atoms with corresponding orbital composition of 14.99%, 13.32%, 11.02%, 9.40%. Likewise, HOMO orbital of compound H4 is greatly concentrated on the following atoms O11, C3, C5 with respective orbital composition of 17.50%, 6.34%, 4.33% while the LUMO orbital is majorly positioned on O11, C1 atoms depicting orbital composition of 11.97%, 11.89% respectively. Moreover, compound H5 HOMO orbital was found to be highly circulated on the subsequent atoms O13, O9, O11, C3, C4 having orbital composition 18.72%, 13.59%, 11.58%, 8.01%, 5.64% while the LUMO orbital is predominantly localized on O9, O17, C5, C3, O10 with individual orbital composition of 19.40%, 17.11%, 12.81%, 10.71%, 6.32%. In summary, it was observed that mostly, oxygen atoms with same atomic labelling contributed both in the HOMO and LUMO orbital of the studied cyclohexanone derivatives as a result of lone pair on the oxygen atoms and negative inductive effect of oxygen atom respectively.
3.1.2 Natural bond orbital (NBO) analysis
NBO is a computational approach that gives the most plausible method for elucidating interactions between Lewis valence orbital (donor) and non-Lewis valence orbital (acceptor) in a molecule (Undiandeye et al., 2022; Patrick-Inezi et al., 2022; Nemykin et al., 2021). It is a powerful tool used to decipher complex Schrödinger wave equation into a simple and more assimilating chemical bonding concept (Patrick-Inezi et al., 2022). NBO analysis was conducted by employing DFT/PBE0/6-31+G(d,p) level of theory in order to comprehend intramolecular and intermolecular hyperconjugation, charge transfer, delocalization of electron within the candidate molecules (Unimuke et al., 2022). The most interacting Lewis valence orbitals and non-Lewis valence orbitals of the studied compounds were computed by the second order perturbation theory analysis of the Fock matrix (Undiandeye et al., 2022; Golding Sheeba et al., 2021). From the second order perturbation theory analysis of the Fock matrix calculation, the interaction of the Lewis valence orbital (donor) and non-Lewis valence orbital (acceptor) were measured as a function of stabilization energy
or second order perturbation energy
. Large stabilization energy
reflects stronger interaction between the donor and acceptor orbital. The stabilization energy
involved with the delocalization of electron between Lewis valence orbital and non-Lewis valence orbital is predicted using equation (1) (Undiandeye et al., 2022; Patrick-Inezi et al., 2022; Nemykin et al., 2021; Shahab et al., 2021).
3.1.3 Electrochemical properties
Electrochemical properties such as redox potential, energy density and theoretical charge capacity of the investigated cyclohexanone derivatives was computed to ascertain their performance as a possible organic cathode (positive) electrode material for lithium-ion battery (Chen et al., 2018; Lopez et al., 2019; Meng and Arroyo-de Dompablo, 2013). Estimated electrochemical properties of free optimized cyclohexanone derivatives is laid out in Table 1. In view from the table, it can be seen that redox potential of the studied free optimized structure of cyclohexanone derivatives is of the trend H2>H5 > H4 > H1 > H3. From the trend, it was observed that compound H2 exhibited the highest redox potential of 1.868 V and highest energy density (447.09 W h kg−1) with corresponding theoretical charge capacity of 239.34 mA h g−1. We can infer that the high redox potential of compound H2 is as a result of localized distribution of the two redox-active carbonyl group attached to it with minimum steric effect which increases the concentration of electron density on one side and thus can be reduced easily. From this line of reasoning, we deduced that compound H2 is the best organic positive electrode material for lithium-ion battery application among the studied free optimized structure of cyclohexanone derivatives. We also noticed that compound H3 portrayed the least redox potential of 1.492 V with resultant energy density and theoretical charge capacity of 317 W h kg−1 and 212.74 mA h g−1 respectively. The low redox potential of compound H3 is attributed to uniform distribution of three redox carbonyl moiety on the compounds which in turn result to increased uniform concentration of electron density and thus can’t be reduced easily. However, H5 was observed to have the least energy density of 311.92 W h kg−1 with redox potential of 1.792 V. The low energy density of H5 is due to its poor theoretical charge capacity (174.06 mA h g−1) arising from high molecular weight of the compound contributed by 5 redox-active carbonyl moieties attached to it. In general, it can be seen that the number and distribution of carbonyl group attached to the studied organic carbonyl compounds greatly affect their redox potential (Li et al., 2021).
Compound
Ered (V)
Energy Density (W h kg−1)
Charge capacity (mA h g−1)
H1
1.508
412.48
273.53
H2
1.868
447.09
239.34
H3
1.492
317.41
212.74
H4
1.789
343.54
191.47
H5
1.792
311.92
174.06
3.2 Cyclohexanone derivatives with EDG
3.2.1 Frontier molecular orbital
The result obtained from the computed HOMO – LUMO energy levels of the candidate molecules with EDG (NH2) is exhibited in Table S2. It was observed that the result obtained from their various energy gap follows the trend H1-EDG > H3-EDG > H2-EDG > H4-EDG > H5-EDG with corresponding values of 3.512, 2.957, 2.081, 2.050, 0.772 eV. From the trend of the result, it is revealed that H1-EDG possesses the highest energy gap of 3.512 eV while H5-EDG possesses the least Eg of 0.772 eV. The least Eg of H5-EDG is as a result of combined delocalization of lone pairs of electrons from five oxygen atoms attached to it and the lone pair of electron on EDG into the cyclo ring thus pushing excess electron into the cyclo ring which in turn increases the electron density (Pelzer et al., 2017) of the cyclo ring while the high Eg of H1-EDG can be attributed to delocalization of lone pair of electron from only one oxygen atom attached to it and the lone pair of electron from the EDG into the cyclo ring. Thus, pushing less electron into the cyclo ring.
3.2.2 Natural bond orbital (NBO) analysis
NBO of all the titled molecules fine-tuned with EDG (NH2) were also explored. NBO and stabilization energy of the most interacting orbitals of the investigated compounds is displayed in Table S4. From the table, it was observed that H5-EDG possesses the highest stabilization energy of 39.55 kcal/mol among the investigated compounds. The high value for H5-EDG resulting from π* π* antibonding transition is attributed to intermolecular charge transfer (Wu et al., 2020) from donor C4 – O8 to acceptor C3 – O12 and also from the intense delocalization of electron density contributed by the presence of EDG.
3.2.3 Electrochemical properties
Influence of EDG on electrochemical properties of the studied cyclohexanone derivatives was also considered by fine tuning their chemical structures with EDG (NH2). The trend of the result obtained from their redox potential is as follows H2-EDG > H1-EDG > H3-EDG > H4-EDG > H5-EDG. It is notable that on substitution of the cyclohexanone derivatives with EDG (NH2), the redox potential of H1-EDG, H2-EDG, H3-EDG was found to increase compared to its free optimized counterpart while that of H4-EDG and H5-EDG was found to decrease. Among the studied cyclohexanone derivatives, H2-EDG was also found to have the highest reduction potential of 1.976 V with energy density and theoretical charge capacity of 417.07 W h kg−1 and 211.07 mA h g−1 respectively. This can be attributed to the fact that the electron density of the compound increases due to the EDG (Thomsen et al., 2018) attached to them which in turn reduces their tendency to accept more electron in order to get reduced i.e their electron affinity is low. We also confirmed that H5-EDG displayed the least redox potential of 1.286 V with corresponding low energy density and low theoretical charge capacity of 203.97 W h kg−1, 158.61 mA h g−1 respectively. The low redox potential of H5-EDG is as a result of its high reactivity contributed by five oxygen atoms of redox-active carbonyl moiety group attached to it thus making it to dissolve in the electrolyte quickly. This observation is confirmed from its frontier molecular orbital (FMO) analysis of which it exhibited the least energy gap value. H1-EDG was found to show the highest energy density of 444.79 W h kg−1 among the studied compounds. This result is due to its high theoretical charge capacity of 237.22 mA h g−1 and moderate redox potential of 1.875 V which are the two main properties for computing energy density of an electrode material. Evaluated electrochemical properties of the investigated cyclohexanone derivatives tuned with EDG (NH2) is visible in Table 2.
Compound
Ered (V)
Energy Density
(W h kg−1)Charge capacity
(mA h g−1)
H1-EDG
1.875
444.79
237.22
H2-EDG
1.976
417.07
211.07
H3-EDG
1.792
340.68
190.11
H4-EDG
1.634
282.58
172.94
H5-EDG
1.286
203.97
158.61
3.3 Cyclohexanone derivatives with EWG
3.3.1 Frontier molecular orbital
Table S5 depicts the result obtained from the HOMO and LUMO energy levels of the investigated compounds with electron withdrawing (fluorine) group (EWG). It can be seen that the result obtained from their energy gap follows a decreasing trend of H1-EWG > H2-EWG > H3-EWG > H4-EWG > H5-EWG with respective values of 3.731, 3.302, 2.887, 1.904, 0.532 eV. This result demonstrates that compound H1-EWG possesses the highest energy gap of 3.731 eV while H5-EWG possesses the lowest energy gap of 0.532 eV. The high energy gap of H1-EWG is accredited to the fact that the electron withdrawing (fluorine) group attached to compound H1 containing only one oxygen atom withdraws more electron from the cyclo ring i.e the (−) inductive effect of the EWG dominates the delocalization of lone pair electron (Antonov et al., 2016; Zhu et al., 2017) from only one oxygen atom attached to it into the cyclo ring, thus rendering the cyclo ring to be electron-poor which in turn makes it less reactive with its corresponding high energy gap. On the other hand, H5-EWG having the least energy gap is as a result of high electron density of the cyclo ring contributed by the delocalization of lone pairs of electrons into the cyclo ring from the five oxygen atoms attached to it compared to withdrawal of electron by the EWG (fluorine) attached to it i.e delocalization of lone pair of electrons into the cyclo ring from the five oxygen atoms dominates the (-) inductive effect of the electron withdrawing group (fluorine). Thus, making the cyclo ring to be electron-rich due to excess electrons that is pushed into it which in turn makes H5-EWG the most reactive with least energy gap.
3.3.2 Natural bond orbital (NBO) analysis
NBO of the cyclohexanone derivatives functionalized with EWG was also computed to understand the nature of donor–acceptor interaction persisting within the studied compounds. Table S6 summarizes the result obtained from the second order perturbation theory analysis of the investigated compounds with EWG (fluorine). On substituting EWG (fluorine) to the investigated compounds, it is obvious that the most prominent interaction was observed in H5-EWG resulting from intermolecular hyperconjugative interaction of LP(2) O10 C1 – C2 with stabilization energy of 22.80 kcal/mol which is lesser compared to stabilization energy of H5 (23.31 kcal/mol) in free optimized state. This is due to the fact that delocalization of electron density in H5-EWG is reduced as a result of presence of EWG (fluorine) which tends to withdraw electron from the molecules in order to fill its vacant P-acceptor orbital thus behaving as a non-Lewis valence orbital (acceptor) too.
3.3.3 Electrochemical properties
More interestingly, on tuning the chemical structures of cyclohexanone derivatives with functional groups such as EWG (fluorine), the redox potential of the studied compounds was found to increase for H1-EWG and H2-EWG with respective values of 2.138 V and 2.144 V when compared to both free optimized structures and optimized structures tuned with EDG (NH2). This is as a result of strong (-) inductive effect (Antonov et al., 2016) of fluorine group (EWG) which tends to withdraw more electron density from their cyclo ring into its vacant-orbital from less delocalized electron density contributed from one and two carbonyl group attached to them respectively. However, estimated redox potential was found to decrease for H3-EWG, H4-EWG, H5-EWG having corresponding values of 1.354, 1.553, 1.387 V when compared to that of free optimized structures. This unusual behavior exhibited by H3-EWG, H4-EWG, H5-EWG can be related to the intense delocalization of electron density contributed by increased number of carbonyl groups attached to them with lone pair of electrons. The trend of the redox potential result obtained from the studied cyclohexanone derivatives is as follows H2-EWG > H1-EWG > H4-EWG > H5-EWG > H3-EWG. From this analysis, we can infer that H2-EWG possesses the highest redox potential of 2.144 V among the investigated compounds with energy density and theoretical charge capacity of 442.09 W h kg−1 and 206.20 mA h g−1 respectively. The high redox potential of H2-EWG can also be attributed to its localized distribution of the two redox-active carbonyl group attached to it with minimum steric effect which makes electron density to be concentrated on one side and thus can be reduced easily.
On the other hand, it is of notice that H3-EWG showed the least redox potential of 1.354 V with corresponding energy density and theoretical charge capacity of 252.04 W h kg−1 and 186.15 mA h g−1 respectively. The poor redox potential of H3-EWG is as a result of uniform distribution of three carbonyl group (Kim et al., 2016) with minimum steric effect on the compound leading to uniform concentration of electron density stabilized by strong resonance effect on the compound. Also, H5-EWG demonstrated the least energy density which is attributed to its high reactivity and instability and thus can dissolve in the electrolyte effortlessly. This result is validated from the estimated energy gap of compound H5-EWG aforementioned in Section 3.3.1.
To this end, it was found that H1-EWG exhibited the highest energy density of 494.05 W h kg−1 among all (free optimized, tuned with EDG and EWG) the investigated cyclohexanone derivatives with respective redox potential and theoretical charge capacity of 2.138 V, 231.08 mA h g−1. Computed electrochemical properties of cyclohexanone derivatives functionalized with EWG (fluorine) is pointed out in Table 3.
Compound
Ered (V)
Energy Density
(W h kg−1)Charge capacity
(mA h g−1)
H1-EWG
2.138
494.05
231.08
H2-EWG
2.144
442.09
206.20
H3-EWG
1.354
252.04
186.15
H4-EWG
1.553
263.48
169.66
H5-EWG
1.387
216.16
155.85
3.4 Electrochemical properties of cyclohexanone derivatives with Li – Ion
Electrochemical properties of the cyclohexanone derivatives were also studied on lithiation of the compounds with only one Li atom for free optimized structures and optimized structures structurally engineered with functional groups such as EDG (NH2) and EWG (fluorine).
3.4.1 Lithiated pure cyclohexanone derivatives
Free optimized structures of cyclohexanone derivatives were lithiated by binding one lithium atom to the high electronegative and electron rich oxygen atom of the redox active carbonyl moiety group. We performed electrochemical properties calculation on lithiation of the investigated cyclohexanone derivatives. From the result obtained, it was observed that the redox potential of the studied compounds ranges from 1.097 to 5.168 V vs Li/Li+. As evident from the result presented in Table 4., H3-Li was observed to reflect the highest redox potential of 5.168 V vs Li/Li+ with high energy density of 1041.61 W h kg−1 and theoretical charge capacity of 201.55 mA h g−1. The high redox potential for H3-Li can be attributed to the probability of the high electropositive lithium ion to possibly bind to the three-uniform distributed electronegative oxygen atoms on the redox active carbonyl group as a result of strong electron cloud surrounding the cyclo ring. The high redox potential of compound H3-Li is in harmony with that reported by Kim and coworkers in their DFT work on carbonyl functionalized graphene oxide (5.3 V vs Li/Li+) as cathode material for Li-ion battery using PBE0 hybrid functional (Kim et al., 2016). H2-Li was detected to exhibit the highest energy density (1108.28 W h kg−1) among the studied compound as a result of its high redox potential of 4.92 V vs Li/Li+ and moderate theoretical charge capacity (225.26 mA h g−1). In addition, compound H4-Li demonstrated electrochemical properties of 1.711 V vs Li/Li+, 312 W h kg−1, 182.35 mA h g−1. The redox potential obtained from H4-Li is in excellent agreement with experimental redox potential obtained from Lu and coworkers (Lu et al., 2019) on cyclohexanehexone (1.7 V vs Li/Li+) and also in close range with experimental redox potential observed by Miroshnikov and coworkers (Miroshnikov et al., 2019) on their work on Tetrakislawsone (1.9 V vs Li/Li+) as cathode material in LIBs. Moreover, compound H5-Li displayed electrochemical properties of 1.638 V vs Li/Li+, 272.72 W h kg−1, 166.49 mA h g−1. Our redox potential obtained theoretically from H5-Li is in compliance to that gotten by Yang et al. (Yang et al., 2020) on their experimental work on truxenone-base covalent organic framework (∼1.6 V vs Li/Li+) as carbonyl-based organic cathode material for Lithium-ion batteries.
Compound
Ered (V)
Experimental Ered (V)
Energy density (W h kg−1)
Charge Capacity (mA h g−1)
H1-Li
1.097
280.05
255.29
H2-Li
4.92
1108.28
225.26
H3-Li
5.168
1041.61
201.55
H4-Li
1.711
1.7 [ref Lu et al., 2019], 1.9 [ref Miroshnikov et al., 2019]
312.00
182.35
H5-Li
1.638
∼1.6 [ref Yang et al., 2020]
272.72
166.49
3.4.2 Lithiated cyclohexanone derivatives fine-tuned with EDG
Modification of electrochemical properties of lithiated cyclohexanone derivatives by fine-tuning their chemical structures with EDG (NH2) was also considered. As laid out in Table 5., it is plain that redox potential of the lithiated cyclohexanone derivatives functionalized with EDG was found to decrease as compared to lithiated free optimized structures of which is between the range of 0.575 to 5.011 V vs Li/Li+. From the estimated electrochemical properties of the investigated compounds, it is visible that H2-Li-EDG displayed the highest redox potential of 5.011 V vs Li/Li+ and also highest energy density (1002.4 W h kg−1) among the lithiated compounds modified with EDG having theoretical charge capacity of 200.04 mA h g−1. This high redox potential of H2-Li-EDG can be as a result that the presence of the EDG increases the localized concentration of electron density (Kim et al., 2016) on one side and thus can be reduced by the highly electropositive and electrophilic Li-ion without stress. On the contrary, H1-Li-EDG was observed to exhibit the least redox potential of 0.575 V vs Li/Li+ with energy density of 128.44 W h kg−1 and theoretical charge capacity of 223.38 mA h g−1. In the same light, H3-Li-EDG depicted redox potential of 1.978 V vs Li/Li+ with respective energy density and theoretical charge capacity of 358.26 W h kg−1, 181.12 mA h g−1. The observed redox potential of H3-Li-EDG is in line with experimental redox potential validated by Kim and coworkers (Kim et al., 2016) on their work anthraquinone substituted with EDG (NH2) (2.1 V vs Li/Li+) and also to that obtained by Jung and coworkers using density functional theory method with PBE0 hybrid functional on 2-Amino anthraquinone (2.1 V vs Li/Li+) as organic quinone-based material for possible application as positive electrode material in Lithium-ion batteries (Jung et al., 2020). Furthermore, it is clearly observed that H4-Li-EDG exhibited redox potential of 1.718 V vs Li/Li+ having individual energy density and charge capacity of 284.28 W h kg−1, 165 mA h g−1. The estimated redox potential of H4-Li-EDG is in close range to that reported experimentally by Kim and coworkers (Kim et al., 2016) on their work on amino group functionalized anthraquinone (2.0 V vs Li/Li+) as cathode material for LIBs and also to the theoretical redox potential (1.9 V vs Li/Li+) observed by Jung et al. using DFT method on amino group substituted anthraquinone-based compound when utilized as organic cathode material for Lithium-ion batteries (Jung et al., 2020). At this end, H5-Li-EDG was found to display electrochemical properties of 1.655 V vs Li/Li+, 252.06 W h kg−1, 152.30 mA h g−1. The observed redox potential of H5-Li-EDG varies a little bit from that reported by Kim and coworkers on lithiation of 2-amino anthraquinone (1.4 V vs Li/Li+) with only one Li atom at DFT/PBE0/6-31+G(d,p) (Kim et al., 2016).
Compound
Ered (V)
Experimental Ered (V)
Energy density (W h kg−1)
Charge Capacity (mA h g−1)
H1-Li-EDG
0.575
128.44
223.38
H2-Li-EDG
5.011
1002.4
200.04
H3-Li-EDG
1.978
2.1[ref Jung et al., 2020]
358.26
181.12
H4-Li-EDG
1.718
2.0 [ref Kim et al., 2016]
284.28
165.47
H5-Li-EDG
1.655
252.06
152.30
3.4.3 Lithiated cyclohexanone derivatives fine-tuned with EWG
Molecular engineering of cyclohexanone derivatives lithiated with only one lithium atom was also conducted by fine tuning their chemical structures with EWG (fluorine) in order to validate its influence on their electrochemical properties when utilized as an organic cathode material for lithium-ion battery. Evaluated electrochemical properties of lithiated cyclohexanone derivatives fine-tuned with EWG is unveiled in Table 6. From the result obtained, it is crystal clear that the trend of redox potential of the studied cyclohexanone derivatives lithiated by only one lithium atom in the presence of EWG is as follows H2-Li-EWG > H5-Li-EWG > H3-Li-EWG > H4-Li-EWG > H1-Li-EWG with individual values of 3.313, 3.145, 2.133, 1.654, 1.108 V vs Li/Li+. From the result, it is comprehendible that H2-Li-EWG manifested the highest redox potential of 3.313 V vs Li/Li+ with specific energy density of 648.22 W h kg−1 and theoretical charge capacity of 195.66 mA h g−1. This can be as a result of presence of EWG which tends to withdraw the locally concentrated electron density within the compound easily by negative inductive effect and thus making the bounded lithium atom to also reduce less available electron easily which in turn leads to the compound having high electron affinity which relates with redox potential as reported by (Kim et al., 2019; Sood et al., 2018; Allam et al., 2018). This redox potential exhibited by H2-Li-EWG is in tandem with that reported by Wang and coworkers on their experimental work on CF3 substituted quinone based conductive redox polymer (3.2 V vs Li/Li+) (Wang et al., 2019) as a cathodic material for LIBs. Also, H1-Li-EWG and H5-Li-EWG displayed higher redox potential than its corresponding counterpart in free optimized state and structurally engineered with EDG. This can be attributed to inductive effect of the EWG attached to it which withdraws more electron from the compounds and thus raising their electron affinity leading to their high redox potential (Kim et al., 2016). In the same line, redox potential of 2.133 V vs Li/Li+ manifested by H3-Li-EWG is in agreement with the experimental redox potential obtained by Kim et al. (Kim et al., 2016) on their work on anthraquinone functionalized with EWG (–COOH) (2.36 V vs Li/Li+) as an organic cathode material for LIBs and also on theoretical calculation conducted by Jung and coworkers using DFT/PBE0/6-31+G(d,p) level of theory on Anthraquinone-2-Carboxylic acid (2.4 V vs Li/Li+) for potential application as organic cathode material in Lithium-ion batteries (Jung et al., 2020). To this end, H4-Li-EWG exhibited redox potential of (1.654 V vs Li/Li+) which is in relation to that obtained by Kim and coworkers on one Li atom bounded to anthraquinone substituted with an EWG (–COOH) at DFT/PBE0/6–31+(d,p) theory level (Kim et al., 2016).
Compounds
Ered (V)
Experimental Ered (V)
Energy density
(W h kg−1)Charge Capacity
(mA h g−1)
H1-Li-EWG
1.108
241.47
217.93
H2-Li-EWG
3.313
3.2 [ref Wang et al., 2019]
648.22
195.66
H3-Li-EWG
2.133
2.36 [ref Kim et al., 2016]
378.65
177.52
H4-Li-EWG
1.654
268.71
162.46
H5-Li-EWG
3.145
470.87
149.72
3.5 Comparison between investigated cyclohexanone derivatives with and without lithium – Ion
In comparison between the electrochemical properties of the investigated cyclohexanone derivatives with and without bounded lithium atom for the free optimized structures, it is recognizable that H2-Li and H3-Li exhibited redox potential (4.92 V vs Li/Li+ and 5.168 V vs Li/Li+) and energy density (1108.28 W h kg−1, 1041.61 W h kg−1) higher than that of those without lithium atom (1.868 V, 1.492 V) with their specific energy density of 447.09 W h kg−1, 317.41 W h kg−1. This result shows that presence of Li atom aids in improving redox potential of the outlined compounds which in turn increases its energy density since redox potential is a crucial parameter used for computing its energy density of which is in total agreement with the work of Kim and coworkers on quinone derivatives (Kim et al., 2016).
More importantly, for cyclohexanone derivatives with and without one bounded lithium atom structurally modified with EDG, we observed that redox potential and energy density of H2-Li-EDG to H5-Li-EDG were higher for the lithiated compounds compared to its counterpart without lithium atom while H1-Li-EDG redox potential (0.575 V vs Li/Li+), energy density (128.44 W h kg−1) and theoretical charge capacity (223.38 mA h g−1) is lower compared to that without bounded Li atom. This result implies that presence of more than one redox-active carbonyl group aids in improving the redox potential of lithiated compounds in the sense that they provide more electron density for the highly electropositive and electrophilic Li atom to bind and get reduced easily and at such improves its energy density as well which was also confirmed by Kim and coworkers on their study on functionalization of graphene oxide for cathode material in LIBs (Kim et al., 2016). Among the aforementioned results, H2-Li-EDG and H2-EDG i.e with and without bounded Li atom was found to exhibit the highest values for its electrochemical properties with corresponding redox potential, energy density and theoretical charge capacity of 5.011 V vs Li/Li+, 1002.4 W h kg−1, 200.04 mA h g−1 and 1.976 V, 417 W h kg−1, 211.07 mA h g−1.
Furthermore, we also liken the results from the structurally designed cyclohexanone derivatives with EWG in the presence and absence of bounded one Li atom. As clearly expressed from their respective results, it was observed that redox potential of H2-Li-EWG, H3-Li-EWG, H4-Li-EWG, H5-Li-EWG were higher in contrast to its fellow compound without bounded Li atom. This result also validates that presence of Li atom increases redox potential which is also confirmed by ref. (Kim et al., 2016; Xu et al., 2020). However, reverse is the case for H1-Li-EWG to its relative structure without Li atom. Low redox potential of H1-Li-EWG (1.108 V vs Li/Li+) can be stressed to be as a result of poor availability of electron density from only one redox-active carbonyl moiety for highly electropositive and electron-loving Li atom to attack properly.
In general, we totally observed that among all the studied compounds, H2 with and without functional groups for unlithiated compounds revealed the highest redox potential and also for its corresponding lithiated compound with exception for its free optimized structure. Also, the theoretical charge capacity of all the investigated cyclohexanone derivatives without Li atom was found to be higher than those with Li atom. This result is verified from the additional molecular weight contributed by the added one Li atom and theoretical charge capacity varies inversely with molecular weight of the studied compounds (Jeong et al., 2020; Li et al., 2020; Xu et al., 2012; Yang et al., 2017).
3.6 Binding energy
Binding energy was also computed for the lithiated cyclohexanone derivatives in free optimized state, with EDG and EWG in order to confirm the use of cyclohexanone derivatives as an organic positive electrode material in LIBs. During the lithiation of cyclohexanone derivatives, the electropositive Li atom preferentially binds to the redox active and electronegative oxygen atom of the carbonyl group by Lewis acid – Lewis base interaction leading to the formation of Li–O chemical bond (Kim et al., 2016). Results obtained from the calculated binding energies of the studied cyclohexanone derivatives is represented in Table S7. From the table, it is evident that all the studied cyclohexanone derivatives (in free optimized state, with EDG and EWG) demonstrated negative value of binding energy indicating that binding of the electron-deficient Li atom to the electron-rich redox active carbonyl group present in the cyclohexanone derivatives is favourable. Moreover, a more negative binding energy indicates a strong Li–O chemical bond and this was observed mostly on lithiation of cyclohexanone derivatives fine-tuned with EWG.
4 Conclusions
In conclusion, we have theoretically investigated five set of selected cyclohexanone derivatives with redox-active carbonyl moiety as prospective organic cathode material for rechargeable Li-ion battery using density functional theory (DFT) model. Electrochemical properties of the titled compounds with and without one lithium atom and also on modification with functional groups was thoroughly screened. The results revealed that the distribution and number of carbonyl groups attached to the studied compounds played a greater role in their stability and electrochemical properties. H2 with 2 locally distributed carbonyl group exhibited the highest redox potential among its fellow counterparts both in the absence and presence of one lithium atom with an exception of the free optimized structure with lithium atom. The investigated cyclohexanone derivatives demonstrated appreciating redox potential and energy density in the presence of one lithium atom than those without lithium atom. On modification of the studied compounds (lithiated and unlithiated) with functional groups EWG (fluorine) and EDG (NH2), the structurally designed compounds with EWG tends to exhibit better redox potential and energy density due to the fact that EWG withdraws electron from the compound and thus increases their electron affinity which correspond to their high redox potential compared to EDG counterpart. Among the lithiated cyclohexanone derivatives, compound H2 and H3 in free optimized state with one bounded Li atom manifested better electrochemical property of 4.92 V vs Li/Li+, 1108.28 W h kg−1, 225.26 mA h g−1 and 5.168 V vs Li/Li+, 1041.61 W h kg−1, 201.55 mA h g−1 respectively, thus making them the most suitable organic carbonyl-based cathode material for LIBs among the studied cyclohexanone derivatives.
Funding
This work was not funded by any agency.
Author Contributions
Hitler Louis: Project conceptualization, design, supervision, and administration. ThankGod C. Egemonye: Writing, editing, analysis, and manuscript draft. Henry O. Edet, Terkumbur E. Gber: Analysis, writing, editing. Tomsmith O. Unimuke: Methodology and analysis. Victoria M. Bassey: Resources, review, and editing. Adedapo S. Adeyinka: Validation, review, and editing.
Acknowledgements
The authors would like to acknowledge the center for high performance computing (CHPC), South Africa for providing computational resources for this research project. Also, the authors will like to acknowledge the University of Johannesburg and the Research Center for Synthesis and Catalysis for prividing funding for the APC charges.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Application of DFT-based machine learning for developing molecular electrode materials in Li-ion batteries. RSC Adv.. 2018;8(69):39414-39420.
- [Google Scholar]
- A new class of ionically conducting fluorinated ether electrolytes with high electrochemical stability. J. Am. Chem. Soc.. 2020;142(16):7393-7403.
- [Google Scholar]
- Characterization of electronic and ionic transport in Li1-xNi0. 33Mn0. 33Co0. 33O2 (NMC333) and Li1-xNi0. 50Mn0. 20Co0. 30O2 (NMC523) as a function of Li content. J. Electrochem. Soc.. 2016;163(8):A1512.
- [Google Scholar]
- A carbonyl compound-based flexible cathode with superior rate performance and cyclic stability for flexible lithium-ion batteries. Adv. Mater.. 2018;30(4):1703868.
- [Google Scholar]
- 2-iminopyridine nickel (II) complexes bearing electron-withdrawing groups in the ligand core: Synthesis, characterization, ethylene oligo-and polymerization behavior. J. Organomet. Chem.. 2016;822:241-249.
- [Google Scholar]
- Antimalarial potential of naphthalene-sulfonic acid derivatives: Molecular electronic properties, vibrational assignments, and in-silico molecular docking studies. J. Mol. Struct. 2022:133298.
- [Google Scholar]
- Conjugated microporous polymer based on star-shaped triphenylamine-benzene structure with improved electrochemical performances as the organic cathode material of Li-ion battery. Electrochim. Acta. 2018;286:187-194.
- [Google Scholar]
- Solid polymer electrolytes incorporating cubic Li7La3Zr2O12 for all-solid-state lithium rechargeable batteries. Electrochim. Acta. 2017;258:1106-1114.
- [Google Scholar]
- Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater.. 2002;1(2):123-128.
- [Google Scholar]
- A perspective on organic electrode materials and technologies for next generation batteries. J. Power Sources. 2021;482:228814
- [Google Scholar]
- NBO, version 3.1, Gaussian. PA: Inc.: Pittsburgh; 2003.
- Molecular structure, vibrational spectroscopic, frontier molecular orbital and natural bond orbital analysis of anti-cancer drug 6-chloro-3-pyridine carbonitrile. Spectroscopy Lett.. 2021;54(6):419-436.
- [Google Scholar]
- Noninvasive in situ NMR study of “dead lithium” formation and lithium corrosion in full-cell lithium metal batteries. J. Am. Chem. Soc.. 2020;142(49):20814-20827.
- [Google Scholar]
- Strategies towards enabling lithium metal in batteries: interphases and electrodes. Energy Environ. Sci.. 2021;14(10):5289-5314.
- [Google Scholar]
- Ambiguities in solvation free energies from cluster-continuum quasichemical theory: lithium cation in protic and aprotic solvents. PCCP. 2021;23(30):16077-16088.
- [Google Scholar]
- Electrochemical characteristics of cyanoquinones as organic cathodes for high-potential sodium-ion batteries. ACS Sustainable Chem. Eng.. 2020;8(30):11328-11336.
- [Google Scholar]
- Synergistic effect of incorporating intra-and inter-molecular charge transfer in nonfullerene acceptor molecules for highly-efficient organic solar cells. J. Mater. Chem. A. 2021;9(31):16834-16840.
- [Google Scholar]
- Conjugacy of organic cathode materials for high-potential lithium-ion batteries: Carbonitriles versus quinones. Energy Storage Mater.. 2020;24:237-246.
- [Google Scholar]
- practical challenges hindering the development of solid state Li ion batteries. J. Electrochem. Soc.. 2017;164(7):A1731.
- [Google Scholar]
- Thermodynamic and redox properties of graphene oxides for lithium-ion battery applications: a first principles density functional theory modeling approach. PCCP. 2016;18(30):20600-20606.
- [Google Scholar]
- First-principles density functional theory modeling of Li binding: thermodynamics and redox properties of quinone derivatives for lithium-ion batteries. J. Am. Chem. Soc.. 2016;138(7):2374-2382.
- [Google Scholar]
- Unveiled correlations between electron affinity and solvation in redox potential of quinone-based sodium-ion batteries. Energy Storage Mater.. 2019;19:242-250.
- [Google Scholar]
- A review on the stability and surface modification of layered transition-metal oxide cathodes. Mater. Today. 2021;46:155-182.
- [Google Scholar]
- Engineering redox potential of lithium clusters for electrode material in lithium-ion batteries. J. Cluster Sci.. 2017;28(5):2779-2793.
- [Google Scholar]
- Using high-HFP-content cathode binder for mitigation of heat generation of lithium-ion battery. Int. J. Energy Res.. 2017;41(14):2430-2438.
- [Google Scholar]
- Kinetic phase evolution of spinel cobalt oxide during lithiation. ACS Nano. 2016;10(10):9577-9585.
- [Google Scholar]
- In-situ construction of stable cathode/Li interfaces simultaneously via different electron density azo compounds for solid-state lithium metal batteries. Energy Storage Mater. 2021
- [Google Scholar]
- An aromatic carbonyl compound-linked conjugated microporous polymer as an advanced cathode material for lithium-organic batteries. Mater. Chem. Front.. 2020;4(9):2697-2703.
- [Google Scholar]
- Designing polymers for advanced battery chemistries. Nat. Rev. Mater.. 2019;4(5):312-330.
- [Google Scholar]
- Understanding the lithiation mechanisms of pyrenetetrone-based carbonyl compound as cathode material for lithium-ion battery: Insight from first principle density functional theory. Mater. Chem. Phys. 2021:125518.
- [Google Scholar]
- Cyclohexanehexone with ultrahigh capacity as cathode materials for lithium-ion batteries. Angew. Chem.. 2019;131(21):7094-7098.
- [Google Scholar]
- An Insoluble Benzoquinone-Based Organic Cathode for Use in Rechargeable Lithium-Ion Batteries. Angew. Chem.. 2017;129(41):12735-12739.
- [Google Scholar]
- Organic Cathode Materials for Lithium-Ion Batteries: Past, Present, and Future. Adv. Energy Sustainability Res.. 2021;2(1):2000044.
- [Google Scholar]
- Recent advances in first principles computational research of cathode materials for lithium-ion batteries. Acc. Chem. Res.. 2013;46(5):1171-1180.
- [Google Scholar]
- Made from henna! A fast-charging, high-capacity, and recyclable tetrakislawsone cathode material for lithium ion batteries. ACS Sustainable Chem. Eng.. 2019;7(16):13836-13844.
- [Google Scholar]
- Accurate Prediction of Mossbauer Hyperfine Parameters in Bis-Axially Coordinated Iron (II) Phthalocyanines Using Density Functional Theory Calculations: A Story of a Single Orbital Revealed by Natural Bond Orbital Analysis. Inorg. Chem.. 2021;60(6):3690-3706.
- [Google Scholar]
- Onori, S., Serrao, L., Rizzoni, G., 2016. Hybrid electric vehicles: Energy management strategies.
- Systematic molecular design of ketone derivatives of aromatic molecules for lithium-ion batteries: First-principles DFT modeling. ChemSusChem. 2017;10(7):1584-1591.
- [Google Scholar]
- Analeptic activity of 2-Hydroxyl-5-Nitrobenzaldehyde: Experimental, DFT studies, and in silico molecular docking approach. Healthcare Analytics. 2022;2:100030
- [Google Scholar]
- Effects of functional groups in redox-active organic molecules: A high-throughput screening approach. J. Phys. Chem. C. 2017;121(1):237-245.
- [Google Scholar]
- Geometry Optimization, UV/Vis, NBO, HOMO and LUMO, Excited State and Antioxidant Evaluation of Pyrimidine Derivatives. Lett. Org. Chem.. 2021;18(6):465-476.
- [Google Scholar]
- Revealing practical specific capacity and carbonyl utilization of multi-carbonyl compounds for organic cathode materials. PCCP. 2021;23(23):13159-13169.
- [Google Scholar]
- Electrochemical and electronic properties of nitrogen doped fullerene and its derivatives for lithium-ion battery applications. J. Energy Chem.. 2018;27(2):528-534.
- [Google Scholar]
- Sreenidhi, P.R., SD, B. S., 2021, August. Study on Positive Electrode material in Li-ion Battery. In: 2021 Second International Conference on Electronics and Sustainable Communication Systems (ICESC), IEEE, pp. 246–251.
- Probing Cyclic π-Electron Delocalization in an Imidazol-2-ylidene and a Corresponding Imidazolium Salt. Chem.–A Eur. J.. 2018;24(19):4973-4981.
- [Google Scholar]
- Energy storage emerging: A perspective from the Joint Center for Energy Storage Research. Proc. Natl. Acad. Sci.. 2020;117(23):12550-12557.
- [Google Scholar]
- Gaussian09, Revision D. 01. Wallingford CT: Gaussian, Inc.; 2013.
- Reactive azo compounds as a potential chemotherapy drugs in the treatment of malignant glioblastoma (GBM): Experimental and theoretical studies. J. Photochem. Photobiol.. 2022;10:100116
- [Google Scholar]
- Spectroscopic, conformational analysis, structural benchmarking, excited state dynamics, and the photovoltaic properties of Enalapril and Lisinopril. J. Indian Chem. Soc. 2022:100500.
- [Google Scholar]
- Meta-Hybrid Density Functional Theory Prediction of the Reactivity, Stability, and IGM of Azepane, Oxepane, Thiepane, and Halogenated Cycloheptane. ACS Omega 2022
- [Google Scholar]
- Spectroscopic and molecular electronic property investigation of 2-phenylpyrimidine-4, 6-diamine via 1H-NMR, UV-vis, FT-Raman, FT-IR, and DFT approach. J. Mol. Struct. 2022
- [Google Scholar]
- Redox-state-dependent interplay between pendant group and conducting polymer backbone in quinone-based conducting redox polymers for lithium ion batteries. ACS Appl. Energy Mater.. 2019;2(10):7162-7170.
- [Google Scholar]
- Recent progress in carbonyl-based organic polymers as promising electrode materials for lithium-ion batteries (LIBs) J. Mater. Chem. A. 2020;8(24):11906-11922.
- [Google Scholar]
- Organotrisulfide: a high capacity cathode material for rechargeable lithium batteries. Angew. Chem.. 2016;128(34):10181-10185.
- [Google Scholar]
- An empirical model for the design of batteries with high energy density. ACS Energy Lett.. 2020;5(3):807-816.
- [Google Scholar]
- Capacitive charge storage enables an ultrahigh cathode capacity in aluminum-graphene battery. J. Energy Chem.. 2020;45:40-44.
- [Google Scholar]
- Recent progress in cathode materials research for advanced lithium ion batteries. Mater. Sci. Eng.: R: Reports. 2012;73(5–6):51-65.
- [Google Scholar]
- Olivine LiMn x Fe 1–x PO 4 cathode materials for lithium ion batteries: restricted factors of rate performances. J. Mater. Chem. A. 2021;9(25):14214-14232.
- [Google Scholar]
- A Truxenone-based Covalent Organic Framework as an All-Solid-State Lithium-Ion Battery Cathode with High Capacity. Angew. Chem. Int. Ed.. 2020;59(46):20385-20389.
- [Google Scholar]
- A density functional theory study on the thermodynamic and dynamic properties of anthraquinone analogue cathode materials for rechargeable lithium ion batteries. PCCP. 2017;19(19):12480-12489.
- [Google Scholar]
- A dynamic stability design strategy for lithium metal solid state batteries. Nature. 2021;593(7858):218-222.
- [Google Scholar]
- Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy. 2020;5(7):526-533.
- [Google Scholar]
- Functionalized NbS2 as cathode for Li-and Na-ion batteries. Appl. Phys. Lett.. 2017;111(4):043903
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104026.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




