

Translate this page into:
COx -free H2 Production via Catalytic Decomposition of CH4 over Fe Supported on Tungsten oxide-activated Carbon Catalyst: Effect of Tungsten Loading
⁎Corresponding authors. kr.rawesh@gmail.com (Rawesh Kumar), aalfatesh@ksu.edu.sa (Ahmed S. Al-Fatesh)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Production of COx-free H2 from CH4 (a major global warming contributor) over cheap catalysts is a dominant task for the scientific community to accomplish environmental-friendly clean H2 energy sources. Herein, a tungsten oxide-activated carbon-supported Fe catalyst is prepared by impregnation method, characterized by X-ray diffraction, surface area-porosity measurement, temperature programmed reduction/oxidation and thermogravimetry analysis. 30wt.%Fe supported tungsten oxide incorporated activated carbon catalyst is found superior to 30 wt% Fe supported on activated carbon incorporated tungsten oxide due to higher surface area and high concentration of reducible catalytic active sites. 30wt.%Fe impregnated over 25 wt%WO3-75 wt%activated carbon support catalyst has the highest concentration of reducible surface-active species and it had excellent performance among other tungsten oxide incorporated catalysts. The catalyst showed 66.04% CH4 conversion, 63.12% H2 yield and / > 0.9 initially which didn’t fall below 35 % up to 160-minutes. Improper matching between the rate of carbon formation and the rate of diffusion over a highly crystalline 30Fe50W50Ac catalyst resulted in rapid deactivation.
Keywords
CH4 decomposition
Tungsten oxide-activated carbon support
Iron catalyst
YCH4/CCH4

1 Introduction
CH4 is a major contributor to global warming. A reaction like the decomposition of methane has great environmental importance as CH4 is consumed and its concentration is depleted in the environment. Again, this reaction has great economic feature as it produces clean energy source H2 and high-quality carbon without the formation of CO/CO2 (CH4 → C + 2H2). The separation of H2 gas from solid carbon is easier than the separation of two gases in other reforming processes (stream reforming; CH4 + H2O ⇌ CO + 3H2, dry reforming; CH4 + CO2 ⇌ 2CO + 2H2). However, the decomposition of methane through C–H cleavage occurs at very high reaction temperature (up to 1200 °C). To decrease the reaction temperature or bond dissociate energy of C–H prominently, the high temperature sustainable Fe, Co, Ni, Cu-based catalyst is required (Gajewski and Pao, 2011; Wu et al., 2009). When the electronic promotor La2O3 was added with physical mixture of Ni-Cu alloy with a mass ratio of 0.107, the rate of C–H dissociation was increased (Figueiredo et al., 2010). The interaction of deposited carbon (after CH4 decomposition) with lattice oxygen at high temperatures cannot be neglected which forms CO in low quantity (Choudhary et al., 2001). The catalyst community is trying to develop a catalyst for COx-free H2 production through CH4 decomposition.
Among the supported catalyst systems, Ni supported on activated carbon had drawn attention due to the in-situ generation of the active site (metallic Ni) through the reducibility of activated carbon in the carbonization process. Coal char derived from lignite coal showed minute CH4 decomposition in micropores. C–H bond dissociation energy of CH4 over coal char was found four times (89–105 kJ/mol) less than in uncatalyzed reaction (Bai et al., 2006). Prasad et al. (Sarada Prasad et al., 2011) prepared activated carbon from a coconut shell and impregnated Ni over it. Ni supported on activated carbon-showed an initial decrease of CH4 conversion in the first two hours and thereafter an increase in CH4 conversion. 10 wr% Ni supported on carbon (derived from coal liquefaction residue) (Zhang et al., 2013) showed a continuous rise of CH4 conversion (13 to 60%) at 850 °C during 9 h time on stream. Another thermally stable support that can hold Ni during high temperature reaction was tried. Nanosized Ni (prepared by citric acid at pH controlled condition) supported over high silica ZSM (Si/Al = 300) showed about 45% CH4 conversion for a small time (14 min) at 700 °C (Michalkiewicz and Majewska, 2014). Among Ni/HY, Ni/SiO2, Ni/H-ZSM; CO contamination was found lowest on Ni/SiO2. Ni supported on showed ∼ 10% CH4 conversion during the entire range of reaction temperatures 500 °C – 800 °C for 5 h (Dong et al., 2015). 30 wt% Ni loading over SiO2 support showed 15% CH4 conversion up to 8 h time (Venugopal et al., 2007). Longevity of Ni-supported catalyst was found in the following order Ni/MgO > Ni/SiO2 > Ni/LiAlO2 > Ni/ZrO2 (Bonura et al., 2006) in which Ni supported on MgO showed>30% CH4 conversion up to 210-minutes. Ni/MgO catalyst can be regenerated in the O2 stream and utilized again for the reaction without any prior reduction step. 10-40 wt% Ni-MgO catalyst prepared by hydrothermal method showed the presence of NiO-MgO solid solution with mesoporosity (Bai et al., 2021). CH4 and N2 gas feed (1:2 vol ratio) over 30–40% Ni-MgO reached above 45% H2-yield within 3 h at 600 °C. Karimi et al. studied the decomposition of CH4 (in CH4: N2 = 3: 17) over Ni supported on MgSiO3-(prepared by the coprecipitation method) (Karimi et al., 2021). The catalyst showed 64% CH4 conversion at 600 °C. Low La/Ni ratio in LaNiO3, La4Ni3O10, La3Ni2O7 and La2NiO4 was known for bulk carbon decomposition with high degree of graphitisation (Li et al., 2001). Ni-incorporated hydrotalcite was derived from 2: 0.7: 0.3 mol ratio of Ni: Al: La metal precursors respectively. It had strong metal support interaction and showed ∼ 30% CH4 conversion up to 24 h (Anjaneyulu et al., 2015).
The Co-Al mixed oxide had Co3O4 phase and Co2AlO4 (spinel) phases (Calgaro and Perez-Lopez, 2019; Zardin and Perez-Lopez, 2017). The catalyst reduced under CH4 (than under H2) had lower particle size and showed 75% CH4 conversion at 750 °C reaction temperature. In Co-Al mixed oxide, the Co3O4 phase favoured graphene formation. 20 wt% Co-impregnated Al2O3-coated silica fabric has strong metal support interaction and showed 90% CH4 conversion up to 11.6 h at 700 °C (Italiano et al., 2010). Cu and Ni supported on Alumina-was found better than Cu supported onalumina-catalyst because of the formation of Ni-Cu alloy. After reduction, 70%Ni–10%Cu–10%Fe/Al2O3 catalysts showed the formation of Ni-Cu-Fe alloy (Chesnokov and Chichkan, 2009). Alloy formation caused a decrease in the number of contacts between metal particles and thus sintering was prevented. Upon iron addition in 70%Ni–10%Cu/Al2O3 catalyst, H2 concentration remained between 71 and 77% and the diffusion coefficient of the carbon atom was increased three times (carbon nanofiber yield 136 g/g). At 15 ml/min methane flow rate, 65%Ni-10%Fe-25%SiO2 catalyst showed 20% CH4 conversion at 550 °C reaction temperature (Wang et al., 2012). However, the presence of Ni-Fe redox (in Ni2-xFexAl; x = Fe/Al) also functions as oxygen carrier (Huang et al., 2018) which can mitigate the target of COx-free H2 production.
In the mean of Ni, Co, and Cu free catalyst, a mechanochemical activation of LaFeO3 and CeO2 mixture had drawn attention. It caused an accumulation of oxygen vacancy about Fe+3 which became the sites of oxygen exchange between O2 form air to surface to bulk CeO2 (Pinaeva et al., 2013). However, in presence of oxygen; COx-free hydrogen production from CH4 was not possible over mechanochemical mixture of LaFeO3 and CeO2. If 60 wt% Fe supported on alumina catalyst was reduced under the H2 stream, iron oxide was reduced into metallic Fe (Ibrahim et al., 2015). The metallic Fe is an active site for CH4 decomposition. Fe supported on Al2O3- generated multiwalled nanotube and 77.2 % H2 yield up to 4 h at 700 °C. Decomposition of CH4, C2H4, and C2H2 over Iron-based catalysts was reported (Maroto Valiente et al., 2000; Qian et al., 2008). Jin et al. prepared activated carbon from coconut shell and impregnated the 40 wt% iron oxide and alumina (Fe/Al = 24/16) over activated carbon (Jin et al., 2013). Here, activated carbon brought in-situ reduction of Fe(NO3)3 to metallic. During N2 pre-treatment process at 870 °C, the carbon wall was burned off by Fe and created mesopores. The catalyst showed 35% CH4 conversion up to 100 h.
By literature review, we come to know that the widely available and cheap Fe can be utilized for the generation of COx-free H2 through CH4 decomposition. The activated carbon as support had the additional benefits as it had in-situ generation capacity of catalytic active sites (metallic Fe) by carbon reducibility. Tungsten had appealing redox chemistry and WC had high thermal stability (Mounfield et al., 2019). In the presence of W, additional CH4 decomposition sites were previously claimed also (Patel et al., 2021). Herein, waste date pits were utilized for the preparation of activated carbon. The WO3-activated carbon support was prepared by hydrothermal method and thereafter iron was impregnated over the WO3-activated carbon support. It is expected that if tungsten oxide is used as support along with activated carbon, Ni supported on WO3-activated carbon catalyst system would be benefited by high thermal stability, in-situ reducibility, and enhanced CH4 dissociation. The prepared catalyst was investigated for CH4 decomposition reaction and characterized through X-ray diffraction, N2-physiosorption, and porosity measurement, H2-temperature programmed reduction, thermogravimetric analysis, O2-temperature programmed oxidation and X-ray photoelectron spectroscopy. The fine correlation of catalytic activity and characterization results will add a step up in the development of an industrially suited catalyst for CH4 dissociation.
2 Experimental
2.1 Materials and methods
The following materials were used in the preparation of the newly designed catalysts; Sodium tungstate dehydrate (Na2WO4·2H2O, ≥ 99% Sigma Aldrich), sodium chloride (NaCl; ≥ 99.0%, Sigma Aldrich), hydrated iron nitrate (Fe(NO3)3·9H2O; 99%; Loba Chemie), hydrochloric acid (HCl; 37%, Sigma Aldrich) and waste of date pits (collected from Albaha region, Saudi Arabia).
2.2 Catalyst preparation
2.2.1 Preparation of activated carbon (Ac)
The waste of date pits was cleaned, sieved, and washed several times by deionized water. Further, it is carbonized on heating at 250 °C under an electrical oven for 24 h. The black carbonized pits were obtained, ground and sieved. Finally, black carbon powder is obtained. To activate the black powder, concentrated H2SO4 was added and the mixture was heated at 250 °C in an oven for 24 h. The obtained material was washed several times with deionized water until pH 7 is not attained. The activated carbon material was abbreviated as “Ac”.
2.2.2 Preparation of WO3 nanoparticles
The support WO3 nanoparticles were synthesized by hydrothermal process. 1.067 g of Na2WO4·2H2O and 0.038 g of pure NaCl were dissolved in 20 ml distilled water in stainless steel autoclave and stirred the solution in the dark for 30 min. Further, 5 ml HCl solution was added dropwise in this solution. The mixture (in an autoclave) was placed in the oven at 150 °C for 10 h. The precipitate in the autoclave was washed several times with distilled water until pH 7 was not reached. Finally, sample was calcined in air at 450 °C for 5 h. The material was used for support further and abbreviated as W.
2.2.3 Preparation of Ac doped WO3 nanoparticles (WO3/Ac)
The support Ac-doped WO3 nanoparticles were prepared by the following procedure. Appropriate amounts of “x” wt% Ac and 100-x wt% WO3 (x = 5–95) were added in 20 ml distilled water under the stirring conditions in the autoclave. Further, HCl solution was added to the solution, kept for 30 min at room temperature, and then placed in an autoclave under the oven at 150 °C for 12 h. The precipitate in the autoclave was washed several times with distilled water until pH 7 was not reached. Finally, the sample was calcined in air at 450 °C for 5 h. The material was used as support further and abbreviated as xW(100-x) Ac (x = 0–100).
2.2.4 Preparation of Fe supported on “AC doped WO3 nanoparticles” catalyst
30 wt% Fe loading was obtained from dissolving the specified amount of hydrated iron nitrate in 30 ml water and followed by impregnated of this solution over Ac or W or xW(100-x)Ac (x = 0–100) support at 80 °C for 3 h. Further, the slurry was dried overnight at 120 °C and calcined at 600 °C for 3 h sequentially. Fe supported on activated carbon, Fe supported on tungsten oxide, Fe supported on “tungsten oxide-activated carbon” catalysts were abbreviated as 30Fe100AC, 30Fe100WO3,and 30FexW(100-x) Ac (x = 0–100) respectively.
2.3 Catalyst characterization
X-ray diffraction (XRD) study of catalyst samples was carried out by Rigaku diffractometer using Cu Kα radiation source operated at 40 kV and 40 mA. 0.01 step size and 5–100 scanning range were set for analysis. Phase analysis was carried out by using X’pert high score plus software and JCPDS database. N2-physiosorption isotherms study of catalyst sample was carried over Micromeritics Tristar II 3020. Surface area was estimated by Brunauer-Emmet Teller (BET) method whereas pore volume and pore diameter were estimated by Barrett-Joyner-Halenda (BJH) method. The reducibility of the catalyst sample was studied by H2-temperature-programmed reduction (TPR) over Micromeritics Auto Chem II 2920, USA. 70 mg of the sample was subjected to a heat treatment at 10 °C/min up to 900 °C under 30 ml/min gas flow of 10% H2/Ar mixture gas. The thermogravimetric analysis (TGA) was carried out over 0.015 g of spent catalyst sample in the temperature range (room temperature to 1000 °C) at heating ramp 20 °C by using Shimadzu TGA-51. The TGA analysis was carried out under oxidizing gas O2. The weight loss/weight gain of catalyst sample against temperature was monitored continuously. O2-Temperature programmed oxidation (TPO) was carried out over spent catalyst system in 50–800 °C temperature range by using a 10% O2/He mixture through by Micromeritics AutoChem II. Before analysis, the spent catalyst was treated under high purity Argon at 150 °C for 30 min and subsequently cooled to room temperature. The morphology of the catalyst sample was investigated by using a field emission scanning electron microscope (FE-SEM, model: JEOL JSM-7100F) and transmission electron microscope (TEM, model: 120 kV JEOL JEM-2100F). Element valance state and binding energy of electron were determined by X-ray photoelectron spectroscopy (XPS) (Themo Fisher Scientific, USA) operated through AlKα excitation source and 20 eV pass energy.
2.4 Catalyst activity test
The detailed reaction set up for the CH4 decomposition reaction is shown in Fig. 1. Catalytic decomposition of methane was carried out over 0.15 g catalyst packed in fixed-bed stainless steel tubular micro-reactor (PID Eng & Tech micro activity reference company; L = 30 cm, I.D = 9.1 mm) at atmospheric pressure. The reactor temperature was monitored by an axially positioned K-type stainless steel sheathed thermocouple at the centre of the catalyst bed. Prior to the reaction, the catalyst was activated under 40 ml/min flow of H2 for 60 min at 600 °C. Futher reactor is purged by N2 for 15 min to remove the remnant of H2. Now, the temperature of the reactor was raised to 800 °C under flow of N2. 15 ml/min CH4 and 5 ml/min N2 (total flow rate of feed gas 20 ml/ min) was allowed to pass through the catalyst bed at 800 °C with 8000 ml/hgcat space velocity of. GC-2014 SHIMADZU (Column: Shin carbon C20380 for gases and Haysepe Q AC0209 column for water analysis; carrier gas: Argon) equipped with conductivity detector was used to analyse the feed and output gas composition. The expression for CH4 conversion, H2 yield and Carbon yield (%) are given as
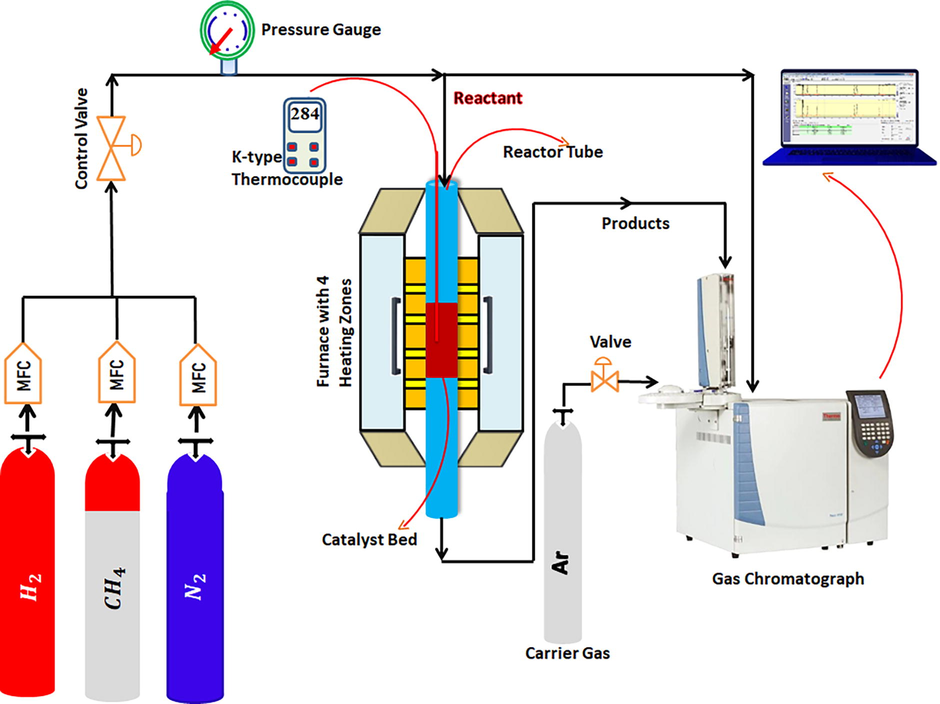
Reaction set up for CH4 decomposition reaction.
(Where Wp is the weight of the product after reaction and Wcat is the weight of the fresh catalyst).
3 Result
3.1 Characterization results
The X-ray diffraction pattern of 30FexW(100-x) Ac (x = 0–100%) catalysts are shown in Fig. 2 and Fig. 3. 30 wt% Fe supported over activated carbon had only phases related to iron oxide at Bragg angle (2θ) 24.07°, 33.12°, 35.60°, 39.19°, 40.80°, 49.38°, 54.10°, 57.56°, 62.33°, 63.99°, 69.55°, 71.80° (JCPDS reference number 00–024-0072) (Fig. 2 A-C). As activated carbon amount is substituted by tungsten oxide up to 5–10 wt% W, the diffraction peak intensity for iron oxide is suppressed greatly in 30FexW(100-x) Ac (x = 5–10) catalyst. However, upon the 1:3 ratio of W and Ac (25 wt%WO3-75%Ac), the many peaks related to Fe2O3 again appeared with variation in intensity over 30Fe25W75Ac catalyst (Fig. 2 D). The 30Fe25W75Ac catalyst also shows crystalline carbon phases at 2θ values of 27.38°, 36.10°, and 62.7° (JCPDS reference number: 00–018-0311) (Fig. 2 B-C). When “50 wt% WO3-50 wt% Ac” support is prepared for 30 wt% Fe dispersion, orthorhombic tungsten oxide phases at 22.97°, 24.02°, 33.22°, 35.70°, 49.55°, 54.24°, 62.59° (JCPDS reference number: 00–020-1324), tungsten carbide phase at 35.70°, 64.11° (JCPDS reference number 01–073-0471) and Fe2(WO4)3 phases at 20.33°, 22.45°, 22.97°, 25.45°, 29.98° (JCPDS reference number 00–038-0200) are appeared additionally (Fig. 2 D). Upon 3:1 ratio of W and Ac respectively, the 30Fe75W25Ac catalyst shows the most intense diffraction peak patterns along with additional diffraction peaks for tungsten oxide (monoclinic phase) at 23.61°, 28.70°, 30°, 34.11°, 38.94°, 56.29°, 68.81° (JCPDS reference number 01–072-0677) (Fig. 2 E-F). It may be expected that on further increasing the weight ratio of W and Ac (W/Ac = 75/25, 90/10, 95/5), the peak intensity of tungsten-related phases should be increased but the opposite diffraction results are noticed (Fig. 3 A-C). It indicates either the addition of “5-10 wt% activated carbon in tungsten oxide matrix” or “addition of 5-10 wt% WO3 in activated carbon” brings a drop of the crystallinity of the catalyst sample. Finally, on complete substitution of activated carbon by tungsten oxide, 30Fe100W catalyst shows iron oxide, tungsten oxide and the intense peak intensity for Fe2WO6 mixed oxide (at 20.50°, 27.41°, 31.08°, 33.21°, 35.99°, 39.09°, 49.59°, 54.19°; JCPDS reference number 00–015-0688) (Fig. 3 D-F).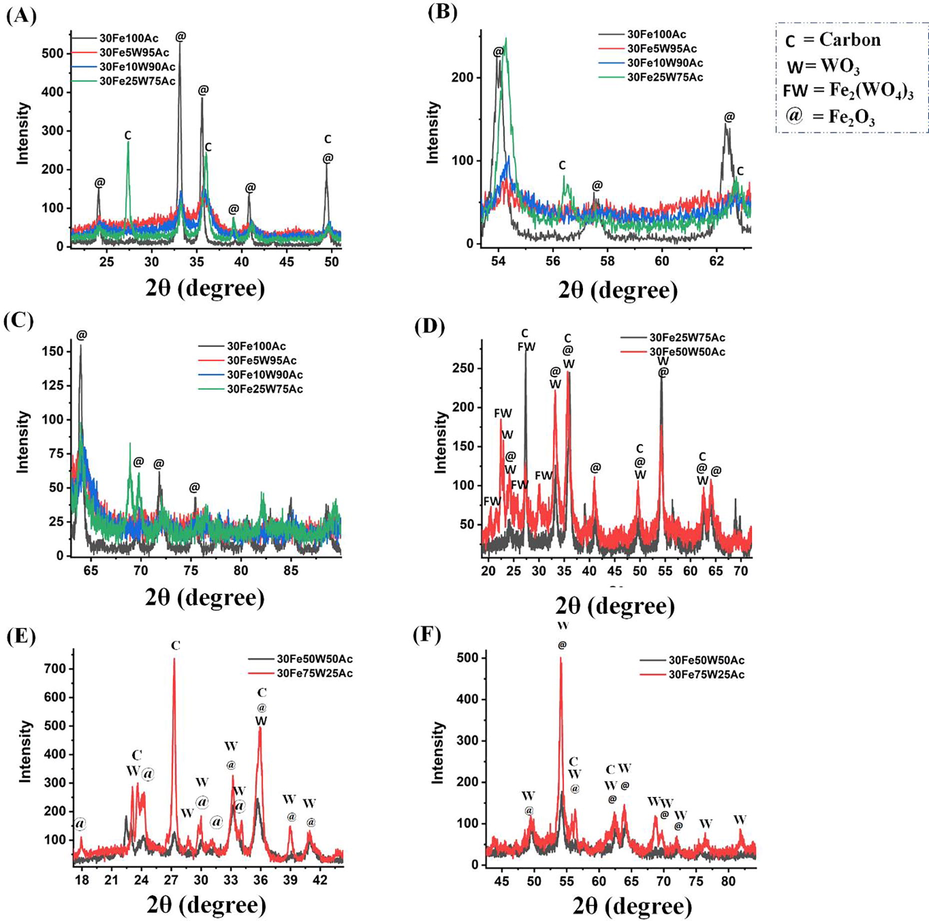
X-ray diffraction of (A-C) 30FexW(100-x) Ac; (x = 0, 5, 10, 25) catalyst (D-) 30FexW(100-x) Ac; (x = 25, 50) catalyst (E-F) 30FexW(100-x) Ac (x = 50, 75) catalyst; C = Carbon, W = WO3, @ = Fe2O3, FW = Fe2(WO4)3.
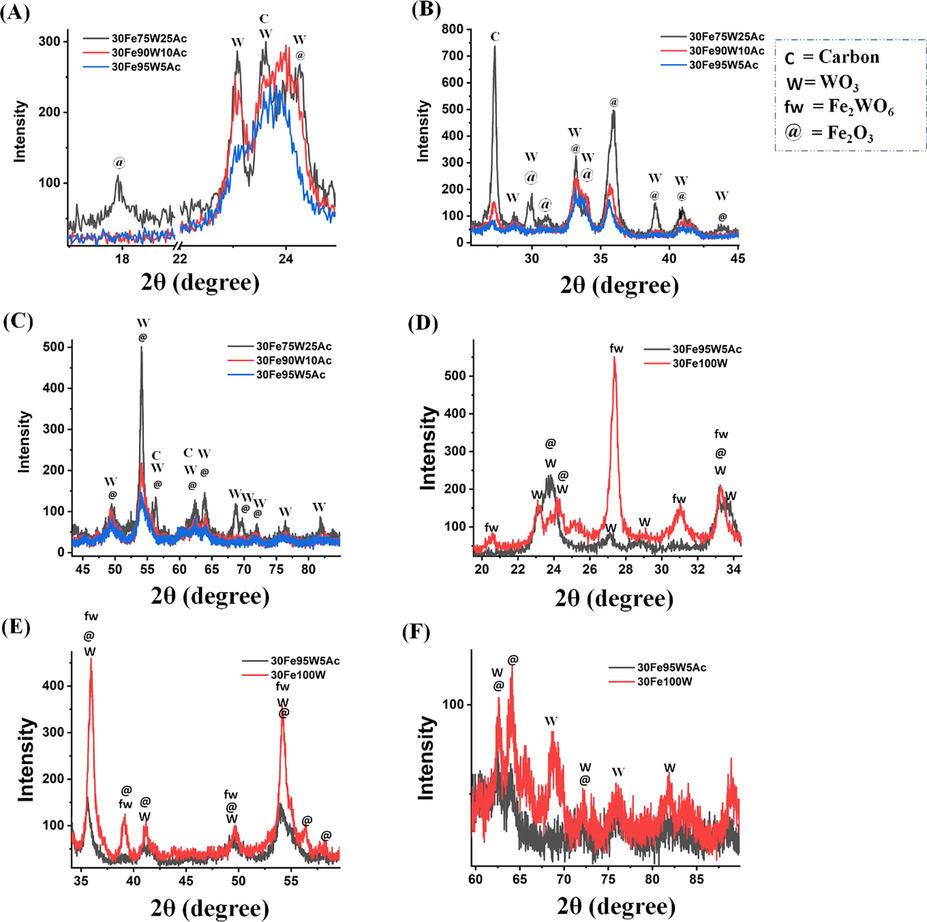
X-ray diffraction of (A-C) 30FexW(100-x) Ac (x = 75, 90, 95) catalyst (D-F) 30FexW(100-x) Ac (x = 95, 100); C = Carbon, W = WO3, fw = Fe2WO6, @ = Fe2O3.
The support “activated carbon” has 0.8348 m2/g surface area, 0.002550 cm3/g pore volume, and 393 Å pore diameter. For 30FexW(100-x) Ac (x = 0–100) catalyst system, adsorption isotherm, pore size distribution, surface area, pore volume, and pore diameter are shown in Fig. 4 and Fig. S2. The catalyst system belongs to the type IV isotherm having an H3 hysteresis loop. It indicates the presence of non-rigid aggregate-like mesopores. Upon incorporation of 5-10 wt% tungsten oxide, the surface area of 30FexW(100-x)Ac (x = 5, 10) catalyst is 2.5 times than 30Fe100Ac indicating expansion of framework (Kumar et al., 2016). Upon 25 wt% tungsten oxide incorporation, the surface area is noticed to decrease to 30% but pore volume is increased by 46%. However, upon further loading up to 50 wt% W; the surface area and pore volume of 30Fe50W50Ac are increased to 3 times and 1.8 times (with respect to 30Fe100Ac) respectively. The pore size distribution plot (dV/dlogW vs W) indicates that up to 50 % incorporation of tungsten oxide, pore size distributions remain bimodal. In the 30Fe50W50Ac catalyst, the intensity of the low pore-width range is more pronounced than the higher pore-width range. In 30FexW(100-x)Ac (x = 0–100) catalyst systems, when support is made up by major WO3 than Ac (upon > 50 wt% W incorporation), the pore size distribution becomes multimodal. In 30FexW(100-x)Ac (x = 75–100),the surface area decreases suddenly to 11–22 m2/g (against 59.15 m2/g in 30Fe50W50Ac) due to deposition of various crystallite inside the pore (Rahman et al., 2015).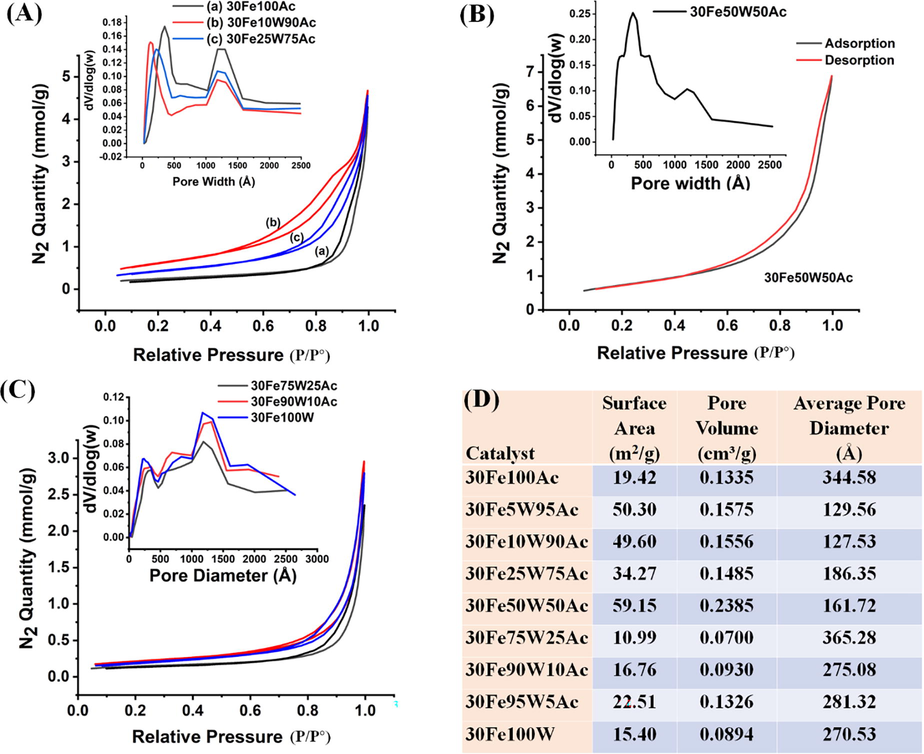
N2-adsorption isotherm and porosity distribution profile of (A) 30FexW(100-x)Ac(x = 0–25) catalyst (B) 30FexW(100-x)Ac(x = 50) catalyst (C) 30FexW(100-x)Ac(x = 75–100) catalyst (D) Surface parameters of 30FexW(100-x)Ac(x = 0–100) catalyst.
The H2-Temperatured programmed reduction profile of 30FexW(100-x) Ac (x = 0–100) catalyst systems are shown in Fig. 5 A and Fig. S3. The total H2-consumption during the H2-TPR experiment is shown in Table S4. H2-TPR of Fe2O3 is constituted by two sharp peaks at 355 °C and 577 °C and a broad peak between 624 °C and 934 °C (Fig. S3). The three peaks are correlated with the sequential reductions Fe2O3 → Fe3O4 → FeO → Fe respectively (Ibrahim et al., 2015; Jozwiak et al., 2007). The activated carbon-supported Fe (30Fe100Ac) catalyst shows shifting of lower temperature reduction peak to relatively higher temperature (at 385 °C) indicating interaction of Fe2O3-species with support. The peak at 385 °C belongs to reduction of interacted-Fe2O3-species into Fe3O4. Upon incorporation of just 5 wt% WO3, a merged peak maximum at 420 °C for reduction of Fe2O3 → Fe3O4 → FeO and a broad peak at a higher temperature for the reduction of FeO → Fe are observed. Further incorporation of 10 wt% WO3, the reduction peak of 30Fe10W90Ac catalyst is shifted towards more higher temperature (444 °C). It is noticeable that the amount of reducible iron species had decreased upon providing support as well as increasing the proportion of tungsten oxide (up to 10 wt%) in the support. It indicates that the total reducible quantity has decreased due to the interaction of Fe-species with the new support composed of xW(100-x) Ac (x = 0, 5, 10) catalyst. As well as WO3 incorporation is increased to 25 wt%, the reduction peak maxima of 30Fe25W75Ac catalyst are shifted to a higher temperature and the amount of reducible iron-species is increased to ∼ 34% with respect to 30Fe10W90Ac catalyst (Table S4). Shifting of reduction peak to a higher temperature also indicates increased metal support interaction upon tungsten oxide loading. 30Fe50W50Ac catalyst has the lower temperature reduction peak (for reduction of Fe2O3 → Fe3O4 → FeO at 470 °C and broad higher temperature reduction peak with comparable amount of reduceable species than 30Fe25W75Ac catalyst. 30FexW(100-x) Ac (x = 90–100 wt%) showed the reduction peak about 530–560 °C. It again shows a general trend of increasing metal-support interaction upon tungsten oxide loading. The peak pattern of 30FexW(100-x)Ac (x = 90–100 wt%) has also an additional peak in the temperature region of 593 to 720 °C for reduction of WO3 crystallite or “WO3 interacted species”(Ramanathan et al., 2013). It is noticeable that the total concentration of reducible species at the catalyst surface is decreased sharply above 50 wt% tungsten oxide incorporation. 30Fe50W50Ac, 30Fe90W10Ac, 30Fe95W5Ac, and 30Fe100W catalysts had 174.2 cm3/g, 72.28 cm3/g, 68.24 cm3/h, and 46.32 cm3/g consumption of hydrogen. The H2-TPR pattern of the 30Fe25W75Ac catalyst is needed to address separately. It has the highest amount of reducible species over the surface (183 cm3/g H2 consumption in H2-TPR result) among other tungsten oxide incorporated catalysts. The H2-TPR peak pattern is constituted by five peaks enveloping each other at 433 °C, 506 °C, 664 °C, 816 °C and 929 °C. It indicates the 30Fe25W75Ac catalyst had the highest concentration of “Fe-related” reducible species which interacted with the support to different extents.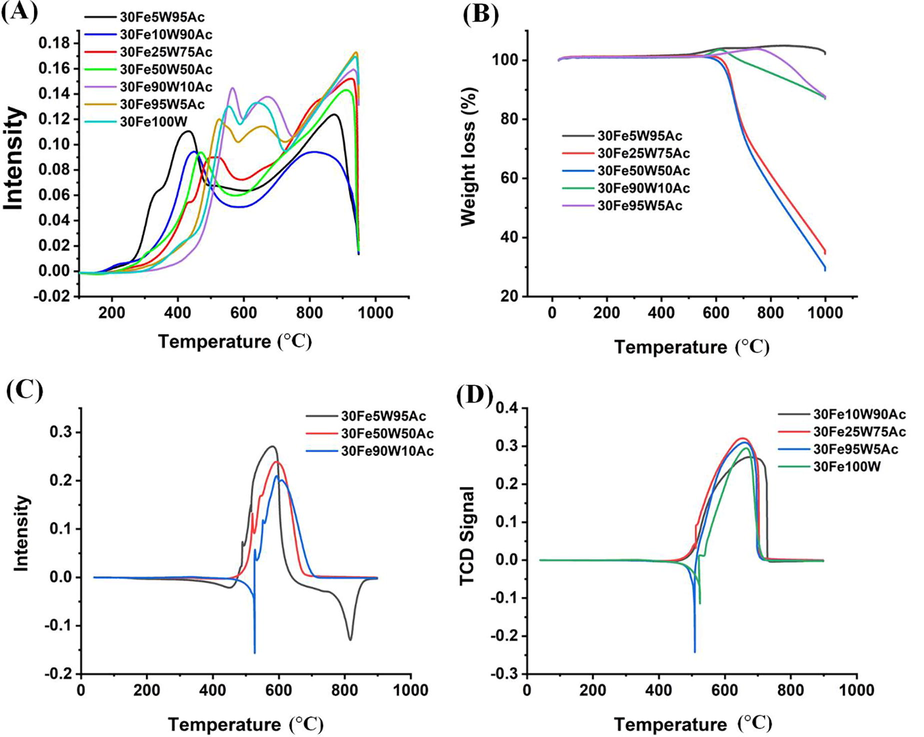
(A) H2-TPR profile of 30FexW(100-x)Ac(x = 5–100) catalyst (B) TGA profile of spent 30FexW(100-x)Ac(x = 5–95) catalyst (C) O2-TPO of 30FexW(100-x)Ac(x = 5, 50, 90) (D) O2-TPO of 30FexW(100-x)Ac(x = 10, 25, 95, 100).
The thermogravimetric analysis (TGA) of spent catalysts is shown in Fig. 5B. The weight gain over the catalyst system in TGA analysis may be due to oxidation of “reduced metal species” (like iron or tungsten-related metal oxide) over the catalyst surface (Ibrahim et al., 2015). In the case of spent 30Fe25W75Ac and spent 30Fe50W50Ac, weight loss due to oxidation of deposit carbon is optimum and so weight gain due to oxidation of “reduced metal species” over these catalyst systems is not evident on TGA analysis. The carbon yield % over the different catalysts is found in the following order; 30Fe25W75Ac (140%) > 30Fe50W50Ac (107%) > 30Fe10W90Ac (1 2 0) 30Fe95W5Ac (93.3%) > 30Fe90W10Ac (66.6%) > 30Fe5W95Ac (13.3 %) > 30Fe75W25Ac (6.7%) (Table S5). Clearly, carbon yield % over 30Fe25W75Ac and 30Fe50W50Ac are higher than other catalysts. Previously, tungsten species were claimed to generate additional CH4 decomposition sites (Patel et al., 2021). It seems that the presence of 25-50 wt% of WO3 in the catalyst system cultivates the optimum amount of catalytic active sites which leads potential dissociation of CH4 into carbon and H2. It resulted in an excellent carbon yield and severe weight loss. These findings also give the sign of higher activity toward CH4 decomposition reaction over 30Fe25W75Ac and 30Fe50W50Ac catalysts. Severe weight loss over spent 30Fe25W75Ac and spent 30Fe50W50Ac catalysts also indicates that the carbon deposits over these catalysts are not inert, it is oxidizable under O2 stream. Inert carbon deposit may shade the catalytic active site permanently and causes fast deactivation. The non-inert carbon deposit over 30Fe25W75Ac and spent 30Fe50W50Ac catalysts may cause slower deactivation than other catalysts.
O2-TPO of spent 30FexW(100-x) Ac (x = 0–100) catalyst system is shown in Fig. 5C-5D. In the literature, the O2-TPO peak profile is differentiated into three regions 300–500 °C for easily oxidizable α-carbon (amorphous carbon) species (Al-Fatesh et al., 2021), 500–600 °C for moderately oxidizable β-carbon species (Patel et al., 2021) and > 600 °C for higher crystallization degree of carbon species (Zhang et al., 2015). In our catalyst system, TPO peak maxima is found about ∼ 600 °C in spent-30Fe5W95Ac, spent-30Fe50W50Ac, and spent-30Fe90W10Ac catalysts whereas, for spent-30Fe10W90Ac, spent-30Fe25W75Ac, spent-30Fe95W5Ac and spent-30Fe100W catalysts, TPO peaks are at about ∼ 650 °C. This observation indicates that a particular amount of tungsten oxide (spent-30Fe5W95Ac, spent-30Fe50W50Ac, and spent-30Fe90W10Ac catalyst) in the support induces less crystallization degree of carbon (than30Fe10W90Ac, 30Fe25W75Ac, and 30Fe100W catalyst).
The morphology of catalyst samples is shown by SEM images in Fig. S6. The catalyst morphology of fresh low tungsten-containing samples (30Fe10W90Ac) or high tungsten-containing samples (30Fe90W10Ac) is not differentiable. In the case of spent 30Fe10W90Ac catalyst, carbon tubes are easily observed than in 30Fe90W10Ac catalyst. The morphology of the catalyst and carbon tube is evident in TEM images under Fig. 6. TEM image indicates particle size over the 30Fe25W75Ac has grown from 5.57 nm to 5.8 nm after the reaction (Fig. 6 A-D). Fig. 6 E shows the presence of carbon nanotubes of varying diameters. A typical multiwalled carbon tube having wall width of 3.81–4.74 nm and total tube width of 13.13 nm is evident in Fig. 6F.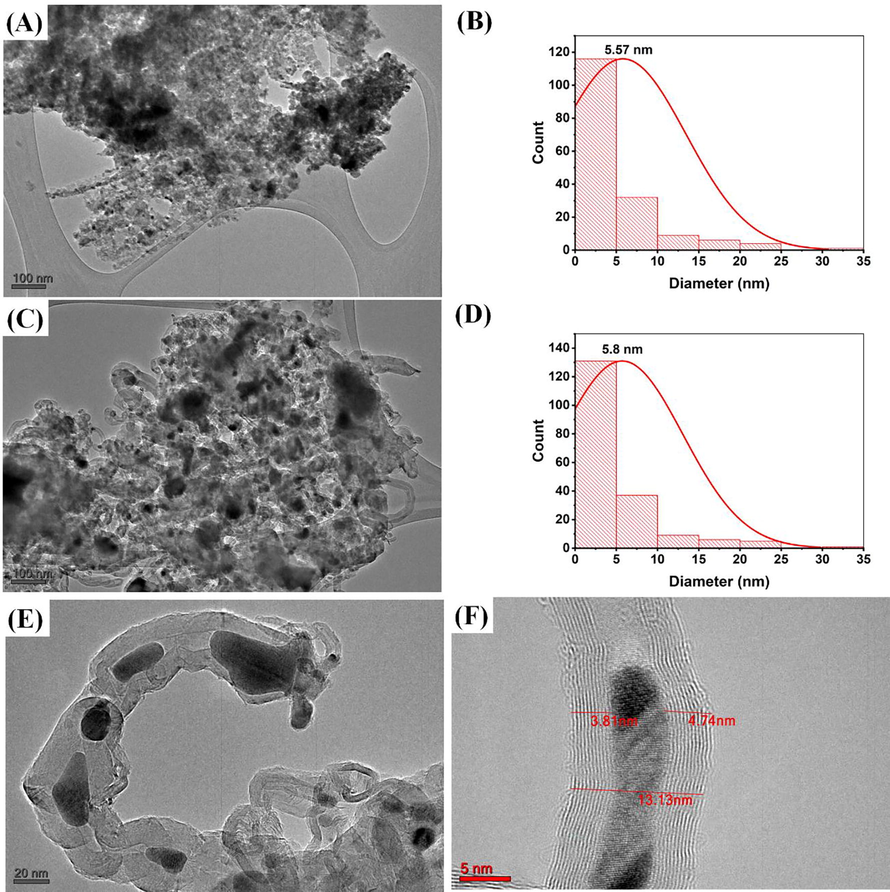
(A) TEM image of fresh 30Fe25W75Ac at 100 nm scale (B) Particle size distribution curve of fresh 30Fe25W75Ac (C) TEM image of spent 30Fe25W75Ac at 100 nm scale (D) Particle size distribution curve of spent 30Fe25W75Ac (E) TEM image of carbon nanotubes in spent 30Fe25W75Ac catalyst at 20 nm scale (D) Single multiwalled carbon nanotube in spent 30Fe25W75Ac catalyst at 5 nm scale.
The X-ray photo-electron spectra of 30Fe25W74Ac catalyst is shown in Fig. 7. Fe (2p3/2) peak at 711 eV and Fe (2p1/2) peak at 725 eV and O (1 s) peak at 530.1 confirms the presence of Fe+3 oxidation state (Allen et al., 1974; Konno and Nagayama, 1980) (Fig. 7A- B). The presence of W(4f7/2) peak at 35.4 eV and W(4f5/2) peak at 37.6 eV confirm the presence of WO3 or W+6 oxidation state (Fig. 7C) (Barreca et al., 2001). The C(1 s) XPS spectra is observed at 284.7 (Barreca et al., 2001; Grünert et al., 1987) (Fig. 7). The 30Fe25W75Ac catalyst has both carbon and WO3 but absence of carbidic carbon peak at 282.7 eV indicates that WC like species are not formed over the catalyst surface (Katrib et al., 1994). Overall, from the XPS spectra presence of Fe (III) (as Fe2O3), W (IV) (as WO3) species are confirmed. Fe2O3 and WO3 phases are already confirmed during the XRD analysis of sample.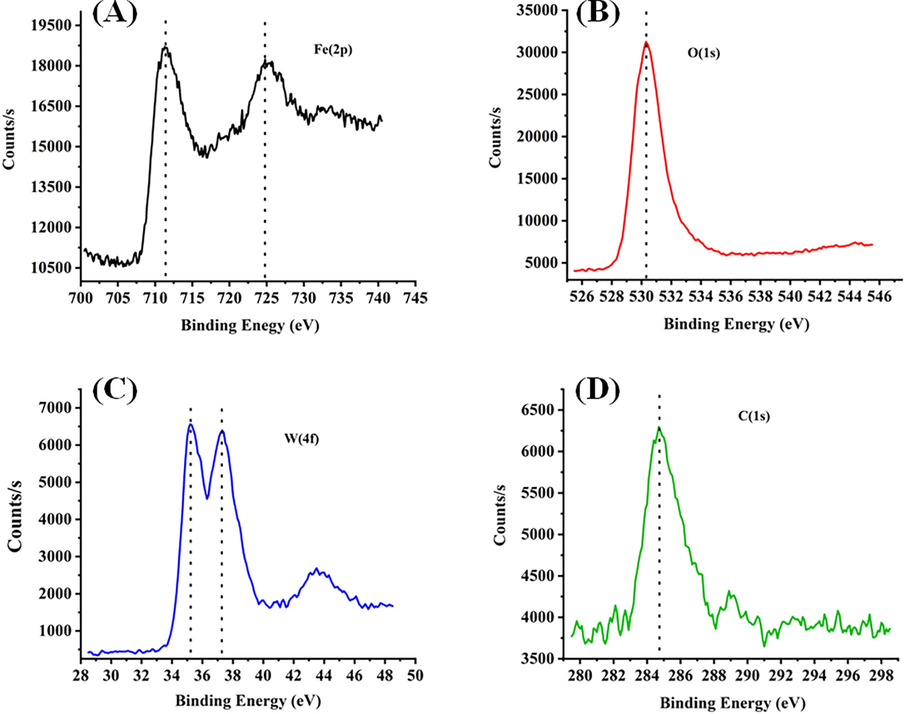
X-ray photo-electron spectra of 30Fe25W74Ac catalyst (A) Fe(2p) spectra (B) O (1 s) spectra (C) W (4f) spectra (D) C (1 s) spectra.
3.2 Results and discussion
The CH4 conversion, H2-yield and ratio of H2-yield/CH4 conversion (
/
) are shown in Fig. 8A-8C. The initial conversion of CH4 and H2-yield at 30wt.%Fe supported over activated carbon (30Fe100Ac) is just 4.43% and 1.96% respectively whereas 30wt.%Fe supported over tungsten oxide (30Fe100W) shows 20.5% initial CH4 conversion and 19.43% initial H2-yield. Markedly tungsten-oxide supported iron has a higher catalytic activity as well as ratio of
/
is > 0.94 whereas activated-carbon-supported iron has low catalytic activity and a ratio of
/
is just half (Fig. 8C). During the entire time on stream (420 min),
/
ratio remains close to 0.9 over 30Fe100W whereas
/
ratio drops to ∼ 0.2 over 30Fe100Ac at the end of 420-minutes time on stream. A high
/
ratio indicates a higher accumulation of CH4 or CHx over the catalyst surface followed by higher H2 release to the gas phase during the reaction (Łamacz and Łabojko, 2019). The higher activity of 30Fe100W than 30Fe100Ac catalyst toward methane decomposition reaction can be explained by X-ray diffraction and H2-TPR results. 30Fe100Ac catalyst had only reducible iron oxide as surface active species whereas 30Fe100W has iron oxide, tungsten oxide, and Fe2WO6 mixed oxide phases. Patel et. al found “additional CH4 decomposition sites” over tungsten oxide-zirconia in CH4-temperature programmed surface reaction experiment (Patel et al., 2021). It was reported that Fe2WO6 may also be reduced to respective Fe and W under the hydrogen stream at reaction temperature (Pak et al., 2009). In H2-TPR results, we found the reduction peak for WO3 at 593 °C to 720 °C over 30Fe100W catalyst. That means 30Fe100W has a variety of reducible surface-active species that markedly influence the CH4-decomposition reaction. Overall, it can be said that tungsten oxide is promising support for Fe-based catalyst for CH4 decomposition reaction.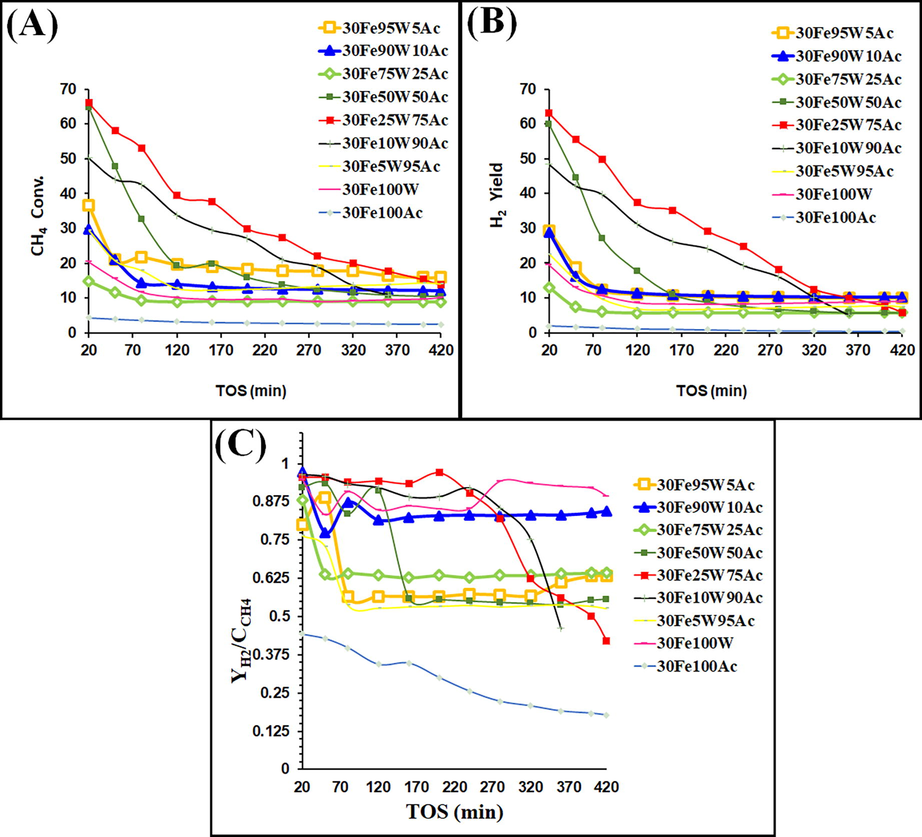
Catalytic activity results of 30FexW(100-x) Ac (x = 0–100) catalyst (A) CH4 conversion (B) H2-yield (C)
/
ratio.
Up to incorporation of 5–10 wt% WO3 in 30FexW(100-x) Ac (x = 5–10) catalyst, X-ray diffraction peaks are suppressed and surface area is increased up to 2.5 times (with respect to 30Fe100Ac catalyst). It indicates the suppression of crystallinity upon expansion of the surface (Khalid et al., 2013). H2-TPR result of 30Fe10W95Ac showed increased metal-support interaction over than 30Fe5W95Ac catalyst. Increased metal-support interaction over an expanded surface is the ideal condition for exposing more active sites for CH4 decomposition. Overall, it can be said that 30Fe5W95Ac catalyst has an expanded surface (than 30Fe100Ac) and 30Fe10W90Ac catalyst has an expanded surface (than 30Fe100Ac) as well as increased interaction of reducible Fe-species (Fe2O3, Fe3O4, FeO) with support (than 30Fe5W95Ac). 30Fe5W95Ac catalyst has 29.5% initial CH4 conversion (against 4.43% in 30Fe100Ac) and 22.5% initial H2-yield (against 1.96% in 30Fe100Ac). 30Fe10W90Ac catalyst shows 50.2% initial CH4 conversion and 48.39% initial H2-yield. The / ratio over 30Fe10W90Ac also remains ∼ 0.9 up to 250 min whereas, over 30Fe5W95Ac, / ratio falls to 0.5 within 70 min time on stream. Here, the role of tungsten in CH4 decomposition, higher surface area, and more “surface interacted reducible Fe-species” are evident over 30Fe10W90Ac catalyst which achieves higher CH4 conversion and higher / ratio.
Up to 25 wt% W incorporation; prominent iron oxide phase is evident over 30Fe25W75Ac catalyst. The presence of Fe2O3 and WO3 phases are also confirmed by Fe(2p), W(4f) and O (1 s) XPS spectra. The decrease in surface area (up to 30%) of 30Fe25W75Ac (with respect to 3010W90Ac) is compensated by an increase in average pore diameter (up to 46%). 30Fe50W50Ac has again expanded surface area (three times than 30Fe100Ac) and other tungsten-related phases like tungsten oxide, tungsten carbide, and Fe2(WO4)3 over the surface. 30Fe25W75Ac catalyst has the highest amount of reducible species (183 cm3/g H2 consmption in H2-TPR result) over the surface among the rest tungsten oxide incorporated catalysts. These reducible iron species have interacted with the support along a wide range of temperatures as per the extent of interaction with support. 30Fe50W50Ac catalyst has also good number of reducible species (174 cm3/g H2 consumption in H2-TPR result) after 30Fe25W75Ac catalyst. Previously, tungsten species were claimed to generate additional CH4 decomposition sites (Patel et al., 2021). 30Fe25W75Ac and 30Fe50W50Ac catalysts show severe weight loss and higher carbon yield. The catalyst activity of 30Fe50W50Ac and 30Fe25W75Ac towards the CH4 decomposition reaction are also close to each other initially. It indicates that the presence of 25-50 wt% of WO3 in the catalyst induces an optimum amount of catalytic active sites leading to potential CH4 dissociation, excellent carbon yield, severe weight loss and optimum H2-yield over 30Fe25W75Ac and 30Fe50W50Ac catalysts. The initial CH4 conversion of 30Fe25W75Ac and 30Fe50W50Ac catalysts are 66.04% and 64.82 % respectively. Again, the initial H2-yield over 30Fe25W75Ac and 30Fe50W50Ac catalyst is found 63.12% and 59.81 % respectively.
Fe supported over “xW(100-x)Ac (x = 10–50)” are able to show > 50% CH4 conversion, ≥50% H2-yield and ∼ 0.9 / ratio initially. Severe weight loss in TGA profile is obtained over 30Fe25W75Ac and spent 30Fe50W50Ac catalysts. It indicates that carbon deposits over these catalysts are oxidizable/not inert/active. The non-inert carbon deposit deactivates 30Fe25W75Ac and spent 30Fe50W50Ac catalyst slowly than the rest catalysts. After 160 min, CH4 conversion and H2 yield of the 30Fe25W75Ac catalyst drop to 37.61% (against 66% initial CH4 conversion) and 35.2% (against 63.12% initial H2 yield) respectively. 30Fe10W90Ac catalyst is found the second best as the CH4 conversion and H2-yield don’t fall below 25% after 160 min time on stream. The / ratio of both 30Fe25W75Ac and 30Fe10W90Ac catalyst is also ≥ 0.9 up to 240 min. At the end of 160 min 30Fe50W50Ac catalysts showed ∼ 19% CH4 conversion, ∼11% H2 yield and 0.56 / ratio. Overall, at the end of 420 min time on stream, 30Fe25W75Ac is found best. It has ∼ 14% CH4 conversion, ∼6% H2-yield and > 0.4 / ratio at 420 min time on stream.
TGA results indicate severe coke decomposition over 30FexW(100-x) Ac (x = 25, 50) catalyst. O2-TPO result indicates that coke over the 30Fe25W75Ac catalyst has a higher crystallization degree than the 30Fe50W50Ac catalyst. The carbon yield calculation of the spent 30FexW(100-x) Ac (x = 5, 10, 25, 50, 90, 95) catalyst system is shown in Table S5. Here also, the carbon yield over the spent 30Fe25W75Ac catalyst is greater than the 30Fe50W50Ac catalyst. Interestingly, the catalytic activity of the 30Fe25W75Ac catalyst is less affected by severe coke deposition but the activity of 30Fe50W50Ac drops suddenly on increasing time on stream. Initially, the activity of both catalysts is close to each other but after the end of 200 min, 30Fe50W50Ac has only ∼ 16% CH4 conversion (against 30% in 30Fe25W75Ac), 9% H2-yield (against 29% in 30Fe25W75Ac) and 0.56 / ratio (against 0.97 in 30Fe25W75Ac). It indicates that coke decomposition affects the performance of 30Fe50W50Ac to a great extent but not the performance of 30Fe25W75Ac catalyst. It seems that over 30Fe25W75Ac, rate of CH4 decomposition (carbon formation) is well matched with the rate of diffusion of carbon species from metal-gas interface (where decomposition of CH4 took place) to the metal-nanofiber interface (where carbon precipitates to form carbon nanofibers). Over highly crystalline 30Fe50W50Ac catalyst (than 30Fe25W50Ac), the rate of carbon formation may not properly match the rate of carbon diffusion. So, carbon species isn’t able to be transferred away in time and would cover the catalyst’s active sites leading to catalyst deactivation (Chen et al., 1997).
Tungsten oxide incorporation of>50 wt% in 30FexW(100-x)Ac (x = 75, 90, 95) causes a fast drop of surface area due to the deposition of various crystallite inside the pore (Rahman et al., 2015) constituted by major-tungsten oxide and minor-activated carbon. These catalysts systems have also low density of reducible reducible-species over the catalyst surface. Low surface area catalyst and few catalytic active sites on the surface conveys less initial CH4 conversion. 30Fe75W25Ac has the highest crystallinity among rest catalyst systems. 30FexW(100-x) Ac (x = 75–95) catalysts has low initial CH4 conversion (14–36%) and initial H2-yield (13–29%).
4 Conclusion
Tungsten oxide incorporated activated carbon is found to be an excellent support for Fe based catalyst towards CH4 decomposition reaction (than activated carbon incorporated tungsten oxide catalyst) due to enhanced surface area, a higher concentration of various types of reducible surface-active species as iron oxide, tungsten oxide, and Iron tungstate. The research outcome over 30FexW(100-x) Ac (x = 0–50) catalyst can be pointed as follow:
-
30Fe10W90Ac catalyst has a comparable surface area but higher metal support interaction than the 30Fe5W95Ac catalyst. So, the earlier one has higher activity than latter.
-
30FexW(100-x) Ac (x = 10–50) catalyst shows > 50% initial CH4 conversion, ∼50% initial H2-yield and ∼ 0.9 initial / ratio.
-
30Fe25W75Ac catalyst has the highest concentration of reducible surface-active species (compared to the rest tungsten incorporated catalysts). It shows 66.04% initial CH4 conversion and 63.12% initial H2 yield and > 0.9 initial / .
-
30Fe50W5pAc catalyst has a comparable concentration of reducible surface-active to 30Fe25W75Ac catalyst. Both catalysts have severe carbon deposits, higher carbon yield, higher initial CH4 conversion and higher initial H2 yield than other catalysts due to the potential dissociation of CH4 into carbon and H2.
-
On longer time on stream, the activity of the 30Fe50W50Ac catalyst drops fast than 30Fe25W75Ac catalyst due to improper matching between the rate of carbon formation and the rate of diffusion over highly crystalline 30Fe50W50Ac catalyst (compared to 30Fe25W75Ac catalyst). Even after 160 min, CH4 conversion and H2 yield over 30Fe25W75Ac catalyst does not drop below 35%.
-
Inferior catalytic activity over 30FexW(100-x)Ac (x = 75, 90, 95) is due to low surface area catalyst and few catalytic active sites.
Acknowledgements
The authors extend their appreciation to the Deanship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project no. (IFKSURG-2-055).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- In situ auto-gasification of coke deposits over a novel Ni-Ce/W-Zr catalyst by sequential generation of oxygen vacancies for remarkably stable syngas production via CO2-reforming of methane. Appl. Catal. B: Environ.. 2021;280:119445
- [CrossRef] [Google Scholar]
- Allen, B.G.C., Curtis, M.T., Hooper, A.J., Tucker, P.M., Generat-, C.E., Laboratories, B.N., Gloucestershire, G.L., Parkinson, R.F.L.D., Mallett, S., Dalton, J.C.S., 1974. 1974.
- Influence of la on reduction behaviour and Ni metal surface area of Ni-Al2O3 catalysts for COx free H2 by catalytic decomposition of methane. Int. J. Hydrogen Energy. 2015;40:3633-3641.
- [CrossRef] [Google Scholar]
- Hydrogen production by methane decomposition over coal char. Int. J. Hydrogen Energy. 2006;31:899-905.
- [CrossRef] [Google Scholar]
- Bai, X., Xie, G., Guo, Y., Tian, L., El-Hosainy, H.M., Awadallah, A.E., Ji, S., Wang, Z. jun, 2021. A highly active Ni catalyst supported on Mg-substituted LaAlO3 for carbon dioxide reforming of methane. Catalysis Today 368, 78–85. https://doi.org/10.1016/j.cattod.2019.12.033
- A study of nanophase Tungsten oxides thin films by XPS. Surf. Sci. Spectra. 2001;8:258-267.
- [CrossRef] [Google Scholar]
- A basic assessment of the reactivity of Ni catalysts in the decomposition of methane for the production of “COx-free” hydrogen for fuel cells application. Catal. Today. 2006;116:298-303.
- [CrossRef] [Google Scholar]
- Graphene and carbon nanotubes by CH4 decomposition over Co–Al catalysts. Mater. Chem. Phys.. 2019;226:6-19.
- [CrossRef] [Google Scholar]
- Growth of carbon nanotubes by catalytic decomposition of CH4 or CO on a Ni-MgO catalyst. Carbon. 1997;35:1495-1501.
- [CrossRef] [Google Scholar]
- Production of hydrogen by methane catalytic decomposition over Ni-Cu-Fe/Al2O3 catalyst. Int. J. Hydrogen Energy. 2009;34:2979-2985.
- [CrossRef] [Google Scholar]
- Hydrogen production via catalytic decomposition of methane. J. Catal.. 2001;199:9-18.
- [CrossRef] [Google Scholar]
- The effect of CH4 decomposition temperature on the property of deposited carbon over Ni/SiO2 catalyst. Int. J. Hydrogen Energy. 2015;40:9670-9676.
- [CrossRef] [Google Scholar]
- Hydrogen production via methane decomposition on Raney-type catalysts. Int. J. Hydrogen Energy. 2010;35:9795-9800.
- [CrossRef] [Google Scholar]
- Ab initio calculations of the reaction pathways for methane decomposition over the Cu (111) surface. J. Chem. Phys.. 2011;135
- [CrossRef] [Google Scholar]
- Reduction behavior and metathesis activity of WO3 Al2O3 catalysts. I. an XPS investigation of WO3 Al2O3 catalysts. J. Catal.. 1987;107:522-534.
- [CrossRef] [Google Scholar]
- High-performance Ni-Fe redox catalysts for selective CH4 to syngas conversion via chemical looping. ACS Catal.. 2018;8:1748-1756.
- [CrossRef] [Google Scholar]
- Methane decomposition over iron catalyst for hydrogen production. Int. J. Hydrogen Energy. 2015;40:7593-7600.
- [CrossRef] [Google Scholar]
- Methane decomposition over Co thin layer supported catalysts to produce hydrogen for fuel cell. Int. J. Hydrogen Energy. 2010;35:11568-11575.
- [CrossRef] [Google Scholar]
- Preparation of activated carbon supported Fe-Al2O3 catalyst and its application for hydrogen production by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013;38:10373-10380.
- [CrossRef] [Google Scholar]
- Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres. Appl. Catal. A. 2007;326:17-27.
- [CrossRef] [Google Scholar]
- Thermocatalytic decomposition of CH4 over Ni/SiO2.MgO catalysts prepared via surfactant-assisted urea precipitation method. Fuel. 2021;284:118866
- [CrossRef] [Google Scholar]
- XPS studies of supported tungsten carbide(s) J. Electron Spectrosc. Relat. Phenom.. 1994;68:589-595.
- [CrossRef] [Google Scholar]
- Effect of surfactant and heat treatment on morphology, surface area and crystallinity in hydroxyapatite nanocrystals. Ceram. Int.. 2013;39:39-50.
- [CrossRef] [Google Scholar]
- X-ray photoelectron spectra of hexavalent iron. J. Electron Spectrosc. Relat. Phenom.. 1980;18:341-343.
- [CrossRef] [Google Scholar]
- Highly stable In-SBA-15 catalyst for vapor phase Beckmann rearrangement reaction. Micropor. Mesopor. Mater.. 2016;234:293-302.
- [CrossRef] [Google Scholar]
- CNT and H2 production during CH4 decomposition over Ni/CeZrO2. II. catalyst performance and its regeneration in a fluidized bed. Chem. Eng.. 2019;3:1-18.
- [CrossRef] [Google Scholar]
- Catalytic production of carbon nanotubes by decomposition of CH4 over the pre-reduced catalysts LaNiO3, La4Ni3O10, La3Ni2O7 and La2NiO4. Catal. Lett.. 2001;74:185-188.
- [CrossRef] [Google Scholar]
- In situ study of carbon nanotube formation by C2H2 decomposition on an iron-based catalyst. Carbon. 2000;38:2003-2006.
- [CrossRef] [Google Scholar]
- Diameter-controlled carbon nanotubes and hydrogen production. Int. J. Hydrogen Energy. 2014;39:4691-4697.
- [CrossRef] [Google Scholar]
- Impact of morphological effects on the activity and stability of tungsten carbide catalysts for dry methane reforming. Energy Fuel. 2019;33:5544-5550.
- [CrossRef] [Google Scholar]
- Synthesis of nanocrystalline Fe-W composite through hydrogen reduction of thermally synthesized iron tungstate, Fe2WO6. J. Alloy. Compd.. 2009;477:357-363.
- [CrossRef] [Google Scholar]
- Impact of ceria over WO3–ZrO2 supported Ni catalyst towards hydrogen production through dry reforming of methane. Int. J. Hydrogen Energy. 2021;46:25015-25028.
- [CrossRef] [Google Scholar]
- La-Fe-O/CeO2 based composites as the catalysts for high temperature N2O decomposition and CH4 combustion. Catal. Lett.. 2013;143:1294-1303.
- [CrossRef] [Google Scholar]
- Enhanced activation and decomposition of CH4 by the addition of C2H4 or C2H2 for hydrogen and carbon nanotube production. J. Phys. Chem. C. 2008;112:7588-7593.
- [CrossRef] [Google Scholar]
- Mesoporous TUD-1 supported indium oxide nanoparticles for epoxidation of styrene using molecular O2. RSC Adv.. 2015;5:46850-46860.
- [CrossRef] [Google Scholar]
- Tungsten-incorporated cage-type mesoporous silicate: W-KIT-5. Micropor. Mesopor. Mater.. 2013;175:43-49.
- [CrossRef] [Google Scholar]
- Production of hydrogen and carbon nanofibers through the decomposition of methane over activated carbon supported Ni catalysts. Int. J. Hydrogen Energy. 2011;36:11702-11711.
- [CrossRef] [Google Scholar]
- Hydrogen production by catalytic decomposition of methane over Ni / SiO2. Int. J. Hydrogen Energy. 2007;32:1782-1788.
- [CrossRef] [Google Scholar]
- Ni-SiO2 and Ni-Fe-SiO2 catalysts for methane decomposition to prepare hydrogen and carbon filaments. Int. J. Hydrogen Energy. 2012;37:9058-9066.
- [CrossRef] [Google Scholar]
- Wu, Z., Yang, Æ.Y., Gu, Æ.D., Tu, B., Webley, Æ.P.A., Yuan, Æ.D., 2009. Synthesis of Ordered Mesoporous Carbon Materials with Semi-Graphitized Walls via Direct In-situ Silica-Confined Thermal Decomposition of CH4 and Their Hydrogen Storage Properties 12–26. https://doi.org/10.1007/s11244-008-9134-8.
- Hydrogen production by methane decomposition over Co-Al mixed oxides derived from hydrotalcites: effect of the catalyst activation with H2 or CH4. Int. J. Hydrogen Energy. 2017;42:7895-7907.
- [CrossRef] [Google Scholar]
- Zhang, X., Zhang, Q., Tsubaki, N., Tan, Y., Han, Y., 2015. Influence of Zirconia Phase on the Performance of Ni/ZrO2 for Carbon Dioxide Reforming of Methane 135–153. https://doi.org/10.1021/bk-2015-1194.ch006.
- Ni doped carbons for hydrogen production by catalytic methane decomposition. Int. J. Hydrogen Energy. 2013;38:3937-3947.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104781.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




