

Translate this page into:
Design, synthesis, crystal structures and biological evaluation of some 1,3-thiazolidin-4-ones as dual CDK2/EGFR potent inhibitors with potential apoptotic antiproliferative effects
⁎Corresponding author. bgyoussif@ju.edu.sa (Bahaa G.M. Youssif),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
A series of thiazolidine-4-one derivatives as dual EGFR/CDK2 inhibitors has been developed. Compounds 5d, 5e, and 5f were the most active antiproliferative agents. Compounds 5d, 5e, and 5f display potent antiproliferative activity with strong inhibition of EGFR/CDK2. Docking study revealed high binding affinities toward EGFR and CDK2.
Abstract
A series of novel thiazolidine-4-one derivatives was synthesized by reacting 1,4-disubstituted hydrazine carbothioamides with diethyl azodicarboxylate. The structures were confirmed by spectroscopic data as well as single-crystal X-ray analyses. The antiproliferative activity of the synthesized compounds was investigated against four human cancer cell lines using an MTT assay. Compounds 5d, 5e, and 5f revealed the most potent antiproliferative activity with GI50 values ranging from 0.70 µM to 1.20 µM, compared to doxorubicin GI50 value = 1.10 µM. Compounds 5d, 5e, and 5f were further investigated for their inhibitory activities against CDK2 and EGFR as potential targets for their molecular mechanism. Compounds 5e and 5f have showed potent inhibitory activity to CDK2 enzyme with IC50 values of 18 and 14 nM, which is more potent than the reference dinaciclib (IC50 = 20 nM). Moreover, compounds 5e and 5f were the most potent EGFR inhibitors, with IC50 values of 93 and 87 nM, respectively, compared to the reference erlotinib (IC50 = 70 nM). In addition, the most potent derivatives were tested for their apoptotic activity against caspases 3, 8, and 9, and the results showed that compounds 5d, 5e, and 5f revealed a greater increase in active caspases 3,8 and 9 than doxorubicin. Also, compounds 5d, 5e, and 5f elevated cytochrome C levels in the MCF-7 human breast cancer cell line by about 15.5, 15.8, and 16.5 times, respectively. Finally, a molecular docking study was performed to investigate the binding sites of these compounds within the active sites of CDK2 and EGFR targets, and the results confirmed that the most potent CDK2 and EGFR inhibitor 5h also have showed the highest docking score.
Keywords
Huisgen cycloaddition
1,3-Thiazolidin-4-ones
CDK2
EGFR
Diethyl azodicarboxylate

- MTT assay
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- CDK2
-
Cyclin-dependent kinase 2
- EGFR
-
Epidermal growth factor receptor
- HER2
-
Human epidermal growth factor receptor 2
Abbreviations
1 Introduction
The syntheses and the biological activities of thiazolidinone derivatives have been the subject of substantial research (Zhou et al., 2020, Santos et al., 2018). The thiazolidinone derivatives are privileged heterocyclic compounds owing to their contribution as biologically active chromophores along with pharmaceutical application in disease treatment such as anticancer (Zhou et al., 2020; Sigalapalli et al., 2021, Rani et al., 2020), anti-inflammatory (Shawky et al., 2019), antimicrobial activities (Arshad and Ahmad, 2020; Beniwal and Jain, 2019), antioxidant activities (Zhang et al., 2018) and antileishmanial (Bhat et al., 2020). Also, thiazolidinone derivatives have been utilized as a hybrid drug in medicines, and their activities were compared with marketed drugs.
Reasonably, the chemistry of azodicarboxylate compounds was determined by their behavior as a mediator and building blocks in organic synthesis. They reacted as a mediator in the conversion of alcohols to the corresponding carbonyl compounds as reported (Hayashi et al., 2012), dehydrogenation reactions (oxidation reactions) (Jung et al., 2016), and the oxidative cleavage of S—S and Se—Se bonds were mediated by azodicarboxylates (An et al., 2018). Also, Bräse et al. have reported the N-amination of α,α-disubstituted aldehydes using L-proline as an asymmetric catalyst with azodicarboxylate to afford α-amino aldehydes (Baumann et al., 2007). Amination of arenes has been achieved using azodicarboxylate in the presence of potassium bisulfate as a catalyst (Tang et al., 2019). Furthermore, azodicarboxylates underwent C—H amination at the para-position of 1-naphthylamides under silver-catalysis (Li et al., 2019). They are used as a key for the synthesis of different organic heterocyclic compounds (Benavent et al., 2018; Zhang et al., 2018; Cheng et al., 2017; Yang et al., 2016; Selvakumar et al., 2015; Leng et al., 2015; Varlet et al., 2019; Jiang et al., 2013; Shao et al., 2013). Many thiazolidinone-based compounds with antiproliferative and apoptotic actions have been reported (Lv et al., 2010; Yousef et al., 2020; Jia et al., 2016). Compound I (Fig. 1) exhibited potent inhibitory anticancer activity (IC50 = 0.09 µM for EGFR and IC50 = 0.42 µM for HER-2), comparable to the positive control erlotinib (IC50 = 0.03 µM). According to the EGFR molecular docking model, the nitrogen atom of the thiazolidinone ring establishes a hydrogen bond with the side chain mercapto group of Cys 751, improving binding to the active sites of the tested enzyme (Lv et al., 2010). A new series of isatin-thiazolidine derivatives were synthesized and evaluated for their antiproliferative activity. The newly synthesized compounds have varying inhibitory effects on three cancer cell lines with IC50 values ranging from 3.29 µM to 100 µM. Compound II (Fig. 1) showed potent CDK inhibitory activity with IC50 of 0.38 µM and good apoptotic activities against caspases 3 and 9 (Yousef et al., 2020).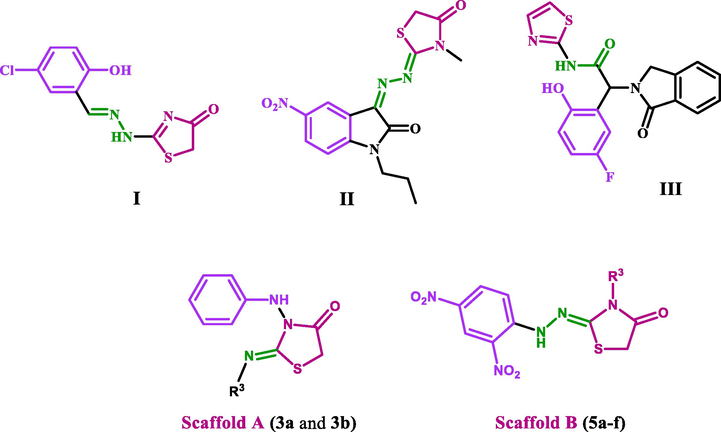
Structure of compounds I-III and new compounds 3a, 3b, and 5a-f.
Another study on compound III (Fig. 1), a thiazolidine derivative, suggests that compound III or a related compound in combination with cetuximab (EGFR dimer-disrupting antibody) would be an effective strategy for treating lung cancers driven by the L858R/T790M mutation, as well as those driven by the triple L858R/T790M/C797S mutation, which is resistant to all current EGFR-targeted therapies (Jia et al., 2016).
We continued our studies on N‐Substituted hydrazine carbothioamides, which are considered one of the most important classes of compounds containing nitrogen, sulfur, and oxygen used for heterocyclization and formation of different heterocyclic rings such as thiadiazole and thiadiazepine, and from the reaction with several π‐deficient compounds (Hassan et al., 2003, 2006, 2007, 2011, 2020; Aly et al., 2021).
Thus, we report the synthesis of thiazolidine-4-one derivatives through the reaction of 1,4-disubstituted hydrazine carbothioamides and diethyl azodicarboxylate under different conditions. In addition, we discuss the antiproliferative activity of the two new scaffolds of thiazolidine-4-one derivatives, Scaffold A (compounds 3a and 3b) and Scaffold B (5a-f). The new derivatives were investigated against a panel of cancer cell lines using an MTT assay. The most potent derivatives from the MTT assay were further investigated for their inhibitory activities against CDK2 and EGFR as potential targets for their molecular mechanism. Also, the most potent derivatives were tested for their apoptotic activity against caspases 3, 8, and 9. Finally, a molecular docking study was performed to investigate the binding sites of these compounds within the active sites of EGFR and CDK2 targets.
2 Experimental
2.1 Chemistry
General Details: See Appendix A.
Diethyl azodicarboxylate was purchased from Sigma-Aldrich chemicals. The hydrazine carbothioamides 1a-h have been prepared according to references (Hassan et al., 2019a,b) by refluxing the appropriate hydrazine with the corresponding isothiocyanates in ethanol for interval time.
General procedure for the synthesis of compounds 3a, 3b, 4, and 5a-f
Dissolving 0.174 gm (1.0 mmol) of diethyl azodicarboxylate 2 with triphenylphosphine (Ph3P) 0.262 gm (1.0 mmol) in 15 ml ethanol and allowed to stir under reflux for 30 min, then the appropriate hydrazine carbothioamide 1 (1.0 mmol) in 5 ml ethanol with two drops of triethylamine (Et3N) was added. The mixture was then refluxed for 6 hrs. The reaction was monitored by TLC after completion of the reaction; the solvent was dried through vacuum evaporation. The reaction mixture was extracted three times with methylene chloride (CH2Cl2), the extract was added to anhydrous calcium chloride (CaCl2) filtered, then subjected to plc chromatography using toluene: ethyl acetate (5:1) as eluent. The separated zones were collected and eluted with acetone to give the thiazolidinones 5a-f as major products (orange-red zones), and the products were recrystallized from methanol. While in the case of N-substituted-2-phenylhydrazine carbothioamide 1a,b compounds 3a, and b were observed as dark zones and was recrystallized from acetonitrile to obtained as a colorless crystal. The side product also was observed as a dark zone and was recrystallized from acetonitrile to obtain the calcium chloride complex (CaCl2(PPh3O)4-H2O). The oxidized form (E)-N-cyclohexyl-2-phenyldiazene-1-carbothioamide (4) was obtained as an orange zone.
General procedure for the synthesis of compounds 5a-f and 7
To a solution of hydrazine carbothioamides 1b-h (1 mmol) in 15 ml dry ethyl acetate, chloroacetyl chloride 6 (0.124 gm, 1.1 mmol) (slightly excess) was added with the addition of two drops of triethylamine. The mixture was stirred at room temperature for an hour and left to stand overnight. The red–orange precipitate was filtered, washed with ethyl acetate several times, and recrystallized from methanol to obtain the target compounds 5a-f with high purity and high yields. On the other hand, a colorless precipitate of (Z)-2-chloro-N-(2-(cyclohexylimino)-4-oxothiazolidin-3-yl)-N-phenyacetamide 7 was obtained when 1b was reacted in the same manner with chloroacetyl chloride, and the product was recrystallized from acetonitrile.
2.1.1 (Z)-2-(Benzylimino)-3-(phenylamino)thiazolidin-4-one (3a)
Colorless crystals (acetonitrile); yield (178 mg, 60%), mp. 182–183 °C; IR (KBr): ν 3223 (NH), 3036 (Ar—CH), 2937 (ali—CH), 1731 (C⚌O), 1639 (C⚌N), 1568 (Ar—C⚌C) cm−1; 1H NMR (400 MHz, DMSO‑d6): δ 4.43 (s, 2H, thiazolidinone-CH2), 4.96 (s, 2H, benzyl-CH2), 7.06–7.47 (m, 10H, Ar—H), 9.29 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, DMSO‑d6): δ 46.62 (C-5), 48.10 (benzyl-CH2), 127.21, 127.48, 128.04, 128.45 (Ar—CH), 136.32, 138.02 (Ar—C), 157.48 (C2), 168.95 (C4) ppm; MS (70 eV): m/z = 297 (M+, 3); 269 (3); 257 (7); 206 (3); 164 (6); 149 (8); 108 (7); 91 (100). Anal. Calcd. for C16H15N3OS: C, 64.62; H, 5.08; N, 14.13; S, 10.78; Found: C, 64.57; H, 4.95; N, 14.02; S, 10.69.
2.1.2 (Z)-2-(Cyclohexylimino)-3-(phenylamino)thiazolidin-4-one (3b)
Colorless crystals (acetonitrile); yield (187 mg, 65%), mp. 185–186 °C; IR (KBr): ν 3233 (NH), 3086 (Ar—CH), 2947 (ali—CH), 1736 (C⚌O), 1649 (C⚌N), 1566 (Ar—C⚌C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.25–1.67 (m, 10H, Cyclohexyl-CH2); 4.09–4.21 (m, 1H, Cyclohexyl-H); 4.33 (s, 2H, thiazolidinone-CH2), 7.23–7.49 (m, 5H, Ar—H), 9.23 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, CDCl3): δ 24.64, 25.41, 33.14 (Cyclohexyl-CH2); 43.85 (C-5), 54.40 (Cyclohexyl-CH), 124.27, 129.30, 130.02 (Ar—CH), 136.48 (Ar—C), 152.42 (C2), 169.10 (C4) ppm; MS (70 eV): m/z = 289 (M+, 100); 249 (33); 198 (25); 156 (81); 91 (67). Anal. Calcd. for C15H19N3OS: C, 62.25; H, 6.62; N, 14.52; S, 11.08; Found: C, 62.14; H, 6.53; N, 14.38; S, 11.00.
2.1.3 (E)-N-cyclohexyl-2-phenyldiazene-1-carbothioamide (4)
Orange crystals (acetonitrile); yield (40%), mp. 126–127 °C; IR (KBr): ν 3347 (NH), 3110 (Ar—CH), 2950 (ali—CH), 1582 (Ar—C⚌C), 1440 (N⚌N), 1139 (C⚌S) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.22–2.46 (m, 10H, Cyclohexyl-CH2), 4.22–4.46 (m, 1H, Cyclohexyl-CH), 7.46–7.52 (m, 3H, Ar—H) 7.78–8.18 (3, 3H, Ar—H and NH) ppm; 13C NMR (100 MHz, CDCl3): δ 24.46, 25.41, 33.14 (cyclohexyl-CH2), 45.40 (cyclohexyl-CH), 124.27, 129.34, 133.38 (Ar—CH), 150.69 (Ar—C), 190.37 (C⚌S) ppm; MS (70 eV): m/z (%) = 247 (M+, 43), 141 (32), 105 (100), 77 (81). Anal. Calcd. for C13H17N3S: C, 63.12; H, 6.93; N, 16.99; S, 12.96; Found: C, 63.05; H, 6.88; N, 16.92; S, 12.89.
2.1.4 (Z)-2-(2-(2,4-Dinitrophenyl)hydrazono)-3-ethylthiazolidin-4-one (5a)
Red crystals (methanol); yield (80% and 95%)), mp. 198–199 °C; IR (KBr): ν 3267 (NH), 3095 (Ar—CH), 2937 (ali—CH), 1722 (C⚌O), 1638 (C⚌N), 1585 (Ar—C⚌C), 1420 (NO2) cm−1, 1H NMR (400 MHz, DMSO‑d6): δ 1.22–1.26 (t, 3H, J = 7.77 Hz, CH3), 3.76–3.80 (q, 2H, J = 7.77 Hz, CH2), 4.25 (s, 2H, thiazolidine-CH2), 7.59–7.62 (d, 1H, Ar—H), 8.38–8.41 (d, 1H, Ar—H), 8.91 (s, 1H, Ar—H), 10.53 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, DMSO‑d6): δ 12.25 (CH3), 33.46 (CH2), 37.73 (C5), 115.14, 122.81, 130.13 (Ar—CH), 129.02, 136.35, 144.93 (Ar—C),156.97 (C2), 170.71 (C4) ppm;; MS (70 eV): m/z (%) = 325 (M+, 100), 285 (28), 168 (57), 158 (71), 137 (55), 123 (46), 87 (52). Anal. Calcd. for C11H11N5O5S: C, 40.61; H, 3.41; N, 21.53; S, 9.86; Found: C, 40.46; H, 3.37; N, 21.45; S, 9.78.
2.1.5 (Z)-3-Allyl-2-(2-(2,4-dinitrophenyl)hydrazono)thiazolidin-4-one (5b)
Red crystals (methanol); yield (68% and 93%), mp. 225–227 °C; IR (KBr): ν 3347 (NH), 3120 (Ar—CH), 2952 (ali—CH), 1724 (C⚌O), 1618 (C⚌N), 1558 (Ar—C⚌C), 1407 (NO2) cm−1; 1H NMR (400 MHz, DMSO‑d6): δ 3.90–3.93 (m, 2H, allyl—CH2), 4.28 (s, 2H, thiazolidine-CH2), 5.12–5.53 (m, 2H, allyl—CH2⚌), 5.82–5.99 (m, 1H, allyl—CH⚌), 7.56–7.59 (d, 1H, Ar—H), 8.16–8.19 (d, 1H, Ar—H), 8.80 (s, 1H, Ar—H), 11.36 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, DMSO‑d6): δ 37.34 (C-5), 45.12 (allyl—CH2), 116.10 (allyl—CH2⚌), 115.77, 123.47, 126.88, 129.66 (Ar—CH), 134.99 (allyl⚌CH), 142.50 (Ar—C),150.10 (C2), 169.18 (C4) ppm; MS (70 eV): m/z (%) = 337 (M+, 100), 297 (36), 171 (57), 168 (41), 137 (65), 123 (71), 99 (82). Anal. Calcd. for C12H11N5O5S: C, 42.73; H, 3.29; N, 20.76; S, 9.51; Found: C, 42.63; H, 3.18; N, 20.65; S, 9.46.
2.1.6 (Z)-3-Benzyl-2-(2-(2,4-dinitrophenyl)hydrazono)thiazolidin-4-one (5c)
Red crystals (methanol); yield (72% and 91%), mp. 213–215 °C; IR (KBr): ν 3246 (NH), 3103 (Ar—CH), 2995 and 2942 (ali—CH), 1718 (C⚌O), 1628 (C⚌N), 1587 (Ar—C⚌C), 1415 (NO2) cm−1, 1H NMR (400 MHz, DMSO‑d6): δ 4.31 (s, 2H, thiazolidine-CH2), 4.96 (s, 2H, benzyl-CH2), 7.36–7.42 (m, 6H, Ar—H), 8.26–8.34 (d, 1H, Ar—H), 8.84 (s, 1H, Ar—H), 10.52 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, DMSO‑d6): δ 37.93 (C-5), 45.77 (benzyl-CH2), 115.96, 122.98, 127.62, 127.80, 128.91 129.97 (Ar—CH), 129.77, 129.94, 135.72, 144.79 (Ar—C), 156.45 (C2), 171.07 (C4) ppm; MS (70 eV): m/z (%) = 387 (M+, 100), 347 (25), 221 (58), 168 (40), 149 (75), 137 (33), 123 (32). Anal. Calcd. for C16H13N5O5S: C, 49.61; H, 3.38; N, 18.08; S, 8.28; Found: C, 49.55; H, 3.27; N, 17.98; S, 8.19.
2.1.7 (Z)-2-(2-(2,4-Dinitrophenyl)hydrazono)-3-phenylthiazolidin-4-one (5d)
Red crystals (methanol); yield (67% and 88%), mp. 234–236 °C; 1H NMR (400 MHz, DMSO‑d6): δ 4.34 (s, 2H, thiazolidine-CH2), 7.46–7.68 (m, 5H, Ar—H), 8.23–8.27 (d, 1H, Ar—H), 8.82–8.87 (d, 1H, Ar—H), 9.42 (s, 1H, Ar—H), 11.61 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, DMSO‑d6): δ 37.46 (C-5), 115.69, 123.53, 127.72, 128.42, 128.77, 130.41 (Ar—CH), 129.74, 134.31, 135.65, 145.82 (Ar—C),158.49 (C2), 171.05 (C4) ppm; MS (70 eV): m/z (%) = 373 (M+, 100), 333 (28), 207 (37), 168 (71), 137 (85), 135 (83), 123 (34). Anal. Calcd. for C15H11N5O5S: C, 48.26; H, 2.97; N, 18.76; S, 8.59; Found: C, 48.19; H, 2.88; N, 18.67; S, 8.46.
2.1.8 (Z)-3-Cyclohexyl-2-(2-(2,4-dinitrophenyl)hydrazono)thiazolidin-4-one (5e)
Red crystals (methanol); yield (80%, 97%), mp. 223–225 °C; 1H NMR (400 MHz, CDCl3): δ 1.40–1.76 (m, 10H, Cyclohexyl-CH2), 3.09–3.20 (m, 1H, Cyclohexyl-CH), 3.92 (s, 2H, thiazolidine-CH2), 7.52–7.56 (d, 1H, Ar—H), 8.25–8.29 (d, 1H, Ar—H), 9.05 (s, 1H, Ar—H), 10.51 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, CDCl3): δ 24.65, 25.53, 27.80 (cyclohexyl-CH2), 36.73 (C5), 45.83 (cyclohexyl-CH), 115.33, 123.31, 129.43 (Ar—CH), 130.12, 137.35, 144.95 (Ar—C), 156.97 (C2), 169.80 (C4) ppm. Anal. Calcd. for C15H17N5O5S: C, 47.49; H, 4.52; N, 18.46; S, 8.45; Found: C, 47.40; H, 4.47; N, 18.35; S, 8.38.
2.1.9 (Z)-2-(2-(2,4-Dinitrophenyl)hydrazono)-3-(p-tolyl)thiazolidin-4-one (5f)
Red crystals (methanol); yield (67% and 95%), mp. 232–234 °C; 1H NMR (400 MHz, CDCl3): δ = 2.5 (s, 3H, CH3); 4.24 (s, 2H, thiazolidine-CH2), 7.21–7.31 (m, 4H, Ar—H), 7.40–7.45 (d, 1H, Ar—H), 8.21–8.25 (d, 1H, Ar—H), 9.10 (s, 1H, Ar—H), 10.61 (s, 1H, hydrazono-NH) ppm; 13C NMR (100 MHz, CDCl3): δ 21.36 (CH3); 33.30 (C-5), 115.94, 123.41, 127.26, 129.26, 130.12 (Ar—CH), 130.31, 131.25, 137.84, 139.73 144.92 (Ar—C),152.81 (C2), 169.54 (C4) ppm. Anal. Calcd. for C16H13N5O5S: C, 49.61; H, 3.38; N, 18.08; S, 8.28; Found: C, 49.57; H, 3.29; N, 17.98; S, 8.17.
2.1.10 (CaCl2(Ph3PO)4·H2O)
Colorless crystals (acetonitrile), yield (10–12%), mp. 172–173 °C; IR (KBr): ν 362 (Ar—CH), 1588 (Ar—C⚌C) cm−1; 1H NMR (400 MHz, DMSO‑d6): δ 7.54–7.65 (m, 60H, Ar—H) ppm; 13C NMR (100 MHz, DMSO‑d6): δ 128.66, 128.78, 131.40, (Ar—CH); 131.50 (Ar—C) ppm. MS (70 eV): m/z = 611 (5), 278 (78), 201 (14). Anal. Calcd. for C72H62CaCl2O5P4: C, 69.62; H, 5.03; Cl, 5.71. Found: C, 69.77; H, 4.98; Cl, 5.65.
2.1.11 (Z)-2-Chloro-N-(2-(cyclohexylimino)-4-oxothiazolidin-3-yl)-N-phenyacet-amide (7)
Colorless crystals (acetonitrile); yield (336 mg, 93%), mp.152–153 °C. Anal. Calcd. for C17H20ClN3O2S: C, 55.81; H, 5.51; Cl, 9.69; N, 11.48; S, 8.76; Found: C, 55.66; H, 5.40; Cl, 9.81; N, 11.33; S, 8.69.
2.2 Biology
Details of all biological assay tests: See Appendix A.
Molecular Docking Simulations: See Appendix A.
Supplementary Information:
CCDC 2177163 (5a, SB1441_HY_HA396), 2177164 (5b, SB1486_HY_HA395), 2177165 (5c, SB1502_HY_HA394), 2177166 (5f, SB1502_HY_HA392), 1939590 (Experimental Crystal Structure Determination, 2019, DOI:0.5517/ccdc.csd.cc2339fz; complex (CaCl2(PPh3O)4-H2O) [complex_ha117]), 2177167 (4, SB1471_HY_HA345) and 2177168 (7, SB1442_HY_HA398) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
3 Results and discussion
3.1 Chemistry
Scheme 1 depicts the reaction of hydrazine carbothioamides 1a-h with diethyl azodicarboxylate (2). First, to optimize the reaction conditions, the reaction of hydrazine carbothioamide 1a,b with diethyl azodicarboxylate (2) were carried in a different solvents such (EtOH, CH3CN, CH2Cl2, and AcOEt) with a free catalyst to investigate our idea. But unfortunately, no products were identified. So, we repeated the reactions of hydrazine carbothioamides 1a,b with compound 2 again in the presence of triphenylphosphine (Ph3P) and triethylamine (Et3N) in absolute ethanol (abs. EtOH) under refluxing conditions for 6 h. Fortuntely, the reactions proceeded to form (Z) 2-(substituted imino)-3-(phenylamino)thiazolidin-4-ones 3a,b and the oxidized structure of the hydrazine carbothioamide 4 (in case 1b, R1 = R2 = H, R3 = cyclohexyl) in addition to a colorless crystals of calcium chloride complex with triphenylphosphine oxide (CaCl2(PPh3O)4·H2O). Based on the previous results, we perform the rest of the reactions betwee hydrazine carbothioamides 1c-h with diethyl azodicarboxylate (2) under the same conditions to complete the chain. But, the unexpected products hydrazonothiazolidin-4-ones 5a-f were obtained (in case 1c-h) in addition to a colorless crystals (CaCl2(PPh3O)4·H2O) (Scheme 1). We attributed the formation of these isomers 5a-f according to our reported literature (Hassan et al., 2019a,b).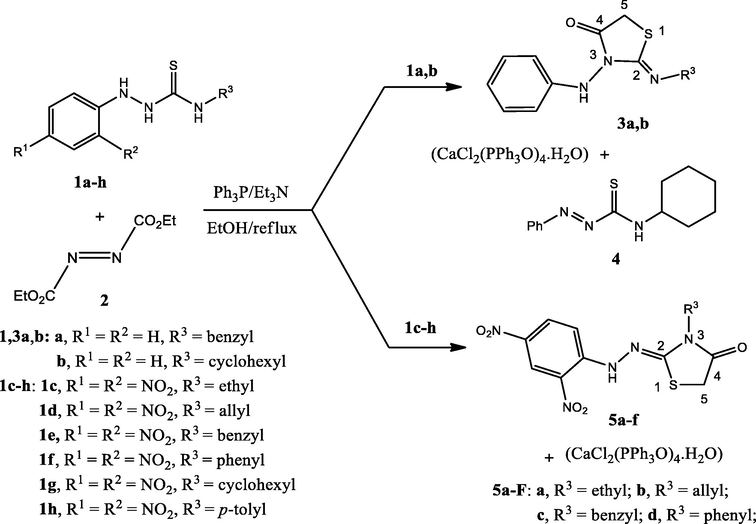
Reactions between hydrazine carbothioamides 1a-h and diethyl azodicarboxylate (2) to form the thiazolidinone derivatives 3a,b, and 5a-f.
The structure of compounds 3a,b, and 4 were also confirmed with spectral data and X-ray crystallography, as shown in Fig. 2. In compound 3a, as example which was assigned as 2-(Benzylimino)-3-(phenylamino)thiazolidin-4-one. The IR spectra of compound 3a showed strong absorptions at 3223, 1731 1639 cm−1 for NH, carbonyl group (C⚌O) and (C⚌N), respectively. The presence of carbonyl group and (C⚌N) was further confirmed by 13C NMR specta which give signals at 168.30 and 156.42 ppm, respectively. The 1H NMR spectra of 3a revealed three broad singlet signals with the ratio (1:2:2) at δH = 4.43, 4.96 and 9.29 ppm, due to thiazolidinon-CH2, benzyl-CH2 and NH, respectively. The benzyl-CH2 give singlet signals with downfield at δH = 4.96 ppm, due to the benzyl group and the imnino-structure (Aly et al., 2007; Beya Haouas et al., 2018). Also, the presence of two CH2 groups will further confirmed from the 13C NMR spectra with downfield signals at δC = 46.62 and 48.10 ppm, for thiazolidinone-CH2 and benzyl-CH2, respectively.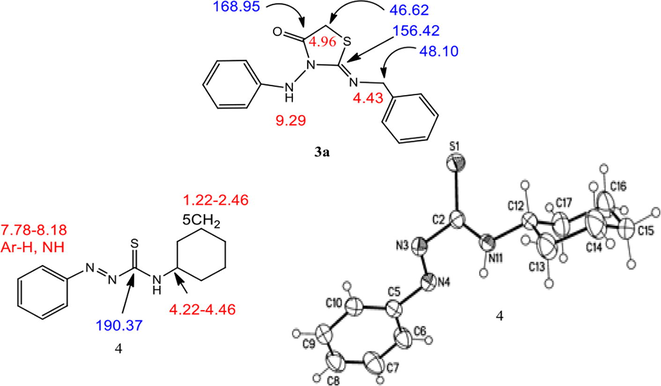
Spectral data of 3a and molecular structure of compound 4 identified according to IUPAC nomenclature as N-cyclohexyl-2-phenyldiazene carbothioamide.
The structures of unexpected products hydrazonothiazolidin-4-ones 5a-f were elucidated by spectroscopic analyses IR, NMR (1H and 13C), mass spectrometry, and elemental analyses in addition to the X-ray crystallographic analyses. IR spectra of (Z)-2-(2-(2,4-dinitrophenyl)hydrazono)-3-ethylthiazolin-4-one (5a, R3 = ethyl) showed a broad band for NH-stretching at 3267 cm−1, other absorptions at ν = 1722, 1638, 1585, and 1420 cm−1 which characteristic for CO, exo-C⚌N, Ar—C⚌C and NO2, respectively. The carbonyl group and exo-C⚌N were further confirmed from 13C NMR spectra, which gave signals at δC = 170.71 and 156.97 ppm, respectively. The 1H NMR spectra of 5a revealed two singlets at δH = 4.25 and 10.53 ppm, corresponding to thiazolidinone-CH2 and NH protons, respectively. Additionally, the triplet-quartet signals for ethyl group at δH = 1.22–1.26 (t, 3H, J = 7.77 Hz) and 3.76–3.80 ppm (q, 2H, J = 7.77 Hz), which were further confirmed from 13C NMR spectra which gave signals at δC = 33.46, 12.25 ppm, for (CH2) and 12.25 (CH3), respectively. On the other hand, single crystal X-ray crystallographic analysis of compound 5a unambiguously supported the structure of the thiazolidinone derivatives. From the tables (S1-7) of the supplementary data of compound 5a and their measurements, it was clear that it possesses molecular formula = C11H11N5O5S. The bond lengths of S1—C2 (1.762 Å), S1—C5 (1.812 Å), C2—N3 (1.383 Å), N3—C4 (1.378 Å), and C4—C5 (1.510 Å) were closed to the single bond lengths. The sum of the bond angles around the atoms in the thiazolidinone ring N6—C2—N3 (121.15°); N6—C2—S1 (126.89°), and N3—C2—S1 (111.92°) equal to 359.96°; C4—N3—C2 (116.13°), C4—N3—C20 (121.16°), C2—N3—C20 (122.70°) equal to (359.99°) and O4—C4—N3 (123.57°), O4—C4—C5 (123.75°), N3—C4—C5 (112.68°) equal to (360.00°), these confirm the planarity of the thiazolidinone ring. Whereas the C2—N6 (1.282 Å) was closed to the C⚌N bond length, the geometry around the C2—N6 double bond was confirmed with X-ray structure to be in the cissoid-geometry concerning the sulfur of the thiazolidinone ring. The structure of compound 5a was further confirmed with X-ray crystallography, as shown in Fig. 3.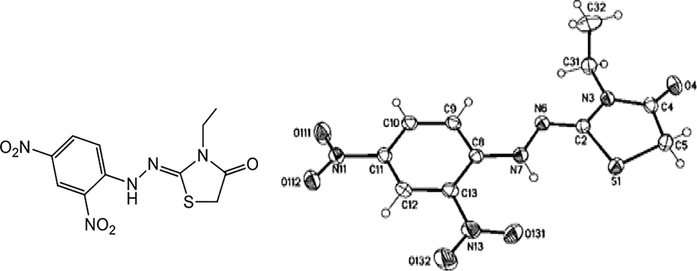
Molecular structure of compound 5a identified according to IUPAC nomenclature as (Z)-2-(2-(2,4-dinitrophenyl)hydrazono)-3-ethylthiazolin-4-one.
To enhance our investigation, the reaction of substituted hydrazine carbothioamides 1b-h with chloroacetyl chloride (6) has been carried out in ethyl acetate as a solvent catalyzed with triethyl amine, resulting in the formation of hydrazonothiazolidinone 5a-f in case of 2-(2,4-dinitrophenyl)-N-substituted hydrazinecarbothioamide 1c-h. While (Z)-2-chloro-N-(2-(cyclohexylimino)-4-oxo-thiazolidin-3-yl)-N-phenyacet-amide 7 was obtained via interaction between hydrazine carbothioamide 1b and 6 (Scheme 2).
Reactions of hydrazine carbothioamides 1b-h with chloroacetyl chloride (6) and formation of 4-thiazolidinone derivatives 5a-f and 7.
The structure of compound 7, which is assigned as (Z)-2-chloro-N-(2-(cyclohexylimino)-4-oxo-thiazolidin-3-yl)-N-phenyacetamide by single crystal X-ray crystallographic analysis has the transoid-geometry, and the cyclohexyl moiety has the most stable chair form conformation (Fig. 4).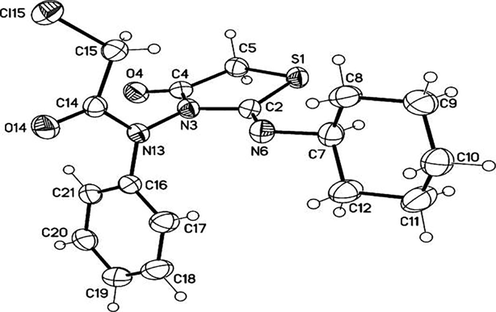
Molecular structure of compound 7 identified according to IUPAC nomenclature as (Z)-2-chloro-N-(2-(cyclohexylimino)-4-oxo-thiazolidin-3-yl)-N-phenylacetamide.
Accordingly, four isomeric structures may be formed via interactions between hydrazine carbothioamides 1b-h and diethyl azodicarboxylate (2) or chloroacetyl chloride (6) as (E/Z)-hydrazonothiazolidinones (A and B) and (E/Z)-iminothiazolidinones (C and D) (Fig. 5).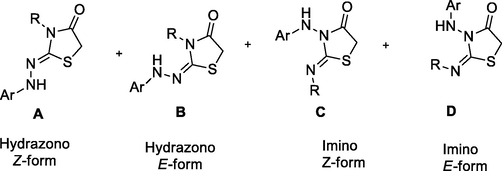
The expected alternative structures formed from interactions between hydrazinecarbothioamides 1b-h and diethyl azodicarboxylate (2) or chloroacetyl chloride (6).
A plausible mechanism for forming the thiazolidinone derivatives 5a-f through the transformation of triethylamine upon the reaction between thiosemicarbazides with diethyl azodicarboxylate in triphenylphosphine/triethylamine catalyst in absolute ethanol as depicted in Scheme 3.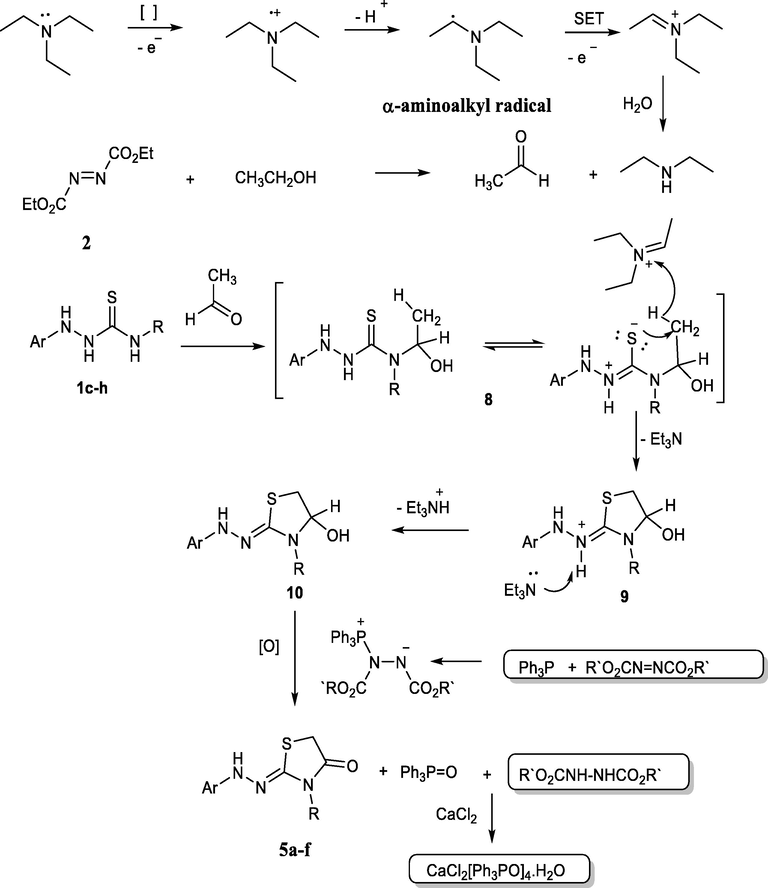
The proposed mechanism for the formation of thiazolidinone 5a-f.
Here, the obtained thiazolidinones were formed by access of both triphenylphosphine and triethylamine, in which the triphenylphosphine reacted with azodicarboxylate to form the zwitterion (Huisgen-Zwitterion) (Brunn and Huisgen, 1969; Huisgen, 1996; Nair et al., 2007). This zwitterion plays a role in the oxidation and formation of the thiazolidinones 3a,b, and 5a-f. The triethylamine played an important role in the formation of acetaldehyde according to the studies carried out by Ye et al. in which the triethylamine was oxidized to acetaldehyde via the single-electron-transfer (SET) process (Ye et al., 1999), as outlined in Scheme 3, and for the progress of the reaction to form the thiazolidinone derivatives. Otherwise, the ethanol may be oxidized in the presence of azodicarboxylate and triphenylphosphine (Mitsunobu reagent) (Hayashi et al., 2012; Yoneda et al., 1966). We attempted to carry out the reaction without these reagents, but none were successful. Furthermore, when ethanol is exchanged with other solvents, no products are generated that assist the oxidation of ethanol to the carbonyl molecule.
Reasonably, the proposed mechanism attributed the reactions of hydrazine carbothioamides with chloroacetyl chloride were heterocyclization via nucleophilic substitution reactions, as depicted in Scheme 4. The suggested mechanism starts with the nucleophilic addition of the thiol lone pair to the electrophilic CH2 in chloroacetyl chloride (6) to give the salt 11 (Scheme 4). Addition of Et3N would then ehance the removal of triethylammonium chloride and gave the intermediate 12. Subsequently, the nitrogen lone pair would attack to the polar carbon in the carbonyl group to give the intermediate 13. Repeating of the previous step, which would show the effect Et3N, compounds 5a-f would then be formed (Scheme 4).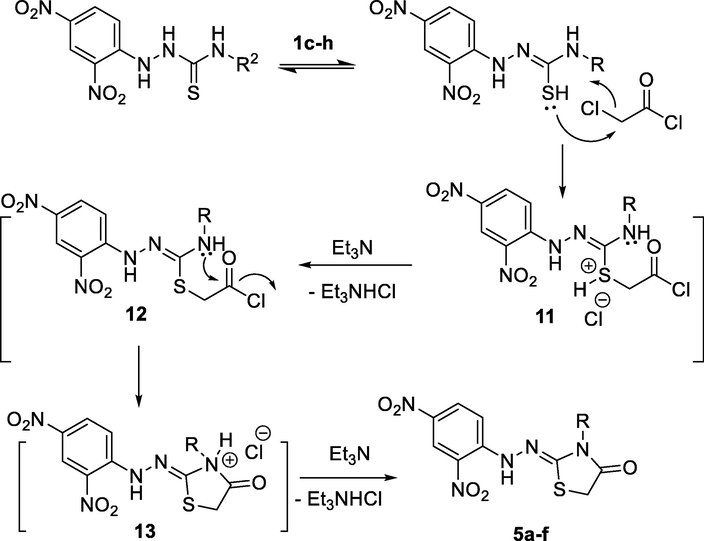
Proposed mechanism for the reaction between 1c-h and 6.
However, the reaction between 1b and 6 behaved differently compared with the other substituents of compounds 1 as shown in Scheme 2. As the NH-1 and the thiol group comptetes each other in the acetylation process (route a or b). Acetylation process via either route a or route b, which would be followed by the nucleophilic attack of N-2 to attempt the cyclization process. Route a describes the formation of the intermediate 15, which on second acetylation process would led to the formation of compound 7 (Scheme 5).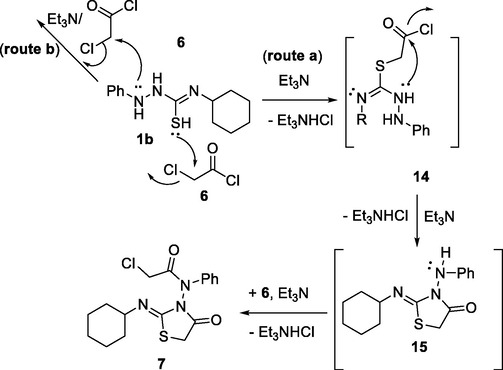
Proposed mechanism for the formation of compound 7.
3.2 Biology
3.2.1 In vitro anticancer activity
3.2.1.1 Cell viability assay
The viability of novel compounds 3a,b, and 5a-f was tested using the human mammary gland epithelial (MCF-10A) cell line (Al-Wahaibi et al., 2020; Abdelbaset et al., 2018). MCF-10A cells were treated with 3a,b, and 5a-f for four days before being evaluated for viability using the MTT assay (Abou-Zied et al., 2019; Hisham et al., 2019). Table 1 demonstrates that none of the compounds tested had cytotoxic effects, and cell viability was greater than 87% for the compounds tested at 50 µM.
Compd. No.
Cell Viability
Antiproliferative activity IC50 ± SEM (μM)
(50µM)
Panc-1
MCF-7
HT-29
A-549
Average
3a
87
2.60 ± 0.20
2.30 ± 0.20
3.10 ± 0.30
3.20 ± 0.30
2.80
3b
89
2.10 ± 0.20
1.80 ± 0.20
2.50 ± 0.20
2.90 ± 0.20
2.30
5a
90
3.40 ± 0.40
3.10 ± 0.30
3.70 ± 0.30
3.70 ± 0.40
3.50
5b
91
1.50 ± 0.10
1.20 ± 0.10
1.70 ± 0.10
1.80 ± 0.20
1.55
5c
87
1.20 ± 0.10
1.10 ± 0.10
1.40 ± 0.10
1.30 ± 0.10
1.25
5d
91
1.05 ± 0.10
0.90 ± 0.40
1.10 ± 0.10
1.20 ± 0.10
1.05
5e
87
0.80 ± 0.10
0.65 ± 0.10
0.90 ± 0.10
0.95 ± 0.10
0.80
5f
89
0.70 ± 0.10
0.60 ± 0.30
0.80 ± 0.10
0.80 ± 0.10
0.70
Doxorubicin
–
1.40 ± 0.10
0.90 ± 0.10
1.00 ± 0.10
1.20 ± 0.10
1.10
3.2.1.2 Antiproliferative activity
Using the MTT assay (Abdelrahman et al., 2017; Youssif et al., 2018) and doxorubicin as the reference drug, compounds 3a,b, and 5a-f were investigated for antiproliferative activity against four human cancer cell lines: Panc-1 (pancreas cancer cell line), MCF-7 (breast cancer cell line), HT-29 (colon cancer cell line), and A-549 (epithelial cancer cell line). The median inhibitory concentration (IC50) is shown in Table 1.
In general, 2,4-dinitrophenyl-hydrazono-thiazolidin-4-ones 5a-f outperformed 3-(phenylamino)thiazolidin-4-ones 3a,b in terms of antiproliferative activity (Table 1). Compared to doxorubicin (GI50 = 1.10 µM), the three most active compounds 5d, 5e, and 5f, all have the backbone 2,4-dinitrophenyl-hydrazono-thiazolidin-4-one in their structure, demonstrated potent antiproliferative activity with GI50 values ranging from 0.70 µM to 1.20 µM.
The 2,4-dinitrophenyl-hydrazono-thiazolidin-4-one derivative 5f (R3 = p-tolyl) had the highest antiproliferative activity of the eight new derivatives, with a GI50 value of 0.70 µM against the four cell lines, comparable to the reference doxorubicin (GI50 = 1.10 µM) and is more potent than doxorubicin against all cancer cell lines tested.
Compound 5e of cyclohexyl moiety (R3 = cyclohexyl) showed potent antiproliferative activity (GI50 = 0.80 µM) against the four cancer cell lines with antiproliferative efficacy higher than that of doxorubicin. Table 1 shows that compounds 5d (GI50 = 1.05 µM) with a phenyl moiety (R3 = phenyl) and 5c (GI50 = 1.25 µM) with a benzyl moiety (R3 = benzyl) had about the same antiproliferative activity as the reference doxorubicin (GI50 = 1.10 µM).
3-(phenylamino)thiazolidin-4-ones 3a (R3 = benzyl) and 3b (R3 = cyclohexyl) demonstrated moderate antiproliferative activity (GI50 = 2.80 µM and 2.30 µM, respectively) against the four cancer cell lines, being 2.5-folds less potent than doxorubicin.
Compound 5e (R3 = cyclohexyl) exhibited potent antiproliferative activity and had a 2,4-dinitrophenyl-hydrazono-thiazolidin-4-one backbone (Scaffold B). In contrast, compound 3b had the same substitution pattern as 5e, with the difference being a 3-(phenylamino)thiazolidin-4-one moiety (Scaffold A) but showed almost three times less activity and that the same pattern holds for 5c versus 3a. Finally, all other derivatives demonstrated moderate to weak inhibitory action against the proliferation of cancer cell lines.
3.2.2 CDK2 inhibitory assay
Previous studies (Youssif et al., 2018) demonstrate the anti-CDK2 activity of thiazolidin-4-one derivatives; the most potent antiproliferative derivatives 5d, 5e, and 5f were further studied for their ability to inhibit the CDK2 enzyme (Mekheimer et al., 2022). Table 2 displays the IC50 values. According to the results, compounds 5d, 5e, and 5f inhibited CDK2 with IC50 values ranging from 14 nM to 23 nM. In cases of compounds 5f (IC50 = 14 nM) and 5e (IC50 = 18 nM), being more potent than the reference dinaciclib (IC50 = 20 nM), which is consistent with the antiproliferative assay results, see Table 2. Compound 5d exhibited strong anti-CDK2 activity, with an IC50 value of 23 nM, and was found to be equivalent to dinaciclib. The results of this experiment indicate that CDK2 could be a possible target for these drugs. ND: Not Determined.
Code No.
CDK2
IC50 ± SEM (nM)EGFR
IC50 ± SEM (nM)
5d
23 ± 2
103 ± 10
5e
18 ± 1
93 ± 8
5f
14 ± 1
87 ± 6
Dinaciclib
20 ± 1
ND
Erlotinib
ND
70 ± 5
3.2.3 EGFR inhibitory assay
Several prior investigations have demonstrated the efficiency of many thiazolidin-4-one derivatives as EGFR inhibitors (Lv et al., 2010; Jia et al., 2016). The EGFR-TK assay (Mohamed et al., 2021) was used to assess the inhibitory potency of compounds 5d, 5e, and 5f against EGFR; the findings are shown in Table 2. The results of this assay supplement the findings of the cancer-cell-based investigation. All compounds tested inhibited EGFR, with IC50 values ranging from 87 nM to 103 nM. In all cases, the tested compounds were at least 1.2-fold less potent than the reference erlotinib (IC50 = 70 nM). Once again, the 2,4-dinitrophenyl-hydrazono-thiazolidin-4-one derivatives 5f and 5e were the most potent derivatives, with IC50 values of 87 and 93 nM, respectively. Based on the findings of this investigation, we may conclude that CDK2 and EGFR are attractive targets for this class of chemical compounds. In the future, a more in-depth mechanistic study may be required.
3.2.4 Apoptosis assay
Previous research has demonstrated that thiazolidin-4-one derivatives can induce apoptosis (Yousef et al., 2020). To determine our new compounds' proapoptotic potential, we tested the most active compounds, 5d, 5e, and 5f, for their ability to activate apoptosis flow in the MCF-7 breast cancer cell line.
3.2.4.1 Activation of proteolytic caspases cascade
The effects of 5d, 5e, and 5f on caspase 3 were investigated and compared to doxorubicin (Slee et al., 2001). The results showed that when compared to control cells, the tested compounds increased the level of active caspase 3 by 7.75–9.3 folds and that 5d, 5e, and 5f had outstanding overexpression of caspase-3 protein levels (507.50 ± 6.0, 515.50 ± 5.0, and 610.50 ± 4.5 pg/mL, respectively) when compared to doxorubicin (503.50 ± 4.0 pg/mL). All the compounds tested showed a greater increase in active caspase 3 than doxorubicin, Table 3.
Comp. Code
Caspase-3
Caspase-8
Caspase-9
Cytochrome C
Conc (pg/ml)
Fold change
Conc (ng/ml)
Fold change
Conc (ng/ml)
Fold change
Conc (ng/ml)
Fold change
5d
507.50 ± 4.5
7.75
1.80
9.00
16.80
17.70
0.77
15.50
5e
515.50 ± 5.0
7.90
1.85
9.25
17.80
18.75
0.79
15.80
5f
610.50 ± 6.0
9.30
1.98
9.90
18.30
19.25
0.85
16.50
Doxorubicin
503.50 ± 4.0
7.70
1.75
8.75
16.20
17.05
0.60
12.00
Control
65.50
1
0.20
1
0.95
1
0.05
1
The effect of compounds 5d, 5e, and 5f on caspases 8 and 9 were further investigated to underline the significance of intrinsic and extrinsic apoptotic pathways in the antiproliferative activity. Compound 5f raises caspase 8 and 9 levels by 10 and 19 folds, respectively, while compounds 5d and 5e raise caspase 8 and 9 levels by 9 and 18 folds, respectively, compared to the control cells, Table 3. Once again, all of the compounds tested showed a greater rise in active caspases 8 and 9 levels than the control doxorubicin.
3.2.4.2 Cytochrome C assay
Cytochrome C levels within the cell are important for activating caspases and initiating the apoptotic process (Mahmoud et al., 2022). The results of testing derivatives 5d, 5e, and 5f as cytochrome C activators in the MCF-7 human breast cancer cell line are shown in Table 3. Compared to untreated control cells, compounds 5d, 5e, and 5f elevated cytochrome C levels in the MCF-7 human breast cancer cell line by about 15.5, 15.8, and 16.5 times, respectively. The findings add to the evidence that apoptosis can be attributable to Cytochrome C overexpression and activation of the intrinsic apoptotic pathway caused by the examined compounds.
3.3 Molecular docking simulations
As discussed before in Sections 3.2.2 and 3.2.3, how effective are the thiazolidin-4-ones as inhibitors for both EGFR and CDK-2, we decided to explore their possible interaction modes within active sites of both of these two targets, also, as shown in Table 4 that compounds 5d, 5e, and 5f are the best candidates to achieve such target. Molecular docking simulations of these compounds within the EGFR active site revealed their good interaction profile, as summarized in Table 4. Compound 5f showed the highest docking score (S; kcal/mol) among the three test compounds.
5d
5e
5f
Ref. 1d
Ref. 2e
EGFR (PDB ID: 1M17)
S (kcal/mol)
−6.17
−6.54
−6.86
−7.30
NA
RMSD (Å)
1.72
0.90
1.78
1.28
Amino acids residues' binding interactions & their bond length (Å)
Met769
(3.45)a
Cys751
(3.57)a
Asp831
(3.92)c
Gln767
(3.15)c
Lys721
(3.10)a
Met742
(3.62)c
Leu694
(3.73)b
Gly772
(3.67)b
Met769
(2.70)a
CDK2 (PDB ID: 4KD1)
S (kcal/mol)
−6.40
−6.21
−6.50
NA
−8.66
RMSD (Å)
1.93
1.93
1.99
1.28
Amino acids residues' binding interactions & their bond length (Å)
Leu83
(3.46)a
Leu83
(3.39)a
Leu83
(3.25)a
Leu83
(2.65)c
Leu83
(3.43)a
Asp86
(3.64)c
Val18
(4.15)b
Lys33
(2.99)a
Leu83
(3.24)a
Lys33
(3.82)a
Lys33
(2.57)a
Visual inspections of binding interactions of best docking pose of each of the three test compounds and co-crystallized ligand (Erlotinib), showed stabilization of their molecules inside cavity of active site with number of H-bonds and pi-H hydrophobic interactions with various amino acid residues lining active site, as shown in Fig. 6. Furtherly, as a possible explanation for better inhibitory activity of 5f over its congeners 5d and 5e, we examined deeply their best docking poses, and we found that even 5d and 5f have more binding interactions over 5e, compound 5e still showing better docking score over 5d because of its closer distance to amino acid residues lining active site indicated by continuity of its proximity contour compared to that of 5d (as shown in Fig. 6a and b), additionally, compound 5f still having better docking score over 5e because of its extra H-donor and pi-H binding interactions, as shown in Fig. 6c, and this why we took compound 5f as a model compound to examine the possible binding interactions of such class of compounds with EGFR protein as illustrated by the generated electrostatic map shown in Fig. 7.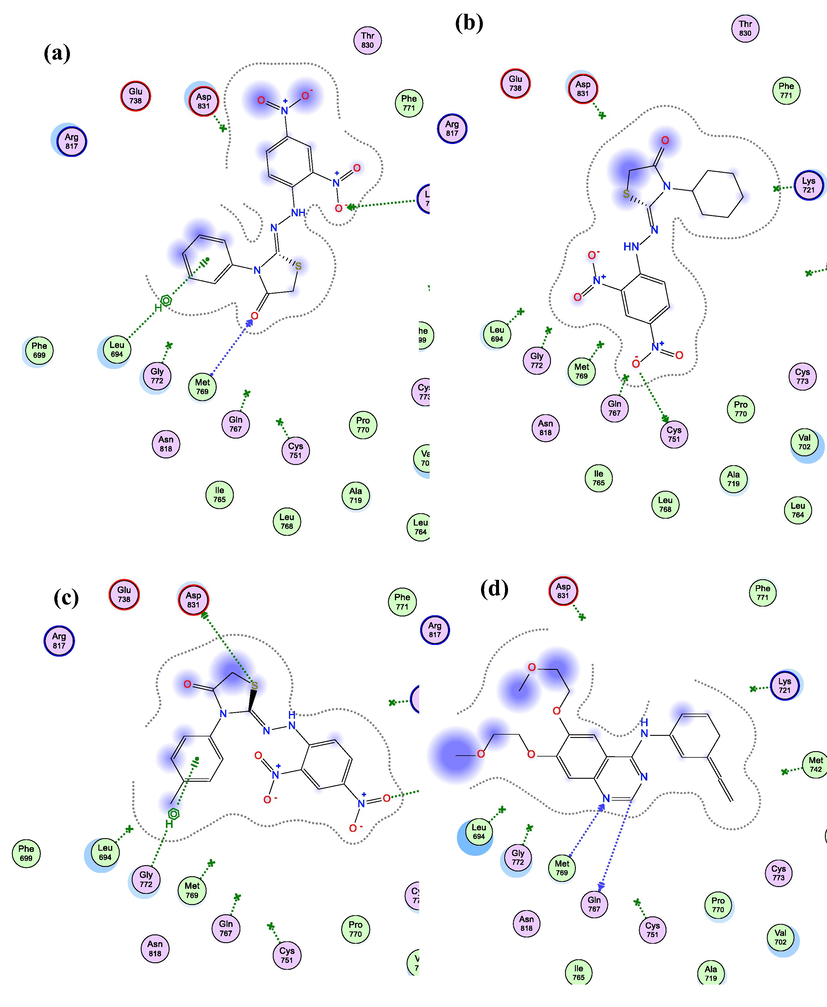
2D Interaction diagram of 5d (a), 5e (b), 5f (c), & Erlotinib (d) within EGFR (PDB ID: 1M17) active site showing H-bonding (green and blue arrows), pi-H (green dotted-line), and proximity contour around each molecule (grey dotted-line).
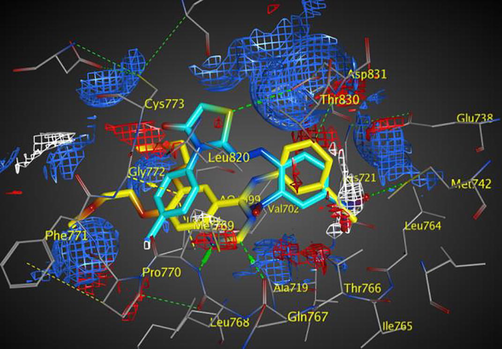
Electrostatic map of EGFR showing a good overlay of compound 5f (cyan) with erlotinib (yellow) and three binding hot spots of the active site: Blue and red contours of H-donor and acceptor favorable region, respectively (H-donors shown in green dotted-lines and H-acceptor shown in yellow dotted-lines), in addition to the white contour of hydrophobic interactions (notice perfect overlay of the nitro group of 5f with this binding hot spot).
The electrostatic isoenergy contours around the EGFR receptor with erlotinib “as a co-crystallized ligand” illustrated the binding hot spots within EGFR active site and revealed the good overlay of compound 5f atoms and its functional groups with the three favorable regions of H-donor, H-acceptor, and hydrophobic interactions, which characterize the EGFR active site, as shown in Fig. 7.
Additionally, molecular docking simulations of compounds 5d-f within CDK2 (PDB ID 4KD1) active site revealed docking poses with docking scores (S; kcal/mol) equipotent to the ones obtained within EGFR active site, indicating possible multi-target action of this class of compounds, as shown in Table 4. In addition, the best docking poses of all three compounds showed binding interactions with key amino acid residues; Lys 33 and Leu 83; as do the co-crystallized ligand (Dinaciclib), as listed in Table 4. Interestingly, visual inspection of docking poses of all three tested compounds showed a common H-bonding between Leu 83 and/or Lys 33 and either of nitro groups of phenyl hydrazine ring as shown in the interaction diagrams of Fig. 8. Interestingly, compound 5e showed better inhibitory activity over 5d against CDK2 protein, which opposite of we was found during molecular docking virtual simulations, compound 5d have a little better docking score compared to 5e, and from this point we tried to explain at least molecular docking results, since both of them showed single H-binding interaction with either Leu 83 or Lys 33, using the VWD interaction map of both of these molecules within CDK2 protein, and the results were largely realistic; compound 5d because of its aromatic ring on N3 showed VWD interactions with Gln 131 which in turns made the molecule closer to have another VWD interaction with Val 18 anchoring the molecule tightly within CDK2 active site which either were missing with compound 5e as shown in Fig. 9.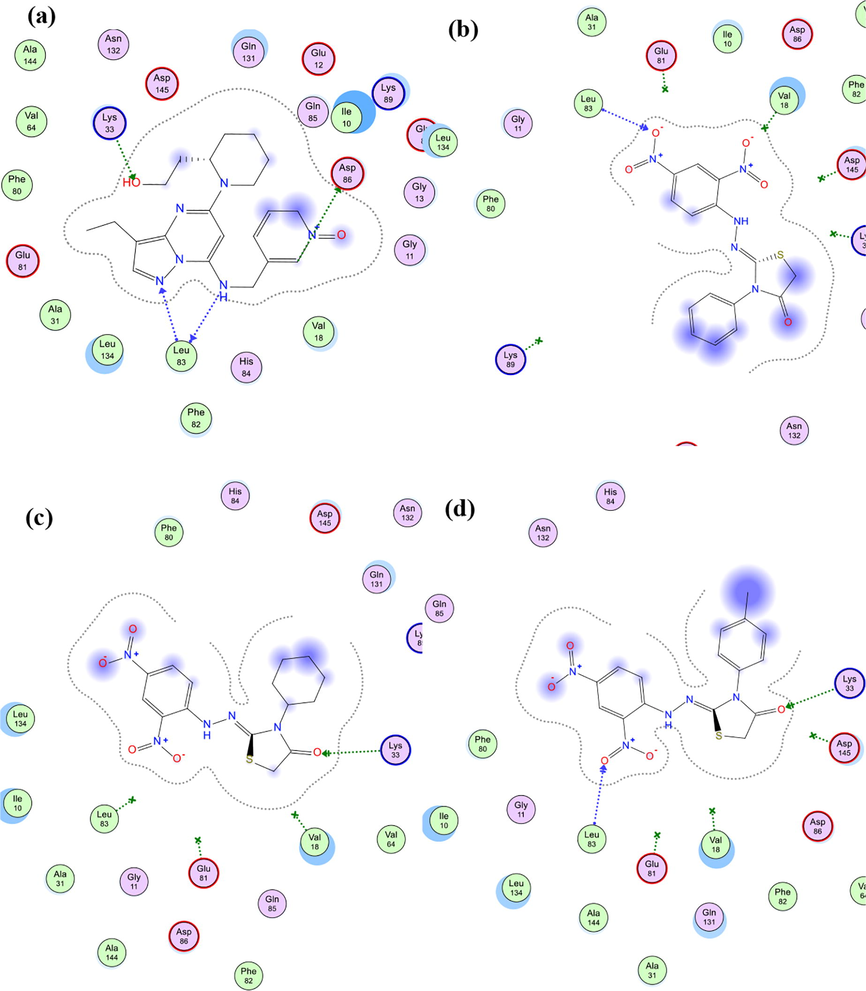
Schematic 2D diagram of binding interactions of 5e within CDK2 (PDB ID: 4KD1) active site showing H-bonding (green and blue arrows) and pi-H interactions (green dotted line).
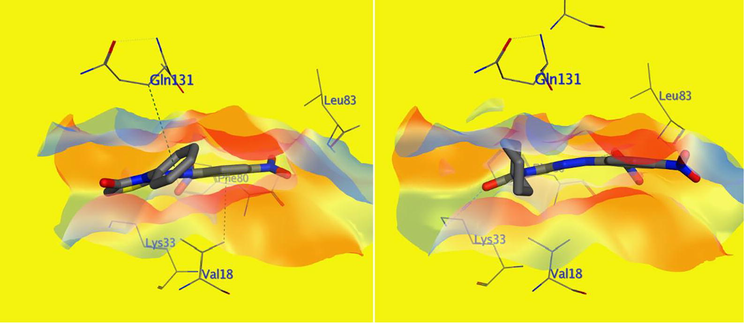
Schematic diagram of VWD interaction surface of both 5d (right) and 5e (left) within CDK2 (PDB ID: 4KD1) active site: showing compound 5d hydrophobic interactions with Gln131 and Val18 (green dotted line), which both are missing with compound 5e (N.B. most of the active site amino acids have been hidden to simplify and clarify the diagram).
3.4 Structure activity relationship (SAR) analysis
The results show that our synthetic Scaffold A and B targeted molecules have the following structure–activity relationship (Fig. 10):
-
The antiproliferative action of the 2,4-dinitrophenyl-hydrazono-thiazolidin-4-one backbone (Scaffold B) is better tolerated than the 3-(phenylamino)thiazolidin-4-one moiety (Scaffold A)
-
The antiproliferative activity of scaffold B compounds appears to be influenced by the type of the R3 group, with activities rising in the following order: p-tolyl > cyclohexyl > phenyl > benzyl, allyl, and ethly.
-
The type of the R3 group appears to impact both CDK2 and EGFR inhibitory activities in scaffold B compounds (5d-f), with activities rising in the order p-tolyl > cyclohexyl > phenyl.
-
For antiproliferative activity, the cyclohexyl group appeared to be more tolerated in Scaffold A compounds than the benzyl group.
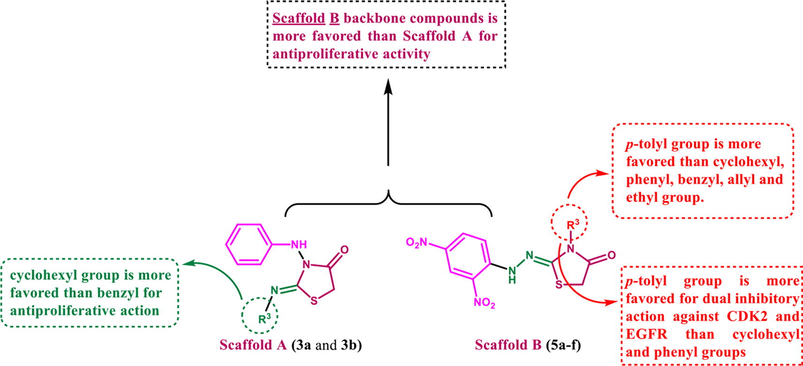
- SAR analysis of compounds 5a-f and 3a,b.
4 Conclusion
A series of novel thiazolidine-4-one derivatives was synthesized through the reaction of 1,4-disubstituted hydrazine carbothioamides with diethyl azodicarboxylate in the presence of triphenylphosphine and triethylamine, the structures were confirmed by spectroscopic data as well as single-crystal X-ray analyses. The role of the electronic effect of the aromatic substitution controls the formation of thiazolidine-4-one derivatives in either Scaffold A with H-substitution or Scaffold B with NO2-substitution; this effect is further confirmed through the reaction of 1,4-disubstituted hydrazine carbothioamides with chloroacetylchloride. The antiproliferative activity of the synthesized compounds was investigated against four human cancer cell lines where compounds 5d, 5e, and 5f revealed the most potent antiproliferative activity. Compounds 5e and 5f showed potent inhibitory activity against EGFR and CDK2 enzymes. Moreover, 5d, 5e, and 5f revealed a greater increase in active caspase 3, 8, and 9 than doxorubicin. Also, compounds 5d, 5e, and 5f elevated cytochrome C levels in the MCF-7 human breast cancer cell line by about 15.5, 15.8, and 16.5 times, respectively. Additionally, compound 5h showed the best docking score (S) within active sites of both EGFR and CDK2 proteins which matches its antiproliferative activity against four cancer lines used and inhibitory activity against EGFR and CDK2 proteins.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Novel pyrrol-2 (3H)-ones and pyridazin-3(2H)-ones carrying quinoline scaffold as antiproliferative tubulin polymerization inhibitors. Bioorg. Chem.. 2018;80:151-163.
- [Google Scholar]
- Design, Synthesis and Pharmacophoric Model Building of New 3-Alkoxymethyl/3-Phenyl indole-2-carboxamides with Potential Antiproliferative Activity. Chem. Biol. Drug Des.. 2017;90:64-82.
- [Google Scholar]
- EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity, and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem.. 2019;89:102997.
- [Google Scholar]
- Design, and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem.. 2020;104:104260-.
- [Google Scholar]
- Unusual reactivity of thiosemicarbazides towards 2,3-diphenylcyclopropenone: synthesis of new pyridazinethiones and 1,2,4-triazolo[4,3-b]pyridazinethiones. Arkivoc. 2007;xiv:1-11.
- [Google Scholar]
- Design and synthesis of hydrazinecarbothioamide sulfones as potential antihyperglycemic agents. Arch Pharm.. 2021;354:e2000336.
- [Google Scholar]
- Acid-catalyzed oxidative cleavage of S-S and Se–Se bonds with DEAD: efficient access to sulfides and selenides. Org. Chem. Front.. 2018;5:3557-3561.
- [Google Scholar]
- 1,3-thiazolidin-4-one derivatives bearing pyrimidine moieties: Design, computational studies, synthesis, characterization, antimicrobial, MTT assessment and molecular docking assessment. Chem. Data Collect.. 2020;28:100405.
- [Google Scholar]
- The Proline-Catalyzed Asymmetric Amination of Branched Aldehydes. Eur. J. Org. Chem.. 2007;2007:266-282.
- [Google Scholar]
- Chiral 2-Aminobenzimidazole as Bifunctional Catalyst in the Asymmetric Electrophilic Amination of Unprotected 3-Substituted Oxindoles. Molecules. 2018;23:1374-2133.
- [Google Scholar]
- Microwave Assisted Synthesis, Characterization and Antimicrobial Screening of Thiazolidin-4-one Substituted Pyrazole Derivatives. Curr. Microw. Chem.. 2019;6:44-53.
- [Google Scholar]
- Efficient synthetic procedure to new 2-imino-1,3-thiazolines and thiazolidin-4-ones promoted by acetonitrile electrogenerated base. New J. Chem.. 2018;42:11776-11781.
- [Google Scholar]
- Synthesis and characterization of quinoline-carbaldehyde derivatives as novel inhibitors for leishmanial methionine aminopeptidase. Eur. J. Med. Chem.. 2020;186:111860-111873.
- [Google Scholar]
- Structure and Reactivity of the Betaine Derived from Triphenylphosphine and Dimethyl Azodicarboxylate. Angew. Chem. Int. Ed. Engl.. 1969;8:513-515.
- [Google Scholar]
- A new approach to aryl hydrazides via the reaction of the Mitsunobu reagent with arynes: further application to access diverse nitrogen-containing heterocycles in one pot. Org. Chem. Front.. 2017;4:1636-1639.
- [Google Scholar]
- Reaction of 4-substituted thiosemicarbazides with (2,4,7-trinitro-9H-fluoren-9-ylidene) propanedinitrile. J. Heterocycl. Chem.. 2006;43:849-854.
- [Google Scholar]
- Indazole, oxathiadiazole, and thiadiazine derivatives from thiosemicarbazides. J. Sulfur Chem.. 2007;28:211-222.
- [Google Scholar]
- Synthesis of Novel Spiro(indolone-3,2′ -[1,3,4]thiadiazol)-2-ones and Evaluation of Their Antidepressant and Anticonvulsant Activities. J. Heterocycl. Chem.. 2011;48:1050-1055.
- [Google Scholar]
- Stereoselective synthesis of 2-(2,4-dinitrophenyl)hydrazone and (2-tosylhydrazono)-4-oxo-thiazolidine derivatives and screening of their anticancer activity. Monatshefte für Chem. 2020;151:1453-1466.
- [Google Scholar]
- Convenient diastereoselective synthesis of annulated 3-substituted-(5S*,6S*, Z)-2-(2-(2,4-dinitrophenyl)hydrazono)-5,6-diphenyl-1,3-thiazinan-4-ones. Mol. Diver.. 2019;23:821.
- [Google Scholar]
- Pyrazole, pyrazolo[1,2-c]-1,3,4-thiadiazole and thiadiazepine derivatives from thiosemicarbazides. Arkivoc. 2003;i:118-128.
- [Google Scholar]
- A convenient and efficient synthesis of thiazolidin-4-ones via cyclization of substituted hydrazinecarbothioamides. Arab. J. Chem.. 2019;12:289-294.
- [Google Scholar]
- Oxidation of Alcohols to Carbonyl Compounds with Di isopropyl azodicarboxylate Catalyzed by Nitroxyl Radicals. J. Org. Chem.. 2012;77:3005-3009.
- [Google Scholar]
- Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019;176:117-128.
- [Google Scholar]
- Am. Chem. Soc, Washington, DC. 1996;62
- Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129-132.
- [Google Scholar]
- Enantioselective Synthesis of Spirooxindoles: Asymmetric [3+2] Cycloaddition of (3-Isothiocyanato)oxindoles with Azodicarboxylates. Eur. J. Org. Chem.. 2013;35:7895-7901.
- [Google Scholar]
- Cu-Catalyzed Aerobic Oxidation of Di-tert-butyl Hydrazodicarboxylate to Di-tert-butyl Azodicarboxylate and Its Application on Dehydrogenation of 1,2,3,4-Tetrahydroquinolines under Mild Conditions. Org. Lett.. 2016;18:6300-6303.
- [Google Scholar]
- Core-Scaffold-Inspired Asymmetric Synthesis of Polysubstituted Chiral Hexahydropyridazines that Potently Inhibit Breast Cancer Cell Proliferation by Inducing Apoptosis Chemistry. Eur. J.. 2015;21:18100-18108.
- [Google Scholar]
- Silver-Catalyzed para-Selective C-H Amination of 1-Naphthylamides with Azodicarboxylates at Room Temperature. Synthesis. 2019;51:2697-2704.
- [Google Scholar]
- Design, synthesis, and biological evaluation of thiazolidinone derivatives as potential EGFR and HER-2 kinase inhibitors. Bioorg. Med. Chem.. 2010;18:314-319.
- [Google Scholar]
- New 1,3,4-oxadiazoles linked 1,2,3-triazole moiety as antiproliferative agents targeting EGFR-TK. Archiv Der Pharmazie 2022:e2200009.
- [Google Scholar]
- Discovery of new pyrimido[5,4-c]quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and ADME studies. Bioorg. Chem.. 2022;121:105693.
- [Google Scholar]
- Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem.. 2021;112:104960.
- [Google Scholar]
- A Novel Reaction of the “Huisgen Zwitterion” with Chalcones and Dienones: An Efficient Strategy for the Synthesis of Pyrazoline and Pyrazolopyridazine Derivatives. Ang. Chem. Int. Ed.. 2007;46:2070-2073.
- [Google Scholar]
- Synthesis, Antimicrobial, and Molecular Docking Studies of 1,3-Thiazolidin-4-one Analogs Bearing Benzothiazole. Ind. J. Heterocyclic Chem.. 2020;30:325-335.
- [Google Scholar]
- 1,3-Thuazolidin-4-ones: Biological Potential, History, Synthetic Development and Green Methodologies. Curr. Org. Synth.. 2018;15:1109-1123.
- [Google Scholar]
- An efficient and facile synthesis of divergent C-3/C-5 bis-functionalized 2-oxindoles from 5-formyl-Morita-Baylis-Hillman adducts of oxindole. J. Chem. Sci.. 2015;127:1417-1426.
- [Google Scholar]
- Enantioselective synthesis of 1,2,4-triazolines catalyzed by a cinchona alkaloid-derived organocatalyst. Organocatalyst. Chem. Comm.. 2013;49:11098-11100.
- [Google Scholar]
- Optimization of pyrrolizine-based Schiff bases with 4-thiazolidinone motif: Design, synthesis and investigation of cytotoxicity and anti-inflammatory potency. Eur. J. Med. Chem. 2019:111780.
- [Google Scholar]
- Novel chromenyl-based 2-iminothiazolidin-4-one derivatives as tubulin polymerization inhibitors: Design, synthesis, biological evaluation, and molecular modelling studies. J. Mol. Struct.. 2021;1225:128847.
- [Google Scholar]
- Executioner Caspase-3, -6, and -7 Perform Distinct, Non-redundant Roles during the Demolition Phase of Apoptosis. J. Biol. Chem.. 2001;276:7320-7326.
- [Google Scholar]
- Direct Amination of Arenes with Azodicarboxylates Catalyzed by Bisulfate Salt/HFIP Association. ACS Omega. 2019;4:8960-8966.
- [Google Scholar]
- Catalyst-free cycloaddition of 1,3-diene-1-carbamates with azodicarboxylates: A rapid click reaction. Bioorg. Med. Chem.. 2019;27:2438-2443.
- [Google Scholar]
- Divergent Reactivity of Nitrocyclopropanes with Huisgen Zwitterions and Facile Syntheses of 3-Alkoxy Pyrazolines and Pyrazoles. Org. Lett.. 2016;18:4936-4939.
- [Google Scholar]
- Syntheses of 2-hydroxypyrano[3,2-c]quinolin-5-ones from 4-hydroxyquinolin-2-ones by tandem Knoevenagel condensation with aldehyde and Michael addition of enamine with the quinone methide-thermo- and photochemical approaches. J. Chem. Soc. Perkin Trans.. 1999;1:2017-2024.
- [Google Scholar]
- A New Hydrogen-Abstracting Reaction with Diethyl Azodicarboxylate. J. Am. Chem. Soc.. 1966;88:2328-2329.
- [Google Scholar]
- Design and Synthesis of Novel Isatin-Based derivatives Targeting Cell Cycle Checkpoint Pathways as Potential Anticancer Agents. Bioorg. Chem.. 2020;105:104366-.
- [Google Scholar]
- Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino [1,2-a] indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur. J. Med. Chem.. 2018;146:260-273.
- [Google Scholar]
- Phosphine-Free [3+2] Cycloaddition of Propargylamines with Dialkyl Azodicarboxylates: An Efficient Access to Pyrazole Backbone. Synthesis. 2018;50:3499-3505.
- [Google Scholar]
- Identification of novel quinoline analogues bearing thiazolidinones as potent kinase inhibitors for the treatment of colorectal cancer. Eur. J. Med. Chem.. 2020;204:112643.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary material to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104280.
Appendix A
Supplementary material
The following are the Supplementary material to this article:Supplementary data 1
Supplementary data 1




