

Translate this page into:
Unraveling the molecular structure, spectroscopic properties, and antioxidant activities of new 2,4-dinitrophenylhydrazone derivatives through a comprehensive investigation
⁎Corresponding authors. azim@alum.imr.ac.cn (Azim. Khan), mominkhan@awkum.edu.pk (MominKhan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The synthesis and characterization of novel 2,4-dinitrophenylhydrazone (2,4-DNPH) derivatives were investigated using a comprehensive combination of experimental and theoretical approaches. Spectroscopic techniques such as 1H NMR, IR, Raman, and UV–Vis were employed to probe the structural and chemical properties of the synthesized compounds. These compounds were tested for their in vitro anti-oxidant activity. All the synthesized derivatives showed good to excellent activities. Density functional theory (DFT) calculations at the B3LYP functional with 6-31G and 6–311++G(d,p) basis sets were performed to reveal the molecular nature of the synthesized derivatives. Conformational analysis confirmed the stability of the optimized structures. The DFT simulations revealed the presence of hydrogen bonding between the N–H and nitro groups and further explored the charge transfer within the molecule as well as the sites that are favorable for electrophilic and nucleophilic attacks. The frontier molecular orbital (FMO) calculations provided insights into various reactivity parameters, such as ionization potential, electron affinity, electronegativity, chemical hardness, and chemical softness. Overall, this study provides a deeper understanding of the molecular structure, spectroscopic properties, and reactivity of these new 2,4-dinitrophenylhydrazone derivatives, which could have potential applications in the field of biochemistry.
Keywords
Schiff bases
Synthesis
Antioxidant activities
DFT Calculations
Spectroscopic properties

1 Introduction
Hydrazones have been identified as promising entities for the preparation of new drugs owing to their versatile pharmacological properties (Abu-Dief et al., 2022, 2021; Monfared et al., 2007). The synthesis of hydrazones typically involves the reaction between substituted hydrazines or hydrazides with ketones or aldehydes, often in solvents like methanol, ethanol, tetrahydrofuran, or butanol. Furthermore, hydrazones can serve as important intermediates in the synthesis of hydrazine derivatives through reduction with sodium borohydride (Deng and Mani, 2008; Gil, 2020). The unique structural feature makes hydrazones important for their physical and chemical properties. Hydrazones feature an imine functional group, with nitrogen being electrophilic while carbon has both electrophilic and nucleophilic properties. These unique properties make hydrazones versatile and reactive in various chemical reactions (Anoop et al., 2010). The distinctive characteristics of the imine group support the unique physical and chemical properties of hydrazones (Mohamad et al., 2019; Qasem et al., 2022; Sumrra et al., 2022). Azomethines (–HC = N-) or imines were first introduced by Hugo Schiff in 1864 (Görgülü, 2018; Hameed et al., 2017). Incoordination chemistry, these compounds play a significant role (Kou et al., 2004; Patange et al., 2015; Rosenberg et al., 1965; Rani et al., 2017) and have the capability to make complexes with transition metals. They have various activities for biological systems including anti-cancer, anti-fungal, anti-viral, anti-malarial, anti-HIV, anti-bacterial, and anti-leishmanial properties (Guo et al., 2007; Mohamad et al., 2019; Rajasekar et al., 2010; Shivakumar et al., 2008; Taha et al., 2013; Tarafder et al., 2002, 2000, Khalid et al., 2020). These compounds exhibit a notable capability as corrosion inhibitors, making them a valuable tool for preventing corrosion effectively (Agrawal et al., 2004; Ashassi-Sorkhabi et al., 2006). Inhibitor molecules act as nucleophiles that interact with the metal center by sharing electron pairs from oxygen and nitrogen atoms. This process forms a protective layer over the metal surface and reduces corrosion rates, making these compounds valuable in various industries for preventing metal degradation and extending equipment lifespan. Different moieties on the benzene ring provide multiple absorption sites for the inhibitor molecule (Abdel-Rahman et al., 2017; Guerrab et al., 2023; Mortada et al., 2023; Williams, 1972).
Hydrazone Schiff bases have been observed to demonstrate tautomerism, where the keto and phenyl tautomers have been extensively investigated, particularly for hydrazones derived from o-hydroxy aromatic aldehydes and ketones. The presence of these tautomeric forms is dependent on the nature of substituents and the medium in which they exist (Martínez et al., 2011). In addition, the equilibrium between the keto and phenol hydrazone tautomersis influenced by different anions present in the solution (Shang et al., 2012). Moreover, it has been reported that only the salicyl-hydrazone form, which is a derivative of 2,4-DNPH, exists in crystalline form and is stabilized by hydrogen bonding (Monfared et al., 2007).
2,4-dinitrophenylhydrazine is a nitro-substituted phenyl hydrazine compound that is characterized by its reddish appearance. It is commonly utilized in the analysis of carbonyl derivatives (aldehydes & ketones) via measurement of their melting point (Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020)(Gil, 2020, Anoop et al., 2010, Mohan et al., 1988). The electronic environment of hydrazone derivatives (–C = N–) is significant, and NO2 functional group enhances its biological activity (Kurt et al., 2018; Muğlu et al., 2023, 2022; Sonmez et al., 2019; Zhao et al., 2007). Hydrazones possess a dual nature, characterized by the presence of both electron-donating and electron-withdrawing groups. This feature is of significant importance in material chemistry (Naseema et al., 2010). The -N-N- linkage present in hydrazones acts as a spacer, imparting unique physical and chemical properties to these compounds. The DFT analysis has been reported by different researchers to understand the chemical nature (Avcı et al., 2023; Dege et al., 2021; Özge et al., 2023, Noreen and Sumrra (2022), Sumrra et al. (2021)).
To ensure deep insight into the electronic environment of hydrazone derivatives, quantum chemical calculations are required. In this study, a series of reactions were employed to successfully synthesize novel derivatives of 2,4-dinitrophenylhydrazone. The newly synthesized compound structures were determined through the use of spectroscopic techniques, which were further validated by employing density functional theory (DFT) calculations. Furthermore, an in-depth analysis of the electrophilic and nucleophilic sites was carried out by examining the electronic excitation/de-excitation of the synthesized compounds. The theoretical analysis results aligned well with the experimental findings, and screened them for their in vitro anti-oxidant inhibitory activity compared with the reference compounds vitamin C and EDTA.
1.1 Experimental
1.1.1 General
The novel 2,4-DNPH compounds were synthesized in a series of reactions. During the experimental procedure, a solution containing equimolar amounts of 2,4-dinitrophenylhydrazine (1 ml) and terephthalaldehyde (1 ml) was refluxed in ethanol for 2.5 h in presence of acetic acid. The stepwise progress of the reaction was observed by TLC. Upon completion of the reaction, the resulting product was purified via recrystallization. These experimental steps are illustrated in Scheme 1.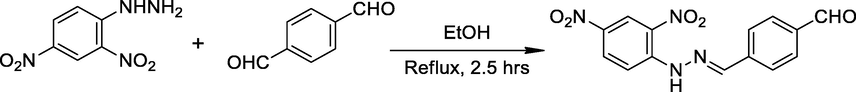
Reaction of 2,4-DNPH and terephthalaldehyde.
Scheme 1: Schiff base formation from 2,4-dinitrophenylhydrazine and terephtaldehyde.
In the second step, different substituted aromatic aldehydes were refluxed with hydrazine hydrate in the presence of ethanol for 1 hr (Scheme 2).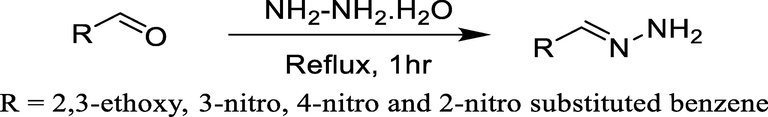
Reaction of hydrazine hydrate and substituted aldehydes.
In the last step, different aromatic aldehydes reacted with 4((2-(2,4-dinitrophynl)hydrazono)methyl)benzaldehydes in the presence of ethanol for 3 to 5 hrs at a refluxed temperature, resulting in the formation of different compounds (Scheme 3). The compounds obtained from the synthesis were subjected to purification and recrystallization with the use of ethanol. Spectroscopic methods such as 1H NMR and EI-Spectrometry were utilized to identify the structures of the samples, which were subsequently validated through DFT calculations. Four different compounds were successfully synthesized through the incorporation of different substituents, as detailed in Table 1. *Et = Ethoxy group.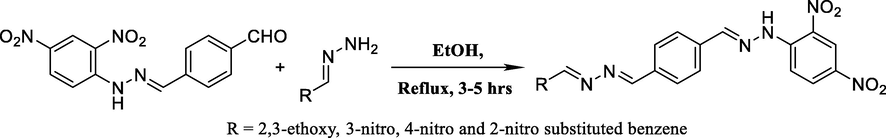
Formation of new derivatives of 2,4-DNPH.
Derivatives
Substituents
Reaction time (hrs)
Derivatives
Substituents
Reaction time (hrs)
2,3-diEt-DNPH
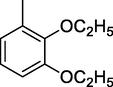
5.0
3-NO2-DNPH
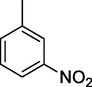
4.0
4-NO2-DNPH
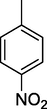
3.0
2-NO2-DNPH
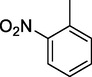
3.5
1.2 Material and measurements
All the solvents, reagents, and chemicals (2,4-dinitrophenyl hydrazine CAS = 119–26-6, 97 % pure, 2,3-diethoxybenzaldehyde, 3-nitrobenzaldehyde, 4-nitrobenzaldehyde, and 2-nitrobenz- aldehyde 97–99 % pure) used in this work, were of analytical grade and brought from Sigma Aldrich, BDH, and Merck. An examination of the structures of the synthesized derivatives was conducted by utilizing Nuclear Magnetic Resonance (NMR) and Electron Impact Mass Spectra (EIMS) techniques. These analyses were performed in Germany using the Advance Bruker AM 500 MHz and Finnigan MAT-311A instruments, respectively. In the determination of the mass spectrum, an energy value of approximately 70 eV was utilized. The melting points of the synthesized derivatives were found to be in the range of 239–241 °C, with an average value. Furthermore, all the synthesized compounds were examined using the Density Functional Theory (DFT) method.
1.3 Spectral interpretation
1.3.1 1-(2,3-diethoxybenzylidene)-2-(4-(2-(2,4-dinitrophenyl)hydrazono) methyl)benzylidene)hydrazine (2,3-diEt-DNPH)
The 2,3-diEt-DNPH was synthesized with a reaction time of 5 h by using the chemical reaction as given in Scheme 3. Mass fragmentation was performed by a mass spectrometer with an energy of 70 eV for evaluating the molecular ion (M+) and other fragments. All the fragments were detected by the detector regarding their m/z ratio. The fragmentation was observed in polar region of the compound. The experimental findings, which were based on 1H-NMR and mass spectrometry (MS), are as follows: Yield: 0.25 g (65%); 1H NMR (500 MHz, DMSO‑d6) δ 11.57 (s, 1H), 8.89 (d, J = 1.9 Hz, 1H), 8.84 (s, 1H), 8.51 (s, 1H), 8.38 (dd, J = 7.5, 2.0 Hz, 1H), 7.98–7.87 (m, 2H), 7.68 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.23 – 7.10 (m, 3H), 4.10 (q, J = 8.0 Hz, 4H), 1.45 (t, J = 8.0 Hz, 3H), 1.34 (t, J = 8.0 Hz, 3H); 13C NMR (125 MHz, DMSO‑d6) δ 14.3 (2C), 65.5 (2C), 112.8 (1C), 114.0 (1C), 116.5 (1C), 117.7 (1C), 120.1 (1C), 128.8 (4C), 129.6 (1C), 131.5 (1C), 133.9 (2C), 140.6 (3C), 144.1 (1C), 145.2 (1C), 147.5 (1C), 153.9 (1C), 163.8 (1C); MS: m/z (rel. abund. %) 504 (M+, 03), 490 (31), 311 (34), 163 (1 0 0), 148 (31); Analysis calculated for C25H24N6O6 (504.18): C, 59.52; H, 4.80; N, 16.66; O, 19.03; Found: C, 59.50; H, 4.83; N, 16.69.
1.3.2 1-(2,4-dinitrophenyl)-2-(4-(3-nitrobenzylidene)-hydrazono)methyl) benzylidene)hydrazine (3-NO2-DNPH)
The 3-NO2-DNPH was obtained with a reaction time of 4 h according to Scheme 3. The results obtained from the experimentation based on 1H NMR and mass spectrometry (MS) are as follows: Yield: 0.21 g (63%); 1H NMR (500 MHz, DMSO‑d6) δ 11.53 (s, 1H), 8.86 (d, J = 1.9 Hz, 1H), 8.79 (s, 1H), 8.57 (s, 1H), 8.35–8.20 (m, 4H), 7.85 (d, J = 7.5 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, DMSO‑d6) δ 114.0 (1C), 116.1 (1C), 117.7 (2C), 121.3 (1C), 128.5 (5C), 129.7 (1C), 134.1 (2C), 136.7 (1C), 140.8 (4C), 147.2 (1C), 163.5 (2C); MS: m/z (rel. abund. %) 461.11 (M+, 100), 282 (24), 159 (14); Analysis calculated for C21H15N7O6 (461.39): C, 54.67; H, 3.28; N, 21.25; O, 20.81; Found: C, 54.70; H, 3.26; N, 21.28.
1.3.3 1-(2,4-dinitrophenyl)-2-(4-(4-nitrobenzylidene)hydrazono)methyl) benzylidene)hydrazine (4-NO2-DNPH)
Similarly, the 4-NO2-DNPH was synthesized with a reaction time of 3 h. The experimental outcomes based on 1H NMR and mass spectrometry (MS) are as follows: Yield: 0.18 g (59%); 1H NMR (500 MHz, DMSO‑d6): 1H NMR (500 MHz, DMSO‑d6) δ 11.57 (s, 1H), 8.86 (d, J = 1.9 Hz, 1H), 8.57 (s, 2H), 8.39–8.23 (m, 5H), 7.68 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H); 13C NMR (1250 MHz, DMSO‑d6) δ 115.5 (1C), 116.3 (1C), 117.5 (3C), 128.6 (4C), 129.8 (2C), 134.9 (3C), 140.3 (4C), 145.2 (1C, s), 161.8–162.3 (2C); MS: m/z (rel. abund. %) 461.11 (M+, 04), 185 (1 0 0), 171 (1 0 0), 157 (1 0 0); Analysis calculated for C21H15N7O6 (461.39): C, 54.67; H, 3.28; N, 21.25; O, 20.81; Found: C, 54.68; H, 3.25; N, 21.27.
1.3.4 1-(2,4-dinitrophenyl)-2-(4-(2-nitrobenzylidene)-hydrazono)methyl) benzylidene)hydrazine (2-NO2-DNPH)
Finally, the 2-NO2-DNPH was prepared with a reaction time of 3.5 h according to Scheme 3. The outcomes of the experimentation based on 1H NMR and mass spectrometry (MS) are as follows: Yield: 0.27 g (54 %); 1H NMR (500 MHz, DMSO‑d6) δ 11.52 (s, 1H), 8.86 (d, J = 1.9 Hz, 1H), 8.83 (s, 1H), 8.57 (s, 1H), 8.29–8.69 (m, 3H), 7.72 (td, J = 7.5, 2.0 Hz, 1H), 7.67 – 7.55 (m, 5H); 13C NMR (1250 MHz, DMSO‑d6) δ 114.0 (1C), 116.5 (1C), 117.8 (2C), 128.2 (1C), 128.6 (5C), 129.3 (1C), 131.8 (1C), 133.6 (2C), 141.6 (4C), 146.2 (1C), 147.5 (1C), 161.8 (1C); MS: m/z (rel. abund. %) 461.11 (M+, 04), 297 (29), 165 (66), 148 (65); Analysis calculated for C21H15N7O6 (461.39): C, 54.67; H, 3.28; N, 21.25; O, 20.81; Found: C, 54.64; H, 3.24; N, 21.31.
1.4 Antioxidant activities
The antioxidant activity of the obtained derivatives was determined using the stable DPPH free radical as reported. Different concentrations (50, 100, 200, and 400 µM) of the synthesized compounds were combined with an ethanolic solution of 85 µM DPPH radical. The mixture was incubated at room temperature for 30 min, and the decrease in absorbance was measured at 518 nm using a UV spectrophotometer. Ascorbic acid was used as a positive control at the same concentrations. The experiment was repeated three times, and the percentage inhibition of the compounds and ascorbic acid was calculated using the relation (1);
The DPPH inhibition effect was represented by “X*”, while the control and sample absorbance readings were labeled as “AC” and “AS”, respectively. The ferrous ion chelating activity and ferric ion reducing power of the synthesized derivatives were determined using a standard method previously reported (Puntel et al., 2005). The compounds were mixed with 0.2 ml of 3.6 mM ferrous sulfate, 0.3 ml of 100 mM Tris-HCl (pH = 7.4), and 0.1 ml of 9 mM O-Phenanthroline at various concentrations (50, 100, 200, and 400 µM) and diluted up to 3.0 ml with ultra-pure distilled water. The mixture was then incubated at room temperature for 10 min. The absorbance decrease and increase of the synthesized derivatives for ferrous ion chelating activity and ferric ion reducing power were measured at 510 nm using a UV spectrophotometer. EDTA and ascorbic acid were used as reference standards for ferrous and ferric ions, respectively. The chelating capacity of Fe2+ and reducing power comparable with ascorbic acid were calculated using relation (1) with “X*” representing the chelating effect and reducing power, respectively.
The overall antioxidant capacities of the four compounds were assessed through a phosphomolybdenum assay (Saha et al., 1970). The reagent solution with various concentrations of compounds (50, 100, 200, and 400 µM) in ethanol and ultra-pure distilled water, along with 0.7 ml of 0.6 M sulphuric acid, 1.0 mM ammonium molybdate, and 1.0 ml of 28 mM potassium phosphate was incubated at 95 °C for 90 min. The increase in absorbance of the mixture was measured at 695 nm using a UV spectrophotometer to evaluate the total antioxidant capacities of the compounds. The reducing power was calculated using the same relation (1) as mentioned earlier. The scavenging activity for hydroxyl radicals was measured using the Fenton reaction mechanism (Li et al., 2011).
The reaction mixture for all compounds contained 0.1 ml of 7.5 mM O-phenanthroline, 0.5 ml of 0.2 M phosphate buffer (pH 6.6), 0.1 ml of 7.5 mM ferrous sulfate, and 0.1 ml of 0.1% H2O2 and was diluted up to 3 ml with distilled water. The reaction mixture was incubated at room temperature for 30 min, and the absorbance was measured at 510 nm using a UV spectrophotometer. The reaction mixture with Schiff bases complexes was used as the control and a blank was prepared without Schiff bases complexes and H2O2. The DPPH radical scavenging activity of the synthesized compounds and ascorbic acid was calculated using the following relation:
Where “As” and “Ac” are the absorbance reading of sample and control. While “Ab” is the absorbance reading of the blank.
1.5 Computational methods
The DFT method was employed for the computational studies using the G09 of Gaussian. The geometry optimizations were carried out at B3LYP (Lee et al., 1988) with a 6–311++G(d,p) basis set (E. T. Aljohani et al., 2021b, 2021a; F. S. Aljohani et al., 2021; Rassolov et al., 1998). Grimme’s D3 method was included for the account of dispersion forces(Grimme et al., 2010).The fundamental vibrational analysis was used to confirm true minima without imaginary frequency. A potential energy surface scan was carried out for all derivatives to confirm the lowest energy structure with a transition structure. The Gauge-Invariant Atomic Orbital (GIAO) method was utilized to carry out theoretical NMR calculations in DMSO solvent at B3LYP/311 + G(2d,p).The TD-DFT method was employed to determine the HOMO and LUMO energies with 6-31G and 6–311++G(d,p) basis sets, using the B3LYP functional. The energy gap between HOMO and LUMO was then calculated as,
2 Result and discussion
2.1 Geometry optimization
To understand the chemical characteristics of the synthesized compounds, all the structures were optimized using the g09 package of Gaussian (Frisch, 2009). The optimized structures are shown in Fig. 1. The structure optimization was verified through frequency calculation, with no imaginary frequency. The optimized structures reveal that the bond lengths of sp2 carbons (C = C) fall within the range of 1.39 to 1.41 Å, while the bond length of sp3 carbons (C–C) is 1.51 Å. Additionally, the C–H bond lengths for both aromatic and non-aromatic carbons were computed to be 1.08 Å and 1.09 Å, respectively. The presence of nitro groups at the 2nd and 4th positions leads to the elongation of the C2-C3 and C3-C4 bond lengths to 1.43 Å.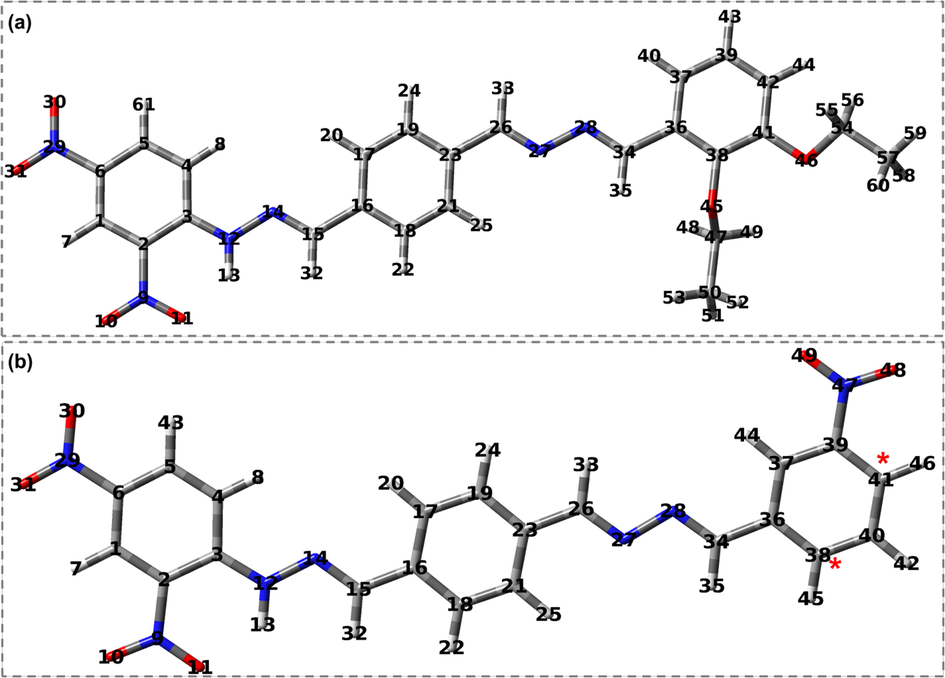
Optimized structures of 2,4-DNPH derivatives at B3LYP/6–311++G(d,p).The * sign indicates the position of the NO2 group.
The synthesized compounds exhibit a nitrogen atom positioned at varying locations, resulting in differing carbon–nitrogen (C-N) bond lengths. The C-N bond length between carbon and nitrogen of NO2 was found to be 1.45 Å, consistent with reported values (Ji et al., 2010).The calculated length of the C = N bond is 1.28 Å, which is less than the length of a C-N single bond, which is 1.35 Å. Additionally, the nitrogen-nitrogen single bonds were calculated in a range of 1.36 Å, as presented in Table 2. To evaluate the discrepancy between the computed and experimental values, the root mean square deviation (RMSD) was determined for the computed bond lengths, and the results are shown in Table 2. A good correlation between the experimental and calculated data, as evidenced by an RMSD value of 0.019 at the 6–311++G(d,p) basis set. The calculated data was found well matched with the existing experimental data.
Bond length (Å)
Calculated
Experimental
C1-C2
1.39
1.39a
N27-N28
1.38
1.35b
N28-C34
1.28
1.28b
C36-C37
1.40
1.45b
C2-N9
1.46
1.47a
C6-N29
1.47
1.47a
C39-N47
1.48
1.47a
C41-N46
1.47
1.47a
C38-N47
1.48
1.47a
N9-O10
1.22
1.21a
RMSD
0.019
The bond angles between sp2 carbons were calculated to range from 119.0° to 121.9°. The inclusion of substituents resulted in a minor modification of the bond angle. However, the experimental value of the bond angle between sp2 hybridized carbons is 120°. Furthermore, the dihedral angles for C3-N12-N14-C15 and C26-N27-N28-C34 were computed to be 179.9°.
2.2 Potential energy surface scan (PES)
The evaluation of conformation is essential because different conformations of a compound are correlated with different properties such as physical, chemical, and medicinal properties. Conformational stability is also useful in determining the reaction mechanism and rate (Barton and Cookson, 1956). Herein, to identify the most stable conformation and transition structure, the PES scan was carried out around the dihedral angles D(C26-N27-N28-C34) by rotating the torsional angle every 10° for all compounds. The shapes of the potential energy curves as a function of torsional angles are illustrated in Fig. 2.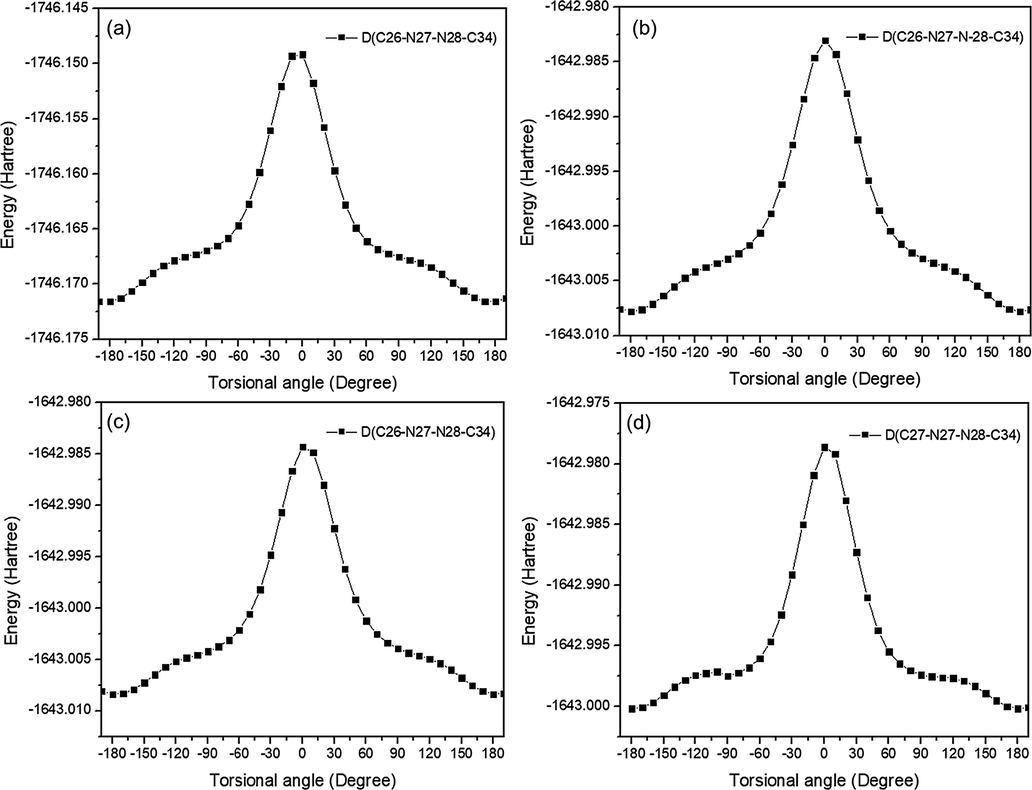
Potential energy surface (PES) scan of selected torsional angle D(C26-N27-N28-C34). Plots (a-d) correspond to 2,3-diEt-DNPH, 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH, respectively.
The PES curve of 2,3-diEt-DNPH shows the two stable conformers at dihedral angles ± 180.0° with the energy of −1746.17 Hartree (Ha). However, the transition structure appears asan energy barrier of −1746.14 Ha at an angle of 0°. The energy barrier to this rotation is determined to be 0.03 Ha, which is the energy difference between the transition state and the stable conformers. Similarly, for 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH, the two stable conformers were found with the same energy of −1643.00 Ha and with dihedral angles of −179.42° and 180.0°, respectively. The transition structure for these three derivatives was found at an energy barrier of −1642.98 Ha at dihedral angles of 0°, as shown in Fig. 2. It has been calculated that the energy difference between the stable conformers and transition structure is 0.02 Ha. Figure S1 in the supporting information file displays all of the stable conformers that have corresponding transition structures.
The similarity of the potential energy surfaces (PES) of nitro compounds is in line with the similarities in their chemical structures. The only distinguishing factor among them is the position of NO2. The presence of the -OC2H5 group in 2,3-diEt-DNPH results in its distinct electronic properties compared to other derivatives. Despite this difference, the PES was conducted at the same dihedral angle for all compounds, indicating that the structures were stable. These stable structures were then utilized for subsequent electronic calculations and spectroscopic analysis.
2.3 NMR studies
The 1H NMR was calculated at B3LYP/6–311 + G (2d,p) GIAO method. The 1H NMR chemical shifts (δ) values were compared with experimental 1H NMR as given in Table 3. The shielding and deshielding of a proton are crucial in 1H NMR. The electronegativity of an atom is directly linked to the deshielding of neighboring protons which results in downfield chemical shifts.
Atom No
2,3-diEt-DNPH
3NO2-DNPH
4NO2-DNPH
2NO2-DNPH
Exp
Calc
Exp
Calc
Exp
Calc
Exp
Calc
Ar-H
7.10–8.89
7.32–8.56
7.60–8.86
7.04–8.57
7.60–8.86
6.73–8.59
7.55–8.86
6.77–8.54
H-33
8.57
7.42
8.57
7.44
8.57
7.36
8.57
7.36
N = CH-35
8.28
7.56
8.79
7.05
8.57
7.06
8.83
8.06
H-13
11.57
10.13
11.57
10.16
11.57
10.17
11.57
10.16
Herein, the most downfield chemical shift value noted for H-13, both experimentally and theoretically. This was attributed to the presence of electronegative atoms and hydrogen bonding, resulting in values of 11.57 and 10.88 ppm, respectively. Further investigation revealed that H-13 was a deshielded proton, likely satirically hindered by the neighboring NO2 group, with a possible hydrogen bonding between the oxygen atom of the NO2 group as shown in Figure S2. The 1H NMR peak for H-33 was found experimentally as a singlet peak at 8.57 ppm, while the theoretically calculated value ranged from 7.36 to 7.44 ppm downfield from TMS. Furthermore, the experimental 1H NMR spectrum for the H-C = N (H-35) proton was confirmed by a singlet peak appearing within 8.28–8.83 ppm range, while the computed peak was found within 7.05–8.06 ppm.
The chemical shift values for the aromatic protons appeared as multiplets within the range of 7.10–8.57 ppm, experimentally. The computed values for the same protons fell within the range of 7.04–8.54 ppm. The experimental chemical shift (δ) value for the CH2 proton was 4.10 ppm, while the computed value fell between 3.7 and 4.22 ppm. Similarly, the triplet peak observed in the experimental spectrum for the CH3 proton appeared at 1.34 and 1.45 ppm, while the calculated chemical shift range for CH3 was 1.08–1.60 ppm. These findings provide insights into the shielding and deshielding of protons, contributing to a well understanding the nature of synthesized compounds.
The shielding values for 13C NMR were determined for the ring carbons and appeared in the range of 118.50–158.0 ppm due to the different chemical positions of 13C in the molecule. The chemical shift for the C-N carbon was found to be downfield in the range of 151.50 to 169 ppm. Additionally, the 13C chemical shift for –HC = N- was explored and found to be at 169.53 ppm downfield. A detailed analysis of the 13C chemical shift values with root mean square deviation (RMSD) is given in Table S1.
2.4 Vibrational analysis
The B3LYP/6–311++G (d,p) method was utilized to calculate the infrared (IR) spectroscopic analysis of the synthesized compounds.The wave numbers of observed and calculated spectra with tentative assignments are provided in Table S2. Frequencies below 4000 cm−1 in the calculated spectra were adjusted by a scaling factor of 0.958 (Scott and Radom, 1996).The existing experimental data was used as a reference to assign the vibrational spectra (Bakir, 2018). Figure S18 shows the calculated spectra of synthesized compounds. The observed vibration modes were identified as C–H (aromatic and non-aromatic), C = C, C = N, N-N, N–H, and NO2 by comparison with known experimental values.
2.4.1 C–H vibrations
Literature suggests that the C–H stretching modes of aromatic rings typically appears in the 3000–2800 cm−1 region (Hobson, 2012). The presence of an electronegative atom may cause a blue shift. In this study, strong bands for the stretching modes of aromatic C–H were observed within 3190–3184 cm−1 region. While for non-aromatic, the C–H stretching vibrations were found within 3148–3182 cm−1 region. The stretching mode of N = C–H appeared at 3288 cm−1. Regarding the aromatic compound, the in-plane bending vibrations for C–H were investigated within the 1400–850 cm−1 range, while the out-of-plane bending vibrations were detected within the 950–600 cm−1 range (Arunagiri et al., 2015). The bending modes of vibrations observed in the 1488–1148 cm−1 region were assigned to in-plane bending vibrations, as listed in Table S2.
2.4.2 N–H vibrations
The stretching vibration of the N–H group is anticipated to take place within the region of 3500–3300 cm−1 (“A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures (Roeges, Noel P. G.),” 1995). In our study, the stretching mode for the N–H functional group was observed at 3500 cm−1. The broadening of this band was attributed to intermolecular interactions via hydrogen bonding with the neighboring oxygen of the nitro group, which were found in the synthesized derivatives. The N–H bending vibrations were assigned to the band observed at 1379 cm−1 in the IR spectra, which exhibited good conformity with the experimental data.
2.4.3 NO2 vibrations
Thecharacteristic bands present in the IR spectra for NO2 are linked to the symmetric and asymmetric stretching modes of vibration, which are found within the region of 1550–300 cm−1 (Anoop et al., 2010). The asymmetric stretching vibrational modes of N = O were observed at a higher wave number than the symmetric stretching modes. The IR bands for asymmetric stretching were detected in the region of 1538 cm−1, whereas the symmetric band was identified at 1351 cm−1, exhibiting a close resemblance to the reported bands at 1600 and 1349 cm−1 (Silverstein and Bassler, 1962). The in-plane bending mode of the nitro group is allocated to the weak band at 866 cm−1, whereas the out-of-plane mode is observed at 675 cm−1.
2.4.4 C = N & N–N vibrations
A medium-intensity band observed in the range of 1582 cm−1 is frequently used to identify the C = N stretching mode in IR spectroscopy. In the synthesized compounds, the presence of a hydrazine linkage was indicated by the appearance of the C = N stretching mode within the range of 1583–1557 cm−1. Furthermore, the observed stretching vibration at 1017 cm−1 was assigned to N-N stretching. A detailed analysis of the vibrational frequencies is provided in Table S2. These findings demonstrate the importance of IR spectroscopy in characterizing the chemical structure of the synthesized compounds.
2.5 Frontier molecular orbitals (FMO) and electronic properties
To obtain a better understanding of the electronic transitions of the synthesized compounds, TD-DFT calculations were conducted utilizing the B3LYP functional with 6-31G and 6–311++G (d,p) basis sets. The calculated spectra were assigned to the compounds shown in Figure S19. The absorption band for HOMO-LUMO transition was observed at a wavelength of 480.25, 454.90, 417.16, and 419.31 nm for 2,3-diEt-DNPH, 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH, respectively. A detailed analysis of the HOMO-LUMO transition with absorption spectra and oscillator strength is presented in Figure S19. These results provide valuable information about the electronic properties of the synthesized compounds and can aid in understanding their potential applications.
TD-DFT calculations were performed to obtain various reactivity parameters, which are important for understanding the chemical nature of the synthesized compounds (Parr et al., 1999; Parr and Yang, 1983). The HOMO and LUMO energies are important descriptors for determining the electronic density in a compound, and their energies correspond to the ionization potential and electron affinity, respectively (Rahmani et al., 2018). The HOMO and LUMO energy gap is a significant factor that influences the electronic properties of a molecule. Molecules exhibiting a larger HOMO-LUMO energy gap tend to be less reactive compared to those molecules with a smaller energy gap. The computed HOMO and LUMO energies for the synthesized compounds are presented in Figures S3 and S4.
Reactivity parameters were determined through FMO analysis and are presented in Table 4. These parameters include the HOMO and LUMO gap (ΔE), ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical hardness (η), and chemical softness (σ) (Blanchard and Brüning, 2015; Parr and Pearson, 1983; Pearson, 1986). The ΔEgaps for the synthesized compounds in the gas phase were calculated as 2.518, 3.083, 3.156, and 3.111 eV at B3LYP/6-31G.Whereas, in the aqueous phase, the gaps were 2.582, 2.887, 2.940, and 2.938 eV, respectively (see Figure S3). However, the HOMO-LUMO gaps calculated using 6–311++G(d,p) basis sets were 2.908, 3.099, 3.418, and 3.397 eV, as shown in Figure S4. A molecule has a larger HOMO-LUMO gap, it is typically less reactive than one with a smaller gap so the transfer of an electron from the HOMO to the LUMO becomes energetically unfavorable. The smaller energy gap observed for 2,3-diEt-DNPH indicates a higher reactivity compared to the other synthesized derivatives.
Parameter
2,3-diEt-DNPH
3NO2-DNPH
4NO2-DNPH
2NO2-DNPH
Gas
Aqueous
Gas
Aqueous
Gas
Aqueous
Gas
Aqueous
EHOMO
−0.2150
−0.2182
−0.2407
−0.2296
−0.2451
−0.2316
−0.2405
−0.2315
ELUMO
−0.1225
−0.1233
−0.1274
−0.1235
−0.1291
−0.1235
−0.1262
−0.1235
ΔE
0.093
0.095
0.113
0.106
0.116
0.108
0.114
0.108
IP
0.2150
0.2182
0.2407
0.2296
0.2451
0.2316
0.2405
0.2315
EA
0.1225
0.1233
0.1274
0.1235
0.1291
0.1235
0.1262
0.1235
χ
0.1687
0.1707
0.1841
0.1765
0.1871
0.1775
0.1833
0.1775
Η
0.0462
0.0474
0.0566
0.0530
0.0580
0.0540
0.0571
0.0540
Σ
16.111
16.002
15.457
15.964
15.240
15.963
15.607
15.962
The ionization potential (IP) refers to the minimum energy needed to remove an electron from a molecule and is associated with the EHOMO, as per equation (4).
The present study indicates that low ionization potential and electron affinity (EA) values suggest a higher reactivity of 2,3-diEt-DNPH relative to the other derivatives.. The electron affinity the amount of energy released by the addition of an electron to an atom or a molecule and it is calculated from equation (5).
Moreover, knowledge of the ionization potential and electron affinity values enables one to determine electronegativity (x), which reflects the ability of an atom or group of atoms to attract electrons toward itself and it is calculated according equation (6)
The calculated electronegativity for 2,3-diEt-DNPH is lower than that of the other derivatives. The FMO analysis clearly indicates the high reactivity of 2,3-diEt-DNPH compared to the other synthesized compounds.
In addition, the softness (Σ) and hardness (H) values of a molecule can provide insight into its polarizability, with high softness and low hardness indicating increased polarizability. In this study, the calculated data for all the derivatives showed nearly the same level of polarizability as given in Table 4. The hardness and softness is calculated according equation (7) and (8).
The reactivity information obtained from the TD-DFT calculations provides a significant implication for the potential applications of the synthesized compounds. By changing the nature and position of substituent brings the changes in the chemical reactivity of compounds (Avcı et al., 2023). The relation between SAR and TD-DFT analysis lies in the understanding of how changes in the molecular structure (as revealed in the SAR analysis) influence the electronic properties (as obtained from TD-DFT calculations) and subsequently impact the observed activities of the compounds. SAR analysis indicates that the change in nature/position of substituent exhibited change in the antioxidant activity as discussed in the section 2.7.
2.6 Molecular electrostatic potential (MEP)
By analyzing the distribution of charged particles, the MEP calculation provides a useful method for identifying the electrophilic and nucleophilic sites of a molecule (Luque et al., 2000). The same DFT method employed for FMO calculations was used to perform MEP calculations. The colorized figures presented in Figure S5 offer a clear description of the nucleophilic and electrophilic sites located on the molecular surface. The blue region of the molecular surface indicates the absence of electrons, making it a good site for nucleophilic attack. In contrast, the red region shows the availability of electrons, indicating a favorable site for electrophilic attack.
The red region in Figure S5 is occupied by oxygen atoms of the nitro group, confirming the availability of electrons and a more favorable site for an electrophilic attack. The blue color of the molecule shows the unavailability of electrons, indicating the suitable region for a nucleophilic attack.
2.7 Antioxidant activity
In vitro, free radical scavenging studies were conducted on all the synthesized compounds, and varying degrees of scavenging activity was observed, with IC50 values ranging from 160.66 ± 13.75 µM to 192.40 ± 14.14 µM (see Table 5). The standard Vitamin C exhibited an IC50 value of 181.43 ± 18.59 µM. 2,3-diEt-DNPH showed activity values that were close to the standard Vitamin C, with an IC50 value of 160.66 ± 13.75 µM. However, the other synthesized compounds, 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH, were comparatively less active than the standard Vitamin C, with IC50 values of 183.92 ± 13.71 µM, 184.61 ± 13.97 µM, and 192.40 ± 14.14 µM, respectively.
Derivatives
% Inhibition means
IC50(µM) + SEM
50 (µM)
100 (µM)
200 (µM)
400 (µM)
2,3-diEt-DNPH
14.75
29.80
52.07
77.86
160.66 ± 13.75
3NO2-DNPH
10.56
23.86
41.98
73.87
183.92 ± 13.71
4NO2-DNPH
10.65
24.80
42.07
75.44
184.61 ± 13.97
2NO2-DNPH
9.42
20.97
40.11
74.08
192.40 ± 14.14
Vitamin C
14.89
29.45
60.33
98.88
181.43 ± 18.59
Table 6 displays the analyzed values for the ferrous ion chelation activity of all compounds. According to the results, the 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH exhibited activity values of 187.70 ± 7.77 µM, 189.68 ± 8.20 µM, and 181.41 ± 9.09 µM, respectively. However, 2,3-diEt-DNPH showed a value of 199.95 ± 6.42 µM as shown in Table 6.
Derivatives
% Inhibition means
IC50(µM) + SEM
50 (µM)
100 (µM)
200 (µM)
400 (µM)
2,3-diEt-DNPH
5.48
9.49
15.39
34.47
199.95 ± 6.42
3NO2-DNPH
8.39
14.35
21.08
43.90
187.70 ± 7.77
4NO2-DNPH
7.65
14.80
22.07
45.44
189.68 ± 8.20
2NO2-DNPH
9.76
15.97
28.11
50.98
181.41 ± 9.09
EDTA
11.26
22.15
40.23
78.25
193.57 ± 14.69
Table 7 presents the results of the ferric ion-reducing activity of the synthesized compounds. Among all compounds, 3NO2-DNPH exhibited higher ferric ion-reducing activity, with an IC50 value of 164.06 ± 13.46 µM. The other three compounds showed almost similar values compared to the standard vitamin C, with IC50 values ranging from 170.10 ± 17.10 µM to 171.26 ± 15.79 µM.
Derivatives
% Inhibition means
IC50(µM) + SEM
50 (µM)
100 (µM)
200 (µM)
400 (µM)
2,3-diEt-DNPH
11.75
23.54
42.39
69.76
175.31 ± 12.65
3NO2-DNPH
13.87
24.30
51.24
73.47
164.06 ± 13.46
4NO2-DNPH
11.45
23.45
47.57
70.49
169.48 ± 13.11
2NO2-DNPH
11.54
22.45
45.25
72.45
177.67 ± 13.48
Vit.C
16.45
33.56
60.35
94.88
169.80 ± 17.10
The synthesized compounds were subjected to molybdate ion reduction potential analysis. Among the synthesized compounds, 3NO2-DNPH showed comparable potential to the standard vitamin C, with an IC50 value of 178.46 ± 13.13 µM. However, the other compounds exhibited less reduction potential compared to the standard values as given in Table 8.
Derivatives
% Inhibition means
IC50(µM) + SEM
50 (µM)
100 (µM)
200 (µM)
400 (µM)
2,3-diEt-DNPH
9.59
19.23
39.30
74.97
196.67 ± 14.46
3NO2-DNPH
10.54
22.32
43.54
70.32
178.46 ± 13.13
4NO2-DNPH
9.48
17.76
37.65
73.65
199.37 ± 14.29
2NO2-DNPH
8.45
15.55
32.45
65.55
201.34 ± 12.72
Vitamin C
13.44
27.34
55.47
89.08
179.12 ± 16.72
The hydroxyl radical scavenging activity for all the compounds are presented in Table 9. Among the synthesized compounds, 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH showed higher activity than the standard vitamin C, with IC50 values of 170.18 ± 12.27 µM, 196.27 ± 13.19 µM, and 186.82 ± 13.89 µM, respectively. However, 2,3-diEt-DNPH showed activity close to the standard Vitamin C, with an IC50 value of 197.84 ± 18.02 µM. 2,3-diEt-DNPH was found to be less active than the standard Vitamin C as shown in Table 9.
Derivatives
% Inhibition means
IC50(µM) + SEM
50 (µM)
100 (µM)
200 (µM)
400 (µM)
2,3-diEt-DNPH
6.79
11.23
23.30
44.56
194.92 ± 8.45
3NO2-DNPH
11.27
23.26
43.54
67.23
170.18 ± 12.27
4NO2-DNPH
10.38
18.36
35.15
69.87
196.27 ± 13.19
2NO2-DNPH
10.35
21.45
42.24
73.55
186.82 ± 13.89
Vit.C
11.44
23.34
48.47
92.88
197.84 ± 18.02
3 Structure activity relationship (SAR)
In this study, we conducted a comprehensive Structure-Activity Relationship (SAR) analysis to assess the radical scavenging and metal chelation activities of the synthesized compounds. The IC50 values for each compound were determined in four different assays, including radical scavenging activity, ferrous ion chelation activity, ferric ion-reducing activity, and molybdate ion reducing activity, and were compared to that of Vitamin C (Vit.C) and EDTA as a reference.
For radical scavenging activity, we observed that all compounds, including 2,3-diEt-DNPH, 3NO2-DNPH, 4NO2-DNPH, and 2NO2-DNPH, demonstrated potent scavenging capabilities, with IC50 values ranging from 160.66 ± 13.75 to 192.40 ± 14.14. Notably, Vitamin C also exhibited significant radical scavenging activity with an IC50 value of 181.43 ± 18.59.
Moving on to ferrous ion chelation activity, all compounds displayed efficient chelation abilities with IC50 values ranging from 181.41 ± 9.09 to 199.95 ± 6.42. EDTA, the positive control, exhibited robust ferrous ion chelation activity with an IC50 value of 193.57 ± 14.69.
Regarding ferric ion-reducing activity, the compounds demonstrated promising abilities to reduce ferric ions, with IC50 values varying from 164.06 ± 13.46 to 177.67 ± 13.48. Again, Vitamin C exhibited significant ferric ion-reducing activity with an IC50 value of 169.80 ± 17.10.
In the molybdate ion reducing activity assay, the compounds exhibited notable molybdate ion reducing properties, with IC50 values ranging from 178.46 ± 13.13 to 201.34 ± 12.72. Vitamin C also demonstrated considerable molybdate ion reducing activity with an IC50 value of 179.12 ± 16.72.
Finally, in the hydroxyl radical scavenging activity assay, all compounds showed potent scavenging activity against hydroxyl radicals, with IC50 values ranging from 170.18 ± 12.27 to 201.34 ± 12.72. Vitamin C displayed significant hydroxyl radical scavenging activity with an IC50 value of 197.84 ± 18.02.
Overall, the SAR analysis revealed that the tested compounds exhibited diverse radical scavenging and metal chelation activities, making them potential candidates for therapeutic applications targeting oxidative stress-related conditions. These findings provide valuable insights into the structure–activity relationship of these compounds and pave the way for the development of novel antioxidant and chelating agents with enhanced biological activities. Further studies are required to elucidate the underlying molecular mechanisms responsible for the observed variations in the tested activities among the compounds, contributing to a deeper understanding of their potential roles in oxidative stress mitigation and metal homeostasis.
4 Conclusions
The present study involved the synthesis of novel derivatives of 2,4-DNPH and their characterization using various modern spectroscopic techniques. A combined experimental and theoretical approach was employed to gain insights into the molecular structure and chemical reactivity of these compounds. The calculated NMR and vibrational wave numbers were found to be in good agreement with the experimental data. The presence of H-bonding was confirmed using the DFT-D3 method, and the stable conformers were identified through conformational analysis. The TD-DFT calculation was performed to explore different reactivity parameters, such as the HOMO-LUMO gap, electronegativity, ionization energy, chemical hardness, and softness. Based on these parameters, it was concluded that 2,3-diEt-DNPH exhibited high reactivity.
Furthermore, the synthesized derivatives were evaluated for their antioxidant potential by comparing them with standard vitamin C and EDTA. The compounds exhibited good DPPH radical scavenging activity, ferrous ion chelation activity, ferric ion reducing activity, molybdate ion reducing activity, and hydroxyl radical scavenging activity. Overall, this study provides valuable insights into the molecular structure, chemical reactivity, and antioxidant potential of novel derivatives of 2,4-DNPH, which could be useful for the development of new antioxidants with enhanced properties.
Acknowledgment
This research received financial support from several sources including the Research Funds for Young Scientist National Natural Science Foundation of China (No. 52150410429), and Key Laboratory of Green Fabrication and Surface Technology of Advanced Metals Materials (No. GFST2022ZR12) China, Thanks to Dr. Uzma Habib from the National University of Sciences and Technology (NUST), H-12, Islamabad, Pakistan for her assistance with DFT calculations. Authors are thanksful to researchers supportings project number (RSP2023R235), king Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- DNA binding ability mode, spectroscopic studies, hydrophobicity, and in vitro antibacterial evaluation of some new Fe (II) complexes bearing ONO donors amino acid Schiff bases. Arab. J. Chem.. 2017;10:S1835-S1846.
- [Google Scholar]
- Tailoring, structural elucidation, DFT calculation, DNA interaction and pharmaceutical applications of some aryl hydrazone Mn (II), Cu (II) and Fe (III) complexes. J. Mol. Struct.. 2021;1244:131017
- [Google Scholar]
- Synthesis, structural elucidation, DFT calculation, biological studies and DNA interaction of some aryl hydrazone Cr3+, Fe3+, and Cu2+ chelates. Comput. Biol. Chem.. 2022;97:107643
- [Google Scholar]
- Schiff bases of ethylenediamine as corrosion inhibitors of zinc in sulphuric acid. Corros. Sci.. 2004;46:633-651.
- [CrossRef] [Google Scholar]
- Structural inspection for novel Pd(II), VO(II), Zn(II) and Cr(III)- azomethine metal chelates: DNA interaction, biological screening and theoretical treatments. J. Mol. Struct.. 2021;1246:131139
- [CrossRef] [Google Scholar]
- Design, synthesis, structural inspection of Pd2+, VO2+, Mn2+, and Zn2+ chelates incorporating ferrocenyl thiophenol ligand: DNA interaction and pharmaceutical studies. Appl. Organomet. Chem.. 2021;35:e6169.
- [Google Scholar]
- Development and structure elucidation of new VO2+, Mn2+, Zn2+, and Pd2+ complexes based on azomethine ferrocenyl ligand: DNA interaction, antimicrobial, antioxidant, anticancer activities, and molecular docking. Appl. Organomet. Chem.. 2021;35:e6154.
- [Google Scholar]
- Anoop, M.R., Binil, P.S., Suma, S., Sudarsanakumar, M.R., Y, S.M., Varghese, H.T., Panicker, C.Y., 2010. Vibrational spectroscopic studies and computational study of ethyl methyl ketone thiosemicarbazone. J. Mol. Struct. 969, 48–54. https://doi.org/10.1016/j.molstruc.2010.01.041.
- Synthesis, crystal structure and theoretical studies of a Schiff base 2-[4-hydroxy benzylidene]-amino naphthalene. Spectrochim. Acta - Part A Mol. Biomol. Spectrosc.. 2015;135:307-316.
- [CrossRef] [Google Scholar]
- The effect of some Schiff bases on the corrosion of aluminum in hydrochloric acid solution. Appl. Surf. Sci.. 2006;252:4039-4047.
- [CrossRef] [Google Scholar]
- Synthesis, crystal structures, and DFT calculations: novel Mn (II), Co (II) and Ni (II) complexes of N-(pyridin-2-ylmethylene) methanamine with isothiocyanate as promising optical materials. Opt. Quant. Electron.. 2023;55:408.
- [Google Scholar]
- X-ray crystallographic, spectroscopic and electrochemical properties of a bi-stable di-2-thienyl ketone 2,4-dinitrophenyl hydrazone (dtkdnph) J. Mol. Struct.. 2018;1173:942-950.
- [CrossRef] [Google Scholar]
- The principles of conformational analysis. Q. Rev. Chem. Soc.. 1956;10:44-82.
- [CrossRef] [Google Scholar]
- Blanchard, P., Brüning, E., 2015. Density functional theory of atoms and molecules. Prog. Math. Phys. https://doi.org/10.1007/978-3-319-14045-2_37.
- Concentration effects on optical properties, DFT, crystal characterization and α-glucosidase activity studies: Novel Zn (II) complex. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2021;262:120072
- [Google Scholar]
- Regioselective synthesis of 1,3,5-tri- and 1,3,4,5-tetrasubstituted pyrazoles from N-arylhydrazones and nitroolefins. J. Org. Chem.. 2008;73:2412-2415.
- [CrossRef] [Google Scholar]
- Frisch, M.J., 2009. gaussian09. http//www. gaussian. com/.
- Synthesis, molecular structure, spectroscopic and theoretical investigation of 5-chlorosalicylaldehyde-2,4-dinitrophenylhydrazone. J. Mol. Struct.. 2020;1205
- [CrossRef] [Google Scholar]
- Experimental and theoretical study of a novel naphthoquinone Schiff base. Open Chem.. 2018;16:1115-1121.
- [CrossRef] [Google Scholar]
- A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys.. 2010;132
- [CrossRef] [Google Scholar]
- Synthesis, molecular docking, ADMET evaluation and in vitro cytotoxic activity evaluation on RD and L20B cell lines of 3-substituted 5, 5-diphenylimidazolidine-2, 4-dione derivatives. J. Biomol. Struct. Dyn.. 2023;41:4592-4600.
- [Google Scholar]
- Antifungal properties of Schiff bases of chitosan, N-substituted chitosan and quaternized chitosan. Carbohydr. Res.. 2007;342:1329-1332.
- [CrossRef] [Google Scholar]
- Hameed, A., al-Rashida, M., Uroos, M., Abid Ali, S., Khan, K.M., 2017. Schiff bases in medicinal chemistry: a patent review (2010-2015). Expert Opin. Ther. Pat. https://doi.org/10.1080/13543776.2017.1252752.
- Hobson, J., 2012. Socrates., Occupational medicine (Oxford, England). https://doi.org/10.1093/occmed/kqs085.
- Synthesis, crystal structure and quantum chemistry of a novel Schiff base N -(2,4-dinitro-phenyl)-N′-(1-phenyl-ethylidene)-hydrazine. Bull. Kor. Chem. Soc.. 2010;31:881-886.
- [CrossRef] [Google Scholar]
- Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino)benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct.. 2020;1219
- [CrossRef] [Google Scholar]
- Isatin endowed metal chelates as antibacterial and antifungal agents. Sains Malays.. 2020;49:1891-1904.
- [Google Scholar]
- A cyano-bridged molecule-based magnet containing manganese(III) Schiff base and octacyanotungstate(V) building blocks. Inorg. Chem. Commun.. 2004;7:1150-1153.
- [CrossRef] [Google Scholar]
- Synthesis, anticholinesterase, antioxidant, and anti-aflatoxigenic activity of novel coumarin carbamate derivatives. ChemistrySelect. 2018;3:3978-3983.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [CrossRef] [Google Scholar]
- Free radical-scavenging capacity, antioxidant activity and phenolic content of Pouzolzia zeylanica. J. Serbian Chem. Soc.. 2011;76:709-717.
- [CrossRef] [Google Scholar]
- Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 2000
- [CrossRef] [Google Scholar]
- Tautomerism in Schiff bases. the cases of 2-hydroxy-1-naphthaldehyde and 1-hydroxy-2-naphthaldehyde investigated in solution and the solid state. Org. Biomol. Chem.. 2011;9:8268-8275.
- [CrossRef] [Google Scholar]
- Antimicrobial and anticancer activities of cobalt (III)-hydrazone complexes: Solubilities and chemical potentials of transfer in different organic co-solvent-water mixtures. J. Mol. Liq.. 2019;290:111162
- [CrossRef] [Google Scholar]
- Synthesis, characterization and antitumour properties of some metal(II) complexes of 2-pyridinecarboxaldehyde 2′-pyridylhydrazone and related compounds. Inorg. Chim. Acta. 1988;151:61-68.
- [CrossRef] [Google Scholar]
- Synthesis and spectral characterization of hydrazone schiff bases derived from 2,4-dinitrophenylhydrazine. Crystal structure of salicylaldehyde-2,4- dinitrophenylhydrazone. Zeitschrift fur Naturforsch. - Sect. B J. Chem. Sci.. 2007;62:717-720.
- [CrossRef] [Google Scholar]
- New styrylquinoxaline: synthesis, structural, biological evaluation, ADMET prediction and molecular docking investigations. J. Biomol. Struct. Dyn.. 2023;41:2861-2877.
- [Google Scholar]
- Preparation, antioxidant activity, and theoretical studies on the relationship between antioxidant and electronic properties of bis (thio/carbohydrazone) derivatives. J. Phys. Chem. Solid. 2022;164:110618
- [Google Scholar]
- New Schiff bases based on isatin and (thio)/carbohydrazone: preparation, experimental–theoretical spectroscopic characterization, and DFT approach to antioxidant characteristics. Res. Chem. Intermed.. 2023;49:1463-1484.
- [Google Scholar]
- Synthesis, characterization and studies on the nonlinear optical parameters of hydrazones. Opt. Laser Technol.. 2010;42:741-748.
- [CrossRef] [Google Scholar]
- Correlating the charge transfer efficiency of metallic sulfa-isatins to design efficient NLO materials with better drug designs. Biometals. 2022;35:519-548.
- [CrossRef] [Google Scholar]
- Synthesis, DFT calculations, α-glucosidase inhibitor activity, and docking studies on Schiff base metal complexes containing isothiocyanate. Appl. Organomet. Chem.. 2023;37:e7084.
- [Google Scholar]
- Parr, R.G., Yang, W., 1983. Density-Functional Theory of Atoms and (3) Parr, R. G. Pearson, R. G. J. Am. Chem. Soc 105, 7512–7516.
- Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc.. 1983;105:7512-7516.
- [CrossRef] [Google Scholar]
- Synthesis and antimicrobial activities of novel Palladium (II) complexes of active Schiff’s base ligand derived from 5-bromo isatin. Int. Lett. Chem. Phys. Astron.. 2015;52:22-27.
- [CrossRef] [Google Scholar]
- Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci.. 1986;83:8440-8441.
- [CrossRef] [Google Scholar]
- Krebs cycle intermediates modulate thiobarbituric acid reactive species (TBARS) production in rat brain in vitro. Neurochem. Res.. 2005;30:225-235.
- [CrossRef] [Google Scholar]
- Tailoring of some novel bis-hydrazone metal chelates, spectral based characterization and DFT calculations for pharmaceutical applications and in-silico treatments for verification. J. Mol. Struct.. 2022;1264:133263
- [CrossRef] [Google Scholar]
- On the molecular structure, vibrational spectra, HOMO-LUMO, molecular electrostatic potential, UV–Vis, first order hyperpolarizability, and thermodynamic investigations of 3-(4-chlorophenyl)-1-(1yridine-3-yl) prop-2-en-1-one by quantum chemistry calculati. J. Mol. Struct.. 2018;1155:484-495.
- [CrossRef] [Google Scholar]
- Synthesis, characterization, and antimicrobial activities of nickel(II) and copper(II) Schiff-base complexes. J. Coord. Chem.. 2010;63:136-146.
- [CrossRef] [Google Scholar]
- Metal based sulfanilamides: A note on their synthesis, spectral characterization, and antimicrobial activity. Russ. J. Gen. Chem.. 2017;87:1834-1842.
- [Google Scholar]
- 6–31G* basis set for atoms K through Zn. J. Chem. Phys.. 1998;109:1223-1229.
- [CrossRef] [Google Scholar]
- A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures (Roeges, Noel P. G.), 1995. . J. Chem. Educ. 72, A93. https://doi.org/10.1021/ed072pa93.4.
- Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode [17] Nature. 1965;205:698-699.
- [CrossRef] [Google Scholar]
- In vitro free radical scavenging activity of methanol extract of the leaves of Mimusops elengi Linn. Bangladesh J. Vet. Med.. 1970;6:197-202.
- [CrossRef] [Google Scholar]
- Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem.. 1996;100:16502-16513.
- [CrossRef] [Google Scholar]
- The tautomerization between keto- to phenol-hydrazone induced by anions in the solution. J. Mol. Struct.. 2012;1010:52-58.
- [CrossRef] [Google Scholar]
- Shivakumar, K., Shashidhar, Vithal Reddy, P., Halli, M.B., 2008. Synthesis, spectral characterization and biological activity of benzofuran Schiff bases with Co(II), Ni(II), Cu(II), Zn(II), Cd(II) and Hg(II) complexes. J. Coord. Chem. 61, 2274–2287. https://doi.org/10.1080/00958970801905239.
- Spectrometric identification of organic compounds. J. Chem. Educ.. 1962;39:546-553.
- [CrossRef] [Google Scholar]
- Synthesis, antioxidant activity and SAR study of novel spiro-isatin-based Schiff bases. Mol. Divers.. 2019;23:829-844.
- [Google Scholar]
- Facile synthesis, spectroscopic evaluation and antimicrobial screening of metal endowed triazole compounds. Biometals. 2021;34:1329-1351.
- [Google Scholar]
- A review on the biomedical efficacy of transition metal triazole compounds. J. Coord. Chem.. 2022;75:293-334.
- [Google Scholar]
- Taha, M., Baharudin, M.S., Ismail, N.H., Khan, K.M., Jaafar, F.M., Samreen, Siddiqui, S., Choudhary, M.I., 2013. Synthesis of 2-methoxybenzoylhydrazone and evaluation of their antileishmanial activity. Bioorganic Med. Chem. Lett. 23, 3463–3466. https://doi.org/10.1016/j.bmcl.2013.03.051.
- Complexes of a tridentate ONS Schiff base. Synthesis and biological properties. Transit. Met. Chem.. 2000;25:456-460.
- [CrossRef] [Google Scholar]
- S-methyldithiocarbazate and its Schiff bases: Evaluation of bondings and biological properties. J. Biochem. Mol. Biol. Biophys.. 2002;6:85-91.
- [CrossRef] [Google Scholar]
- 2-Phenyl-4H-3,1-benzoxazin-4-one. Acta Crystallogr. Sect. E Struct. Reports Online. 2008;65
- [CrossRef] [Google Scholar]
- Zhao, X., Song, D.K., Radbil’, A.B., Radbil’, B.A., 2007. Synthesis and biological activity of Schiff bases derived from dehydroabietylamine and benzaldehyde derivatives. Russ. J. Appl. Chem. 80, 1373–1375. https://doi.org/10.1134/S1070427207080216.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105259.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




