

Translate this page into:
New synthetic quinaldine conjugates: Assessment of their anti-cholinesterase, anti-tyrosinase and cytotoxic activities, and molecular docking analysis
⁎Corresponding authors. falgethami@imamu.edu.sa (Faisal K. Algethami), Hichem.BenJannet@fsm.rnu.tn (Hichem Ben Jannet)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Synthesis of new quinoline-succinimide derivatives. Synthesized derivatives were screened for their anti-tyrosinase and cytotoxic activities. Compounds 3d, 3e and 3 g showed interesting tyrosinase inhibitory activities. Compounds 3f, 3 g and 3a displayed the highest activity against HeLa cell lines. Molecular docking analysis reinforced the experimental results.
Abstract
Despite all the progress made to enrich the existing bank of drugs used to treat and cure Alzheimer and cancer patients, there is still a need to research and develop new bioactive candidates with superior efficacy but minimal side effects. In this context, a new series of anti-butyrylcholinesterase (anti-BChE), anti-tyrosinase and cytotoxic succinimide linked quinaldine conjugates 3a-i was designed and synthesized starting from 8-hydroxyquinaldine. The condensation of quinoleine-hydrazide 2 with electrophilic species such as aromatic and nonaromatic anhydrides provided the new compounds 3a-i. These synthesized heterocycles were characterized by spectroscopic means (1H NMR, 13C NMR and ESI-HRMS). Their anti-butyrylcholinesterase, anti-tyrosinase and cytotoxic (cervical cancer cell (HeLa) and lung cancer cell (A549)) activities have been evaluated in vitro. Compounds 3e and 3 g were found to be more anti-BChE than Galanthamine. Compounds 3d, 3e and 3 g exerted better anti-tyrosinase activity than kojic acid. Also, 3a, 3f and 3 g showed interesting cytotoxic potential towards HeLa cell lines. These results were supported by the molecular docking analysis (structure–activity relationship (SAR)) to estimate and discuss possible interactions between these derivatives and active sites of proteins butyrylcholinesterase (PDB: 4B0P), tyrosinase (PDB: 2Y9X) and cytotoxic (topoisomerase IIα enzyme (PDB: 5GWK)).
Keywords
Quinaldine linked succinimides
Anti-butyrylcholinesterase
Anti-tyrosinase
Cytotoxic
Molecular docking
SAR analysis

1 Introduction
Cancer is a major disease, the cases of which have not ceased to increase in recent decades all over the world (Rodrigues et al., 2016; Teo et al., 2017). Thus, the discovery of new synthesized molecules for the treatment of cancer remains an important challenge for the healthcare community. However, human cervical cancer is representing the second most common cancer disease, after breast cancer, in the world with an annual incidence of five hundred and twenty-eight thousand (528.000) new cases in developed countries (Wang et al., 2018).
Current treatments for cervical cancer have many disadvantages, such as resistance to drugs loss of efficacy, side-effects and pharmacokinetic tolerability (Dasari et al., 2015; Sawaya et al., 2001). Therefore, development of new drugs effective against cervical cancer is essential. Moreover, among the different types of malignant tumors, lung cancer is the deadliest one in worldwide, causing approximately 1.69 million deaths in 2015 (Baek et al., 2018). For the discovery of potential agents targeting cervical and lung cancers, many efforts have been focused. Previous research indicates that besides its essential presence in the central and peripheral nervous systems, cholinesterase (ChE) also has different isoforms that constitute several other cells in the body, such as rat kidney cells, erythrocytes, vascular endothelial, leukocytes, nerve endings, brain and lungs cells (Patocka et al., 2004). It has been shown that in addition to their role in the hydrolysis of butyrylcholine, ChEs have other non-classical biological functions that influence apoptotic sensitivity, cell proliferation and differentiation, thus showing a possible involvement in tumorigenesis (Xi et al., 2015).
On the other hand, ChE has been shown to be involved in the regulation of tumor development. Indeed, abnormal expression and structural alteration of ChE have been observed in several tumors, such as brain and lung cancers (De Castro et al., 2008; Montenegro et al., 2006; Muñoz-Delgado et al., 2010; Vidal, 2005).
Melanoma is a threatening tumor expressing tyrosinase, the immunogenic pigmentation enzyme, and spreads from cells originating from the neural crest. The increase in tyrosinase activity during tumorigenesis allows for selective treatment of this kind of tumor (Vargas et al., 2011). Anterior studies revealed positive results in the treatment of melanoma with the induction of a cytotoxic T cell response using tyrosinase as the tumor-associated antigen (Colella et al., 2000; Meyer et al., 2005).
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder with the defects in cognitive function and central nervous system (Yurtta et al., 2017). In AD, there is a thinking loss, memory impairment, neuroinflammation and cognitive decline (Graham, et al., 2017; Ringman, 2017). At the present time, the total number of people affected by this disease was over fifty millions (Alzheimer’s Association, 2015). The exact pathogenesis of this ailment is not fully known and this disease could be termed as a neurological disorder caused by multiple factors, such as oxidative stress, hyperphosphorylated T-protein, and extracellular β-amyloid plaques (Ringman, 2017; Tang et al., 2012; Tumiatti et al., 2010).
On the other hand, inhibiting the enzymatic activity of butyrylcholinesterase BChE, also called pseudocholinesterase or plasma (cholin)esterase, offers a strategy for the treatment of neurodegenerative diseases by preventing the degradation of choline, an important neurotransmitter associated with memory (Makhaeva et al., 2016). The determination of plasma butyrylcholinesterase activity can also be used to assess liver function since both hypercholinesterasemia and hypocholinesterasemia indicate a pathological process.
It has been reported that a dual anti-cancer and cholinesterase inhibitory activity is possible and can be considered as a valuable combination for the treatment of both cancer and AD prevention (Jakub et al., 2020; Temel, 2017).
Tyrosinase is a copper containing oxidase that extremely exists in animals, microorganisms and plants and it is responsible for biosynthesis of melanin pigment in skin, eyes and hair by catalyzing the bio-oxidation of phenol to o-quinone (Fan et al., 2017; Ramsden and Riley, 2014; Yoshimori et al., 2014). Lately, unregulated tyrosine action has also been reported to be linked with neurodegenerative diseases (Asanuma et al., 2003; Ghani and Ullah, 2010; Komori et al., 2014). Inhibiting the tyrosinase activity may reduce the browning of foods, eliminates hyperpigmentation spots of human skin and causes a skin-whitening, by preventing the enzymatic oxidation and melanin production (Zhang et al., 2011).
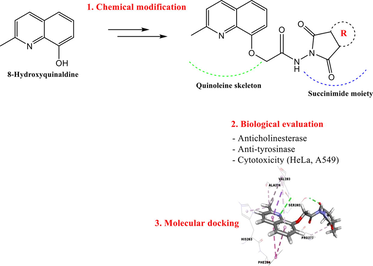
Recent studies have demonstrated that a variety of natural products, such as alkaloids exhibit excellent anti-tyrosinase (Piechowska et al., 2020), anticholinesterase (Cespedes et al., 2017) and anticancer effects (Filali et al., 2016; Luo and Song, 2021; Pompeo et al., 2019). Among the alkaloids of pharmacological interest, quinoline derivatives constitute a major class well known for their various biological and pharmacological activities such as: anticholinesterase (Fig. 1-A) (Bazine et al., 2020), antimicrobial (Fig. 1-B) (Katariya et al., 2020), antioxidant (Fig. 1-C) (El-Saghier et al., 2021), anticancer (Fig. 1-D) (Afzal et al., 2015), antidiabetic (Fig. 1-E) (Ganesan, et al., 2020) and anti-tyrosinase activities (Fig. 1-F) (Gardelly et al., 2021).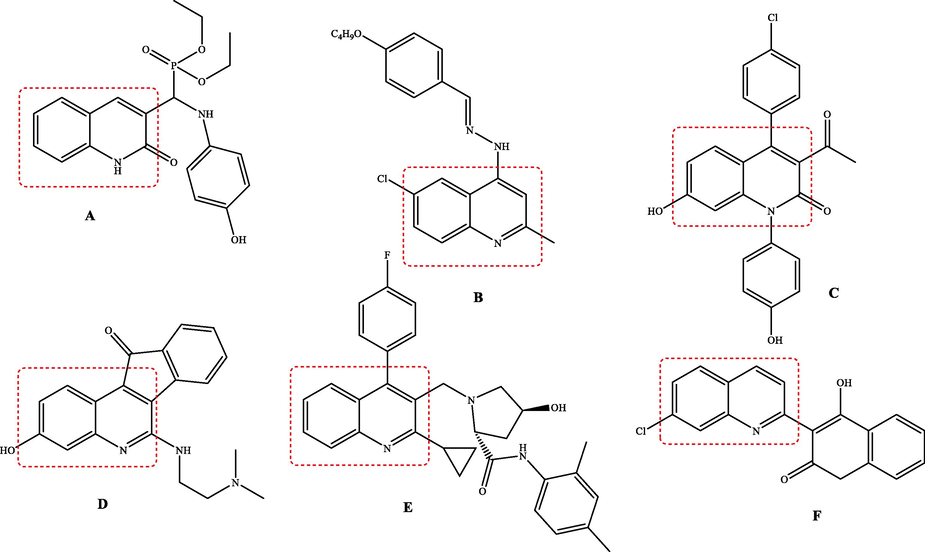
Structures of bioactive quinoline derivatives.
Moreover, the pharmacophore succinimide fragment has received important attention due to its broad spectrum of biological and pharmacological applications. Likewise, succinimides are known as anticancer (Fig. 2-A) (Han et al., 2016), anti-diabetic (Fig. 2-B) (Hussain et al., 2019), antimicrobial (Fig. 2-C) (Cvetković et al., 2019), anticholinesterase (Fig. 2-D) (Ahmad et al., 2019) and anti-tyrosinase (Fig. 2-E) (Ha et al., 2011) agents.
Structures of bioactive succinimide derivatives.
Encouraged by the data above, we thought of designing and synthesizing new derivatives by combining the 8-hydroxyquinaldine and succinimide fragments in order to obtain hybrid molecules with interesting biological activities. Such a combination has never been described before where we find in the same molecule these two moieties, both known for their various biological activities. The evaluation of anticholinesterase, anti-tyrosinase and cytotoxic activities against the human cell lines: cervical cancer cell (HeLa) and lung cancer cell (A549) of all the synthesized compounds has been studied and reported here. The structure–activity relationship (SAR) was discussed and further supported by molecular docking prediction.
2 Experimental
2.1 General experimental methods
All chemicals were obtained from commercial suppliers and used without further purification. 1H (300 MHz) and 13C (75 MHz) NMR spectra were recorded on a DPX 300 spectrometer (Bruker BioSpin, USA), using CDCl3 and DMSO‑d6 as solvents and a nondeuterated residual solvent as an internal standard. Melting points were measured on a DSC-50 Shimadzu apparatus (Kyoto, Japan). ESI-HRMS were recorded with a Q-TOF Agilent mass spectrometer (Agilent Technologies, Inc).
2.2 Chemical synthesis
2.2.1 General procedure synthesis of ethyl 2-((2-methylquinolin-8-yl)oxy)acetate (1)
8-Hydroxyquinaldine (1 equiv.) was dissolved in anhydrous DMF to which ethyl chloroacetate (1.5 equiv.) was added in the presence of potassium carbonate (2 equiv.). The mixture was stirred for 12 h at room temperature, then poured into ice-cold water, and extracted with ethyl acetate. After removal of solvent, the resulting residue was purified by silica gel column chromatography (Petroleum ether (EP)/EtOAc) followed by the recristallization in ethanol to give compound 1.
2-((2-methylquinolin-8-yl)oxy)acetate (1). White powder, yield: 92 %; m.p.: 80–82 °C. NMR 1H (300 MHz, CDCl3) δ: 8.01 (d, J = 8.4 Hz, 1H), 7.40 (dd, J = 8.2, 1.5 Hz, 1H), 7.36 (d, J = 7.4 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 6.96 (dd, J = 7.3, 1.5 Hz, 1H), 4.94 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 2.79 (s, 3H), 1.29 (t, J = 7.1 Hz, 3H). NMR 13C (75 MHz, CDCl3) δ: 169.1, 158.5, 153.2, 139.7, 136.2, 127.9, 125.4, 122.8, 120.9, 110.1, 66.5, 61.5, 25.5, 14.2. HRMS (ESI) m/z: 246.1126 [M + H]+ calcd. For (C14H16NO3)+: 246.1130.
2.2.2 General procedure synthesis of 2-((2-methylquinolin-8-yl)oxy)acetohydrazide (2)
Hoping the preparation of hydrazide 2, compound 1 was stirred with hydrazine hydrate (NH2NH2·H2O, 1 equiv.) in ethanol under reflux for 5 h. When the reaction is completed, the precipitate is filtered. The hydrazide 2 was obtained as a white solid.
2-((2-methylquinolin-8-yl)oxy)acetohydrazide (2). White powder, yield: 94 %; m.p.: 88–90 °C. NMR 1H (300 MHz, CDCl3) δ: 10.57 (s, 1H, NH), 8.06 (d, J = 8.4 Hz, 1H), 7.48 (dd, J = 8.1, 1.2 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.17 (dd, J = 7.4, 1.1 Hz, 1H), 4.82 (s, 2H), 3.29 (s, 2H, NH2), 2.80 (s, 3H). NMR 13C (75 MHz, CDCl3) δ: 169.1, 158.9, 154.0, 140.1, 136.8, 128.1, 126.1, 123.1, 122.2, 114.3, 71.1, 25.4. HRMS (ESI) m/z: 254.0917 [M + Na]+ calcd. For (C12H13N3O2Na)+: 254.0905.
2.2.3 General procedure synthesis of pyrrolidinedione derivatives (3a-i)
A mixture of hydrazide 2 (1 equiv.), cyclic anhydride (1 equiv.) and 10 mL of glacial acetic acid was refluxed for 24 h. After completion of the reaction, the solid product formed was collected by filtration and washed twice with water to give 3a-i as white powders in 50–87 % yields. Their perfect purity has been well confirmed through their 1H and 13C NMR spectra given as Supplementary materials.
N-(1,3-dioxoisoindolin-2-yl)-2-((2-methylquinolin-8-yl)oxy)acetamide (3a). White powder, yield: 71 %; m.p.: 138–140 °C. NMR 1H (300 MHz, CDCl3) δ: 12.73 (s, 1H, NH), 8.11 (d, J = 8.5 Hz, 1H), 7.91 (dd, J = 5.4, 3.1 Hz, 2H), 7.78 (dd, J = 5.4, 3.0 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.34 (dd, J = 10.7, 7.9 Hz, 2H), 5.02 (s, 2H), 2.62 (s, 3H). NMR 13C (75 MHz, CDCl3) δ: 168.8, 165.1, 158.5, 137.3, 134.6, 130.5, 128.2, 126.4, 124.0, 123.1, 123.0, 116.8, 72.6, 25.0. HRMS (ESI) m/z: 362.1141 [M + H]+ calcd. For (C20H16N3O4)+: 362.1141.
N-(1,3-dioxo-1,3,3a,4,7,7a-hexahydro-2H-4,7-methanoisoindol-2-yl)-2-((2-methylquinolin-8-yl)oxy)acetamide (3b). White powder, yield: 76 %; m.p.: 160–162 °C. NMR 1H (300 MHz, CDCl3) δ: 12.12 (s, 1H, NH), 8.09 (d, J = 8.5 Hz, 1H), 7.54 (dd, J = 8.2, 1.3 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.33 (d, J = 6.0 Hz, 1H), 7.30 (dd, J = 6.3, 1.1 Hz, 1H), 6.21 (s, 2H), 4.92 (s, 2H), 3.45 (dt, J = 4.5, 1.6 Hz, 2H), 3.37 (dd, J = 2.8, 1.6 Hz, 2H), 2.65 (s, 3H), 1.78 (dt, J = 8.8, 1.7 Hz, 1H), 1.56 (d, J = 8.7 Hz, 1H). NMR 13C (75 MHz, CDCl3) δ: 173.8, 167.7, 158.5, 137.3, 134.9, 128.2, 126.4, 123.1, 122.8, 116.4, 72.3, 52.0, 48.5, 46.6, 45.1, 44.5, 25.1. HRMS (ESI) m/z: 378.1456 [M + H]+ calcd. For (C21H20N3O4)+: 378.1454.
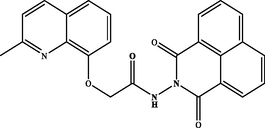
N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-2-((2-methylquinolin-8-yl)oxy)acetamide (3c). White powder, yield: 87 %; m.p.: 184–186 °C. NMR 1H (300 MHz, DMSO‑d6) δ: 11.56 (s, 1H, NH), 8.66 – 8.45 (m, 4H), 8.27 (d, J = 8.4 Hz, 1H), 7.97 – 7.89 (m, 3H), 7.62 (dd, J = 8.1, 1.2 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.46 (d, J = 8.5 Hz, 1H), 7.42 (dd, J = 7.6, 1.2 Hz, 1H), 5.11 (s, 2H), 2.64 (s, 3H). NMR 13C (75 MHz, DMSO‑d6) δ: 167.5, 161.4, 157.7, 136.3, 135.4, 135.2, 132.4, 131.5, 127.5, 127.4, 125.8, 122.6, 121.6, 121.4, 119.0, 113.5, 68.8, 24.8. HRMS (ESI) m/z: 412.1296 [M + H]+ calcd. For (C24H18N3O4)+: 412.1297.
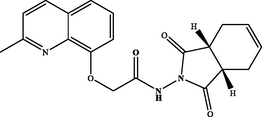
N-((3aR,7aS)-1,3-dioxo-1,3,3a,4,7,7a-hexahydro-2H-isoindol-2-yl)-2-((2-methylquinolin-8-yl)oxy)acetamide (3d). White powder: 70 %; m.p.: 134–136 °C. NMR 1H (300 MHz, CDCl3) δ: 12.75 (s, 1H, NH), 8.09 (d, J = 8.5 Hz, 1H), 7.55 (dd, J = 8.1, 1.1 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.34 – 7.29 (m, 2H), 6.02 – 5.90 (m, 2H), 4.94 (s, 2H), 3.33 – 3.14 (m, 2H), 2.70 – 2.61 (m, 2H), 2.59 (s, 3H), 2.36 – 2.23 (m, 2H). NMR 13C (75 MHz, CDCl3) δ: 176.3, 158.4, 137.3, 128.2, 127.9, 126.4, 125.4, 123.1, 122.9, 116.6, 72.4, 40.0, 37.8, 26.1, 24.9, 23.6. HRMS (ESI) m/z: 366.1455 [M + H]+ calcd. For (C20H20N3O4)+: 366.1454.
N-((3aR,7aS)-1,3-dioxooctahydro-2H-isoindol-2-yl)-2-((2-methylquinolin-8-yl)oxy)acetamide (3e). White powder, yield: 72 %; m.p.: NMR 1H (300 MHz, CDCl3) δ: 12.40 (s, 1H, NH), 8.10 (d, J = 8.5 Hz, 1H), 7.56 (dd, J = 8.2, 1.3 Hz, 1H), 7.51 – 7.43 (m, 1H), 7.33 (dd, J = 7.9, 1.6 Hz, 2H), 4.96 (s, 2H), 3.06 – 2.95 (m, 2H), 2.94 – 2.85 (m, 2H), 2.64 (s, 3H), 2.14 – 1.98 (m, 2H), 1.96 – 1.86 (m, 2H), 1.86 – 1.69 (m, 2H). NMR 13C (75 MHz, CDCl3) δ: 180.1, 176.3, 168.3, 158.4, 137.3, 128.2, 126.4, 123.1, 122.9, 116.4, 72.3, 42.6, 38.9, 26.2, 24.9, 24.1, 23.8, 21.9. HRMS (ESI) m/z: 368.1614 [M + H]+ calcd. For (C20H22N3O4)+: 368.1610.
N-(5-methyl-1,3-dioxoisoindolin-2-yl)-2-((2-methylquinolin-8-yl)oxy)acetamide (3f). White powder, yield: 83 %; m.p.: 150–152 °C. NMR 1H (300 MHz, DMSO‑d6) δ: 11.48 (s, 1H, NH), 8.27 (d, J = 8.5 Hz, 1H), 7.90 – 7.79 (m, 2H), 7.74 (d, J = 7.4 Hz, 1H), 7.65 – 7.58 (m, 1H), 7.54 – 7.43 (m, 2H), 7.35 (dd, J = 7.6, 1.2 Hz, 1H), 5.08 (s, 2H), 3.29 (s, 3H), 2.64 (s, 3H). NMR 13C (75 MHz, DMSO‑d6) δ: 168.01, 164.9, 164.9, 157.8, 153.4, 146.4, 139.4, 136.4, 135.6, 129.7, 127.8, 126.7, 125.8, 124.2, 123.7, 122.7, 121.6, 113.8, 68.9, 24.8, 21.4. HRMS (ESI) m/z: 376.1308 [M + H]+ calcd. For (C21H18N3O4)+: 376.1297.
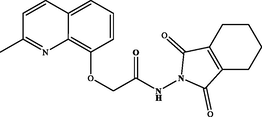
N-(1,3-dioxo-1,3,4,5,6,7-hexahydro-2H-isoindol-2-yl)-2-((2-methylquinolin-8 yl)oxy)acetamide (3 g). White powder: 84 %; m.p.: 144–146 °C. NMR 1H (300 MHz, DMSO‑d6) δ: 11.25 (s, 1H, NH), 8.26 (d, J = 8.4 Hz, 1H), 7.59 (dd, J = 8.1 Hz, 1H), 7.50–7.45 (m, 2H), 7.29 (d, J = 7.8 Hz, 1H), 5.01 (s, 2H), 2.64 (s, 3H), 2.29 (s, 4H), 1.70 (s, 4H). NMR 13C (75 MHz, DMSO‑d6) δ: 168.1, 167.6, 157.8, 153.4, 140.4, 139.4, 136.3, 127.5, 125.7, 122.6, 121.5, 113.7, 68.8, 66.3, 24.8, 20.6, 19.5. HRMS (ESI) m/z: 366.1459 [M + H]+ calcd. For (C20H20N3O4)+: 366.1454.
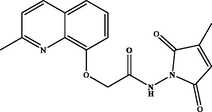
N-(3-methyl-2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-2-((methylquinolin-8-yl)oxy)acetamide (3 h). White powder: 81 %; m.p.: 130–132 °C. NMR 1H (300 MHz, CDCl3) δ: 12.39 (s, 1H, NH), 8.13 (d, J = 8.4 Hz, 1H), 7.56 (dd, J = 8.2, 1.1 Hz, 1H), 7.49 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 8.5 Hz, 1H), 7.34 (dd, J = 7.5, 1.2 Hz, 1H), 6.45 (dd, J = 3.4, 1.6 Hz, 1H), 4.96 (s, 2H), 2.67 (s, 3H), 2.14 (d, J = 1.8 Hz, 3H). NMR 13C (75 MHz, CDCl3) δ: 168.7, 167.4, 158.5, 154.1, 145.2, 137.6, 128.2, 126.9, 126.5, 123.2, 122.8, 116.2, 72.0, 24.8, 11.6. HRMS (ESI) m/z: 326.1142 [M + H]+ calcd. For (C17H16N3O4)+: 326.1141.
N-(3,4-dimethyl-2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-2-((methylquinolin-8 yl)oxy)acetamide (3i). White powder: 78 %; m.p.: 138–140 °C. NMR 1H (300 MHz, DMSO‑d6) δ: 11.86 (s, 1H, NH), 8.27 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.50–7.45 (m, 2H), 7.29 (d, J = 7.6 Hz, 1H), 4.97 (s, 2H), 2.65 (s, 3H), 1.95 (s, 6H). NMR 13C (75 MHz, DMSO‑d6) δ: 168.8, 168.0, 157.8, 153.3, 139.3, 136.5, 127.5, 125.8, 122.7, 121.5, 113.6, 68.7, 24.8, 8.6. HRMS (ESI) m/z: 340.1295 [M + H]+ calcd. For (C18H18N3O4)+: 340.1297.
2.3 Biological evaluation
2.3.1 Anti‑butyrylcholinesterase activity
Anticholinesterase (BChE) activity was assayed as described by Ellman et al. method with some modifications (Znati et al., 2020). 50 μL of buffer phosphate solution (pH = 8, 0.1 M), 25 µL of BChE (equine serum, Sigma-Aldrich), 25 µL of the synthesized compounds (100 µM in DMSO 1 % dissolved in in the same buffer) and 125 μL of 5,5-dithiobis-2-nitrobenzoic acid (DTNB) (3 mM in buffer, pH 8.1). The mixtures were incubated for 15 min at room temperature. The reaction was then initiated with the addition of 25 μL of butyarylthiocholine iodide BTCI and measured at 412 nm by Thermo-fisher scientific mutliscan GO®. Anti-BChE activity was calculated as a percentage compared to an assay using a buffer without test sample using nonlinear regression. IC50 values are means ± SD. Galanthamine was used as a positive control.
2.3.2 Anti-tyrosinase activity
Anti-tyrosinase activity was studied according to literature protocol with slight modifications (Chortani et al., 2019). 880 μL of substrate (l-Dopa dissolved in 50 mM phosphate buffer, pH 6.8, 2 mM) was mixed with 100 μL of the synthesized compounds (in DMSO) at 25 °C for 2 min. Next, 20 μL of mushroom tyrosinase enzyme (1000 U/mL in phosphate buffer (0.1 M)) was added to initiate the mixture. The assay reaction was incubated at 25 °C for 10 min. The increase in absorbance at 475 nm due to the formation of amount of dopachrome was monitored by spectrophotometry. Anti-tyrosinase activity was calculated as a percentage compared to an assay using a buffer without test sample using nonlinear regression. IC50 values are means ± SD. Kojic acid was used as a positive control. All measurements were conducted in triplicate.
2.3.3 Cytotoxic activity
The synthesized compounds were tested for their cytotoxic activity towards two human cells lines; cervical cancer cell (HeLa) and a human lung cancer cell (A549), as well as on normal mouse fibroblast cell line (L929) (Zardi-Bergaoui et al., 2019). Cells were seeded at 5*103 cells/well in 100 µL of growth medium and incubated at 37 °C for 24 h to adhere. The cells were treated by synthesized derivatives (1 μL at different concentrations) and incubated for 48 h; then 10 µL of (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) (5 mg/mL) was added to each well and the incubation for 2 h. Then, 100 µL of DMSO were added to each well. The absorbance was measured at 540 nm by spectrophotometry. Ellipticine (A549) and Doxorubucin (HeLa) were used as positive controls and the assays were performed in triplicate. The percentages of cell growth were calculated as follow: Cell growth (%) = [A (sample)/ A (control)] × 100 and IC50 (µM) were obtained by logarithmic curve.
2.4 Molecular docking procedure
Structures of the studied products were generated and optimized using ACD (3D viewer) software. The enzymatic crystal structures of tyrosinase (PDB: 2Y9X), cholinesterase (PDB: 4B0P) and DNA topoisomerase IIα (PDB: 5GWK) were downloaded from the protein data bank (https://www.rcsb.org). Before starting, water molecules and the complexed inhibitor ligands were removed. Then, the polar hydrogens were added to the protein structures. Hence, the grid box in the three target enzymes are with spacing of 0.375 Å. The grid box is of dimension 40 × 45 × 40 points and centered with the coordinates x: −8.684, y: −29.177, and z: −44.399 for anti-tyrosinase activity (Tyrosinase, PDB: 2Y9X). For anticholinesterase activity (Butyrylcholinesterase, PDB: 4B0P), it is of 50 × 40 × 40 points and centered with the coordinates x: 40.415, y: 20.400, and z: 23.480 and 40 × 40 × 40 points and centered with the coordinates x: 23.350, y: −38.580, and z: −59.570 for cytotoxic activity (DNA topoisomerase IIα, PDB: 5GWK).
The molecular docking analysis of the potent active products at enzymatic binding sites was performed using AutoDock Vina software (Trott and Olson, 2010). Binding poses and ligand-enzyme interactions were construed with Python Molecule Viewer and Biovia Discovery Studio Visualizer.
2.5 Statistical analysis
Statistical analysis was carried out with the help of Graph Pad Prism 7.0 (Graph Pad Software Inc., CA, and USA). All the experimental data are given as the mean ± standard error of the mean (SEM). The difference between two groups was evaluated using Student's t-test. Significant difference among three or more groups was determined by one-way ANOVA with a post hoc analysis.
3 Results and discussion
3.1 Chemistry
Our approach to the target compounds 3a-i, firstly started by the construction of the ester skeleton 1, via condensation reaction of 8-hydroxyquinaldine with ethyl chloroacetate to give the corresponding ester. The starting compound 1 was obtained in good yield (92 %) (Scheme 1). The structure of compound 1 was established according to its spectral data (1H and 13C NMR spectra and ESI-HRMS). In fact, in addition to the signals corresponding to the protons introduced by the 8-hydroxyquinaldine, new signals of the ester moiety (2-ethoxy-2-oxoethyl) were observed in the 1H NMR spectrum, which also showed a singlet at δ 4.94 (2H) assigned to the methylene group, a quadruplet at δ 4.29 (q, J = 7.1 Hz, 2H) attributable to the methylene group, and a triplet at δ 1.29 (t, J = 7.1 Hz, 3H) relative to the methyl protons. The 13C NMR spectrum confirmed the introduction of the ester group by the observation of new signals at δ 14.2, 66.5 and 169.1 attributable to methyl, methylene and carbonyl groups, respectively. Furthermore, the quinoline hydrazide 2 was prepared by condensation of the compound 1 with hydrazine hydrate in ethanol at room temperature (Scheme 1). The 1H NMR spectrum of compound 2 showed the presence of new signals, attributable to NH and NH2 protons of the hydrazide moiety at δ 10.57 (s, 1H, NH) and 3.29 (s, 2H, NH2), respectively.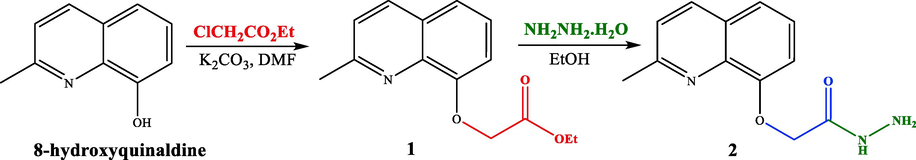
Synthetic pathway to quinoline hydrazide 2.
Hydrazide 2 constitutes an important intermediate for the formation of diverse heterocyclic derivatives known for their high reactivity and their bioactivity (Gan et al., 2017; Sławiński et al., 2017). This has aroused our attentiveness for its use to design and synthesize the new target compounds 3a-i by using the hydrazide function in 2 to attach a succinimide moiety to the quinaldine (Table 1).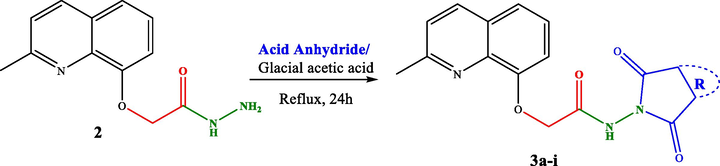 .
.
Entry
Product
Entry
Product
3a
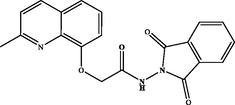
71 %3f
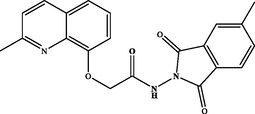
83 %
3b
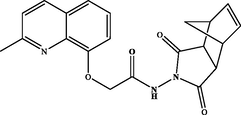
76 %3 g
84 %
3c
87 %3 h
81 %
3d
70 %3i
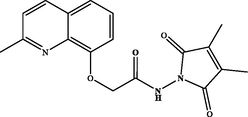
78 %
3e
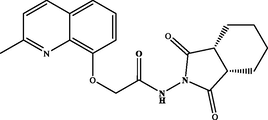 72 %
72 %
The hydrazide 2 was reacted with a series of cyclic anhydrides under refluxing glacial acetic acid for 24 h to give compounds 3a-i in good yields (70–87 %) (Table 1). The newly formed derivatives 3 were characterized by their 1H and 13C NMR spectra and ESI-HRMS. The 1H NMR spectra of compounds 3a-i showed, in addition to the signals of the quinaldine moiety, the appearance of a new singlet at δ 11.25–12.75 due to the NH group. The 13C NMR spectra of the same derivatives essentially showed the appearance of a new signal relative to the carbonyl group at δ 158.4–180.1.
3.2 Biological evaluation
3.2.1 Anti‑butyrylcholinesterase activity
The butyrylcholinesterase inhibitory of compounds 3a-i and their precursors 1 and 2 has been assayed and the IC50 (µM) values are indicated in Table 2. The results showed that compound 2 (IC50 = 33.2 ± 2.0 µM) was found to be more active than its precursor 1 (IC50 = 69.2 ± 3.4 µM). This finding can be explained the incorporation of the hydrazide group (Bingul et al., 2020; Sıcak et al., 2019). On the other hand, the compounds 3a-i displayed higher activity than 1 and 2 with IC50 values ranging from 1.1 ± 0.2 to 58.0 ± 2.0 µM. These results reveal the importance of the association between succinimide and quinoline to inhibit the BChE enzyme (Ahmad et al., 2020; Sadiq et al., 2015). Indeed, the compound 3 g with a succinimide moiety bearing a 3,4,5,6-tetrahydrophthalic group displayed the highest anti-BChE effect with an IC50 value of 1.1 ± 0.2 µM, followed by 3e (IC50 = 3.1 ± 0.5 µM) with a succinimide moiety bearing a 4-cyclohexene-cis-1 2-dicarboxylic system. These two compounds exerted better activities than Galanthamine used as a positive control (IC50 = 3.7 ± 0.3 µM). These findings show the importance of the non aromatic moieties introduced by the used anhydrides (case of 3 g and 3e) in improving this activity compared to aromatic systems attached to the corresponding anhydrides used to prepare 3a, 3c and 3f. The comparison of the anti-BChE effect of 3d (IC50 = 9.1 ± 1.0 µM) bearing a 1,2,3,6-tetrahydrophthalic (Δ4,5) with that of 3 g (IC50 = 1.1 ± 0.2 µM) with a 3,4,5,6-tetrahydrophthalic (Δ1,2), allows to conclude that the position of the double bond in the tetrahydrophthalic moiety intervenes to increase/decrease the BChE inhibitory. It has been noticed, on the other hand, that the additional methyl group on the pyrrole-2,5-dione ring in the compound 3i compared to its analogue 3 h with only one methyl group in the same ring, contributed, via its donor inducing effect, to enhance this activity, but while remaining less active compared to compounds 3a, 3c, 3d, 3e, 3f and 3 g. Values in a column followed by the same superscript letter are not significantly different at P < 0.05 (LSD test).
Sample
Anticholinesterase IC50 (μM)*
Anti-tyrosinase IC50 (μM)*
Cytotoxicity IC50 (μM)**
HeLa
A549
1
69.2 ± 3.4 k
˃ 100 h
44.6 ± 3.0j
˃ 100j
2
33.2 ± 2.0 h
˃ 100 h
12.1 ± 1.5 h
˃ 100j
3a
15.1 ± 1.1f
23.2 ± 2.1f
2.0 ± 0.2c
44.0 ± 3.7i
3b
40.4 ± 2.7i
37.2 ± 2.2 g
13.2 ± 1.3f
19.1 ± 2.0 g
3c
07.0 ± 1.2d
˃ 100 h
13.9 ± 1.7i
˃ 100j
3d
09.1 ± 1.0e
4.7 ± 1.0b
10.0 ± 1.5e
11.5 ± 1.0c
3e
03.1 ± 1.2c
6.9 ± 1.9c
14.0 ± 1.0 g
18.2 ± 1.8e
3f
07.6 ± 1.3d
15.9 ± 1.2e
0.7 ± 0.1b
39.2 ± 3.0 h
3 g
01.1 ± 1.2b
8.1 ± 1.9d
1.7 ± 0.4d
5.1 ± 0.8b
3 h
58.0 ± 2.0j
˃ 100 h
˃ 100 h
25.3 ± 1.0f
3i
29.3 ± 1.8 g
˃ 100 h
˃ 100 h
19.7 ± 1.1d
Doxorubicin
0.3 ± 0.02a
Ellipticine
0.3 ± 0.07a
Galanthamine
3.7 ± 0.3a
Kojic acid
13.6 ± 0.5a
The contribution of the succinimide and the quinoline moieties to the anti-BChE activity is supported by the data of the literature which show the importance of both moieties in improving the anti-BChE potential (Ahmad et al., 2020; Gardelly et al., 2021; Mo et al., 2019).
3.2.2 Anti-tyrosinase activity
All the synthesized derivatives 1, 2 and 3a-i, were evaluated for their in vitro anti-tyrosinase activity. According to the IC50 values shown in Table 2, it was found that derivatives 3a-i, except compounds 3c, 3 h and 3i, were more active than their precursors 1 and 2, demonstrating the clear important contribution of the introduced succinimide moiety, which appears to be involved in the anti-tyrosinase activity. Compounds 3d, 3e and 3 g were found to display the highest tyrosinase inhibiting potential (IC50 = 4.7 ± 1.0, 6.9 ± 1.9 and 8.1 ± 1.9 µM, respectively) compared to the positive control kojic acid (IC50 = 13.6 ± 0.5 μM). Indeed, the compound 3d with a cis-1,2,3,6-tetrahydrophthalic system exhibited the highest activity. By comparing the structures and activities of derivatives 3d and 3e, it was found that the presence and the position of the double bond in the succinimide system in 3d is certainly at the origin of the improvement of this activity. On the other hand, the higher activity of compound 3f with a 4-methylphtalic system (IC50 = 15.9 ± 1.2 µM) compared with that of 3a bearing an unsubstituted phthalic system (IC50 = 23.2 ± 2.1 µM) shows that the methyl group in C-4 position in 3f is clearly at the origin of the improvement of the anti-tyrosinase activity.
The results showed the net contribution, even the responsibility, of the introduced succinimide fragment to improve the activity of precursors 1 and 2. This finding was found in good agreement with the literature data showing the contribution of the succinimide fragment in some anti-tyrosinase molecules (Chortani et al., 2019; Zhao et al., 2020).
3.2.3 Cytotoxic activity
The cytotoxic effects of synthesized heterocyclic 3a-i and their precursors 1 and 2 were evaluated towards HeLa and A549 cancer cell lines, as well as on L929 (normal mouse fibroblast cell line) using the standard MTT assay. The obtained results of the cytotoxic activity are presented in Table 2, and all the tested compounds did not show any toxicity against L929 cell line (IC50 > 500 μM). It was found that the hydrazide 2 is more active than the ester 1 against HeLa with IC50 values of 12.1 ± 1.5 and 44.6 ± 3.0 µM, respectively. This finding showed that the NH-NH2 system is more effective than the ethoxy group against HeLa cell line (Çıkla et al., 2013; Roullier et al., 2010). On the other hand, the succinimide derivatives 3a-i, exhibited an interesting activity against cervical cancer cell (HeLa) and lung cancer cell (A549) with IC50 values ranged from 0.7 ± 0.1 to 44.0 ± 3.7 µM, except 3 h and 3i which showed no effect against HeLa, and 3c towards A549 (IC50 > 100 µM). Indeed, the high activity of most derivatives 3 confirmed the contribution of the succinimide moiety to this activity. This result was supported by some previous works showing the cytotoxic activity of several succinimide derivatives against various cancer cell lines. In fact, the manner of attachment of this group in the molecule certainly affects the cytotoxic activity (Luo et al., 2019; Milosevic et al., 2017). The results showed that the derivative 3f displayed the most potent cytotoxic effect against HeLa cell line with an IC50 value of 0.7 ± 0.1 µM, followed by 3 g and 3a (IC50 = 1.7 ± 0.4 and 2.0 ± 0.2 µM, respectively). The increase in the cytotoxic activity of compound 3f compared to that of the precursor 2 was surely due to the simple 4-methylphtalic moiety introduced by the corresponding anhydride.
The two cancer cell lines used (HeLa and A549) showed a particular sensitivity to the derivative 3 g (3,4,5,6-tetrahydrophthalic group) compared to the rest of the synthesized compounds. In fact, the derivative 3 g showed an interesting activity against the two human cancer cell lines (IC50 = 1.7 ± 0.4 µM, HeLa) and (IC50 = 5.1 ± 0.8 µM, A549). The comparison of the activity of 3a (phtalic) with that of 3f (4-methylphtalic), demonstrated that the methyl group in the structure of 3f is responsible for the increase in cytotoxic potency. In addition, the compound 3a (phtalic; IC50 = 7.7 ± 1.2 µM) was found to be five time more active against HeLa cell line than its analogue 3c (napthalic; IC50 = 13.9 ± 1.7 µM). The loss in activity can be explained by the extent of conjugation in the naphalic system as well as its larger dimension compared to the phtalic one. It has been noticed that compounds 1, 2 and 3c showed a selective cytotoxic power against the cell lines used, they exerted an activity towards HeLa but they were inactive against A549, while compounds 3 h and 3i were only cytotoxic towards A549.
3.3 Molecular docking studies
The in silico experiments of molecular docking can show the binding mode and the ability of molecules to fit well into the active binding site of enzymes, which rationalize and explain in vitro biological activities (Saidi et al., 2020; Saidi et al., 2022).
3.3.1 Molecular docking analysis for anti-butyrylcholinesterase activity
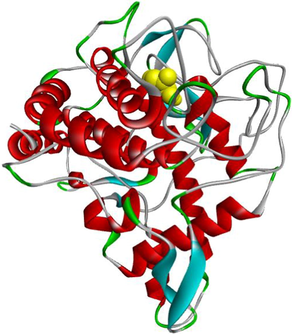
Butyrylcholinesterase (EC 3.1.1.8) is a non-specific cholinesterase enzyme. It hydrolyzes several choline-based esters (Chen et al., 2018). Butyrylcholinesterase crystal structure with PDB: 4B0P is composed of a chain A with sequence length of 529 amino acids (https://www.rcsb.org/structure/4B0P). In depth, molecular docking analysis was executed to investigate the binding mode of the most anti-butyrylcholinesterase compounds (3e and 3 g) within the active binding site of MF5 (1-(1-methylpyridin-1-ium-2-yl)-N-[[2,3,4,5,6-pentakis(fluoranyl)phenyl]methoxy]methanimine: the standard complexed ligand) in the protein crystal structure of the enzyme (https://www.rcsb.org/structure/4B0P).
The binding modes of MF5 and compounds 3e and 3 g within the active site of butyrylcholinesterase enzyme are compared below.
The standard complexed ligand (MF5) and the two potent anticholinesterase compounds (3e and 3 g) share an important number of interactions, which suggests that compounds 3e and 3 g are in the perfect bonding pose (Table 3).
Code
3D Pocket Positioning
3D representation
3e
3 g
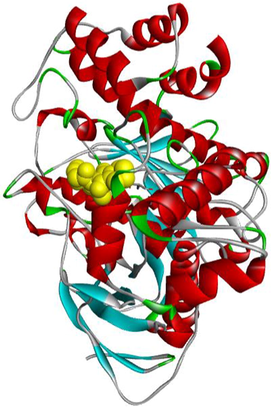
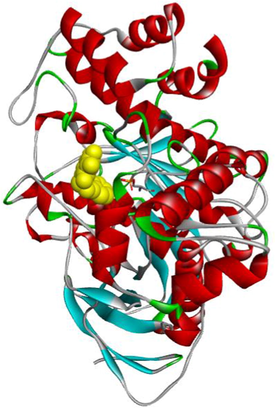
MF5
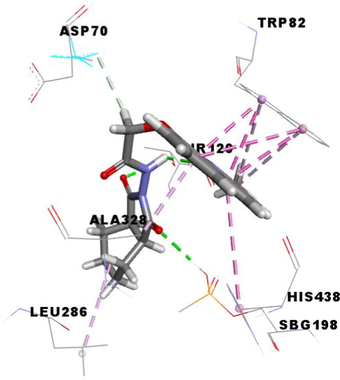
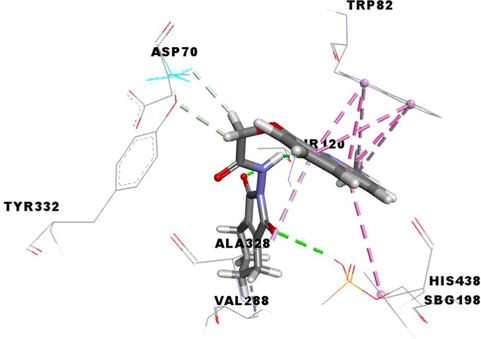
From the molecular docking results (Table 3), it was observed that MF5, the standard complexed ligand with the butyrylcholinesterase enzyme (PDB: 4B0P), exhibited a good binding energy of −8.8 kcal/mol and implicated in nine non-covalent interactions with six amino acids. On the other hand, the mode of binding showed that MF5 is implicated in three halogen (Fluorine) bonds with ASP70, PRO285 and ALA328 residues, a Pi-Pi Stacked bond with TYR332, two carbon-hydrogen bonds with ASP70 and HIS438, and three Pi-Alkyl interactions with TRP82 and HIS438.
The compound 3 g, the potent in vitro anti-butyrylcholinesterase derivative, exhibited an excellent binding energy of −10.0 kcal/mol, which will allow it to fit better than MF5 into the active site of the butyrylcholinesterase enzyme. The mode of binding shows that it is implicated in thirteen non-covalent interactions with eight amino acids. Accordingly, molecular insight of docking analysis suggests that it is involved in two conventional hydrogen bindings with THR120 and SBG198 residues, five Pi-Pi Stacked bonds with the TRP82 and HIS438, two carbon-hydrogen bindings with ASP70 and TYR332, three Pi-Alkyl interactions with TRP82 and ALA328, and an alkyl bond with VAL288 (Table 3).
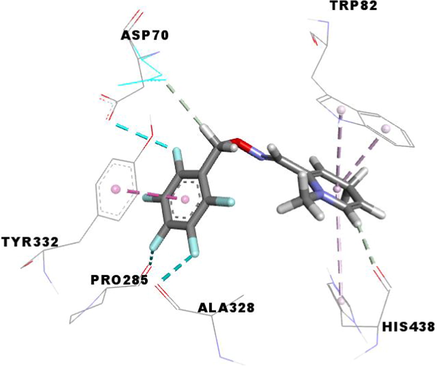
The compound 3e carried out an important binding energy of −10.4 kcal/mol. It fits favorably into the enzymatic active site, better than 3 g and MF5. The binding mode of 3e shows that it is implicated in twelve non-covalent interactions with seven amino acids. Consequently, it is involved in two conventional hydrogen bindings with THR120 and SBG198 residues, five Pi-Pi Stacked bonds with the TRP82 and HIS438, a carbon-hydrogen binding with ASP70, three Pi-Alkyl interactions with TRP82 and ALA328, and an alkyl bond with LEU286 (Table 3).
3.3.2 Molecular docking analysis for anti-tyrosinase activity
The tyrosinase (EC 1.14.18.1) is a polyphenoloxidase enzyme frequently associated with pigmentation. It is the key enzyme of the first step in the melanogenesis (Ismaya et al., 2011; Zhang et al., 2011). The structure of tyrosinase enzyme is composed of tetrameric subunits, chains A-D, with a sequence length of 391 amino acids (https://www.rcsb.org/structure/2Y9X). Tyrosinase is a binuclear copper-containing enzyme catalyzes the conversion of l-tyrosine to l-DOPA (L-3,4-dihydroxyphenylalanine) and l-DOPA to l-DOPA quinone, which is further polymerized to constitute melanins (Ismaya et al., 2011; Zhang et al., 2011). Extensive molecular docking analysis was carried out to elucidate the interactions of the potent anti-tyrosinase compounds (3d and 3e) within the binding pocket of 0TR (tropolone: the standard complexed ligand) in PDB: 2Y9X ( https://www.rcsb.org/structure/2Y9X).
The binding modes of the tropolone (0TR) and compounds (3d and 3e) within the tyrosinase enzyme are compared below.
The standard complexed ligand (0TR) and the two potent anti-tyrosinase compounds (3d and 3e) share important interactions with the two residues SER282 and VAL283. Therefore, compounds 3d and 3e are in the perfect bonding pose (Table 4).
Code
3D Pocket Positioning
3D representation
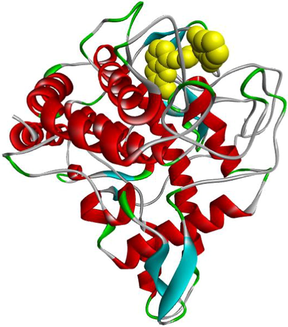
3d
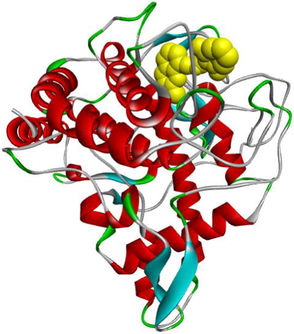
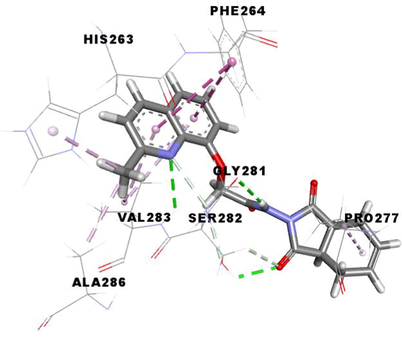
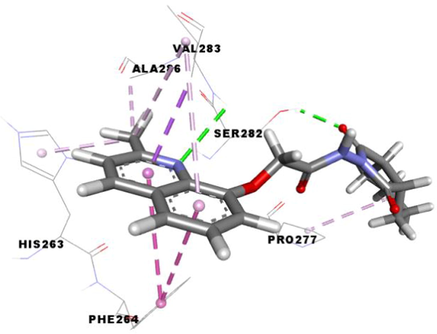
3e
0TR
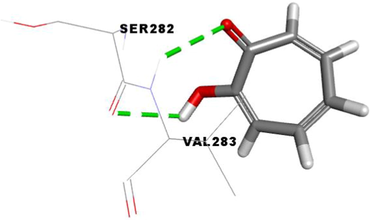
From the in silico docking results (Table 4), it was observed that the tropolone, the standard complexed ligand with the tyrosinase enzyme (PDB: 2Y9X), exhibited a binding energy of −5.9 kcal/mol. On the other hand, their binding mode showed that it is involved in two conventional hydrogen bonds with SER282 and VAL283 residues.
The compound 3d, the potent in vitro inhibitor of tyrosinase, exhibited a good binding energy of −7.8 kcal/mol, which will allow it to fit more favorably than the tropolone (0TR) into the active site of the tyrosinase enzyme. Its binding mode shows that it is implicated in twelve non-covalent interactions with seven amino acids. Accordingly, molecular insight of docking analysis suggests that it is involved in three conventional hydrogen bindings with GLY281, SER282 and VAL283 residues, a Pi-Sigma bond with the VAL283, two Pi-Pi T-shaped bindings with the PHE264, a carbon-hydrogen binding with SER282, three alkyl interactions with PRO277, VAL-283 and ALA286, and two Pi-Alkyl bonds with HIS263 and VAL283 (Table 4).
The compound 3e carried out an important binding energy of −7.3 kcal/mol. It fits favorably into the enzymatic active site. The binding mode of this compound shows that it is implicated in ten non-covalent interactions with six amino acids. Consequently, the molecular docking analysis suggests that it is involved in two conventional hydrogen bindings with SER282 and VAL283 residues, a Pi-Sigma bond with the VAL283, two Pi-Pi T-shaped with the PHE264, three alkyl interaction with PRO277, VAL-283 and ALA286, and two Pi-Alkyl bonds with HIS263 and VAL283 (Table 4).
3.3.3 Molecular docking analysis for cytotoxic activity
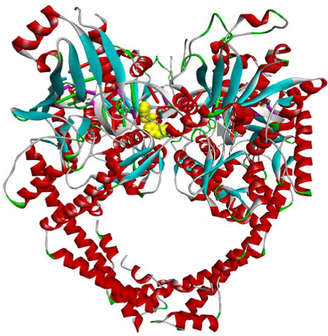
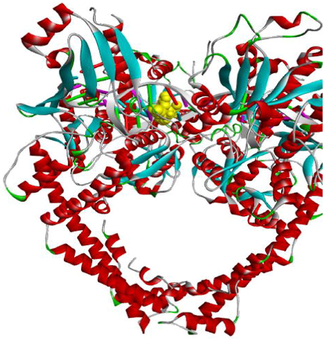
The enzyme topoisomerase IIα is preferentially secreted in proliferating cells. It plays a key role in transcription, replication, repair and catalysis of DNA cleavage. Cancer cells depend on topoisomerase IIα enzyme more than healthy cells, since they divide more rapidly. Therefore, topoisomerase IIα inhibition considered as a target approach for killing cancer and highly proliferative cells. Consequently, they are of particular interest (Arencibia et al., 2020; Cowell et al., 2012; Lynch et al., 1997). The structure of the topoisomerase IIα enzyme (PDB: 5GWK) is composed of two chains, A and B, with a sequence length of 806 amino acids (https://www.rcsb.org/structure/5GWK).
In addition, molecular docking analysis were executed to rationalize the observed in vitro cytotoxicity and to explore the binding mode of the potent active compounds (3a, 3f and 3 g) within the active site of etoposide (EVP) of the protein crystal structure of the human topoisomerase IIα enzyme (PDB: 5GWK) (https://www.rcsb.org/structure/5GWK).
The binding modes of EVP and compounds 3a, 3f and 3 g within the active site of topoisomerase IIα enzyme are compared below.
The standard complexed ligand (EVP) and the three potent cytotoxic compounds (3a, 3f and 3 g) share many interactions with the same residues, which suggests that the docked compounds are in the perfect bonding pose (Table 5).
Code
3D Pocket Positioning
3D representation
3a
3f
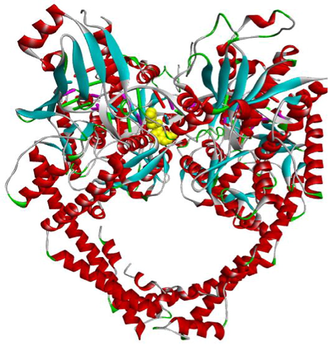
3 g
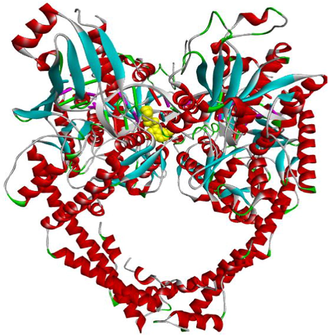
EVP
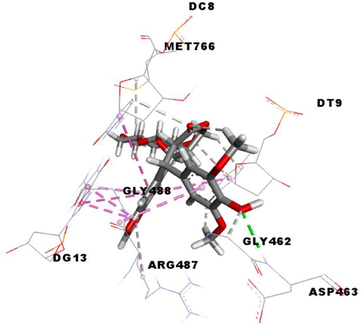
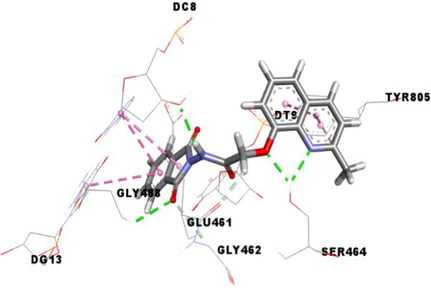
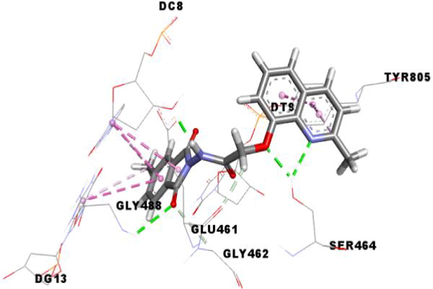
Etoposide (EVP), the standard complexed ligand with the DNA topoisomerase IIα enzyme, exhibited an excellent binding energy of −11.1 kcal/mol and was involved in fifteen non-covalent interactions with seven residues. It interacts by a conventional hydrogen bond with the residue ASP463, four carbon-hydrogen bonds with DC8, DT9 and GLY462, six Pi-Pi Stacked bonds with DC8, DT9 and DG13, an amide-Pi Stacked binding with ARG487, an alkyl bond with MET766 and two Pi-Alkyl bonds with ARG487 and DT9 residues (Table 5).
Compound 3f, the most in vitro cytotoxic product, exhibited an excellent binding energy of −10.6 kcal/mol. The binding mode shows that it is implicated in fifteen non-covalent interactions with eight residues. Therefore, the molecular insight of docking analysis suggests that it is interacts by four conventional hydrogen bindings with DC-8, SER464 and GLY488, three carbon-hydrogen interactions with the GLU461, GLY462 and DT9, five Pi-Pi Stacked bonds with DC8, DG13 and TYR805, and three Pi-Alkyl bonds with DC8, DG13 and TYR805 residues (Table 5).
Compound 3a exhibited a good binding energy of −10.2 kcal/mol and involved in thirteen non-covalent interactions with eight amino acids. Therefore, it forms four conventional hydrogen bindings with DC-8, SER464 and GLY488, three carbon-hydrogen interactions with the GLU461, GLY462 and DT9, five Pi-Pi Stacked bonds with DC8, DG13 and TYR805, and a Pi-Alkyl bond with the TYR805 residue (Table 5).
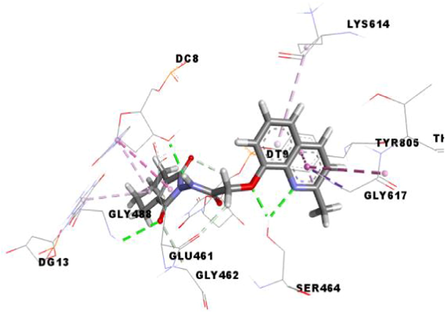
Compound 3 g carried out a significant binding energy of −9.8 kcal/mol. The mode of binding shows that it is involved in fifteen non-covalent interactions with ten residues. Therefore, the molecular docking analysis suggests that it is implicated in four conventional hydrogen bonds with DC-8, SER464 and GLY488, three carbon-hydrogen interactions with the GLU461, GLY462 and DT9, two Pi-Pi Stacked bonds with DC8 and TYR805, an amide-Pi Stacked interaction and a Pi-Sigma bond with GLY617, and four Pi-Alkyl bonds with the DC8, DG13, LYS614 and TYR805 residues (Table 5).
4 Conclusion
In this study, we developed an easy and simple method to access to new hybrid molecules, in good yields, in which quinaldine was linked to various succinimides. This new combination was achieved by including 8-hydroxyquinaldine and electrophilic species (cyclic anhydrides). We, also, scrutined the anti-butyrylcholinesterase, anti-tyrosinase and cytotoxic activities of all the synthesized compounds and some of them exhibited promising anti-butyrylcholinesterase, anti-tyrosinase and cytotoxic activities, showing for the first time the importance of having the two quinaldine and succinimide fragments in the same molecule for biological purposes. Molecular docking analyses led to conclude that the succinimide moiety is essential for the build-up and improvement of anti-butyrylcholinesterase, anti-tyrosinase and cytotoxic activities of quinaldine conjugates 3a-i. In silico Structure activity-Relationship (SAR) studies were found to be in well agreement with experimental biological results, showing that the nature of the anhydride moiety is essential to give significant binding (Hydrogen bond) interaction with amino acids of enzymes.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University for funding this work through Research Group no. RG-21-09-69.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships which might appear to influence the work reported in this article.
References
- Afzal, O., Kumar, S., Haider, M.R., Ali , M.R., Kumar, R., Jaggi , M., Bawa, S.,2015. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 97, 871–910. htpps ://doi.org/10.1016/j.ejmech.2014.07.044.
- Ahmad, A., Ullah, F., Sadiq, A., Ayaz, M., Rahim, H., Rashid, U., Ahmad, S., Muhammad Jan, S., Ullah, R., Shahat, A.A., 2019. Pharmacological Evaluation of Aldehydic-Pyrrolidinedione Against HCT-116, MDA-MB231, NIH/3T3, MCF-7 Cancer Cell Lines, Antioxidant and Enzyme Inhibition Studies. Drug Des. Devel. Ther. 13, 4185–4194.https://doi.org/10.2147/DDDT.S226080.
- Comparative cholinesterase, α-glucosidase inhibitory, antioxidant, molecular docking, and kinetic studies on potent succinimide derivatives. Drug Des. Devel. Ther.. 2020;14:2165-2178.
- [CrossRef] [Google Scholar]
- Alzheimer’s Association, 2015. 2015 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 11, 332–384. https://doi.org/10.1016/j.jalz.2015.02.003
- Design, synthesis, dynamic docking, biochemical characterization, and in vivo pharmacokinetics studies of novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem.. 2020;63:3508-3521.
- [CrossRef] [Google Scholar]
- Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res.. 2003;5:165-176.
- [CrossRef] [Google Scholar]
- Bioactivity-based analysis and chemical characterization of cytotoxic constituents from Chaga mushroom (Inonotus obliquus) that induce apoptosis in human lung adenocarcinoma cells. J. Ethnopharmacol.. 2018;224:63-75.
- [CrossRef] [Google Scholar]
- Synthesis, antioxidant and anticholinesterase activities of novel quinoline-aminophosphonate derivatives. J. Heterocycl. Chem.. 2020;57:2139-2149.
- [CrossRef] [Google Scholar]
- The design of novel 4,6-dimethoxyindole based hydrazide-hydrazones: molecular modeling, synthesis and anticholinesterase activity. J. Mol. Struct.. 2020;1213:128202
- [CrossRef] [Google Scholar]
- Inhibition on cholinesterase and tyrosinase by alkaloids and phenolics from Aristotelia chilensis leaves. Food Chem. Toxicol.. 2017;109:984-995.
- [CrossRef] [Google Scholar]
- Redox-controlled fluorescent nanoswitch based on reversible disulfide and its application in butyrylcholinesterase activity assay. Anal. Chem.. 2018;90:1643-1651.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation and molecular docking analysis of novel benzopyrimidinone derivatives as potential anti-tyrosinase agents. Bioorg. Chem.. 2019;92:103270
- [CrossRef] [Google Scholar]
- Synthesis, cytotoxicity, and pro-apoptosis activity of etodolac hydrazide derivatives as anticancer agents. Arch. Pharm. (Weinheim). 2013;346:367-379.
- [CrossRef] [Google Scholar]
- Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J. Exp. Med.. 2000;191:1221-1231.
- [CrossRef] [Google Scholar]
- Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. U. S. A.. 2012;109:8989-8994.
- [CrossRef] [Google Scholar]
- Synthesis, antimicrobial activity and quantum chemical investigation of novel succinimide derivatives. J. Mol. Struct.. 2019;1181:148-156.
- [CrossRef] [Google Scholar]
- Cervical cancer: Biomarkers for diagnosis and treatment. Clin. Chim. Acta. 2015;445:7-11.
- [CrossRef] [Google Scholar]
- Cancer-associated differences in acetylcholinesterase activity in bronchial aspirates from patients with lung cancer. Clin. Sci.. 2008;115:245-253.
- [CrossRef] [Google Scholar]
- Eco-friendly synthesis, biological evaluation, and in silico molecular docking approach of some new quinoline derivatives as potential antioxidant and antibacterial agents. Front. Chem.. 2021;9:1-14.
- [CrossRef] [Google Scholar]
- Quercetin as a tyrosinase inhibitor: Inhibitory activity, conformational change and mechanism. Food Res. Int.. 2017;100:226-233.
- [CrossRef] [Google Scholar]
- Filali, I., Romdhane, A., Znati, M., B. Jannet, H., Bouajila, J., 2016. Synthesis of New Harmine Isoxazoles and Evaluation of their Potential Anti-Alzheimer, Anti-inflammatory, and Anticancer Activities. Med. Chem. (Los. Angeles). 12, 184–190. https://doi.org/10.2174/157340641202160209104115
- Synthesis and antiviral evaluation of novel 1,3,4-oxadiazole/thiadiazole-chalcone conjugates. Bioorg. Med. Chem. Lett.. 2017;27:4298-4301.
- [CrossRef] [Google Scholar]
- Design, synthesis, α-amylase inhibition and in silico docking study of novel quinoline bearing proline derivatives. J. Mol. Struct.. 2020;1208:127873
- [CrossRef] [Google Scholar]
- Anti-tyrosinase and anti-butyrylcholinesterase quinolines-based coumarin derivatives: synthesis and insights from molecular docking studies. Chem. Afr.. 2021;4:491-501.
- [CrossRef] [Google Scholar]
- New potent inhibitors of tyrosinase: novel clues to binding of 1,3,4-thiadiazole-2(3H)-thiones, 1,3,4-oxadiazole-2(3H)-thiones, 4-amino-1,2,4-triazole-5(4H)-thiones, and substituted hydrazides to the dicopper active site. Bioorg. Med. Chem.. 2010;18:4042-4048.
- [CrossRef] [Google Scholar]
- Update on Alzheimer’s Disease therapy and prevention strategies. Annu. Rev. Med.. 2017;68:413-430.
- [CrossRef] [Google Scholar]
- Synthesis and biological activity of hydroxybenzylidenyl pyrrolidine-2,5-dione derivatives as new potent inhibitors of tyrosinase. Med. Chem. Comm.. 2011;2:542-549.
- [CrossRef] [Google Scholar]
- Synthesis of succinimide-containing chromones, naphthoquinones, and xanthones under Rh(III) catalysis: evaluation of anticancer activity. J. Org. Chem.. 2016;81:12416-12425.
- [CrossRef] [Google Scholar]
- Synthesis, in-vitro α-glucosidase inhibition, antioxidant, in-vivo antidiabetic and molecular docking studies of pyrrolidine-2,5-dione and thiazolidine-2,4-dione derivatives. Bioorg. Chem.. 2019;91:103128
- [CrossRef] [Google Scholar]
- Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry. 2011;50:5477-5486.
- [CrossRef] [Google Scholar]
- Dual action of dipyridothiazine and quinobenzothiazine derivatives—anticancer and cholinesterase-inhibiting activity. Molecules. 2020;25:2604.
- [CrossRef] [Google Scholar]
- Anticancer, antimicrobial activities of quinoline based hydrazone analogues: synthesis, characterization and molecular docking. Bioorg. Chem.. 2020;94:103406
- [CrossRef] [Google Scholar]
- Effect of p-aminophenols on tyrosinase activity. Bioorg. Med. Chem.. 2014;22:3994-4000.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of novel benzylidene-succinimide derivatives as noncytotoxic antiangiogenic inhibitors with anticolorectal cancer activity in vivo. Eur. J. Med. Chem.. 2019;179:805-827.
- [CrossRef] [Google Scholar]
- A comprehensive overview of β-carbolines and its derivatives as anticancer agents. Eur. J. Med. Chem.. 2021;224:113688
- [CrossRef] [Google Scholar]
- Human DNA topoisomerase II-alpha: A new marker of cell proliferation in invasive breast cancer. Hum. Pathol.. 1997;28:1180-1188.
- [CrossRef] [Google Scholar]
- Synthesis, molecular docking and biological evaluation of N, N-disubstituted 2-aminothiazolines as a new class of butyrylcholinesterase and carboxylesterase inhibitors. Bioorg. Med. Chem.. 2016;24:1050-1062.
- [CrossRef] [Google Scholar]
- A phase I vaccination study with tyrosinase in patients with stage II melanoma using recombinant modified vaccinia virus Ankara (MVA-hTyr) Cancer Immunol. Immunother.. 2005;54:453-467.
- [CrossRef] [Google Scholar]
- Evaluation of in silico pharmacokinetic properties and in vitro cytotoxic activity of selected newly synthesized N-succinimide derivatives. J. Pharm. Biomed. Anal.. 2017;137:252-257.
- [CrossRef] [Google Scholar]
- Design, synthesis, biological evaluation, and molecular modeling studies of quinoline-ferulic acid hybrids as cholinesterase inhibitors. Bioorg. Chem.. 2019;93:103310
- [CrossRef] [Google Scholar]
- Cholinesterases are down-expressed in human colorectal carcinoma. Cell. Mol. Life Sci.. 2006;63:2175-2182.
- [CrossRef] [Google Scholar]
- Expression of cholinesterases in human kidney and its variation in renal cell carcinoma types. FEBS J.. 2010;277:4519-4529.
- [CrossRef] [Google Scholar]
- Acetylcholinesterase and butyrylcholinesterase-important enzymes of human body. Acta Med. (Hradec Kralove). 2004;47:215-228.
- [CrossRef] [Google Scholar]
- Tropinone-derived alkaloids as potent anticancer agents: synthesis, tyrosinase inhibition, mechanism of action, DFT calculation, and molecular docking studies. Int. J. Mol. Sci.. 2020;21:1-24.
- [CrossRef] [Google Scholar]
- Total synthesis and anti-cancer activity of all known communesin alkaloids and related derivatives. J. Am. Chem. Soc.. 2019;141:14411-14420.
- [CrossRef] [Google Scholar]
- Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem.. 2014;22:2388-2395.
- [CrossRef] [Google Scholar]
- RCSB-Protein Data Bank, National Science Foundation, the US Department of Energy (https://www.rcsb.org).
- RCSB-Protein Data Bank, National Science Foundation, the US Department of Energy (https://www.rcsb.org/structure/4B0P).
- RCSB-Protein Data Bank, National Science Foundation, the US Department of Energy (https://www.rcsb.org/structure/5GWK).
- RCSB-Protein Data Bank,National Science Foundation, the US Department of Energy (https://www.rcsb.org/structure/2Y9X).
- Update on Alzheimer’s and the dementias: introduction. Neurol. Clin.. 2017;35:171-174.
- [CrossRef] [Google Scholar]
- Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents. Chem. Soc. Rev.. 2016;45:6130-6137.
- [CrossRef] [Google Scholar]
- A novel aryl-hydrazide from the marine lichen Lichina pygmaea: isolation, synthesis of derivatives, and cytotoxicity assays. Bioorg. Med. Chem. Lett.. 2010;20:4582-4586.
- [CrossRef] [Google Scholar]
- Synthesis, anticholinesterase and antioxidant potentials of ketoesters derivatives of succinimides: A possible role in the management of alzheimer’s. Chem. Cent. J.. 2015;9:31-39.
- [CrossRef] [Google Scholar]
- Iridoid glycosides from the Tunisian Citharexylum spinosum L.: isolation, structure elucidation, biological evaluation, molecular docking and SAR analysis. Ind. Crops Prod.. 2020;151:112440
- [CrossRef] [Google Scholar]
- Synthesis of new halogenated flavonoid-based isoxazoles: in vitro and in silico evaluation of α-amylase inhibitory potential, a SAR analysis and DFT studies. J. Mol. Struct.. 2022;1247:131379
- [CrossRef] [Google Scholar]
- Current approaches to cervical-cancer screening. N. Engl. J. Med.. 2001;344:1603-1607.
- [CrossRef] [Google Scholar]
- Novel fluorine-containing chiral hydrazide-hydrazones: design, synthesis, structural elucidation, antioxidant and anticholinesterase activity, and in silico studies. Chirality. 2019;31:603-615.
- [CrossRef] [Google Scholar]
- Novel 2-benzylthio-5-(1,3,4-oxadiazol-2-yl)benzenesulfonamides with anticancer activity: synthesis, QSAR study, and metabolic stability. Eur. J. Med. Chem.. 2017;132:236-248.
- [CrossRef] [Google Scholar]
- Novel oxoisoaporphine-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation. Bioorg. Med. Chem. Lett.. 2012;22:2257-2261.
- [CrossRef] [Google Scholar]
- Novel tetrazole derivatives : synthesis, anticholinesterase activity and cytotoxicity evaluation. Turk. J. Biochem.. 2017;42:169-180.
- [CrossRef] [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [CrossRef] [Google Scholar]
- Tacrine derivatives and Alzheimer’s Disease. Curr. Med. Chem.. 2010;17:1825-1838.
- [CrossRef] [Google Scholar]
- Exploiting tyrosinase expression and activity in melanocytic tumors : quercetin and the central role of p53. Intgr. Cancer Ther.. 2011;10:328-340.
- [CrossRef] [Google Scholar]
- Expression of cholinesterases in brain and non-brain tumours. Chem. Biol. Interact.. 2005;158:227-232.
- [CrossRef] [Google Scholar]
- Role of acetylcholinesterase in lung cancer. Thorac. Cancer. 2015;6:390-398.
- [CrossRef] [Google Scholar]
- Novel 1,3,5-triazine derivatives exert potent anti-cervical cancer effects by modulating Bax, Bcl2, and caspases expression. Chem. Biol. Drug Des.. 2018;91:728-734.
- [CrossRef] [Google Scholar]
- Structure – activity relationships of the thujaplicins for inhibition of human tyrosinase. Bioorg. Med. Chem.. 2014;22:6193-6200.
- [CrossRef] [Google Scholar]
- Novel tetrazole derivatives: synthesis, anticholinesterase activity and cytotoxicity evaluation. Turk. J. Biochem.. 2017;42:169-180.
- [CrossRef] [Google Scholar]
- Caryophyllene Sesquiterpenes from Pulicaria vulgaris Gaertn.: isolation, structure determination, bioactivity and structure−activity relationship. Chem. Biodivers.. 2019;16:e1800483.
- [Google Scholar]
- Methyl Lucidenate F isolated from the ethanol-soluble-acidic components of Ganoderma lucidum is a novel tyrosinase inhibitor. Biotechnol. Bioprocess Eng.. 2011;16:457-461.
- [CrossRef] [Google Scholar]
- Research progress in biological activities of succinimide derivatives. Bioorg. Chem.. 2020;108:104557
- [CrossRef] [Google Scholar]
- Semi-synthesis, antibacterial, anticholinesterase activities, and drug likeness properties of new analogues of Coumarins isolated from Ferula lutea (Poir.) Maire. Chem. Afr.. 2020;3:635-645.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijtst.2018.11.001.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




