

Translate this page into:
New benzimidazole derivatives: Design, synthesis, docking, and biological evaluation
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The aim of this study was to synthesize novel enaminonitrile derivatives starting from 2-aminobenzimidazole and utilize this derivative for the preparation of novel heterocyclic compounds and assess their function for biological activity screening. The key precursor N-(1H-benzo[d]imidazol-2-yl)carbonohydrazonoyl dicyanide (2) was prepared in pyridine by coupling of diazotized 2-aminobenzimidazole (1) with malononitrile. Compound 2 was subjected to react with various secondary amines such as piperidine, morpholine, piperazine, diphenylamine, N-methylglucamine, and diethanolamine in boiling ethanol to give the acrylonitriles (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(piperidin-1-yl)acrylonitrile (3), (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-morpholinoacrylonitrile (4), (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(piperazin-1-yl)acrylonitrile (5), (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(diphenylamino)acrylonitrile (6), (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)acrylonitrile (7), and (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(bis(2-hydroxyethyl)amino)acrylonitrile (8), respectively. It has been found that the behaviour of nitrile derivative 2 towards hydrazine hydrate to the creation of 4-((1H-benzo[d]imidazol-2-yl)diazenyl)-1H-pyrazole-3,5-diamine (9). The reaction of malononitrile with compound 2 in an ethanolic solution catalyzed with sodium ethoxide afforded 4-amino-1-(1H-benzo[d]imidazol-2-yl)-6-imino-1,6-dihydropyridazine-3,5-dicarbonitrile (11). Moreover, malononitrile reacted with 7 in a boiling ethanolic sodium ethoxide solution to give 2-(5-((1H-benzo[d]imidazol-2-yl)diazenyl)-4-amino-6-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)pyrimidin-2-yl)acetonitrile (14). Heating 7 in boiling acetic anhydride and pyridine afforded (2R,3R,4R,5S)-6-(((1E)-2-((1-acetyl-1H-benzo[d]imidazol-2-yl)diazenyl)-1-(N-acetylacetamido)-2-cyanovinyl)(methyl)amino)hexane-1,2,3,4,5-pentayl pentaacetate (15). When compound 15 is heated for a long time in refluxing DMF including a catalytic of TEA, cyclization occurs to give the corresponding (2R,3R,4R,5S)-6-((1-acetyl-3-((1-acetyl-1H-benzo[d]imidazol-2-yl)diazenyl)-4-amino-6-oxo-1,6-dihydropyridin-2-yl)(methyl)amino)hexane-1,2,3,4,5-pentayl pentaacetate (16). In addition, triethyl orthoformate was reacted with compound 7 in the presence of acetic anhydride to afford the corresponding ethoxymethyleneamino derivative (2R,3R,4R,5S)-6-(((1E)-2-((1-acetyl-1H-benzo[d]imidazol-2-yl)diazenyl)-2-cyano-1-(((E) ethoxymethylene)amino)vinyl)(methyl)amino)hexane-1,2,3,4,5-pentayl pentaacetate (17). Also, it has been found that heating a mixture of 7 with DMF/DMA in anhydrous xylene yielded compound (1E)-N'-((1E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-2-cyano-1-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)vinyl)-N,N-dimethylformimidamide (18). In addition, compound 7, when reacted with several acid anhydrides, allowed the matching phthalimide derivatives 19–26. The results showed that compound 14 has significantly higher ABTS and antitumor activities than the other compounds. Molecular modelling was also studied for compounds 22 and 24. The viability of four many cell lines—the African green monkey kidney epithelial cells (VERO), human breast adenocarcinoma cell line (MCF-7), human lung fibroblast cell line (WI-38), and human hepatocellular liver carcinoma cell line (HepG2) was examined to determine the antitumor activities of the newly synthesized compounds. Also, it was found that compounds 9, 11, 15, 16, 22, 23, 24 and 25 are strong against HepG2 cell lines, while 16, 22, and 25 are strong against WI-38 cell lines. Moreover, it was also found that compounds 16 and 22 are strong against VERO cell lines. On the other hand, compounds 7, 14, 15, 16, and 22 are strong while the rest of the other compounds are moderate against the MCF-7 cell line. The result of docking showed that compound 24 got stabilized inside the pocket with a very promising binding score of − 8.12 through hydrogen bonds with Arg184 and Lys179, respectively.
Keywords
Enaminonitrile
N-Methylglucamine
Azo compounds
Pyrazole
Pyrimidine
N-phthalimide

1 Introduction
Most of the azo dye compounds are characterized by several biological activities such as azo reduction monoamine oxidase inhibition mutagenic, carcinogenesis, and other medicinal and industrial uses (Lohse, 1986; Pati, 1975). Additionally, azo dye derivatives are biologically interesting compounds well known for their therapeutic value and their usage as antineoplastics (Child and Wilkinson, 1977), antidiabetics (Garg and Praksh, 1972), antiseptics (Browing et al., 1926); antibacterials (Khalid et al., 2008; Pagga and Brown, 1986), and antitumor agents (Thoraya, 2008). The suppression of protein, RNA, and DNA production, nitrogen fixation, and carcinogenesis are only a few of the biological processes they are known to be involved with (Park et al., 2007). It has been published many works associated with the chemistry of azo dyes by Fadda and coworkers (Fadda and Abbas, 2016; Fadda and Elattar, 2012; Fadda et al., 2006; Erton, 2000; Dakiky and Nemcov, 2000; Fadda et al., 1994; Fadda and Etman, 1995; Fadda et al., 1995; Fadda et al., 1995; Fadda et al., 1998). Additionally, N-methyl-d-glucamine (meglumine, MG) possesses structural characteristics that are similar to glycosides and is made up of sorbitol and fragments of tertiary amines. Its capacity to generate supramolecular adducts with lipophilic chemical molecules in water is its most interesting future (Cassimiro et al., 2013). This characteristic is particularly helpful in the pharmaceutical sector for making medications more soluble in aqueous solutions and stabilizing them there (Cassimiro et al., 2013; Aloisio et al., 2014). Potential medicinal compounds have been found using meglumine derivatives as the amine component in the Mannich reaction (Afsah et al., 2018). The crucial factor of boosting system stability led to the selection of N-methyl-d-glucamine, for example, the N-Mannich base. For instance, replacement with N-methyl-d-glucamine makes cyclic peptide dimers and nanotubes more stable (Khavani et al., 2017). N-methyl-d-glucamine was used successfully to create water-soluble macrocycles (Nifantiev and Yashunsky, 7 April 2004.), such as porphyrin derivatives that can localize in cancer tissues (Nakajima et al., 1990). It was as well demonstrated that chlorin e6 noncovalently complexed with N-methyl-d-glucamine groups might be used for photodynamic therapy (Pegaz et al., 2005). Furthermore, the presence of N-methyl-d-glucamine and azo dye moiety may develop several permeation ways at receptors (Jiang et al., 2005) and hence increase the biological activity of newly synthesized compounds. Enaminonitriles are important as a key step to prepare heterocyclic biological active compounds. They are very promising mediates for the preparation of interesting heterocyclic compounds (El-Sayed and Althagafi, 2016). Furthermore, the presence of N-methyl-d-glucamine and azo dye moiety may use many permeation ways at receptors (Fadda and Elattar, 2013) and hence increase the biological activity of newly synthesized compounds. The present study is a part of our research program (Abuo-Melha and Fadda, 2012; Madkour et al., 2009; Shaaban et al., 2009; Elkholy et al., 2008) directed toward the preparation of novel pyrazole, pyridine and pyrimidine derivatives containing azo dye moiety using β-enaminonitrile derivative and its use for example building blocks in the preparation of novel promising biologically active compounds.
Many heterocyclic compounds are characterized by different biological activities and synthesized by using enaminonitriles as key intermediates (Salaheldin et al., 2008; Madkour et al., 2008; Azab, 2008; Dyachenko and Dyachenko, 2008; [38]; Wang et al., 2000; Zaki et al., 2004). A variety of heterocyclic compounds with pharmacological applications, such as anti-inflammatory (Bondock et al., 2008), antitumor (Fadda and Elattar, 2015; El Ashry and ElKilany, 1997), antibacterial and antifungal activity (Makara and Keseru, 1997), and used as analgesic agents (Fadda et al., 2012), can be formed through reactions between these compounds' amino and cyano functions and common reagents.
It is well known that sugar and its related compounds have a broad spectrum of biological activities like antitumor, antiviral and antibiotic activities (Fadda et al., 2013; Bischoff and Hoffmann, 2002).
Accordingly, in order to enhance the biological activity resulting from the attachment of sugar moieties with different heterocyclic rings, we studied the synthesis of some new heterocyclic compounds incorporating glucaminoid moiety.
In this work, we aimed to synthesize some new enaminonitrile derivatives starting from 2-aminobenzimidazole and utilize this derivative in the synthesis of new heterocyclic compounds, and assess their function for biological activity screening.
2 Results and discussion
The key precursor N-(1H-benzo[d]imidazol-2-yl)carbonohydrazonoyl dicyanide (2) was prepared by coupling of diazotized 2-aminobenzimidazole (1) with malononitrile in pyridine at 0–5 °C. Compound 2 was subjected to react with various secondary amines such as piperidine, morpholine, piperazine, diphenylamine, N-methylglucamine, and diethanolamine in boiling ethanol to give the acrylonitriles 3–8, respectively (Scheme 1).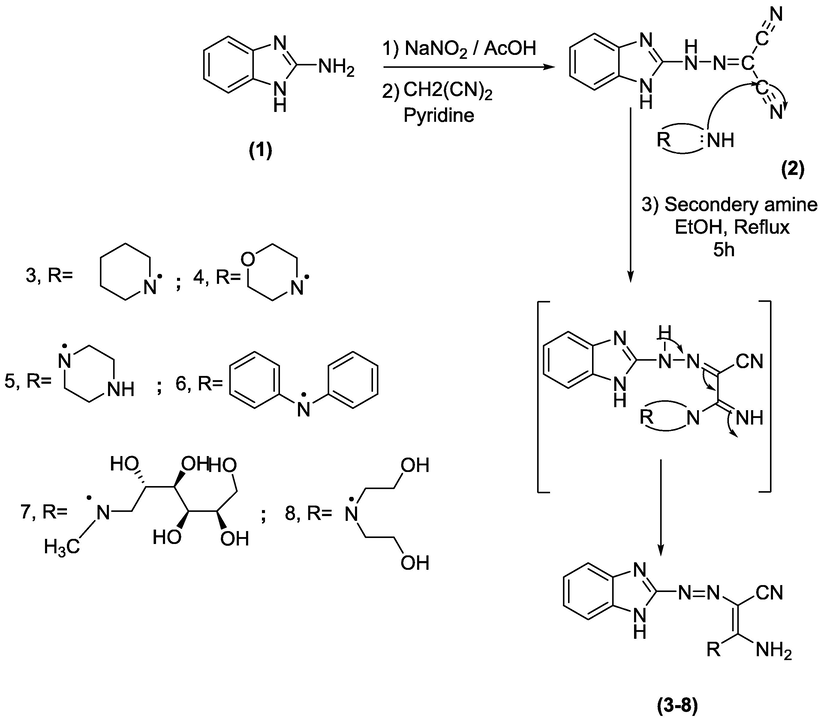
The structure of enaminonitrile derivatives 3–8 was founded on the origin of elemental analyses and spectral data. Their IR spectra showed characteristic bands within ν = 3450–3350 according to the amino group and bands within ν = 2220–2180 corresponding to cyano function, besides the other expected bands. The 1H NMR of 3 showed multiplets at δ 1.55–1.70, 1.72–1.85 ppm due to 3CH2 of piperidine ring besides triplet sharp signal at 3.06 ppm attributable to CH2-N-CH2 of piperidine moiety, respectively, also to one signal at 6.51 ppm corresponding to NH2 protons. The MS of compound 3 showed its molecular ion peak at m/z (%) = 295 (M+, 30 %) attributed to the chemical formula C15H17N7. The 1H NMR of 4 revealed triplets at δ 3.14 and δ 3.67 attributable to CH2-N-CH2 besides CH2-O-CH2 protons, respectively. MS spectrum of compound 4 revealed its M+ at m/z (%) = 297 (M+, 25) corresponding to chemical formula C14H15N7O. While the 1H NMR of 5 showed triplets at 3.16 ppm besides δ 3.69 ppm corresponding to CH2-NH-CH2, CH2-N-CH2, separately. The MS spectrum of compound 5 revealed its M+ at m/z (%) = 296 (M+, 31) attributed to chemical formula C14H16N8. Moreover, MS of 6 displayed its molecular ion peak at m/z (%) = 379 (M+, 30) attributed to chemical structure C22H17N7. The 1H NMR of 7 revealed multiplets at δ 3.56–3.62 corresponding to 4CH + 2CH2, 3.93 singlets for OH proton, and 4.35–4.51 multiplets for 4OH groups. Also, revealed two doublets at δ 7.25 then 7.53 due to four Ar-H and one signal at δ 3.04 ppm of N-CH3 protons. The MS spectrum of compound 7 displayed its M+ at m/z (%) = 405 (M+, 60) attributed to chemical formula C17H23N7O5. While the 1H NMR spectrum of 8 displayed signals at δ 3.16 besides 3.87 corresponding to CH2-N-CH2 and O-CH2 protons, separately. It also, revealed two signals at δ 4.85 and δ 6.50 according to hydroxyl and NH2 protons, separately and multiplets at δ 7.40–7.70 attributed to the aromatic protons. Also, MS of compound 8 showed its molecular ion peak at m/z (%) = 315 (M+, 28) attributed to its chemical structure C14H17N7O2. The 13C NMR of compounds 3–8 displayed signals, in general, at δ 114.5 ppm for CN (nitrile), 141.5 ppm for C⚌N, and 173.5 ppm for C-NH2. For compound 7 its 13C NMR showed signals at δ 50.5 ppm for CH2 and 64.0–72.0 ppm due to C-OH, while signals due to C-OH in compound 8 appeared at δ 59.4 ppm and C—N showed a signal at δ 53.1 ppm and characteristic signals 87.5 and 173.5 ppm for C⚌C.
It has been found that the behavior of nitrile derivative 2 towards hydrazine hydrate is according to the creation of the pyrazole compound 9 (Scheme 2). The chemical structure of compound 9 was deduced based on spectroscopic analysis. The 1H NMR spectrum showed signals at δ 6.20, 12.53, and 12.75 ppm attributable to 2NH2 and 2NH protons, separately, also two doublets signal due to 4H aromatic protons at δ 7.40 and 7.70 ppm. The 13C NMR showed a characteristic signal at δ 151.3 ppm due to C-NH2. Although, it was found that compound 2 was used as a key intermediate to synthesize the pyridazine derivative of pharmacological activity interest. The reaction of malononitrile with compound 2 in ethanolic solution catalyzed with sodium ethoxide afforded pyridazine derivative 11 via through structure 10 which was obtained by the addition of CH2 group of malononitrile to the CN group in compound 2 followed by cycloaddition reaction (Scheme 2).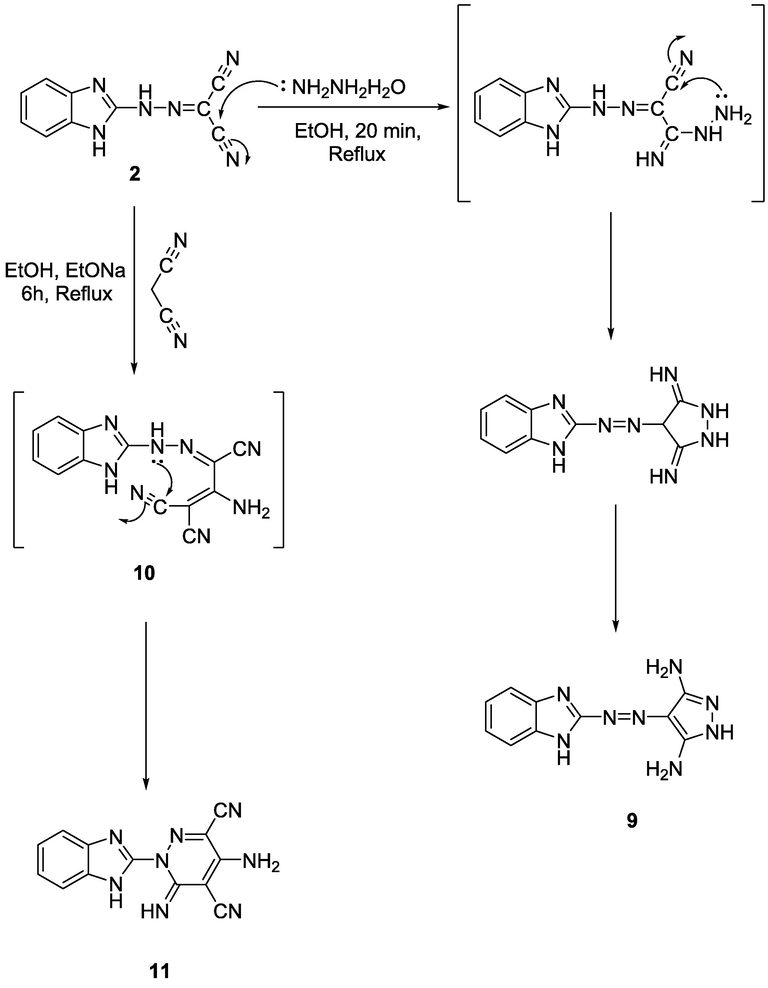
Structure 11 was explained based on its correct spectroscopic data. The infrared spectrum revealed bands at 3450, 3336, and 3214 cm−1 attributed to the NH2 and NH moieties. The MS spectrum showed its M+ at m/z (%) = 276 (M+, 28) due to the chemical structure C13H8N8. The 13C NMR displayed a characteristic signal at δ 115.8 ppm for CN (nitrile) and δ 162.7 ppm for C-NH2.
Moreover, malononitrile reacted with enaminonitrile 7 in boiling ethanolic sodium ethoxide solution to give pyrimidine 14. Structure 14 was deduced by spectroscopic analysis. The 1H NMR displayed singlets at δ 3.04 for N-CH3, 3.93 for OH proton, and 8.45 for NH2 protons. The MS spectrum revealed its M+ at m/z (%) = 470 (M+−1, 20). Based on these data, it seemed that pyrimidine derivative 14 was obtained through the intermediate 13 as shown in Scheme 3.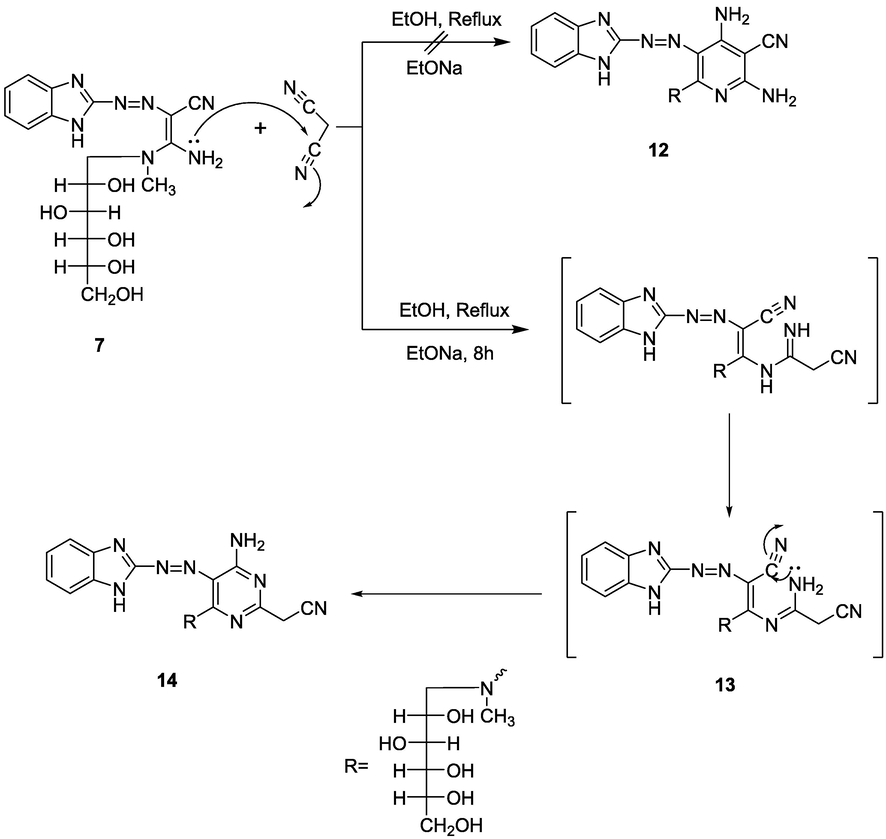
Heating 7 in boiling acetic anhydride and pyridine afforded compound 15 (Scheme 4). The structure of pentaacetyl derivative 15 was deduced based on spectroscopic analysis. The IR spectrum indicated the absence of an amino group. The 1H NMR indicated a multiplet at δ 2.03 ppm according to five OCOCH3 protons besides a singlet at δ 2.29 ppm due to two COCH3 protons. The MS spectrum revealed its M+ at m/z (%) = 741 (M+, 40) attributed to the molecular formula C33H39N7O13. Once compound 15 is heated for a long time in refluxing DMF including a catalytic of TEA undergoes cyclization to give the corresponding pyridine derivative 16.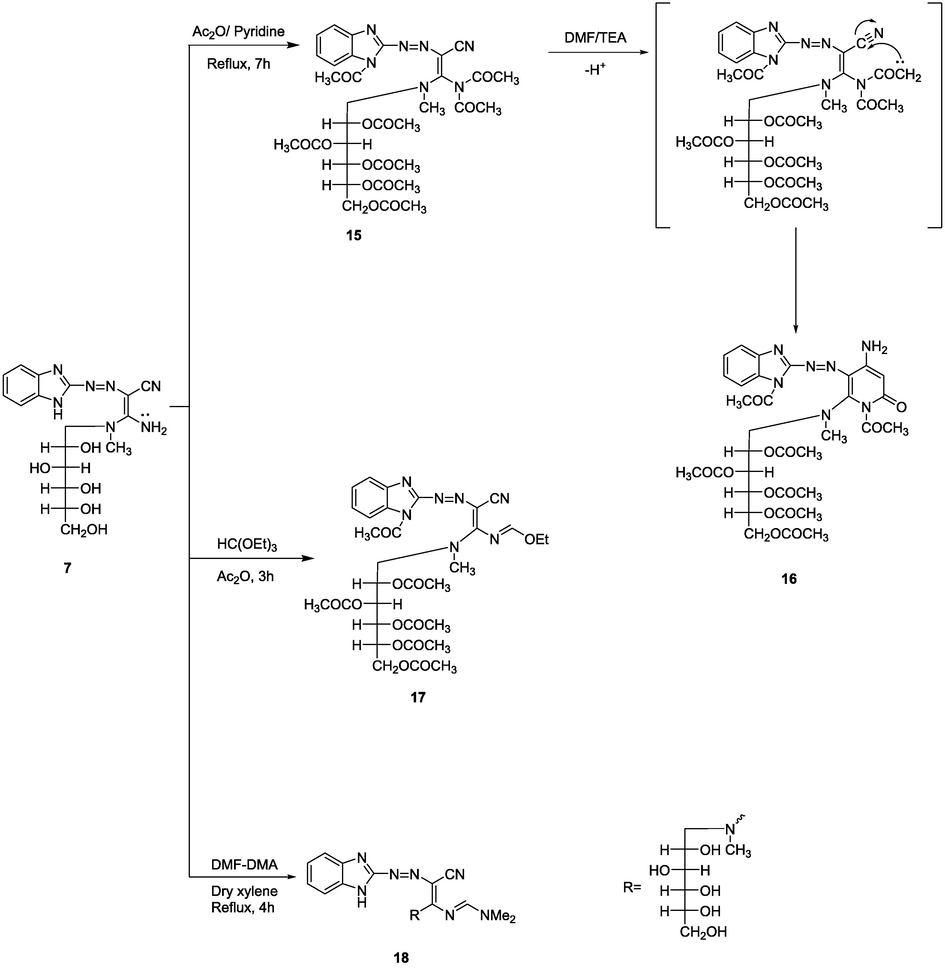
In addition, triethyl orthoformate was reacted with compound 7 including of acetic anhydride allowing the matching ethoxymethyleneamino derivative 17. In addition, it has been found that heating a mixture of 7 with DMF/DMA in anhydrous xylene yielded compound 18 (Scheme 4). The structures of compounds 17 and 18 were explained by spectroscopic analyses. The IR of compounds 17 and 18 revealed characteristic bands at 2179 and 2177 cm−1 due to cyano groups, respectively. In addition, 1H NMR spectra of derivatives 17 and 18 showed singlet signals at δ 8.50 and 8.73 ppm according to CH = N proton, respectively. In addition, compound 17 showed triplets at δ 1.24 ppm attributed to CH3 protons and quartet at δ 3.65 according to CH2 protons, and compound 18 displayed singlets at δ 2.90 attributed to NMe2 protons. Moreover, the MS of 17 and 18 revealed their molecular ion peaks at m/z (%) = 713 (M+, 55), and 460 (M+, 40) attributed to the molecular formula C32H39N7O12 and C20H28N8O5, respectively.
Acid anhydride derivatives as; phthalic anhydride, quinolinic anhydride, pyromellitic anhydride, 1,2,4-benzene tricarboxylic anhydride, 4-nitrophthalic anhydride, 3-nitrophthalic anhydride, and tetrabromophthalic anhydride were refluxed with compound 7 in DMF solution catalyzed with the amount of acetic acid gave the phthalimide derivatives 19–25, respectively (Schemes 5 and 6). The chemical structures of compounds 19–25 were confirmed by spectroscopic data, in which in their IR spectra the NH2 group disappeared, and instead absorption band at ν = 1700–1680 cm−1 attributed to the amidic carbonyl group appeared. The 13C NMR of compounds 19–25 showed, in general, signals at δ 54.1, 141.5, and 185.8 ppm attributed to CH2, C⚌N, and C⚌O, respectively. Also, MS spectra of compounds 19, 20, 22–25 indicated the molecular ion peaks at m/z (%) = 535 (M+, 15), 536 (M+, 30), 579 (M+, 25), 580 (M+, 15), 580 (M+, 15) and 846 (M+−5, 60), respectively, corresponding to the chemical formula of compounds 19–25.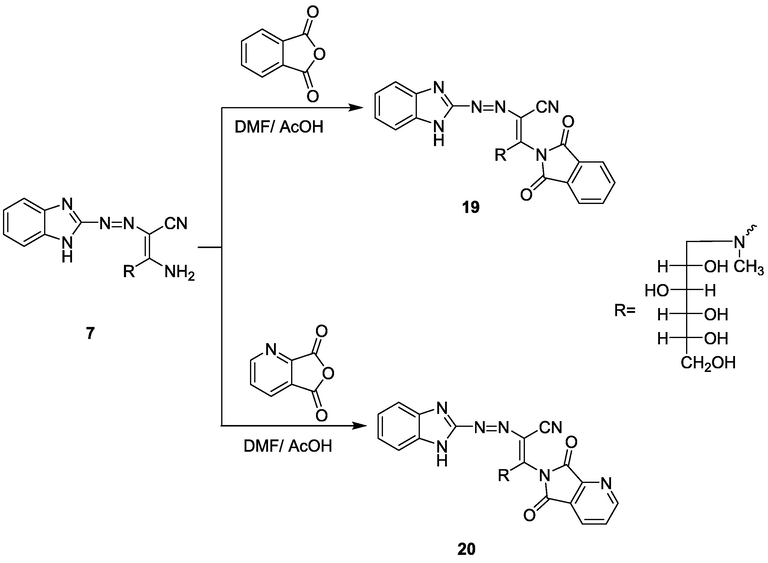
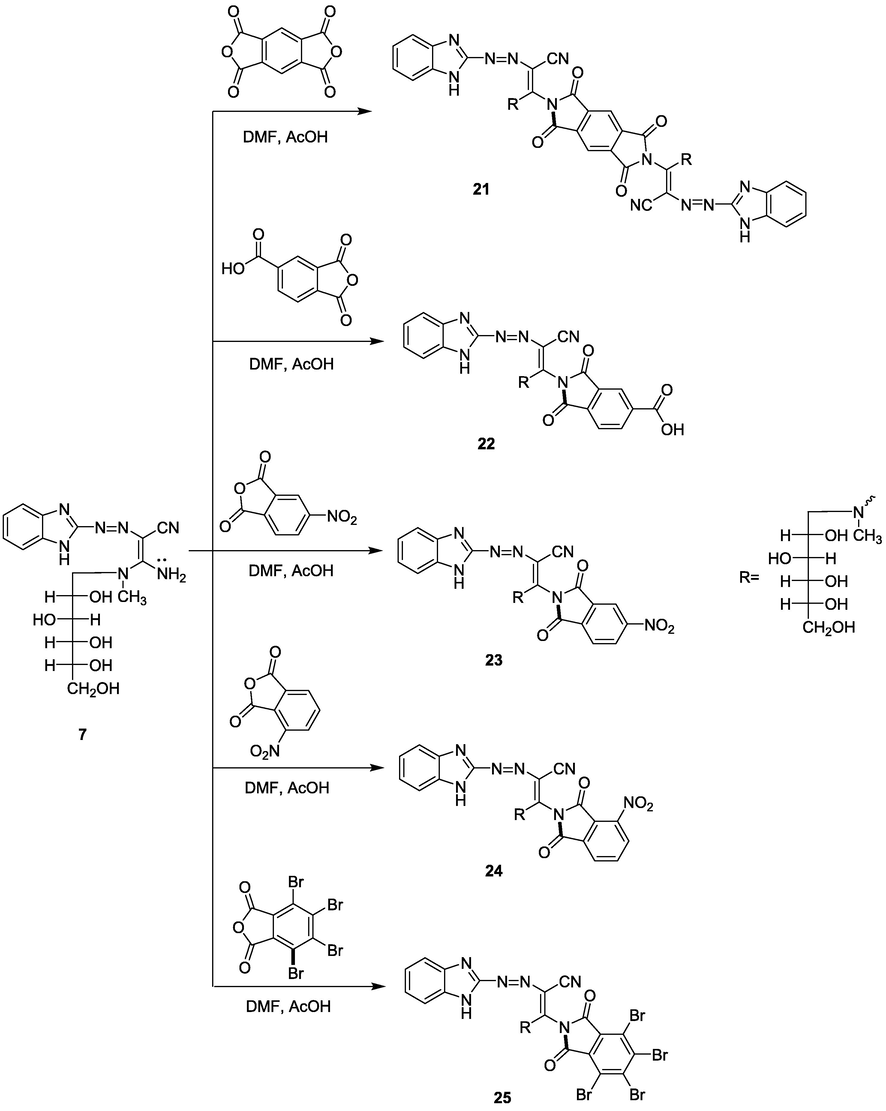
Fusion of enaminonitrile derivative 7 with 3,4,9,10- perylenetetracarboxylic dianhydride including freshly fused sodium acetate afforded compound 26 (Scheme 7).
The structure of compound 26 was established by IR spectrum which indicated absorption bands at 3450–3440, 2190, 2180, 1700–1680 and 1530, 1510 due to 10 OH, 2CN, 4 CO, 2 N⚌N groups, respectively.
The overall mechanistic consideration for the formation of compounds 19–26 is shown in the Scheme 8 below: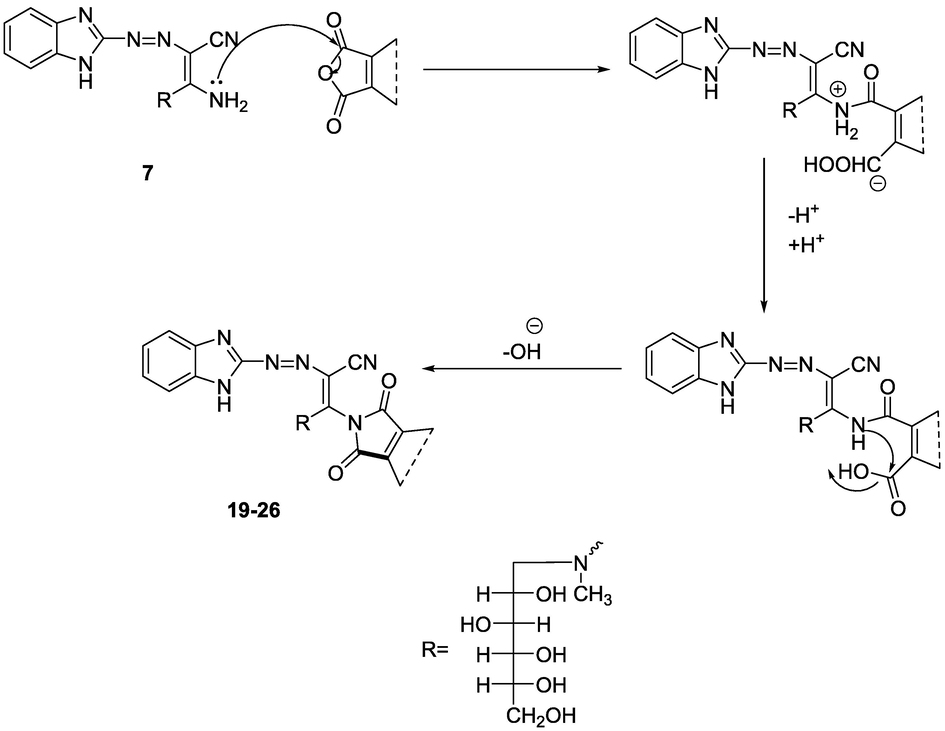
3 Pharmacological evaluation
3.1 Antioxidant activity ABTS
The assay for antioxidant activity was conducted in accordance with the previously published work (Martinez and Chacon-Garcia, 2005). In the present work, the obtainable compounds showed their antioxidant results in Table 1. It was found that compounds 9 and 11, including no glucaminoid moiety, resulted in low activity and showed less than 50 % inhibition of the ABTS radical cation. The other rest of the compounds which having glucaminoid moiety showed good to moderate inhibition of the ABTS radical cation. In spite of compound 21, characterized by containing two glucaminoid moieties, having low activity, it may be due to its large structure, which enters the cell with difficulty. Compound 16 demonstrated that converting the cyano group in compound 15 to an amino group significantly increased antioxidant activity. a Where Ac is the absorbance value of the control and As is the absorbance of the additional samples test solution, ABST Scavenging activity (%) is calculated as [Ac- As/Ac] × 100. b The concentration of the pure compounds was 2 mM. c The concentration showing 50% inhibition is expressed in mM. d The positive control was Ascorbic acid (0.24 mM, and showed 80.81 ± 1.16 % at the same concentration of the tested compounds. e Values are means of 6 replicates ± SD, and significant difference at P <. 0.05 by Student’s test.
Compound No.
ABTS (% of scavenging Inhibition) a,b,c,d,e
Erythrocyte haemolysis (%)
Ascorbic acid
80.81 ± 1.16
0.85
4
36.72 ± 2.38 e
1.90
7
70.91 ± 0.04 e
0.90
8
68.04 ± 0.14 e
1.01
9
46.45 ± 1.15 e
1.90
11
51.50 ± 0.16 e
1.01
14
79.79 ± 1.30 e
0.85
15
48.38 ± 0.35 e
1.91
16
77.10 ± 1.27
0.83
17
61.58 ± 0.25 e
1.01
18
63.58 ± 1.30 e
1.91
19
61.04 ± 1.24
0.84
20
75.00 ± 0.23
0.84
21
65.00 ± 1.34
0.86
22
75.79 ± 0.32
0.86
23
74.82 ± 0.29
0.84
24
70.51 ± 0.12
0.91
25
66.77 ± 1.32
0.80
26
66.01 ± 1.74
0.90
3.2 AAPH-induced RBC haemolysis
Haemolysis was evaluated according to the reported work (Martinez and Chacon-Garcia, 2005).
3.3 Bleomycin-dependent DNA damage
It was evaluated according to the previously published work (Martinez and Chacon-Garcia, 2005). Table 2 showed that compound 14 has the strongest DNA damage prevention activity, which decreases the formation of chromogen between thiobarbituric acid (TBA) and the damaged DNA.
Compound No.
Absorbance
Ascorbic acid
0.00881
7
0.00858
14
0.00882
3.4 Antitumor testing
All the newly synthesized compounds were screened (Fadda et al., 2009) in Vetro for their anticancer effect via the standard MTT method, against a panel of four human tumor cell lines, namely, the African green monkey kidney epithelial cells (VERO), human breast adenocarcinoma cell line (MCF-7), human lung fibroblast cell line (WI-38), and human hepatocellular liver carcinoma cell line (HepG2). The cell lines were obtained from ATCC via the holding company for biological products and vaccines (VACSERA), Cairo, Egypt. The concentration that resulted in a 50 % loss of the cell monolayer (IC50) was used to determine the cytotoxicity (Table 3)Fig. 1. * (IC50, μg/mL): 1–10 (very strong), 11–25 (strong), 26–50 (moderate), 51-. 100 (weak), 100–200 (very weak), and above 200 (non-cytotoxic). # Values are means of 6 replicates ± SD, and significant difference at P less than 0.05 by Student’s test.
Compound No.
IC50, (μg/mL) *
HepG-2
WI-38
VERO
MCF-7
4
61 ± 0.08
84 ± 0.12
72 ± 0.22
50 ± 1.30
7
28 ± 0.16
29 ± 0.16
31 ± 0.33
22 ± 0.08
8
42 ± 0.12
31 ± 0.21
41 ± 0.16
51 ± 0.08
9
25 ± 0.05
32 ± 0.13
33 ± 0.13
39 ± 1.10
11
23 ± 0.13
27 ± 0.21
38 ± 0.13
32 ± 0.45
14
26 ± 0.12
50 ± 1.11
31 ± 1.01
22 ± 0.23
15
25 ± 0.05
41 ± 0.12
27 ± 0.10
20 ± 0.01
16
22 ± 0.25
21 ± 0.25
18 ± 0.04
25 ± 0.22
17
44 ± 1.24
39 ± 0.05
40 ± 0.05
32 ± 0.38
18
47 ± 0.21
39 ± 0.17
54 ± 0.22
34 ± 2.01
19
52 ± 0.02
40 ± 0.14
46 ± 0.15
43 ± 0.12
20
50 ± 0.51
47 ± 0.34
45 ± 0.21
43 ± 0.06
21
61 ± 3.01
52 ± 0.23
61 ± 0.36
57 ± 1.02
22
11 ± 3.02
12 ± 0.05
15 ± 0.04
25 ± 0.01
23
21 ± 0.16
31 ± 0.02
33 ± 0.02
41 ± 0.13
24
19 ± 0.39
27 ± 0.07
26 ± 0.02
27 ± 0.05
25
20 ± 0.16
22 ± 0.03
32 ± 0.06
53 ± 0.05
26
62 ± 0.02
70 ± 0.01
71 ± 0.05
47 ± 0.11
5-Fu
8.6 ± 0.04
3.2 ± 0.11
6.5 ± 0.03
2.3 ± 0.01
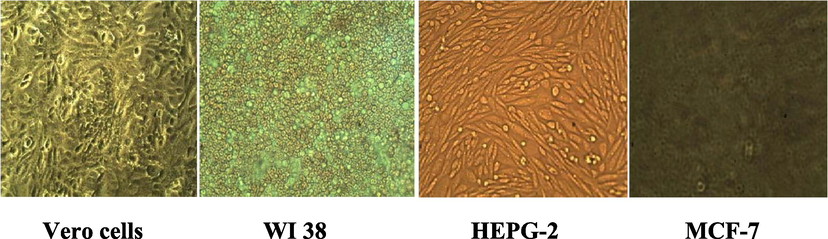
Vero cells: Cells from the kidney of green monkey; WI: fibroblast cells; HEPGII: Hepatoma cells; MCF-7: Cells from breast cancer.
It was found that compounds 9, 11, 15, 16, 22, 23, 24, and 25 are strong against HepG2 cell lines, while 16, 22, and 25 are strong against the WI-38 cell lines. Moreover, it was also found that compounds 16 and 22 are strong against VERO cell lines. On the other hand, compounds 7, 14, 15, 16, and 22 are strong while the rest of the other compounds are moderate against the MCF-7 cell line.
It can get the following conclusion from structure-activity relationships (SARs):
-
Compounds containing pyrazole, pyridine, and/or pyrimidine moieties, which are characterized by basic properties, indicated a wide spectrum of cytotoxicity against the four cell lines.
-
Compound 22 showed high cytotoxic activity against all cell lines, this may be attributed to glucaminoid moiety.
-
Introducing carbonyl groups (electron withdrawing groups) increased the cytotoxicity activity markedly.
3.5 Structure-activity relationship
It is well known that nucleotide moieties serve as the chemical building blocks of DNA. The nucleotide structure has four different forms of nitrogen bases: cytosine (C), guanine (G), thymine (T), and adenine (A). Adenine and cytosine are typically joined by hydrogen bonds. The presence of intramolecular hydrogen bonds with DNA bases and the electrostatic attraction between the investigated compounds and the cell wall are the two parameters (Elmaaty, 2021; Alesawy, 2021) that have an effect on the cytotoxic activity toward the cancer cell lines. The following information can be presented on the structure–activity relationship of the cytotoxicity of the examined compounds:
-
Strong activity was seen in compounds 9, 11, 14, and 16, which may be related to intramolecular hydrogen bonding between NH and NH2 groups and one of the DNA's nucleobases and instances of DNA damage.
-
The presence of two amidic carbonyl groups in compounds 19–26 and acetoxy groups in compounds 15–18 both function as potent electron-withdrawing groups and cause electrostatic interaction with DNA nucleobases. Compounds 22 and 24, which include nitro and carboxylic groups, are similar.
-
All of the examined compounds include an NH group, which can establish an intramolecular hydrogen bond with DNA nucleobases and harm it.
-
The pyrazole ring of compound 9 exhibited high cytotoxic activity because the NH and NH2 of the pyrazole ring have the ability to form a hydrogen bond with DNA nucleobases, and cause its damage.
3.6 Molecular docking
In comparison to ascorbic acid, a standard reference extracted from the cytochrome c peroxidase enzyme, molecular docking analysis of the created data set was used to conduct an investigation of the hypothesized mechanism of action for the recently designed and prepared active compounds like potential antioxidants (PDB code: 2X08). This research was done to learn more about how the produced chemicals bind to the protein-binding site of the cytochrome c peroxidase enzymeFig. 2.![2D and 3D receptor interactions of the reference ligand [Ascorbic acid].](/content/184/2023/16/2/img/10.1016_j.arabjc.2022.104505-fig10.png)
2D and 3D receptor interactions of the reference ligand [Ascorbic acid].
To validate the current docking investigation at the active site, the co-crystallized ligand ascorbic acid was re-docked into the active site using the same set of parameters. The best-docked pose's root means square deviation (RMSD) was 0.43, and the energy score was −5.51 Kcal/mol, indicating that the docking research with MOE software was valid. Ascorbic acid established four hydrogen bonds with the amino acids Asp37, Val45, Gly41, and Arg184.
The proposed binding mode of compound 22 showed an energy score of − 8.45 kcal/mol, whereas compound 22 showed a hydrogen bond with Gly84 and aromatic stacking interactions with Ser185 through pi-H bonds.
Table 4 shows that compound 24 got stabilized inside the pocket with a very promising binding score of − 8.12 through hydrogen bond with Arg184 and Lys179, respectively Fig. 3.
Comp.
Score (Kcal/mol)
RMSD
Interacting residues
Receptor interactions
Distance (Å)
E (Kcal/mol)
Ascorbic acid
−5.51
0.43
Asp37/ H-donor
Val45/ H-acceptor
Arg184/ H-acceptor
Gly41/ H- donor2.58
2.82
2.75
2.67−4.8
−2.9
−4.3
−1.5
22
−8.45
1.61
Gly84/ H- donor
Ser185/ pi-H3.51
3.71−0.8
−0.9
24
−8.12
1.73
Arg184/ H- donor
Lys179/ H- acceptor3.18
3.26−0.7
−1.5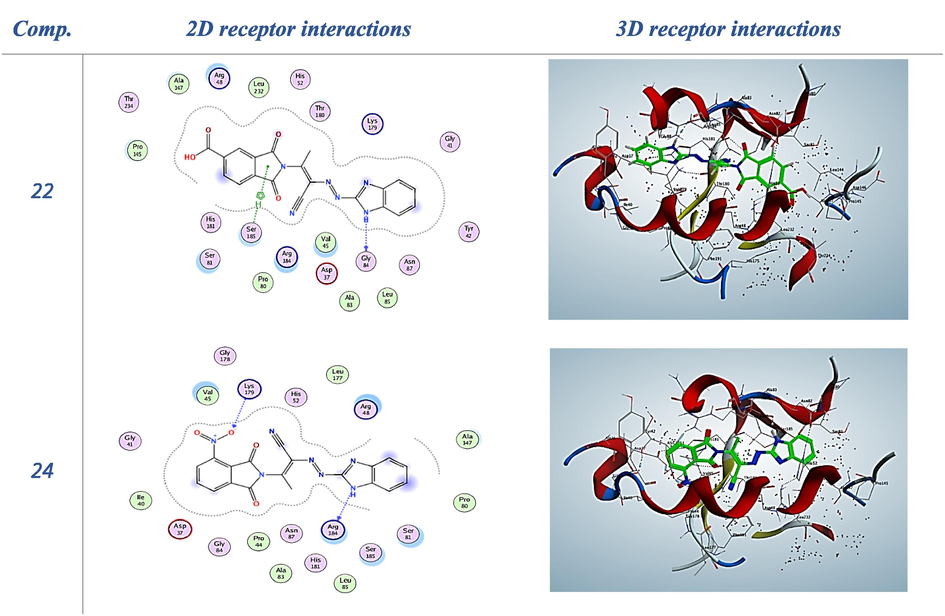
2D and 3D receptor interactions of the promising synthesized compounds.
4 Experimental
4.1 N-(1H-benzo[d]imidazol-2-yl)carbonohydrazonoyl Dicyanide (2)
It was prepared according to the previously reported work (Labute, 2009).
Synthesis of compounds (3–8).
4.2 General procedure
A mixture of N-(1H-benzo[d]imidazol-2-yl)carbonohydrazonoyl dicyanide (2) (5 mmol) and the appropriate secondary amine namely; piperidine (0.50 mL, 5 mmol), morpholine (0.43 mL, 5 mmol), piperazine (0.43 g, 5 mmol), diphenylamine (0.85 g, 5 mmol), N-methylglucamine (0.98 g, 5 mmol), diethanolamine (0.48 mL, 5 mmol), in refluxed EtOH (20 mL) was heated for 6 h. The reaction mixture was then cooled to room temperature and the separated solid material was filtered off and recrystallized from ethanol to give enaminonitrile derivatives 3–8.
4.3 (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(piperidin-1-yl)acrylonitrile (3)
Brown crystals; yield 68 %; mp 220–222 °C. IR (KBr): ν/cm−1 = 3450,3375 (NH2), 3150 (NH), 2960 (C—H, stretching), 2180, (CN), 1535,1530 (2 N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 1.55–1.70 (m, 2H, CH2), 1.72–1.85 (m, 4H, 2CH2), 3.06 (t, 4H, CH2-N-CH2, J = 6.5 Hz), 6.51 (s, 2H, NH2), 7.29 (d, 2H, Ar-H), 7.60 (d, 2H, Ar-H), 12.06 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 24.6, 26.0, 52.7, 87.5, 112.2, 114.6, 123.0, 135.7, 141.5, 173.5. MS (EI, 70 eV): m/z (%) 295 (M+, 30), 197 (11), 169 (15), 163 (76), 150 (10), 120 (45), 105 (78), 84 (35), 77 (1 0 0). Anal. for C15H17N7 (295.35): Calculated.: C, 61.00; H, 5.80; N, 33.20 %. Found: C, 60.88; H, 5.76; N, 33.10 %.
4.4 (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-morpholinoacrylonitrile (4)
Brown crystals; yield 70 %; mp 180–183 °C. IR (KBr): ν/cm−1 = 3455,3370 (NH2), 3155 (NH), 2967 (C—H, stretching), 2185, (CN), 1549, 1530 (2 N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 3.14 (t, 4H, CH2-N-CH2, J = 6.0 Hz), 3.67 (t, 4H, CH2-O-CH2, J = 6.0 Hz), 6.54 (s, 2H, NH2), 7.42 (d, 2H, Ar-H), 7.75 (d, 2H, Ar-H), 11.81 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 51.5, 66.8, 87.6, 112.4, 114.5, 123.2, 135.5, 141.4, 173.6. MS (EI, 70 eV): m/z (%) 297 (M+, 25), 197 (11), 169 (15), 163 (76), 150 (10), 120 (45), 105 (78), 84 (35), 77 (1 0 0). Anal. for C14H15N7O (297.32): Calculated: C, 56.56; H, 5.09; N, 32.98 %. Found: C, 56.38; H, 4.98; N, 32.77 %.
4.5 (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(piperazin-1-yl)acrylonitrile (5)
Brown crystals; 75 % yield; m.p. 248–250 °C. IR (KBr): ν/cm−1 = 3453, 3380 (NH2), 3145 (NH), 2929 (C—H, stretching), 2182, (CN), 1500, 1520 (2 N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 3.16 (t, 4H, CH2-NH-CH2, J = 7.0 Hz), 3.69 (t, 4H, CH2-N-CH2, J = 7.0 Hz), 6.54 (s, 2H, NH2), 7.32 (d, 2H, Ar-H), 7.60 (d, 2H, Ar-H), 11.21 (s, 1H, NH), 11.81 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 47.8, 52.6, 87.4, 112.5, 114.3, 123.1, 135.8, 141.6, 173.4. MS (EI, 70 eV): m/z (%) 296 (M+, 31) 292 (48), 268 (37), 249 (37), 216 (34), 203 (32), 188 (33), 169 (55), 165 (40), 147 (46), 135 (37), 117 (37), 92 (57), 77 (1 0 0). Anal. for C14H16N8 (296.34): Calculated: C, 56.74; H, 5.44; N, 37.81 %. Found: C, 56.66; H, 5.38; N, 37.77 %.
4.6 (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(diphenylamino)acrylonitrile (6)
Brown crystals; yield 80 %; mp 186–189 °C. IR (KBr): ν/cm−1 = 3455,3385 (NH2), 3160 (NH), 2929 (C—H, stretching), 2172, (CN), 1540, 1520 (2 N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 89.5, 112.2, 114.6, 121.9, 123.0, 127.0, 129.6, 135.7, 141.5, 141.9, 165.9. MS (EI, 70 eV): m/z (%) 379 (M+, 30), 368 (12), 357 (20), 320 (14), 292 (12), 274 (8), 215 (21), 187 (28), 169 (11), 144 (9), 117 (15), 105 (16), 91 (37), 77 (1 0 0). Anal. for C22H17N7 (379.43): Calculated: C, 69.64; H, 4.52; N, 25.84 %. Found: C, 69.53; H, 4.33; N, 25.77 %.
4.7 (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)acrylonitrile (7)
Orange crystals; yield 90 %; mp 210–213 °C. IR (KBr): ν/cm−1 = 3410 (OH) + (NH2), 3260 (NH), 2929 (C—H, stretching), 2222 (CN), 1550 (2 N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 3.04 (s, 3H, N-CH3), 3.56–3.62 (m, 8H, 4CH + 2CH2), 3.93 (s, 1H, OH), 4.35–4.51 (m, 4H, 4OH), 6.45 (s, 2H, NH2), 7.25 (d, 2H, Ar-H,), 7.53 (d, 2H, Ar-H), 12.53 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 43.1, 64.4, 70.5, 72.3, 72.6, 72.9, 91.5, 112.2, 114.6, 123.3, 135.6, 141.5, 170.5. MS (EI, 70 eV): m/z (%) 405 (M+, 60), 370 (69), 350 (63), 331 (68), 305 (63), 289 (69), 268 (68), 258 (77), 245 (86), 234 (69), 216 (77), 203 (68), 189 (80), 174 (85), 158 (1 0 0), 144 (77), 136 (76), 112 (71), 109 (82). Anal. for C17H23N7O5 (405.42): Calculated: C, 50.36; H, 5.72; N, 24.18 %. Found: C, 50.21; H, 5.66; N, 24.02 %.
4.8 (2Z)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-amino-3-(bis(2-hydroxyethyl)amino)acrylonitrile (8)
Brown crystals; yield 70 %; mp 184–186 °C. IR (KBr): ν/cm−1 = 3415–3455 (OH), 3170 (NH), 3375,3409 (NH2), 2180, (CN), 1535, 1515 (2 N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 3.16 (t, 4H, CH2-N-CH2, J = 6.5 Hz), 3.87 (t, 4H, 2O-CH2, J = 6.5 Hz), 4.85 (s, 2H, 2OH), 6.50 (s, 2H, NH2), 7.40 (d, 2H, Ar-H,), 7.70 (d, 2H, Ar-H), 12.53 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 53.1, 59.4, 87.5, 112.1, 114.3, 123.1, 135.8, 141.3, 173.5. MS (EI, 70 eV): m/z (%) 315 (M+, 28), 272 (20), 197 (17), 167 (23), 120 (25), 105 (15), 92 (59), 77 (1 0 0). Anal. for C14H17N7O2 (315.34): Calculated: C, 53.33; H, 5.43; N, 31.09 %. Found: C, 53.11; H, 5.30; N, 30.88 %.
4.9 4-((1H-benzo[d]imidazol-2-yl)diazenyl)-1H-pyrazole-3,5-diamine (9)
To a solution of 2 (5 mmol) in refluxing ethanol was added hydrazine hydrate (0.24 mL, 5 mmol) and heating was continued for 30 min then cool to room temperature and filter the solid material and purified by crystallization from ethanol to give pyrazole derivative 9.
Orange powder; yield 80 %; mp 292–295 °C. IR (KBr): ν/cm−1 = 3414–3350 (2NH2), 3190 (NH), 1530, 1550 (N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 6.20 (s, 4H, 2NH2), 7.40 (d, 2H, Ar-H,), 7.70 (d, 2H, Ar-H), 12.53 (s, 1H, NH), 12.75 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 74.4, 112.2, 123.0, 135.7, 141.5, 151.3. MS (EI, 70 eV): m/z (%) 242 (M+, 29), 219 (5), 185 (5), 173 (5), 153 (5), 129 (5), 125 (71), 105 (15), 91 (14), 77 (1 0 0). Anal. for C10H10N8 (242.25): Calculated: C, 49.58; H, 4.16; N, 46.26 %. Found: C, 49.44; H, 4.10; N, 46.12 %.
4.10 4-amino-1-(1H-benzo[d]imidazol-2-yl)-6-imino-1,6-dihydropyridazine-3,5-dicarbonitrile (11)
To a solution of sodium ethoxide “prepared by adding 1.0 g sodium metal into EtOH (20 mL)” in ethanol (25 mL) was added a mixture of 2 (5 mmol) and malononitrile (0.33 g, 5 mmol). The reaction was heated for 8 h. Then cool to room temperature and diluted with water to obtain solid material which is filtered off and purified by crystallization from ethanol to yield derivative 11.
Brown powder; yield 60 %; mp 202–205 °C. IR (KBr): ν/cm−1 = 3450,3336 (NH2), 3214 (NH), 2200, 2180 (2CN). 1H NMR (DMSO‑d6) δ (ppm): 4.80 (s, 2H, NH2), 7.40 (d, 2H, Ar-H,), 7.70 (d, 2H, Ar-H), 9.40 (s, 1H, NH), 12.53 (s, 1H, NH). 13C NMR (DMSO‑d6) δ (ppm): 81.2, 115.2, 115.3, 115.8, 123.2, 136.6, 141.5, 154.4, 155.0, 162.7. MS (EI, 70 eV): m/z (%) 276 (M+, 28), 321 (5), 292 (5), 274 (24), 244 (5), 216 (9), 197 (19), 187 (14), 169 (15), 142 (5), 117 (5), 105 (25), 91 (30), 77 (1 0 0). Anal. for C13H8N8 (276.26): Calculated C, 56.52; H, 2.92; N, 40.56 %. Found: C, 56.48; H, 2.88; N, 40.44 %.
4.11 2-(5-((1H-benzo[d]imidazol-2-yl)diazenyl)-4-amino-6-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)pyrimidin-2-yl)acetonitrile (14)
To a solution of 7 (5 mmol) in absolute ethanol (25 mL) containing sodium ethoxide “prepared by adding 1.0 g sodium metal into EtOH (20 mL)” was added malononitrile (0.33 g, 5 mmol) and the reaction was heated for 10 h. The obtained solid product on cooling filtered off and recrystallized from ethanol to give compound 14.
Orange crystals; yield 70 %; mp 192–194 °C. IR (KBr): ν/cm−1 = 3450 (OH), 3350 (NH2), 3300 (NH), 2197 (CN), 1620 (C⚌N), 1550, 1521 (N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 3.04 (s, 3H, N-CH3), 3.56–3.62 (m, 8H, 4CH + 2CH2), 3.93 (s, 1H, OH), 4.35–4.51 (m, 4H, 4OH), 7.25 (d, 2H, Ar-H,), 7.53 (d, 2H, Ar-H), 8.45 (s, 2H, NH2), 12.53 (s, 1H, NH). MS (EI, 70 eV): m/z (%) 470 (M+−1, 20), 459 (9), 446 (17), 424 (17), 407 (15), 197 (18), 120 (28), 105 (13), 84 (1 0 0), 77 (59). Anal. for C20H25N9O5 (471.48): Calculated: C, 50.95; H, 5.34; N, 26.74 %. Found: C, 49.99; H, 5.23; N, 26.66 %.
4.12 (2R,3R,4R,5S)-6-(((1E)-2-((1-acetyl-1H-benzo[d]imidazol-2-yl)diazenyl)-1-(N-acetylacetamido)-2-cyanovinyl)(methyl)amino)hexane-1,2,3,4,5-pentayl pentaacetate (15)
Acetic anhydride (25 mL) was added to compound 7 (5 mmol) and heated for 8 h. The reaction is then diluted with ice-cold water to give brown solid material collected by filtration and recrystallized from ethanol to give 15.
Brown powder; yield 70 %; mp 107–109 °C. IR (KBr): ν/cm−1 = 2960 (C—H, stretching), 2174 (CN), 1730–1705 (5CO), 1680 (2CO), 1540, 1530 (N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 2.03 (m, 15H, 5OCOCH3), 2.29 (s, 6H, 2 N-COCH3), 2.71 (s, 3H, N-COCH3), 3.39 (s, 3H, N-CH3), 3.55–3.76 (m, 2H, N-CH2), 4.20–4.42 (m, 2H, O-CH2), 5.20–5.85 (m, 4H, 4CH), 7.33 (d, 2H, Ar-H,), 7.65 (d, 2H, Ar-H). MS (EI, 70 eV): m/z (%) 741 (M+, 40), 554 (24), 553 (42), 536 (32), 524 (34), 512 (33), 494 (27), 484 (34), 465 (37), 452 (40), 438 (42), 425 (37), 412 (36), 395 (36), 391 (50), 370 (42), 350 (32), 340 (35), 239 (35), 162 (31), 149 (36), 134 (75), 113 (7), 105 (19), 92 (22), 77 (1 0 0). Anal. for C33H39N7O13 (741.71): Calculated: C, 53.44; H, 5.30; N, 13.22 %. Found: C, 53.17; H, 5.12; N, 13.10 %.
4.13 (2R,3R,4R,5S)-6-((1-acetyl-3-((1-acetyl-1H-benzo[d]imidazol-2-yl)diazenyl)-4-amino-6-oxo-1,6-dihydropyridin-2-yl)(methyl)amino)hexane-1,2,3,4,5-pentayl pentaacetate (16)
A solution of Compound 15 (5 mmol) in DMF (20 mL) containing a few drops of triethyl amine (TEA) was heated for a prolonged time. Then the reaction cooled to room temperature and diluted with ice-cooled water to give dark solid material collected by filtration and recrystallized from ethanol to yield compound 16.
Black crystals; yield 50 %; mp 148–150 °C. IR (KBr): ν/cm−1 = 3380,3350 (NH2), 2960 (C—H, stretching), 1730–1705 (5CO), 1680 (2 N-CO), 1540, 1530 (N⚌N). MS (EI, 70 eV): m/z (%) 741 (M+, 23), 720 (15), 553 (51), 525 (63), 449 (33), 421 (28), 404 (61), 389 (15), 359 (48), 210 (33), 133 (48), 92 (78), 77 (86).
4.14 (2R,3R,4R,5S)-6-(((1E)-2-((1-acetyl-1H-benzo[d]imidazol-2-yl)diazenyl)-2-cyano-1-(((E)-ethoxymethylene)amino)vinyl)(methyl)amino)hexane-1,2,3,4,5-pentayl pentaacetate (17)
To a solution of compound 7 (5 mmol) in acetic anhydride (10 mL) was added triethyl orthoformate (0.83 mL, 5 mmol). The reaction was heated at 80 °C for 4 h. The reaction was then cooled and diluted with ice-cold water to give a brown precipitate which was collected by filtration and recrystallized from ethanol to give compound 17.
Brown crystals; yield 60 %; mp 113–115 °C. IR (KBr): ν/cm−1 = 2940–2836 (CH stretching aliphatic), 2179 (C≡N), 1730–1710 (5CO), 1543, 1680 (CO), 1515 (N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 1.24 (t, 3H, CH3, J = 6.0 Hz), 2.02 (m, 15H, 5OCOCH3), 2.65 (s, 3H, N-COCH3), 3.45 (s, 3H, N-CH3), 3.65 (q, 2H, CH2), 3.85–4.05 (m, 2H, N-CH2), 4.30–4.50 (m, 2H, O-CH2), 5.20, 5.85 (m, 4H, 4CH), 7.30 (d, 2H, Ar-H,), 7.64 (d, 2H, Ar-H), 8.50 (s, 1H, CH). MS (EI, 70 eV): m/z (%) 713 (M+, 55), 526 (51), 525 (68), 506 (62), 490 (66), 480 (76), 464 (64), 453 (66), 445 (82), 433 (73), 414 (89), 393 (82), 376 (64), 357 (66), 338 (84), 332 (74), 314 (66), 298 (76), 274 (62), 258 (68), 239 (71), 229 (68), 214 (76), 203 (1 0 0), 183 (92), 152 (69), 134 (91), 116 (71), 111 (83), 97 (77). Anal. for C32H39N7O12 (713.70): Calculated: C, 53.83; H, 5.51; N, 13.74 %. Found: C, 53.77; H, 5.46; N, 13.66 %.
4.15 (1E)-N'-((1E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-2-cyano-1-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)vinyl)-N,N-dimethylformimidamide (18)
A mixture of compound 7 (4 mmol) and N,N-dimethylformamide-dimethylacetal (0.53 mL, 4 mmol) was refluxed in xylene (15 mL) for 5 h. The separated brownish crystals filtered off and crystallized from ethanol to yield 18.
Brown crystals; yield 50 %; mp 126–128 °C. IR (KBr): ν/cm−1 = 3455–3443 (OH), 2931–2856 (C—H, aliphatic), 2177 (CN), 1616 (C⚌N), 1550, 1530 (N⚌N). 1H NMR (DMSO‑d6) δ (ppm): 2.90 (s, 6H, N(CH3)2), 3.04 (s, 3H, CH3), 3.45 (d, 2H, CH2, J = 7.0 Hz), 3.65 (m, 6H, 4OH + CH2), 3.93 (s, 1H, OH), 4.35–4.51 (m, 4H, 4CH), 7.25 (d, 2H, Ar-H,), 7.53 (d, 2H, Ar-H), 8.73 (s, 1H, CH), 12.53 (s, 1H, NH). MS (EI, 70 eV): m/z (%) 460 (M+, 40), 456 (55), 437 (20), 427 (15), 405 (15), 391 (15), 371 (6), 346 (15), 323 (5), 312 (6), 285 (21), 279 (5), 161 (15), 137 (15), 85 (55), 71 (60).
Anal. for C20H28N8O5 (460.50): Calculated: C, 52.17; H, 6.13; N, 24.33 %. Found: C, 52.10; H, 6.00; N, 24.11 %.
5 Reaction of compound 7 with activated anhydrides
5.1 General procedure
In DMF solution catalyzed with drops of glacial acetic acid, an equivalent quantity of compound 7 (5 mmol) and acid anhydride derivatives namely; phthalic anhydride (0.74 g, 5 mmol), quinolinic anhydride (0.49 g, 5 mmol), pyromellitic anhydride (1.18 g, 5 mmol), 1,2,4-benzene tricarboxylic anhydride (0.96 g, 5 mmol), 4-nitrophthalic anhydride (0.97 g, 5 mmol), 3-nitrophthalic anhydride (0.97 g, 5 mmol) and tetrabromophthalic anhydride (2.32 g, 5 mmol) was refluxed for 12–16 h. The reaction mixture was poured into ice-cold water. The precipitated solid was filtered off, washed with ethanol, dried, and recrystallized from ethanol to yield derivatives 19–25, respectively.
5.2 (2E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-(1,3-dioxoisoindolin-2-yl)-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)acrylonitrile (19)
Brown powder; yield 30 %; mp 188–190 °C. IR (KBr): ν/cm−1 = 3453–3350 (OH), 2220 (CN), 1712, 1686 (2CO, amide), 1555 (N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 50.5, 54.4, 64.2, 70.6, 72.3, 72.8, 89.5, 112.0, 114.8, 123.2, 123.7, 132.0, 132.2, 135.5, 141.8, 160.8, 165.5. MS (EI, 70 eV): m/z (%) 535 (M+, 15), 512 (25), 500 (25), 480 (25), 470 (30), 452 (1 0 0), 434 (55), 419 (20), 196 (20), 120 (10), 105 (20), 93 (45), 77 (1 0 0), 64 (25). Anal. for C25H25N7O7 (535.52): Calculated: C, 56.07; H, 4.71; N, 18.31 %. Found: C, 55.98; H, 4.66; N, 18.25 %.
5.3 (2E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-(5,7-dioxo-5,7-dihydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6 pentahydroxyhexyl)amino)acrylonitrile (20)
Brown crystals; yield 30 %; mp 162–165 °C. IR (KBr): ν/cm−1 = 3455–3440 (OH), 2195 (CN), 1690, 1680 (2CO, amide), 1530, 1510 (N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 50.5, 54.4, 64.2, 70.6, 72.3, 72.8, 89.5, 112.0, 114.8, 120.0, 123.2, 128.4, 135.2, 135.5, 141.8, 146.1, 152.6, 160.8, 162.3, 165.5. MS (EI, 70 eV): m/z (%) 536 (M+, 30), 536 (30), 494 (60), 469 (10), 452 (20), 429 (15), 405 (25), 389 (25), 377 (25), 361 (30), 341 (30), 319 (30), 306 (25), 285 (25), 276 (60), 259 (40), 196 (30), 181 (15), 167 (15), 143 (20), 128 (1 0 0), 117 (60), 101 (65), 93 (1 0 0), 77 (1 0 0), 59 (85). Anal. for C24H24N8O7 (536.51): Calculated: C, 53.73; H, 4.51; N, 20.89 %. Found: C, 53.66; H, 4.35; N, 20.75 %.
5.4 (2E,2′E)-3,3′-(1,3,5,7-tetraoxo-5,7-dihydropyrrolo[3,4-f]isoindole-2,6(1H,3H)-diyl)bis(2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)acrylonitrile) (21)
Brown powder; yield 20 %; mp 143–145 °C. IR (KBr): ν/cm−1 = 3485–3345 (OH), 2180 (2CN), 1690–1640 (4CO, amide), 1570 (2 N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 50.7, 54.1, 64.4, 70.5, 72.0, 72.6, 89.7, 112.2, 114.2, 123.0, 125.2, 135.5, 135.7, 141.5. 160.5, 165.8. Anal. for C44H44N14O14 (992.92): Calculated: C, 53.23; H, 4.47; N, 19.75 %. Found: C, 53.10; H, 4.35; N, 19.66 %.
5.5 2-((1E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-2-cyano-1-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)vinyl)-1,3-dioxoisoindoline-5-carboxylic acid (22)
Brown powder; yield 20 %; mp 188–191 °C. IR (KBr): ν/cm−1 = 3500–3440 (OH), 2195 (CN), 1690, 1680, (2CO, amide), 1730 (CO), 1530, 1510 (N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 50.8, 54.3, 64.3, 70.5, 72.1, 72.7, 89.6, 112.3, 114.5, 123.0, 123.3, 124.8, 131.9, 133.7, 133.8, 135.6, 137.2, 141.6, 160.4, 165.9, 169.3. MS (EI, 70 eV): m/z (%) 579 (M+, 25), 563 (40), 551 (40), 535 (30), 522 (40), 514 (30), 386 (10), 371 (15), 281 (10), 266 (40), 197 (10), 167 (10), 148 (15), 120 (20), 105 (20), 93 (75), 77 (1 0 0), 64 (60). Anal. for C26H25N7O9 (579.53): Calculated: C, 5.89; H, 4.35; N, 16.92 %. Found: C, 53.77; H, 4.26; N, 16.78 %.
5.6 (2E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)-3-(5-nitro-1,3-dioxoisoindolin-2-yl)acrylonitrile (23)
Brown powder; yield 30 %; mp 179–182 °C. IR (KBr): ν/cm−1 = 3445–3415 (OH), 2180 (CN), 1659 (2CO, amide), 1555 (N⚌N), 1519, 1318 (NO2). 13C NMR (DMSO‑d6) δ (ppm): 50.6, 54.2, 64.5, 70.4, 72.2, 72.5, 89.8, 112.1, 114.3, 121.2, 123.2, 127.4, 128.5, 132.9, 135.7, 138.5, 141.4, 151.4, 160.3, 165.7. MS (EI, 70 eV): m/z (%) 580 (M+, 15), 544 (25), 522 (30), 514 (40), 452 (20), 276 (15), 196 (25), 120 (15), 105 (20), 93 (60), 77 (1 0 0), 64 (25). Anal. for C25H24N8O9 (580.51): Calculated: C, 51.73; H, 4.17; N, 19.30 %. Found: C, 51.65; H, 4.12; N, 19.21 %.
5.7 (2E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)-3-(4-nitro-1,3-dioxoisoindolin-2-yl)acrylonitrile (24)
Brown powder; yield 20 %; mp 145–147 °C. IR (KBr): ν/cm−1 = 3455–3440 (OH), 2195 (CN), 1690, 1680 (2CO, amide), 1550, 1350 (NO2), 1530, 1510 (N⚌N). MS (EI, 70 eV): m/z (%) 580 (M+, 15), 544 (25), 522 (30), 514 (40), 452 (20), 276 (15), 196 (25), 120 (15), 105 (20), 93 (60), 77 (1 0 0), 64 (25). Anal. for C25H24N8O9 (580.51): Calculated: C, 51.73; H, 4.17; N, 19.30 %. Found: C, 51.68; H, 4.15; N, 19.25 %.
5.8 (2E)-2-((1H-benzo[d]imidazol-2-yl)diazenyl)-3-(methyl((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)amino)-3-(4,5,6,7-tetrabromo-1,3-dioxoisoindolin-2-yl)acrylonitrile (25)
Black powder; yield 60 %; mp over 300 °C. IR (KBr): ν/cm−1 = 3455–3440 (5OH), 2198 (CN), 1690, 1680 (2CO, amide), 1530, 1510 (N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 50.7, 54.1, 64.4, 70.5, 72.0, 72.6, 89.7, 112.2, 114.6, 123.0, 124.9, 129.8, 135.8, 139.0, 141.5, 160.5, 165.8. MS (EI, 70 eV): m/z (%) 846 (M+−5, 60), 810 (65), 786 (60), 782 (80), 764 (60), 752 (60), 741 (50), 716 (40), 710 (60), 684 (70), 664 (70), 646 (50), 623 (65), 596 (70), 581 (70), 568 (55), 543 (70), 394 (20), 197 (10), 152 (15), 120 (10), 105 (15), 93 (1 0 0), 77 (95), 64 (55). Anal. for C25H21Br4N7O7 (850.10): Calculated C, 35.28; H, 2.49; N, 11.52 %. Found: C, 35.14; H, 2.23; N, 11.46 %.
5.9 Reaction with of compound 7 with 3,4,9,10-perylenetetracarboxylic dianhydride
A mixture of equivalent quantities of compound 7 (5 mmol), 3,4,9,10-perylenetetracarboxylic dianhydride (1.96 g, 5 mmol) and anhydrous sodium acetate (freshly fused) (5 mmol) was heated for 3 h in a silicon oil bath at 150 °C. Then the reaction was left to cool at room temperature. The separated solid material was washed with conc. HCl and water were filtered and recrystallized from ethanol to give compound 26.
5.10 (2E,2′E)-3,3′-(1,3,8,10-tetraoxo-1,3,8,10-tetrahydroanthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-2,9-diyl)bis(3-(methyl((2R,3S,4S,5S)-2,3,4,5,6-pentahydroxyhexyl)amino)-2-((E)-phenyldiazenyl)acrylonitrile) (26)
Black powder; yield 40 %; mp over 300 °C. IR (KBr): ν/cm−1 = 3450–3440 (10OH), 2190, 2180 (2CN), 1700–1680 (4CO), 1530, 1510 (2 N⚌N). 13C NMR (DMSO‑d6) δ (ppm): 37.5, 52.8, 54.1, 64.4, 70.5, 72.4, 72.5, 72.6, 80.1, 119.5, 122.6, 124.5, 125.7, 126.1, 129.2, 130.9, 135.7, 150.9, 159.3. Anal. for C56H50N10O14 (1087.07): Calculated: C, 61.87; H, 4.64; N, 12.89 %. Found: C, 61.76; H, 4.58; N, 12.77 %.
6 Pharmacology
6.1 Antioxidant screening assay (ABTS method)
It was carried out according to the previously reported work (Martinez and Chacon-Garcia, 2005).
6.2 Antioxidant activity screening assay for erythrocyte haemolysis
It has been also carried out according to the previously published work (Martinez and Chacon-Garcia, 2005).
6.3 Bleomycin-dependent DNA damage assay
It was recorded according to the reported work (Martinez and Chacon-Garcia, 2005).
6.4 Antitumor activity
In vitro Cytotoxic Activity by MTT assay (Fadda et al., 2009).
Samples were prepared for assay by dissolving tested compounds in 50 μL of DMSO and diluting aliquots into a sterile culture medium at 0.4 mg/mL. These solutions were subdiluted to 0.02 mg/mL in a sterile medium and the two solutions were used as stocks to test samples at 100, 50, 20, 10, 5, 2, and 1 mg/mL in triplicate in the wells of microtiter plates. The test compounds 4–26 were assayed in triplicate on monolayers grown in Dulbecco’s modified Eagle’s medium supplemented with 10 % (v/v) calf serum (Hyclone Laboratories, Ogden, UT), 60 mg/mL Penicillin G and 100 mg/mL streptomycin sulfate maintained at 37 ∘C in a humidified atmosphere containing about 15 % (v/v) CO2 in the air. All medium components were obtained from Sigma Chemical Co., St. Louis, MO unless otherwise indicated. Cells stocks were maintained at 34 ∘C in culture flasks filled with medium supplemented with 1 % (v/v) calf serum. Subcultures of cells for screening were grown in the wells of microtiter trays (Falcon Microtest III 96-wells trays, Becton Dickinson Labware, Lincoln Park, NJ) by suspending cells in medium following trypsin-EDTA treatment, counting the suspension with a hemocytometer, diluting in medium containing 10 % calf serum to 2 × 104 cells per 200 mL culture, aliquoting into each well of a tray, and culturing until confluent. Microtiter trays with confluent monolayer cultures of cells were inverted, the medium was shaken out and replaced with serial dilutions of sterile compounds in triplicate in 100 μL medium followed by the tittered virus in 100 μL medium containing 10 % (v/v) calf serum in each well. In each tray, the last row of wells was reversed for controls that were not treated with compounds. The trays were cultured for 96 h. The trays were inverted onto a pad of paper towels, the remaining cells were rinsed carefully with medium and fixed with 3.7 % (v/v) formaldehyde in saline for at least 20 min. The fixed cells were rinsed with water and examined visually. The cytotoxic activity is identified as confluent, relatively unaltered monolayers of stained cells treated with compounds. Cytotoxicity was estimated as the concentration that caused approximately 50 % loss of the monolayer. 5-fluorouracil was used as a positive control.
6.5 Molecular docking
Chemdraw 12.0 was used to create molecular modeling for the most active chemicals that would be docked using the Molecular Operating Environment software (2015). The London DG force and force field energy were used to fine-tune the results. All minimizations were completed with MMFF 94 (Merck molecular force field 94) until a root mean square deviation (RMSD) gradient of 0.1 kcal mol-1A-1 was reached (Aziz, 2021; Singh, 2015), and partial charges were estimated automatically. The ligand's binding affinity was determined using the MOE software's dock function (S, Kcal/mol) scoring function. The enzyme's X-ray crystal structure (PDB ID: 2X08, resolution: 2.01) was retrieved in PDB format from the protein data bank (https://www.rcsb.org/structure/2X08). The enzyme was prepared for docking studies through removed water, added all hydrogen bonds, fixed potential, generated dummy atoms from the resulting alpha spheres [55] then analyzed the ligand's interaction with the active site's amino acids. The best Docking Score is obtained as the most negative value for the active ligands [56, 57].
7 Conclusion
We have synthesized several compounds based on benzimidazole, some of which contain secondary amines. The compounds are synthesized and then characterised using several techniques, such as 1H NMR, 13C NMR, FT-IR, MASS, and CHN elemental analysis. Using ABTS as a stable radical agent, the activity of the synthesized compounds in radical scavenging behaviour was assessed. The compounds were found to have effective radical scavenging abilities. The ABTS method was used to examine the synthetic compounds' capacity to scavenge free radicals. The activity of compounds 14 in the reaction with ABTS was the best of all the produced compounds. Additionally, it was found that most of the synthesized compounds are very active against the tested cell lines. The most effective anti-cancer synthesized compounds were evaluated for their potential capacity to bind to the human cyclin-dependent kinase 2. (CDK2). The compounds under investigation are prospective candidates for the development of innovative anticancer drugs that target CDK2 in the future based on molecular docking outcomes and physicochemical and pharmacokinetic (ADMET) predictions.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (a) Zaki MEA, Saliman HA, Hickal OA, Rashad AE. Z. Naturforsch., C: Biosci. 61 (2006) 1–5; (b) Sheng CK, Li JH, Hideo N. J. Med. Chem. 27 (1984) 539–544.
- Spectrochim. Acta Part A: Mol. Biomol. Spectrosc.. 2012;89:123-128.
- J. Heterocycl. Chem.. 2018;55:736-742.
- Design and discovery of new 1, 2, 4-triazolo [4, 3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors. Archiv. Pharma.. 2021;354(3):2000237.
- [Google Scholar]
- Aloisio, C.; Gomes De Oliveira, A.; Longhi, M. Drug Dev. Ind. Pharm. 2014, 40, 919–928.
- Sulfur Silicon Rel. Elem.. 2008;183:1766-1782.
- Design, synthesis, biological evaluation, 2D-QSAR modeling, and molecular docking studies of novel 1H-3-indolyl derivatives as significant antioxidants. int. J. Mol. Sci.. 2021;22(19)
- [Google Scholar]
- DNA-binding of drugs used in medicinal therapies. Curr. Med. Chem.. 2002;9(3):321-348.
- [Google Scholar]
- Eur. J. Med. Chem.. 2008;43:2122-2129.
- Storage. 1926;100:293-1225.
- J. Pharm. Biomed. Anal.. 2013;73:24-28.
- Child, R.G.; Wilkinson, R.G.; Tomcu-Fucik, Chem. Abstr. 1977, 87, 6031
- Dyes Pigments. 2000;44:181-193.
- Russ. J. Org. Chem.. 2008;44:412-420.
- Adv. Heterocycl. Chem.. 1997;69:129.
- Elkholy A, Al-Qalaf F, Elnagdi MH. Arkivoc xiv (2008) 124–131. 420
- Computational insights on the potential of some NSAIDs for treating COVID-19: priority set and lead optimization. Molecules. 2021;26:3772.
- [Google Scholar]
- J. Oleo Sci.. 2016;65:177-192.
- Dyes Pigments. 2000;44:41-48.
- Pigm. Resin Technol.. 2016;45:10-17.
- Fadda, A.A.; Elattar, Kh. M. J. Chem. 2013, 10 pages, 2013.
- Am. J. Org. Chem.. 2012;2:52-57.
- J. Biosci. Med.. 2015;3:114-123.
- J. Chem. Technol. Biotechnol.. 1994;61:343-349.
- Fadda AA, Etman HA, Ali MM, A., Indian J. Fibre Text. Res. 20 (1995) 34–38
- Fibre Text. Res.. 1995;20:108.
- J. Chem. Technol. Biotechnol.. 1995;62:165-169.
- Boll. Chim. Farmac.. 1998;137:191-194.
- Heterocycl. Commun.. 2006;12:47-52.
- Bioorg. & Med. Chem.. 2009;17:5096-5105.
- Arch. Pharm. Chem. Life Sci (Archiv. Pharma.). 2012;345:378-385.
- Archiv. Pharma.. 2013;346:53-61.
- J. Med. Chem.. 1972;15:435-446.
- Am. J. Physiol. Cell Physiol.. 2005;289:C1295-C1302.
- Appl. Microbiol. Biotech.. 2008;78:361-439.
- J. Mol. Graph. Model.. 2017;71:28-39.
- Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins Struct. Funct. Bioinf.. 2009;75(1):187-205.
- [Google Scholar]
- Lohse E. Pharmazie 41 (1986) 815; C.A. 106 (1987) 95471y.
- Bulg. Chem. Comm.. 2008;40:147-159.
- Phosphorus, Sulfur Silicon Rel. Elem.. 2009;184:719-732.
- J. Med. Chem.. 1997;40:4154.
- The search of DNA-intercalators as antitumoral drugs: what it worked and what did not work. Curr. Med. Chem.. 2005;12(2):127-151.
- [Google Scholar]
- J. Photochem. Photobiol. B Biol.. 1990;7:189-198.
- Nifantiev, N.E.; Yashunsky, D.V. Water-Soluble Porphyrin Derivatives for Photodynamic Therapy, Their Use and Manufacture. European Patent EP1404678B1, 7 April 2004
- Chemosphere. 1986;15:479-491.
- EnzymeMicrob. Tech.. 2007;40:1758-2164.
- The Chemistry of the Hydrazo and Azoxy Groups, Part 1. New York: John Wiley; 1975.
- Photochem. Photobiol.. 2005;81:1505.
- ARKIVOC. 2008;14:180-190.
- Heterocycles. 2009;78:151-159.
- Singh, P., et al., Docking, synthesis and evaluation of antioxidant activity of 2, 4, 5-triaryl imidazole. Clin Med Biochem Open Access, 2015. 1(105): p. 2471-2663.1000105.
- Thoraya, A.; Farghaly, Abdallah ZA. Arkivoc 2008, 17, 295
- Proc. Natl. Acad. Sci. U. S. A.. 2000;97:7124-7129.
- Heterocycl. Commun.. 2004;10:97-102.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104505.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




