

Translate this page into:
An enhanced approach for targeted multi-residue screening of pesticides in complex herbal medicines by ultra high-performance liquid chromatography tandem ion mobility/quadrupole time-of-flight mass spectrometry
⁎Corresponding authors. huqingyjs@163.com (Qing Hu), jishen2021@126.com (Shen Ji)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Effect of collision gas on post-mobility fragmentation was first discussed. A detailed workflow was presented for establishing a pesticide library. Experimental CCS values of 438 pesticides were measured for identification. An enhanced multi-residue qualitative screening approach was developed. Pesticide residue detection in LJFs of different producing regions was conducted.
Abstract
Recently, serious concerns have been raised about potential safety risk posed by exogenous pesticide contaminants in herbal medicines. It is highly desirable to have reliable detection and determination of suspected pesticide residues for understanding of their overall quality. However, inherent metabolites abundant in herbal medicines typically make screening of trace-level pesticide residues arduous. Taking Lonicerae Japonicae Flos (LJF) as an example, the current study investigated the capability of a three-dimensional separation system, ultra high-performance liquid chromatography tandem ion mobility/quadrupole time-of-flight mass spectrometry (UPLC-IM/Q-TOF MS), for qualitative detection of numerous pesticides when suffering from complex matrix interferences. The comparison of collision gases was first performed to improve unwanted dissociations of pesticides, particularly labile ones, and nitrogen was ultimately chosen instead of argon. Further optimization of key MS parameters resulted a data-independent high-definition MSE (HDMSE) method was developed. Then, an in-house pesticide scientific library (PSL) containing 438 pesticides was constructed following a four-step workflow. Based on multi-dimensional information in this library, different identification criteria were compared and an enhanced qualitative screening approach for 432 targeted pesticides was finally proposed and validated. By its application to 104 batches of LJFs, the approach was demonstrated effective in resolving complex matrix interferences. Moreover, collision cross section (CCS) for identification purpose was favorable to improve detection sensitivity while maintain ideal accuracy.
Keywords
Pesticide residues
Qualitative screening
Ion mobility spectrometry
Collision cross section
Lonicerae Japonicae Flos

1 Introduction
In the large-scale cultivation of medicinal plants, a type of special agricultural product for medicinal materials, it is unavoidable to use various pesticides, such as herbicides, fungicides, insecticides, plant growth regulators, and so on, to prevent natural damages and ensure productivity and quality (Gahukar, 2018). While, terminal pesticide residues in plant-derived medicines may pose some risk to human health (Williamson et al., 2015; Wang et al., 2015). Reliable detection and determination of suspected pesticide residues in herbal medicines is highly desirable for their holistic quality control and safety risk assessment (Luo et al., 2021; WHO, 2017). However, the presence of numerous inherent ingredients in herbal medicines hampered it by complex matrix interferences (Parrilla Vázquez et al., 2019). When compared to water-rich vegetables and fruits, accurate screening and identification of pesticide residues in dried herbal medicines is far more challenging.
Because of its superior sensitivity and specificity, mass spectrometry combined with gas or liquid chromatography (GC/LC-MS) has gained popularity in pesticide residue analysis (Afify et al., 2022; Lehotay et al., 2015; Masiáet al., 2016). Among these, triple quadrupole mass spectrometry in selected or multiple reaction monitoring scan mode is currently considered as the “gold standard” for targeted qualitative and quantitative pesticide residue determination (Lehotay et al., 2015; Miao et al., 2019). However, the instrument’s compound-specific data acquisition, inherent unit-mass resolution and scan speed limit its application in untargeted multi-residue screening of numerous pesticides. Moreover, reference standard is required to individually optimize parameters to achieve optimal sensitivity and selectivity (Huang et al., 2021; Regueiro et al., 2016). In comparison, high-resolution mass spectrometry (HRMS) could provide excellent mass-resolving power and mass accuracy, as well as heterogeneous quasi-molecular ions through a sensitive full scan, allowing to outperform in residue detection of an unlimited number of pesticides and retrospective analysis (Lehotay et al., 2015; Wong et al., 2018). Without the need of reference standard materials for method development, LC-HRMS-based screening of pesticide residues could be performed by searching against a compound database that contains orthogonal identification points on targeted pesticides, such as retention time, quasi-molecular ion m/z and so on (Wong et al., 2018). Additionally, fragment ions provided by hybrid HRMS, such as the widely used Q-TOF and Q-Orbitrap, are commonly utilized for identification purposes. Data dependent acquisition (DDA) and data independent acquisition (DIA) modes are typically used to trigger dissociation and obtain qualitative fragments. DIA mode, such as MSE (elevated energy MS), AIF (All Ion Fragmentation) and MS/MSALL acquisition methods separately offered by commercial Q-TOF, Q-Orbitrap and Triple TOF mass spectrometers, could acquire high-throughput fragmentation information and support untargeted identification and retrospective analysis (Knolhoff and Croley, 2016; Wong et al., 2018). However, in this mode, the correlation of fragment ions and their corresponding precursor ion is difficult. Matrix interferences and co-elution of isomers inevitably complicate post-data interpretation, thus resulting in false-positive and false-negative assignments in multi-residue screening of trace-level pesticides in highly complex herbal medicines.
In this regard, the introduction of ion mobility spectrometry (IMS) into a UPLC-HRMS system provides an additional separation dimension and is favorable to improve qualitative performance of both targeted and untargeted analyses (Ewing et al., 2016; Hernández-Mesa et al., 2017; Kaufmann, 2020a; Levy et al., 2019). Different from conventional mass spectrometry, ion separation in IMS is related to not only size and charge but also shape, which makes isomeric or isobaric compounds revolving promising. For the most employed travelling wave ion mobility spectrometry (TWIMS), the device comprises a series of ring electrodes. Charged ions migrate in a drift tube under confinement of radio-frequency voltage of opposite phases applied to adjacent ring electrodes and transient direct current voltage, meanwhile suffer from collisions with buffer gas filled in the drift tube (Lanucara et al., 2014). Ion mobility separation is achieved based on drifting speeds of ions with different mass-to-charge ratios and stereo-chemical structures, namely enjoying different cross-sectional areas. In general, compact ions with lower mass-to-charge ratios take shorter drift times through the drift tube (Hernández-Mesa et al., 2017). According to drift time of the charged ion, TWIMS, for example, a commercial Vion IMS system can directly afford its collision cross section (CCS) value, a physicochemical parameter for compound identification (Goscinny et al., 2019; Regueiro et al., 2016). Furthermore, in this system, the ion mobility drift tube positions in the front of a quadrupole mass analyzer and a collision cell. From the view of mobility separation dimension, fragment ions produced in the collision cell theoretically possess the same drift time with the corresponding precursor. Thus, by aligning drift time in an operating and processing UNIFI software of the instrument, fragments in the DIA mode could be assigned to their parent ion, while interferences from co-eluted analytes reduce. It ultimately results in a cleaner MS and high-quality MS/MS spectra beneficial to data interpretation (Regueiro et al., 2016). So far, an IMS-integrated multi-dimensional separation system has been widely used for targeted contaminants screening in various matrix (Goscinny et al., 2019; Mosekiemang et al., 2021; Regueiro et al., 2016; Zainudin et al., 2022). And, CCS value with a deviation of 2% has shown promises in enhancing identification confidence. However, its application for large-scale pesticide residue screening in complex herbal medicines has still remained unexplored.
The dried flower buds of Lonicera japonica Thunb. are traditionally used as the herbal medicine Lonicerae Japonicae Flos (LJF), recorded in the latest edition Chinese Pharmacopoeia (2020 edition), and are also widely used for cosmetics and functional tea due to their extensive therapeutic and health functions (Chinese Pharmacopoeia Commission, 2020; Shang et al., 2011). Herbicides, fungicides, insecticides and plant growth regulators are commonly used in the large-scale cultivation of L. japonica to prevent insect pests and plant diseases, meanwhile improve the yield and quality. Multi-residue screening and quantitative assay of LJFs have revealed serious overuse and misuse of pesticides (Li et al., 2018; Zhou et al., 2019). On the one hand, terminal pesticide residues undoubtedly raise safety risk on human health. On the other hand, we also found that overuse of insecticides would give rise to ingredient variations in LJF, which may undermine its pharmacological effects (Pan et al., 2021). Hence, extensive detection and targeted determination of pesticide residues in LJF are urgent for its holistic quality control.
In the present study, we systematically investigated qualitative performance of a three-dimensional separation system, ultra high-performance liquid chromatography tandem ion mobility/quadrupole time-of-flight mass spectrometry (UPLC-IM/Q-TOF MS), for the large-scale screening of pesticide residues in complex herbal medicine. An enhanced approach for targeted screening of 432 pesticide residues in LJF was developed and validated by data acquisition optimization, an in-house library construction and different identification criteria comparison. Herein, effect of collision gas on post-mobility fragmentation was discussed for the first time to reduce unwanted dissociations of pesticides. And, a detailed workflow for creating a database containing a large set of compounds was proposed. The applicability of the approach we proposed was finally assessed by analyzing 104 batches of the real LJF samples.
2 Experimental
2.1 Chemicals and reagents
Analytical standards of pesticides used in this study were purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany) and Sigma-Aldrich (Seelze, Germany). Stock solutions of pesticide standards were individually prepared in acetonitrile at concentrations of 1000 μg/mL. Some with poor solubility in acetonitrile were dissolved in methanol. Eight intermediate mix solutions were prepared in acetonitrile at concentrations of 10 μg/mL from the individual stock solution. Working solutions of two concentration levels at 100 and 1000 μg/L were separately obtained by spiking 1.0 and 10.0 mL of each intermediate mix solution into two 100 mL of volumetric flasks and diluting to volumes with acetonitrile. The stock, intermediate mix solutions and working solutions were all stored in brown glass bottles at −20 °C prior to use.
LC-MS grade methanol and water purchased from Merck KGaA (Merck, Darmstadt, Germany) were employed for chromatographic separation. HPLC-grade acetonitrile (J.T. Baker, Phillipsburg, USA), ultra-pure water prepared by a Millipore Alpha-Q water purification system (Millipore, Bedford, USA), analytical acetic acid (Shanghai Lingfeng Chemical Reagent Co., Ltd., Shanghai, China) were used for sample preparation. Formic acid (Thermo Fisher Scientific, Shanghai, China; FA) and 10 M ammonium formate (Sigma-Aldrich Co., MO, USA; AF) of LC/MS grade were used as chromatographic additives. Leucine enkephalin solution and major mix calibration solution were purchased from Waters (Milford, MA, USA) for instrumental calibsample containing eight standards was purchased from Waters for monitoring instrumental performance. The information of the eight standards was showed in Table S1 (Supporting Information).
Cleanert MAS-Q 50 mL centrifuge tubes embodying 6.0 g anhydrous magnesium sulfate (MgSO4) and 1.5 g anhydrous sodium acetate (NaOAc) and Cleanert MAS-Q 15 mL centrifuge tubes containing 900 mg MgSO4, 900 mg primary secondary amine (PSA), 300 mg C18, 300 mg silica and 90 mg graphitized carbon black (GCB) were purchased from Bonna-Agela Technologies (Tianjin, China) for sample extraction and purification.
LJF samples, totaling 104 batches, were collected from nine Chinese provinces and the detailed information was provided in Table S2. These samples were crushed into powders and stored at −20◦C before analysis.
2.2 Sample preparation
A modified QuEChERS method fully optimized and validated in our previous works was used for sample preparation (Miao et al., 2019; Zhou et al., 2019). In detail, an aliquot of 3.0 g fine powder of each material was accurately weighed and immersed for 30 min in a 50 mL centrifuge tube with 15 mL water containing 1% acetic acid. After being added 15 mL acetonitrile, the sample was shaken for 5 min using the IKA®KS 260 control shaker (Germany). Then, 6.0 g anhydrous MgSO4 and 1.5 g anhydrous (NaOAc) were added into the mixture, and another shaking was conducted for 5 min. The obtained extract was cooled in an ice bath for 10 min and centrifuged at 4000 rpm for 5 min. Subsequently, 9 mL of the supernatant was transferred into 15 mL centrifuge tube and cleaned up with a mixture of 900 mg MgSO4, 900 mg PSA, 300 mg C18, 300 mg silica and 90 mg GCB by shaking for 5 min. After centrifugation of the mixture at 4000 rpm for 5 min, 5 mL of the supernatant was transferred into another 15 mL centrifuge tube and concentrated to about 0.4 mL in a water bath at 40 °C with a gentle stream of nitrogen. The residues were resolved with acetonitrile and made up to 1 mL. After vortexing, the test solution was obtained by filtering the cleaned-up extract through a 0.22 μm membrane and stored at 4 °C before analysis.
2.3 UPLC-IM/Q-TOF MS analysis
Chromatographic separation was conducted on an ACQUITY UPLC I-Class system (Waters, Milford, MA, USA). And the system was equipped with a binary solvent manager, a column manager and a sample manager. An Agilent Poroshell EC-C18 column (3.0 × 150 mm, 2.7 μm) was used at 35 °C. The gradient elution was performed with a binary mobile phase system consisted of water (A) and methanol (B), both containing 0.05% FA and 5 mM AF (v/v), at the flow rate of 0.4 mL/min. The elution program was followed as below: 0–1 min, 5% (B); 1–4 min, 5%-60% (B); 4–8 min, 60%-64% (B); 8–8.5 min, 64%-68% (B); 8.5–9 min, 68%-75% (B); 9–16 min, 75%-95% (B); 16–20 min, 95% (B); 20–20.1 min, 95%-5% (B); 20.1–25 min, 5% (B). The injection volume was set to 5 μL.
A VionTM IM/Q-TOF hybrid mass spectrometer (Waters, Milford, MA, USA) was coupled to UPLC system via a ZprayTM ESI source to acquire high-resolution MS data in the positive mode. The instrument tuning parameters were set as given in Table S3. The optimized source parameters were set as follows: capillary voltage, 0.5 kV; cone voltage, 40 V; source temperature, 120 °C; desolvation temperature, 550 °C; Cone gas flow, 50 L/h; desolvation gas flow, 800 L/h. Desolvation and cone gas were nitrogen (greater than99.99%). The IMS buffer gas was nitrogen (greater than99.999%). A high-definition MSE (HDMSE) scan method was applied for data acquisition. The analyzer in sensitivity mode scanned over a mass range of m/z 100–1000 and the scan time was 0.2 s. Two independent scan events, so-called full scan and pseudo-MS/MS scan, were separately performed at a low collision energy of 4 eV and a high collision energy ramping from 25 to 60 eV. Acquired MS data contained a low-energy function and a high-energy function, separately representing full scan MS and pseudo-MS/MS. Nitrogen (greater than99.999%) was used for collision induced dissociation (CID). Real-time mass calibration (LockSprayTM) was conducted using a 50 pg/μL external reference of leucine encephalin. To assess instrumental stability, the QC sample was analyzed every six injections of the test solutions during data acquisition. An UNIFI v1.9 software (Waters, Milford, MA, USA) was utilized for instrument control, data acquisition and processing.
2.4 Automatic targeted screening
Qualitative screening was performed using a solution of accurate mass screening on HDMSE in UNIFI v1.9 software. Referring to the guidelines from the United States Food and Drug Administration (United States Food and Drug Administration, 2015) and European Union Reference Laboratories for Residues of Pesticides (European Commission, 2021), different identification criteria were compared based on the screening detection limits of the tested pesticides, the false-positive rate and the false-negative rate. The retention time tolerance was set to ± 0.1 min. The mass accuracy for the quasi-molecular ions detected in the low-energy function was within ± 5 ppm. CCS tolerance was defined as ± 2%. Fragments within a mass tolerance of ± 10 ppm were also discovered.
2.5 Qualitative method validation
The qualitative screening method was validated in accordance with the EU guidance document SANTE/11312/2021 (European Commission, 2021). The qualitative performance of the proposed approach was evaluated by a screening detection limit (SDL), which is the lowest level at which an analyst can be detected. A 5% false-negative rate was considered acceptable. In other words, a pesticide’s SDL should be detected in at least 19 out of 20 samples. Herein, ten different samples of LJF were spiked with pesticide standard working solutions and prepared in duplicate to yield a total of 20 spiked samples at three concentration levels (0.01, 0.05 and 0.20 mg/kg). To detect false positives, non-spiked samples were also prepared and analyzed.
3 Results and discussion
3.1 Effect of collision gas on post-mobility fragmentation
Dissociation of analysts, especially labile ones, occurred before the mass analyzer in full scan, has been widely discussed in some previous IMS-based qualitative analyses (Celma et al., 2020; Regueiro et al., 2016). This will inevitably decrease detection sensitivity of the method. So far, effective solutions have remained unexplored. Our preliminary analysis showed that serious unwanted fragmentations were detected in the low-energy function for about 150 out of more than 500 tested pesticides. Taking a labile pesticide paraoxon-ethyl as an example (Figure S1), a negligible improvement was observed by decreasing the settings of capillary voltage (2.5 eV to 0.5 eV) and sampling cone voltage (40 V to 10 V) that have been indicated to correspond to in-source fragmentations (Yang et al., 2016). For dissociation taking place in the mobility cell, although a more massive mobility gas is unfavorable, it has been reported that the best compromise of ion mobility resolution, relative distribution over the drift time range and signal response was obtained with nitrogen, as used in this study (Goscinny et al., 2015). In addition, given that a low-energy channel voltage was applied in full scan of HDMSE mode, there was likely dissociating of ions during transferring in the collision cell. Unfortunately, visual improvements were also not detected by changing this voltage from 6 eV to 4 eV (Figure S1). Thus, we focused on impact of collision gas on post-mobility fragmentations of pesticides. Two collision gases, argon (Ar) and nitrogen (N2), were compared from two aspects, abundance of fragments in the low-energy channel and intensity of the targeted quasi-molecular ions. After tuning the instrument with the same parameters as given in Table S3, data acquisition was conducted with a HDMSE method holding consistent acquiring settings for both collision gases.
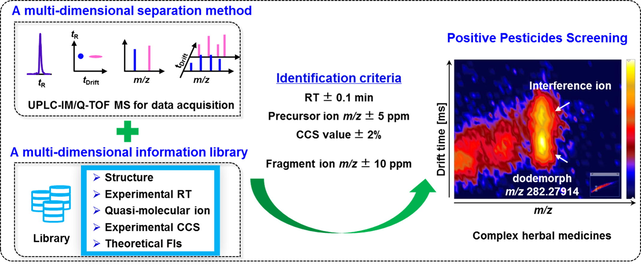
From the view of fragments abundance in the low-energy channel, the dissociations of most labile pesticides were significantly reduced with nitrogen as the collision gas compared to argon. The reduced fragmentations mainly occurred in the collision cell, namely after IMS separation, thus called “post-mobility fragmentation”. For illustration, Fig. 1 gave an example of a comparison for furalaxyl with argon and nitrogen as collision gases, respectively. The protonated ion (m/z 302.13868) of furalaxyl tended to successively eliminate a molecule of CH3OH and a unit of CO to produce two fragments at m/z 270.11247 (Fragment 1) and m/z 242.11756 (Fragment 2) (Fig. 1A). When argon was used, the two fragments predominated in its low-energy channel spectrum. After resolving the spectra using drift, Fragment 2 was unobserved, whereas Fragment 1 still remained abundant (Fig. 1B). It was indicative that the dissociation for Fragment 1 occurred after the IMS separation, few possibly in the ion source or the mobility cell, while Fragment 2 mainly produced in the ion source or the mobility cell (Celma et al., 2020; Regueiro et al., 2016). Some evidences were also found in the overlapped mobiligram of the extracted ions of m/z 302.13868, 270.11247 and 242.11756 (Fig. 1B). Fragment 1 presented a similar drift time to that of its precursor ion, whereas Fragment 2 obviously showed a shorter drift time. Moreover, the asymmetrical mobility peak shapes of Fragments 1 and 2 were also suggestive of partial dissociations of the quasi-molecular ion in the mobility cell (Regueiro et al., 2016). Comparatively, when using nitrogen, the relative abundance of Fragments 1 and 2 remarkably decreased, and the two fragments were absent in the drift time-aligned low-energy spectrum, which filtered out fragments that do not match the precursor’s drift time (Fig. 1C). Additionally, both Fragments 1 and 2 visibly showed shorter drift times than the protonated ion in the overlapped mobiligram. The nitrogen collision gas appeared to reduce the post-mobility dissociations of the labile pesticides while presenting the simplified drift time-aligned spectra. As a supplement, examples of two fragile pesticides, paraoxon-ethyl (Figure S2) and aminocarb (Figure S3), were also gave and explained in Supporting Information to support this conclusion.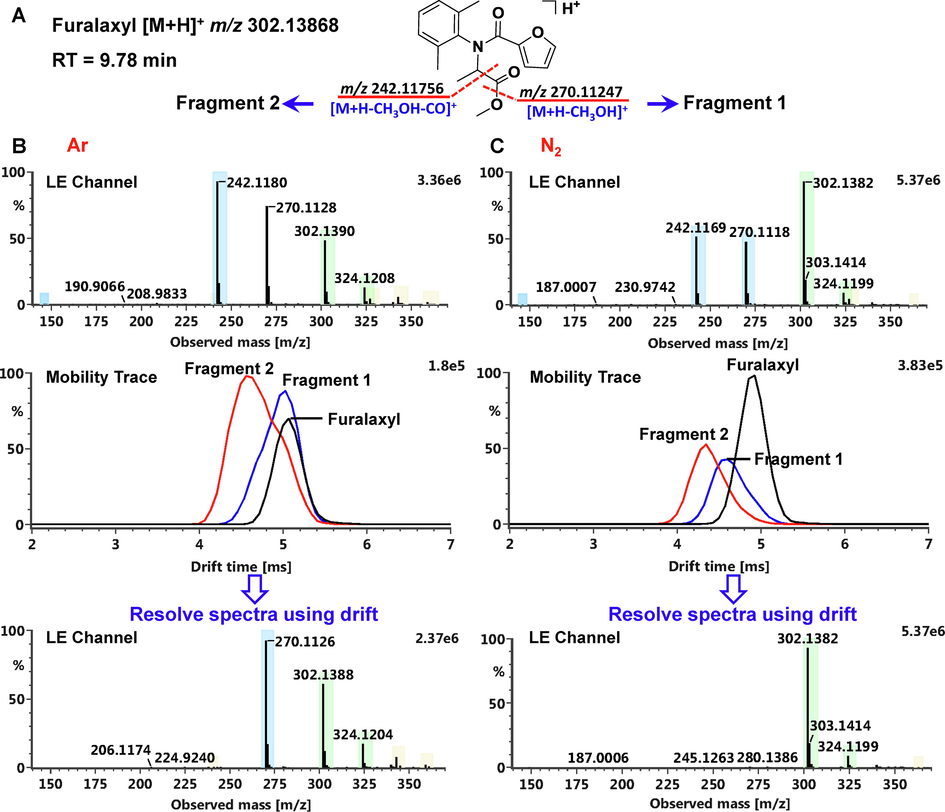
The proposed fragmentation of furalaxyl to produce Fragment 1 (m/z 270.11247) and Fragment 2 (m/z 242.11756) (A), the low-energy (LE) spectra of m/z 302.13868 and the overlapped mobiligram of the extracted ions of m/z 302.13868, 270.11247 and 242.11756 with argon (B) and nitrogen (C) as collision gas, respectively.
On the other hand, the effect of collision gas on intensity of the targeted quasi-molecular ions was also assessed. The response ratio of the quasi-molecular ions detected with nitrogen as the collision gas to argon (RN/A) was calculated, and a total of 438 pesticides (443 items) at the concentration of 100 μg/L were examined in the six consecutive injections. As summarized in Figure S4, about 93.9% of the pesticide items (4 1 6) acquired higher responses with nitrogen than argon, namely the obtained RN/A value greater than 1. Among them, 54.2% of the items (2 4 0) had the RN/A value greater than 1.2, for which the responses were considered remarkably increased when using nitrogen. Furthermore, 6.1% of pesticide items (27) reduced responses with nitrogen. And only 1.35% (6) presented the significant decrease (0.4 < RN/A < 0.8). The results demonstrated that the post-mobility transmission efficiency of the quasi-molecular ions was distinctly improved with nitrogen as the collision gas.
Taken together, under the same parameter settings of mass spectrometer, a less massive collision gas, nitrogen compared to argon, possibly provided less energy when crashing with the quasi-molecular ions, leading to the decreased fragmentations (Florencio et al., 1994). Moreover, less scattering of the ion beam for collision with nitrogen than argon likely resulted in the greater efficiency of ion transmission (Hoteling et al., 2003). Thus, nitrogen was finally chosen as the collision gas in this study. It was valuable to improve detection sensitivity and selectivity of trace-level pesticide residues in complex matrix.
3.2 Establishment of a UPLC-IM/Q-TOF MS method
To facilitate complementary investigation and mutual confirmation of different methods in our laboratory, the same chromatographic separation method developed in our previous works (Miao et al., 2019; Zhou et al., 2019) was adopted in this study. For MS data acquisition, even though the HDMSE mode has been widely applied for qualitative screening of pesticide residues, a detailed report on optimization of MS parameters was unavailable hitherto. In order to get the optimal response and abundant fragment ions for multi-class pesticide residues, the current work systematically optimized the key MS parameters including capillary voltage, sampling cone voltage, and ramp collision energy, and a total of 438 pesticides were tested.
Capillary voltage, one of the major ESI source parameters effecting ionization of analytes, was first optimized. The effects of different capillary voltage settings (0.5, 1.0, 1.5, 2.0, 2.5, 3.0 and 3.5 kV) on the responses of 438 pesticides (443 items) were investigated. As summarized in Fig. 2A, 93.9% (4 1 6) pesticides obtained the highest responses at the capillary voltage of 0.5 kV. Among them, the responses of 292 items obtained separately at 0.5 kV and 1.0 kV had statistical differences (p value < 0.05) by the Student’s t-test. Using representative pesticides of eight sub-structure types as examples (Figure S5), their responses tended to decrease with increasing capillary voltage. Thus, the capillary voltage was ultimately set to 0.5 kV. Then, sampling cone voltage, a parameter affecting adduct ions and possibly causing in-source fragmentation (Yang et al., 2016), was optimized. The responses of 438 pesticides (443 items) at different cone voltages (10, 20, 30, 40, 50 and 60 V) were compared. As given in Fig. 2B, there were 27.5% (1 2 2), 35.7% (1 5 8), and 30.7% (1 3 6) pesticides getting the highest responses at the cone voltage of 40, 50 and 60 V, respectively. Furthermore, the in-source fragmentations of the labile pesticides were assessed by separately setting the cone voltage to 40, 50 and 60 V. It was implied that the in-source fragmentations of the labile pesticides enhanced with the increase of cone voltage. For instance, demeton-S-methyl-sulfoxide and metrafenone were separately dissociated into the fragment ions of m/z 169.0088 ([C4H10O3PS]+) and m/z 209.0814 ([C11H13O4]+) at 60 V (Figure S6). And the two fragments had the similar intensities with the protonated ions. When the cone voltage was lower (40 V or 50 V), their intensities significantly decreased and were about one-third of their protonated ions. Considering the two factors above, the sampling cone voltage was finally determined to set at 40 V.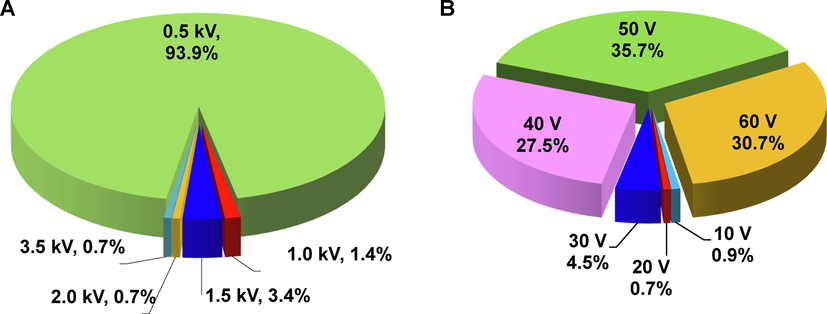
The statistics of pesticides with the highest response at different capillary voltages (A) and cone voltages (B).
In addition, a mass-dependent ramp collision energy (RCE) for collision-induced dissociation (CID) of quasi-molecular ions was optimized to acquire more balanced pseudo-MS/MS spectra for multi-class pesticides. Herein, different low mass energy (LME) and high mass energy (HME) settings were separately compared for fragile and stable pesticides. Two representative pesticides, tralkoxydim and pirimiphos-methyl, were used as examples for illustration (Figure S7). Firstly, with the LME setting at 20 eV, data acquisition was operated in the HME at 55, 60 and 65 eV, respectively. As a result, significant differences of the high-energy channel spectra were not observed for two pesticides with the increase of HME. Thus, the voltage of HME was set to 60 eV. Holding this setting, different LME voltages separately at 20, 25 and 30 eV were further compared. Comparison of the high-energy channel spectra showed that collision-induced dissociations of both fragile and stable pesticides remarkably enhanced with the increase of LME voltage. For stable pirimiphos-methyl, the optimal strength of the main fragment ion at m/z 164.1182 was 30 eV. While, under the same setting, fragile tralkoxydim easily dissociated into a series of fragments with lower masses. The abundance of the diagnostic fragment ions with higher masses, such as an intense ion at m/z 284.1634, dramatically reduced with the higher LME, which was considered unfavorable for qualitative identification of those fragile pesticides. By comprehensive consideration, the LME was determined as 25 eV. An optimized UPLC-IM/Q-TOF HDMSE method was thus developed.
3.3 Construction of an in-house pesticide scientific library
A database is a prerequisite for targeted matching and screening of pesticide residues with a LC-HRMS-based methodology. Up to 280 pesticide standards have been measured for experimental CCS values so far (Bauer et al., 2018). And few reports were available on developing an applicable IMS-based database. Hence, the current work is intended to introduce a detailed workflow for construction of an in-house pesticide scientific library (PSL) with a large set of pesticides.
As presented in Fig. 3, the overall workflow was mainly composed of four steps. Step 1, chemical structures of the 438 pesticides were firstly downloaded through the free website ChemSpider (https://www.chemspider.com/), generating a compound list containing accurate masses (AMs). Then, the targeted pesticides were divided into 18 AM-staggered groups by sorting AMs and generating repetitive serial Group IDs 1–18. As a result, isobaric and isomeric pesticides were distributed in different groups. Step 2, the basic information of these pesticides, including item name, CAS number, item tag “Pesticide Classification”, item tag “Group ID” and structure in the format of.mol, was listed in an Excel template (Table S4) and imported into the UNIFI software by selecting ESI + to develop a basic structural library. Step 3, according to AM-staggered grouping, 18 mixed solutions of pesticide standards at a concentration of 1000 μg/L were prepared and analyzed with the optimized UPLC-IM/Q-TOF HDMSE method. Then, targeted screening of each group of pesticides was performed using an individual accurate mass screening method. Retention time-based screening was not enabled. Adducts including + H+, +Na+, +K+, +Li+, and + NH4+ were allowed, and an acceptable mass tolerance of quasi molecular ions was defined as ± 5 ppm. The theoretical fragmentation detected in the high-energy spectra was predicted and explained based on the imported structures and a mass accuracy of ± 10 ppm was set for fragments. The maximum number of fragment ions was kept to 5 and in-source fragments were also sought. Step 4, by targeted match with AM-staggered pesticide groups, four-dimensional identification information, including retention time, accurate quasi-molecular ion m/z, 1–5 theoretical fragment ions with accurate m/z and elemental composition, and experimental CCS values of quasi-molecular ions, was attributed to a unique AM, namely a specific pesticide. Integrating this information into the library in step 1 ultimately resulted in the construction of an in-house PSL.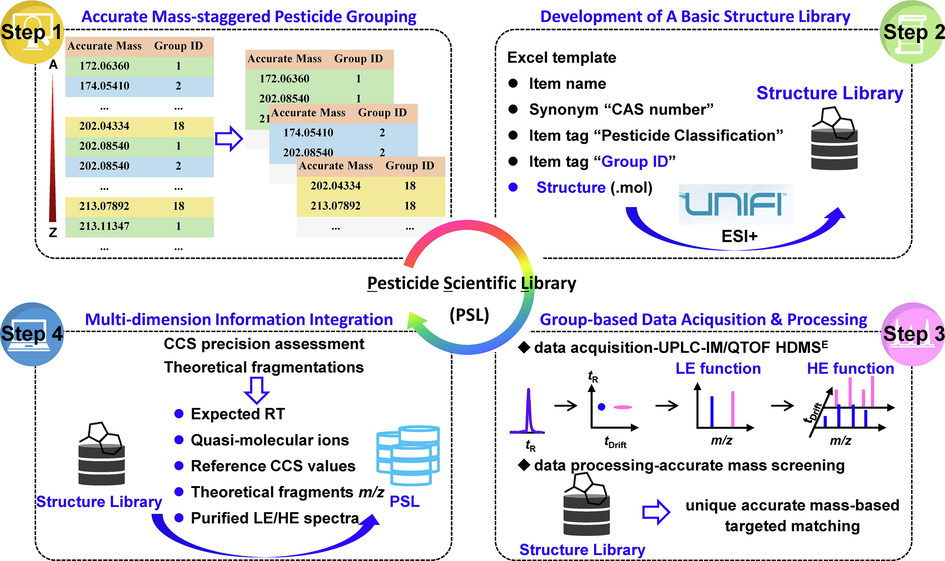
A workflow for the establishment of database including a large set of targeted analytes using UPLC-IM/Q-TOF MS.
For the majority of pesticides, the protonated ions were predominately detected in the low-energy spectra. There were around 125 pesticides mainly in the sodium or ammonium adducts. It was hardly necessary to predefine a targeted pesticide as a specific adduct form for this PSL. On the basis of the imported chemical structures, automatic searching of the quasi-molecular ions could be made by allowing all feasible adduct forms in the qualitative screening method. Particularly, some attention should be given to those labile pesticides, such as azoxystrobin, difenzoquat-methyl-sulfate, dioxathion, isofenphos, pretilachlor, quinmerac, and so on. The pre-mobility fragmentations leaded to their molecular ions being either absent or detected with low intensity in the low-energy function. Thus, structures of the abundant fragments were theoretically deduced to replace their original structures. Herein, a “chopping” bond disconnection algorithm offered by the UNIFI software played an important role in explaining fragment ions in both the low-energy and high-energy functions (Kaufmann et al., 2017). In silico fragmentation based on the pesticide structures could predict theoretical structures of the fragment ions, thus affording their theoretical accurate m/z and elemental composition, which were considered superior to average measured or corrected masses for identification purposes (Kaufmann et al., 2017; Zhou et al., 2019). Additionally, although the HDMSE mode was a DIA method, product ions observed in the high-energy spectra could be highly associated with corresponding precursor ions by drift time alignments (Regueiro et al., 2017). Therefore, intense fragment ions in the high-energy spectra (a maximum number of 5) were also integrated into the PSL.
As a molecular descriptor, CCS value is mainly obtained from experimental measurement of a reference standard (Regueiro et al., 2016). The investigation of the measurement precision of CCS values is essential to ensure their transferability and robustness for identification purposes. Thus, the study assessed the intra-day precision with six successive analyses on one day and the inter-day precision with three replicates over five consecutive days. Standard mixture solutions at three concentration levels (10, 100, and 1000 μg/L) were applied. In order to monitor and evaluate the acquired data quality in real time, the sequence included analyses of a commercial QC sample containing eight components covering a wide mass range from m/z 100 to 700 and CCS values ranging from 100 to 300 Å2 (Table S1). As a consequence, for the intra-day CCS measurement of 438 pesticides (443 items) at three concentration levels, the relative standard deviations (RSDs) were less than 0.8%, of which more than 93% were less than 0.2% (Figure S8). And for the inter-day CCS measurement, the RSDs were also less than 0.8%, and above 97% were less than 0.4% (Figure S8). The excellent intra-day and inter-day precision demonstrated the robustness of the CCS measurements at different detectable concentrations. Ultimately, the average CCS values measured at the concentration of 100 μg/L (n = 15) were attached to the PSL.
Following this workflow, an in-house PSL integrating the multiple features of 438 pesticides (443 items), including chemical structure, molecular formula, experimental retention time (RT), accurate mass of the quasi-molecular ions and the corresponding expected CCS values, theoretical fragment ions with accurate m/z, as well as the simplified low- and high-energy spectra, was finally established. And the essential four-dimensional descriptors, namely expected retention time and CCS value, accurate m/z of the abundant quasi-molecular ion, and 1–5 theoretical fragment ions, were given in Table S5.
3.4 An enhanced strategy for targeted multi-residue screening of pesticides
Data acquisition and processing are two essential aspects for a targeted screening methodology based on LC-HRMS. In this study, the optimized UPLC-IM/Q-TOF HDMSE method was developed for data acquisition. And its superiority was evidenced in the following aspects. I) IMS separation orthogonal to chromatographic retention was favorable for increasing peak capacity. On the basis of the established PSL, the separation difference between IMS and LC was estimated with the linearity regression correlation coefficient (R2). A two-dimensional distribution of CCS values against retention times for all pesticide entries was plotted in Figure S9. A good linearity, namely the value of R2 close to one, means a strong correlation. Conversely, the R2 value of 0.2371 implied the excellent orthogonality of CCS values and retention times in accordance with the previously published work (Regueiro et al., 2016). Namely, IMS-derived CCS was promising in enhancing identification confidence. II) IMS facilitated differentiation of isomeric compounds. In line with the previous findings (Regueiro et al., 2016), the tough correlation between CCS value and ion m/z was suggested via a two-dimensional correlogram with an R2 value of 0.8935 (Figure S10). Nonetheless, a few differences still remained since IMS separation was also related to molecular shape in addition to ion m/z. This allowed to resolve isomeric or isobaric pesticides, as well as matrix interferences. For instance, fenoxycarb and furalaxyl have an identical molecular formula of C17H19NO4. As depicted in Fig. 4A, furalaxyl possessed a more compact structure in comparison with fenoxycarb, thus suffering from less resistance from mobility gas (Regueiro et al., 2016). This accounted for the faster migration of furalaxyl in the drift tube and thus a smaller CCS value (162.04 vs 178.85 Å2). Interestingly, the potentiality of IMS for differentiation of stereoisomers was also revealed by an illustrative example in Fig. 4A. The 3D structure of trans-permethrin was relatively folded, thus presenting a smaller CCS value (184.88 Å2) for its sodium adduct ([M + Na]+) than that of cis-permethrin (190.96 Å2). On the other hand, it also evidenced that IMS was promising in reducing matrix interferences from complex herbal medicines. As shown in Fig. 4B, the targeted pesticide dodemorph at m/z 282.27914 in the spiked sample (200 μg/L) could be successfully distinguished with an unknown interference ion from the blank matrix by IMS separation. III) IMS separation integrated into a DIA method of MSE, namely HDMSE, increased selectivity while acquiring high-throughput fragmentation information. Since the mobility cell locates in the front of the collision cell, fragments could be correlated to the corresponding precursor ion by drift time alignment. From this perspective, the additional IMS separation was capable of avoiding false assignments in the case of fragment ions as an identification point. Additionally, as highlighted in the analogous works (Mosekiemang et al., 2021; Regueiro et al., 2017), the high-energy spectra were simultaneously purified, which was rewarding for both targeted and untargeted data interpretation. IV) For “stiff” pesticides that are poorly fragmented and those existing chiefly as sodium adducts, although a higher response can be detected in the low-energy channel, nonexistent or low-intensity diagnostic fragments in the high-energy channel hampered confirmation of the positive detections. While IMS-derived CCS values could facilitate their identification.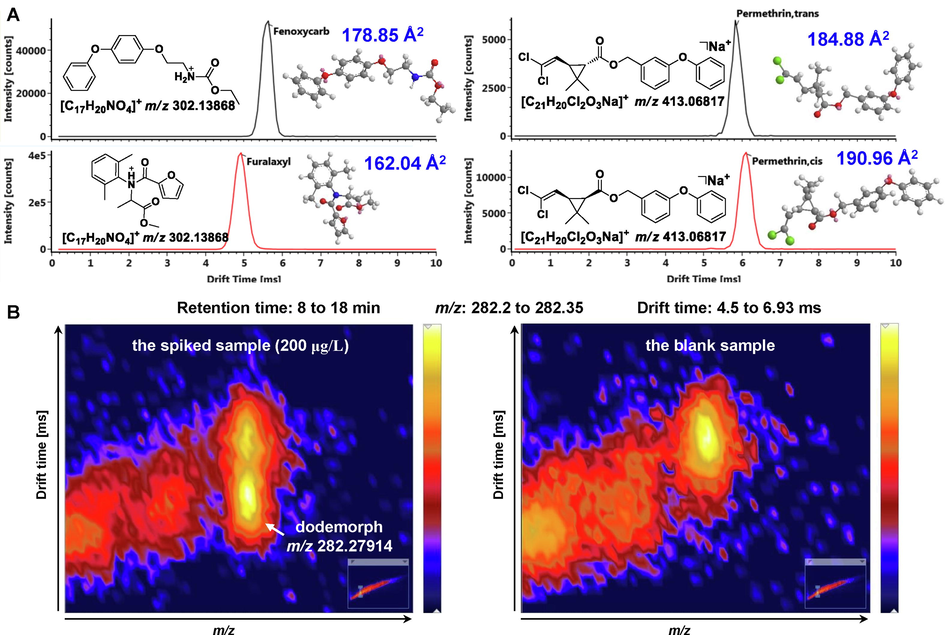
Comparison of the mobiligrams and structures of two pairs of isomeric pesticides, fenoxycarb vs. furalaxyl and trans vs. cis permethrin (A), and illustration of IMS capability on resolving interferences from complex matrix (B).
Once the qualitative data has been acquired, data processing began with a four-dimensional peak picking. And then targeted searching against an in-house database with a targeted accurate mass screening method within the UNIFI platform was operated (Regueiro et al., 2017). Automatic matching of experimental values and the expected pesticide entries recorded in the PSL was achieved by predefining specific qualitative parameters and identification criteria. Referring to the guidelines from the United States Food and Drug Administration (United States Food and Drug Administration, 2015), and European Union Reference Laboratories for Residues of Pesticides (European Commission, 2021), the current work examined the applicability of different customized identification criteria with chromatographic retention time, precursor ions, at least one fragment ion and CCS value as identification points for suspect screening and identification of pesticides in LJF. Herein, ten different LJF samples spiked with a mixed standard working solution, including 438 pesticides, were prepared in duplicate, generating a total of 20 spiked samples at three concentration levels of 0.01, 0.05 and 0.20 mg/kg. With an acceptable false-negative rate of 5% (European Commission, 2021), the lowest detectable concentrations of 438 pesticides in the LJF samples were determined when different screening criteria were utilized. Moreover, 20 non-spiked samples, namely the 10 LJF samples prepared in duplicate, were analyzed for false-positive determination. It was worth noting that pesticides present in the 10 samples, which have been confirmed in our previous work (Zhou et al., 2019), were not of concern here. For the entire qualitative analysis process, manual verification was conducted to exclude misattributions of false positives and false negatives.
The detection rate, false-positive and false-negative rates, which reflect the sensitivity and selectivity of a screening method to a certain extent, were utilized for comparison of four qualitative screening modes. Given the excellent mass resolving power of Q-TOF MS, mass tolerance of precursor ion in four modes was all set to ± 5 ppm. In addition, the retention time deviation setting was investigated. As shown in Table 1, the number of detections at the concentration levels of 0.01 and 0.05 mg/kg slightly increased by setting the retention time tolerance to ± 0.1 min instead of ± 0.2 min, while the number of false positives remarkably reduced from 114 to 85. This implied that the large retention time window (± 0.2 min) was prone to merging detection of pesticides, particularly isomeric pesticides with similar chromatographic behaviors (Bauer et al., 2018). For instance, the retention time difference of sebuthylazine and propazine was 0.15 min, less than 0.2 min (Figure S11). Propazine failed to be resolved with the retention time tolerance of 0.2 min, leading to its false-negative detection. From this perspective, the retention time tolerance of ± 0.1 min enhanced the selectivity of suspect screening, thus simultaneously reduced false assignments due to complex matrix interferences. With regard to this, there were some concerns about the increasing possibility of missing detection due to chromatographic instability (Zainudin et al., 2022). Nonetheless, real-time correction of expected retention time (Zhou et al., 2019) or a filter function offered by the UNIFI software (Goscinny et al., 2019) appeared promising, which was not the focus of this study. Furthermore, the extra requirements on the CCS value and at least one fragment ion were separately supplemented on the basis of the screening criteria with the retention time tolerance ± 0.1 min and the mass accuracy tolerance of the precursor ion ± 5 ppm. As displayed in Table 1, when adding the identification criteria of fragment ions with the mass accuracy of ± 10 ppm, there were 162 and 101 detected pesticides separately at the levels of 0.01 and 0.05 mg/kg. It was indicative of an exceedingly decreased sensitivity, especially at 0.01 mg/kg. While its excellent selectivity was demonstrated by only 15 false positives in the 20 spiked samples. In comparison, adding the CCS value with an acceptable tolerance of ± 2% resulted in a slight decrease in detection rates, from 107 to 105 at 0.05 mg/kg and 59 to 55 at 0.2 mg/kg, but a reduction in false positives to 65 (Table 1). It suggested that CCS was valuable in reducing false positives, meanwhile achieving considerable sensitivity comparable to that obtained in the case of only defined retention time and precursor ions. Regarding the results, the poor detectability of fragment ions at low concentration levels, even absence for some “stiff” pesticides, seriously hampered positive findings. Whereas CCS values of precursor ions, independent of matrix interference (Bauer et al., 2018; Goscinny et al., 2019), could be accurately measured even at low concentrations, providing a valuable identification criterion and preventing false assignments. However, limited IMS resolution determined CCS values insufficiently selective against complex matrix interferences compared to fragment ions (Kaufmann et al., 2020b).
Detection constraints
No. detections
false positives
false negatives
0.01 mg/kg
0.05 mg/kg
0.2 mg/kg
>0.2 mg/kg
± 0.2 min and ± 5 ppm (parent ion m/z)
241
104
60
31
114
7
± 0.1 min and ± 5 ppm (parent ion m/z)
244
107
59
28
85
5
± 0.1 min and ± 5 ppm (parent ion m/z) and ± 2% (CCS value)
243
105
55
29
65
11
± 0.1 min and ± 5 ppm (parent ion m/z) and ± 10 ppm (≥1 fragments m/z)
162
101
61
37
15
82
In conclusion, an enhanced screening approach as given in Fig. 5 was finally proposed for qualitative detection and identification of a large set of pesticide residues in complex herbal medicine. Sample preparation was initially conducted with a modified QuEChERS method (Miao et al., 2019; Zhou et al., 2019). Then, multi-dimensional data was acquired by using the optimized UPLC-IM/Q-TOF HDMSE method. Subsequently, the data was imported into the UNIFI software for information extraction and targeted database searching. Four-dimensional (4D) peak picking generated a compound list that involved experimental values of descriptors, such as retention time, precursor ion m/z (adducts), CCS, response, and so on. By predefining identification criteria with a retention time tolerance ± 0.1 min, and a mass accuracy of precursor ion ± 5 ppm, and a ΔCCS value ± 2%, PSL-based target match was performed for detection and identification of suspected pesticides. In addition, fragment ions with a mass accuracy ± 10 ppm were also utilized to exclude false positives for auxiliary identification purposes.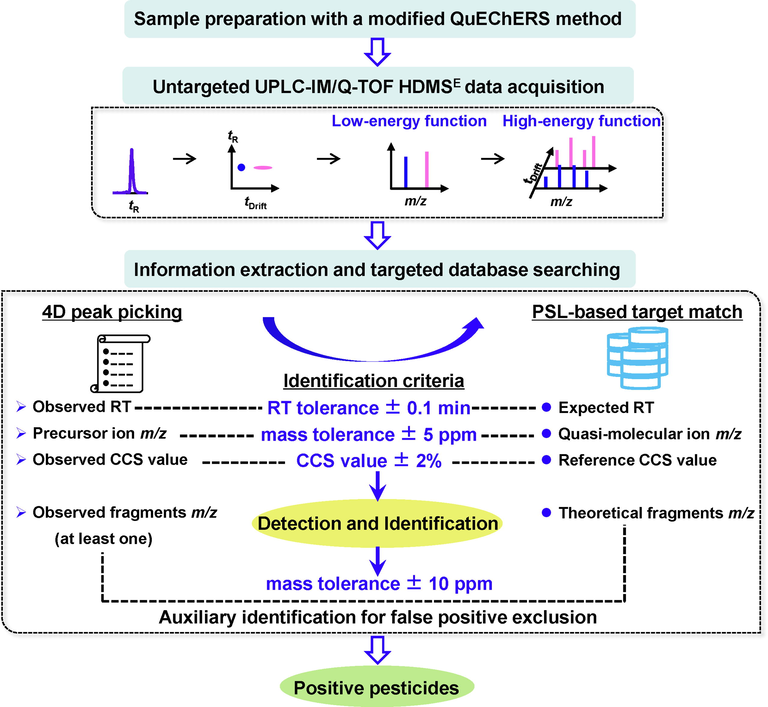
A workflow of the enhanced approach for targeted screening of pesticide residues in complex herbal medicines based on UPLC-IM/Q-TOF MS.
3.5 Method validation
According to the EU guidance document SANTE/11312/2021 (European Commission, 2021), the SDL is recommended for validation of a HRMS-based qualitative screening method. In this regard, an acceptable false negative rate of 5% is set, namely the pesticide being detected in at least 95% of the samples. Herein, the SDLs of the 438 tested pesticides were determined by the analyses of the 20 spiked LJF samples at three concentration levels of 0.01, 0.05, and 0.20 mg/kg, respectively. Table S6 showed the number of positive detections for each pesticide at the three levels. The concentration, at which 19 or 20 positive detections were obtained, was defined as the SDL. As a consequence, the SDLs of 230 pesticide items were 0.01 mg/kg, and 103 pesticides had the SDLs of 0.05 mg/kg. There were 53 pesticides only detectable at concentrations of 0.2 mg/kg. In addition, 29 pesticides could only be detected in 10 of the 20 spiked samples. Their SDLs were considered greater than 0.2 mg/kg. Unfortunately, dipropetryn and secbumeton could not be screened out even in the standard solution at a concentration of 200 μg/L. Because they enjoyed the similar chromatographic retention and ion mobility drift with their isomeric compounds, dimethametryn and prometon. And for the other nine undetectable pesticides, including cinido-ethyl, dimoxystrobin, methoprene, cis-permethrin, trans-permethrin, propoxycarbazone, sulfentrazone, thiencarbazone-methyl, and triazophos, a proper sample preparation may be needed to further eliminate the ion suppression effect from the co-eluted matrix ingredients.
3.6 Application to analyses of real samples
Once the qualitative approach was established and validated, its applicability was further examined by multi-residue pesticide screening of 104 LJF samples which were extensively collected from nine different regions in China. Prior to the sample investigations, it should be noted that the analyses of the standard QC sample were also integrated into the sample sequence for monitoring and evaluating the data quality in real time. By matching with the expected in Table S1, the retention times and the CCS values of the QC components satisfactorily met the predefined acceptable thresholds (retention time ± 0.1 min, ΔCCS value ± 2%). It ensured no false negatives due to the chromatographic separation instability or inadequate calibration of the IM/Q-TOF instrument (Figure S12). Additionally, manual exclusion of false assignments was also supplemented.
As a consequence, a total of 44 pesticide residues were detected from 104 batches of LJF samples, mainly involving fungicides and insecticides (Fig. 6). Among them, 20 pesticides were found in more than 10 batches of the LJF samples. The detection frequencies of six pesticides were more than 50%, including thiophanate-methyl (83.7%), chlorpyrifos (65.4%), cyhalothrin (54.8%), difenoconazole (52.9%), thiamethoxam (52.9%) and tebuconazole (51.9%). From the view of regional distribution (Fig. 6), the LJF samples originating from the three main producing areas of Henan, Hebei, and Shandong provinces have relatively more pesticide residues, indicating a more serious contamination of LJFs in these three regions. With regard to this situation, in addition to the abuse of fungicides and insecticides during the large-scale cultivation of LJF, the growing environmental pollution originating from the cultivation of other crops, such as water and soil, was also one of the reasons. The findings provided crucial information for further establishment of a targeted quantitative method to economically monitor pesticide residues with a high detection frequency in LJF. It was significant to guide the comprehensive treatment of pesticide pollution of LJF at the source of cultivation and finally improve its holistic quality.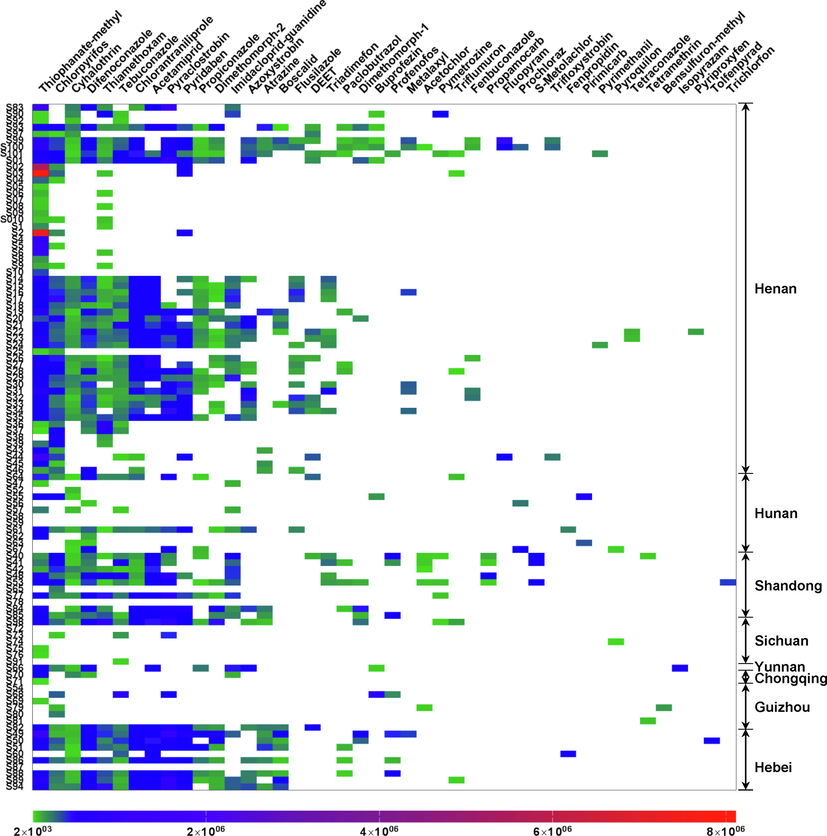
A heatmap of 44 pesticides in 104 batches of LJFs based on responses.
4 Conclusions
In this study, an enhanced approach for qualitative screening of 432 targeted pesticides was proposed and validated by a multi-dimensional separation system of UPLC-IM/Q-TOF MS. Unwanted dissociation of labile pesticides in full scan was first discussed. Compared with argon, nitrogen as a collision gas showed remarkable superiorities in improving post-mobility fragmentation and ion transmission. A sensitive HDMSE method was developed by optimizing the key MS parameters based on comprehensive evaluation of 438 multi-class pesticides. By this method, an in-house library containing multi-dimensional information of 438 pesticides (443 items) was constructed following a detailed workflow. Furthermore, different identification criteria were compared for targeted searching against the library. It manifested that IMS-derived CCS value as an identification point could effectively enhance detection and identification of pesticide residues in complex herbal medicines. Finally, analyses of the real LJF samples further validated applicability of the approach. In the future, its application may be extended to other herbal medicines.
Acknowledgement
This work sponsored by National Natural Science Foundation of China (82104390), Shanghai Sailing Program (21YF1442100), China Postdoctoral Science Foundation (2019M651551), Shanghai Science and Technology Commission Research and Development Platform (21DZ2290200) and The Development Fund for Shanghai Talents.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Development of GC–MS/MS method for environmental monitoring of 49 pesticide residues in food commodities in Al-Rass, Al-Qassim region. Saudi Arabia. Arab. J. Chem.. 2022;15:104199
- [CrossRef] [Google Scholar]
- Evaluation and validation of an ion mobility quadrupole time-of-flight mass spectrometry pesticide screening approach. J. Sep. Sci.. 2018;41(10):2178-2187.
- [CrossRef] [Google Scholar]
- Improving target and suspect screening high-resolution mass spectrometry workflows in environmental analysis by ion mobility separation. Environ. Sci. Technol.. 2020;54(23):15120-15131.
- [CrossRef] [Google Scholar]
- Chinese Pharmacopoeia Commission, 2020. Pharmacopoeia of the People’s Republic of China, vol. 1, Chinese Medical Science and Technology Press, Beijing, pp. 230–231.
- European Commission, 2021. Analytical quality control and method validation for pesticide residues analysis in food and feed (SANTE/11312/2021). Sante/11312/2021 1–57.
- Hybrid ion mobility and mass spectrometry as a separation tool. J. Chromatogr. A. 2016;1439:3-25.
- [CrossRef] [Google Scholar]
- Collision gas effects in the collision-induced decomposition of protonated and cationized molecules of carbohydrate antibiotics. Org. Mass Spectrom.. 1994;29:483-490.
- [Google Scholar]
- Management of pests and diseases of important tropical/subtropical medicinal and aromatic plants: a review. J. Appl. Res. Med. Aromat. Plants. 2018;9:1-18.
- [CrossRef] [Google Scholar]
- Travelling-wave ion mobility time-of-flight mass spectrometry as an alternative strategy for screening of multi-class pesticides in fruits and vegetables. J. Chromatogr. A. 2015;1405:85-93.
- [CrossRef] [Google Scholar]
- Towards the use of ion mobility mass spectrometry derived collision cross section as a screening approach for unambiguous identification of targeted pesticides in food. Rapid Commun. Mass Spectrom.. 2019;33(S2):34-48.
- [CrossRef] [Google Scholar]
- Current applications and perspectives of ion mobility spectrometry to answer chemical food safety issues. Trac-Trends Anal. Chem.. 2017;94:39-53.
- [CrossRef] [Google Scholar]
- Optimization of matrix-assisted laser desorption/ionization time-of-flight collision-induced dissociation using poly (ethylene glycol) Rapid Commun. Mass Spectrom.. 2003;17:1671-1676.
- [Google Scholar]
- Nontarget and high-throughput screening of pesticides and metabolites residues in tea using ultra-high-performance liquid chromatography and quadrupole-orbitrap high-resolution mass spectrometry. J. Chromatogr. B. 2021;1179:122847
- [CrossRef] [Google Scholar]
- The use of UHPLC, IMS, and HRMS in multiresidue analytical methods: a critical review. J. Chromatogr. B. 2020;1158:122369
- [CrossRef] [Google Scholar]
- Using in silico fragmentation to improve routine residue screening in complex matrices. J. Am. Soc. Mass Spectrom.. 2017;28(12):2705-2715.
- [CrossRef] [Google Scholar]
- Does the ion mobility resolving power as provided by commercially available ion mobility quadrupole time-of-flight mass spectrometry instruments permit the unambiguous identification of small molecules in complex matrices? Anal. Chim. Acta. 2020;1107:113-126.
- [CrossRef] [Google Scholar]
- Non-targeted screening approaches for contaminants and adulterants in food using liquid chromatography hyphenated to high resolution mass spectrometry. J. Chromatogr. A. 2016;1428:86-96.
- [CrossRef] [Google Scholar]
- The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem.. 2014;6:281-294.
- [Google Scholar]
- Current issues involving screening and identification of chemical contaminants in foods by mass spectrometry. Trac-Trends Anal. Chem.. 2015;69:62-75.
- [CrossRef] [Google Scholar]
- Recent progress in metabolomics using ion mobility-mass spectrometry. Trac-Trends Anal. Chem.. 2019;116:274-281.
- [CrossRef] [Google Scholar]
- Dietary exposure risk assessment of flonicamid and its effect on constituents after application in Lonicerae Japonicae Flos. Chem. Pharm. Bull.. 2018;6(6):608-611.
- [CrossRef] [Google Scholar]
- Detection and risk assessments of multi-pesticides in 1771 cultivated herbal medicines by LC/MS-MS and GC/MS-MS. Chemosphere. 2021;262:127477
- [CrossRef] [Google Scholar]
- Determination of pesticides and veterinary drug residues in food by liquid chromatography-mass spectrometry: a review. Anal. Chim. Acta. 2016;936:40-61.
- [CrossRef] [Google Scholar]
- Simultaneous determination of 508 pesticide residues in Panax notoginseng by liquid chromatography-tandem mass spectrometry. J. Instrum. Anal.. 2019;38(7):761-774.
- [Google Scholar]
- Ultra-high pressure liquid chromatography coupled to travelling wave ion mobility-time of flight mass spectrometry for the screening of pharmaceutical metabolites in wastewater samples: application to antiretrovirals. J. Chromatogr. A. 2021;1660:462650
- [CrossRef] [Google Scholar]
- Plant metabolomics for studying the effect of two insecticides on comprehensive constituents of Lonicerae Japonicae Flos. Chin. J. Nat. Med.. 2021;19(1):70-80.
- [CrossRef] [Google Scholar]
- Pesticide residues in spices and herbs: Sample preparation methods and determination by chromatographic techniques. Trac-Trends Anal. Chem.. 2019;115:13-22.
- [CrossRef] [Google Scholar]
- Ion-mobility-derived collision cross section as an additional identification point for multiresidue screening of pesticides in fish feed. Anal. Chem.. 2016;88(22):11169-11177.
- [CrossRef] [Google Scholar]
- Targeted approach for qualitative screening of pesticides in salmon feed by liquid chromatography coupled to traveling-wave ion mobility/quadrupole time-of-flight mass spectrometry. Food Control. 2017;78(2017):116-125.
- [CrossRef] [Google Scholar]
- Lonicera japonica Thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol.. 2011;138(1):1-21.
- [CrossRef] [Google Scholar]
- United States Food and Drug Administration (U.S. FDA), 2015. Acceptance criteria for confirmation of identity of chemical residues using exact mass data within the office of foods and veterinary medicine program. U.S. FDA: Silver Spring, MD.
- Safety issues and new rapid detection methods in traditional Chinese medicinal materials. Acta Pharm. Sin. B. 2015;5(1):38-46.
- [CrossRef] [Google Scholar]
- WHO, 2017. WHO guidelines for assessing the quality of herbal medicines with reference to contaminants and residues. Geneva: World Health Organization.
- Evaluating the safety of herbal medicines: integrated toxicological approaches. Science. 2015;347(6219):S47-S49.
- [Google Scholar]
- Perspectives on liquid chromatography-high resolution mass spectrometry for pesticide screening in foods. J. Agric. Food Chem.. 2018;66(37):9573-9581.
- [CrossRef] [Google Scholar]
- Method development and application of offline two-dimensional liquid chromatography/quadrupole time-of-flight mass spectrometry-fast data directed analysis for comprehensive characterization of the saponins from Xueshuantong Injection. J. Pharm. Biomed. Anal.. 2016;128:322-332.
- [CrossRef] [Google Scholar]
- Comprehensive strategy for pesticide residue analysis in cocoa beans through qualitative and quantitative approach. Food Chem.. 2022;368:130778
- [CrossRef] [Google Scholar]
- Qualitative screening and quantitative determination of 569 pesticide residues in honeysuckle using ultrahigh-performance liquid chromatography coupled to quadrupole-Orbitrap high resolution mass spectrometry. J. Chromatogr. A. 2019;1606:460374
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105007.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




