

Translate this page into:
New coumarin derivative with potential antioxidant activity: Synthesis, DNA binding and in silico studies (Docking, MD, ADMET)
⁎Corresponding author. skecel@istanbul.edu.tr (Serda Kecel-Gunduz)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
A new coumarin derivative, 7-((8-(4-benzylpiperidin-1-yl)octyl)oxy)-4-methyl-2H-chromen-2-one (C3), was synthesized by two-step alkylation reaction of 7-hydroxy-4-methyl coumarin. The structure and purity of the compound were characterized by its 1H and 13C NMR, FT-IR and LC-MS spectral data. The DNA binding interaction of C3 was evaluated using UV–vis spectrophotometric and viscosimetric methods. These experiments showed that C3 was bound in intercalative mode. The antioxidant activity of C3 was evaluated by the DPPH method, the antioxidant activity results displayed that C3 had DPPH radical scavenging effect. The possible mechanism of antioxidant and anticancer activity of C3 was investigated via molecular docking by using two enzymes CYP450 and EGFR as receptors. The C3 also tended a good antioxidant ability based on the result of the molecular docking analysis, with good binding affinity values (-7.82 kcal/mol) and binding site interactions. Molecular Dynamics (MD) simulation was implemented to elucidate the interactions with the protein–ligand complex in 20 ns. The ADMET analyzes which paved the way for us to predict C3 as a drug candidate were also performed. All experimental and theoretical results showed that the compound C3 was a potential drug candidate as an antioxidant and anticancer agent.
Keywords
Antioxidants
Synthesis design
DNA
Molecular dynamics
ADMET

1 Introduction
Coumarin and its derivatives constitute an important natural and synthetic heterocyclic compound class with their simple benzopyran core ring system. Since the isolation of coumarin from its natural source, the coumarin derivatives have attracted the researcher’s attention due to their important biological activities to use in applications for medicine. They have different kinds of biological activities such as anticancer, anticoagulant, antiviral, anti-inflammatory, antimicrobial, antifungal, antibacterial, antithrombotic, antioxidant, anti-Alzheimer, etc (Sashidhara et al., 2011, Keri et al., 2015, Thakur et al., 2015, Dandriyal et al., 2016, Hassan et al., 2016, Bang et al., 2019, Bouhaoui et al., 2021, Carneiro Viqueira et al., 2021, Li et al., 2021, Xu et al., 2021). Warfarin, Phenprocoumon and Acenocoumarol as an anticoagulant, and Hymecromone as a choleretic and antispasmodic agent are some of the marketed coumarin drugs with important clinical applications (Salehian et al., 2021). The easy synthesis and the possibility to functionalize at different positions of the coumarin ring provide a wide range of synthetic derivatives with important promising biological activity. Not only coumarin but also other heterocyclic compounds have attractive biological activities and their introduction to a structure provides stronger or different biological activities to the systems (Gomtsyan 2012). Therefore, the formation of hybrid molecules by combining the heterocyclic ring systems in a structure is an important strategy for the synthesis of new biologically active synthetic compounds with enhanced or new biological activities. Up to now, several coumarin hybrid molecules have been synthesized and their biological activities have been investigated successfully. The heterocycles can be introduced to the coumarin ring as a substituent or as a fused ring (Gomtsyan 2012, Paul et al., 2013, Sandhu et al., 2014, Medina et al., 2015, Kraljević et al., 2016, Bang et al., 2019, Zhang and Xu 2019, Ostrowska 2020, Basappa et al., 2021, Husain et al., 2021, Mamidala et al., 2021, Mustafa 2021, Pasala et al., 2021, Reddy et al., 2021, Salehian et al., 2021).
In this study, we designed and synthesized a novel piperidine-substituted coumarin hybrid compound (3) with promising biological activities as a part of our ongoing research on biological activities of novel coumarin derivatives (Budama‐Kilinc et al., 2020, Kecel-Gunduz et al., 2021). In this hybrid molecule, the combination of the coumarin ring with the piperidine ring with a long alkyl chain linker was expected to improve the antioxidant and anticancer properties of the coumarin ring. Because the piperidine ring has an important role in biological activity and is an important pharmacophore in many antioxidant and anticancer compounds (Das et al., 2018, Goel et al., 2018, Rk et al., 2018). In the literature, the antioxidant activities of some coumarin derivatives have been investigated by several methods. Various synthetic (Ghalehshahi et al., 2018, Li et al., 2020, Naik et al., 2020, Konidala et al., 2021, Mohammed and Ahamed 2022) or natural (Vera et al., 2007, Venugopala et al., 2013, J Matos et al., 2017, Zhang et al., 2020) coumarin derivatives have been found as potential antioxidant agents (Pedersen et al., 2007, Kostova et al., 2011, Al-Majedy et al., 2017). A few coumarin hybrid molecules also have been investigated for their antioxidant activity, and it was found that the coumarin hybrids with different heterocyclic rings showed antioxidant activities (Arora et al., 2014, Nagamallu et al., 2016, Kumar K et al., 2018, Bensalah et al., 2020, Basappa et al., 2021). But as far as we know, any coumarin hybrid with a piperidine ring has not been investigated as an antioxidant agent. The antioxidant activity is another important feature to reduce the risk of chronic diseases by neutralizing free radicals and minimizing oxidative damage. Therefore, after the synthesis of this piperidinyloctyloxy substituted coumarin (3) by alkylation reactions of coumarin ring on C7, its antioxidant activity was investigated.
Theoretical calculations performed with density functional theory (DFT) are very important for molecular stabilization and interactions of bioactive molecules (Sumrra et al., 2018, Sumrra et al., 2021). In this study, the most stable molecular structure of the C3 molecule was obtained by optimizing the DFT/B3LYP/6–311 ++ G(d,p) theory level in a vacuum environment. The characteristic vibrational wavenumbers of the molecule were compared with the experimentally observed wavenumbers and all vibrational modes of the molecule were assigned by determining the potential energy distribution (PED). The molecular structure was defined by performing 1H- and 13C NMR, UV–vis and FT-IR experimental and theoretical spectral analyzes.
Theoretical and/or experimental investigation of the interactions of small molecules defined as ligands with macromolecules such as DNA and protein defined as receptors may be useful for anticancer studies (Joksimović et al., 2019, Petronijević et al., 2021, Milović et al., 2022). In this study, the anticancer activity of the newly developed piperidine-substituted coumarin hybrid compound was also investigated. The anticancer activity of the compound has been demonstrated by studies involving its interaction with DNA because DNA is an important pharmacological target of molecules developed for cancer diseases. Besides this, the molecular docking approach has been used to explain the atomic-level interaction between the synthesized compound with corresponding receptors.
Optimization of the structure–activity relationship was achieved by molecular interactions and biochemical activity with molecular-level analyzes. Investigation of active sites in molecules was provided by molecular descriptive analyzes such as HOMO-LUMO, and Molecular Docking (Nazar et al., 2015, Ashraf et al., 2021, Hassan et al., 2022). The molecular docking analysis, the elucidation of specific molecular descriptors obtained from the results of quantum chemical calculations such as the energies of molecular orbitals (HOMO, LUMO), electron acceptor or donor characteristics, and the analysis of their physical and chemical properties provide important information on the antioxidant and anticancer activity of the molecules. To evaluate the antioxidant and anticancer potential of C3 via molecular docking, the two enzymes CYP450 and EGFR receptors were chosen. Molecular dynamics (MD) simulations allowed the elucidation of amino acid residues and interactions in the binding region of the CYP450 enzyme that interacted with C3, taking into account the time dynamics for 20 ns. The lipophilicity parameter, which determines the solubility of drug candidate molecules in different body fluids, is related to the behavior of the molecule in the biological environment and its ADMET profile. It is known that lipophilicity is associated with important parameters such as permeability in the gastrointestinal tract, passage through the blood–brain barrier, and binding to plasma proteins (Ristovski et al., 2022). ADMET analyzes were also performed to predict the pharmacological profile of the synthesized C3 molecule.
2 Materials and methods
2.1 Materials and equipment
Reagent-quality solvents were used without further purification. Column chromatography was conducted on silica gel 60 (40–63 μM) (Merck). TLC was carried out on aluminium sheets precoated with silica gel 60F254 (Merck). IR spectra were determined on a Thermo Scientific NICOLET IS10 FT-IR spectrometer. NMR spectra were recorded on a Varian UNITY INOVA 500 MHz NMR spectrometer. Chemical shifts, δ are reported in ppm with TMS as the internal standard and the solvents are CDCl3 and CD3OD. The synthesis of compounds 1 and 2 was carried out according to the literature procedure (Manidhar et al., 2012, Jiang et al., 2018).
Calf thymus DNA (CT-DNA), 1,1-Diphenyl-2-picril-hydrazine (DPPH) and ethanol were purchased from Sigma Aldrich. Dimethyl sulfoxide (DMSO), tris base, ethylene diamine tetraacetic acid (EDTA) and hydrochloric acid (HCl) were purchased from Merck Millipore.
2.2 Methods
2.2.1 Synthesis of C3
Compound 2 (1.0 mmol) and N-benzylpiperidine (1.0 mmol) were dissolved in dry acetone, and then K2CO3 (3.0 mmol) was added to the solution. The mixture was refluxed for 24 h. After filtration, the solvent was evaporated. The crude product was purified by column chromatography on silica gel (ethyl acetate-hexane 5:1). The purity of the compound was controlled by LC-MS (QTOF) data (Fig. S3) and it was seen that the compound was pure. White solid, m.p.60–64 °C, yield 77 %. 1H NMR (500 MHz, MeOD): δ = 1.68–2.01 (m, 12H, CH2), 2.16–2.34 (m, 7H, CH2 and CH), 2.67–2.83 (m, 5H, CH3 and N-CH2), 2.87–2.95 (m, 2H, N-CH2), 3.32 (bs, 2H, N-CH2), 4.41–4.46 (m, 2H, OCH2), 6.50–6.54 (m,1H, =CH), 7.22–7.31 (m, 2H, ArH), 7.50–7.65(m, 5H, ArH), 7.99–8.01 (m, 1H, ArH); 13C NMR (125 MHz, MeOD):δ = 17.61 (CH3), 25.91 (CH2), 26.28 (CH2), 27.57 (CH2), 29.04 (CH2), 29.22 (CH2), 29.43 (CH2), 31.49 (2 × CH2), 37.9 (CH), 42.86 (CH2, PhCH2), 53.7 (2 × N-CH2), 58.7 (N-CH2), 68.65 (O-CH2), 101.16 (CaroH), 110.93 (CaroH), 112.9 (Caro), 113.59 (=CH), 125.87 (CaroH), 126.21 (CaroH), 128.17 (CaroH), 129.07 (CaroH), 140.58 (Caro), 154.66 (Caro), 155.42 (Caro), 162.50 (Caro), 162.95 (C⚌O) ppm. FTIR (ATR): ν = 3059 (aromatic C—H stretching), 2921, 2852 and 2802 (aliphatic C—H stretching), 1719 (C⚌O stretching), 1470 and 1385 (aliphatic C—H bending) cm−1; LC-MS (ESI-QTOF): m/z [M + H]+ calc for C30H39NO3, 462.3008; found, 462.3009.
2.2.2 Computational method
The characteristic wavenumbers of the C3 molecule were obtained from the experimental vibrational (FT-IR) spectra and together with the fundamental vibrational wavenumbers calculated at the DFT-RB3LYP / 6–311++G (d, p) (Lehtola 2015) level of theory using Gaussian 09 program package (Frisch and Clemente 2009) Wavenumbers calculated with harmonic approximation with the theory of level DFT/B3LYP/6–311++G(d,p) were scaled with a dual scaling factor (Halls et al., 2001) to fit the experimental wavenumbers. The assignments of all vibrational modes of the synthesized C3 molecule were performed using the GAR2PED program, based on the potential energy distribution of the calculated vibrational modes (Martin and Van Alsenoy 2007).
2.2.3 DNA binding assay
2.2.3.1 UV–vis spectrometric method
The experiment was performed using CT-DNA by UV–vis absorption titration method. CT-DNA solution in Tris-HCl/NaCl (pH 7.2) buffer confirmed a 1.9 UV absorbance ratio at A260/A280 at wavelengths of 260 and 280 nm. This ratio indicates that the CT-DNA is protein-free (Kecel-Gunduz et al., 2021). The DNA solution was prepared using doubly distilled water. The stock solution of C3 was dissolved in DMSO and prepared by appropriate dilution with buffer corresponding to the concentration required for the experiment. The UV–vis absorption titration assay was performed by keeping the C3 concentration (60 µM) constant and adding increasing concentrations (0–240 µM) of CT-DNA. After each successive addition of DNA solution, it was incubated for 5 min, and absorbance values were recorded. The percent ratio of changes in absorbance density of C3 was calculated using Eq. (1).
In an equation, Ai indicates the free absorbance intensity of the C3, and As indicates the absorbance intensity of the C3 after adding DNA at the maximum concentration.
The intrinsic binding constant (Kb) used to investigate the binding strength of the C3 molecule to DNA was calculated using Eq. (2).
In the formula, εf; molar damping coefficient of free C3, εB; molar damping coefficient of fully bound C3, εA; It expresses the ratio of absorbance to C3 concentration. In the [DNA]/(εA − εf) versus [DNA] plot, the ratio of the slope to the point of intersection gives Kb (Liu et al., 2011, Kecel-Gunduz et al., 2021).
2.2.3.2 Fluorescence spectroscopy method
Besides the electronic absorption titration technique, the interactions of the complexes with DNA were performed using fluorescence spectroscopy based on the principle of substitution with ethidium bromide (EB) (Dehkhodaei et al., 2018). The analyzes were performed with a QM-4/2003 QuantaMaster Steady-State Fluorescence System. For analysis, the solution containing 50 µM CT-DNA and 5 µM EB was dissolved in a buffer, and the first spectrum was obtained. C3 solution was added to the CT DNA-EB solution in increasing concentrations (5–22.5 µM) and incubated for 3 min at 25 °C in the dark for each measurement. The slits of the monochromators were adjusted to 2 nm. The excitation was performed at 330 nm, and the emission spectra were obtained from two different regions between 340 and 500 nm and 550–650 nm. The fluorescence spectra were obtained.
2.2.4 Viscosimetric method
Viscosity measurements were performed by using the Ubbelohde viscometer held in a thermostatic bath at 30.0 ± 0.1 °C (Coban et al., 2013). The experiment was conducted keeping the CT-DNA concentration (0.5 µM) constant and adding increasing concentrations (0–6.125 µM) of C3. It was incubated for 5 min after each successive addition of C3 solution and flow times were measured in triplicate with a digital chronometer and mean flow time was calculated. Data are presented as (ŋ /ŋ0) 1/3 versus the ratio of C3 concentration to CT-DNA (Coban et al., 2013, Shankaraiah et al., 2016). Here, ŋ is the viscosity of CT-DNA in the presence of C3 and ŋ0 is the viscosity of CT-DNA alone.
2.2.5 Antioxidant activity
The antioxidant activity of C3 was determined by diphenyl-2-picrylhydrazyl (DPPH) radical scavenger test (Shanty et al., 2017, Özdemir 2020). In the experiment, solutions (2 mL) containing different concentrations (0.03125–1 mg/mL) were added to 0.5 mL ethanolic DPPH solution (25 µg/mL). It was then mixed by vortex and incubated at 27 °C for 30 min in the dark. Absorbances were measured at 517 nm against ethanol used as a blank (Ejidike and Ajibade 2015, Shanty et al., 2017). BHT (Butyl HydroxyToluene) was used as a standard antioxidant (Kecel-Gunduz et al., 2021). Percent inhibition of DPPH radical was calculated using Eq. (3) (Brand-Williams et al., 1995).
2.2.6 Statistical analysis
Statistical analysis of DPPH antioxidant activity results was analyzed with SPSS (Statistic 22). The differences between the C3 and BHT groups were determined by t-test. In the graph, the data were expressed as mean ± standard error. The α-significance level was determined as p < 0.05.
2.2.7 In silico analysis
2.2.7.1 HOMO-LUMO and UV analysis
HOMO-LUMO and theoretical UV analyses were realized in a vacuum and DMSO environments at Gaussian09 program (Frisch and Clemente 2009) using time-dependent density functional theory (TD-DFT) (Runge and Gross 1984) approach with B3LYP / 6–311 ++ G (d, p) basis set (Lehtola 2015). With the determined HOMO LUMO energy values, important information about the synthesized compound such as chemical stability, ionization potential, electron affinity, and chemical hardness was obtained. Information on HOMO-LUMO transitions that contribute to the wavelengths and energy values determined in the theoretical UV analysis obtained with the Gauss Sum program (O'boyle et al., 2008). The experimental UV–Vis absorption spectra of the C3 in DMSO solution were recorded using the Shimadzu UV-1280 UV–Vis recording spectrometer in the spectral region of 200–700 nm.
2.2.7.2 Molecular docking and ADME analysis
For molecular docking simulation, the Glide SP module of Schrödinger Software Maestro version 11.4 was chosen (Friesner et al., 2004, Halgren et al., 2004). Membrane-associated heme-protein cytochrome P450 proteins (CYP450s), which contribute to the oxidative metabolism of more than 90 % of drugs in current clinical use was preferred for the first receptor. In this study, we used the crystal structure of a human CYP450, CYP2C9, in complex with the anticoagulant drug warfarin as a receptor (Williams et al., 2003). The crystal structure of the receptor structure (PDB ID: 1OG5), which has A and B chains and a sequence length of 475 amino acids, determined by X-ray Diffraction at 2.55 Å resolution in the protein data bank, was downloaded from the Swiss model online site with no missing residues and has only the A chain form (Bienert et al., 2017). There are 19 differences between the amino acid sequence numbers of the receptors downloaded from the protein data bank and the Swiss model site (iprotein data bank= iswiss model amino acid number + 19). Using the protein preparation wizard tool, all water and ions were removed to prepare all receptors for docking analysis, only polar hydrogens were added, preprocess was performed after the bond orders assigning and defective parts in the receptor structures were analyzed, then using PROPKA (Søndergaard et al., 2011) at pH 7.0, the receptor structures were optimized with the OPLS3force field and then minimized (Sastry et al., 2013, Harder et al., 2016). The optimized geometries were preferred for docking analysis using Gaussian 09 software for the synthesized C3 molecule (Frisch and Clemente 2009). Using the ligand preparation tool, 32 possible stereoisomers of the selected ligand were obtained by selecting the OPLS3 force field (Harder et al., 2016). By using the receptor grid generation tool, a cubic box-shaped grid was created, releasing the residues in the binding region of the receptors which contains especially thiol and hydroxyl groups in a rotational position, docking calculation analysis of the synthesized C3 molecule to the active binding site of the selected receptors were performed. The epidermal growth factor receptor (ErbB1, Her1) is a transmembrane receptor tyrosine kinase that transmits and transforms critical signals for cell proliferation, differentiation and motility. It has been shown that irregularities and abnormal behaviors of EGFR signaling such as overexpression or mutation play an important role in the development and growth of tumor cells (Yun et al., 2007). The crystal structures of EGFR (PDB ID: 1 M17) with 2.60 Å resolution, A chain and 333 sequence length were used in this work for another receptor structure (Stamos et al., 2002). The shift between the amino acid numbers of the receptors downloaded from the protein data bank and the Swiss model site (iprotein data bank= iswiss model amino acid number + 665) was determined, too. All pharmacokinetic and physicochemical properties of the synthesized C3 molecule due to its geometry were determined by using the Qik-Prop module of the same software (Ioakimidis et al., 2008, Singh et al., 2013). The properties of a molecule that affect drug susceptibility, such as molecular weight (MW), percent human oral absorption, predicted octanol/water distribution coefficient (QPlogPo/w), total surface area (TSA), and Lipinski's five-rule conformity, were also calculated, and revealed by ADME analysis for the first time.
Molecular docking analysis was also performed using B-DNA Dodecamer as a receptor (PDB Code: 1BNA) (Drew et al., 1981) with 1.9 Å resolution. After all water molecules were removed and polar hydrogen atoms were added to the receptor, molecular docking analysis was implemented by AutoDockTools 1.5.6 software. The grid box was adjusted as a 40x40x40 unit with 0.375 Å grid spacing. The calculated binding affinities and RMSD values of the C3-DNA complex were given by AutoDock Vina 1.1.2 (Trott and Olson 2010). The receptor-ligand interactions were visualized by PyMOL (DeLano 2002) and Schrodinger, software programs.
2.2.7.3 Molecular dynamics (MD) analysis
The MD simulations were carried out using the Desmond simulation package of the Schrödinger program to indicate the stability of the system by monitoring interactions of the C3 with antioxidant protein (PDB: 1OG5). The NPT ensemble with a temperature of 300 K and a pressure of 1 bar was applied in all runs. The simulation length was 20 ns. Interactions between ligands and proteins and categorizations of these interactions were analyzed using the Simulation Interaction Diagram tool implemented in the Desmond MD package. The stability of the MD simulations was revealed by considering the RMSD of the atom positions of the ligand and protein over time (Bowers et al., 2006, Release 2017).
3 Results and discussion
3.1 Synthesis of C3
In this study, 7-((8-(4-benzylpiperidin-1-yl)octyl)oxy)-4-methyl-2H-chromen-2-one (C3) was prepared by two-step alkylation reaction of 7-hydroxy-4-methylcoumarin (1) (Fig. 1). Firstly, 1 was synthesized from the Pechmann reaction of resorcinol with equmolar amounts of ethyl acetoacetate in trifluoroacetic acid under ultrasonic effect. Then, the alkylation of 1 was accomplished by the reaction with 1,8-dibromooctane in dry acetone in the presence of dry K2CO3 under reflux to give 2. Compounds 1 and 2 were known in the literature (Manidhar et al., 2012, Jiang et al., 2018). Their characteristic and spectral data were following the literature data, and they were pure enough to go to the next steps of the synthesis. The last step which was the alkylation of 2 with N-benzypiperidine gave pure C3 after the purification of the crude product by column chromatography. The structure and purity of the compound were determined by its spectral data (IR, NMR and LC-MS (ESI-QTOF)) and it was concluded that they are in accordance with the structure.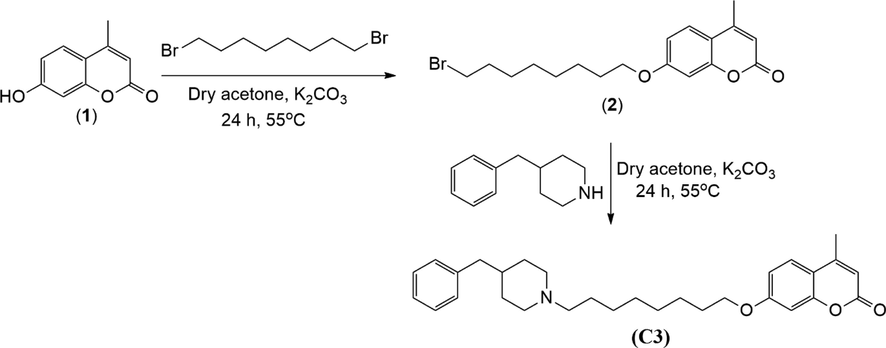
Synthesis of compound C3.
3.2 Structure characterization of C3
The most stable molecular structure of the C3 molecule which was optimized in a vacuum environment using the DFT/B3LYP/6–311 ++ G(d, p) theory level was given in Fig. 2, and the optimized geometry parameters of C3 were also tabulated in Table S1.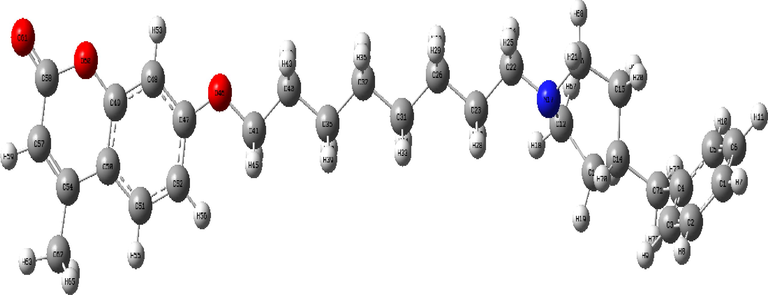
The optimized molecular structure of C3, calculated at the DFT/B3LYP/6–311 ++ G(d,p) level of theory.
3.2.1 Structure characterization of C3 with vibrational analysis
The calculated theoretical vibrational wavenumbers and corresponding experimental IR wavenumbers of the C3 molecule (C30H39NO3), which has 73 atoms and 213 vibrational modes, together with the corresponding reference values and potential energy distributions (PED) of all vibrational modes, and the assigned modes of C3 were also given in Table S2. The experimental FT-IR and calculated IR absorption spectra in the region 3800–2000 cm−1 (a), 1800–1000 cm−1 (b), and 1200–400 cm−1 (c) were also seen in Fig. 3.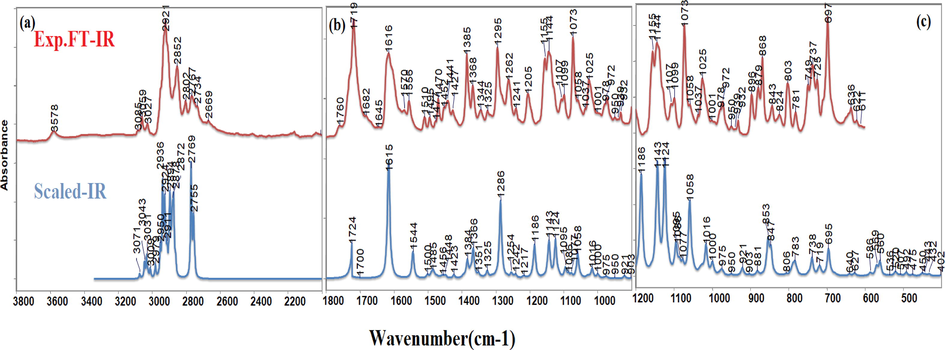
The experimental FT-IR and calculated IR spectra of C3 with 3800–2000 cm−1 (a), 1800–1000 cm−1 (b), and 1200–400 cm−1 (c) regions.
3.2.1.1 3800–2000 cm−1 wavenumber region
In this region, the CH stretching of the rings is generally defined. It was reported in the literature that CH stretching of the rings is observed in the range of 3101–3024 cm−1 (Ceylan et al., 2015, Ceylan et al., 2021). The peaks observed at 3085, 3059 cm−1 and 3027 cm−1 in the IR spectrum are represented by the CH asymmetric and symmetric stretching vibrations of the coumarin and cyclohexane ring, respectively. Corresponding peaks were recorded at 3079 cm−1 and 3071 cm−1, 3068 cm−1 and 3046 cm−1, 3029 cm−1 for coumarin ring (Bahgat 2006) and 3073 cm−1 and 3039 cm−1 (Janani et al., 2021) for cyclohexane ring. The experimental peaks were marked at 2921 cm−1, and 2852 cm−1 corresponding to CH asymmetric and symmetric stretching, respectively.
3.2.1.2 1800–1200 cm−1 wavenumber region
In this region, generally, C⚌O, CC, and CO stretching and bending movements of CH2 and CH3 groups are characterized. The peak of 1719 cm−1, which was observed very strongly in the FTIR spectrum, was calculated at 1724 cm−1 and assigned as the C⚌O bond stretching on the coumarin ring based on the PED analysis. The 1718 cm−1 and 1727 cm−1 values corresponding to this peak were also seen in the references (Avdović et al., 2018, Kiraz et al., 2022). 1616 cm−1 peak in the experimental spectrum calculated at 1618 and 1615 cm−1 and was assigned as the CC stretching of the coumarin ring following the literature (Bahgat 2006, Arivazhagana et al., 2010, Kiraz et al., 2022, Malacaria et al., 2022). The peaks recorded at 1570 cm−1, 1556 cm−1 and 1510 cm−1 in the experimental spectrum were assigned as the CC stretching on the cyclohexane ring and the coumarin ring, respectively. Peaks at 1495 cm−1, 1477 cm−1 and 1470 cm−1 represented bending vibrations of CH2 groups assigned to scissoring. The vibrations found as a small intensity peak at 1452 cm−1 and 1441 cm−1 in the experimental spectrum were referred to as CH3 rocking and CH3 asymmetric bending, respectively. The peak observed with high intensity at 1385 cm−1 in the FT-IR spectrum was also characterized by the symmetric bending movement of the CH3 group. The peaks observed at 1368 cm−1 and 1344 cm−1 defined by the wagging movement of CH2 groups in the piperidine ring in C3 are in agreement with the literature values at 1366 cm−1 (Chandra et al., 2011), 1364 cm−1 and 1340 cm−1 (Mahalakshmi and Balachandran 2014). The 1325 cm−1 peak observed in the experimental FTIR and calculated with the same value was assigned as the CC bond stretching of the coumarin ring. The peak observed at 1295 cm−1 in the FT-IR spectrum, assigned as the CO bond stretching according to the PED analysis due to the 42 percent contribution of CO bond stretching of the coumarin ring was calculated at 1286 cm−1. The assignment of the peak observed at 1205 cm−1 and calculated at 1186 cm−1, and similarly observed in the literature at 1211 cm−1 and scaled at 1189 cm−1, were defined as the rocking movement of the CH2 group in the coumarin ring structure following the literature (Bahgat 2006).
3.2.1.3 1200–400 cm−1 wavenumber region
In this region, stretching, bending and deformations were observed mostly in the three-ring structure. The stretching of the CN modes for piperidine is observed in the region of 1180–1100 cm−1. The band obtained at 1144 cm−1 was assigned to CN stretching and also referred to as 1144 cm−1 (Janani et al., 2021). The peak observed at 1107 cm−1 with low intensity was assigned as the twisting movement of the CH2 group, and the peak observed at 1099 cm−1 was named CC stretching. The peak, which appeared very strong at 1073 cm−1, was identified as CC stretching with cyclohexane ring rocking by the PED analysis. For the coumarin ring, the CH2 rocking together with CC stretching was observed in the experimental spectrum at 1058 cm−1, which is consistent with the literature value of 1050 cm−1 (Arivazhagana et al., 2010). The CO stretching of coumarin was also defined as the observed peak at 1025 cm−1 and recorded at 1026 cm−1 in the literature value (Arivazhagana et al., 2010). The CC bond stretching of the piperidine ring was recorded at 972 cm−1 and its theoretical value was calculated at 971 cm−1, which is in agreement with the previously found value of 971 cm−1 (Chandra et al., 2011) and 953 cm−1 (Mahalakshmi and Balachandran 2014). In addition, the 932 cm−1 peaks represented the CC together with CN stretching for the piperidine ring concerning the reference value at 933 cm−1 (Janani et al., 2021). The peaks at 896 cm−1, 879 cm−1 and 868 cm−1 observed in the FTIR spectrum, characterized the deformations of the cyclohexane ring, piperidine ring and coumarin rings, respectively. In addition to these, 843 cm−1 and 824 cm−1 peaks explained the coumarin and cyclohexane ring deformations. The peak observed at 803 cm−1 in the experimental spectrum is also observed in the literature (Chandra et al., 2011) with the same value and was defined as the CH2 bending motion of the cyclohexane group mixed with CC stretching. The peak at 697 cm−1, which is observed very strongly in the spectrum, was defined as the deformation of the cyclohexane ring in accordance with the literature (Chandra et al., 2011, Janani et al., 2021).
3.2.2 Structure characterization of C3 with NMR
The 1H- and 13C NMR spectra of C3 were used to confirm its structure. In the 1H NMR spectrum, CH2 groups of the octyl chain located at 1.68–2.01 ppm as multiplets, N-CH2 groups belonging to the octyl chain and piperidine ring were seen at 2.83–3.32 ppm as multiplet or broad singlet. The multiplet at 4.41–4.46 was assigned to the OCH2 group of the octyl chain. The alkenic = CH proton of the coumarin ring was seen at 6.50–6.54 as a multiplet. The aromatic protons of the benzyl group and coumarin ring are located between 7.22 and 8.01 as multiplets.
The 13C NMR spectrum showed the CH3 group of the coumarin ring at 17.61 ppm, the CH2 groups of the octyl chain between 25.91 and 31.49 ppm, CH2 group of the benzyl group at 42.86 ppm. The signals at 53.7 and 58.7 were assigned to N-CH2 groups of the octyl chain and piperidine ring, and the OCH2 group of the octyl chain was seen at 68.65 ppm. The alkenic = CH carbon of the coumarin ring was seen at 113.59 ppm. The aromatic carbon signal was seen at 125.87–162.50 ppm as expected. The carbonyl carbon of the coumarin ring was located at 162.95 ppm. To confirm the molecular structure of the C3 molecule, the theoretical analysis of 1H- and 13C NMR and chemical shift calculations in the methanol solution were also performed using the GIAO method and B3LYP/6–311 ++ G(d, p) level of theory and given Table S3 and Fig. S1and Fig.S2 together with experimental NMR spectra. The LC-MS (ESI-QTOF) spectra of C3 are also given in Fig.S3.
3.3 DNA binding assay results
One of the first targets of anticancer drugs is DNA. Therefore, investigating the interactions of DNA and small molecules plays a key role in the design of new drugs with anticancer activities (Zhang et al., 2012, Pages et al., 2015).
3.3.1 Spectroscopic results
3.3.1.1 DNA binding study via UV-Vis spectroscopy
UV–vis spectroscopy is one of the fundamental techniques for studying small molecule-DNA interactions (Strekowski and Wilson 2007, Zhou et al., 2007, Kumar et al., 2015, Shi et al., 2015, Dehkhodaei et al., 2018, Mahmood et al., 2019, Phadte et al., 2019). In this technique, the molecule-DNA interaction is observed as a shift in absorbance (hypochromic, hyperchromic) (Doshi et al., 2010, Shi et al., 2015, Phadte et al., 2019) or maximum wavelength (bathochromic, hypsochromic) (Shi et al., 2015, Mahmood et al., 2019). The interaction of the C3 molecule with DNA was determined by UV spectroscopy using calf thymus DNA CT-DNA. The spectra of C3 were recorded under varying concentrations of CT-DNA (Fig. 4). With successively added CT-DNA, 35.6 % hypochromism at 329 nm and a blue shift of 5 nm were observed in the UV–vis absorption of C3.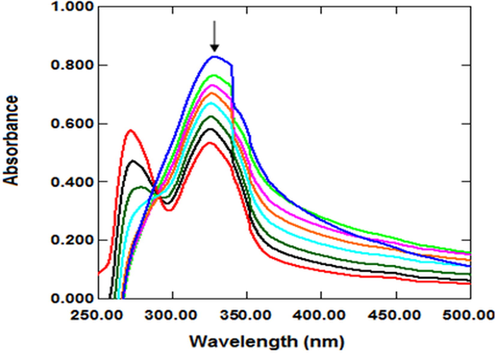
Absorption spectra of C3 in the absence (blue peak) and presence of CT-DNA.
With the addition of DNA to the molecule, the absorption peak intensities decreased, as indicated by the arrow (Zafar et al., 2017). The decrease in absorbance indicates that the DNA structure has become compact. This compactness can cause hypochromism (Li et al., 1996) Shah et al. (2010) reported hypochromism due to DNA contraction (Shah et al., 2010). Hypochromism is observed due to a strong π → π* stacking interaction that reduces the distance between the bases of DNA (Liu et al., 2003). The π → π* stacking interaction in the interaction of DNA with the molecule is a typical indicator of the intercalative binding mode (Shah et al., 2008). In addition, the blue or red shift in hypochromism indicates that the molecule's binding mode to DNA is intercalation, which involves the partial insertion of the aromatic ring between DNA base pairs (Liu and Yang 2009, Asadi and Nasrollahi 2017).
Hypochromism is observed because of a strong π → π* stacking interaction, reducing the distance between the bases of the DNA after the molecule has bound to the DNA (Liu et al., 2003). This mode of interaction is known as intercalation. There may be a red-or blue-shifted hypochromism on intercalation (Liu and Yang 2009, Asadi and Nasrollahi 2017). The molecules that bind to DNA through intercalation are used in cancer treatment because they inhibit DNA replication in cancer cells (Frederick et al., 1990, Wang 1992).
The Kb ensures a measure of molecule-CT-DNA stability, while the sign of ΔG indicates the spontaneous/non-spontaneous nature of the molecule-CT-DNA interaction. Negative values of ΔGb identify spontaneous binding of the molecule with DNA (Zafar et al., 2021).
Our results showed that C3 bound by the intercalation pathway. The Kb value was calculated to explain the binding strength of C3. The Kb value of C3 was found to be 1.16x105 M-1. This value indicates that C3 binds at medium strength. It was found that the Kb value is consistent with other studies reporting moderate binding of the molecule to DNA (Gao et al., 2006, Liu et al., 2009, Coban et al., 2013, Durgapal et al., 2017, Pivetta et al., 2017, Mahmood et al., 2019, Bora et al., 2021). The spontaneous binding between C3 and DNA was also proved by the negative value of ΔG (-28.8 kJ/m).
In literature, AMC –DNA intercalation was also considered by the shift in peak potential and decrease in peak current and hypochromic accompanied with bathochromic shift was noticed in UV–vis absorption spectroscopy with the binding constant (Kb) with a value of 6.15 × 105 M−1 which resulting in a stable AMC-DNA complex (Shah et al., 2008). In our study, the calculated Kb value 1.16x105 M−1 is in close agreement with that obtained by (Shah et al., 2008, Zafar et al., 2021).
3.3.1.2 DNA binding study via fluorescence spectroscopy
EB is a DNA intercalator that shows fluorescence emission when bound to DNA. Therefore, the decrease in DNA/EB fluorescence intensity by the tested compound is indicative of EB displacement from the DNA/EB complex (Amin et al., 2018). Competitive EB binding studies were performed to determine the DNA binding interaction of C3 at two different regions between 340 and 500 nm and 550–650 nm.
With the addition of increasing concentrations of C3 to DNA/EB, there was no significant change in fluorescence intensity in the range of between 550 and 650 nm (Fig. S10a). This result indicates that C3 cannot compete with EB for DNA binding sites. Studies in the literature reporting that compounds cannot compete with EB for DNA binding sites have reported that the interactions of compounds with DNA may be in the form of external binding (Sarwar et al., 2015, Anjomshoa and Torkzadeh-Mahani 2016).
As a result of UV–vis measurements for the DNA interaction of the C3 compound, the Kb value was calculated as 1.16x105 M−1. In Fig. 4, the small peaks seen around 275 nm in the UV–vis spectra support our interpretation. The Kb value of EB (7 × 107 M−1) (Waring 1965) is high compared to C3. In addition, as can be seen in Fig. 5, it is understood that the C3 compound has a lower interaction with DNA than EB in the viscosity experiment results. These results support the idea that C3 cannot be replaced by EB.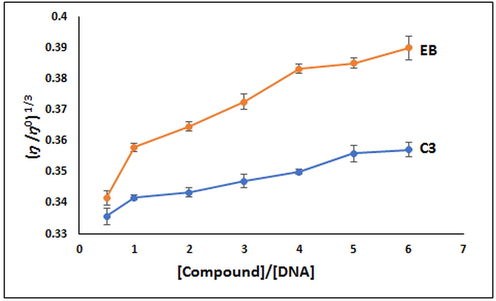
Changes in relative viscosity of CT-DNA due to the addition of C3 and EB (n = 3).
Ramana et al. (2015) evaluated the DNA interaction of the anti-retroviral drug nelfinavir spectroscopically with two different methods: UV–vis and fluorescence spectroscopy. In the study, it was reported that the drug exhibited groove bonding or electrostatic interaction as a result of UV–vis analysis, while it was reported that it exhibited intercalative bonding as a result of analyzes performed with fluorescence spectroscopy (Ramana et al., 2015). Our study determined that different interactions were observed in the analysis results performed with UV–vis spectroscopy and in the analysis results performed with fluorescence spectroscopy (Fig. S10b). Our molecular docking results showed that DNA interacted with adenine and guanine regions. Although the literature has reported that the molecules do not have DNA sequence selectivity, studies are known as intercalative binding when they bind more to guanine and cytosine-dense regions (Geierstanger and Wemmer 1995) and groove binders when they bind to adenine and thymine-rich groove regions (Kecel-Gunduz et al., 2021). This situation indicates that C3 interacts with both minor and significant grooves of DNA.
3.4 Results of viscosimetric analysis
The viscosimetric analysis provides valuable information about the mode of the binding interaction between the CT-DNA and the compound (Maiti et al., 2020). Therefore, a viscosity test was performed to better elucidate the interaction of C3 with DNA, besides the UV–vis absorption titration experiment. Compounds that bind intercalatively to DNA strongly increase viscosity by elongating the DNA double helix (Suh and Chaires 1995, Abdel-Rahman et al., 2016, Shankaraiah et al., 2016, Mahaki et al., 2019, Kiwaan et al., 2021). It was found that the viscosity experiment resulted in a gradual increase in viscosity indicating intercalation of C3 with DNA (Fig. 5). Data from the viscosity study support the result from the UV–vis absorption titration study (Shankaraiah et al., 2016, Maiti et al., 2020, Perka et al., 2021).
In our study, the effect of increasing the amount of synthesized C3 on the relative viscosity of DNA is not linear. This may be due to the ratio of CT-DNA and the amount of C3 to each other or the incubation time needed for the interaction of CT-DNA and C3. It has been reported in the literature that there is a continuous increase in the viscosity of DNA after the interaction of DNA and compound, as in our study. However, this increase is not linear (Reddy et al., 2011, Abu-Dief and Nassr 2015, Abdel-Rahman et al., 2016, Shankaraiah et al., 2016, Maiti et al., 2020).
3.5 Antioxidant activity
Oxidative reactions of biological molecules cause many diseases such as cancer, ageing, inflammation, atherosclerosis, immunosuppression, diabetes and neuro degenerative disorders (Alzheimer's and Parkinson's diseases, etc.) (Montine et al., 2005, Wu et al., 2010, Kumar and Rawat 2013), and these damages are caused by free radicals (Lai et al., 2001, Kumar and Arunachalam 2009). Therefore, there is great interest in the discovery of natural or synthetic antioxidants to prevent free radical damage in the body (Burton et al., 1985, Olanlokun and Akomolafe 2013, Ejidike and Ajibade 2015). The antioxidant activity of C3 was evaluated by the DPPH radical scavenging assay (Burits and Bucar 2000, Chen et al., 2020, Özdemir 2020, Shankar et al., 2020). The DPPH radical scavenging ability of C3 and standard drug (BHT) was given in Fig. 6. Although the antioxidant activity of C3 was lower than BHT (p < 0.05), it was found that all used concentrations owed DPPH radical scavenging ability. This ability can be attributed to the stable radical formation by hydrogen transfer from different parts of C3 to DPPH as proposed in Fig. 7. There are similar studies in the literature in which the synthesized molecules do not show antioxidant activity as high as the standard drug (Ejidike and Ajibade 2015, Božić et al., 2017, Shanty et al., 2017, Bakır and Lawag 2020, Bingöl and Turan 2020).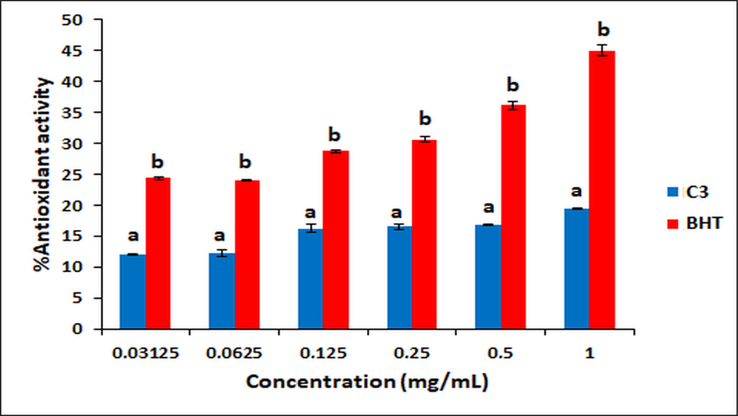
Antioxidant activity of C3 and BHT. Different letters mean significant differences between the C3 and BHT groups (mean ± SE).
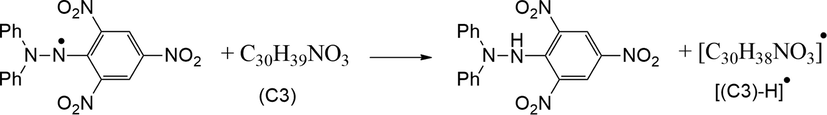
A proposed mechanism for antioxidant activity of C3.
3.6 In silico analysis results
3.6.1 HOMO-LUMO analysis results
The reactivity of a molecule is proportional to the HOMO LUMO energy gap (ΔE), and low ΔE indicates the high reactivity of the molecule. In addition, the ionization potential, electron affinity, electronegativity, chemical potential, and chemical hardness of a molecule can also be obtained by determining the HOMO LUMO energies (Govindarajan and Karabacak 2012, Ünver et al., 2018).
The HOMOs (HOMO and HOMO-1) and LUMOs (LUMO and LUMO + 1) shown in Table 1 and Fig. 8 were obtained with the Gaussian09 package program using TDDFT/B3LYP/6–311++G(d,p) basis set for vacuum and DMSO environments. It was seen that the HOMO orbitals were located on the piperidine ring while the LUMO orbitals were located on the coumarin ring. That is, HOMO-LUMO transitions were piperidine ring to coumarin ring of C3 for both environments. HOMO-1, HOMO, LUMO and LUMO + 1 energy values were determined as −6.34026 eV, −5.84746 eV, −1.91323 eV and −0.67158 eV for vacuum environment, respectively. HOMO-1, HOMO, LUMO and LUMO + 1 energy values were also determined as −6.40611 eV, −5.81943 eV, −2.01092 eV and −0.69797 eV for DMSO environment, respectively. In a study with coumarins, different LUMO values from −4.73 eV to −0.80 eV were obtained, and in our study, −2.01 eV and −1.91 eV values were obtained for DMSO and vacuum environments within the range of literature values (Novak and Kovač 2000).
TD-B3LYP/6–311++G(d,p)
Energy(kcal/mol)
ELUMO+1
ELUMO
EHOMO
EHOMO-1
ΔEHOMO-LUMO
ΔE(HOMO-1)-(LUMO)
ΔE(HOMO-1)-(LUMO+1)
Vacuum
−0.67158
−1.91323
−5.84746
−6.34026
3.93423
4.42703
5.66868
DMSO
−0.69797
−2.01092
−5.81943
−6.40611
3.80851
4.39519
5.70814
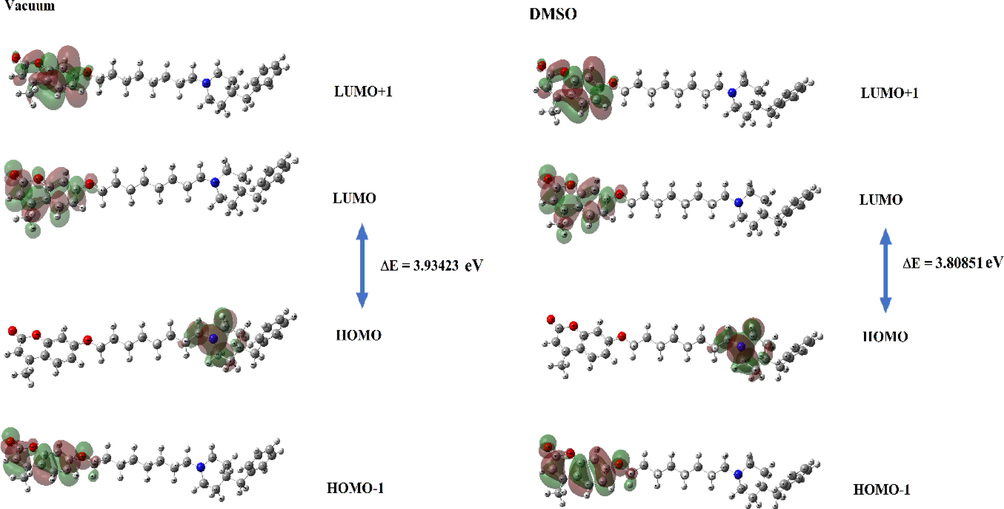
The molecular orbitals HOMO-1, HOMO, LUMO and LUMO + 1 of C3.
When the energy gaps between HOMO and LUMO were examined, it was seen that the vacuum medium has a larger ΔE gap with a value of 3.93423 eV compared to the DMSO environment. Information such as ionization potential, electron affinity, and chemical hardness calculated with the help of HOMO and LUMO values were obtained for the C3 molecule in DMSO and vacuum environments and were listed in Table 2. In many studies with coumarins, it was seen that parameters such as HOMO, LUMO, ionization potential, and chemical hardness were quite variable due to the structure (Ishihara et al., 2006, Al-Amiery et al., 2017).
Vacuum
TD-DFT/6311++G(d,p)
Energy (a.u.)
Energy (eV)
HOMO Energy
EHOMO
−0.21489
−5.84746
LUMO Energy
ELUMO
−0.07031
−1.91323
Ionization potential
I = -EHOMO
0.21489
5.84746
Electron affinity
A= - ELUMO
0.07031
1.91323
Electronegativity
χ= (I + A)/2
0.14260
3.88035
Chemical potential
µ = -(I + A)/2
−0.14260
−3.88035
Chemical hardness
η = (I-A)/2
0.07229
1.96711
Δ E(gap)
ELUMO- EHOMO
0.14458
3.93423
DMSO
TD-DFT/6311++G(d,p)
Energy (a.u.)
Energy (eV)
HOMO Energy
EHOMO
−0.21386
−5.81943
LUMO Energy
ELUMO
−0.07390
−2.01092
Ionization potential
I = -EHOMO
0.21386
5.81943
Electron affinity
A= - ELUMO
0.07390
2.01092
Electronegativity
χ= (I + A)/2
0.14388
3.91518
Chemical potential
µ = -(I + A)/2
−0.14388
−3.91518
Chemical hardness
η = (I-A)/2
0.06998
1.90425
Δ E(gap)
ELUMO- EHOMO
0.13996
3.80851
In studies with coumarins, it was observed that HOMO energy values were obtained in different values from −5 eV to −10 eV (∼-5.8 eV that we obtained), and accordingly ionization potential, chemical hardness, and electronegativity values changed. For example, in one study in which the ionization potential was calculated, a 5.116 eV value was obtained (Al-Amiery et al., 2017), while in another study it was obtained ∼ 9 eV (Ishihara et al., 2006). In this study, it was determined as ∼ 5.85 eV in a vacuum environment and ∼ 5.82 eV in a DMSO environment. When we compare the chemical hardness values, it was determined that different values from 1 eV to 4 eV were reported in the literature (Ishihara et al., 2006, Al-Amiery et al., 2017) and our values were within the range of these literature values.
In the theoretical study carried out by Preat et al. in 2006, it was reported that the first absorption band of coumarin was at 330 nm, the second absorption band contained an excitation from HOMO-1 to LUMO and was observed in the 300 nm region (Preat et al., 2006). In this study, according to obtained theoretical and experimental absorption spectra (see Fig. 9 and Fig. S4), the maximum absorption wavelengths were calculated as 341.91 nm and 330.55 nm for DMSO and vacuum environments, respectively. It was seen that these wavelengths correspond to the electronic transition from the HOMO to LUMO with 100 % contribution. The second absorption wavelengths were calculated at 312.05 nm and 302.95 nm for DMSO and vacuum environments, it has been observed that the contribution to this absorption comes from an excitation from HOMO-1 to LUMO. The coumarin derivative, which has a peak at 311 nm in the literature (Preat et al., 2005, Khemakhem et al., 2018), synthesized in this study was observed at 328 nm in the experimental UV spectrum (see Fig. 9). All these obtained excitation and absorption values were found to be compatible with the literature. The obtained all wavelength, excitation energies, oscillator strength (f) and major contributions were listed in Table 3.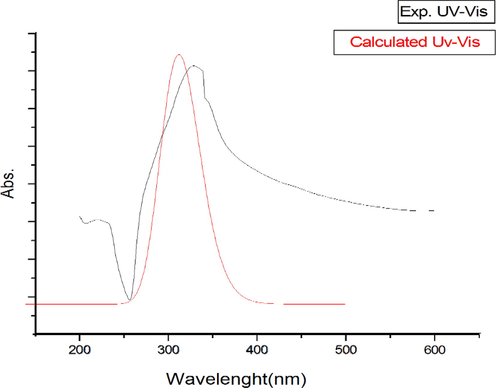
The experimental and calculated UV–vis spectra of C3 in DMSO.
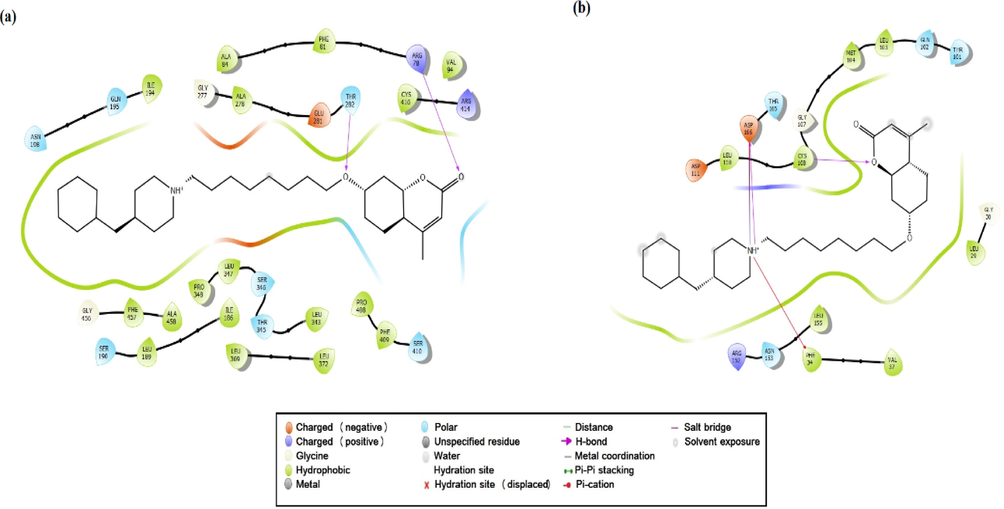
The 2D ligand C3-receptors interactions diagrams, CYP450 (a), EGFR (b).
TD-B3LYP/6–311++G(d,p)
E (eV)
λ (nm)
f major contribs symmetry.
DMSO
3.6262
341.91
0.0
H → L
(100 %)
Singlet-A
3.9732
312.05
0.5103
H-1 → L
(97 %)
Singlet-A
4.3787
283.15
0.0042
H-4 → L
(88 %)
Singlet-A
H-1 → L + 1
(11 %)
Vacuum
3.7508
330.55
0.0
H → L
(100 %)
Singlet-A
4.0926
302.95
0.4067
H-1 → L
(93 %)
Singlet-A
4.4022
281.64
0.0117
H-3 → L
(84 %)
Singlet-A
H-1 → L + 1
(13 %)
DMSO
E (eV)
λ (nm)
Abs.
DMSO
3.7777
328.20
0.828
5.6459
219.60
0.011
3.6.2 Molecular docking (CYP450, EGFR and DNA) and ADME analysis results
As a result of the possible binding poses of the synthesized C3 molecule with CYP450, it can be said that only 16 possible stereoisomers of the 32 performed CYP450-appropriate coupling pose in Table S4. The possible binding poses and possible interactions with the lowest energy docking score of the C3 molecule located in the active binding site of CYP450 were given in Fig. 10a and Table 4. The active site of CYP450 structure with S-warfarin were given by ARG97, GLY98, ILE99, PHE100, LEU102, ALA103, VAL113, PHE114, ASN217, THR364, SER365, LEU366, PRO367 and PHE476 residues in (Costa et al., 2018). In this study, many similar residues such as hydrophobic residues (ARG97, PHE100, ALA103, VAL113, LEU 366, PRO367 and PHE476) and polar residues (ASN217, THR364 and SER365) were in the site where C3 binds and had some special interactions with C3 in Fig. 10a and Table 4. The interactions between C3 and the active binding sites of CYP450 and EGFR were given in Fig. 11. The positive charged ARG78(97) and polar THR282(301) residues made the hydrogen bonding interactions (2.02 Å and 2.66 Å) with O atoms of the C3 molecule, respectively in Fig. 10a, and 11a. The binding affinity values were given −7.8 kcal/mol and −7.5 kcal/mol for the tested two molecules (Costa et al., 2018). The binding affinity of the synthesized C3 molecule was calculated as −7.82 kcal/mol and shown in Fig. S5., similar to the binding affinity values of these two tested molecules. Based on the similarity of the binding affinity values and the binding site interactions, it can be said that the C3 molecule also tends a good antioxidant ability.
Receptors
Docking Score
(Kcal/mol)
H.Bonding interaction (Angstrom)
Salt Bridge interaction
Pi-cation interaction
Hydrophobic Residues
Polar Residues
Charged (positive) Residues
Charged (negative) Residues
Glycine
PDB ID: 1OG5
−7.28
ARG78(97)
(2.02)THR282
(3 0 1)
(2.66)–
–
PHE81(1 0 0), ALA84(1 0 3),VAL94
(1 1 3),ILE186
(2 0 5),LEU189
(2 0 8),ILE194
(2 1 3),ALA278
(2 9 7)LEU343
(3 6 2),
LEU347(3 6 6),PRO348
(3 6 7)LEU369
(3 8 8),LEU372
(3 9 1),PRO408
(4 2 7)PHE409
(4 2 8)CYS416
(4 3 5)PHE457
(4 7 6)ALA458
(4 7 7)SER190(2 0 9),GLN195
(2 1 4),ASN198
(2 1 7),THR282
(3 0 1),THR345
(3 6 4),SER346
(3 6 5),SER410
(4 2 9)ARG78(97),ARG414
(4 3 3)GLU281(3 0 0)
GLY277
(2 9 6)
GLY456
(4 7 5)
PDB ID:
1 M17
−5.24
ASP166(8 3 1)
(1.80)CYS108
(7 7 3)
(2.05)ASP166
(8 3 1)
(2.79)PHE34(6 9 9)
LEU29(6 9 4),PHE34
(6 9 9),VAL37
(7 0 2),ALA54
(7 1 9),
LEU103(7 6 8)MET104
(7 6 9),CYS108
(7 7 3),LEU110
(7 7 5),LEU155
(8 2 0)THR101(7 6 6),GLN102
(7 6 7),THR165
(8 3 0),ASN153
(8 1 8)LYS56(7 2 1),ARG152
(8 1 7)ASP111(7 7 6),ASP166
(8 3 1)GLY30(6 9 5)GLY107
(7 7 2)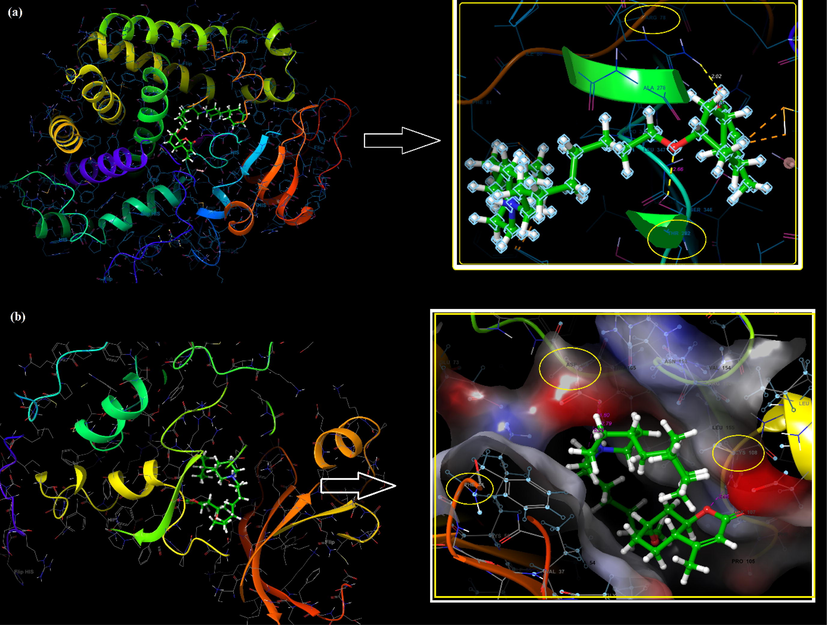
The active binding sites of receptors CYP450 (a), EGFR (b), and the interactions with C3.
Based on the similarity of the binding affinity values and the binding site interactions, it can be said that the C3 molecule also tends a good antioxidant ability. The C3 molecule was made to dock into the active site of the EGFR enzyme, and the docking score was found to be −5.24 kcal/mol (see Fig. 10b). Molecular docking studies revealed that the binding site of C3 is alike the binding site of the Pyrazofurin and antifol molecules to EGFR. The C3 molecule indicated hydrogen bonding and salt bridge interactions with the negatively charged ASP166(831) residue with 1.80 Å and 2.79 Å, respectively. The protonated NH+ part of C3 had an electrostatic interaction with negatively charged ASP shown in Fig. 10b, 11b and Table 4. The same part in C3 made pi-cation interaction with PHE34(699) residue. Another hydrogen bonding interaction occurred between the O atom in C3 and the hydrophobic CYS108(773) residue with 2.05 Å was seen in Fig. 10b. The interacted residue ASP166(831) was also a residue that Pyrazofurin interacts within (Arthur et al., 2018). Also, the pi-cation interaction with PHE34(6 9 9) contributed to the high affinity of the molecule in the binding site by stabilizing its structure like an antifol in (Arthur et al., 2018).
According to the result of molecular docking analysis, it can be seen that the adenines and guanines belonging to DNA and the C3 molecule have interacted with each other. The DG16 (deoxyguanosines), DA17, DA18, DA5 and DA6 (deoxyadenosine), which have close interactions with C3, have been seen to be capable of hydrogen bonding with C3 (cf. FigS9 and Table S8). The hydrogen bonding interactions of C3 bounded by B-DNA dodecamer were given in TableS8, and the binding affinity energies and RMSD values of C3 bounded by B-DNA dodecamer were also given in TableS9. The best-docked pose, close interaction and hydrogen bonding interaction between C3 and the B-DNA was seen in Fig.S9.
The hydrogen bonds revealed that the O atoms of C3 and the hydrogen atom bound to the DA5, DA6, DG16, DA17 and DA18 come to the fore. In this study, it was seen that the O55 atom of the C3 molecule made hydrogen bonds with H7 (2.6 Å) and H61 (2.3 Å). The O74 atom of C3 molecule made hydrogen bond with DG16 and H7 (3.1 Å) and H61 (1.8 Å) and H7 (2.2 Å) of DA17. Additionally, it was seen that the O61 atom of the C3 molecule made three hydrogen bonding with H61 (3.0 Å), H62 (2.9 Å) and H61 (3.0 Å) of DA17 and DA18, respectively.
Molecules that bind to the groove, as well as molecules that bind to DNA by intercalation, have clinical importance as anticancer agents. It was reported that molecules bind to the groove regions rich in adenine and thymine on the DNA, however, molecules bind to the DNA by intercalation where guanine and cytosine are dense (Geierstanger and Wemmer 1995, Neidle 1997, Takenaka and Takagi 1999).
In this study, molecular docking analysis was performed by using B-DNA as a target and evaluating the minor and major grooves of DNA. According to the visual data of the docking pose to B-DNA, it was seen that C3 was attached to the B-DNA with a major groove. Adenine and guanine bases of DNA more interacted with the C3 molecule and this interaction was achieved through the hydrogen bonds that provide high stability and strong binding (−5.5 kcal/mol in Table S9) energy. The DNA-double helix has many hydrogens bonding sites accessible in the minor and major grooves. The electronegative atoms (O and N) of the C3 molecule are in a position to form hydrogen bonds with DNA.C3 molecule binded to the groove regions with adenine (DA17, DA18, DA5 and DA6), however, it was provided to bind to the DNA by intercalation with guanine (DG16).
Its UV–vis and viscosimetric results confirmed an intercalative mode of binding with high affinity for DNA as is clear from its binding constant of 1.16 × 105 M−1. The Gibbs energy change (ΔG = -RTlnK) of −28.88 kJ/mol at 25 °C indicates the spontaneity of the binding interaction.
ADME (absorption, distribution, metabolism, and excretion) analysis was performed by the QikProp tool of the same software program to define the pharmacological profile of the synthesized C3 molecule, which paves the way for predicting it as a drug candidate. The molecular structure properties of the docked C3 molecule and the calculated ADME properties as a result of docking with preferred receptors are given in Table 5.
Receptors
PDB ID: 1OG5
PDB ID: 1 M17
Docking Score(Kcal/mol)
−7.28
−5.24
Principal Descriptors:
(Range 95 % of Drugs)
Solute Molecular Weight
473.738
473.738
(130.0 / 725.0)
Solute Total SASA
962.207
766.930
(300.0 /1000.0)
Solute Hydrophobic SASA
839.912
681.968
(0.0 / 750.0)
Solute Hydrophilic SASA
88.316
57.656
(7.0 / 330.0)
Solute Carbon Pi SASA
33.978
27.306
(0.0 / 450.0)
Solute Weakly Polar SASA
0.000
0.000
(0.0 / 175.0)
Solute Molecular Volume (A^3)
1755.176
1570.473
(500.0 /2000.0)
Solute vdW Polar SA (PSA)
53.084
52.367
(7.0 / 200.0)
Solute No, of Rotatable Bonds
12.000
12.000
(0.0 / 15.0)
Solute as Donor - Hydrogen Bonds
0.000
0.000
(0.0 / 6.0)
Solute as Acceptor - Hydrogen Bonds
6.700
6.700
(2.0 / 20.0)
Solute Globularity (Sphere = 1)
0.731
0.852
(0.75 / 0.95)
Predictions for Properties:
QP Polarizability (Angstroms^3)
56.707 M
49.252 M
(13.0 / 70.0)
QP log P for hexadecane/gas
15.974 M
13.172 M
(4.0 / 18.0)
QP log P for octanol/gas
21.694 M
20.331 M
(8.0 / 35.0)
QP log P for water/gas
6.819 M
6.095 M
(4.0 / 45.0)
QP log P for octanol/water
6.385
5.384
(−2.0 / 6.5)
QP log S for aqueous solubility
−7.770
−4.069
(−6.5 / 0.5)
P log S - conformation independent
−5.195
−5.195
(−6.5 / 0.5)
QP log K hsa Serum Protein Binding
1.412
0.875
(−1.5 / 1.5)
QP log BB for brain/blood
−0.862
−0.291
(−3.0 / 1.2)
No, of Primary Metabolites
3
3
(1.0 / 8.0)
Predicted CNS Activity (-- to ++)
+/-
+
HERG K + Channel Blockage: log IC50
−6.768
−4.783
(concern below −5)
Apparent Caco-2 Permeability (nm/sec)
359
701
(<25 poor, greater than500 great)
Apparent MDCK Permeability (nm/sec)
180 M
373 M
(<25 poor, greater than500 great)
QP log Kp for skin permeability
−3.938
−3.396
(Kp in cm/hr)
Jm, max transdermal transport rate
0.000
0.016
(micrograms/cm^2-hr)
Lipinski Rule of 5 Violations
1
1
(maximum is 4)
Jorgensen Rule of 3 Violations
1
0
(maximum is 3)
% Human Oral Absorption in GI (+-20 %)
100
96
(<25 % is poor)
Qual, Model for Human Oral Absorption
LOW
HIGH
(greater than80 % is high)
Log P is widely used as a measure of the chemical's hydrophobicity or lipophilicity, defined as the ratio of the concentration of a chemical in the n-octanol phase to its concentration in the aqueous phase of a biphasic system. The log P for octanol/water was given by 6.385 and 5.384 values in the range of recommended 95 % of drug molecules for CYP450 and EGFR receptor, respectively. This value has also a very important indicator in describing many properties of the chemical such as drug action, cellular uptake, metabolism, bioavailability, and toxicity (Hou and Xu 2003). There is a relation between brain-blood concentration ratio (BB) and logP.
The calculated log BB for brain/blood was −0.862 for CYP450, and −0.291 for EGFR receptor and found in the drug range. The total surface area of a molecule affects its solubility and molecular interactions due to its surface contact with other molecules. By making similar analyzes for molar volume (MV), the interaction level of molecules with larger TSA and MV can be higher. Molecular volumes calculated for C3 were 1755.176 and 1570.473 for CYP450, and EGFR receptors, respectively. The total surface area and molecular volume of interaction with CYP450 for C3 are greater than for the EGFR receptor. In other words, it can be said that the C3 molecule interacts more with CYP 450 than the EGFR receptor. The total surface area of the C3 molecule (SASA) which is accessible to a solvent, its hydrophobic component and the hydrophilic component of the SASA values also were found as 962.207, 839.912 and 88.316 for CYP450, 766.930, 681.968 and 57.656 for EGFR, respectively. For 95 % of drug molecules, the HSA Serum Protein Binding values are given from the −1.5 to 1.5 range. The prediction of binding to human serum albumin values was calculated at 1.412 and 0.875 for CYP450 and EGFR. The Caco-2 Permeability and MDCK Permeability (nm/sec) values of C3 were obtained at normal and great levels. It is possible to predict that the C3 molecule, which cannot comply with only 1 of the Lipinski 5 violation rules and has a high percentage of human oral absorption, can also be used in oral applications, and has the ability to be a drug. The probability of a molecule being an oral drug in terms of its bioavailability can be explained by demonstrating drug-likeness properties. For this reason, drug-likeness properties of the C3 molecule were evaluated by Molinspiration (Ertl et al., 2000) and SwissADME (Daina et al., 2017) web tools. Swiss ADME tool; applies filtering of chemical libraries to distinguish molecules with an acceptable pharmacokinetic profile from those without. In particular, the Lipinski (Pfizer) filter is used to characterize the drug similarity of a molecule. According to the Lipinski 5 s rule; low absorption or permeability is more likely if more than 5H-bond donors, 10H-bond acceptors, molecular weight (MWT) greater than 500, and LogP (CLogP) greater than 5 (or MlogP greater than 4.15) has been shown (Lipinski et al., 1997). The drug-likeness properties using Molinspiration and SwissADME tool for C3 were given in Table S5.
According to the results, the C3 molecule had 4H-bond acceptors and no H-bond donor, and its molecular weight was given 473.73 g/mol, but its calculated LogPO/W(MLOGP) value is given 4.97. This value was calculated as a little greater than 4.15 value. Although the calculated MlogP value was slightly higher than 4.15, the Molinspiration and SwissADME tools showed that the synthesized C3 confirmed Lipinski and that it had drug-likeness properties. The Bioavailability Score also estimates the probability that a compound may have at least 10 % oral bioavailability or measurable Caco-2 permeability, which contributes to its drug-likeness property(Daina et al., 2017). The bioavailability score of C3 was calculated at 0.55 in Table S5.The toxicity of the C3 molecule was also investigated using the PreADMET tool (Lee et al., 2003) and given in Table S6. The results indicated that the synthesized C3 molecule was non-mutagenic as a result of the Ames test, and the values of the carsino mouse and carsino rat were also negative. This means that C3 may not cause mutagenic and carcinogenic effects. In other words, the C3 structure is not reactive to cause any DNA damage or mutation in cells. The pkCSM web another web server (Pires et al., 2015) which has outperformed well-established tools and the AMES test result has been shown to have an accuracy of 83.8 %, was also performed to reevaluate the toxicity of C3. The calculated toxicity by pkCSM was also given in Table S7. According to the results, there was no AMES toxicity for C3, too.
3.6.3 MD analysis results
MD simulations for 20 ns were obtained using the Desmond package of the Schrodinger program for the C3 molecule docked to the potential binding site of the CyP450 enzyme (PDB: 1OG5). The MD system, which was generated by adding 13,740 water molecules, 42 CI- and 38Na+ ions, stabilized at 300 K temperature and 1 atm pressure, was shown in Fig. S6.
Monitoring the RMSD of the protein can provide insight into its structural harmony throughout the simulation and show whether the simulation is stabilizing. The changes at the 1–3 Å level are perfectly acceptable for small, globular proteins. The ligand RMSD also shows how stable the ligand is relative to the binding pocket of the protein. This RMSD value also shows the internal fluctuations of the ligand atoms. The Root Mean Square Deviation (RMSD) for Cα (blue line) and side chains (green line) of protein together with C3 molecule ligand (purple line) were given in Fig. 12.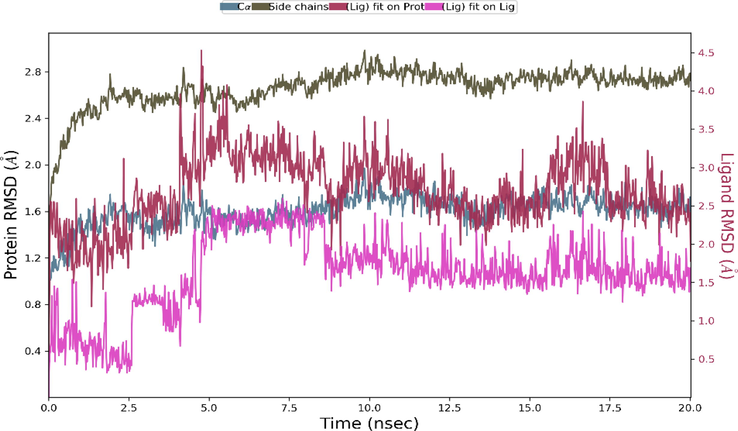
The Root Mean Square Deviation (RMSD)of the system for 20 ns.
Changes in the protein RMSD value between 1 and 3 Å are within the acceptable range. The RMSD of the protein remained stable throughout the 20 ns simulation without significant deviations in its structural conformation. The ligand RMSD remained stable during the 20 ns simulation, varying at most between 5 ns and 10 ns. Root Mean Square Fluctuation (RMSF) refers to local changes along the protein chain. The most fluctuating protein residues during the simulation are indicated by the RMFS peaks. Residues of protein interacting with the ligand are indicated by vertical lines in green and also RMSF of protein by experimental X-ray B factor (right Y-axis) are given with red lines in Fig. 13a. Ligand RMSF also shows ligand fluctuations occurring in the atoms of the ligand corresponding to the 2D structure. Ligand RMSF also provides information about how ligand fragments interact with the protein and thus its role in binding. Ligand RMSF was shown in Fig. S7. The most fluctuating atoms in the ligand are Oxygen atoms with 32 and 31 numbers and C atoms with 26 and 6 number appeared. In interactions with protein, the 6th C atom and the 31st and 32nd Oxygen atoms play an active role. By monitoring the ligand and protein interactions throughout the simulation, it can be seen that the interactions can be categorized into four types Hydrogen Bonds (green column), Hydrophobic (lilac column), Ionic (pink column) and Water Bridges (blue column) and that some protein residues can also make multiple contacts with the ligand. The interaction fraction between ligand and protein was given in Fig. 13c and Fig. 14a. As can be seen from the figures (Fig. 13b and 13c, and Fig. 14a), the hydrogen bonding occurred between ARG78(97) and the Oxygen atom defined by the atomic number 32, and this interaction was also revealed in the docking analysis results with the 2.02 Å length hydrogen bond interaction (see Fig. 10a and Table 4).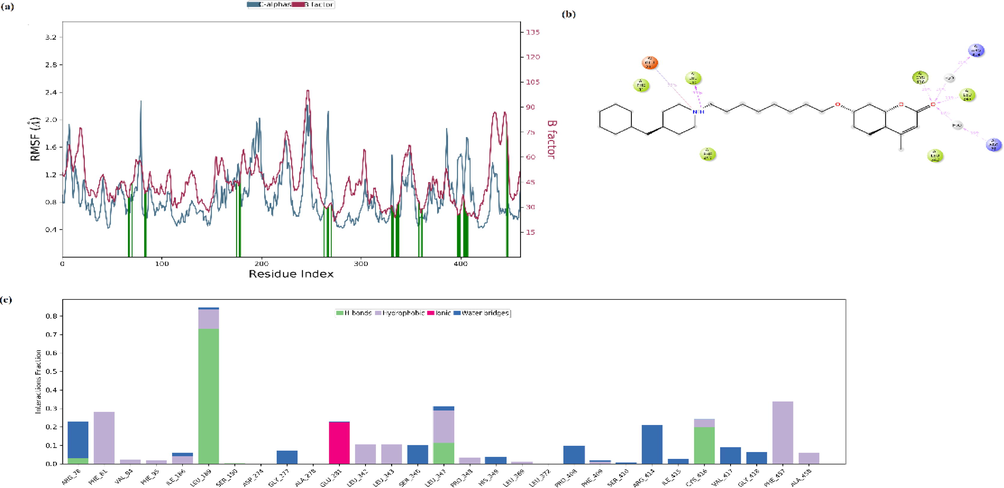
The Root Mean Square Fluctuation (RMSF) of protein (a), ligand interaction diagram (b), and protein–ligand contacts (c) in the MD system for 20 ns.
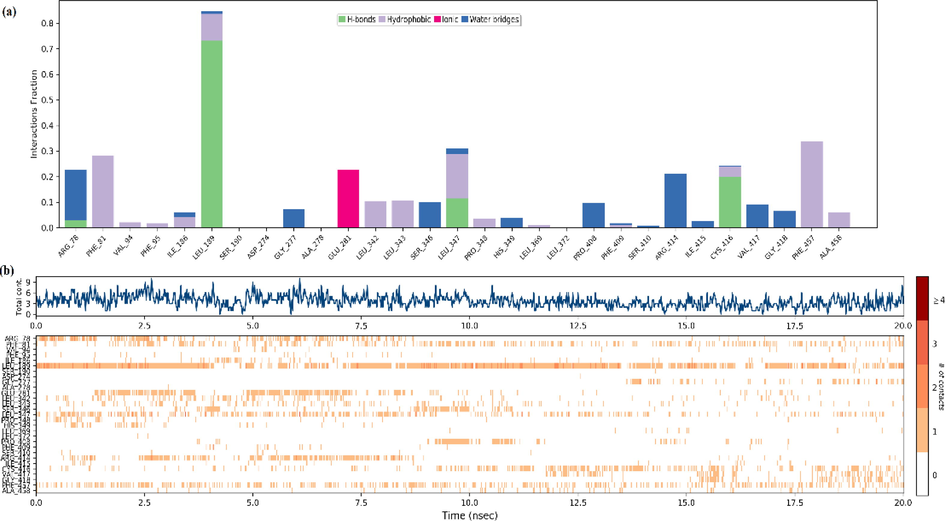
The interactions and contacts between protein and ligand (a), timeline representation of a total number of specific contacts(b) in the MD system for 20 ns.
In addition, hydrogen bonding interactions were performed with the residues LEU189, LEU347 and CYS416 and the C3 molecule during the simulation process. While the residues LEU347 and CYS416 interacted with the Oxygen atom with 32 numbers, LEU189 interacted with the Nitrogen atom with 12 numbers. An ionic interaction has occurred between GLU281 and the same Nitrogen atom. These residues (LEU 189(2 0 8), LEU 347(3 6 6) and CYS 416(4 3 5)) are also located in the binding region of the protein and are defined as hydrophobic residues in docking analysis. These residues also found hydrophobic interactions with the ligand see Fig. 13a, 13b, 13c. The schedule of interactions and contacts (H-bonds, Hydrophobic, Ionic, Water bridges) between ligand and protein over 20 ns was also shown in Fig. 14b. The upper panel shows the total number of specific contacts the protein makes with the ligand along the trajectory, while the lower panel indicates which residues interact with the ligand and for how long. As can be seen here, the residues that interacted most with the ligand within 20 ns are LEU189, GLU281, LEU 347, CYS 416 and PHE457. Ligand RMSD, Radius of Gyration, Intramolecular Hydrogen Bonds, Molecular Surface Area (MolSA), Solvent Accessible Surface Area (SASA) and Polar Surface Area (PSA) of C3 was also given in Fig. S8.
4 Conclusion
In this study, a coumarin hybrid compound (C3) was designed and synthesized as a new drug molecule with anticancer and antioxidant properties. Synthesis of C3 was accomplished by simple two-step reaction sequences, and the structure and purity of the compound were characterized by FT-IR, MS, 1H and 13C NMR. The interaction studies of C3 with DNA were revealed by the UV–vis absorption titration method, viscosimetric method and fluorescence emission spectroscopy. The results indicated that the presence of noncovalent interactions between C3 and DNA. The UV–vis and viscosimetric results confirmed an intercalative mode of binding with the Kb binding constant of 1.16 × 105 M−1. The Gibbs energy change (ΔG = -RTlnK) of −28.88 kJ/mol remarks the spontaneity of the binding interaction. The negative values of Gibbs free energy change (ΔG) is also indicative of spontaneous C3-DNA binding interaction. The result of fluorescent emission spectroscopy showed that C3 could not compete with EB for DNA binding sites. Studies in the literature reporting that coumarin compounds cannot compete with EB for DNA binding sites have reported that the interactions of compounds with DNA may be in the form of external binding. Our study determined that different interactions were observed in the analysis results performed with UV–vis spectroscopy and in the analysis results performed with fluorescence spectroscopy. As a results of viscosity experiment C3 compound has a lower interaction with DNA than EB. These results support the idea that C3 cannot be replaced by EB.
DNA interaction studies showed that C3 is a good anticancer drug candidate. In addition, the antioxidant activity of C3 was evaluated experimentally and theoretically. Experimental results showed that C3 has radical scavenging properties.
Molecular docking analysis was performed by using B-DNA as a target to evaluate the minor and major groove binding of DNA. According to the results, it was seen that C3 was attached to the B-DNA with a major groove with hydrogen bondings. Adenine and guanine bases of DNA more interacted with the C3 molecule and this interaction was achieved through the hydrogen bonds that provide high stability and strong binding energy. Our results showed that C3 can exhibit both forms of binding.
As a result of molecular docking analysis, the possible binding poses and possible interactions with the lowest energy docking score of C3 (-7.82 kcal/mol) in the active binding site of CYP450 were clarified. C3 interacted with similar residues such as hydrophobic residues (ARG97, PHE100, ALA103, VAL113, LEU 366, PRO367 and PHE476) and polar residues (ASN217, THR364 and SER365), which are also found in the S-warfarin binding site. The binding of positively charged residues ARG78(97) and polar THR282(3 0 1) with the O atoms of C3 by hydrogen bonding interaction (2.02 Å and 2.66 Å) gave it good binding affinity. Based on the similarity of the binding affinity values and the binding site interactions, it can be said that the C3 molecule also tends a good antioxidant ability. Computational molecular docking studies on CYP450 endorsed the experimental antioxidant activity of C3 which was evaluated by the DPPH radical scavenging assay results.
In addition, based on the ADMET analysis results, it is possible to predict that the C3 molecule, which has a high percentage of human oral absorption, can also be used in oral applications, and has the ability to be a drug. According to the drug-likeness assessment, the C3 molecule obeys Lipinski rules and has drug-likeness properties. According to the toxicity evaluation, it was shown that the synthesized C3 molecule was not mutagenic as a result of the Ames test and the values of the carsino mouse and carsino rat were also negative. This means that C3 may not cause mutagenic and carcinogenic effects.
Acknowledgement
The authors are also very thankful to Anupriya Kumar for allowing using the docking program with Schrödinger’s Small-Molecule Drug Discovery Suite. This work was supported by Yildiz Technical University Scientific Research Foundation (project number FYL-2021-4077). In this study, the infrastructure of the Applied Nanotechnology and Antibody Production Laboratory established with TUBITAK support (project numbers: 115S132 and 117S097) was used. The authors would also like to thank TUBITAK for their support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Sonochemical synthesis, DNA binding, antimicrobial evaluation and in vitro anticancer activity of three new nano-sized Cu (II), Co (II) and Ni (II) chelates based on tri-dentate NOO imine ligands as precursors for metal oxides. J. Photochem. Photobiol. B Biol.. 2016;162:298-308.
- [Google Scholar]
- New Cd (II), Mn (II) and Ag (I) Schiff base complexes: synthesis, characterization, DNA binding and antimicrobial activity. IJNC.. 2016;2:83-91.
- [Google Scholar]
- Tailoring, physicochemical characterization, antibacterial and DNA binding mode studies of Cu (II) Schiff bases amino acid bioactive agents incorporating 5-bromo-2-hydroxybenzaldehyde. J. Iran. Chem. Soc.. 2015;12:943-955.
- [Google Scholar]
- Comparative molecular modelling studies of Coumarin derivatives as potential antioxidant agents. Free Radicals Antioxid.. 2017;7:31-35.
- [Google Scholar]
- Synthesis, antitumor activity evaluation, and DNA-binding study of coumarin-based agents. Arch. Pharm.. 2018;351:1700199.
- [Google Scholar]
- Competitive DNA-binding studies between metal complexes and GelRed as a new and safe fluorescent DNA dye. J. Fluoresc.. 2016;26:1505-1510.
- [Google Scholar]
- Arivazhagana, M., Sambathkumarb, K. and Jeyavijayanc, S. 2010. Density functional theory study of FTIR and FT-Raman spectra of 7-acetoxy-4-methyl coumarin.
- Novel coumarin–benzimidazole derivatives as antioxidants and safer anti-inflammatory agents. Acta Pharm. Sin. B. 2014;4:368-375.
- [Google Scholar]
- Structure-based optimization of tyrosine kinase inhibitors: a molecular docking study. Network Modeling Analysis in Health Informatics and Bioinformatics.. 2018;7:1-18.
- [Google Scholar]
- The effect of metal and substituent on DNA binding, cleavage activity, and cytotoxicity of new synthesized Schiff base ligands and Zn (II) complex. J. Mol. Struct.. 2017;1147:582-593.
- [Google Scholar]
- Exploring 3-hydroxyflavone scaffolds as mushroom tyrosinase inhibitors: synthesis, X-ray crystallography, antimicrobial, fluorescence behaviour, structure-activity relationship and molecular modelling studies. J. Biomol. Struct. Dyn.. 2021;39:7107-7122.
- [Google Scholar]
- Synthesis, spectroscopic characterization (FT-IR, FT-Raman, and NMR), quantum chemical studies and molecular docking of 3-(1-(phenylamino) ethylidene)-chroman-2, 4-dione. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2018;195:31-40.
- [Google Scholar]
- Scaled quantum chemical studies of the structural and vibrational spectra of acetyl coumarin. Open Chem.. 2006;4:773-785.
- [Google Scholar]
- Preparation, characterization, antioxidant properties of novel Schiff bases including 5-chloroisatin-thiocarbohydrazone. Res. Chem. Intermed.. 2020;46:2541-2557.
- [Google Scholar]
- Synthesis and in vivo evaluation of new coumarin conjugates as potential indirect-action anticoagulants. Pharm. Chem. J.. 2019;53:419-422.
- [Google Scholar]
- Synthesis, characterization and antioxidant activity studies of new coumarin tethered 1, 3, 4-oxadiazole analogues. J. Chem. Sci.. 2021;133:1-8.
- [Google Scholar]
- Synthesis and antioxidant properties of some new thiazolyl coumarin derivatives. Green Chem. Lett. Rev.. 2020;13:155-163.
- [Google Scholar]
- Bienert, S., Waterhouse, A., de Beer, T. A. et al., 2017. The SWISS-MODEL Repository—new features and functionality. Nucleic acids research. 45, D313-D319.
- Schiff base and metal (II) complexes containing thiophene-3-carboxylate: Synthesis, characterization and antioxidant activities. J. Mol. Struct.. 2020;1205:127542
- [Google Scholar]
- Studies on the effect of remote substituents on the DNA binding activity of novel chiral Schiff bases. J. Mol. Struct.. 2021;1234:130179
- [Google Scholar]
- Synthesis and Biological Properties of Coumarin Derivatives. A Review. ChemistrySelect.. 2021;6:5848-5870.
- [Google Scholar]
- Bowers, K. J., Chow, D. E., Xu, H. et al., 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. SC'06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, IEEE.
- Synthesis, antioxidant and antimicrobial activity of carbohydrazones. J. Serb. Chem. Soc.. 2017;82:495-508.
- [Google Scholar]
- Use of a free radical method to evaluate antioxidant activity. LWT-Food science and Technology.. 1995;28:25-30.
- [Google Scholar]
- New nanodrug design for cancer therapy: Its synthesis, formulation, in vitro and in silico evaluations. Arch. Pharm.. 2020;353:2000137.
- [Google Scholar]
- Antioxidant activity of Nigella sativa essential oil. Phytother. Res.. 2000;14:323-328.
- [Google Scholar]
- Autoxidation of biological molecules. 4. Maximizing the antioxidant activity of phenols. J. Am. Chem. Soc.. 1985;107:7053-7065.
- [Google Scholar]
- Carneiro Viqueira, A., Matos, M. J. C. P. C. d., Uriarte Villares, E. et al., 2021. Trending Topics on Coumarin and Its Derivatives in 2020.
- Theoretical and experimental investigation of 4-[(2-hydroxy-3-methylbenzylidene) amino] benzenesulfonamide: Structural and spectroscopic properties, NBO, NLO and NPA analysis. J. Mol. Struct.. 2015;1089:222-232.
- [Google Scholar]
- Design, spectroscopy, quantum chemical study and Hirshfeld analysis of single crystal ferrocene-based boronate ester. J. Mol. Struct.. 2021;1243:130767
- [Google Scholar]
- FT-IR, FT-Raman spectra and scaled quantum mechanical study of 4-amino-1-benzylpiperidine. J. Mol. Struct.. 2011;998:69-78.
- [Google Scholar]
- Synthesis of Schiff bases modified inulin derivatives for potential antifungal and antioxidant applications. Int. J. Biol. Macromol.. 2020;143:714-723.
- [Google Scholar]
- Synthesis, characterization, and DNA binding of complexes [Pt (bpy)(pip)] 2+ and [Pt (bpy)(hpip)] 2+. J. Biol. Inorg. Chem.. 2013;18:461-471.
- [Google Scholar]
- An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules.. 2018;23:2801.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:1-13.
- [Google Scholar]
- Recent developments of C-4 substituted coumarin derivatives as anticancer agents. Eur. J. Med. Chem.. 2016;119:141-168.
- [Google Scholar]
- Highly functionalized piperidines: Free radical scavenging, anticancer activity, DNA interaction and correlation with biological activity. J. Adv. Res.. 2018;9:51-61.
- [Google Scholar]
- DNA and HSA interaction of Vanadium (IV), Copper (II), and Zinc (II) complexes derived from an asymmetric bidentate Schiff-base ligand: multi spectroscopic, viscosity measurements, molecular docking, and ONIOM studies. J. Biol. Inorg. Chem.. 2018;23:181-192.
- [Google Scholar]
- DNA and HSA interaction of Vanadium (IV), Copper (II), and Zinc (II) complexes derived from an asymmetric bidentate Schiff-base ligand: multi spectroscopic, viscosity measurements, molecular docking, and ONIOM studies. J. Biol. Inorg. Chem.. 2018;23:181-192.
- [Google Scholar]
- Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr.. 2002;40:82-92.
- [Google Scholar]
- Spectrophotometric analysis of nucleic acids: oxygenation-dependant hyperchromism of DNA. Anal. Bioanal. Chem.. 2010;396:2331-2339.
- [Google Scholar]
- Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Natl. Acad. Sci.. 1981;78:2179-2183.
- [Google Scholar]
- Anticancer Activity and DNA Binding Studies of Novel 3, 7-Disubstituted Benzopyrones. ChemistrySelect. 2017;2:147-153.
- [Google Scholar]
- Synthesis, characterization, and in vitro antioxidant and anticancer studies of ruthenium (III) complexes of symmetric and asymmetric tetradentate Schiff bases. J. Coord. Chem.. 2015;68:2552-2564.
- [Google Scholar]
- Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem.. 2000;43:3714-3717.
- [Google Scholar]
- Structural comparison of anticancer drug-DNA complexes: adriamycin and daunomycin. Biochemistry. 1990;29:2538-2549.
- [Google Scholar]
- Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem.. 2004;47:1739-1749.
- [Google Scholar]
- Frisch, M. and Clemente, F. 2009. Gaussian 09, Revision A. 01, MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, MA Robb, JR Cheeseman, G. Scalmani, V. Barone, B. Mennucci, GA Petersson, H. Nakatsuji, M. Caricato, X. Li, HP Hratchian, AF Izmaylov, J. Bloino, G. Zhe.
- DNA interactions of a functionalized ruthenium (II) mixed-polypyridyl complex [Ru (bpy) 2 ppd] 2+. J. Inorg. Biochem.. 2006;100:1487-1494.
- [Google Scholar]
- Complexes of the minor groove of DNA. Annu. Rev. Biophys. Biomol. Struct.. 1995;24:463-493.
- [Google Scholar]
- Synthesis of 4-N-α-coumaryl amino acids and investigation of their antioxidant, antimicrobial activities and fluorescence spectra. Amino Acids. 2018;50:1461-1470.
- [Google Scholar]
- Recent advancement of piperidine moiety in treatment of cancer-A review. Eur. J. Med. Chem.. 2018;157:480-502.
- [Google Scholar]
- Spectroscopic properties, NLO, HOMO–LUMO and NBO analysis of 2, 5-Lutidine. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2012;96:421-435.
- [Google Scholar]
- Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem.. 2004;47:1750-1759.
- [Google Scholar]
- Harmonic frequency scaling factors for Hartree-Fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set. Theor. Chem. Acc.. 2001;105:413-421.
- [Google Scholar]
- OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput.. 2016;12:281-296.
- [Google Scholar]
- Therapeutic potential of coumarins as antiviral agents. Eur. J. Med. Chem.. 2016;123:236-255.
- [Google Scholar]
- New organosulfur metallic compounds as potent drugs: synthesis, molecular modeling, spectral, antimicrobial, drug likeness and DFT analysis. Mol. Divers.. 2022;26:51-72.
- [Google Scholar]
- ADME evaluation in drug discovery. 2. Prediction of partition coefficient by atom-additive approach based on atom-weighted solvent accessible surface areas. J. Chem. Inf. Comput. Sci.. 2003;43:1058-1067.
- [Google Scholar]
- Coumarin linked heterocyclic hybrids: A promising approach to develop multi target drugs for Alzheimer's disease. J. Mol. Struct.. 2021;130618
- [Google Scholar]
- Benchmarking the reliability of QikProp. Correlation between experimental and predicted values. QSAR Comb. Sci.. 2008;27:445-456.
- [Google Scholar]
- Quantitative structure-cytotoxicity relationship analysis of coumarin and its derivatives by semiempirical molecular orbital method. Anticancer Res. 2006;26:2883-2886.
- [Google Scholar]
- Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR), HOMO-LUMO, chemical reactivity, AIM, ELF, LOL and Molecular docking studies on 1-Benzyl-4-(N-Boc-amino) piperidine. J. Mol. Struct.. 2021;1230:129657
- [Google Scholar]
- Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer's disease. Eur. J. Med. Chem.. 2018;146:287-298.
- [Google Scholar]
- Synthesis, characterization, anticancer evaluation and mechanisms of cytotoxic activity of novel 3-hydroxy-3-pyrrolin-2-ones bearing thenoyl fragment: DNA, BSA interactions and molecular docking study. Bioorg. Chem.. 2019;88:102954
- [Google Scholar]
- Computer-aided anticancer drug design: In vitro and in silico studies of new iminocoumarin derivative. J. Mol. Struct.. 2021;1239:130539
- [Google Scholar]
- Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem.. 2015;100:257-269.
- [Google Scholar]
- Hydrolysis and substitution effects on the optical properties of coumarin derivatives studied by vibrational spectroscopy and DFT calculation. J. Mol. Struct.. 2018;1168:65-72.
- [Google Scholar]
- Synthesis and computational studies on a coumarin derivative: 4-chloromethyl coumarin-7-yl-methacrylate. J. Mol. Struct.. 2022;132702
- [Google Scholar]
- Synthesis, spectral characterization, DNA binding, catalytic and in vitro cytotoxicity of some metal complexes. J. Mol. Liq.. 2021;326:115381
- [Google Scholar]
- Design, multistep synthesis and in-vitro antimicrobial and antioxidant screening of coumarin clubbed chalcone hybrids through molecular hybridization approach. Arab. J. Chem.. 2021;14:103154
- [Google Scholar]
- Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1, 2, 3-triazole–coumarin hybrids. Eur. J. Med. Chem.. 2016;124:794-808.
- [Google Scholar]
- DNA binding and antimicrobial studies of polymer–copper (II) complexes containing 1, 10-phenanthroline and L-phenylalanine ligands. Eur. J. Med. Chem.. 2009;44:1878-1883.
- [Google Scholar]
- Synthesis and antioxidant activity of thymol and carvacrol based Schiff bases. Bioorg. Med. Chem. Lett.. 2013;23:641-645.
- [Google Scholar]
- Synthesis, crystal structure, DNA binding and cleavage studies of copper (II) complexes with isoxazole Schiff bases. Polyhedron. 2015;102:111-120.
- [Google Scholar]
- Synthesis and in-vitro antioxidant activities of some coumarin derivatives containing 1, 2, 3-triazole ring. Phosphorus Sulfur Silicon Relat. Elem.. 2018;193:294-299.
- [Google Scholar]
- Studies on the antioxidative activities of Hsian-tsao (Mesona procumbens Hemsl) leaf gum. J. Agric. Food Chem.. 2001;49:963-968.
- [Google Scholar]
- Lee, S., Lee, I., Kim, H. et al., 2003. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR 2002 Designing Drugs and Crop Protectants: processes, problems and solutions. 2003, 418-420.
- Automatic algorithms for completeness-optimization of G aussian basis sets. Wiley Online Library; 2015.
- Pharmacological perspectives and molecular mechanisms of coumarin derivatives against virus disease. Genes & Diseases. 2021
- [Google Scholar]
- Synthesis and antioxidant activity of conjugates of hydroxytyrosol and coumarin. Bioorg. Chem.. 2020;105:104427
- [Google Scholar]
- Diorganotin (IV) antitumor agent. (C2H5) 2SnCl2 (phen)/nucleotides aqueous and solid-state coordination chemistry and its DNA binding studies. J. Inorg. Biochem.. 1996;64:181-195.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23:3-25.
- [Google Scholar]
- Cellular uptake, cytotoxicity, apoptosis, antioxidant activity and DNA binding of polypyridyl ruthenium (II) complexes. J. Organomet. Chem.. 2011;696:2728-2735.
- [Google Scholar]
- Effect of substituents on DNA-binding behaviors of ruthenium (II) complexes:[Ru (dmb) 2 (dtmi)] 2+ and [Ru (dmb) 2 (dtni)] 2+. J. Coord. Chem.. 2009;62:1701-1708.
- [Google Scholar]
- Synthesis, crystal structure, antioxidation and DNA binding properties of binuclear Ho (III) complexes of Schiff-base ligands derived from 8-hydroxyquinoline-2-carboxyaldehyde and four aroylhydrazines. J. Organomet. Chem.. 2009;694:3091-3101.
- [Google Scholar]
- Interaction of macrocyclic copper (II) complexes with calf thymus DNA: effects of the side chains of the ligands on the DNA-binding behaviors. Dalton Trans. 2003:114-119.
- [Google Scholar]
- Cytotoxicity and antioxidant activity of Kamolonol acetate from Ferula pseudalliacea, and studying its interactions with calf thymus DNA (ct-DNA) and human serum albumin (HSA) by spectroscopic and molecular docking techniques. Process Biochem.. 2019;79:203-213.
- [Google Scholar]
- Molecular structure, vibrational spectra (FTIR and FT Raman) and natural bond orbital analysis of 4-Aminomethylpiperidine: DFT study. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2014;131:587-598.
- [Google Scholar]
- Synthesis, DNA binding and antibacterial activity of metal (II) complexes of a benzimidazole Schiff base. Polyhedron. 2019;157:326-334.
- [Google Scholar]
- Investigation of DNA binding and bioactivities of thioether containing Schiff base Copper (II), Cobalt (II) and Palladium (II) complexes: Synthesis, characterization, spectrochemical study, viscosity measurement. Polyhedron. 2020;184:114559
- [Google Scholar]
- Experimental insights on the coordination modes of coumarin-3-carboxilic acid towards Cr (III)-, Co (II)-, Ni (II)-, Cu (II)-and Zn (II): A detailed potentiometric and spectroscopic investigation in aqueous media. J. Mol. Liq.. 2022;346:118302
- [Google Scholar]
- Microwave irradiated one pot, three component synthesis of a new series of hybrid coumarin based thiazoles: Antibacterial evaluation and molecular docking studies. J. Mol. Struct.. 2021;1225:129114
- [Google Scholar]
- Synthesis of new 8-formyl-4-methyl-7-hydroxy coumarin derivatives. J. Korean Chem. Soc.. 2012;56:459-463.
- [Google Scholar]
- GAR2PED, a program to obtain a potential energy distribution from a Gaussian archive record. Belgium: University of Antwerp; 2007.
- Coumarin heterocyclic derivatives: chemical synthesis and biological activity. Nat. Prod. Rep.. 2015;32:1472-1507.
- [Google Scholar]
- Anticancer evaluation of the selected tetrahydropyrimidines: 3D-QSAR, cytotoxic activities, mechanism of action, DNA, and BSA interactions. J. Mol. Struct.. 2022;1257:132621
- [Google Scholar]
- Synthesis and Characterization of New Substituted Coumarin Derivatives and Study Their Biological Activity. Chem. Methodol.. 2022;6:813-822.
- [Google Scholar]
- F2-isoprostanes in Alzheimer and other neurodegenerative diseases. Antioxid. Redox Signal.. 2005;7:269-275.
- [Google Scholar]
- Synthesis, characterization, and biomedical assessment of novel bisimidazole–coumarin conjugates. Appl. Nanosci. 2021:1-12.
- [Google Scholar]
- Synthesis of novel coumarin appended bis (formylpyrazole) derivatives: Studies on their antimicrobial and antioxidant activities. Bioorg. Med. Chem. Lett.. 2016;26:690-694.
- [Google Scholar]
- Naik, M. D., Bodke, Y. D. and R. BC, 2020. An efficient one-pot synthesis of coumarin-amino acid derivatives as potential anti-inflammatory and antioxidant agents. Synthetic Communications. 50, 1210-1216.
- Synthesis, structure–activity relationship and molecular docking of cyclohexenone based analogous as potent non-nucleoside reverse-transcriptase inhibitors. J. Mol. Struct.. 2015;1086:8-16.
- [Google Scholar]
- Crystallographic insights into DNA minor groove recognition by drugs. Biopolymers: Original Research on Biomolecules.. 1997;44:105-121.
- [Google Scholar]
- UV photoelectron spectroscopy of coumarins. J. Electron Spectrosc. Relat. Phenom.. 2000;113:9-13.
- [Google Scholar]
- O'boyle, N. M., Tenderholt, A. L. and Langner, K. M. 2008. Cclib: a library for package‐independent computational chemistry algorithms. Journal of computational chemistry. 29, 839-845.
- Olanlokun, J. O. and Akomolafe, S. F. 2013. Antioxidant potentials of various solvent extracts from stem bark of Enantia chlorantha. Journal of Biomedical Science and Engineering. 2013,
- Coumarin-piperazine derivatives as biologically active compounds. Saudi Pharmaceutical Journal.. 2020;28:220-232.
- [Google Scholar]
- Bis-azo-linkage Schiff bases—Part (II): Synthesis, characterization, photoluminescence and DPPH radical scavenging properties of their novel luminescent mononuclear Zn (II) complexes. J. Photochem. Photobiol. A Chem.. 2020;392:112356
- [Google Scholar]
- Design, synthesis and biological evaluation of selective hybrid coumarin-thiazolidinedione aldose reductase-II inhibitors as potential antidiabetics. Bioorg. Chem.. 2021;114:104970
- [Google Scholar]
- Synthesis of new conjugated coumarin–benzimidazole hybrids and their anticancer activity. Bioorg. Med. Chem. Lett.. 2013;23:3667-3672.
- [Google Scholar]
- Antioxidant activity of 4-methylcoumarins. J. Pharm. Pharmacol.. 2007;59:1721-1728.
- [Google Scholar]
- Influence of Co (III) Polypyridyl Complexes on Luminescence Behavior, DNA Binding, Photocleavage, Antimicrobial Activity and Molecular Docking Studies. J. Fluoresc.. 2021;31:1009-1021.
- [Google Scholar]
- Antitumor activity, DNA and BSA interactions of novel copper (II) complexes with 3, 4-dihydro-2 (1H)-quinoxalinones. Chem. Biol. Interact.. 2021;348:109647
- [Google Scholar]
- Spectroscopic and viscometric determination of DNA-binding modes of some bioactive dibenzodioxins and phenazines. Biochem. Biophys. Rep.. 2019;18:100629
- [Google Scholar]
- pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58:4066-4072.
- [Google Scholar]
- Novel coumarins and related copper complexes with biological activity: DNA binding, molecular docking and in vitro antiproliferative activity. J. Inorg. Biochem.. 2017;177:101-109.
- [Google Scholar]
- Theoretical investigations of the UV spectra of coumarin derivatives. Chem. Phys. Lett.. 2005;415:20-24.
- [Google Scholar]
- TD-DFT investigation of the UV spectra of pyranone derivatives. Chem. A Eur. J.. 2006;110:8144-8150.
- [Google Scholar]
- In vitro DNA binding studies of antiretroviral drug nelfinavir using ethidium bromide as fluorescence probe. J. Photochem. Photobiol. B Biol.. 2015;151:194-200.
- [Google Scholar]
- Coumarin hybrid derivatives as promising leads to treat tuberculosis: Recent developments and critical aspects of structural design to exhibit anti-tubercular activity. Tuberculosis. 2021;102050
- [Google Scholar]
- Synthesis, structure, DNA binding and cleavage properties of ternary amino acid Schiff base-phen/bipy Cu (II) complexes. J. Inorg. Biochem.. 2011;105:1603-1612.
- [Google Scholar]
- 3: Desmond molecular dynamics system. New York, NY: DE Shaw Research; 2017.
- The evaluation of the anticancer activity of the Biginelli hybrids and pharmacokinetic profiling based on their retention parameters. J. Mol. Struct.. 2022;1254:132373
- [Google Scholar]
- Antioxidant potential of piperidine containing compounds-a short review. Atherosclerosis. 2018;10:12.
- [Google Scholar]
- Density-functional theory for time-dependent systems. Phys. Rev. Lett.. 1984;52:997.
- [Google Scholar]
- A review: Biologically active 3, 4-heterocycle-fused coumarins. Eur. J. Med. Chem.. 2021;212:113034
- [Google Scholar]
- Coumarin hybrids as novel therapeutic agents. Bioorg. Med. Chem.. 2014;22:3806-3814.
- [Google Scholar]
- Interaction of coumarin with calf thymus DNA: deciphering the mode of binding by in vitro studies. Int. J. Biol. Macromol.. 2015;73:9-16.
- [Google Scholar]
- Discovery and synthesis of novel 3-phenylcoumarin derivatives as antidepressant agents. Bioorg. Med. Chem. Lett.. 2011;21:1937-1941.
- [Google Scholar]
- Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des.. 2013;27:221-234.
- [Google Scholar]
- Redox behavior of anticancer chalcone on a glassy carbon electrode and evaluation of its interaction parameters with DNA. Int. J. Mol. Sci.. 2008;9:1424-1434.
- [Google Scholar]
- Voltammetric and spectroscopic investigations of 4-nitrophenylferrocene interacting with DNA. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2010;75:1082-1087.
- [Google Scholar]
- Evaluation of DNA interaction, free radical scavenging and biologically active compounds of thermally stable p-tolylmethanamine Schiff bases and their binary Co (II) and Ni (II) complexes. Chem. Data Collect.. 2020;28:100439
- [Google Scholar]
- Design and synthesis of C3-tethered 1, 2, 3-triazolo-β-carboline derivatives: Anticancer activity, DNA-binding ability, viscosity and molecular modeling studies. Bioorg. Chem.. 2016;64:42-50.
- [Google Scholar]
- Synthesis, characterization and biological studies of Schiff bases derived from heterocyclic moiety. Bioorg. Chem.. 2017;70:67-73.
- [Google Scholar]
- Binding interaction between sorafenib and calf thymus DNA: spectroscopic methodology, viscosity measurement and molecular docking. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2015;136:443-450.
- [Google Scholar]
- Comparative docking and ADMET study of some curcumin derivatives and herbal congeners targeting β-amyloid. Network Modeling Analysis in Health Informatics and Bioinformatics.. 2013;2:13-27.
- [Google Scholar]
- Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J. Chem. Theory Comput.. 2011;7:2284-2295.
- [Google Scholar]
- Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem.. 2002;277:46265-46272.
- [Google Scholar]
- Noncovalent interactions with DNA: an overview. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis.. 2007;623:3-13.
- [Google Scholar]
- Criteria for the mode of binding of DNA binding agents. Bioorg. Med. Chem.. 1995;3:723-728.
- [Google Scholar]
- Synthesis, crystal structure, spectral and DFT studies of potent isatin derived metal complexes. J. Mol. Struct.. 2018;1166:110-120.
- [Google Scholar]
- Computational investigation of molecular structures, spectroscopic properties, cholinesterase inhibition and antibacterial activities of triazole Schiff bases endowed metal chelates. J. Mol. Struct.. 2021;1238:130382
- [Google Scholar]
- Threading intercalators as a new DNA structural probe. Bull. Chem. Soc. Jpn.. 1999;72:327-337.
- [Google Scholar]
- Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem.. 2015;101:476-495.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- New bis 1, 3, 4-oxadiazole derivatives: syntheses, characterizations, computational studies, and antioxidant activities. Can. J. Chem.. 2018;96:1047-1059.
- [Google Scholar]
- Venugopala, K. N., Rashmi, V. and Odhav, B. 2013. Review on natural coumarin lead compounds for their pharmacological activity. BioMed research international. 2013.
- Vera, N., Zampini, C., Isla, M. I. et al., 2007. Antioxidant and XOD inhibitory coumarins from Pterocaulon polystachyum DC. Natural Product Communications. 2, 1934578X0700200508.
- Complex formation between ethidium bromide and nucleic acids. J. Mol. Biol.. 1965;13:269-282.
- [Google Scholar]
- Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464-468.
- [Google Scholar]
- Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci.. 2010;107:7479-7484.
- [Google Scholar]
- Coumarin-based derivatives with potential anti-HIV activity. Fitoterapia. 2021;104863
- [Google Scholar]
- Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217-227.
- [Google Scholar]
- Solvent-free Synthesis of Water-Soluble New Pyridinium Amines: Spectroscopic Characterization, Biological Screening, and Interaction Study with DNA. J. Chin. Chem. Soc.. 2017;64:822-832.
- [Google Scholar]
- Pd (II) complexes with chelating N-(1-alkylpyridin-4 (1H)-ylidene) amide (PYA) ligands: Synthesis, characterization and evaluation of anticancer activity. J. Inorg. Biochem.. 2021;224:111590
- [Google Scholar]
- Coumarin-pi, a new antioxidant coumarin derivative from Paxillus involutus. Nat. Prod. Res.. 2020;34:1246-1249.
- [Google Scholar]
- Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem.. 2019;181:111587
- [Google Scholar]
- Recent progress and future potential for metal complexes as anticancer drugs targeting G-quadruplex DNA. Curr. Med. Chem.. 2012;19:2957-2975.
- [Google Scholar]
- Synthesis, characterization and studies on DNA-binding of a new Cu (II) complex with N1, N8-bis (l-methyl-4-nitropyrrole-2-carbonyl) triethylenetetramine. J. Inorg. Biochem.. 2007;101:10-18.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104440.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




