

Translate this page into:
Design, synthesis, biological evaluation and in silico studies of novel 1,2,3-triazole linked benzoxazine-2,4-dione conjugates as potent antimicrobial, antioxidant and anti-inflammatory agents
⁎Corresponding authors at: Department of Chemistry, Faculty of Science and Arts of Baljurashi, Albaha University, Saudi Arabia. ansariiqrar50@gmail.com (Iqrar Ahmad), lukadel@yahoo.fr (Adel Kadri)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In an attempt to rationalize the search for new potential anti-inflammatory and anti-infection agents, a new series of 1,4-and 1,5-disubstituted 1,2,3-triazoles linked benzoxazine conjugates have been synthesized via “Click Chemistry” reaction, were designed, synthesized and characterized by means of spectral and elemental data. The newly synthesized compounds have been assessed for their antimicrobial, antioxidant and anti-inflammatory potential. Results revealed that all synthesized compounds display superior activities to the standard drug against different bacterial strains especially S. aureus, M. luteus, and P. aeruginosa, with good to moderate activity towards the tested E. coli bacteria, in respect to the commercial antibiotic, tetracycline. Moreover, the antifungal activity was screened against C. albicans and C. krusei yeasts and results demonstrate potent activity as compared to the standard drug, ampicillin. The antioxidant activity was evaluated using 2,2-diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS) radical scavenging assays, whose results indicate that analogues 4a (IC50 1.88 ± 0.07 µM and 2.17 ± 0.02) followed by 4b (IC50 2.19 ± 0.09 µM and 2.38 ± 0.43 µM), 4d (IC50 2.30 ± 0.01 µM and 4.07 ± 0.57 µM), and 4f (2.98 ± 0.02 µM and 3.80 ± 0.01 µM), respectively, exhibited the strongest activity when compared to the standard reference, butylated hydroxytoluene (BHT) (3.52 ± 0.08 µM and 4.64 ± 0.11 µM). In addition, their anti-inflammatory activity was assessed using the xylene-induced ear edema standard technique and the results demonstrated the potency of 4a, 4b and 4d as excellent anti-inflammatory agents. Preliminary structure–activity relationship studies (SARs) provide those biological activities can be modulated by the presence of unsubstituted aromatic ring as well as the position of substituents on the phenyl moiety via electron withdrawing groups (EWGs) or electron donating groups (EDGs) effects. Docking studies on the most promising compounds 4a, 4b, and 4d into the active sites of S. aureus tyrosyl-tRNA synthetase, Candida albicans N-Myristoyltransferase, Human COX-2 enzyme, and Human Peroxiredoxin 5 revealed good binding profiles with the target proteins. The interaction's stability was further assessed using a conventional atomistic 100 ns dynamic simulation study. Hence, our results recommended the rationalized targets 4a, 4b and 4d, to be promising lead candidates for the discovery of novel dual anti-inflammatory and anti-infection agents.
Keywords
1,4- and 1,5-disubstituted 1,2,3-triazoles
Click chemistry
Antimicrobial
Antioxidant
Ant-inflammatory
In silico studies

1 Introduction
The high emergence of multidrug-resistant (MDR) due to the invasion of human pathogenic microbes represents an urgent threat to global public health, which was associated with serious and costly problem, high morbidity and mortality level accounting for approximately 700,000 individuals being killed by drug-resistant bacteria worldwide (Catalano et al., 2022, https://www.cdc.gov/drugresistance/). The Staphylococcus aureus strain well known for its high antibiotic-resistant rate, is covering about 65–85% of nosocomial S. aureus infections associated with a beta-lactam-resistant strain (Santajit and Indrawattana, 2016). Also, fungal infections have recently risen and are responsible for 1–2 million fatalities annually, especially caused by Aspergillus and Candida species (Berkow and Lockhart, 2017, Calderone et al., 2014). Candida albicans and Candida krusei strains (responsible for 75–88% of fungal infections) are resistant to the most common azole drug fluconazole (Rodrigues et al., 2016). The biochemical resistance mechanisms used by bacteria include antibiotic inactivation through the activation of drug efflux pumps that deliberately remove the drug from the cell, target modification, and the alteration of cell-wall proteins that inhibit drug uptake (Varsha et al., 2018). Considering the increase in AMR, a growing awareness on the search for new antibacterial agents that play a key role in controlling and curing infectious disease is essential. On the other side, antibiotics exerted accelerated selective pressure that helped bacterial resistance via mutation, and acquisition external genes (Hasan et al., 2022).
Nitrogen-containing heterocycles are a highly important class of drug compounds, with diverse targets and therapeutic properties. Due to their applications in medicinal chemistry with multivarious pharmaceutical activities and therapeutic index, the pharmacophores, triazoles considered as one of the important N-heterocyclic building blocks, have received a special interest (Kerru et al., 2020). Among them, 1,2,3-triazole as a major pharmacophore, is one of the most important classes of nitrogen-rich heterocyclic scaffolds, which attracted considerable attention using the ‘click chemistry’ approach owing to the two popular contributions to the synthesis of 1,4-disubstituted 1,2,3-triazoles followed shortly thereafter by the ruthenium-catalyzed synthesis of the 1,5-disubstituted isomer (Oubella et al., 2022, Celik et al. 2018). The Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) offers a versatile root to the 1,2,3-triazoles scaffolds, that could form diverse non-covalent interactions, such as van der Waals forces, dipole–dipole bonds and hydrogen bonds with various proteins, enzymes, and receptors with high resistance to enzymatic degradation, which enables their potential use in medicinal chemistry. They have been widely recognized for their diverse biological and pharmaceutical activities such as antitubercular (Pradeep Kumar et al., 2021, Badar et al., 2020), antimicrobial (Badar et al., 2020, Bitla et al., 2021), antituberculosis (Hervin et al., 2020), antiproliferative (Vanaparthi et al., 2020), antibacterial (Xu, 2020), antioxidant (Sahin et al., 2021), anticancer (Sahin et al., 2021), α-glucosidase (Shareghi-Boroujeni et al., 2021) and antifungal (Joolakanti et al., 2021) agents. In addition, some medications containing 1,2,3-triazole as an active moiety are clinically used as antimicrobial agents such as Radezolid, Cefatrizine and Tazobactam (Fig. 1).
Marketed drugs having 1,2,3-triazole moiety.
Inflammation remains a hallmark of many metabolic diseases and plays a central role in the pathophysiology of infections, being one of the principal body defensive mechanisms. Besides, therapeutic compounds that generate both antibacterial as well as anti-inflammatory effects are therefore likely to be most effective at treating bacteria-induced inflammatory diseases based on that inflammation is the body’s natural response associated with infection, irritation, or tissue damage. Likewise, due to the side effects related with commercial anti-inflammatory drugs, medicinal chemists are promoted to develop alternative scaffolds. The anti-inflammatory activity has also been previously associated with 1,2,3-triazole derivatives (Shafi et al., 2012). The anti-inflammatory effect of 1,4-disubstituted 1H-1,2,3-triazoles and ferrocene-1H-1,2,3-triazole hybrids as well as the high potential of phenyl-1H-1,2,3-triazole derivatives than the existing anti-inflammatory drug diclofenac have been reported (Kim et al., 2015, Haque et al., 2017).
On the other hand, due to their ability of electrophiles, nucleophiles and to undergo various cyclization reactions, isatoic anhydride (2H-3,1-benzoxazine-2,4(1H)-dione) remains as an extremely versatile heterocycle that attracted more interest. Its coupling with other molecules has resulted in compounds with anti-inflammatory, analgesic and antimicrobial properties. Inspired by our previous work on the discovery of bioactive antioxidant (Badraoui et al., 2020, Noumi et al., 2020, Alminderej et al., 2020, Felhi et al., 2017, Mseddi et al., 2020, Bakari et al., 2018, Ben Mefteh et al., 2018), antibacterial (Felhi et al., 2017) and antimicrobial agents (Haddaji et al., 2021, Hajlaoui et al., 2021, Alminderej et al., 2021), as well as and in continuation to our effort to design and synthesize a pharmacologically active heterocyclic compounds (Aouadi et al., 2013, Brahmi et al., 2016, Ghannay et al., 2017, 2020a, 2020b, 2020c, Kadri and Aouadi, 2020, Othman et al., 2020a, 2020b, 2020c, 2021a, 2021b), the current study aimed to investigate the antibacterial, antifungal antioxidant and anti-inflammatory activity of novel 1,2,3- triazole linked isatoïc anhydride derivatives. Further, molecular docking and dynamic simulation studies were carried out to explore their efficiency and target interactions in the active site of S. aureus tyrosyl-tRNA synthetase (PDB, ID: 1JIJ), C. albicans N-Myristoyltransferase (PDB, ID: 1IYL), human COX-2 (PDB, ID: 1PXX), and human Peroxiredoxin 5 (PDB, ID: 1HD2) enzymes. The study was achieved with SAR and ADMET investigation.
2 Experimental section
2.1 General experimental methods
2.1.1 General
1H and 13C NMR were recorded using a Bruker spectrometer operating at 400 and 100 MHz, respectively. The chemical shifts were recorded in ppm relative to tetramethylsilane and with the solvent resonance as the internal standard. Data were reported as follows: chemical shift, multiplicity (s = singlet, m = multiplet), coupling constants (Hz), integration. 13C NMR data were collected with complete proton decoupling. Chemical shifts were reported in ppm with respect to TMS with the solvent resonance as internal standard. Elemental analyses were performed on a Perkin Elmer 2400 Series II Elemental CHNS analyzer. Column chromatography (cyclohexane/ethyl acetate (5:5)) was carried out on silica gel (300–400 mesh, Qingdao Marine Chemical ltd., Qingdao, China). Thin layer chromatography (TLC) was performed on TLC silica gel 60 F254 plates 0.2 mm 200x200 nm; The spots were visualized using UV light at 254 nm and 360 nm.
2.1.2 General procedure for the synthesis of compounds 4(a-i): Cu-catalyzed azide-alkyne cycloaddition
A mixture of dipolarophile 1 (0.2 mmol), copper (I) iodide (0.5 eq) and trimethylamine (1 eq) were added at room temperature. To this mixture, aryl azide 1 (0.4 mmol) was added in toluene and the reaction mixture was subjected to microwave irradiations at 250 W completed within 10–15 min. the crude mixture was extracted with EtOAc (3x25mL) and the combined organic layer was dried over sodium sulfate, concentrated under reduced pressure and purified through a column chromatography (cyclohexane/ethyl acetate (5:5)) or recrystallization (ethanol), to give pure 4a-i in 60–86% (percentage yields).
2.1.2.1 4a: N-(1-(phenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (56 mg, 77%); melting point (mp) 217–219 °C; IR (FTIR (Fourier transform infrared), cm−1): ν = 1773 (C⚌O), 1H NMR (400 MHz, CDCl3) δ (ppm): 5.44 (s, 2H, N—CH2), 7.32–8.17(m, 9H, Ar-H), 8.23 (s, 1H, C—Htriazole). 13C NMR (100 MHz, CDCl3) δ 40.3(C11), 122.0 (C16), 141.0 (C12), 111.6–147.4 (Caromatique), 148.3 (C2), 158.1 (C4). ESI-MS [C17H12N4O3 + H]+ m/z: 332.3. Anal. Calcd. For C17H12N4O3: C, 63. 04; H, 3.74; N, 17.50. Found. C, 64.0; H, 3.75; N, 17.49.
2.1.2.2 4b: N-(1-(4-methylphenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (60 mg, 86%); mp 202–204 °C; IR (FTIR, cm−1): ν = 1777 (C⚌O); 1H NMR (400 MHz, CDCl3) δ (ppm): 2.42 (s, 3H, CH3), 5.43 (s, 2H, N—CH2), 7.30–8.16 (m, 8H, Ar-H), 8.18 (s, 1H, C—Htriazole). 13C NMR (100 MHz, CDCl3) δ 40.3(C11), 122.0 (C16), 141.0 (C12), 111.6–147.4 (Caromatique), 148.3 (C2), 158.1 (C4). ESI-MS [C18H14N4O3 + H]+ m/z: 335.2. Anal. Calcd. For C18H14N4O3: C, 64.65; H, 4.18; N, 16.76. Found. C, 64.66; H, 4.20; N, 16.81.
2.1.2.3 4c: N-(1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (63 mg, 74%); mp 222–224 °C; IR (FTIR, cm−1): ν = 1781 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 3.80 (s, 3H, OCH3), 5.39 (s, 2H, N—CH2), 7.09–8.05 (m, 8H, Ar-H), 8.67 (s, 1H, C—Htriazole). 13C NMR (100 MHz, DMSOd6) δ 40.4 (C11), 56.0 (OCH3), 122.1(C16), 112.2–137.8(Caromatique), 141.5 (C12), 148.2 (C2), 159.2 (C4). ESI-MS [C18H14N4O4 + H]+ m/z: 351.1. Anal. Calcd. For C18H14N4O4: C, 61.70; H, 3.99; N, 15.99. Found. C, 61.71; H, 4.01; N, 16.03.
2.1.2.4 4d: N-(1-(2-chlorophenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (56 mg, 73%); mp 198–200 °C; IR (FTIR, cm−1): ν = 1777 (C⚌O); 1H NMR (400 MHz, CDCl3) δ (ppm): 5.45 (s, 2H, N—CH2), 7.31–8.15 (m, 8H, Ar-H), 8. 20 (s, 1H, C—Htriazole). 13C NMR (100 MHz, CDCl3) δ 40.2 (C11), 124.4 (C16), 111.6–137.6 (Caromatique), 141.0 (C12), 148.1(C2), 158.2(C4). ESI-MS [C17H11ClN4O3 + H]+ m/z: 355.1. Anal. Calcd. For C17H11ClN4O3: C, 57.55; H, 3.10; N, 15.78. Found. C, 58.01; H, 3.11; N, 15.77.
2.1.2.5 4e: N-(1-(3-chlorophenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (43 mg, 68%); mp 212–214 °C; IR (FTIR, cm−1): ν = 1774 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 5.43 (s, 2H, N—CH2), 7.35–8.07 (m, 8H, Ar-H), 8.89 (s, 1H, C—Htriazole). 13C NMR (100 MHz, DMSOd6) δ 40.6 (C11), 124.4 (C16), 112.3–137.7 (Caromatique), 141.5 (C12), 148.2(C2), 159.2 (C4). ESI-MS [C17H11ClN4O3 + H]+ m/z: 355.1. Anal. Calcd. For C17H11ClN4O3: C, 57.55; H, 3.10; N, 15.78. Found. C, 57.57; H, 3.11; N, 15.80.
2.1.2.6 4f: N-(1-(2, 4, 5-trichlorophenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (40 mg, 67%); mp 225–227 °C; IR (FTIR, cm−1): ν = 1770 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 5.45 (s, 2H, N—CH2), 7.34–8.44 (m, 6H, Ar-H), 9.01 (s, 1H, C—Htriazole). 13C NMR (100 MHz, DMSOd6) δ 40.5(C11), 124.4 (C16), 141.5 (C12), 112.2–147.2, (Caromatique),
148.2 (C2), 159.2 (C4). ESI-MS [C17H9Cl3N4O3 + H]+ m/z: 424.7. Anal. Calcd. For C17H9Cl3N4O3: C, 48.19; H, 2.12; N, 13.22. Found. C, 48.2; H, 2.13; N, 13.21.
2.1.2.7 4g: N-(1-(3, 4-dichlorophenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (39.5 mg, 60%); mp 201–203 °C; IR (FTIR, cm−1): ν = 1774 (C⚌O),1H NMR (400 MHz, CDCl3) δ (ppm): 5.44 (s, 2H, N—CH2), 7.32–8.16 (m, 8H, Ar-H), 8.19 (s, 1H, C—Htriazole). 13C NMR (100 MHz, DMSOd6) δ 40.6 (C11), 124.6 (C16), 111.9–135.7 142.6 (C12), 150.6 (C2), 157.5 (C4). Anal. Calcd. For C17H10Cl2N4O3: C, 52.4; H, 2.56; N, 14.39. Found. C, 52.46; H, 2.55; N, 14.42.
2.1.2.8 4h: N-(1-(3-nitrophenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (45 mg, 72%); mp 219–221 °C; IR (FTIR, cm−1): ν = 1775 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 5.45 (s, 2H, N—CH2), 7.36–8.64 (m, 8H, Ar-H), 9,03 (s, 1H, C—Htriazole). 13C NMR (100 MHz, DMSOd6) δ 40.5 (C11), 122.8 (C16), 112.2–143.8 (Caromatique), 141.5(C12), 148.9(C2), 159.2(C4). ESI-MS [C17H11N5O5 + H]+ m/z: 366.1. Anal. Calcd. For C17H11N5O5: C, 55.89; H, 3.01; N, 19.17. Found. C, 55.9; H, 3.03; N, 19.20.
2.1.2.9 4i: N-(1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methylisatoïc anhydride
White solid (35 mg, 63%); mp 211–213 °C; IR (FTIR, cm−1): ν = 1774 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 5.43 (s, 2H, N—CH2), 7.38–8.44 (m, 8H, Ar-H), 8.92 (s, 1H, C—Htriazole). 13C NMR (100 MHz, DMSOd6) δ 40.5(C11), 122.0 (C16), 141.4 (C12), 110.2–143.5 (Caromatique), 147.1(C2), 157.2(C4). ESI-MS [C17H11N5O5 + H]+ m/z: 366.1. Anal. Calcd. For C17H11N5O5: C, 55.89; H, 3.01; N, 19.17. Found. C, 55.96; H, 3.01; N, 19.21.
2.1.3 General procedure for the synthesis of compounds 5(a-f, j): Ru-catalyzed azide-alkyne cycloaddition
The dipolarophile 3 was reacted with various aromatic azides and 5 mol% Cp ∗ RuCl(PPh3)2 catalyst. All the reactions were performed under microwave conditions in toluene and completed within 10–15 min. The desired new compounds 5(a–f, j) were obtained in yields ranging from 44% to 60%.
2.1.3.1 5a: N-(1-(phenyl)-1H-1,2,3-triazol-5-yl)methylisatoïc anhydride
White solid (32 mg, 52%); mp 179–181 °C; IR (FTIR, cm−1): ν = 1773 (C⚌O); 1H NMR (400 MHz, CDCl3) δ (ppm): 4.65 (s, 2H, N—CH2), 6.59–7.79 (m, 10H, Ar-H and C—H triazole).13C NMR (100 MHz, CDCl3) δ 36.4 (C11), 133.5(C16), 111.4–134.8 (Caromatique), 136.8(C12), 150.1(C2), 170.2(C4). Anal. Calcd. For C17H12N4O3: C, 63.78; H, 3.74; N, 17.50. Found. C, 63.79; H, 3.76; N, 17.51.
2.1.3.2 5b: N-(1-(4-methylphenyl)-1H-1,2,3-triazol-5-yl)methylisatoïc anhydride
White solid (39 mg, 61%); mp 209–211 °C; IR (FTIR, cm−1): ν = 1777 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 2.41 (s, 3H, CH3), 4.60 (s, 2H, N—CH2), 6.59–7.79 (m, 9H, Ar-H and C—H triazole). 13C NMR (100 MHz, DMSOd6) δ 21.3 (CH3), 36.4 (C11), 133.1(C16), 111.6–134.9 (Caromatique), 136.4 (C12), 150.1(C2), 170.2(C4). Anal. Calcd. For C18H14N4O3: C, 64.65; H, 4.18; N, 16.74. Found. C, 64.64; H, 4.20; N, 16.75.
2.1.3.3 5c: N-(1-(4-methoxyphenyl)-1H-1,2,3-triazol-5-yl)methylisatoïc anhydride
White solid (47 mg, 66%); mp 199–201 °C; IR (FTIR, cm−1): ν = 1781 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 3.84 (s, 3H, OCH3), 4.59 (s, 2H, N—CH2), 6.59–8.03 (m, 9H, Ar-H and C—H triazole). 13C NMR (100 MHz, DMSOd6) δ 36.3 (C11), 56.1 (OCH3), 133.8 (C16), 111.9–136.7 (Caromatique), 137.6(C12), 150.1 (C2), 170.2 (C4). Anal. Calcd. For C18H14N4O4: C, 61.70; H, 3.99; N, 15.99. Found. C, 61.73; H, 3.98; N, 16.01.
2.1.3.4 5d: N-(1-(2-chlorophenyl)-1H-1,2,3-triazol-5-yl)methylisatoïc anhydride
White solid (25 mg, 52%); mp 176–178 °C; IR (FTIR, cm−1): ν = 1777 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 4.50 (s, 2H, N—CH2), 6.58–8.04 (m, 9H, Ar-H and C—H triazole). 13C NMR (100 MHz, DMSOd6) δ 35.8(C11), 133.2(C16), 111.7–134.7(Caromatique), 137.5(C12), 149.5(C2), 170.1(C4). Anal. Calcd. For C17H11ClN4O3: C, 57.55; H, 3.10; N, 15.78. Found. C, 57.59; H, 3.09; N, 15.75.
2.1.3.5 5e: N-(1-(3-chlorophenyl)-1H-1,2,3-triazol-5-yl)methylisatoïc anhydride
White solid (21 mg, 42%); mp 169–171 °C; IR (FTIR, cm−1): ν = 1774 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 4.72 (s, 2H, N—CH2), 6.59–7.78 (m, 9H, Ar-H and C—H triazole). 13C NMR (100 MHz, DMSOd6) δ 36.3(C11), 133.6(C16), 111.4–134.8(Caromatique), 137.6(C12), 150.1(C2), 170.2 (C4). Anal. Calcd. For C17H11ClN4O3: C, 57.55; H, 3.10; N, 15.78. Found. C, 57.56; H, 3.12; N, 15.79.
2.1.3.6 5f: N-(1-(2, 4, 5-trichlorophenyl)-1H-1,2,3-triazol-5-yl)methylsatoïc anhydride
White solid (37 mg, 58%); mp 217–219 °C; IR (FTIR, cm−1): ν = 1770 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 4.62 (s, 2H, N—CH2), 6.57–8.06 (m, 7H, Ar-H and C—H triazole). 13C NMR (100 MHz, DMSOd6) δ 35.5(C11), 133.1(C16), 111.2–134.9(Caromatique), 138.3(C12), 149.7(C2), 170.1(C4). Anal. Calcd. For C17H9Cl3N4O3: C, 48.19; H, 2.12; N, 13.21. Found. C, 48.20; H, 2.13; N, 13.20.
2.1.3.7 5j: N-(1-(4-chlorophenyl)-1H-1,2,3-triazol-5-yl)methylisatoïc anhydride
White solid (23 mg, 44%); mp 173–175 °C; IR (FTIR, cm−1): ν = 1774 (C⚌O); 1H NMR (400 MHz, DMSOd6) δ (ppm): 4.69 (s, 2H, N—CH2), 6.59–7.78 (m, 9H, Ar-H and C—H triazole). 13C NMR (100 MHz, DMSOd6) δ 36.4(C11), 132.1(C16), 111.4–135.2 (Caromatique), 137.0(C12), 150.1(C2), 170.2(C4). Anal. Calcd. For C17H11ClN4O3: C, 57.55; H, 3.10; N, 15.78. Found. C, 57.54; H, 3.12; N, 15.79.
2.2 Pharmacological study
2.2.1 Antimicrobial screening
First, the optical density of each microorganism suspensions was adjusted to 0.1 at OD600 for bacteria and 0.4 at OD540 for yeasts. Then, 500 μL of inoculums were dropped onto adequate agar plates. Sterile filter discs (diameter 6 mm, Biolife, Italy) were placed at the surface of the appropriate agar mediums and 10 mg/disc of the product dissolved in 10% of dimethyl sulfoxide was dropped onto each disc. Tetracycline (10 mg/mL; 10 μL/disc) and Ampicillin (10 mg/mL; 10 μL/disc), were used as reference drugs. After incubation for 18–24h at 37 °C, the antibacterial activities were evaluated by measuring an inhibition zone formed around the disc. Each assay was performed in triplicate. For this, the microbial inocula were prepared from 12 h broth cultures and spectrophotometrically adjusted to 107 CFU/mL. Serial twofold dilutions of the different amounts of the compounds (1000 to 0.09 μg/mL) were prepared in adequate broth. Then, 10 μL of the inocula of each reference strain were added to the plates containing the serial dilution and were incubated aerobically at 37 ◦C for 24h. MIC for minimum inhibition concentration was defined as the lowest concentration that completely inhibited visible cell growth during 24h incubation period at 37 °C. MBC for minimum bactericidal concentration and MFC for minimum fungicidal concentration values were determined by inoculating 10 μL of each well medium with no visible growth on Müller Hinton or Sabouraud Chloramphenicol agar plates and were defined as the lowest concentration at which 99% of the tested strains were killed after 24h of incubation at 37 °C.
2.2.2 Antioxidant assays
DPPH• radical ABTS•+ radical scavenging assays have been achieved based on the same protocol done by with slight modification (Abdelhamid et al., 2018). The samples, at different concentrations (0.078, 0.15, 0.31, 0.62, 1.25 and 2.50 mg/mL), were pipetted in separate test tubes and then mixed with DPPH• radical or ABTS•+ radical solutions. After 30 min of incubation in the darkness and at a temperature of 25 °C. The absorbance of the resulting solution was measured at 520 nm with a spectrophotometer.
Inhibition of free radicals in percent (PI%) was calculated by following equation:
PI% = 100 × (AControl – ASample)/AControl.
2.2.3 Anti-inflammatory test
2.2.3.1 Animals
Experiments were conducted using adult Wistar Swiss mice (20–25 g) of both sex. Animals were obtained from the Pasteur Institute (Tunis, Tunisia). They were kept in polypropylene cages at 25 ± 2 °C. Balanced pellet diet and water were supplied ad libitum. All animals were treated in accordance with guidelines established by the European Union regarding the Use and the Animal Care (CCE Council 86/609).
The investigation of anti-inflammatory properties was performed as done by Abdelhamid et al., (2018) with some modifications. Mice were divided into several batches, each consisting of 6 mice.
-
–
A first batch (negative control) received nothing,
-
–
A second batch (positive control - reference) that receives Lysine AcetylSalicylate (LAS).
-
–
A third batch that receives 4a, 4b and 4d at different doses.
30 min after intraperitoneal administration of the synthesized compounds or LAS, 30 µL of xylene (phylogenic agent) is applied to the inner and outer sides of the right ear of each mouse. The left ear is considered as a control. The thickness of the ear is measured with a digital caliper three hours after the induction of inflammation. The difference in thickness between the two ears was determined. Thus, we can calculate the percentage of edema inhibition compared to the control group according to the following formula:
Percentage inhibition (%) = [1 − (Δe (test)/ Δe (negative control)] × 100.
-
–
-Δe (test): Average of the differences in thickness between the two ears in the treated batch.
-
–
-Δe (negative control): Average of the differences in thickness between the two ears in the untreated batch.
2.3 Computational study
2.3.1 Molecular docking study
The 3D-coordinates of S. aureus tyrosyl-tRNA synthetase, C. albicans N-Myristoyltransferase, human COX-2 enzyme, and human Peroxiredoxin 5 crystals were retrieved from the Protein Data Bank by selecting 1JIJ, 1IYL, 1PXX, and 1HD2 entries, respectively. For the preparation of protein crystal structures, the Protein Preparation Wizard (PPW) tool was used, which involves a three-step procedure: preprocessing, optimization, and protein minimization. Initially, the proteins were preprocessed by introducing hydrogen atoms to the structure and removing crystallographic water molecules beyond 5 Å. The H-bond network was adjusted in PPW's optimize tab in order to fix overlapping hydrogens and the most likely positions of thiol and hydroxyl hydrogen atoms. Finally, using the force field Optimized potentials for liquid simulation 3e (OPLS3e), restrained minimization was attempted until the mean root mean square deviation (RMSD) of the non-hydrogen atoms converged at 0.30 Å (Zrieq et al., 2021). Based on the promising biological activity compounds, 4a,4b, and 4d were selected for docking, which were treated by using the LigPrep tool to explore the chirality, ionization states, ring conformations, and tautomers of each input structure. At the site of a co-crystallized ligand, a grid was generated utilizing the Receptor grid generation tool, which describes the targeted protein properties as well as the shape used to provide more precise ligand pose scoring. A standard precision docking methodology was used with a default force field, and a detailed investigation of ligand binding affinities was done by computing docking score and binding free energies (ΔG) between protein and ligands.
2.3.2 Molecular dynamics (MD) simulation study
The MD simulation studies were carried out using the Desmond tool of Schrödinger molecular modelling, which allows us to comprehend the binding of a ligand–protein complex in simulated physiological conditions (Lee et al., 2021, Ayipo et al., 2022, Acar Çevik et al., 2022). A molecular dynamic simulation study was performed for the promising compound 4b in a complex with the human COX-2 enzyme (PDB ID: 1PXX). The ligand–protein docked (4b-1PXX) complex system was developed using a simple point charged (SPC) solvent system with an orthorhombic box of 10 ˚A on each side. To maintain the system eclectically neutral, the built system charge was neutralized with appropriate Na+ and Cl– ions (Ghosh et al., 2021, Ahmad et al., 2022). After building the solvated system, the 4b-1PXX complex was minimize and relaxed using Desmond's default protocol, which relaxed the system into a local energy minimum. The model system is minimized using a hybrid approach of the steepest decent and the limited-memory Broyden-Fletcher-Goldfarb-Shanno (LBFGS) algorithms (Abdelhamid et al., 2018). The simulation was run in NPT ensemble mode with a 300 K temperature and 1 bar of pressure for 100 ns. For isothermal-isobaric conditions, the 'Nose-Hoover chain thermostat' and 'Martyna-Tobias-Klein barostat' algorithms were ensembled at 100 and 200 ps, respectively, using the Ensemble panel (Patel et al., 2018, 2022). Throughout the simulation, the recording interval trajectory was taken every 100 ps and 1000 frames. The Simulation Interactions Diagram Panel was used, which graphically displayed information regarding protein and ligand behavior and interactions throughout a simulation.
2.3.3 ADME study
Pharmacokinetic properties of the titled compounds were predicted using ADME (absorption, distribution, metabolism and excretion) descriptors by a SwissADME online server (https://www.swissadme.ch/).
3 Results and discussion
3.1 Chemistry
A straight forward one-step route to the targeted 1,2,3-triazole linked isatoïc anhydride derivatives 4 and 5, was performed via the “click chemistry” based on a Cu(I) or Ru(II)-catalyzed dipolar cycloaddition between N-propargylisatoïc anhydride 2 and arylazides 3a-j (Chouaïb et al., 2019). The Huisgen 1,3-dipolar cycloaddition of alkynes and azides (AAC) has appeared as a powerful linking reaction to give substituted-1,2,3-triazoles. This reaction when catalyzed with Copper (I) (CuAAC) leads exclusively to the 1,4-regioisomer (Amosova et al., 2022). On the other hand, the ruthenium-catalyzed version of the reaction (RuAAC), affords mainly the 1,5-regioisomer (Hosseinnejad and Mahdavian, 2018).
We have initiated the work by the treatment of isatoïc anhydride 1 with propargyl bromide using NaH as catalyst under refluxing anhydrous DMF for 2 h. The reaction provided the propargylated anhydride 2 in excellent yield (98%). The latter was transformed into a series of 1,4-disubstituted 1,2,3-triazole 4a-i (Scheme 1 and Table 1) by CuAAC reaction under microwave activation (200 W, 30 min). Under the same operating conditions using Ru(II) instead of Cu(I) and in the absence of trimethylamine promotes the formation of 1,5-disubstituted triazoles 5(a-f,j) (Scheme 1 and Table 1).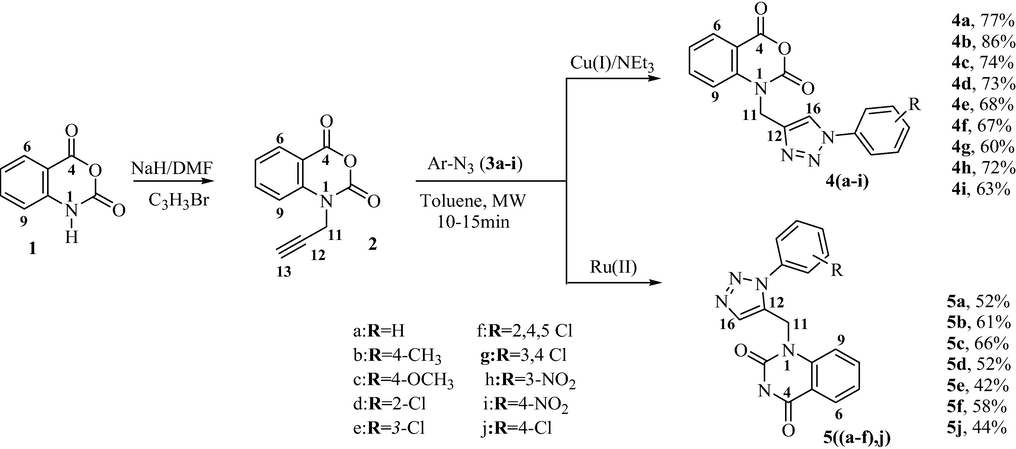
Synthesis of the regioisomers 4(a-i) and 5(a-f, j).
Entry
Compounds
Ar
Yields (%)a
1
4a
C6H5
77
2
4b
4-MeC6H4
86
3
4c
4-OMeC6H4
74
4
4d
2-ClC6H4
73
5
4e
3-ClC6H4
68
6
4f
2,4,5-Cl3C6H4
67
7
4g
3,4-Cl2C6H4
60
8
4h
3-NO2C6H4
72
9
4i
4-NO2C6H4
63
10
5a
C6H5
52
11
5b
4-MeC6H4
61
12
5c
4-OMeC6H4
66
13
5d
2-ClC6H4
52
14
5e
3-ClC6H4
42
15
5f
2,4,5- Cl3C6H4
58
16
5j
4-ClC6H4
44
Unprecedented structures of regioisomers 4(a-i) and 5(a-f,j) have been demonstrated by means of NMR experiments (1D and 2D), mass spectrometry and elemental analysis.
For example, the 1H NMR spectrum of compound 4a showed a singlet at δH 8.23 attributable to the proton H-16 of the triazole ring and signals at the level of the aromatic region (δH 7.32–8.17) attributable to the aromatic protons introduced by the used arylazides. The structure was supported by the 13C NMR spectrum which showed all the expected carbon signals corresponding to the anhydride-triazolyl derivatives, essentially the aromatic carbons resonating at (δC 111.6–158.1) and the C-16 carbon (δC 142.8). The ʋ(C⚌O) absorption at 1773 cm−1 in the IR spectrum of 4a is due to the carbonyl group of the anhydride.
In addition, the interpretation of the 2D NOESY spectrum of 1,4-disubstituted 1,2,3-triazole 4a shows strong correlations between the H11-H16 and H16-Harylazide protons and the absence of NOE effect between the H11-Harylazide protons (Fig. 2). On the other hand, the 2D NOESY spectrum of 1,5-disubstituted 1,2,3-triazole 5a shows strong correlations between H11-H16 and H11-Harylazide protons and the absence of NOE effect between H16-Harylazide protons (Fig. 1). Finally, the collected observations and the literature data (Pawara et al., 2021a, 2021b, D. E. Shaw Research, 2021, Girase et al., 2022) corroborate the structures proposed for compounds 4a and 5a.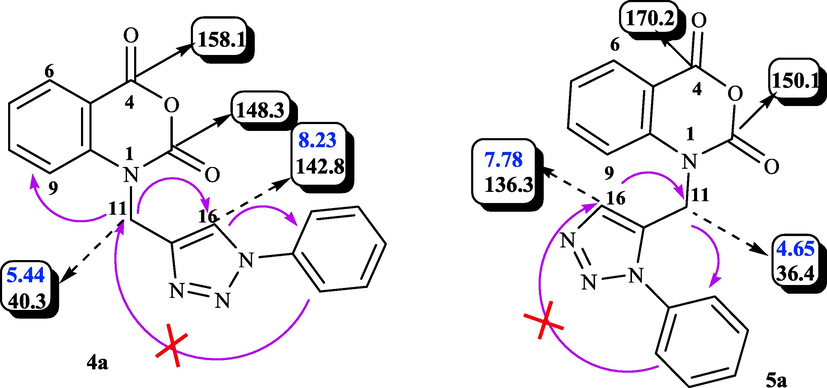
NOESY correlations of compounds 4a (left) and 5a (right).
3.2 Pharmacological study
3.2.1 Antimicrobial activity
The antimicrobial potential of the novel synthesized compounds was evaluated towards six different pathogenic species including 2 g-positive bacteria (S. aureus ATCC 25923, M. luteus NCIMB 81660), 2 g-negative bacteria (E. coli ATCC 25922, P. aeruginosa ATCC 27853), and 2 yeasts (C. albicans ATCC 90028, C. krusei ATCC 6258). As shown in Table 2, all synthesized analogues were found to be more active against highly resistant S. aureus MRSA with 4a (MIC 0.25 mM), 4b (MIC 0.23 mM) and 4d (MIC 0.21 mM) had MIC values less than the standard drug, tetracycline (MIC 0.28 mM). Towards M. luteus and P. aeruginosa, a potent activity was ascribed for all compounds in respect to the standard, tetracycline, however significant to moderate activity was achieved with E. coli. Interestingly, all tested compounds exhibited excellent antifungal properties against the fungal strains, C. albicans and C. krusei with MIC values lower than the standard drug, ampicillin (MIC 0.36 mM). The remaining compounds exhibited good to moderate antimicrobial properties.
MIC (mM)
Gram positive bacteria
Gram negative bacteria
Fungi
Entry
S. aureus ATCC 25923
M. luteus NCIMB 8166
E. coli ATCC 25922
P. aeruginosa ATCC 27853
C. albicans ATCC 90028
C. krusei ATCC 6258
4a
0.25
0.51
0.51
0.51
0.25
0.25
4b
0.23
0.47
0.47
0.47
0.47
0.23
4c
0.86
0.86
0.43
0.43
0.43
0.21
4d
0.21
0.84
0.42
0.21
0.21
0.21
4e
0.42
0.84
0.42
0.42
0.21
0.21
4f
0.29
0.58
0.29
0.58
0.29
0.21
4g
0.35
0.71
0.35
0.35
0.35
0.14
4h
0.40
0.80
0.40
0.40
0.20
0.20
4i
0.40
0.20
0.80
0.40
0.40
0.40
5a
0.51
1.03
1.03
0.51
0.25
0.25
5b
0.47
0.94
0.47
0.47
0.23
0.23
5c
0.43
0.86
0.86
0.43
0.21
0.21
5d
0.42
0.84
0.84
0.21
0.84
0.21
5e
0.42
0.84
0.84
0.42
0.21
0.21
5f
0.58
0.58
0.58
0.29
0.14
0.14
5j
0.42
0.84
0.42
0.42
0.21
0.42
Tetracycline
0.28
1.15
0.28
1.15
–
–
Ampicillin
–
–
–
–
0.36
0.36
3.2.2 Antioxidant activity
The antiradical potential of our synthesized compounds was evaluated spectrophotometrically as the capacity to scavenge DPPH• and ABTS•+.
-
DPPH• radical scavenging analysis
The capacity of the tested compounds and BHT to scavenge DPPH• is outlined in Table 3. From the results, it can be observed that out of the synthesized series 1,4-disubstituted 1,2,3-triazole (4a-i), compounds 4a (IC50 1.88 ± 0.07 µM) followed by 4b (IC50 2.19 ± 0.09 µM), 4d (IC50 2.30 ± 0.01 µM), and 4f (IC50 2.98 ± 0.02 µM), respectively, exhibited excellent DPPH• radical scavenging properties with lower IC50 values than BHT (IC50 3.52 ± 0.08 µM), used as standard antioxidant. Meanwhile, compounds 4c, 4e, 4g-i showed the weaker DPPH• radical scavenging ability than BHT. In regards to 1,5-disubstituted 1,2,3-triazole analogues, their DPPH• radical scavenging ability is weaker than those of 1,4-disubstituted 1,2,3-triazole with only 5c (IC50 2,59 ± 0.34 µM) had potent activity than the standard, BHT, however the remaining compounds had lesser activity.
-
ABTS•+ radical scavenging analysis
| Antioxidant activity (IC50 µM) | ||
|---|---|---|
| Entry DPPH ABTS | ||
| 4a | 1.88 ± 0.07 | 2.17 ± 0.02 |
| 4b | 2.19 ± 0.09 | 2.38 ± 0.43 |
| 4c | 4.59 ± 0.02 | 4.82 ± 0.14 |
| 4d | 2.30 ± 0.01 | 4.07 ± 0.57 |
| 4e | >7 | >7 |
| 4f | 2.98 ± 0.02 | 3.80 ± 0.01 |
| 4g | >7 | >7 |
| 4h | >7 | >7 |
| 4i | >7 | >7 |
| 5a | 5.58 ± 0.04 | 6.76 ± 0.32 |
| 5b | >7 | >7 |
| 5c | 2,59 ± 0.34 | 6.73 ± 0.32 |
| 5d | 4.17 ± 0.10 | 6.82 ± 0.17 |
| 5e | 5.90 ± 0.10 | 5.97 ± 0.02 |
| 5f | >7 | >7 |
| 5j | >7 | >7 |
| BHT | 3.52 ± 0.08 | 4.64 ± 0.11 |
The antioxidant results related to the capture of ABTS•+ radical in comparison to reference compounds are summarized in Table 3. As shown, data revealed that out of the synthesized compounds, 4a (IC50 2.17 ± 0.02 µM), 4b (IC50 2.38 ± 0.43 µM), 4f (IC50 3.80 ± 0.01 µM) and 4d (IC50 4.07 ± 0.57 µM) displayed relatively high ABTS•+ scavenging activities, in relation to BHT (IC50 4.64 ± 0.11 µM). The rest of compounds appeared to possess good to moderate scavenging potential. The same trend has been observed for compounds 4e, 4g-i, 5b, 5f and 5g displaying the weakest activities.
Our results generally indicate that the scavenging ability of our compounds seems to be even more potent in DPPH assay than ABTS test suggesting that the positive contribution of electron-donating substituents is more pronounced with DPPH test.
3.2.3 Anti-inflammatory activity
Inflammation is a physiological response of immune system to infections injuries, that involves many enzymatic and cellular processes to protect the body from all kinds of trauma. The acute anti-inflammatory potentiality of the prepared samples was assessed using xylene-induced ear edema in mice. The increase or decrease ear thickness of each group was measured after 3 h of xylene application. Results showed that the sample 4a was the most active against the inflammation. The percentages values showed a dose-dependent effect on the ear inflammation. At 100 mg/kg and after 3 h of xylene application, the ear edema was almost totally suppressed (84.41%) which had the same results as the drug reference “aspegic 300 mg/Kg” (PI = 87.53%). On the other hand, at only 25 mg/kg of 4a, the anti-inflammatory activity exhibited a potentiality exceed 50% of edema inhibition (61.03%) which is a strong activity compared to our reference (the dose is almost twelve times more; 300 mg/kg vs 25 mg/kg)). The samples 4b had a medium activity compared to 4a, but always more active than the reference. However, the inhibition percentage of the tested 4d showed an ineffective value at the highest tested dose (100 mg/kg; PI = 42.84%). As previously demonstrated, the central carbocyclic or heterocyclic ring system as found in many COX-2 inhibitors can be replaced by a central 1,2,3-triazole unit without losing COX-2 inhibition potency and selectivity. The high anti-inflammatory (via COX-2) inhibition potency of some 1,2,3-triazoles having a vicinal diaryl substitution pattern along with their ease in synthesis through versatile Ru(II)-catalyzed click chemistry make this class of compounds interesting candidates for further design and synthesis of highly selective and potent COX-2 inhibitors (Othman et al., 2021b) (Table 4). The letters (a–e) indicate significant differences between the tested samples (Duncan’s multiple range test, p < 0.05).
Entry
Dose mg/kg
Oedema inhibition (%)
4a
12.5
25
50
10021.66 ± 0.55a
61.03 ± 7.79cd
71.68 ± 4.29de
84.41 ± 7.79e
4b
25
50
10024.78 ± 3.86ab
63.62 ± 4.50d
74.01 ± 9.00de
4d
50
10015.43 ± 0.55a
42.84 ± 1.35bc
Aspegic
300
87.53 ± 6.97e
3.3 Structure-activity relationships (SARs)
The SARs of the synthesized compounds (Fig. 3) were analyzed on the basis of substitution on aryl ring and the type-disubstituted 1,2,3-triazoles. The biological results showed that they play a decisive role in controlling the activity. Of all the synthesized, the overall assessment revealed that among the 1,4-disubstituted 1,2,3-triazoles series (4a-j), compound 4a which did not have any substituents on the phenyl ring followed by 4b sharing a 4-CH3 as an EDG on the aromatic ring, were found to possess the strongest activity decrease both antioxidant and anti-inflammatory activities. Upon replacing 4-CH3 (4b) by a more powerful EDG 4-OCH3 (4C), both antioxidant and anti-inflammatory activities decreased, due to the existence of resonance conjugating structures in 4c. The presence of an EWG (2-Cl) on the phenyl ring (4d) seems to decrease both antioxidant (more pronounced on ABTS scavenging ability) and anti-inflammatory activities, however when the phenyl ring was monosubstituted with 3-Cl group (4e), or disubstituted with tow Cl groups at 3- and 4-positions (4g), a dramatically decrease of the activity was observed. Besides, when the phenyl ring was trichloro-substituted (4f), the ABTS scavenging ability was not affected but a slightly decrease of the DPPH scavenging and the anti-inflammatory activities was observed, though, the activity remains still higher than the reference, BHT. We note the effectiveness of 1,2,3-triazole (aggressive pharmacophore) and phenyl (hydrophobic pharmacophore) moieties in the activity. The drastically decreased activity observed in 4c bearing the methoxy group (4-OCH3) as an EDG, attached to the phenyl ring may be explained by the existence of resonance conjugating structures when compared to 4a containing the 4-CH3. The presence of strongly EWG attached to the phenyl ring (4h with 3-NO2 and 4i with 4-NO2) seems to reduce the activity. In regards to the 1,5-disubstituted 1,2,3-triazoles series (5a-f, 5j), results showed lower potency than 1,4-disubstituted 1,2,3-triazoles series (4a-j) suggesting the implication of the substitution type 1,4 vs1,5 of the 1,2,3-triazoles scaffold on the activity.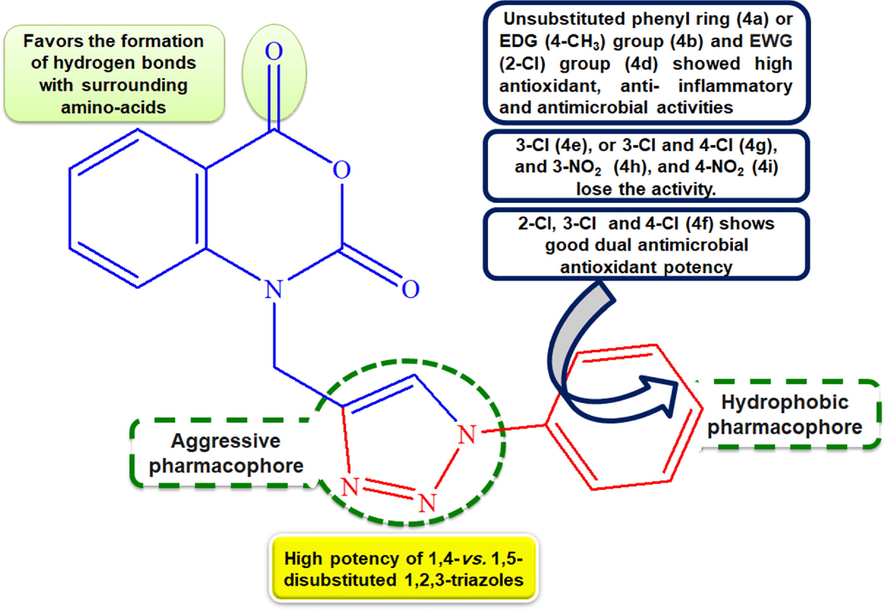
Generalized SARs of the synthesized compounds.
3.4 Computational study
3.4.1 Drug-likeness assessment
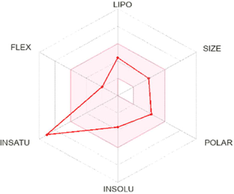
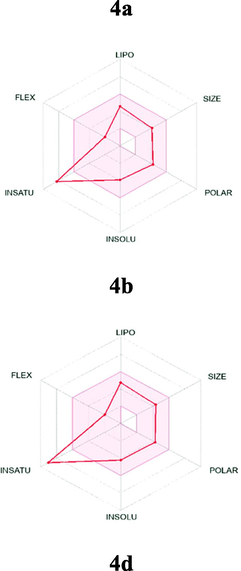
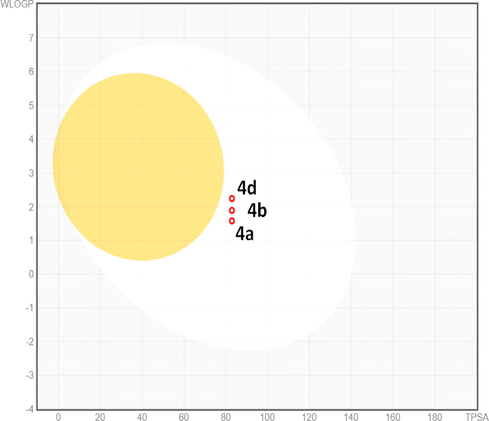
The most active analogues have been further predicted for their druglikeness and pharmacokinetic properties including absorption, distribution, metabolism and excretion toxicity (ADME) parameters to verify that the designed molecules are a viable drug (Table 5). The selected ligands were predicted to comply with Lipinski's rule-of-five, as well as Ghose, Veber, Egan and Muegge filters. They displayed good lipophilic property with consensus log Po/w value of 4.28 good bioavailability score (55%), high gastrointestinal absorption (GI) and were predicted to be not blood–brain-barrier (BBB) permeant as well as not P-gp substrate. Simultaneously, predictive data showed that the designed compounds are inhibitors of cytochrome P450 isoenzymes CYP 1A2 and CYP2C19, but not inhibitor of CYP2D6 and particularly CYP3A4, known as the most abundant in the liver and an active participant in the metabolism of approximately 60% of known drugs. The skin permeation ability of the selected compounds (LogKP), was in the range −6.72 cm/s to −6.02 cm/s. As per as medicinal chemistry properties, all compounds showed none PAINS alert and the majority of them with also none brenk alert and generally good synthetic accessibility. The bioavailability radar plot in which the following properties were taken into account such as flexibility, lipophilicity, saturation, size, polarity and solubility indicate that the selected analogues exceed the pink area zone by one parameter, signifying its good predicted oral bioavailability. The BOILED-Egg graph (Table 5) clearly specifies that compound 4a, 4b and 4d are not a substrate for P-glycoprotein (PGP-) which diminished the possibility of their resistance by tumor cell lines through efflux.
Entry
4a
4b
4d
Entry
4a
4b
4d
Physicochemical Properties /Lipophilicity/Druglikeness
Pharmacokinetics
Molecular weight
320.30
334.33
354.75
GI absorption
High
High
High
Num. heavy atoms
24
25
25
BBB permeant
No
No
No
Num. arom. heavy atoms
21
21
21
P-gp substrate
No
No
No
Fraction Csp3
0.06
0.11
0.06
CYP1A2 inhibitor
Yes
Yes
Yes
Num. rotatable bonds
3
3
3
CYP2C19 inhibitor
Yes
Yes
Yes
Num. H-bond acceptors
5
5
5
CYP2C9 inhibitor
No
Yes
No
Num. H-bond donors
0
0
0
CYP2D6 inhibitor
No
No
No
Molar Refractivity
87.41
92.38
92.42
CYP3A4 inhibitor
No
No
No
TPSA (Å2)
82.92
82.92
82.92
Log Kp (cm/s)
−6.72
−6.55
−6.48
Consensus Log Po/w
2.06
2.47
2.54
Radar chart
Lipinskiˈs Rule
Yes
Yes
Yes
Veber
Yes
Yes
Yes
Egan
Yes
Yes
Yes
Ghose
Yes
Yes
Yes
Muegge
Yes
Yes
Yes
Bioavailability Score
0.55
0.55
0.55
Medicinal Chemistry
PAINS
0
0
0
Brenk
0
0
0
Leadlikeness
Yes
Yes
No
Synthetic accessibility
3.36
3.42
3.35
BOILED-Egg graph
3.4.2 Molecular docking: Receptor-ligands interactions
After identifying the activity and the pharmacokinetics proprieties of the most active compounds, they also were chosen for a virtual screening by molecular docking and dynamic simulation for predicting their interactions mode in order to elucidate their possible mechanism of action. The different targets were selected as follows: S. aureus tyrosyl-tRNA synthetase enzyme was selected as an emerging and attractive enzyme for finding new antibacterial agents. The monomeric enzyme, N-Myristoyltransferase (NMT) that catalysis the transfer of the myristoyl group of myristoyl-CoA to the N-terminal glycine of various eukaryotic cellular proteins have been demonstrated for its viability of pathogenic fungi, such as C. albicans and C. neoformans. Thus, NMT would be a potential target for the discovery of novel fungicidal drugs. On the other hand, cyclooxygenase (COX-2) is widely known for its crucial role in converting arachidonic acid to prostaglandins (PG), that play an important role in the process of inflammation. Therefore, COX-2 has been validated as a molecular target for treating inflammatory diseases. Concerning the enzyme Human Peroxiredoxin 5, known as a novel thioredoxin peroxidase, which was widely expressed in mammalian tissues suggesting its important role as antioxidant in organelles that are major sources of ROS, has been selected as a potent antioxidant target. The molecular docking results (Table 6) on the bacterial target S. aureus tyrosyl-tRNA synthetase suggested that compound 4a had been shown to have the highest binding energy at −5.9 kcal/mol; from which conventional hydrogen bonding interactions of the amino acid residues Asp40, and Gln190, of the target with the ligand 4a. Furthermore, hydrophobic interactions were also observed between His50 of the bacterial target and the same compound. DS, Docking score in kcal/mol; ΔG, Prime MMGBSA Binding free energies in kcal/mol.
Protein targets
S. aureus tyrosyl-tRNA synthetase
(PDB ID: 1JIJ)C. albicans N-Myristoyltransferase
(PDB ID: 1IYL)Human COX-2 enzyme
(PDB ID: 1PXX)Human Peroxiredoxin 5
(PDB ID: 1HD2)
DS
ΔG
Interacted residues
DS
ΔG
Interacted residues
DS
ΔG
Interacted residues
DS
ΔG
Interacted residues
4a
−5.90
−53.54
His50a
Asp40b (1.89 Å)
Gln190b (2.29 Å)−7.06
−69.93
Tyr225a
Tyr354a
−9.10
−59.92
Trp387a
−3.68
−46.63
Cys47b
(2.25 Å)
Arg127b (2.16 Å)
Arg127c
4b
−5.85
−55.53
His50a
Gln190b (2.23 Å)−7.98
−56.7
Tyr225a
Tyr354a
Tyr335b
(2.46 Å)−9.14
−64.17
Trp387a
−3.67
−48.03
Cys47b
(2.25 Å)
Arg127b (2.17)
Arg127c
4d
−5.85
−52.17
Lys84b (2.53 Å)
−7.95
−56.18
Phe240a
Phe115a
Phe117a
Asn392b
(1.78 Å)
His227b
(1.80 Å)−8.94
−72.5
Ser530b (2.11 Å)
−3.76
−48.52
Cys47b
(2.20 Å)
Arg127b (2.10 Å)
Arg127c
Another potent compound, 4b, interacted with significant hydrogen and hydrophobic interactions, which accounts for its binding affinity towards both the targets, C. albicans N-Myristoyltransferase and human COX-2 enzymes; the docking scores were −7.98 kcal/mol and −9.142 Kcal/mol with 1IYL and 1PXX, respectively.
The central triazole ring of compound 4b and the phenyl groups of Tyr225 and Tyr354 residues generate three hydrophobic interactions in the C. albicans N-Myristoyltransferase protein; another hydrophobic interaction exists between the 2H-benzo[d][1,3]oxazine-2,4(1H)-dione and the phenyl group of Tyr354. In compound 4b, the carbonyl oxygen in the 2H-benzo[d][1,3]oxazine-2,4(1H)-dione scaffold is implicated in hydrogen bonding with Tyr335 (Fig. 4). Hydrophobic interactions with Tyr225 is also seen in the co-crystallized ligand, which supports the potency of compound 4b towards the C. albicans N-Myristoyltransferase protein. With a binding affinity of −9.14 kcal/mol (ΔG = -64.17 kcal/mol), compound 4b has the second-highest binding affinity with the human COX-2 enzyme. The docking scores and ΔG of the same molecule with human Peroxiredoxin 5 are −3.67 and −48.03Kcal/mol, respectively. In the human COX-2 enzyme, compound 4b made one hydrophobic interaction with the 2H-benzo[d][1,3]oxazine-2,4(1H)-dione ring and the phenyl ring of residue Trp387 (Fig. 5). Compound 4d, which comprises a 2-chlorophenyl group in the 1,2,3-triazole structure, has a binding affinity of −5.85 kcal/mol with the bacterial S. aureus tyrosyl-tRNA synthetase target, and −7.95, −8.94, and −3.76 kcal/mol with the fungal and human targets 1IYL, 1PXX, and 1HD2, respectively.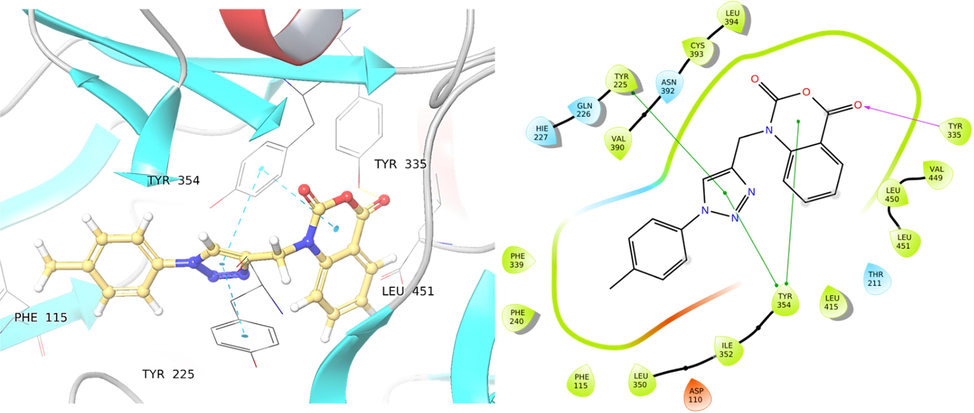
Binding of compound 4b into the cavity of C. albicans N-Myristoyltransferase (PDB ID: 1IYL).
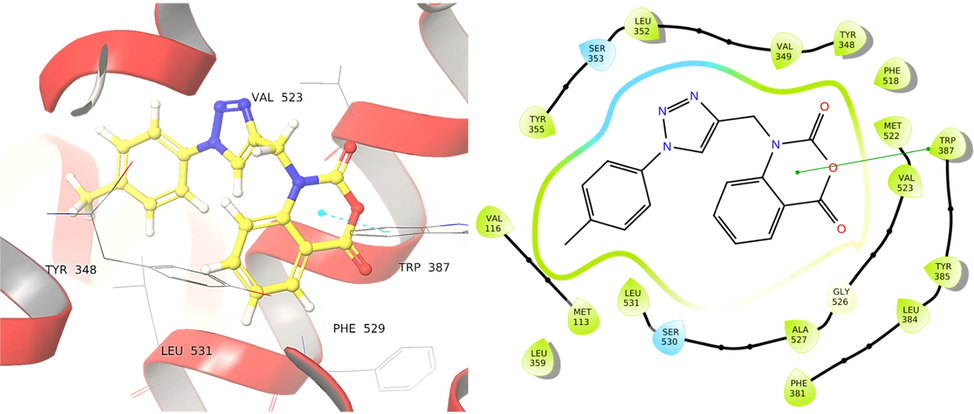
Binding of compound 4b to into the cavity of Human COX-2 enzyme (PDB ID: 1PXX).
3.4.3 Molecular dynamic (MD) simulation
MD is a comprehensive analysis tool for determining the structural stability and dynamics of proteins caused by ligand/inhibitor binding. In this study, we have conducted a 100 ns MD simulation of the best scored dock complex (4b-Human COX-2 enzyme). The MD simulation trajectory was used to assess key parameters, including the protein Cα atoms RMSD, RMSF, ligand atoms RMSD, and protein ligand interactions.
RMSD is defined as a difference in the structure of a protein or protein–ligand complex from a reference structure, typically the beginning frame. Assessing the protein's RMSD over the simulation can provide details on its structural conformation, indicating the protein's stability and whether the simulation has stabilized. Ligand RMSD can reveal the ligand's stability with respect to the protein, as well as the variation of its intrinsic conformation. The RMSD is a key quantitative measurement in which small deviations suggest stable conformation and vice versa (Acar Çevik et al., 2022, Abdelhamid et al., 2018, Ahmad et al., 2021, 2022, Boulaamane et al., 2022, D. E. Shaw Research, 2021, Girase et al., 2022, Ghosh et al., 2021, Malani et al., 2021, Patel et al., 2018, 2022, Pawara et al., 2021a, 2021b). Fig. 6A shows the RMSD of Cα atoms of 1PXX (protein) in teal color and compound 4b (ligand) RMSD in a brown line. Throughout the simulation time, no major changes in RMSD values of protein and ligand were noticed as compared with the initial frame. The average RMSD values for 1PXX alone and for the ligand in relation to protein were 2.64 Å and 2.89 Å, respectively. The formation of a stable 4b-1PXX complex is expected because the variance in RMSD values of the protein and ligand with respect to protein was substantially lower than the acceptable limit of 3.0 Å.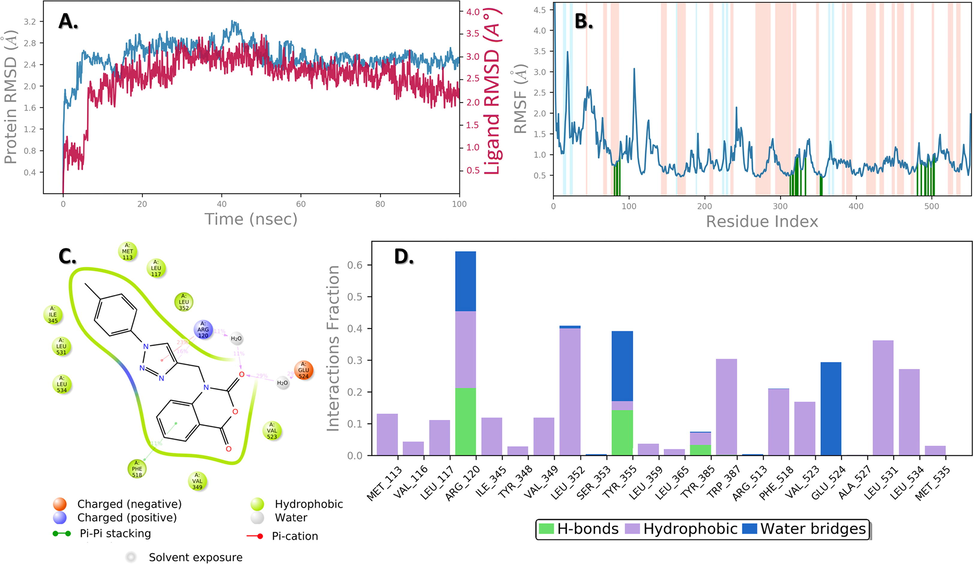
MD simulation analysis of compound 4b in complex with Human COX-2 enzyme (PDB ID: 1PXX) (A) RMSD (Protein RMSD is shown in teal while RMSD of compound 4b are shown in brown) (B) Protein RMSF (C) 2d Interaction diagram and (D) Protein–ligand contact analysis of MD trajectory.
During simulation, the stability of the protein–ligand complex is influenced by the individual amino acid residues of the protein. The RMSF parameter can be used to investigate the residue fluctuations during the simulation. The RMSF value was derived from the MD simulation trajectory and is presented in Fig. 6B. With the exception of the loop region and the C-terminal Overall, the amino acids were stable during the simulation window with an RMSF range of between 0.60 and 3.50 Å. The amino acids that have made contact with the ligand during the simulation trajectory include: Met113, Val116, Leu117, Arg120, Ile345, Tyr348, Val349, Leu352, Ser353, Tyr355, Leu359, Leu365, Tyr385, Trp387, Arg513, Phe518, Val523, Glu524, Ala527, Leu531, Leu534, and Met535. The amino acids that fluctuate more are Asp53 with an RMSF of 3.49 Å, and Trp139 with an RMSF of 3.074 Å and Ile274 with an RMSF of 2.144 Å. It's believed that such high fluctuations in RMSF are acceptable since there are no significant molecular interactions with bound ligand in that region, and the structure is highly flexible. As a result, the stability of the protein–ligand complex was certainly explained by low RMSF values at ligand-contact residues.
Fig. 6C and 6D show the ligand 2D interaction and the histogram depicting the interactions type of compound 4b with 1PXX throughout the simulation, respectively. Carbonyl oxygen of 2H-benzo [d][1,3]oxazine-2,4 (1H)-dione is involved in amino acid mediated (Arg120 and Glu524) hydrogen bonding. On the other hand, charged positive residue Arg120, makes an π-cation contact with the core triazole ring for 23% of the simulation time. Major hydrophobic interactions are observed with the amino acids Met113, Leu117, Ile345, Val349, Leu352, Trp387, Phe518, Val523, Leu531, and Leu534. Three hydrogen bonds are observed between the Arg120, Tyr355, and Tyr385 residues in the 4b-1PXX complex. Overall, the simulation revealed more hydrophobic interactions, and amino-acid mediated water bridges stabilized the compound 4b in the cavity of the 1PXX protein.
4 Conclusions
We have successfully designed and achieved the synthesis a novel series of 1,4-and 1,5-disubstituted 1,2,3-triazoles linked isatoïc anhydride derivatives via click chemistry reaction strategy and under microwave irradiation. The newly synthesized compounds were evaluated for their multifunctional biological profile. Antimicrobial study revealed that all synthesized compounds exhibited interesting antimicrobial activity against the tested pathogenic strains with high susceptibility towards S. aureus, E. coli and P. aeruginosa. Analogues 4a, followed by 4b and 4d displayed excellent DPPH and ABTS radical scavenging ability in respect to the standard BHT. The anti-inflammatory effect on 4a, 4b and 4d revealed also their high potency. SAR study revealed that compounds with Unsubstituted phenyl ring (4a), EDG (4-CH3) (4b) and EWG (2-Cl) (4d) showed high antioxidant, anti- inflammatory and antimicrobial activities. Docking analyses disclose that compounds 4a, 4b, and 4d are bound efficiently with the active site, displaying good affinity for different amino acids in the target proteins which corroborate with the in vitro data. The 100 ns simulated trajectories under dynamic conditions revealed strong and consistent interaction stability. Finally, this study proves the importance of the synthesized compounds 4a, 4b and 4d for designing and developing new antioxidant, antimicrobial and anti-inflammatory potential agents. Further studies are encouraged to test these compounds against other activities and other possible modifications can also be done to improve their potency.
Acknowledgments
This research has been funded by Scientific Research Deanship at University of Ha’il - Saudi Arabia through project number MDR-22 021.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Phytochemical Analysis and Evaluation of the Antioxidant, Anti-Inflammatory, and Antinociceptive Potential of Phlorotannin-Rich Fractions from Three Mediterranean Brown Seaweeds. Mar. Biotechnol.. 2018;20:60-74.
- [Google Scholar]
- Design, synthesis, molecular modeling, DFT, ADME and biological evaluation studies of some new 1,3,4-oxadiazole linked benzimidazoles as anticancer agents and aromatase inhibitors. J. Biomol. Struct. Dyn. 2022:1-15.
- [CrossRef] [Google Scholar]
- Computational investigation of phytochemicals from Withania somnifera (Indian ginseng/ashwagandha) as plausible inhibitors of GluN2B-containing NMDA receptors. J. Biomol. Struct. Dyn. 2021:1-13.
- [CrossRef] [Google Scholar]
- Design, synthesis, in vitro anticancer and antimicrobial evaluation, SAR analysis, molecular docking and dynamic simulation of new pyrazoles, triazoles and pyridazines based isoxazole. J. Mol. Struct.. 2022;1251:131972.
- [Google Scholar]
- Antioxidant Activities of a New Chemotype of Piper cubeba L. Fruit Essential Oil (Methyleugenol/Eugenol). In Silico Molecular Docking and ADMET Studies. Plants. 2020;9:1534.
- [Google Scholar]
- Antimicrobial and Wound Healing Potential of a New Chemotype from Piper cubeba L. Essential Oil and In Silico Study on S. aureus tyrosyl-tRNA Synthetase Protein. Plants. 2021;10:205-224.
- [Google Scholar]
- 2-Bromomethyl-1, 3-thiaselenole in click chemistry: Synthesis of 1-(2, 3-dihydro-1, 4-thiaselenin-2-yl)-1H-1,2,3-triazoles via copper-catalyzed and thermal 1, 3-dipolar cycloaddition with alkynes. J. Organomet. Chem.. 2022;977:122442.
- [Google Scholar]
- Stereoselective synthesis of 1,2,3-triazolyl-functionalized isoxazolidines, via two consecutive 1, 3-dipolar cycloadditions, as precursors of unnatural amino acids. Tetrahedron Lett.. 2013;54:1967-1971.
- [Google Scholar]
- Molecular modelling and structure-activity relationship of a natural derivative of o-hydroxybenzoate as a potent inhibitor of dual NSP3 and NSP12 of SARS-CoV-2: in silico study. J. Biomol. Struct. Dyn. 2022:1-19.
- [CrossRef] [Google Scholar]
- Synthesis of isoniazid-1,2,3-triazole conjugates: Antitubercular, antimicrobial evaluation and molecular docking study. J. Heterocycl. Chem.. 2020;57:3544-3557.
- [Google Scholar]
- Allium subhirsutum L. as a Potential Source of Antioxidant and Anticancer Bioactive Molecules: HR-LCMS Phytochemical Profiling, In Vitro and In Vivo Pharmacological Study. Antioxidants. 2020;9:1-22.
- [Google Scholar]
- Phytochemicals, antioxidant and antimicrobial potentials and LC-MS analysis of hydroalcoholic extracts of leaves and flowers of Erodium glaucophyllum collected from Tunisian Sahara. Food Sci. Biotechnol.. 2018;38:310-317.
- [Google Scholar]
- Date Palm Tree’s Root-Derived Endophytes as Fungal Cell Factories for Diverse Bioactive Metabolites. Int. J. Mol. Sci.. 2018;19:1986.
- [Google Scholar]
- Fluconazole resistance in Candida species: a current perspective. Infect Drug Resist.. 2017;10:237-245.
- [Google Scholar]
- Design and synthesis of triazole conjugated novel 2,5-diaryl substituted 1,3,4-oxadiazoles as potential antimicrobial and anti-fungal agents. Bioorg. Med. Chem. Lett.. 2021;41:128004.
- [Google Scholar]
- Structural exploration of selected C6 and C7-substituted coumarin isomers as selective MAO-B inhibitors. J. Biomol. Struct. Dyn. 2022:1-15.
- [CrossRef] [Google Scholar]
- Unprecedented stereoselective synthesis of 3-methylisoxazolidine-5-aryl-1,2,4-oxadiazoles via 1,3-dipolar cycloaddition and study of their in vitro antioxidant activity. Synth. Commun.. 2016;46:2037-2044.
- [Google Scholar]
- Antifungal drug discovery: the process and outcomes. Future Microbiol.. 2014;9:791-805.
- [Google Scholar]
- Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules. 2022;27:616.
- [Google Scholar]
- Synthesis, Characterization and Biological Activities of New Symmetric Bis-1,2,3-Triazoles with Click Chemistry. Med. Chem.. 2018;14:230-241.
- [Google Scholar]
- Regiospecific synthesis by copper-and ruthenium-catalyzed azide–alkyne 1, 3-dipolar cycloaddition, anticancer and anti-inflammatory activities of oleanolic acid triazole derivatives. Arab. J. Chem. 2019;12(8):3732-3742.
- [Google Scholar]
- D. E. Shaw Research, Schrödinger Release (2021-1). Desmond molecular dynamics system. Maestro-Desmond interoperability tools.
- Investigation of phytochemical contents, in vitro antioxidant and antibacterial behavior and in vivo anti-inflammatory potential of Ecballium elaterium methanol fruits extract. Food Sci. Technol. (Campinas). 2017;37:558-563.
- [Google Scholar]
- Stereoselective synthesis of enantiopure N-substituted pyrrolidin-2,5-dione derivatives by 1,3-dipolar cycloaddition and assessment of their in vitro antioxidant and antibacterial activities. Bioorg. Med. Chem. Lett.. 2017;27:2302-2307.
- [Google Scholar]
- Synthesis, in vitro antimicrobial assessment, and computational investigation of pharmacokinetic and bioactivity properties of novel trifluoromethylated compounds using in silico ADME and toxicity prediction tools. Monatsh. Chem.. 2020;151:267-280.
- [Google Scholar]
- Design, synthesis, molecular properties and in vitro antioxidant and antibacterial potential of novel enantiopure isoxazolidine derivatives. Arab J Chem.. 2020;13:2121-2131.
- [Google Scholar]
- Novel enantiopure isoxazolidine and C-alkyl imine oxide derivatives as potential hypoglycemic agents: Design, synthesis, dual inhibitors of α-amylase and α-glucosidase, ADMET and molecular docking study. Bioorg. Chem.. 2020;104:104270.
- [Google Scholar]
- In silico validation of anti-viral drugs obtained from marine sources as a potential target against SARS-CoV-2 Mpro. J. Indian Chem. Soc.. 2021;98:100272.
- [Google Scholar]
- Optimizing cardio, hepato and phospholipidosis toxicity of the Bedaquiline by chemoinformatics and molecular modelling approach. SAR QSAR Environ. Res.. 2022;33:215-235.
- [Google Scholar]
- Bioactivities and in silico study of Pergularia tomentosa L. phytochemicals as potent antimicrobial agents targeting type IIA topoisomerase, TyrRS, and Sap1 virulence proteins. Environ. Sci. Poll. Res.. 2021;28:25349-25367.
- [Google Scholar]
- Antimicrobial, Antioxidant, Anti-Acetylcholinesterase, Antidiabetic, and Pharmacokinetic Properties of Carum carvi L. and Coriandrum sativum L. Essential Oils Alone and in Combination. Molecules. 2021;26:3625.
- [Google Scholar]
- Synthesis, characterization, and pharmacological studies of ferrocene-1H-1,2,3-triazole hybrids. J. Mol. Struct.. 2017;1146:536-545.
- [Google Scholar]
- Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook. Antibiotics. 2022;11:40.
- [Google Scholar]
- Design and Synthesis of Various 5′-Deoxy-5′-(4-Substituted-1,2,3-Triazol-1-yl)-Uridine Analogues as Inhibitors of Mycobacterium tuberculosis Mur Ligases. Molecules. 2020;25:4953.
- [Google Scholar]
- Quantum chemistry study on regioselectivity in ruthenium catalyzed synthesis of 1, 5-disubstituted 1,2,3-triazoles. Comput. Theor. Chem.. 2018;1143:29-35.
- [Google Scholar]
- Synthesis, Docking, and Biological activities of novel Metacetamol embedded [1,2,3]-triazole derivatives. J. Mol. Struct.. 2021;1242:130786.
- [Google Scholar]
- In vitro antimicrobial and α-glucosidase inhibitory potential of enantiopure cycloalkylglycine derivatives: Insights into their in silico pharmacokinetic, druglikeness, and medicinal chemistry properties. J. App. Pharm. Sci.. 2020;10:107-115.
- [Google Scholar]
- A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules. 2020;25:1909.
- [Google Scholar]
- Synthesis and biological evaluation of phenyl-1H-1,2,3-triazole derivatives as anti-inflammatory agents. Bioorg. Chem.. 2015;59:1-11.
- [Google Scholar]
- Mining of a novel esterase (est3S) gene from a cow rumen metagenomic library with organosphosphorus insecticides degrading capability: Catalytic insights by site directed mutations, docking, and molecular dynamic simulations. Int. J. Biol. Macromol.. 2021;190:441-455.
- [Google Scholar]
- Synthesis, molecular docking, DFT study, and in vitro antimicrobial activity of some 4-(biphenyl-4-yl)-1,4-dihydropyridine and 4-(biphenyl-4-yl) pyridine derivatives. J. of biochem. and mol. toxi 2021
- [CrossRef] [Google Scholar]
- Thymus musilii Velen. as a promising source of potent bioactive compounds with its pharmacological properties: In vitro and in silico analysis. Arab. J. Chem.. 2020;13:6782-6801.
- [Google Scholar]
- HR-LCMS-Based Metabolite Profiling, Antioxidant, and Anticancer Properties of Teucrium polium L. Methanolic Extract: Computational and In Vitro Study. Antioxidants. 2020;9:1-23.
- [Google Scholar]
- Design, synthesis ADMET and molecular docking of new imidazo[4,5-b]pyridine-5-thione derivatives as potential tyrosyl-tRNA synthetase inhibitors. Bioorg. Chem.. 2020;102:104105.
- [Google Scholar]
- Design, synthesis ADMET and molecular docking of new imidazo[4,5-b]pyridine-5-thione derivatives as potential tyrosyl-tRNA synthetase inhibitors. Bioorg. Chem.. 2020;102:104105.
- [Google Scholar]
- Novel fused pyridine derivatives containing pyrimidine moiety as prospective tyrosyl-tRNA synthetase inhibitors: Design, synthesis, pharmacokinetics and molecular docking studies. J. Mol. Struct.. 2020;1219:128651.
- [Google Scholar]
- Synthesis, Structure-Activity Relationship and in silico Studies of Novel Pyrazolothiazole and Thiazolopyridine Derivatives as Prospective Antimicrobial and Anticancer Agents. ChemistrySelect. 2021;6:7860-7872.
- [Google Scholar]
- New substituted pyrazolones and dipyrazolotriazines as promising tyrosyl-tRNA synthetase and peroxiredoxin-5 inhibitors: Design, synthesis, molecular docking and structure-activity relationship (SAR) analysis. Bioorg. Chem.. 2021;109:104704.
- [Google Scholar]
- New 1,2,3-Triazoles from (R)-Carvone: Synthesis, DFT Mechanistic Study and In Vitro Cytotoxic Evaluation. Molecules. 2022;27:769.
- [Google Scholar]
- Design and synthesis of novel 2,4-disubstituted aminopyrimidines: reversible non-covalent T790M EGFR inhibitors. J. Recept. Signal Transduct. Res.. 2018;38:393-412.
- [Google Scholar]
- Investigating the Impact of Different Acrylamide (Electrophilic Warhead) on Osimertinib's Pharmacological Spectrum by Molecular Mechanic and Quantum Mechanic Approach. Comb. Chem. High Throughput Screen.. 2022;25:149-166.
- [Google Scholar]
- Novel, selective acrylamide linked quinazolines for the treatment of double mutant EGFR-L858R/T790M Non-Small-Cell lung cancer (NSCLC) Bioorg. Chem.. 2021;115:105234.
- [Google Scholar]
- Computational identification of 2,4-disubstituted amino-pyrimidines as L858R/T790M-EGFR double mutant inhibitors using pharmacophore mapping, molecular docking, binding free energy calculation, DFT study and molecular dynamic simulation. In Silico Pharmacol.. 2021;6:54.
- [Google Scholar]
- Click synthesis of 1,2,3-triazole based imidazoles: Antitubercular evaluation, molecular docking and HSA binding studies. Bioorg. Med. Chem. Lett.. 2021;36:127810.
- [Google Scholar]
- Novel strategies to fight Candida species infection. Crit. Rev. Microbiol.. 2016;42:594-606.
- [Google Scholar]
- Synthesis, Characterization, and Antioxidant and Anticancer Activity of 1,4-Disubstituted 1,2,3-triazoles. J. Mol. Struct.. 2021;1232:130042.
- [Google Scholar]
- Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed. Res.. 2016;2016:2475067.
- [Google Scholar]
- Synthesis of novel 2-mercapto benzothiazole and 1,2,3-triazole based bis-heterocycles: their anti-inflammatory and anti-noceptive activites. Eur. J. Med. Chem.. 2012;49(31):324-333.
- [Google Scholar]
- Synthesis, in vitro evaluation, and molecular docking studies of novel hydrazineylideneindolinone linked to phenoxymethyl-1,2,3-triazole derivatives as potential α-glucosidase inhibitors. Bioorg. Chem.. 2021;111:104869.
- [Google Scholar]
- Synthesis and anti-proliferative activity of a novel 1,2,3-triazole tethered chalcone acetamide derivatives. Bioorg. Med. Chem. Lett.. 2020;30:127304.
- [Google Scholar]
- Inhibiting Bacterial Drug Efflux Pumps via Phyto-Therapeutics to Combat Threatening Antimicrobial Resistance. Front. Microbiol.. 2018;9:2990.
- [Google Scholar]
- 1,2,3-Triazole-containing hybrids with potential antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) Eur. J. Med. Chem.. 2020;206:112686.
- [Google Scholar]
- Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations. Int. J. Mol. Sci.. 2021;22:10693.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary material to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104226.
Appendix A
Supplementary material
The following are the Supplementary material to this article:Supplementary data 1
Supplementary data 1




