

Translate this page into:
Synthesis and biological evaluation of a peptide-remifentanil conjugate as a novel bifunctional mu/delta-opioid receptor agonist for the treatment of pain
⁎Corresponding authors. believe890521@163.com (Tao Zhang), shiwg1988@126.com (Weiguo Shi), voncedar@126.com (Xuesong Feng)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
For the treatment of pain, the design of a bifunctional mu-opioid receptor (MOR)/delta-opioid receptor (DOR) agonist is an effective strategy to seek safer opioids with higher antinociceptive efficacy and diminished adverse side effects. Herein, we describe the design, synthesis, and evaluation of a novel bivalent ligand (SW-WL-2) with a methyl 1-(3-methoxy-3-oxopropyl)-4-(phenylamino) piperidine- 4-carboxylate moiety (remifentanil derivative) covalently linked to a dermorphin-like structure (H-Dmt-N-Me-D-Ala-Aba-Gly-NH2, BVD03) at the C-terminus. Our results showed that SW-WL-2 behaved as a potent dual agonist of MOR and DOR with significant and prolonged antinociceptive effects in acute pain models in vivo. Furthermore, SW-WL-2 exhibited reduced or no opioid-like side effects such as physical dependence or respiratory depression, in contrast to an equipotent analgesic dose of morphine or BVD03. Thus, SW-WL-2 should be used as a new lead compound for the discovery of safer opioid drugs for the treatment of pain.
Keywords
Safer opioids
Peptide-fentanyl conjugate
Bifunctional mu/delta opioid receptor agonists
Antinociceptive
Bivalent ligands

1 Introduction
Opioid drugs, such as morphine and fentanyl, play a critical role in the management of moderate-to-severe pain. The pharmacological functions of these drugs are achieved by their interaction with one or more of the three opioid receptor subtypes (mu, delta, and kappa), which belong to the superfamily of G-protein coupled receptors. Among them, the mu opioid receptor (MOR) is the primary target for most conventional opioid-based drugs. However, the activation of MOR is always associated with various undesirable side effects (e.g., tolerance, respiratory depression, constipation, and physical dependence), which greatly limited their clinical use. Therefore, the discovery of new analgesic drugs that retain their potent analgesic actions but prevent adverse side effects is urgently needed (Waldhoer et al., 2004, Al-Hasani and Bruchas 2011, Darcq and Kieffer 2018, Gunther et al., 2018).
It was proven that there are physical and functional interactions between the MOR and the delta opioid receptor (DOR) (Yekkirala et al., 2010, Yekkirala et al., 2012, Ong and Cahill 2014, Erbs et al., 2015). DOR agonists have a beneficial regulating effect on the pharmacological effects of MOR agonists (Gomes et al., 2000, Ong and Cahill 2014, Stefanucci et al., 2017). Therefore, the development of bivalent ligands targeting both MOR and DOR with synergistic antinociceptive effects is an emerging strategy in the search for safer opioid agonists; it has been hypothesized that these bifunctional MOR/DOR agonists may have a more favorable pharmacodynamic and pharmacokinetic profile, increase the therapeutic index, and cause less-severe side effects than monovalent agonists (Yamazaki et al., 2001, Gengo et al., 2003, Lowery et al., 2011, Metcalf et al., 2012, Podolsky et al., 2013, Matsumoto et al., 2014, Lei et al., 2020).
In our previous studies, we synthesized a new series of novel peptide-fentanyl analog conjugates by the covalent coupling of fentanyl derivatives to the C-terminus or N-terminus of a conformationally constrained dermorphin tetrapeptide analog (BVD03) via a chemical linker (Vandormael et al., 2011, Li et al., 2021). The most potent ligand for both MOR and DOR was SW-LJ-11, displaying distinct binding affinities (Ki = 0.31 nM and 0.65 nM, respectively) and agonist activities (EC50 = 10.42 nM and 4.47 nM, respectively) in vitro and significant antinociceptive effects in vivo. What’s more, compared to an equipotent analgesic dose of morphine or BVD03, SW-LJ-11 did not exhibit any physical dependence or respiratory depression. Therefore, SW-LJ-11 deserves further investigation.
Remifentanil is a well-known MOR-selective synthetic analgesic that is 750 times more potent than morphine and has been approved by US Federal Drug Administration. It is a prominent drug due to its high potency, low cardiovascular toxicity, and fast onset. However, its biological activity is easily lost due to its rapid metabolization (with a half-life of approximately 3–5 min) (Glass et al., 1999). Based on our previous study, we designed and synthesized a novel bivalent ligand for MOR and DOR with the C-terminus of BVD03 linked to a methyl 1-(3-methoxy-3-oxopropyl)-4-(phenylamino) piperidine-4-carboxylate moiety, which is a part of the remifentanil structure, to further study the structure–activity relationship of peptide-fentanyl analog conjugates and to explore whether the use of a safer remifentanil analog in the design of dual MOR/DOR agonists could improve the biological safety and decrease the side effects of opioids.(See Fig. 1)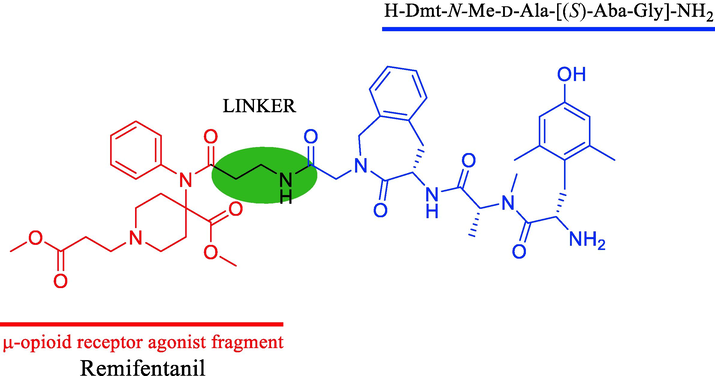
Chemical structure of the bifunctional mu/delta opioid receptor agonist.
2 Results and discussion
2.1 Synthesis and characterization of SW-WL-2
The intermediates 5 and 9 were synthesized according to our previously reported methods (Scheme S1) (Li et al., 2021). Meanwhile, the intermediates 2a, 2b, 2c, 2d, and 2e, as well as the final product, were synthesized according to conventional methods. The compounds were purified by preparative reversed-phase HPLC to afford compounds with ≥ 95.0% purity and overall yields of 10–20% (Scheme 1). The structures and purities were analyzed by 1H NMR, 13C NMR, HR-ESI-MS, and HPLC (Fig. S1A-S6D).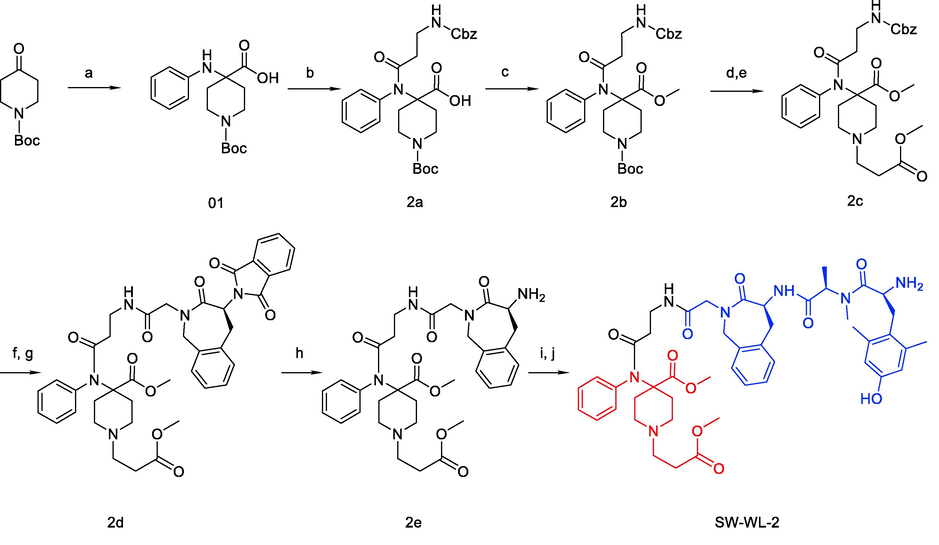
Synthesis of SW-WL-2. Reagents and conditions: (a) PhNH2, NaOH and CHCl3 in THF at 0 °C for 1 h, then rt for 18 h; (b) ClCO(CH2)2COO-Cbz and Et3N in CH2Cl2 at 0 °C, then rt for 4 h; (c) CH3I and Na2CO3 in DMSO at 40 °C for 12 h; (d) 1:1 TFA: CH2Cl2 at rt for 4 h; (e) CH3OOC(CH2)2Br, Et3N, and KI in MeCN at 90 °C (reflux) for 12 h; (f) Pd/C and H2 in MeOH at rt for 3 h; (g) Compound 5 (Scheme S1), HATU, and DIPEA in CH2Cl2 at rt for 4 h; (h) N2H4·H2O in EtOH at 90 °C (reflux) for 1.5 h; (i) Compound 9 (Scheme S1), HATU, and DIPEA in CH2Cl2 at rt for 4 h; (j) 1:1 TFA: CH2Cl2 at rt for 2 h.
2.2 In-vitro opioid receptor binding and efficacy
To characterize the binding affinities (Ki) of the newly synthesized compounds on MOR and DOR, in-vitro competitive radioligand binding assays using [3H] DAMGO or [3H] DADLE were performed, as described previously (Li et al., 2021). As shown in Table 1, SW-WL-2 displayed high opioid affinities in the low nanomolar range for MOR and DOR (Ki = 0.22 and 2.09 nM, respectively), and the values were comparable to those of BVD03 (Ki = 0.24 and 0.29 nM, respectively).
Compound
Binding affinitya
MOR agonist activityb
DOR agonist activityb
MOR (Ki, nM)
DOR (Ki, nM)
EC50 (nM)
Emax (% of ctl)
EC50 (nM)
Emax (% of ctl)
SW-WL-2
0.22
2.09
6.22
81 (100 μM)
1.81
85 (100 nM)
BVD03
0.24
0.29
14.53
60 (100 μM)
1.05
100 (100 μM)
DAMGOc
1.83
—
41.41
100 (100 μM)
—
—
DPDPEd
—
—
—
—
1.00
100 (100 nM)
Morphine
12.14
741.83
1.45
70 (100 μM)
0.57
86 (10 μM)
In addition, an intracellular Ca2+ release assay was carried out to determine the functionality of the compound on MOR and DOR. The observed high opioid affinities of SW-WL-2 were maintained in the intracellular Ca2+ release assays, demonstrating highly efficacious EC50 values. These results merely suggest that the chemical link between the fentanyl-related molecule and the C-terminus of BVD03 is important for the affinity and activity of BVD03-bifunctional peptide derivatives targeting MOR/DOR.
2.3 In-vivo antinociceptive effect
SW-WL-2 was used for the in-vivo antinociceptive evaluation because of its high opioid receptor affinity and in-vitro pharmacological activity. The antinociceptive potency and efficacy of SW-WL-2 were investigated by the formalin paw-licking test, acetic acid-induced writhing test, and hot-water tail withdrawal test. As shown in Fig. 2A and Table S1, SW-WL-2 and BVD03 produced sustained analgesia in phase II of the formalin paw-licking test compared to the vehicle group (P < 0.001), with a percent maximum possible effect (%MPE) of 92.04% and 95.31% in phase II, respectively. By comparison, morphine also showed sustained analgesia and less potency than SW-WL-2 and BVD03, with a %MPE of 80.07% at the same dosage. Fig. 2B shows that SW-WL-2, BVD03, and morphine produced a potent antinociceptive effect in the acetic acid-induced writhing test, with a %MPE of up to 100% at the same dosage of 2.0 mg/kg injected via the tail vein of mice, which is consistent with their formalin assay activities. In addition, the results of the hot-water tail withdrawal test are summarized in Fig. 2C. SW-WL-2 and BVD03 exhibited a higher analgesic potency than morphine. For example, at 30 min, the %MPE values for SW-WL-2 and BVD03 were 1.91 times and 2.34 times greater than that of morphine, respectively, at the same dosage of 2.0 mg/kg. Furthermore, Fig. 2D shows the dose- and time-related antinociception of SW-WL-2 following tail vein administration at doses of 0.5, 1.0, 2.0, and 4.0 mg/kg. Together, these results indicate that SW-WL-2 maintains a greater in-vivo analgesic effect than BVD03.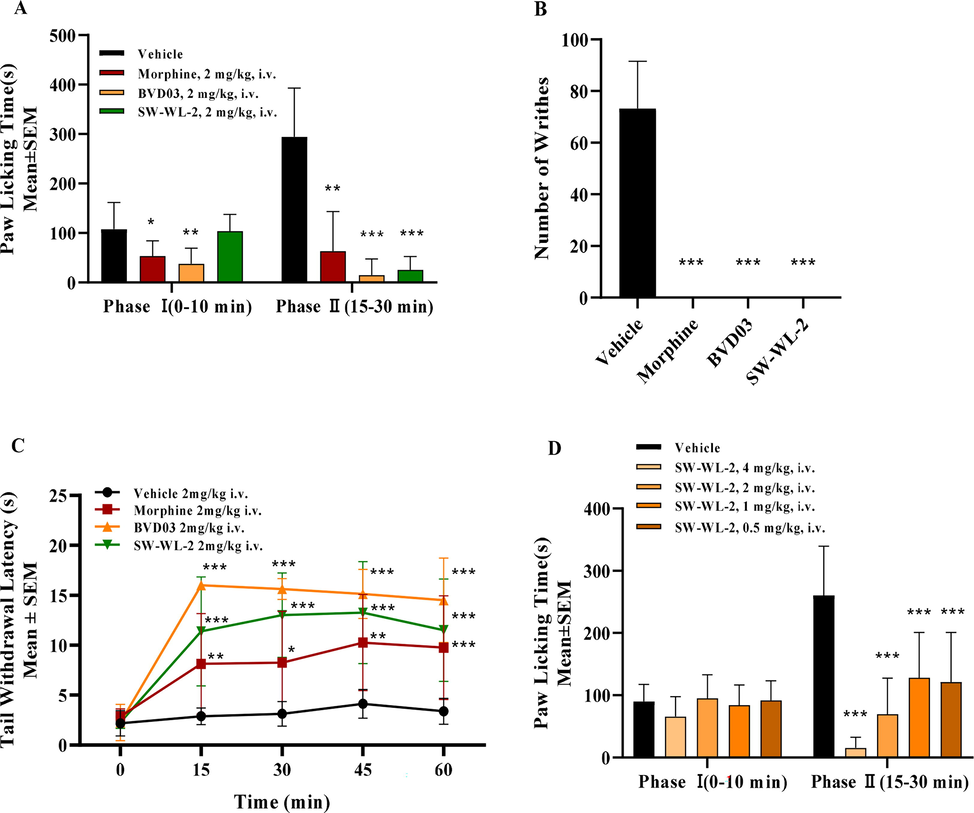
The test compounds produced antinociceptive behavior in an acute pathological pain model assay. The results are expressed as the mean ± standard error of the mean (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle (two-way analysis of variance test). The blank controls consisted of mice treated i.v. with saline for the formalin paw-licking test and injected with saline in the paw for the writhing test. (A) Formalin paw-licking test. (B) Acetic acid-induced writhing test. (C) Hot-water tail withdrawal test. (D) The analgesic activity of SW-WL-2 after i.v. injection at four different doses in the formalin paw-licking test.
2.4 In-vivo adverse reactions studies
The respiratory depression of mice treated with SW-WL-2 was examined by a blood gas analyzer. Compared to the saline group, the SW-WL-2 group did not show a reduction in any respiratory measures (Fig. 3A), while the morphine-exposed mice did present with a significant reduction in pO2 (mmHg) of the blood. These data suggest that SW-WL-2, at the same dose as morphine or remifentanil, does not induce acute respiratory depression. Interestingly, remifentanil only induced a rapid reduction of pO2 (mmHg) in the first 20 min and quickly returned to the same level as that of the saline group; this finding might be due to the rapid metabolization of remifentanil in vivo.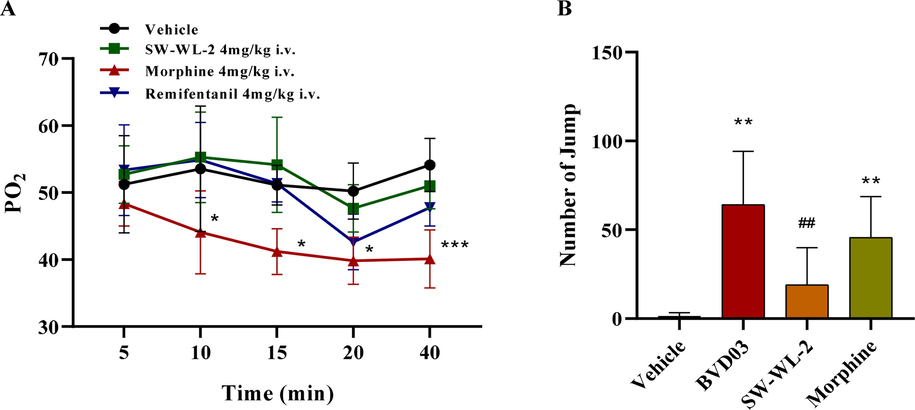
The adverse reactions induced by BVD03 and SW-WL-2 treatment in mice. The results are expressed as the mean ± standard error of the mean (n = 4–8). (A) Respiratory inhibition: Groups of mice (n = 4) were administered (4 mg/kg, ∼EDmax, i.v.) with morphine, BVD03, or SW-WL-2. The pO2 of blood sampled from mouse eyes at various time points was measured. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle (one-way analysis of variance test). (B) Physical dependence: Groups of mice (n = 8) were dosed (4 mg/kg, ∼EDmax, i.v.) once a day (9:00 am) with morphine, BVD03 or SW-WL-2 for 5 days. On day 5, the mice were treated with naloxone (1 mg/kg, i.p.) at 2 h after the administration of morphine, BVD03 or SW-WL-2. The number of jumps was counted over a 20-min period after the injection of naloxone. The symbol * indicates a significant difference from the vehicle, and the symbol # indicates a significant difference from BVD03 (one-way analysis of variance test, *P < 0.05, **P < 0.01, ##P < 0.01).
Moreover, the physical dependence of SW-WL-2 in mice was carried out with a precipitated withdrawal approach, as described previously (Li et al., 2021). Briefly, mice were treated with naloxone (1 mg/kg, i.p.) after the administration of the test compounds for five successive days to assess the physical dependence induced by morphine, BVD03, and SW-WL-2, respectively (Fig. 3B). The mice dosed with morphine or BVD03 jumped significantly more than those treated with the vehicle or SW-WL-2, suggesting that both morphine and BVD03 induced significant physical dependence and withdrawal compared with the vehicle. In contrast, SW-WL-2 did not elicit a significant increase compared with vehicle treatment, indicating that SW-WL-2 did not induce physical dependence or withdrawal in mice.
3 Conclusion
In conclusion, we reported that the novel bifunctional MOR/DOR agonist SW-WL-2, with a dermorphin-like tetrapeptide analog covalently conjugated to a remifentanil moiety, is a safer opioid analgesic with diminished deleterious side effects than morphine and BVD03. SW-WL-2 displayed dual MOR/DOR agonist properties in the low nanomolar range and significant analgesic efficacy in vivo in classic mouse models of pain. Furthermore, SW-WL-2 exhibited a weaker physical dependence and respiratory depression compared to an equipotent analgesic dose of morphine. Thus, SW-WL-2 should be used as a new lead compound for the discovery of safer opioid drugs for the treatment of pain.
4 Experimental section
4.1 Chemistry
All reactions were routinely monitored by thin layer chromatography (TLC) on silica gel plates (GF254) and were visualized using a UV lamp (λ = 254 nm). 1H NMR spectra and 13C NMR spectra were recorded on a 600 MHz spectrometer and a 151 MHz spectrometer, respectively, in which DMSO–d6 was used as the solvent and TMS was used as an internal standard. Coupling constants (J values) and chemical shifts (δ values) are expressed in Hz and ppm, respectively. Peak multiplicity is reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). High-resolution mass spectra (HRMS) were recorded on an Agilent 6210 ESI/TOF mass spectrometer. Accurate masses are reported for the molecular ion [M + H]+. Purification of the target compounds was carried out on a Shimadzu semipreparative HPLC system using a reversed-phase C-18 column (3 cm × 25 cm × 5 μm). All analyses were conducted at an ambient temperature with a flow rate of 10 mL/min. The HPLC eluent conditions were as follows: initially, a mixture of 40% MeCN/60% water (with 0.1% TFA) was used; over a period of 30 min, the gradient of MeCN increased to 45%. The UV detector was set at 215 nm. The sample purity was analyzed on an Agilent HPLC system using a reversed-phase C-18 column (250 mm × 4.6 mm × 5 μm). All analyses were conducted at an ambient temperature with a flow rate of 1 mL/min. The HPLC eluent conditions were as follows: initially, a mixture of 20% MeCN/80% water (with 0.1% TFA) was used; over a period of 30 min, the gradient of MeCN increased to 80%. The UV detector was set at 215 nm. The injection volume was 1 μL. The reagents and solvents were obtained commercially and were used without further purification.
4.2 Animals
Male or female Institute of Cancer Research mice (CD-1), weighing 23–26 g, were used for the current experiments (obtained from SPF Biotechnology, Beijing, China). The mice were housed in groups in a temperature-controlled environment, which was maintained on a 12-h light/dark cycle (with lights on at 07:00–19:00). Food and water were available ad libitum. All animal procedures were performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institute of Health and Animal Care Committee at the Beijing Institute of Pharmacology and Toxicology. Best efforts were made to minimize the number of animals used and their suffering.
4.3 General procedures for the synthesis of BVD03 and SW-WL-2
BVD03 was synthesized according to our previously reported methods (Scheme S1) (Li et al., 2021). SW-WL-2 was synthesized via the following procedure: firstly, compound 2b was deprotected by 1:1 TFA/CH2Cl2 and substituted with PhCH2CH2Br to obtain compound 2c. The Cbz protecting group was removed by catalytic hydrogenation. Finally, the target compound SW-WL-2 was produced by condensation of BVD03 with compound 2d using HATU and DIPEA as the condensation agents.
4.3.1 1-(Tert-butoxy carbonyl)-4-(phenylamino) piperidine-4-carboxylic acid (01)
Compound 01 was synthesized by a previously reported method (Li et al., 2021). Under ice-bath conditions, NaOH (111.1 g, 2.778 mol), N-Boc-4-piperidone (331.5 g, 1.667 mol), and CHCl3 (214.8 mL, 2.778 mol) were dissolved in a solution of aniline (50 mL, 0.567 mol) in THF (4.0 L), the reaction mixture was stirred at 0 ℃ for 1 h, then the ice bath was removed, and the reaction mixture was stirred at room temperature for an additional 18 h. After completion of the reaction was monitored by TLC, the reaction mixture was filtered through a Buchner funnel, and the filter cake was washed with THF. The filter cake was dissolved in 200 mL of water. The aqueous layer was acidified to pH 2–3 with a 1 N HCl solution. The mixture was extracted with EtOAc (500 mL × 3), dried with anhydrous sodium sulfate, and filtered. After concentration under reduced pressure, compound 01 was obtained as a yellow solid in 78.93% yield.
4.3.2 4-(3-(((Benzyloxy)carbonyl)amino)-N-phenylpropanamido)-1-(tert-butoxy carbonyl) piperidine-4-carboxylic acid (2a)
Cbz-NH(CH2)2COOH (10 g, 44.80 mmol) was dissolved in 100 mL of CH2Cl2, and then oxalyl chloride (4.55 mL, 53.76 mmol) and DMF (3 drops) were added under a stream of N2. The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under reduced pressure to afford Cbz-NH(CH2)2COCl. Under ice-bath conditions, Et3N (18.68 mL, 134.39 mmol) was added to a solution of compound 01 (14.35 g, 44.80 mmol) in CH2Cl2 (100 mL) under an atmosphere of N2. The mixture was stirred at room temperature for 4 h after the slow addition of Cbz-NH(CH2)2COCl in CH2Cl2 (monitored by TLC). The solution was concentrated under reduced pressure. Then, EtOAc (100 mL) was added, and the mixture was washed with water (50 mL × 3). The mixture was acidified to pH 2–3 with 1 N HCl, extracted with EtOAc (100 mL × 3), washed with water (100 mL × 1) and saturated aqueous sodium chloride solution (100 mL × 1), and dried over anhydrous Na2SO4. The crude product was purified by column chromatography (1:1 EtOAc: petroleum ether) to afford compound 2a as a clear yellow oil in 62.70% yield. HR-ESI-MS m/z [M + H]+ calculated for C28H35N3O7: 526.2553; found: 526.2548. 1H NMR (600 MHz, DMSO‑d6) δ 12.66 (s, 1H), 7.49–7.43 (m, 3H), 7.36–7.33 (m, 4H), 7.31–7.28 (m, 3H), 7.06 (t, J = 5.7 Hz, 1H), 4.96 (s, 2H), 3.55 (d, J = 11.04 Hz, 2H), 3.11 (dd, J = 13.32, 6.96 Hz, 4H), 2.09 (d, J = 13.56 Hz, 2H), 1.99 (t, J = 7.2 Hz, 2H), 1.41 (td, J = 13.92, 4.32 Hz, 2H), 1.34 (s, 9H).
4.3.3 1-(Tert-butyl) 4-methyl 4-(3-(((benzyloxy)carbonyl) amino)-N-phenylpropanamido) piperidine-1,4- dicarboxylate (2b)
To a solution of compound 2a (7.41 g, 14.09 mmol) in DMSO (100 mL), Na2CO3 (4.48 g, 42.27 mmol) and CH3I (1.75 mL, 28.18 mmol) were added, and the reaction mixture was stirred at 40 ℃ for 12 h. The mixture was cooled down to room temperature and then added to an ice-water mixture. The mixture was extracted with EtOAc (100 mL × 3). The organic phases were washed with water (100 mL × 1) and saturated NaCl solution (100 mL × 1). The organic layer was dried over Na2SO4, filtered, and concentrated by rotary evaporation to afford 7.38 g (97.09%) of compound 2b. HR-ESI-MS m/z [M + H]+ calculated for C29H37N3O7: 540.2710; found: 540.2704. 1H NMR (600 MHz, DMSO‑d6) δ 7.50–7.45 (m, 3H), 7.36–7.33 (m, 4H), 7.30–7.29 (m, 3H), 7.08 (t, J = 5.7 Hz, 1H), 4.95 (s, 2H), 3.67 (s, 3H), 3.58 (d, J = 12.48 Hz, 2H), 3.08 (dd, J = 13.08, 6.84 Hz, 4H), 2.07 (d, J = 13.5 Hz, 2H), 1.98 (t, J = 7.08 Hz, 2H), 1.39 (td, J = 13.8, 4.62 Hz, 2H), 1.33 (s, 9H).
4.3.4 Methyl 4-(3-(((benzyloxy)carbonyl)amino)-N-phenylpropanamido)-1-(3-methoxy-3-oxopropyl)piperidine-4-carboxylate (2c)
Compound 2b (7.38 g, 13.68 mmoL) was dissolved in a mixture of TFA and CH2Cl2 (50 mL, 1/1, v/v), and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated by rotary evaporation, and the residue was dissolved in water and alkalized to pH 10 with a solution of 0.5 N NaOH. The mixture was extracted with CH2Cl2 (100 mL × 3), and the organic phases were washed with saturated NaCl solution (100 mL × 1). The organic layer was dried over Na2SO4, filtered, and concentrated by rotary evaporation. The residue (5.64 g, 12.83 mmol) was dissolved in MeCN (100 mL), and methyl 3-bromopropionate (4.28 mL, 38.49 mmol), Et3N (5.35 mL, 38.49 mmol), and KI (0.05 g, 0.30 mmol) were added to the solution. The reaction mixture was refluxed in an oil bath at 90 ℃ for 12 h. The mixture was cooled down to room temperature and concentrated by rotary evaporation. The residue was dissolved in EtOAc, and the solution was washed with water (100 mL × 2) and saturated NaCl solution (100 mL × 1). The organic layer was dried over Na2SO4, filtered, and concentrated by rotary evaporation. The residue was purified by silica gel column chromatography using MeOH/CH2Cl2 (1/60, v/v) as the eluent to afford 4.41 g (61.36%) of compound 2c. HR-ESI-MS m/z [M + H]+ calculated for C28H35N3O7: 526.2553; found: 526.2548. 1H NMR (600 MHz, DMSO‑d6) δ 7.50–7.44 (m, 3H), 7.36–7.29 (m, 7H), 7.06 (t, J = 11.22 Hz, 1H), 4.95 (s, 2H), 3.65 (s, 3H), 3.54 (s, 3H), 3.08 (dd, J = 13.08, 6.78 Hz, 2H), 2.48 (s, 4H), 2.38 (t, J = 6.36 Hz, 2H), 2.25 (s, 2H), 2.07 (d, J = 12.84 Hz, 2H), 1.98 (dd, J = 7.14, 3.12 Hz, 2H), 1.49 (t, J = 9.6 Hz, 2H).
4.3.5 Methyl-(S)-4-(3-(2-(4-(1,3-dioxoisoindolin-2-yl)-3-oxo-1,3,4,5-tetrahydro-2H-benzo[c]azepin-2-yl) acetamido)-N-phenylpropanamido)-1-(3-methoxy-3-oxopropyl) piperidine-4-carboxylate (2d)
To a solution of compound 2c (2.50 g, 4.76 mmol) in MeOH (100 mL), Pd/C (0.25 g) was added, and the reaction mixture was stirred at room temperature under a H2 atmosphere for 4 h. The reaction mixture was filtered and concentrated by rotary evaporation to afford 1.53 g (82.10%) of brown oil. After vacuum drying, the brown oil (1.53 g, 3.90 mmol) was added to a mixture of compound 5 (Scheme S1) (1.29 g, 3.55 mmol) and HATU (1.48 g, 3.90 mmol) in CH2Cl2 (100 mL), then DIPEA (1.85 mL, 10.65 mmol) was added dropwise, and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was washed with water (100 mL × 2) and saturated NaCl solution (100 mL × 2). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated by rotary evaporation. The residue was purified by silica gel column chromatography using MeOH/CH2Cl2 (1/80; 1/60; 1/40, v/v) as the eluent to afford 2.19 g (83.60%) of compound 2d as a white solid. HR-ESI-MS m/z [M + H]+ calculated for C40H43N5O9: 738.3139; found: 738.3135. 1H NMR (600 MHz, DMSO‑d6) δ 7.93–7.89 (m, 4H), 7.73 (t, J = 5.7 Hz, 1H), 7.49–7.41 (m, 3H), 7.33 (dd, J = 7.14, 5.88 Hz, 2H), 7.28–7.26 (m, 1H), 7.24 (d, J = 3.18 Hz, 2H), 7.19 (t, J = 7.38 Hz, 1H), 5.35 (dd, J = 11.82, 5.04 Hz, 1H), 4.86 (d, J = 15.96 Hz, 1H), 4.40 (d, J = 16.14 Hz, 1H), 4.14–4.13 (m, 1H), 4.10 (d, J = 16.38 Hz, 1H), 3.90 (dd, J = 16.02, 12 Hz, 1H), 3.78 (d, J = 16.26 Hz, 1H), 3.64 (s, 3H), 3.54 (s, 3H), 3.12 (dt, J = 8.94, 7.26 Hz, 2H), 2.45–2.43 (m, 4H), 2.35 (t, J = 6.9 Hz, 2H), 2.21 (t, J = 11.1 Hz, 2H), 2.04 (d, J = 12.42 Hz, 2H), 1.94 (td, J = 7.14, 1.56 Hz, 2H), 1.45 (t, J = 11.58 Hz, 2H).
4.3.6 Methyl-(S)-4-(3-(2-(4-amino-3-oxo-1,3,4,5-tetrahydro-2H-benzo[c]azepin-2-yl) acetamido)-N-phenylpropanamido)-1-(3-methoxy-3-oxopropyl) piperidine-4-carboxylate (2e)
To a solution of 2d (2.19 g, 2.97 mmol) in EtOH (100 mL), hydrazine hydrate (1.44 mL, 29.7 mmol) was added, and the reaction mixture was refluxed in an oil bath at 90 ℃ for 1.5 h. The reaction mixture was cooled down to 0 ℃ by use of an ice bath and was filtered through a Buchner funnel. After filtration, the filtrate was concentrated by rotary evaporation to afford compound 2e as a white solid in 85.37% yield, which was directly used without further purification in the following step. HR-ESI-MS m/z [M + H]+ calculated for C32H41N5O7: 608.3084; found: 608.3081. 1H NMR (600 MHz, DMSO‑d6) δ 7.83 (t, J = 5.7 Hz, 1H), 7.53–7.50 (m, 2H), 7.48–7.45 (m, 1H), 7.37–7.36 (m, 2H), 7.19 (td, J = 7.5, 1.38 Hz, 1H), 7.10 (dt, J = 15.2, 7.7 Hz, 3H), 5.10 (d, J = 16.5 Hz, 1H), 4.42 (dd, J = 12.72, 3.96 Hz, 1H), 4.16 (q, J = 5.16 Hz, 4H), 3.92 (d, J = 16.74 Hz, 1H), 3.66 (s, 3H), 3.55–3.52 (m, 4H), 3.14–3.10 (m, 3H), 2.75 (dd, J = 17.16, 12.9 Hz, 1H), 2.47 (t, J = 7.38 Hz, 3H), 2.37 (t, J = 6.9 Hz, 2H), 2.26–2.22 (m, 2H), 2.08–2.05 (m, 2H), 1.95 (td, J = 7.56, 1.86 Hz, 2H), 1.48 (t, J = 11.4 Hz, 2H).
4.3.7 Methyl 4-(3-(2-((S)-4-((R)-2-((S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-N-methylpropanamido)propanamido)-3-oxo-1,3,4,5-tetrahydro-2H-benzo[c]azepin-2-yl)acetamido)-N-phenylpropanamido)-1-(3-methoxy-3-oxopropyl)piperidine-4-carboxylate (SW-WL-2)
To a solution of compound 9 (Scheme S1) (0.91 g, 2.30 mmol) in CH2Cl2 (50 mL), HATU (0.96 g, 2.53 mmol), 2e (1.54 g, 2.53 mmol), and DIPEA (1.20 mL, 6.90 mmol) were added, and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was washed with water (50 mL × 2) and saturated NaCl solution (50 mL × 2). The organic layer was dried over Na2SO4, filtered, and concentrated by rotary evaporation. The residue was dissolved in a mixture of TFA and CH2Cl2 (1/1, v/v), and the reaction mixture was stirred at room temperature for 2 h. The mixture was concentrated by rotary evaporation, and the residue was purified by preparative HPLC and then lyophilized to afford compound SW-WL-2 as white crystals in 39.37% yield. m.p.: 155–157 °C; HR-ESI-MS m/z [M + H]+ calculated for C47H61N7O10: 884.4558; found: 884.4553. 1H NMR (600 MHz, DMSO‑d6) δ 9.66 (s, 1H), 9.23 (s, 1H), 8.30 (d, J = 3.78 Hz, 3H), 8.00 (d, J = 7.44 Hz, 1H), 7.84 (t, J = 5.52 Hz, 1H), 7.54–7.50 (m, 3H), 7.35 (d, J = 6.36 Hz, 2H), 7.20 (td, J = 7.32, 1.74 Hz, 1H), 7.13–7.09 (m, 3H), 6.44 (s, 2H), 5.29 (ddd, J = 12.48, 7.44, 4.8 Hz, 1H), 5.12–5.08 (m, 2H), 4.48–4.45 (m, 1H), 4.16 (d, J = 16.20 Hz, 1H), 3.96 (d, J = 16.98 Hz, 1H), 3.73 (s, 3H), 3.63 (s, 3H), 3.59 (d, J = 16.32 Hz, 1H), 3.47 (d, J = 10.92 Hz, 2H), 3.14–3.05(m, 5H), 3.02–2.98 (m, 2H), 2.89 (dd, J = 16.86, 13.20 Hz, 1H), 2.78 (t, J = 7.26 Hz, 2H), 2.31 (t, J = 12.18 Hz, 2H), 2.20 (s, 3H), 2.19 (s, 6H), 1.98 (t, J = 7.20 Hz, 2H), 1.74 (t, J = 13.80 Hz, 2H) 1.06 (d, J = 7.26 Hz, 3H). 13C NMR (151 MHz, DMSO‑d6) δ 172.44, 171.69, 171.23, 170.88, 170.37, 170.13, 168.12, 158.61, 158.40, 156.53, 138.96, 138.23, 135.81, 134.42, 130.79, 130.23, 129.79, 129.34, 128.08, 126.36, 121.99, 118.34, 116.36, 115.37, 60.12, 52.98, 52.32, 52.32, 51.58, 50.24, 49.67, 49.67, 48.43, 48.14, 40.52, 36.04, 35.28, 35.28, 31.47, 30.60, 30.60, 30.49, 28.91, 28.91, 20.18, 20.18, 14.99.
4.4 In-vitro pharmacology
4.4.1 Radioligand competition binding assay for MOR and DOR
The binding affinities of SW-WL-2 and reference compound H-Dmt-N-Me-D-Ala-Aba-Gly-NH2 (BVD03) for MOR and DOR were determined in competitive radioligand binding assays using [3H] DAMGO ([D-Ala2, N-MePhe4, Gly-ol5]-enkephalin) and [3H] DADLE ([D-Ala2, D-Leu5]-enkephalin)as the radioligands for MOR and DOR, respectively. Briefly, assay buffer (50 mM HEPES, pH 7.4, 10 mM MgCl2) and test compound (dissolved in 1% DMSO) were added to wells of a 96-well plate and shaken at 500 rpm for 5 min. Then, the assay buffer with [3H] DAMGO or [3H] DADLE (final concentration of 1 nM) was successively added, and the plate was shaken at 500 rpm for 5 min and incubated at 27 ℃ for 1 h. The GF/B filter plate was preincubated with 0.5% polyethylenimine at 4 ℃ for 1 h and then washed twice with 1 mL of wash buffer (50 mM Tris, pH 7.4, 4 ℃). The membrane mix was transferred to a GF/B filter and dried for 10 min at 55 ℃. Finally, 40 μL of the ULTIMA GOLD scintillation cocktail (PerkinElmer, Waltham, MA, USA) was added, and the counts per min was recorded by a TopCount scintillation counter (PerkinElmer).
4.4.2 Measurements of the intracellular Ca2+ release
The methods were carried out as described previously (Li et al., 2021). Briefly, the evaluation of the agonistic effects of the synthesized bifunctional DOR/MOR ligand was performed on cell (Gα16) membranes from cells stably expressing the corresponding receptors and cultured overnight in a 96-well plate containing Ham’s F-12 nutrient medium. After gentle withdrawal of the medium solution, 40 μL of freshly prepared dye (Fluo-4/AM) solution was added to each well, and the plate was incubated at 37 ℃ for 40 min. The samples were diluted into different concentrations (100 μM, 10 μM, 1 μM, 100 nM, 10 nM, 1 nM, 100 pM, DMSO) by calcium buffer and sufficiently mixed. Then, the dye was discarded, followed by cell washing with freshly prepared calcium buffer, and 50 μL of calcium buffer was added to each well. The fluorescence values at 525 nm of the collected solutions were measured on a Flex Station Ⅱ plate reader (Molecular Devices, San Jose, CA, USA). The percent activity (% Response) was calculated and expressed as where LDAMGO/DPDPE is the fluorescence value after stimulation of 100 μM DAMGO or DPDPE. Three well replications were performed for each concentration of the ligand. The EC50 was calculated by nonlinear regression analysis (GraphPad Prism, San Diego, CA, USA).
4.5 In-vivo pharmacology test
The antinociceptive effect of the synthesized compounds was studied by the formalin paw-licking test, acetic acid-induced writhing test, and hot-water tail withdrawal test. All animals were starved for 12 h with free access to water prior to the tests.
4.5.1 Formalin paw-licking test
The pain response induced by formalin was divided into two phases. The acute pain behavior response was assigned to phase I, which started immediately after the injection of formalin and lasted only about 10 min. Phase Ⅱ was induced by inflammatory mediators and lasted about 15 min. Briefly, 32 mice were randomly divided into four groups: control group, BVD03 group, morphine group, and SW-WL-2 group. After pretreatment with the dose of test compound (2 mg/kg, i.v.), 30 μL of 2.7% formalin solution was injected into the right postal paw of each mouse to make the pain model. The tested mice were placed in hyaline boxes with a 45-degree mirror at the bottom. A camera was used to photograph the behavior of the experimental mice in 30 min, and the numbers of times that the mice licked their paw in phase Ⅰ (0–10 min) and phase Ⅱ (15–30 min) were counted. The antinociception rate was calculated as the %MPE, where %MPE = 100 × (PLTVII – PLTTII) / PLTVⅡ; PLTVII and PLTTII refer to the average paw-licking time in phase II for the vehicle group and the test group, respectively.
4.5.2 Acetic acid-induced writhing test
After pretreatment with the dose of test compound (2 mg/kg, i.v.), 1% (v/v) glacial acetic acid was administered by intraperitoneal injection (i.p.) at a dose of 10 mL/kg. Then, the number of writhing times during 20 min was recorded. The antinociception rate was calculated as the %MPE, where %MPE = 100 × (NWV – NWT) / NWV; NWV and NWT refer to the average number of writhing times of the vehicle group and the test group, respectively.
4.5.3 Tail-withdrawal test
The tail-withdrawal test was performed using a water bath with the temperature maintained at 55 ± 0.5 °C. The baseline latency was measured before any injections. The distal 3-cm section of the tail was immersed perpendicularly into hot water, and the mouse rapidly flicked its tail from the bath at the first sign of discomfort. The duration of time that the tail remained in the water bath was counted as the tail withdrawal latency (TWL0). Untreated mice with TWL0 < 5 s were used. After the administration of a single dose of test compound (2 mg/kg, i.v.) or vehicle, the test withdrawal latency (TWL) was obtained at various time points afterward (15, 30, 45, and 60 min). A 16-s maximum cutoff latency was used to prevent any tissue damage. Antinociception was quantified as the %MPE, which was calculated as %MPE = 100 × (TWL – TFL0) / (16 – TWL0).
4.6 Physical dependence and respiratory depression assessments
4.6.1 Physical dependence
The evaluation of physical dependence with continuous use of the compounds was carried out according to methods described previously (Li et al., 2021). Briefly, groups of mice received i.v. administration of either saline or drug at a dose of 4 mg/kg (∼EDmax) once daily for 5 days, between 8 and 12 am. Two hours after the last i.v. administration on day 5, the mice were treated with naloxone (1 mg/kg, i.p.), and over a 20-min period, the number of vertical jumps was counted to assess the physical dependence of the test compound.
4.6.2 Respiratory depression
All mice were fasted for 12 h with free access to water prior to the experiments. After a single i.v. administration of either saline or drug at a dose of 4 mg/kg (∼EDmax), the blood was collected by eyeball extirpation of the mice at various time points afterward (5, 10, 15, 20, and 40 min), and pO2 was measured on an ABL90 FLEX blood gas analyzer (Radiometer, Copenhagen, Denmark).
4.7 Statistical analysis
Prism 8 (GraphPad Software, San Diego, CA, USA) was used for data and statistical analyses. All results are presented as the mean ± standard error of the mean. The EC50 and Emax values were calculated by nonlinear (three parameter) regression analysis. Significant changes induced by substance application were calculated by a two-tailed Student’s t test with the native activity as the common control. P < 0.05 was considered statistically significant.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363-1381.
- [CrossRef] [Google Scholar]
- Opioid receptors: drivers to addiction? Nat. Rev. Neurosci.. 2018;19:499-514.
- [CrossRef] [Google Scholar]
- A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct. Funct.. 2015;220:677-702.
- [CrossRef] [Google Scholar]
- DPI-3290 [(+)-3-((alpha-R)-alpha-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N-(3-fluorophenyl)-N-methylbenzamide]. II. a mixed opioid agonist with potent antinociceptive activity and limited effects on respiratory function. J. Pharmacol. Exp. Ther.. 2003;307:1227-1233.
- [CrossRef] [Google Scholar]
- A review of the pharmacokinetics and pharmacodynamics of remifentanil. Anesth. Analg.. 1999;89:7.
- [CrossRef] [Google Scholar]
- Gomes, I., Jordan, B.A., Gupta, A, et al., 2000. Heterodimerization of mu and delta opioid receptors: a role in opiate synergy. The Journal of Neuroscience 20, RC110 (111-115). https://doi.org/10.1523/jneurosci.20-22-j0007.2000.
- Targeting multiple opioid receptors - improved analgesics with reduced side effects? Br. J. Pharmacol.. 2018;175:2857-2868.
- [CrossRef] [Google Scholar]
- A novel mu-delta opioid agonist demonstrates enhanced efficacy with reduced tolerance and dependence in mouse neuropathic pain models. J. Pain. 2020;21:146-160.
- [CrossRef] [Google Scholar]
- Synthesis and evaluation of peptide–fentanyl analogue conjugates as dual μ/δ-opioid receptor agonists for the treatment of pain. Chin. Chem. Lett. 2021
- [CrossRef] [Google Scholar]
- In vivo characterization of MMP-2200, a mixed delta/mu opioid agonist, in mice. J. Pharmacol. Exp. Ther.. 2011;336:767-778.
- [CrossRef] [Google Scholar]
- Orally active opioid mu/delta dual agonist MGM-16, a derivative of the indole alkaloid mitragynine, exhibits potent antiallodynic effect on neuropathic pain in mice. J. Pharmacol. Exp. Ther.. 2014;348:383-392.
- [CrossRef] [Google Scholar]
- The delta opioid receptor agonist SNC80 selectively activates heteromeric mu-delta opioid receptors. ACS Chem. Neurosci.. 2012;3:505-509.
- [CrossRef] [Google Scholar]
- Molecular perspectives for mu/delta opioid receptor heteromers as distinct, functional receptors. Cells. 2014;3:152-179.
- [CrossRef] [Google Scholar]
- Novel fentanyl-based dual μ/δ-opioid agonists for the treatment of acute and chronic pain. Life Sci.. 2013;93:1010-1016.
- [CrossRef] [Google Scholar]
- Opioid receptor activity and analgesic potency of DPDPE peptide analogues containing a xylene bridge. ACS Med. Chem. Lett.. 2017;8:449-454.
- [CrossRef] [Google Scholar]
- Superpotent [Dmt1]Dermorphin Tetrapeptides containing the 4-Aminotetrahydro-2-benzazepin-3-one Scaffold with mixed μ/δ opioid receptor agonistic properties. J. Med. Chem.. 2011;54:7848-7859.
- [CrossRef] [Google Scholar]
- The opioid peptide analogue biphalin induces less physical dependence than morphine. Life Sci.. 2001;69:1023-1028.
- [CrossRef] [Google Scholar]
- Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective mu-delta agonists. ACS Chem. Neurosci.. 2010;1:146-154.
- [CrossRef] [Google Scholar]
- Clinically employed opioid analgesics produce antinociception via mu-delta opioid receptor heteromers in Rhesus monkeys. ACS Chem. Neurosci.. 2012;3:720-727.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105018.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




