

Translate this page into:
Synthesis, evaluation of the cytotoxicity, apoptosis induction in AGS cell line and gene expression and molecular modeling studies of novel tetrahydropyrimidine derivatives
⁎Corresponding author. saghisepehridr@gmail.com (Saghi Sepehri) s.sepehri@arums.ac.ir (Saghi Sepehri)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract

Abstract
Cancer is a complex disease that remains a leading cause of death worldwide owing to the lack of effective and efficient drugs. Compounds known as tetrahydropyrmidine have shown great potential as anti-cancer agents. Here, novel compounds based on tetrahydropyrimidine scaffold were successfully synthesized by the multicomponent reaction of Biginelli reaction. The biological functions of the selected compounds in AGS cells, including cytotoxicity, apoptosis induction, caspase3/7 activity, and gene expressions such as P53, CDNK2A, and Caspase-8/9, were evaluated. All synthesized compounds displayed moderate to good activity against the AGS cell line. Four compounds 4c, 4d, 4i, and 4 m induced apoptosis and activated caspase-3/7. Among them, compound 4c exhibited the highest cytotoxicity (IC50 = 69.60 µM), apoptosis induction, and caspase-3/7 activity. In comparison to other compounds, 4i showed a higher stimulation in the expression of P53, CDNK2A, and Caspase 9 genes. However, even though there was an increasing trend in the genes related to apoptosis and the cell cycle pathways, and the expression of caspase 9 was higher than caspase 8, these differences were not large enough to be significant. In addition, molecular docking studies and MD simulations were carried out to recognize the likely mechanism of the anti-cancer effect. Findings of docking revealed compounds fitted in the active site with good binding. MD simulations showed that compounds 4c, 4d and 4 m made a steady complex with the Eg5 enzyme. Thus, the possible mechanism of anti-cancer activity of these compounds can be through inhibition of the Eg5 enzyme.
Keywords
Cancer
Kinesin-5
P53
CDNK2A
Flow cytometry
Molecular dynamics simulations

1 Introduction
Cancer is a complex disease in which cells undergo rapid, uncontrolled, and pathological proliferation by disrupting the principles of normal cell division. This alteration usually happens because of changed cellular repair mechanisms monitored in response to contact with definite carcinogens (physical, biological, or chemical) using genetic factors (Senjor et al., 2023). The latter phase is known as metastasizing, and it is a primary cause of cancer death. Cancer is also known as neoplasm and malignant tumor (Gaur et al., 2022). Despite remarkable progress in procedures for cancer diagnosis and treatment (surgery, chemotherapy, radiotherapy) currently, cancer remained one of the most important danger factors for public health in the world (Senjor et al., 2023). Cancer has been the second most important reason of death in people’s lives, affecting an estimated 19.3 million humans around the world in 2020, and was predicted to grow by about 50 % between 2020 and 2040 (Feng et al., 2022). Changes in age-adjusted death rates are the best indicator of cancer progress, however other indicators, such as quality of life, are equally relevant. A rise in incidence can indicate a true increase in disease occurrence, such as when increased exposure to a risk factor leads to an increase in cancer occurrences. In such a case, the increasing incidence would almost certainly result in an increase in cancer fatalities. On the other hand, the prevalence of cancer may increase as a result of a new screening test that discovers many cancer cases that would not have caused a problem throughout someone's lifetime. In this example, the incidence of the cancer would increase, but death rates would not change. Cancer risk rises inexorably with age. It affects approximately 60 % of adults aged 65 and up. Furthermore, around 70 % of cancer fatalities occur at this stage. As a result, cancer is a disease of old age (Marosi and Köller 2016). Modifying or avoiding key risk factors and implementing existing evidence-based prevention strategies could prevent between 30 % and 50 % of cancer deaths (https://www.who.int/health-topics/cancer#tab = tab_2/2023). Despite tremendous efforts in the field of tumor cell biology, the drug treatment of cancers has not changed significantly, and in most cases, the current chemotherapy does not discriminate between normal and tumor cells (Li et al., 2017). Moreover, drug resistance has been identified as a major problem in cancer patients; thus, numerous techniques are being utilized to overcome the former challenge (Mateev et al., 2022).
Gastric cancer, a public virulent tumor of the digestive system, is the fifth in occurrence and the fourth in death. This cancer is a remarkable risk worldwide because almost one million cases and 8.7 million deaths were reported in 2018. Unfortunately, about 30 % of cases are identified in the middle and final phases since the initial symptoms are not obvious (Luo et al., 2022). Various factors, including Helicobacter pylori, environment, lifestyle, nutrition, and genetics, have been recognized as potential danger agents for gastric cancer (Aslany et al., 2020). Chemotherapy and radiation therapy have the lowest results in treating gastric cancer (Aslany et al., 2020). Some drugs, including oteracil, gimeracil, tegafur, capecitabine, and apatinib as anti-VEGFR2 drugs, and trastuzumab against Her2 have been usually used in clinical practice for the treatment of gastric cancer. Though the clinical problems related to chemotherapy, such as severe side effects, overall toxicity, and strong drug resistance, are becoming more protuberant. Furthermore, high costs and substantial care not only persecute patients but also inflict a heavy economic burden on their society and relations. Therefore, the progress of novel and effective drugs for gastric cancer therapy has aroused great interest (Luo et al., 2022).
Multicomponent reaction (MCR) is an eco-friendly alternative to linear synthesis, which usually involves multiple steps and sequential purification processes (Çağlar Yavuz et al., 2020). The Biginelli reaction is a significant MCR with multiple applications in medicinal and pharmaceutical chemistry, particularly for the production of 1,2,3,4-tetrahydropyrimidine (THPM) derivatives. Aryl substituted-THPM and its derivatives are important in medicinal chemistry because of their various pharmaceutical activities, including anti-bacterial, anti-HIV, anti-fungal, anti-tubercular, anti-oxidant, anti-cancer, anti-inflammatory, and anti-urease properties (Sepehri et al., 2017, Asham et al., 2021, Mirzayi et al., 2021, Ahangarzadeh et al., 2022). Cytosine is present in both DNA and RNA, which are pyrimidine analogs (Ahangarzadeh et al., 2022). Compounds known as THPM have shown great potential as a cancer treatment. The most extensively studied compounds in this group, monastrol, has been found to disrupt mitosis by inhibiting the motor activity of the kinesin Eg5 protein, which plays a crucial role in spindle bipolarity. These compounds have served as the basis for the development of novel anti-cancer compounds (Janković et al., 2019). Previous studies have reported that various THPM derivatives have been utilized as anti-cancer agents with different substitutions (Sepehri et al., 2015, Kantankar et al., 2021, Milović et al., 2022). Moreover, our research group recently reported THPM derivatives that exhibited good cytotoxicity toward different cancer cell lines (Razzaghi-Asl et al., 2019, Safari et al., 2020, Asham et al., 2021, Mirzayi et al., 2021). The design rationale of the present research was based on previous reports (Fig. 1).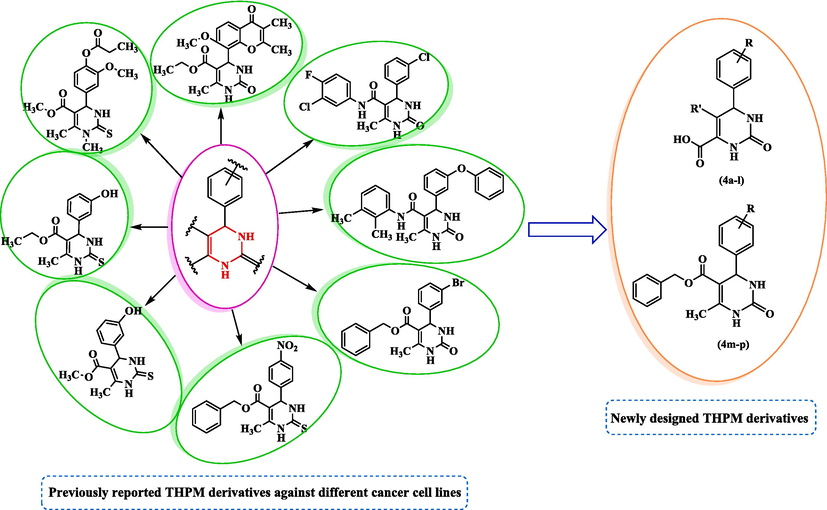
Design strategy of new THPM derivatives as anti-cancer agent.
Based on previous studies of THPM analogs as good anti-cancer agents, a series of new THPM analogs were designed, synthesized, and assessed for cytotoxicity activity against the AGS cell line by the MTT assay. For the most potent compounds, induction of apoptosis and expression levels of P53, CDKN2A, and caspase-8/9 were also assessed. To recognize the stability, interactions, and binding modes of these derivatives in the active site of Eg5, molecular docking, and dynamics simulations were performed.
2 Materials and methods
2.1 Synthesis
2.1.1 Common method of the synthesis of alkyl 4-aryl-6-alkyl-2-thio(oxo)-1,2,3,4-tetrahydropyrimidine-5-alkyl (4a-p)
A combination of α-ketoacid/β-ketoester (3.9 mmol), urea (3.6 mmol), various aldehydes (3 mmol), and Co(HSO4)2 were solved in pure ethanol (5 mL). Compound synthesis was carried out using the technique used in the previous investigation (Ahangarzadeh et al., 2022).
2.1.2 6-(3,4-dimethoxyphenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4a)
White precipitates; yield: 60 %; mp 280–282 °C; Rf = 0.50 (chloroform/methanol 1:0.5), Rf = 0.30 (n-Hexane/EtOAc 1:0.5); FT-IR (KBr, υ): 3304, 3217 (N–H), 2730–3510 (C-OH), 3120 (C–H), 2941 (C–H), 1660 (C = O), 1516, 1464 (C = C), 1273 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.49 (1H, brs, H-O), 7.29 (1H, s, H-N), 7.26 (1H, s, CH-ph), 6.95 (1H, d, J = 8.0 Hz, CH-ph), 6.89 (1H, s, H-N), 6.80 (1H, d, J = 10 Hz, CH-ph), 4.72 (1H, s, H-C4), 3.74 (6H, s, OCH3), 1.86 (3H, s, H-C5); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 164.5, 149.9, 147.1, 146.3, 133.5, 128.4, 127.7, 123.7, 118.2, 113.5, 67.8, 56.6, 55.1, 9.9. MS (m/z, %): 292.29 (1.0), 281.10 (6.1), 260.1 (85.4), 189.20 (1 0 0), 151.10 (20.3); Anal. Calcd for C14H16N2O5: C, 57.53; H, 5.52; N, 9.58 %. Found: C, 57.18; H, 5.61; N, 9.42 %.
2.1.3 6-(4-hydroxy-3-methoxyphenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4b)
White precipitates; yield: 88 %; mp 203–205 °C; Rf = 0.57 (chloroform/methanol 1:0.3), Rf = 0.30 (n-Hexane/EtOAc 1:0.5); FT-IR (KBr, υ): 3525, 3448 (N–H), 2765–3478 (C-OH), 3124 (C–H), 2967 (C–H), 1651 (C = O), 1597, 1454 (C = C), 1285 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.47 (1H, brs, H-O), 9.03 (1H, s, H-N), 7.24 (2H, s, H-N, H-O), 6.69–6.85 (3H, m, CH-ph), 4.66 (1H, s, H-C4), 3.74 (3H, s, OCH3), 1.86 (3H, s, H-C5); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 167.1, 151.5, 149.4, 148.1, 137.3, 128.6, 127.4, 120.8, 118.2, 115.9, 70.5, 55.7, 7.3. MS (m/z, %): 278.26 (1.0), 245.20 (1 0 0), 219.20 (87.1), 171.10 (24.5), 111.10 (50.4); Anal. Calcd for C13H14N2O5: C, 56.11; H, 5.07; N, 10.07 %. Found: C, 55.81; H, 4.83; N, 10.36 %.
2.1.4 5-bromo-6-(3,4-dimethoxyphenyl)-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4c)
Brown precipitates; yield: 56 %; mp 201–203 °C; Rf = 0.57 (chloroform/methanol 1:0.1), Rf = 0.42 (n-Hexane/EtOAc 1:0.25); FT-IR (KBr, υ): 3355, 3136 (N–H), 2689–3618 (C-OH), 3014 (C–H), 2843 (C–H), 1667 (C = O), 1514, 1462 (C = C), 1265 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.36 (1H, brs, H-O), 7.67 (1H, s, H-N), 7.59 (1H, s, CH-ph), 7.18 (1H, d, J = 13.8 Hz, CH-ph), 7.14 (1H, s, H-N), 6.98 (1H, d, J = 8.0 Hz, CH-ph), 4.94 (1H, s, H-C4), 3.74 (6H, s, OCH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 162.5, 152.7, 150.3, 149.6, 134.5, 126.2, 125.8, 114.1, 112.5, 104.4, 64.7, 52.7, 53.6. MS (m/z, %): 359.1 (M + 2), 357.16 (M), 184.0 (20.4), 140.10 (25.7), 92.0 (84.3), 64.0 (1 0 0); Anal. Calcd for C13H13BrN2O5: C, 43.72; H, 3.67; N, 7.84 %. Found: C, 44.03; H, 3.86; N, 7.57 %.
2.1.5 5-bromo-6-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4d)
Brown precipitates; yield: 52 %; mp 214–216 °C; Rf = 0.37 (chloroform/methanol 1:0.1), Rf = 0.25 (n-Hexane/EtOAc 1:0.5); FT-IR (KBr, υ): 3387, 3137 (N–H), 2750–3674 (C-OH), 2986 (C–H), 2859 (C–H), 1668 (C = O), 1515, 1451 (C = C), 1278 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.47 (1H, brs, H-O), 9.36 (1H, s, H-N), 7.45 (2H, s, H-N, O–H), 7.13–7.24 (3H, m, CH-ph), 4.96 (1H, s, H-C4), 3.74 (3H, s, OCH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 163.5, 150.1, 148.2, 147.6, 136.7, 125.8, 126.3, 118.9, 117.5, 114.6, 62.5, 54.7. MS (m/z, %): 345.1 (M + 2), 343.13 (M), 218.1 (20), 171.0 (30), 107.1 (25), 79.0 (1 0 0). 60.1 (12); Anal. Calcd for C12H11BrN2O5: C, 42.00; H, 3.23; N, 8.16 %. Found: C, 42.34; H, 3.03; N, 8.44 %.
2.1.6 5-methyl-6-(4-nitrophenyl)-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4e)
White precipitates; yield: 71 %; mp 277–278 °C; Rf = 0.22 (chloroform/methanol 1:0.25), Rf = 0.62 (n-Hexane/EtOAc 0.5:0.15); FT-IR (KBr, υ): 3421, 3198 (N–H), 2915–3587 (C-OH), 3067 (C–H), 2945 (C–H), 1684 (C = O), 1538, 1461 (C = C), 1283 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.60 (1H, brs, H-O), 8.20 (2H, d, J = 8.0 Hz, CH-ph), 8.16 (1H, s, H-N), 7.78–7.69 (2H, m, CH-ph), 7.56 (1H, s, H-N), 5.04 (1H, s, H-C4), 1.87 (3H, s, H-C5); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 166.4, 152.3, 150.7, 147.8, 128.1, 127.8, 123.5, 119.8, 63.8, 10.1. MS (m/z, %): 277.24 (M), 207.2 (80), 150.1 (1 0 0), 104.1 (40), 76.1 (25); Anal. Calcd for C12H11N3O5: C, 51.99; H, 4.00; N, 15.16 %. Found: C, 52.37; H, 3.78; N, 15.34 %.
2.1.7 6-(4-bromophenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4f)
White precipitates; yield: 60 %; mp 285–286 °C; Rf = 0.60 (chloroform/methanol 1:0.2), Rf = 0.25 (n-Hexane/EtOAc 1:1); FT-IR (KBr, υ): 3252, 3167 (N–H), 2634–3492 (C-OH), 3120 (C–H), 2984 (C–H), 1665 (C = O), 1521, 1462 (C = C), 1253 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.55 (1H, brs, H-O), 7.59 (3H, brs, H-N, CH-ph), 7.27–7.39 (3H, m, H-N, CH-ph), 4.80 (1H, s, H-C4), 1.84 (3H, s, H-C5); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 165.2, 150.1, 140.2, 132.3, 128.3, 127.7, 120.4, 119.8, 62.2, 7.5. MS (m/z, %): 277.24 (M), 313.1 (30), 311.1 (33), 278.1 (1 0 0), 248. 1 (55), 198.2 (15), 170.1 (45); Anal. Calcd for C12H11BrN2O3: C, 46.32; H, 3.56; N, 9.00 %. Found: C, 46.64; H, 3.39; N, 9.28 %.
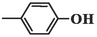
2.1.8 6-(4-hydroxyphenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4g)
White precipitates; yield: 77 %; mp 300–302 °C; Rf = 0.50 (chloroform/methanol 1:0.3), Rf = 0.33 (n-Hexane/EtOAc 1:2); FT-IR (KBr, υ): 3426, 3271 (N–H), 2878–3612 (C-OH), 3086 (C–H), 2893 (C–H), 1683 (C = O), 1594, 1468 (C = C), 1276 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.47 (1H, brs, H-O), 9.45 (1H, s, H-O), 7.23 (1H, s, H-N), 7.09 (2H, d, J = 6.8 Hz, CH-ph), 6.74 (3H, d, J = 8 Hz, H-N, CH-ph), 4.64 (1H, s, H-C4), 1.83 (3H, s, H-C5); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 166.7, 148.5, 139.4, 132.9, 129.3, 128.6, 119.2, 118.5, 63.8, 9.4. MS (m/z, %): 248.2 (0.01), 218.1 (25), 171.0 (35), 106.1 (48), 80.0 (75), 61.1 (1 0 0); Anal. Calcd for C12H12N2O4: C, 58.06; H, 4.87; N, 11.29 %. Found: C, 58.37; H, 5.13; N, 11.03 %.
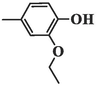
2.1.9 6-(3-ethoxy-4-hydroxyphenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4h)
White precipitates; yield: 70 %; mp 221–223 °C; Rf = 0.62 (chloroform/methanol 1:0.35), Rf = 0.75 (n-Hexane/EtOAc 1:1); FT-IR (KBr, υ): 3456, 3236 (N–H), 2835–3659 (C-OH), 3136 (C–H), 2976 (C–H), 1653 (C = O), 1599, 1453 (C = C), 1254 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.46 (1H, brs, H-O), 8.96 (1H, s, H-O), 7.23 (2H, brs, H-N, CH-ph), 6.69–6.83 (3H, m, H-N, CH-ph), 4.65 (1H, s, H-C4), 3.99 (2H, brs, OCH2CH3), 1.85 (3H, s, H-C5), 1.33 (3H, brs, OCH2CH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 165.7, 150.9, 148.1, 147.5, 135.6, 129.2, 128.6, 117.7, 116.3, 114.4, 66.8, 65.3, 14.7, 8.5. MS (m/z, %): 292.1 (M), 263.1 (9), 233.2 (1 0 0), 204.2 (13), 176.2 (9), 111.1 (50); Anal. Calcd for C14H16N2O5: C, 57.53; H, 5.52; N, 9.58 %. Found: C, 58.73; H, 5.28; N, 9.75 %.
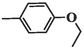
2.1.10 6-(4-ethoxyphenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4i)
White precipitates; yield: 47 %; mp 275–276 °C; Rf = 0.70 (chloroform/methanol 1:0.1), Rf = 0.54 (n-Hexane/EtOAc 1:1); FT-IR (KBr, υ): 3339, 3221 (N–H), 2741–3627 (C-OH), 3128 (C–H), 2965 (C–H), 1721 (C = O), 1633, 1459 (C = C), 1221 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.28 (1H, brs, H-O), 9.46 (1H, s, H-N), 8.66 (1H, s, H-N), 7.17–7.23 (2H, m, CH-ph), 7.02–7.07 (2H, m, CH-ph), 5.24 (1H, s, H-C4), 4.18–4.26 (2H, m, OCH2CH3), 2.27 (3H, s, H-C5), 1.08 (3H, t, J = 7.2 Hz, OCH2CH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 166.5, 158.2, 150.8, 136.1, 128.2, 122.5, 119.3, 114.6, 63.4, 61.8, 14.2, 6.5. MS (m/z, %): 276.2 (M), 231.0 (1 0 0), 186.1 (45), 110.2 (24); Anal. Calcd for C14H16N2O4: C, 60.86; H, 5.84; N, 10.14 %. Found: C, 58.42; H, 5.75; N, 10.41 %.
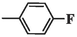
2.1.11 6-(4-fluorophenyl)-5-methyl-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4j)
White precipitates; yield: 33 %; mp 289 °C; Rf = 0.42 (chloroform/methanol 1:0.5), Rf = 0.9 (n-Hexane/EtOAc 1:1.5); FT-IR (KBr, υ): 3316, 3231 (N–H), 2689–3646 (C-OH), 3133 (C–H), 2978 (C–H), 1670 (C = O), 1606, 1469 (C = C), 1221 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.55 (1H, brs, H-O), 7.42–7.37 (3H, m, H-N, CH-ph), 7.26 (3H, t, J = 8 Hz, H-N, CH-ph), 4.86 (1H, s, H-C4), 1.90 (3H, s, H-C5); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 164.2, 160.3, 152.0, 139.5, 128.8, 123.3, 118.1, 116.6, 59.7, 15.5. MS (m/z, %): 250.2 (M), 205.1 (1 0 0), 110.1 (35); Anal. Calcd for C12H11FN2O3: C, 57.60; H, 4.43; N, 11.20 %. Found: C, 57.98; H, 4.16; N, 10.99 %.
2.1.12 6-(3,4-dimethoxyphenyl)-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4k)
Light orange precipitates; yield: 38 %; mp 258–259 °C; Rf = 0.37 (chloroform/methanol 1:0.5), Rf = 0.82 (n-Hexane/EtOAc 1:1.5); FT-IR (KBr, υ): 3353, 3220 (N–H), 2594–3509 (C-OH), 3056 (C–H), 2934 (C–H), 1716 (C = O), 1619, 1446 (C = C), 1258 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.38 (1H, brs, H-O), 7.76 (1H, s, H-N), 7.31 (1H, s, H-N), 7.01 (1H, d, J = 8.0 Hz, CH-ph), 6.96 (1H, d, J = 1.6 Hz, CH-ph), 6.87 (1H, dd, J1 = 2.0 Hz, J2 = 6.4 Hz, CH-ph), 5.83 (1H, d, J = 4.8 Hz, H-C4), 5.16 (1H, dd, J1 = 2.0 Hz, J2 = 2.0 Hz, H-C5), 3.80 (3H, s, OCH3), 3.79 (3H, s, OCH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 162.7, 151.5, 149.1, 147.8, 135.8, 127.2, 118.3, 111.2, 109.7, 108.5, 55.9, 55.4, 54.3. MS (m/z, %): 278.2 (M), 233.0 (1 0 0), 96.1 (47); Anal. Calcd for C13H14N2O5: C, 56.11; H, 5.07; N, 10.07 %. Found: C, 56.45; H, 4.85; N, 9.81 %.
2.1.13 6-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4l)
Light pink precipitates; yield: 41 %; mp 249–250 °C; Rf = 0.50 (chloroform/methanol 1:0.3), Rf = 0.37 (n-Hexane/EtOAc 1:1.3); FT-IR (KBr, υ): 3445, 3240 (N–H), 2521–3643 (C-OH), 3133 (C–H), 2952 (C–H), 1653 (C = O), 1597, 1453 (C = C), 1284 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 13.34 (1H, brs, H-O), 8.46 (1H, s, O–H), 7.33 (1H, s, N–H), 7.28 (1H, s, CH-ph), 6.97 (1H, d, J = 8.0 Hz, CH-ph), 6.91 (1H, s, N–H), 6.82 (1H, d, J = 8.0 Hz, CH-ph), 5.38 (1H, s, H-C4), 4.96 (1H, s, H-C5), 3.74 (3H, s, OCH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 164.5, 150.9, 147.6, 146.4, 136.7, 133.8, 123.5, 118.4, 115.7, 111.8, 57.1, 54.2. MS (m/z, %): 267.2 (M), 219.1 (1 0 0), 188.1 (74), 171.0 (32), 96.2 (28); Anal. Calcd for C12H12N2O5: C, 54.55; H, 4.58; N, 10.60 %. Found: C, 54.21; H, 4.32; N, 10.76 %.
2.1.14 Benzyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4m)
White precipitates; yield: 55 %; mp 168–169 °C; Rf = 0.57 (chloroform/methanol 1:0.5), Rf = 0.71 (n-Hexane/EtOAc 1:1.5); FT-IR (KBr, υ): 3339, 3221 (N–H), 2784–3697 (C-OH), 3128 (C–H), 2967 (C–H), 1721 (C = O), 1633, 1459 (C = C), 1258 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 9.24 (1H, s, N–H), 7.69 (1H, s, N–H), 7.29 (3H, dd, J1 = 1.6 Hz, J2 = 2 Hz, CH-ph), 7.17 (2H, dd, J1 = 4 Hz, J2 = 2 Hz, CH-ph), 6.88 (1H, d, J = 8.4 Hz, CH-ph), 6.78 (1H, d, J = 2.0 Hz, CH-ph), 6.72 (1H, dd, J1 = 2 Hz, J2 = 2 Hz, CH-ph), 5.14 (1H, d, J = 3.2 Hz, H-C4), 5.02 (1H, d, J = 12.8 Hz, CH2C6H5), 5.07 (1H, d, J = 12.8 Hz, CH2C6H5), 3.72 (3H, s, OCH3), 3.60 (3H, s, OCH3), 2.28 (3H, s, H-C6); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 165.5, 151.2, 148.8, 148.5, 147.9, 136.9, 136.1, 127.8, 127.5, 127.3, 117.5, 111.4, 110.6, 98.4, 64.7, 55.5, 54.3, 53.2, 17.1. MS (m/z, %): 382.2 (M), 275.1 (1 0 0), 247.1 (86), 185.0 (67), 110.1 (43); Anal. Calcd for C21H22N2O5: C, 65.96; H, 5.80; N, 7.33 %. Found: C, 65.6; H, 6.01; N, 7.59 %.
2.1.15 Benzyl 4-(3-ethoxy-4-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4n)
White precipitates; yield: 60 %; mp 180–181 °C; Rf = 0.37 (chloroform/methanol 1:0.2), Rf = 0.71 (n-Hexane/EtOAc 1:1.5); FT-IR (KBr, υ): 3416, 3218 (N–H), 2684–3845 (C-OH), 3121 (C–H), 2941 (C–H), 1704 (C = O), 1521, 1481 (C = C), 1256 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 9.20 (1H, s, N–H), 8.84 (1H, s, N–H), 7.65 (1H, s, O–H), 7.29 (3H, dd, J1 = 1.2 Hz, J2 = 2.0 Hz, CH-ph), 7.19 (2H, dd, J1 = 3.6 Hz, J2 = 1.6 Hz, CH-ph), 6.71 (1H, d, J = 2.0 Hz, CH-ph), 6.70 (1H, s, CH-ph), 6.61 (1H, dd, J1 = 2.0 Hz, J2 = 2.0 Hz, CH-ph), 5.09 (1H, d, J = 3.2 Hz, H-C4), 5.05 (1H, s, CH2C6H5), 5.01 (1H, d, J = 12.8 Hz, CH2C6H5), 3.76–3.86 (2H, m, OCH2CH3), 2.27 (3H, s, H-C6), 1.27 (3H, t, J = 6.8 Hz, OCH2CH3); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 164.9, 152.1, 148.6, 146.1, 146.5, 136.8, 135.5, 128.2, 127.6, 126.9, 118.1, 115.3, 112.1, 98.5, 64.6, 63.7, 53.2, 17.7, 13.8. MS (m/z, %): 382.1 (M), 275.1 (1 0 0), 247.1 (77), 218.1 (53), 185.0 (67), 108.0 (21); Anal. Calcd for C21H22N2O5: C, 65.96; H, 5.80; N, 7.33 %. Found: C, 65.66; H, 5.91; N, 7.08 %.
2.1.16 Benzyl 4-(4-bromophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4o)
White precipitates; yield: 70 %; mp 180–181 °C; Rf = 0.71 (chloroform/methanol 1:0.2), Rf = 0.37 (n-Hexane/EtOAc 1:1.3); FT-IR (KBr, υ): 3230, 3115 (N–H), 2931 (C–H), 2895 (C–H), 1707 (C = O), 1599, 1458 (C = C), 1284 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 9.37 (1H, s, N–H), 7.84 (1H, s, N–H), 7.55 (2H, d, J = 8.4 Hz, CH-ph), 7.34–7.35 (3H, m, CH-ph), 7.18–7.22 (4H, m, CH-ph), 5.21 (1H, d, J = 3.2 Hz, H-C4), 5.13 (1H, d, J = 12.8 Hz, CH2C6H5), 5.40 (1H, d, J = 12.8 Hz, CH2C6H5), 2.33 (3H, s, H-C6); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 164.5, 151.6, 149.3, 143.7, 136.6, 131.1, 128.8, 128.3, 127.7, 127.5, 120.1, 97.7, 64.5, 53.2, 17.9. MS (m/z, %): 403.0 (M + 2), 401.1 (M), 309.3 (15), 245.4 (15), 91.3 (1 0 0); Anal. Calcd for C19H17BrN2O3: C, 56.87; H, 4.27; N, 6.98 %. Found: C, 57.22; H, 4.04; N, 7.13 %.
2.1.17 Benzyl 4-(4-hydroxy-3-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4p)
White precipitates; yield: 54 %; mp 171–172 °C; Rf = 0.57 (chloroform/methanol 1:0.3), Rf = 0.66 (n-Hexane/EtOAc 1:1.5); FT-IR (KBr, υ): 3230, 3115 (N–H), 2931 (C–H), 2895 (C–H), 1707 (C = O), 1599, 1458 (C = C), 1284 (C-O) cm−1; 1H NMR (400 MHz, DMSO‑d6) δ/ppm: 9.25 (1H, s, N–H), 8.97 (1H, s, N–H), 7.70 (1H, s, O–H), 7.34–7.35 (3H, m, CH-ph), 7.23–7.24 (2H, m, CH-ph), 6.79 (1H, s, CH-ph), 6.75 (1H, d, J = 8.0 Hz, CH-ph), 6.66 (1H, t, J = 4 Hz, CH-ph), 5.15 (1H, d, J = 2.4 Hz, H-C4), 5.12 (1H, d, J = 12.8 Hz, CH2C6H5), 5.07 (1H, d, J = 12.8 Hz, CH2C6H5), 3.67 (3H, s, OCH3), 2.33 (3H, s, H-C6); 13C NMR (100 MHz, DMSO‑d6) δ/ppm: 165.5, 152.4, 148.2, 147.4, 145.6, 136.2, 135.4, 127.9, 127.6, 118.2, 114.6, 110.7, 98.2, 64.5, 55.5, 53.7, 17.4. MS (m/z, %): 368.3 (M), 261.1 (1 0 0), 233.0 (64), 218.2 (26), 110.1 (15); Anal. Calcd for C20H20N2O5: C, 65.21; H, 5.47; N, 7.60 %. Found: C, 65.53; H, 5.27; N, 7.29 %.
2.2 Biological assay
2.2.1 Cell culture
The AGS (human gastric adenocarcinoma) cell line was purchased from the Pasteur Institute of Iran (Tehran, Iran). The cells were cultured in RPMI 1640 environment and added to 2 mM glutamine at 37 °C in a moistened atmosphere with 5 % CO2 and 10 % fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.).
2.2.2 Cell viability assessment
The MTT test was used to assess the viability of AGS cells. Briefly, the cells were planted in a 96-well plate at a density of 10,000 cells/well and permitted to grow for 24 h in a normal medium. Subsequently, they were exposed to different concentrations of synthetic compounds for 24 h and 48 h. Following treatment intervals, supernatants were removed and cells were treated with a final concentration of 0.5 mg/mL MTT reagent (Sigma, MO, US) and incubated for 3 h. To solubilize the formazan crystals, dimethyl sulfoxide (DMSO) (Sigma, MO, US) solution was added to the plate wells, and after shaking for 15 min, the absorbance was read at 570 nm on an EpochTM microplate spectrophotometer (Agilent Technology, Winooski, VT, US). In the MTT assay, all compounds with seven concentrations ranging from 0 to 200 with 2-fold increments were tested in triplicate.
2.2.3 Flow cytometry analysis of apoptosis and necrosis
The Annexin V test was used to evaluate apoptosis, which is useful for detecting translocated phosphatidylserine (PS), a hallmark of apoptosis (Mariño and Kroemer 2013). According to the manufacturer’s instructions, untreated and treated cells with compounds (after 24 h) were analyzed to distinguish between necrosis and apoptosis by the Annexin V-FITC/propidium iodide assay kit (eBioscience, Fisher Scientific Inc., CA, US). Briefly, the cells were detached and centrifuged at 400–600 × g for 5 min, then the cell pellet was washed with 1X PBS and suspended in 1X binding buffer. The cell pellet was stained with 5 µL of fluorochrome-conjugated Annexin V. The stained cells were incubated in the dark for 10–15 min at room temperature. Then, after adding 2 mL of 1X binding buffer, the cells were centrifuged at 400–600 × g for 5 min at room temperature. The supernatant was discarded, and the cell pellet was re-suspended in 200 μL of 1X binding buffer. Finally, 5 μL propidium iodide was added, and the cells were incubated for another 5–15 min on ice at room temperature. The samples were then diluted with PBS and analyzed by a flow cytometer (Cyflowspace, Partec, Germany). Data analysis was accomplished by FlowJo™ v10 software (BD Biosciences).
2.2.4 Colorimetric detection of caspase-3/7 activity
Caspase-3/7, known as effector caspases, plays a vital role in the intrinsic and extrinsic pathways of apoptosis (Parrish et al., 2013). Based on the manufacturer’s instructions, caspase-3/7 activity was determined via the Kiazist caspase assay kit (Hamadan, Iran). Briefly, following exposure of AGS cells to IC50 concentrations of each compound, including 4c, 4d, 4i, and 4 m, for 24 h, trypsinized cells were exposed to the caspase lysis buffer. Following incubation for 20 min at 4 °C, cells were harvested and centrifuged at 12,000 rpm for 15 min at 4 °C. The supernatant of each well was discarded, then a mixture of caspase buffer: 50 μL, DTT: 0.5 μL, caspase substrate (DEVD-pNA): 5 μL was added to each well and incubated for 1.5–2 h at 37 °C. Finally, the absorbance was measured at 405 nm by an ELISA reader. A standard curve was produced by serial dilutions of the caspase standard (pNA) (final concentrations are 0, 10, 20, 30, 40, and 50 µM). All assays were performed in triplicate.
2.2.5 Analysis of gene transcript levels with RT-qPCR
Overall RNA was isolated from cell samples by RNX-Plus reagent (SincaClon, Tehran, Iran) based on the manufacturer’s instructions. The isolated RNA samples were analyzed by a Nanodrop 2000c spectrophotometer (Thermo Fisher ScientificTM, Wilmington, DE, US) to check quantity and quality. Complementary DNA (cDNA) was constructed by the YektaTajhiz First Strand cDNA Synthesis Kit (YektaTajhiz, Tehran, Iran).
Rotor-Gene Q-Real-Time PCR System (QIAGEN, Hilden, Germany) was used to amplify selected genes. Each 25-μL PCR reaction for each gene amplification was composed of 20 ng of cDNA, 10 μL of RealQ Plus 2x Master Mix Green, high Rox (Odense M, Denmark), and 200 nM of each forward and reverse primers. PCR amplifications were carried out with an original enzyme activation step at 95 °C for 15 min, followed: 40 cycles at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s.
Expression values were corrected for the housekeeping gene B2M. Melting curves were created by checking the fluorescence of the SYBR green signal from 65 °C to 95 °C. Q-PCR data were analyzed by the 2−ΔΔCt method. All reactions were run in duplicate. A no-template control and no-reverse transcriptase controls were included in each run.
2.2.6 Statistical analysis
A one-way analysis of variance (ANOVA) was conducted to compare gene expression levels between the study groups. Following ANOVA further explored and compared the mean of one group with the mean of the control with Fisher's Least Significant Difference (LSD) test. All statistical tests were two-tailed, and statistical P-values below 0.05 (p < 0.05) were considered statistically significant. Statistical analysis and bar graph preparation were performed using GraphPad Prism, ver. 6.07 (Graph Pad Software Inc. La Jolla, CA, U.S.A.).
2.3 Molecular modelling studies
2.3.1 Molecular docking study
Autodock 4.2 was used to calculate the docking of the compounds into the Eg5 active site. The crystal structure of Eg5 (PDB ID: 1X88) was obtained from the protein data bank (PDB). The docking technique for compounds was carried out following the procedure for molecular docking of compounds in the prior study (Asham et al., 2021).
2.3.2 Molecular dynamics simulations study
Molecular dynamics (MD) simulations was carried out by GROMACS 2019.6 computational package. The process was carried out under the MD technique from the prior study (Khalilzadeh et al., 2022). Finally, a 150 ns MD simulation was performed to confirm equilibration by investigating the stability of the system's temperature, energy, and density, as well as the root mean squared deviations (RMSDs) of the backbone atoms.
3 Results and discussion
3.1 Rationale and design
Several research studies have been published since the discovery of monastrol as an antimitotic anticancer THPM derivative to develop more potent kinesin Eg5 inhibitors (El‐Hamamsy et al., 2020). The antiproliferation activity was increased by replacing the ester side chain at position 5 of the THPM ring with a less hydrolyzed amide link (Kumar et al., 2009). Furthermore, THPM with unsubstituted position 5 resulted in more potent analogue cancer cell lines (Kaur et al., 2017). Furthermore, the presence of a benzyl moiety at position 5 increased the activity against cancer cell lines (Razzaghi-Asl et al., 2019, Safari et al., 2020). Furthermore, two monastrol analogues with an unsubstituted position 5 of the THPM ring were created and demonstrated good anticancer activity (El‐Hamamsy et al., 2020). Wright et al. identified N-1 substituted long chain 4-aryl/alkyl-3,4-dihydropyrimidines as a class of MCF-7 cell line cytotoxic agents (Wright et al., 2008). The cytotoxicity of a series of 4H-chromen-3,4-dihydropyrimidine-5-carboxylate analogues against three different human cancer cell lines, A549, CNS (SK-N_SH), and Hela, was explained by Raju et al. (Raju et al., 2011). Yousif et al. synthesized pyrimidine, thioxopyrimidine, and iminopyrimidine derivatives and tested their anticancer activity against HepG-2, PC-3, and HCT-116 cell lines. All compounds demonstrated weak to moderate activity against three cell lines (Yousif et al., 2017). N-alkylation of 3,4-dihydropyrimidin-2 (1H)-one compounds was described by Liu et al. All derivatives demonstrated activity against the U87, U251, Hela, and A549 cell lines. An in vitro cytotoxicity study on all synthesized compounds revealed that the aryl chain in the C4 position, as well as the low electron-donating group in the ester moiety of THPMs, contributed to their anti-proliferative potency (Liu et al., 2019). Several THPM derivatives have been recently tested for their anticancer activity in various cell types. For instance, in a study by Bakavoli et al., they showed that a synthetic series of 6-amino-4-aryl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitriles had cytotoxic effects on MCF-7 and HT-29 cells. They indicated that the biological activity is primarily determined by the different substitutions (Atapour-Mashhad et al., 2016). A researcher’s study also revealed that certain pyrimidine thione derivatives, specifically 6-(1,3-diphenyl-1H-pyrazol-4-yl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile, can induce cytotoxicity in both MCF-7 and HCT-116 cell lines. The authors hypothesized that the presence of the NO2 group, acting as a strong electron-withdrawing group, rendered the molecule positively charged and thus contributed to its cytotoxic effect (Ramadan et al., 2019). Inspired by the findings discussed above, researchers used rational chemical approaches to design and then synthesize monastrol analogues as anticancer agents. Based on the available data on SARs of different THPMs, lead optimization through chemical modification of piperastrol and monastrol structures was used in this study (Prokopcová et al., 2010, de Fátima et al., 2015, de Souza et al., 2017, Liu et al., 2019). A series of ester and acid analogues with lipophilic moiety groups such as alkyl or phenyl groups was created. Furthermore, analogues with an unsubstituted position 5 of the THPM ring were designed.
3.2 Chemistry
In this research, numerous THPM analogs were synthesized through one-pot Biginelli reaction similar to the procedure reported in the previous study (Ahangarzadeh et al., 2022). The proper aldehydes, urea, and various α-ketoacids/β-ketoesters were refluxed in the presence of absolute ethanol as a solvent and Co(HSO4)2 as a catalyst to yield the titled analogs (4a-p). The procedure for titled analogues is shown in Scheme 1. All the titled analogs were established using FT-IR, 1H NMR, 13C NMR, and mass spectra, and elemental analysis was also applied to determine their purity. The FT-IR spectra of compounds (4a-p) identified NHs bonds in 3459–3230 cm−1 and 3271–3115 cm−1 zones. The C = O bond of the acetyl group was detected in the 1721–1660 cm−1 band. Furthermore, the C = C bonds of the aromatic ring were matched in 1633–1514 cm−1 and 1481–1443 cm−1 bands. The compounds' 1H NMR spectra revealed two peaks at 9.46–7.08 and 8.97–6.89 ppm corresponding to ureide N–H bonds, a beak at 13.60–13.13.28 ppm corresponding to COOH bond, and a peak at 5.83–4.64 ppm corresponding to the C4 proton of the THPM ring.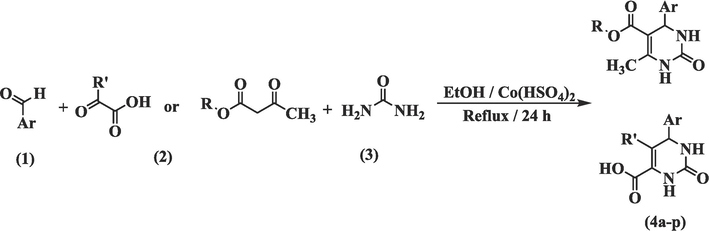
Synthesis procedure of THPM compounds (4a-p).
3.3 Biological assay
3.3.1 In vitro cytotoxic activity and structure-activity relationship
Pyrimidine is commonly used in medicinal chemistry due to its presence in hereditary molecules. Halogenated pyrimidine derivatives were the first substances tested for biological activity (Mohana Roopan and Sompalle 2016). A number of derivatives can be obtained by substituting the aryl ring, derivatizing pyrimidine nitrogen, and substituting carbon at positions 2, 4, 5, and 6 (Xu et al., 2020). A growing body of evidence demonstrates that pyrimidine-based anticancer drugs can affect cancerous cell lines through multiple mechanisms, including cytotoxicity induction, apoptosis, and cell cycle arrest (Mahapatra et al., 2021). In this study, synthesized compounds based on THPM scaffolds were evaluated against the AGS (human gastric adenocarcinoma) cell line using the MTT assay technique, and dose–response charts were plotted based on the survival rate of cells at 24 h and 48 h after exposure to different concentrations. The cytotoxicity results are summarized in Table 1.
Compound
R
R'
Ar
IC50 (µM)
4a
–
CH3
376.68 ± 4.88
4b
–
CH3
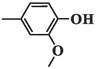
412.92 ± 5.03
4c
–
Br

69.60 ± 5.08
4d
–
Br
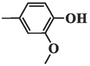
73.80 ± 5.20
4e
–
CH3
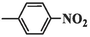
90.39 ± 5.97
4f
–
CH3
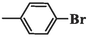
87.23 ± 6.78
4 g
–
CH3
357.59 ± 5.15
4 h
–
CH3
427.29 ± 6.35
4i
–
CH3
86.00 ± 5.72
4j
–
CH3
245.21 ± 4.58
4 k
–
H
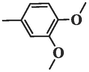
213.75 ± 5.28
4 l
–
H
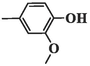
102.56 ± 6.58
4 m

–
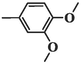
70.08 ± 4.99
4n

–
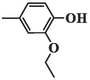
283.94 ± 8.39
4o

–
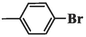
292.08 ± 5.87
4p

–
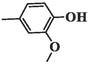
89.49 ± 7.46
Cisplatin
–
–
–
30.78 ± 4.68
All compounds showed moderate to good activity compared to Cisplatin as the standard. Findings demonstrated that all compounds affect the viability of AGS cells. However, among them, four compounds 4c, 4d, 4i, and 4 m (IC50 = 69.60 µM, 73.80 µM, 86.00 µM, and 70.30 µM, respectively) showed the most potent cytotoxicity activity against the AGS cell line (Fig. 2A-D) and were selected for further analysis. Moreover, the results revealed that upon increasing the concentration, all compounds showed a good increase in cytotoxic potency in a dose-dependent manner. Looking at the IC50 of the selected analogs, there was no remarkable variance in their cytotoxicity (Fig. 2A-D).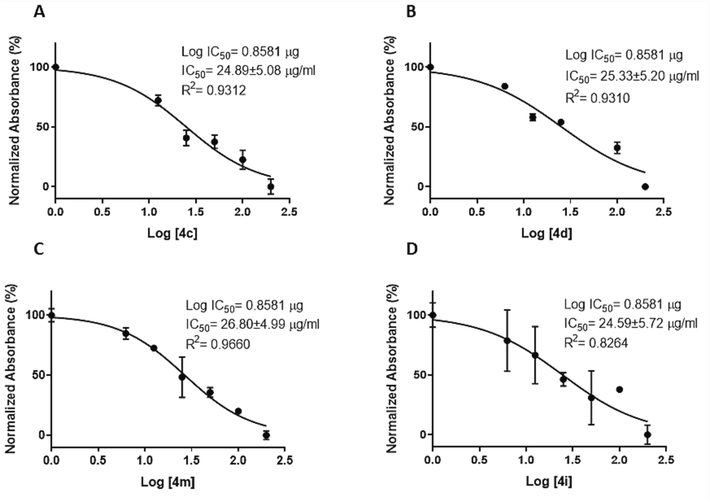
Dose response curve used to generate IC50 after 24 h for selected compounds, A) 4c, B) 4d, C) 4 m, D) 4i. Data are represented as the mean ± SD (n = 3). Note: In each of Figure’s A and B, one concentration was excluded from the IC50 calculation because it was outside the range.
Among the compounds, 4c with a 3,4-dimethoxy phenyl ring, Br, an oxygen atom, and a carboxylic acid at the C4, C5, C2, and C6 positions of the THPM ring, respectively, revealed the highest cytotoxicity effect (IC50 = 69.60 µM). Compounds 4d, 4i, and 4 m after 4c were the most potent compounds. The placing of a diver’s substitution at the C5 position of the THPM ring plays an important role in the activity (Kaur et al., 2017, El‐Hamamsy et al., 2020). The findings indicated that the presence of an electron-withdrawing group (EWG) compared to an electron-donating group (EDG) resulted in higher activity. Compound 4c (IC50 = 69.60 µM) with Br atom in C5 position on THPM ring exhibited higher activity than compound 4a (IC50 = 376.68 µM) having CH3 group and un-substituted compound 4 k (IC50 = 213.75 µM). This result was also observed for 4b, 4d, and 4 l. Compound 4d (IC50 = 73.80 µM) bearing Br atom in C5 position on THPM ring was more potent as compared to compound 4b (IC50 = 412.92 µM) having CH3 group and unsubstituted compound 4 l (IC50 = 102.56 µM). Thus, their cytotoxicity activity against the AGS cell line can be ordered as Br > H > CH3 (Table 1).
Among the mono-substituted compounds on the phenyl ring at the C4 position of the THPM ring, 4i (IC50 = 86.00 µM) with an ethoxy group in the para-phenyl ring was the most potent compound. Replacement of the ethoxy group of compound 4i by other substitutions such as NO2 (4e (IC50 = 90.39 µM)), Br (4f (IC50 = 87.23 µM)), OH (4 g (IC50 = 357.59 µM)), and F (4j (IC50 = 245.21 µM)) in para-phenyl ring attenuated cytotoxicity activity, which suggested that electronic and steric nature of substituted compounds plays an important role in the activity. Increased activity can be achieved via resonance and extended conjugation with the THPM ring. Cytotoxicity activity decreased in the order 4i > 4f > 4e > 4j > 4 g. Moreover, switching the methoxy group in the meta-phenyl ring of compound 4b (IC50 = 412.92 µM) with the ethoxy group in compound 4 h (IC50 = 427.29 µM) showed comparable activity. It seems that increased chain length in the meta-phenyl ring does not play a vital role in the cytotoxicity activity. However, compound 4p having methoxy group (IC50 = 89.49 µM) exhibited approximately 3.5 folds more cytotoxicity activity than 4n having an ethoxy group (IC50 = 283.94 µM). This contradiction might be due to the presence of bulk and a lipophilic group of benzyl ring in the ester moiety at the C5 position of the THPM ring that led to conformation change and its ability to penetrate the cell membrane. Additionally, exchanging the methoxy group in the para-phenyl ring of compound 4 m (IC50 = 70.08 µM) with the hydroxyl group in compound 4p (IC50 = 89.49 µM) slightly decreased activity. The presence of the hydroxyl group may have either intramolecular hydrogen bonding or a reduction electron-donating and lipophilic nature.
The comparison of IC50 values of the ester-substituted compounds with acid-substituted compounds showed that compounds 4 m, 4n, and 4p were more active than compounds 4a, 4 h, and 4b. All of the analogs tested demonstrated less potent cytotoxicity activity against the AGS cell line than Cisplatin. Taken together, our results align with previous studies suggesting that derivatives of THPM cause cytotoxicity in cancer cell lines, which is attributed to their structures.
3.3.2 Apoptotic assay
Apoptosis, or programmed cell death, is important in a variety of activities, including immune system development and function, hormone-dependent atrophy, embryonic development, and chemical-induced cell death (Elmore 2007). Because of the therapeutic potential of modulating cell death in anti-cancer research, it was examined whether the selected compounds induced apoptosis. As shown in Fig. 3, in comparison to the negative control, all selected compounds caused apoptosis under the experimental conditions (Fig. 3B-E). In the detailed inspection of the results, it was observed that after 24 h of treatment with selected compounds 4d, 4i, and 4 m, most of the cells were in the early stage of apoptosis (91.7 %, 90.6 %, and 81.3 %, respectively) (Fig. 3B-E, lower right quadrant). For 4c, most of the cells were in the late phase of apoptosis (55.2 %) (Fig. 3B-E, upper right quadrant). The observed results imply that all compounds can induce apoptosis in AGS cells. However, compound 4c reached the late phase of apoptosis earlier than the other selected compounds. Only a small number of cells underwent necrosis (∼0–0.19 %, Fig. 3B-E, upper left quadrant). In general, the analysis of the results exhibited that there is no observed remarkable variance among the effects of these compounds at IC50 concentration. However, their cytotoxic effects were remarkable compared to those of the negative control group (Fig. 3B-E vs. Fig. 3A). Taken together, the flow cytometry analysis showed that all selected compounds could effectively induce apoptosis in AGS cells. These findings correlate with previous reports showing that THPM derivatives induce apoptosis (Sepehri et al., 2015, Albratty and Alhazmi 2022, Milović et al., 2022), which can be executed by different mechanisms (Reed 2000, Elmore 2007). For instance, Shao et al. showed that pyrimidine derivatives induce apoptosis through caspase-3 activation, and decrease the anti-apoptotic protein Mcl-1 (Shao et al., 2013). In addition, pyrimidine derivatives exhibit anti-cancer activity by inhibiting various targets such as the tyrosine kinase of the Epidermal Growth Factor Receptors, Janus Kinases, Mitotic Checkpoint Protein Kinases (Mps1) (Kilic-Kurt et al., 2018).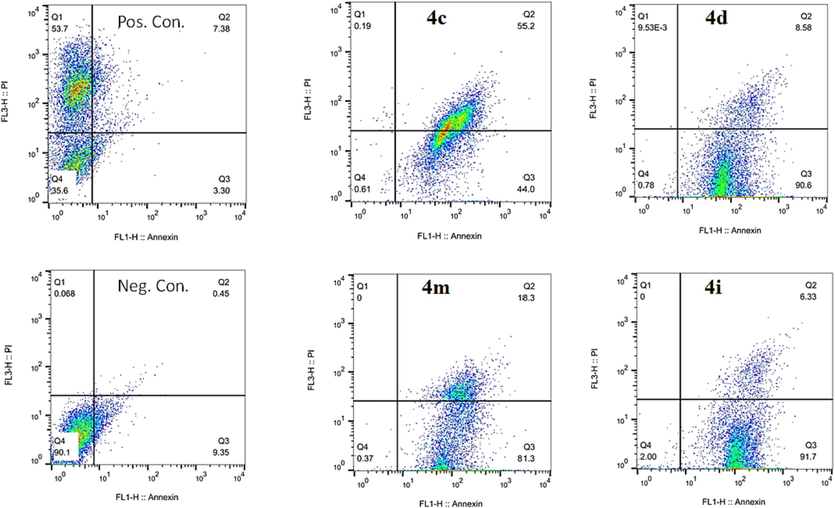
Flow cytometry histograms of AGS cells treated with selected compounds, including 4c, 4d, 4i, and 4 m for 24 h versus positive and negative control.
3.3.3 Caspase-3/7 activity
Caspases are a family of cysteine proteases that play a pivotal role in cell regulatory networks as they are responsible for controlling inflammation and apoptosis (McIlwain et al., 2013). Caspase-3 and caspase-7 are two of the most important members of this family, as they cleave proteins that are involved in programmed cell death events (Riedl and Shi 2004, Walsh et al., 2008). It is well established that currently, available pyrimidine-based anti-cancer agents function by inducing the apoptotic cascade (Sepehri et al., 2015, Sepehri et al., 2018, Razzaghi-Asl et al., 2019, Tylińska et al., 2021, Milović et al., 2022). To determine how the selected compounds could effectively activate effector caspases including caspases-3/7, used ELISA to assess their activity. In general, the results showed that there was a significant difference in mean caspase-3/7 activities (F (4, 5) = 4.5e + 12, P < 0.0001) between selected compounds (Fig. 4). However, the results revealed that compound 4c with a micromolar range of IC50 (69.6 µM) induced the highest caspase-3/7 activation (about 10.30-fold) compared to the negative control. Compounds 4d and 4i increased caspase-3/7 activity by 4.92 and 4.46 folds, respectively. Last, there is compound 4 m, which increased caspase-3/7 activity by about 2.61 folds. Detailed inspection of selected compounds showed that 4c activates caspases-3/7 more than the other complexes (Fig. 4), which may be attributed to the substituents on C4 and C5 positions of the THPM scaffold. A previous study by Mohamed Ahmed et al. showed that synthetic compounds based on the THPM scaffold increased caspase-3/7 activity (Ahmed et al., 2019). In addition, Alblewi et al. showed that the antitumor activity of pyrimidine-based chromenes is achieved by caspase-3/7 activity and cell cycle arrest (Alblewi et al., 2019). Taken together, our findings align with these, although they are generally structurally different. It is worth noting that the results of caspse-3/7 activity corresponded with the apoptotic and cytotoxic effect assays. Among the selected compounds, all assays indicate that the compound 4c is the most effective.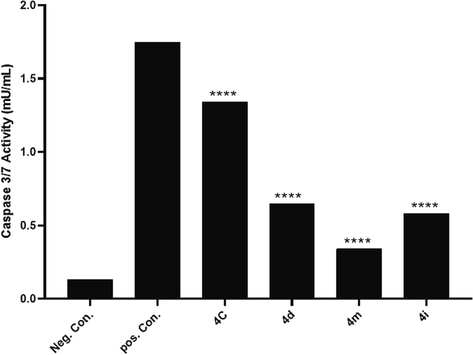
The caspase-3/7 activity resulting from treatment of AGS cells with selected compounds, including 4c, 4d, 4 m, and 4i, and Cisplatin as control positive in IC50 concentrations.
3.3.4 Real-time polymerase chain reaction for P53, CDKN2A, and caspase-8/9
To determine the underlying molecular mechanism of the selected compounds at the genome level, the mRNA expression levels of selected genes, including P53 (apoptosis pathway), CDKN2A (cell cycle-associated gene), and caspase-8/9 (extrinsic and intrinsic pathway-associated genes) were profiled. It is widely accepted that P53 is crucial for controlling cell division and death (Ozaki and Nakagawara 2011). However, the expression of the P53 gene does not necessarily mean that its protein level is increased following gene overexpression. As shown in Fig. 5, although all compounds increased the mRNA expression of P53 in comparison with NTC, this was not statistically significant (F (4,5) = 1.54, P = 0.32). However, there is an increasing trend for compound 4i. In summary, compounds 4d, 4c, 4i, and 4 m increased P53 expression by 2.70-, 1.76-, 6.12-, and 2.68-fold, respectively, relative to NTC. In addition, all selected compounds increased CDKN2A gene expression; however, this increase is not statistically significant (F (4,5) = 1.26, P = 0.39) (Fig. 5). It is determined by a closer look that 4i had the highest effect on CDKN2A gene expression, followed by 4d, 4c, and 4 m. CDKN2A is expressed in various cell types and tissues. The cell cycle is arrested by two proteins encoded by this gene: p16 and p14arf. Retinoblastoma proteins are activated by p16, which inhibits cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) and prevents G1-S progression (Foulkes et al., 1997). Cell cycle arrest is the primary mechanism by which many anti-cancer drugs inhibit tumor cell proliferation (Bai et al., 2017). P53 and CDKN2A expression is not supported by the results of apoptosis and cytotoxicity assays as well as with the caspase activity assay (4c≫ 4 m > 4d > 4i). These contradictory results may be attributed in part to the additional functions of the P53 gene (Feroz and Sheikh 2020). In addition, it is important to understand that although it is commonly believed that an increase in gene expression leads to an increase in protein expression (Jia et al., 2016), however, an increase in gene expression does not necessarily indicate an increase in its protein. Because premature and defective transcripts may eliminated by mRNA decay systems and not delivered to the translational machinery (He and Jacobson 2015). To clarify which apoptosis pathways (extrinsic or intrinsic) were activated by the selected compounds, the expression of caspase-8/9 was analyzed. Fig. 5 demonstrates that these compounds increased caspase-9 expression, whereas caspase-8 showed little change (causing a two-fold increase in caspase-9 expression compared to caspase-8). This suggests that the selected compounds induce apoptosis via the intrinsic pathway. Although the increase of caspase-9 over caspase-8 between compounds is not statistically significant.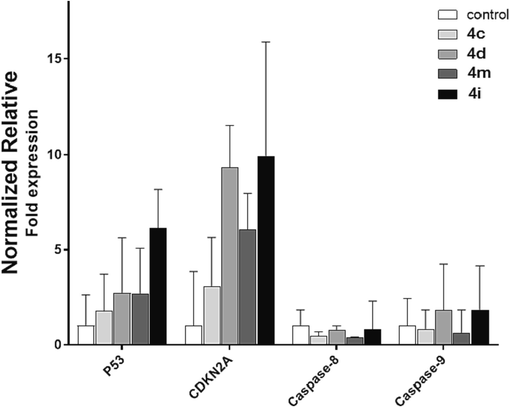
Expression levels of P53, CDKN2A, and Caspase-8/9 upon treatment with selected compounds, including 4c, 4d, 4 m, and 4i at IC50 concentrations. Relative expression (2−ΔCt) normalized to that of the reference gene (B2M) is shown as A panel. The mean-fold change in expression (2−ΔΔCt) for each gene relative to the NTC cells is represented. All data are presented as mean ± SD.
3.4 Molecular modelling studies
3.4.1 Molecular docking study
The kinesin family is a critical family of motor proteins that are required for mitosis and has been identified as a good target for chemotherapeutic intervention (Myers and Collins 2016). Kinesins are in charge of a variety of important functions within the cell. In general, these proteins bind to microtubules and use the energy released by ATP hydrolysis to complete their functions. Mitotic kinesin, a subgroup of kinesin, is expressed during mitotic phase cell division to establish the mitotic spindle, among other things (Bergnes et al., 2005). The mitotic kinesin family contains more than ten members, each of which plays a distinct or overlapping role in mitosis. One of the proteins in the kinesin superclass is Eg5, a member of the kinesin-5 family, which plays a vital role in the creation and maintenance of the bipolar spindle and is overexpressed in almost all tumors and proliferating tissues (Klein et al., 2007) including leukemia as well as solid tumors such as lung, bladder, ovarian, breast, and pancreatic cancers. Whereas there is no Eg5 in non-tumor tissues. Thus, this enzyme is recognized as an attractive target for cancer therapy (El-Hamamsy et al., 2020). Eg5 is a homotetrameric protein (about 320 amino acids). It is composed of two identical light chains and two identical heavy chains. The heavy chain contains a C-terminal tail domain, an α-helical coiled coil stalk domain, and a N-terminal motor domain that binds and hydrolyzes ATP (El-Nassan 2013). The inhibition of Eg5 leads to mitotic cell arrest, the activation of the spindle checkpoint, and subsequent cell death. The inhibition of Eg5 avoids mitotic spindle assembly, causing the creation of monopolar spindles called “monoasters” which this event causes to complete apoptosis (Sarli and Giannis 2006). In opposition, Eg5 inhibitors avoid only cells in mitosis, so normal cells are not affected. Thus, the Eg5 inhibitor might not have serious side effects related to conventional anti-mitotic compounds (Jackson et al., 2007). Lately, numerous inhibitors of Eg5 have arrived in pre-clinical and clinical trials as anti-cancer drugs and have exposed filling results (Nyamaa et al., 2014). Inhibitors of Eg5 and other mitotic kinesins are plausible anti-cancer drugs now under development and testing. They work by disrupting the mitotic spindle, arresting cancer cells in mitosis, and thus triggering apoptosis (Maliga and Mitchison 2006). Monastrol having THPM scaffold was recognized as the first particular allosteric inhibitor of Eg5 in 1999, which it binds to the motor domain of enzyme (Jiang et al., 2021). Monastrol reversibly inhibits microtubule gliding by Eg5 and causes spindle collapse in cells. Moreover, evidence-based studies suggest that monastrol does not struggle for ADP and microtubule binding (Maliga et al., 2002, DeBonis et al., 2003, Nikam and Jain 2022). As per the verified crystal structure, monastrol bind at allosteric position at Eg5 pocket, located 12 Å away from nucleotide-binding site and undergoes structural changes in neck linker and loop 5, situated in a hydrophobic pocket between loop 5 and helix α2 and helix α3 in Eg5 domain (Cochran and Gilbert 2005). Upon binding to loop 5, monastrol locked the loop into ADP bound-like conformation, slowed down the release of ADP and thus inhibited the ATP turnover (Maliga and Mitchison 2006). Residues Asp130 and Leu214 which are located in loop L5 of Eg5 were found to be the key residues for inhibitor resistance. On the other hand, mutations at five points in the loop L5 region of Eg5 (Glu116, Arg119, Ala218, and Tyr211) abolished the inhibitory activity of monastrol (El-Nassan 2013). Moreover, based on previous reports Glu116, Gly117, Gly118, Trp137, Arg119, Arg221, Gly217, and Arg221 (Kaan et al., 2010, El-Hamamsy et al., 2020, Bodun et al., 2023) are main amino acids.
To know the compound-enzyme interactions, molecular docking calculations were carried out by AutoDock 4.2. THPMs inhibit the allosteric site in the N‐terminal motor domain of Eg5. The inhibitors interact with this allosteric area, which possesses hydrophobic pockets (El-Hamamsy et al., 2020). In this research, the crystal structure of the human Eg5 motor domain bound to Mg-ADP and monastrol (PDB ID code 1X88) was selected. First, the docking protocol was valid after re-docking Eg5. Afterward, the RMSD was calculated and showed an acceptable value inside the cut-off of 1.65 Å (<2.0 Å). The superimpose of re-docked monastrol and co‐crystallized monastrol demonstrated that they bind to the same allosteric pocket as revealed in Fig. 6.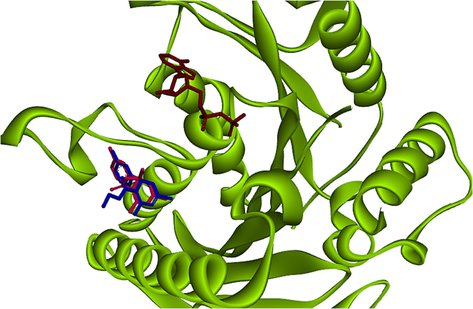
Binding poses of original (Blue stick) and docked (Red stick) cognate ligand in the active site of Eg5 enzyme in presence of adenosine diphosphate (ADP) (Brown) (RMSD: 1.65 Å, 1X88, co-crystallographic ligand: Monastrol compound).
The investigated binding modes of monastrol in the allosteric site of the Eg5 active site extracted main amino acids that interacted with them. Based on the docking studies, the monastrol formed three hydrogen bonds: the hydroxyl group of ligand with the oxygen of the carbonyl group in Glu118, the NH of THPM ring with the oxygen of the carbonyl group in Glu116, and the oxygen of carbonyl group of the monastrol with the hydroxyl group in Tyr211. Monastrol (co-crystal ligand), cognate of the enzyme, made two hydrogen bonds: the NH of THPM ring with the oxygen of carbonyl group in Glu116 and the hydroxy group of ligand with the oxygen of carbonyl group in Glu118.
The molecular docking of the synthesized compounds was performed due to recognizing the best conformation in Eg5 active site and compared with monastrol. The most active analogue, 4c, formed a hydrogen bond through NH and OH of acid moieties of THPM ring with the carbonyl group of the Glu116 binding pocket (Abdel-Aziz et al., 2013, El-Hamamsy et al., 2020, Aremu et al., 2021). In addition, between the aromatic ring and Br atom were formed hydrophobic interactions with the main hydrophobic amino acids as shown in Fig. 7. The re-docked monastrol, compared to compound 4c showed two extra hydrogen bonds while it was similar to monastrol as the co-crystal.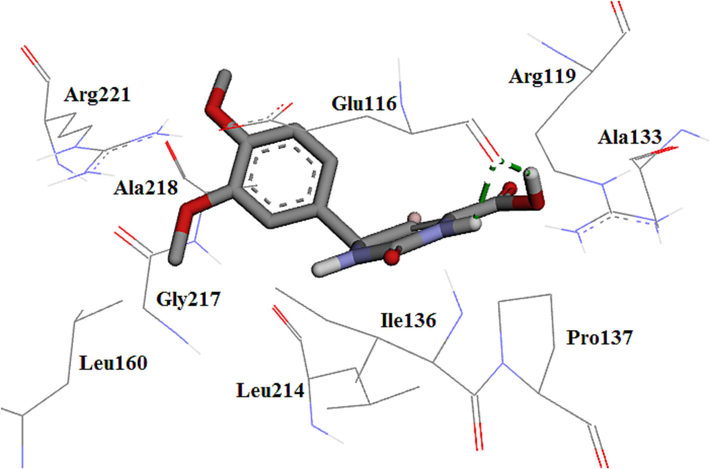
The 3D plot of the conformation of analogue 4c in Eg5 active site.
The high potency of compound 4 m can be attributed to the hydrogen bond made via the carbonyl group of the near amino acids Glu116 (González-Hernández et al., 2019, Tawfik et al., 2019). Hydrophobic interactions are also shown in Fig. 8. Compound 4 m formed a hydrogen bond similar to the monastrol as co-crystal. The decreasing activity of 4 m compared to 4c could lead to the removal of the hydrogen bond of acid moiety in compound 4 m with Glu116 residue. Thus, it seems that the presence of hydrogen bonds with acid plays an important role in the activity.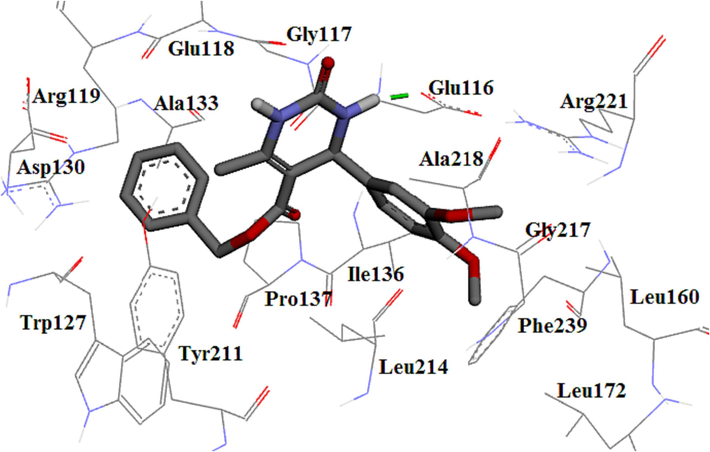
The 3D plot of the binding mode of 4 m in Eg5 active site.
The compound 4d displayed weaker activity than 4c because of the replacement of methoxy with hydroxyl at the para position of the phenyl ring at C4, and consequently missing the hydrophobic interaction with the key amino acids. It displayed three hydrogen bonds with Glu116, Trp127, and Leu214. Compound 4d formed three hydrogen bonds similar to the re-docked monastrol; however, types of two amino acids were different. Compound 4d made a hydrogen bond similar to the monastrol as co-crystal. Contrasting compound 4c, Glu116 formed a hydrogen bond with only NH moiety at position 3 and Leu214 formed a hydrogen bond with the acid moiety of compound 4d. The hydroxyl group at the para position of the phenyl ring also showed a hydrogen bond with Trp127. This decrease in activity could be due to a lack of hydrogen bond acid moiety with Glu116 and the formation of extra hydrogen bonds with the hydroxyl group. Hydrophobic interactions are also shown in Fig. 9.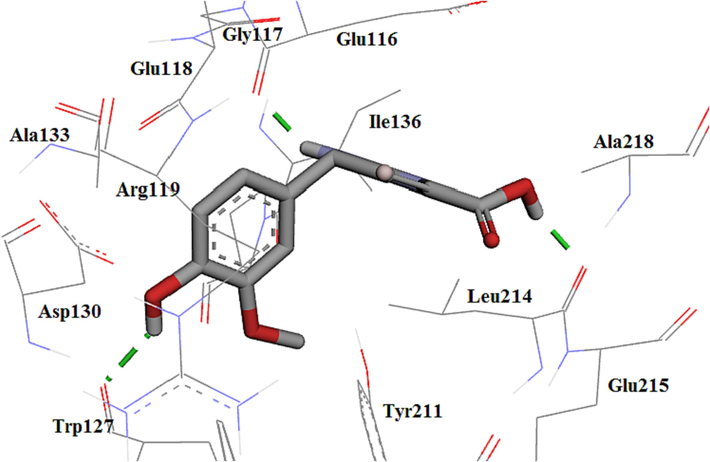
The 3D plot conformation of 4d in the Eg5 active site.
Molecule 4i displayed weaker activity than previous compounds because of the presence of the mono-substituted phenyl ring at the C4 position and the removal of hydrophobic interactions with the main amino acids compared to compounds bearing di-substituted phenyl ring. It displayed two hydrogen bonds with Glu116 and Tyr211 (Fig. 10). The re-docked monastrol showed an extra hydrogen bond compared to compound 4i whereas monastrol as co-crystal made a hydrogen bond with Glu116.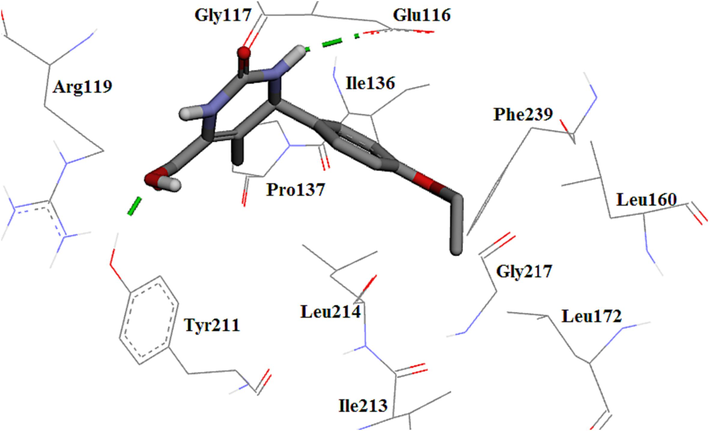
The 3D plot of the binding mode of 4i in the Eg5 active site.
According to the docking calculations, molecules and monastrol made hydrogen bond with Glu116 except compound 4 k. Molecules 4a, 4 h, 4i, 4 k, 4n, and 4p showed a hydrogen bond with Tyr211. Furthermore, 4b, 4d, 4f, 4 g, 4j, 4 l, and 4n formed a hydrogen bond with Leu214. Compounds 4b, 4d, 4 g, 4 k, and 4 l formed a hydrogen bond with Trp127. Among molecules, molecule 4 h displayed π-π interaction. Non-molecules did not demonstrate cation-π interaction. Monastrol did not display these interactions. Based on cytotoxicity results, it seems that π-π and cation-π interactions were not important for the activity.
Reports showed that the key amino acids of the active site are Glu116, Tyr211, Gly117, Arg221, Leu214, Arg119, and Ala218 (Prokopcová et al., 2010, González-Hernández et al., 2019, Tawfik et al., 2019, Safari et al., 2020, Aremu et al., 2021, Asham et al., 2021). Generally, the successful docking investigation has shown that practically all of the derivatives can interact successfully with the key residues in the Eg5 enzyme active site. Ring aromatic, hydrophobicity, and hydrogen bond donors are pharmacophore features that have been identified as important agents in the compound-Eg5 interaction. As a result, a suitable enzyme substrate contains an aromatic ring system, a hydrophobic group, and hydrogen bond acceptors.
3.4.2 Molecular dynamics simulations study
Molecular dynamics (MD) simulations have a significant impact on protein flexibility analysis. It is commonly used to recognize the interaction of compound-enzyme complexes. MD simulations will typically provide a comprehensive analysis of the pattern of conformational configurations of biomolecules, their strengths, characteristics, and interactions with molecules in a certain environment. The trajectory file will be the result of the simulation research, which will take some time (ns) (Cai et al., 2021). The trajectory file represents the system's kinetic and thermodynamic characteristics. The MD simulations were also used to explore the stability and modification of the compound-enzyme complex structures produced by molecular docking. MD simulations lasting 150 ns were performed to investigate the interactions of the complex structures generated with Eg5 and 4c, 4 m, 4d, and monastrol.
3.4.2.1 Conformational stability
The transformations in the structural properties and steadiness of the docked complexes were characterized by the root mean square deviation (RMSD) calculation. Generally, satisfactory evidence for the stability of the docked complex is steady backbone atom RMSD and less fluctuation. Throughout the MD simulations, the molecules 4c, 4d, 4 m, and monastrol (Eg5 inhibitor and co-crystal ligand) achieved equilibrium and remained stable. This indicates that the docked complexes have attained a stable state and interact well with the residues (Fig. 11).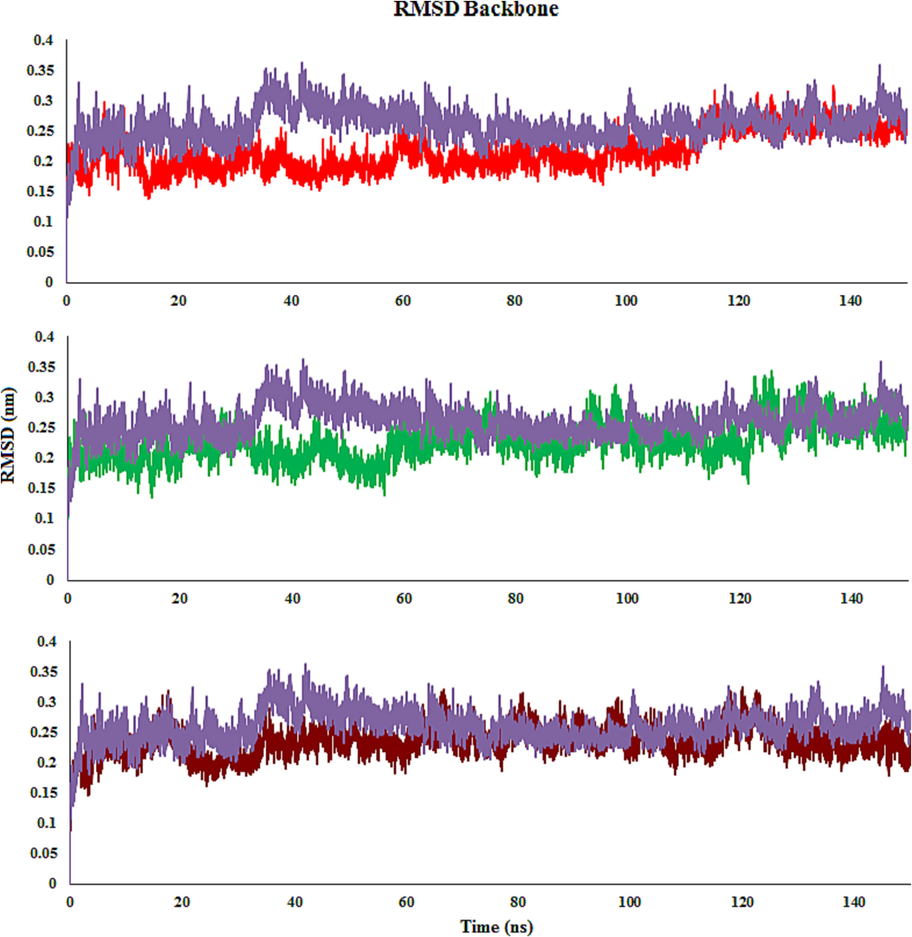
RMSD plot of Eg5 with molecules 4c (Red), 4 m (Green), 4d (Brown) and monastrol (co-crystal) (Purple) throughout the simulation time.
The RMSD graph was created by aligning the complex’s backbone structure during the MD. In the 4c complex, RMSD fluctuated during the first 20 ns before remaining stable for the next 118 ns. Then, a small increase is detected, and the function remains stable until 150 ns. The RMSD graph of 4 m demonstrates that the complex increased stability close to 5 ns and retained it until 35 ns. Minor fluctuations were noticed between 35 ns and 55 ns, while the complex remained stable until the end of the simulation time. The RMSD graph for 4d reveals that the complex steadied at about 20 ns and stayed steady until 150 ns. Though a minor rise in the RMSD graph was observed in comparison with compounds 4c and 4 m, while it was similar to monastrol. It detected was that the 4c-Eg5 complex is slightly more stable than 4d, 4 m, and monastrol, whereas the fluctuation of 4d is less than others in simulation time.
When it comes to the compound-Eg5 complexes, the average RMSD values for 4c- and 4 m-Eg5 were equal an average RMSD value of 0.22 nm. The RMSD values of 4d- and monastrol-Eg5 were 0.23 nm and 0.26 nm, respectively. 4d-Eg5 exhibited the highest RMSD values compared with 4c- and 4 m-Eg5. All screened derivatives indicated average RMSD values nearly like the monastrol (Fig. 11).
As can be seen in the RMSD ligand plots, after 4 ns of simulation time, compound 4c achieved equilibrium. It indicates that compound 4c has a stable binding with the Eg5 enzyme. This compound showed a sharp rise from 112 to 115 ns. RMSD value of 4c is lower than monastrol, with fewer fluctuations. As may be observed from Fig. 12, the RMSD values of 4 m fluctuate about 0.9 Å in the first 60 ns. Following that, there is a rapid spike, and the function remains steady with average RMSD values of 1.4 for roughly 50 ns. A dramatic decline is detected at around 110 ns, and the function subsequently remains stable with average RMSD values of 1 Å for approximately 40 ns. This suggests that the structure of compound 4 m changes significantly during the simulation (Fig. 12). Moreover, the RMSD values of 4 m were lower than monastrol. Compound 4d and monastrol exhibited comparable RMSD values and achieved equilibrium after 10 ns of simulation duration. According to RMSD ligand plots, compound 4c showed the lowest RMSD value among compounds, and it shows a stable binding with Eg5 enzyme. This observation correlates with assay results (Fig. 12).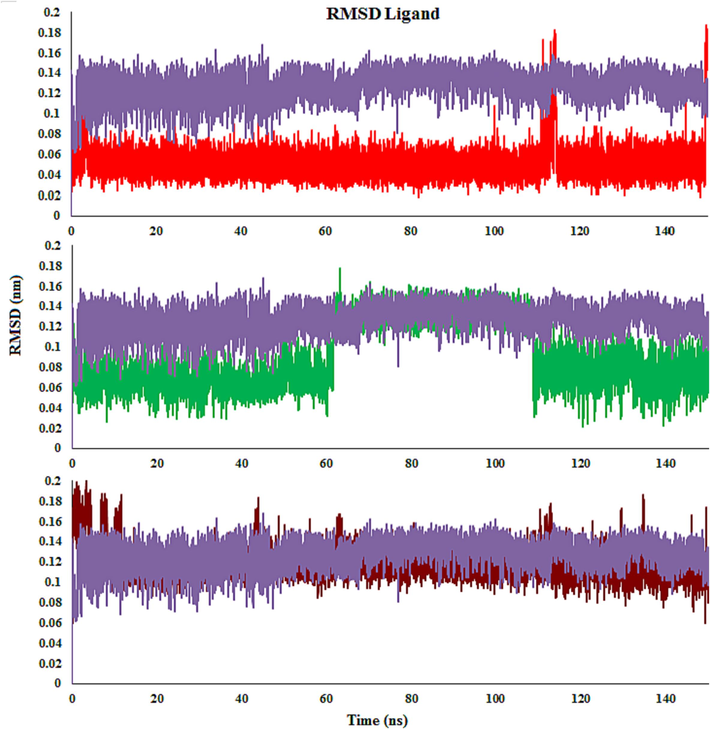
RMSDs of the compounds 4c (Red), 4 m (Green), 4d (Brown) and monastrol (Purple) in complex with Eg5 active site.
3.4.2.2 Residue flexibility analysis
To investigation the influence of compound binds to Eg5, root-mean-square fluctuations (RMSFs) of Cα atoms in Eg5 were computed using the equilibrated MD trajectories. As seen in Fig. 13, the patterns of atomic fluctuations of complexes are comparable, and the RMSF values of complexes exhibit minimal fluctuation when compared to the monastrol-Eg5 complex. The greatest fluctuations (>0.6 nm) are related to amino acids that are distant from the compound’s active site. In contrast, the Eg5 binding pocket residues (e.g., Ala133, Gly117, Arg221, and Glu116) exhibited rigid behavior. Ordinarily, the tails (N- and C-terminals) of enzyme change more than other inner areas. Secondary regions of proteins, such as β-strands and α-helices, are mostly more rigid and inflexible than the unstructured areas, and so they hesitate, not just similar loop-making portions of protein (Patel et al., 2022). To study the effect of compounds on the conformation of Eg5 protein, RMSF analysis for all the side-chain atoms of the protein was carried out, including 4c-, 4i-, 4 m, and monastrol-Eg5 complexes. It could be seen from Fig. 13 that the overall fluctuation trend of 4c- and 4i-Eg5 complexes was smaller than the 4 m-Eg5 complex, showing the 4c- and 4i-Eg5 complexes were more stable than the 4 m-Eg5 complex. In addition, there was the four residue regions marked by the orange, blue, and green boxes. These residue areas were not located in the active pocket or the adjacent regions. Residues with small fluctuations were the active regions the Eg5. The average RMSF in the active site residues region was 0.24 nm, 0.29 nm, and 0.24 nm in 4c-, 4d- and 4 m-Eg5 complexes, respectively. The fluctuations of those regions in the selected compounds-protein complexes were smaller than those in the monastrol-protein complex, indicating that the selected compounds-protein complexes had a more stable structure than the monastrol-protein complex in active regions. For example, the RMSF values of the residues Gly117, Glu116, Ala133, and Pro137 were 0.065, 0.092, 0.053, and 0.064 nm in the 4c-Eg5 complex, 0.082, 0.11, 0.058, and 0.073 nm in the 4d-Eg5 complex, and 0.069, 0.089, 0.061, and 0.063 nm in the 4 m-Eg5 complex, respectively. From the above data, it can be seen that the fluctuation of RMSF in the 4c-Eg5 complex was slightly reduced, which indicates that the active region has become more stable (Fig. 13). The RMSD and RMSF studies found the stability of the complex across the period studied.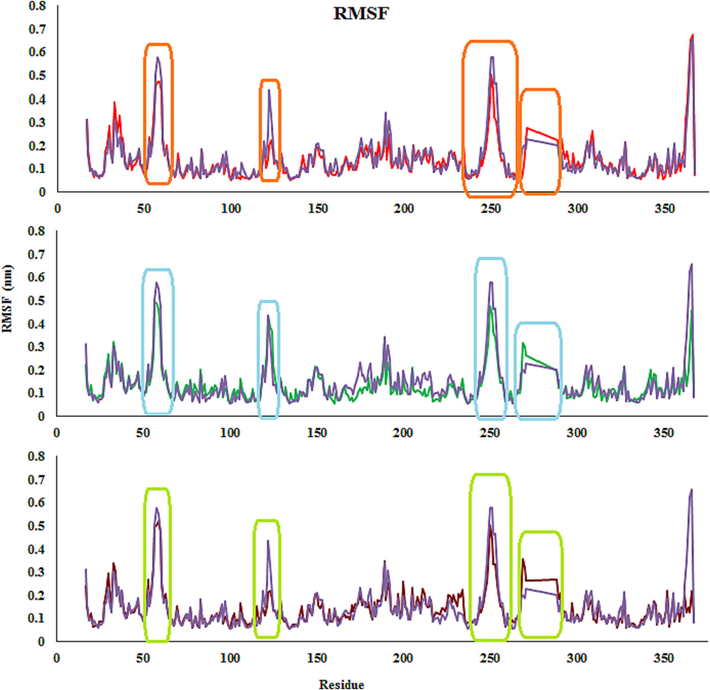
RMSFs of the compounds 4c (Red), 4 m (Green), 4d (Brown) and monastrol (Purple) in complex with Eg5 active site.
3.4.2.3 Compactness analysis
The radius of gyration (Rg) reports the compactness, conformational difference, and tertiary structural volume of the complexes. Overall, an extended structural type of enzyme has a greater Rg value than its spherical or compact counterpart. A higher Rg amount suggests greater expansion, while a lower one reveals a more compact structure. The Rg plots versus time are fairly smooth and nearly constant for complexes. The estimated values are quite homogeneous and remain consistent throughout the experiment. The Rg value of the 4c-Eg5 complex ranges from 2.04 to 2.08 nm and the variance is 0.02 nm. The average Rg values of 4c-, 4d- and 4 m-Eg5 complexes are ∼ 2.06 nm, 2.08 nm, and 2.07 nm, which indicates that the complexes are more compact compared with monastrol-Eg5 complex (∼2.1 nm). Compound 4c has a lower average Rg value and makes a more compact and stable complex with Eg5, owing to a stronger interaction between them than compounds 4d, 4 m, and monastrol (Fig. 14).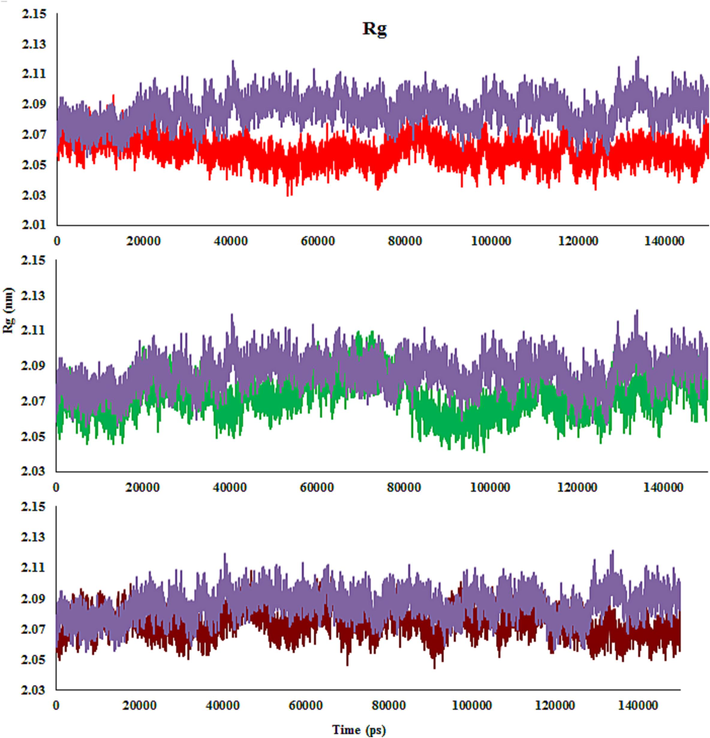
The enzyme backbone Rg throughout the MD, in the presence of 4c (Red), 4 m (Green), 4d (Brown) and monastrol (Purple) in complex with Eg5 active site.
3.4.2.4 Solvent accessible surface area (SASA)
Solvent-accessible surface area (SASA) is the surface area of a complex that interacts directly with solvent molecules. SASA growth indicates relative expansion. The SASA of the complexes of molecules 4c, 4d, 4 m, and monastrol ranges from 165 to 180 nm2, 168 to 181 nm2, 165 to 182 nm2, and 168 to 185 nm2, respectively (Fig. 15). In the first few ps, the SASA value declines abruptly and then progressively decreases for all complexes. A similar pattern of fluctuation is noted in all complexes, which is very small. Comparatively, to the end of 150 ns, the 4c-Eg5 complex has lower SASA values, implying more stability than the 4d-, 4 m-, and monastrol-Eg5 complexes.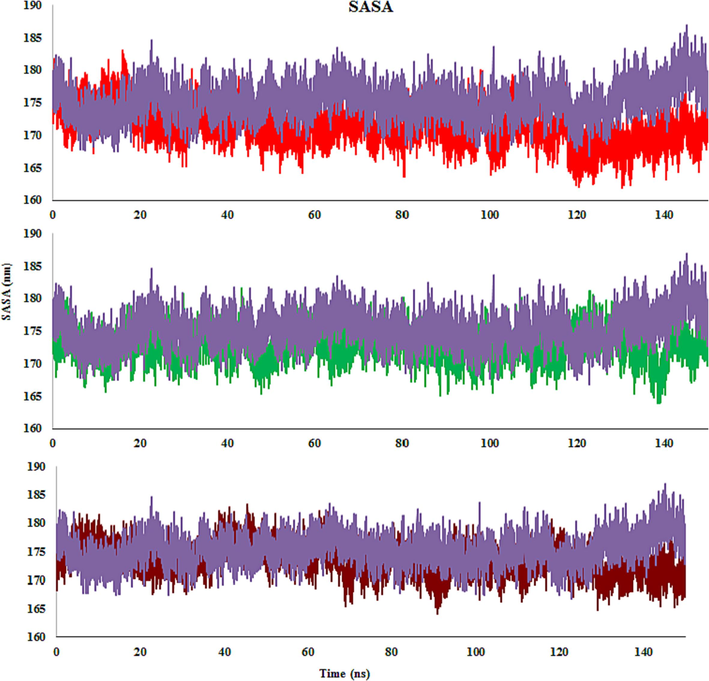
SASA plot of 4c (Red), 4 m (Green), 4d (Brown) and monastrol (Purple) in complex with Eg5 active site.
3.4.2.5 Hydrogen bond occupancy
Hydrogen bonding is a powerful factor in compound-enzyme complexes, dictating the flexibility and stability of the molecules. Electrostatic, Van der Waals, and hydrophobic interactions, as well as non-classical and classical hydrogen bonds, help to stabilize complexes. The number of hydrogen bonds in a complex affects the binding affinity degree, and the more hydrogen bonds there are, the higher the binding affinity level. During the MD simulations, the number of hydrogen bond-maintaining complexes remained constant (Fig. 16). In 4c-, 4d-, 4 m-, and monastrol-Eg5, the average number of hydrogen bonds was 5, 2, 5, and 2, respectively.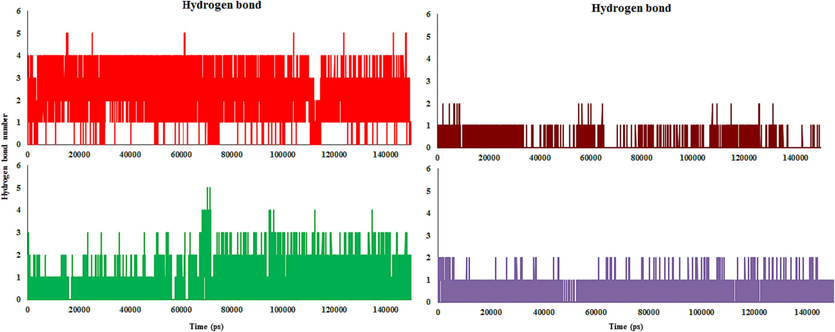
Hydrogen bonds formed number between compound and enzyme throughout simulation (complexes 4c (Red), 4 m (Green), 4d (Brown) and monastrol (Purple) in complex with Eg5 active site)).
4c- and 4 m-Eg5 complexes made the maximum of hydrogen bonds throughout the simulation. The hydrogen bond number was almost constant during the MD run. The majority of the complex conformations in 4c- and 4 m-Eg5 were found to have 2–4 hydrogen bonds. 4d- and monastrol-Eg5 complexes formed at about one hydrogen bond in conformations during the MD simulation. Throughout the 150 ns MD run, all of the complexes were stable, and the compounds maintained their interaction with the active site of Eg5.
The compounds placed in the active site contain bulky amino acids with hydrophobic interactions. Throughout the simulation, only a few hydrogen bonds were recognized and broke regularly because, during the simulation time, hydrophobic interactions were dominating. According to the findings, the important residues that stabilize and boost ligand efficiency in the active site are mostly owing to hydrophobic interactions. The Gly117, Ala118, Arg119, Ile136, Pro137, Arg221, Gly217, Ala218, and Leu214 residues make a hydrophobic main pocket and incorporate the compounds, therefore allowing the compound to investigate into the active site.
Glu116 and Arg221 residues formed hydrogen bonds with 4c about 100 % and 40 % of the throughout MD run. Glu116 residue exhibited a hydrogen bond with 4d at 13 %. Moreover, 4 m formed hydrogen bonds with Glu116, Arg221, Arg119, and Leu214 at about 35, 10, 15, and 12 %. Monastrol formed hydrogen bonds with Glu116 and Glu118 around 7 % and 5 %. For instance, for 4c at 0 ns, three hydrogen bonds were observed with Glu116, Pro137, and Arg119. Throughout 10 ns, 4c formed two hydrogen bonds with Glu116 and Arg221. The direction is altered in this compound. After 20–30 ns, the hydrogen bond was stable with the Glu116 amino acid, while it lacked a hydrogen bond with Arg221. Throughout 40 ns, the binding mode of the molecule in the binding cavity changed, and observed the hydrogen bond with only Arg221. Between 50 and 80 ns, the compound made a hydrogen bond only with Glu116. At 90 ns, observe two hydrogen bonds with Glu116 and Arg221 again. However, at 100 ns removed the hydrogen bond with Arg221 again. 110–150 ns, like others, hydrogen bonds of two amino acids Glu116 and Arg221 with the compound were conserved. Based on MD calculations, Glu116 residue showed a strong hydrogen bond with the molecule because it was in all of the MD simulations. Arg221 residue formed a moderate hydrogen bond with the compound because it was in almost all MD simulations (Fig. 17).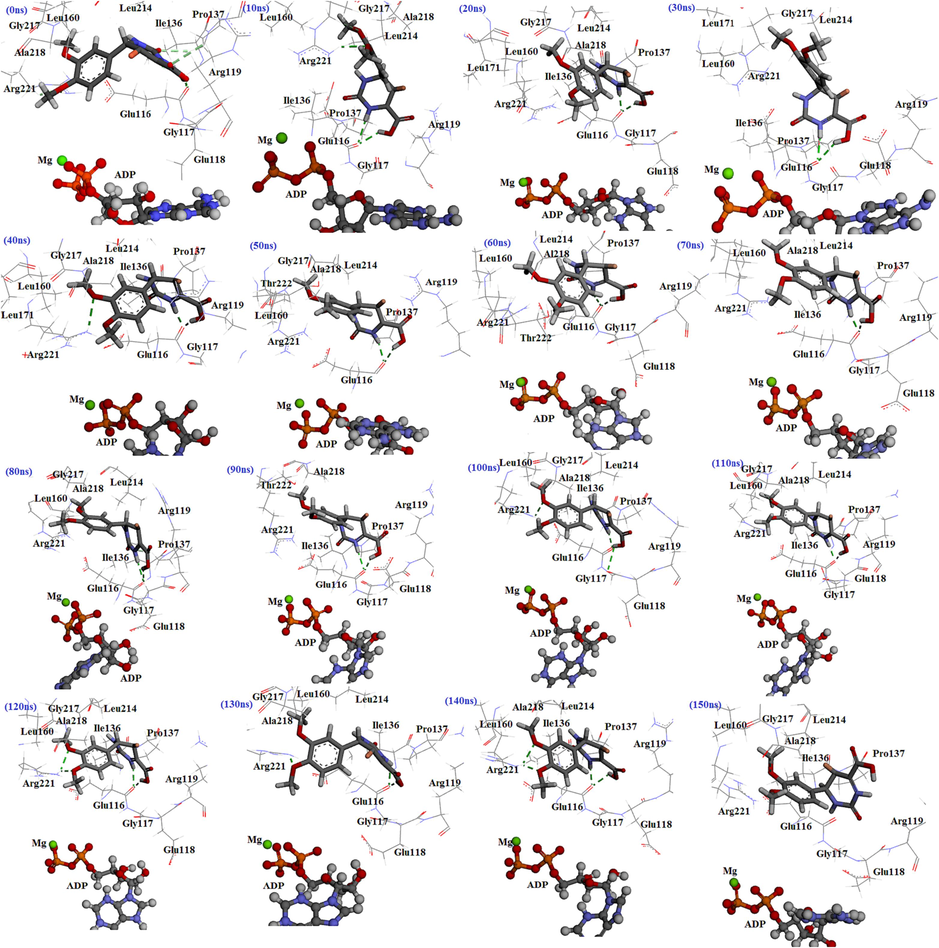
The interactions of molecule 4c with Eg5 active site at diverse times throughout the MD simulations.
4 Conclusion
An effective one-pot method has been established for the synthesis of derivatives with a THPM scaffold. The multicomponent reaction of aldehydes, α-ketoacid/β-ketoester, and urea in the presence of Co(HSO4)2 in absolute ethanol as solvent produced new THPM compounds. Then, their cytotoxicity activity against the AGS cell line was evaluated. Among them, the 5-bromo-6-(3,4-dimethoxyphenyl)-2-oxo-1,2,3,6-tetrahydropyrimidine-4-carboxylic acid (4c) was observed to be the most potent analog. It was discovered that 4b, 4i, and 4 m displayed noteworthy activity against the AGS cell line after 4c. Moreover, similar to cytotoxicity, although all compounds induced apoptosis and stimulated the activity of caspases-3/7, 4c was the best among the selected compounds. In contrast, gene expression studies showed that compound 4i stimulates the expression of P53 and CDNK2A genes more than other compounds. This discrepancy can be partially attributed to the extra functions of P53 and the degradation of non-functional RNAs by the mRNA decay system. Results confirmed that all compounds caused cell cycle arrest in G1 to M phase. Moreover, findings showed that the mechanism of apoptosis induction by THPM derivatives is intrinsic. Moreover, MD simulations of compounds 4c, 4d, and 4 m inside Eg5 showed that hydrogen bonds played a key role in the stability of the selected compounds in the Eg5 active site. Addition, to the hydrogen bond, it also was detected that hydrophobic interactions were also a dominant factor in the binding process of the molecules to the Eg5 active site. Moreover, compound 4c showed the most stability in the Eg5 active site among compounds during MD simulations.
Acknowledgements
This research was supported by Ardabil University of Medical Sciences. Funding Number is IR.ARUMS.REC.1401.185.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity. Eur. J. Med. Chem.. 2013;69:427-438.
- [CrossRef] [Google Scholar]
- Design, synthesis, and in silico studies of tetrahydropyrimidine analogs as urease enzyme inhibitors. Arch. Pharm. 2022:e2200158.
- [Google Scholar]
- Design, synthesis, molecular modelling, and biological evaluation of novel substituted pyrimidine derivatives as potential anticancer agents for hepatocellular carcinoma. J. Enzyme Inhib. Med. Chem.. 2019;34:1110-1120.
- [Google Scholar]
- Design and synthesis of novel heterocyclic-based 4 H-benzo [h] chromene moieties: Targeting antitumor caspase 3/7 activities and cell cycle analysis. Molecules (Basel, Switzerland). 2019;24:1060.
- [Google Scholar]
- Novel pyridine and pyrimidine derivatives as promising anticancer agents: A review. Arab. J. Chem.. 2022;15:103846
- [CrossRef] [Google Scholar]
- Synthesis, molecular docking and anticancer activity of 5,5'-(phenylmethylene)bis(6-amino-2-thiouracil) derivatives. Phosphorus Sulfur Silicon Relat. Elem.. 2021;196:920-928.
- [CrossRef] [Google Scholar]
- Design, synthesis, and biological screening for cytotoxic activity of monastrol analogues. Polycycl. Aromat. Compd.. 2021;1–15
- [CrossRef] [Google Scholar]
- Characterization and evaluation of cytotoxic and apoptotic effects of green synthesis of silver nanoparticles using Artemisia Ciniformis on human gastric adenocarcinoma. Mater. Today Commun.. 2020;24:101011
- [CrossRef] [Google Scholar]
- Synthesis and evaluation of cytotoxicity of 6-amino-4-aryl-2-thioxo-1, 2, 3, 4-tetrahydropyrimidine-5-carbonitriles. Russ. J. Bioorg. Chem.. 2016;42:316-322.
- [Google Scholar]
- Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med.. 2017;14:348-362.
- [CrossRef] [Google Scholar]
- Mitotic kinesins: prospects for antimitotic drug discovery. Curr. Top. Med. Chem.. 2005;5:127-145.
- [CrossRef] [Google Scholar]
- QSAR-based virtual screening of traditional Chinese medicine for the identification of mitotic kinesin Eg5 inhibitors. Comput. Biol. Chem.. 2023;104:107865
- [CrossRef] [Google Scholar]
- Synthesis of novel heterocyclic compounds containing pyrimidine nucleus using the Biginelli reaction: Antiproliferative activity and docking studies. J. Heterocycl. Chem.. 2020;57:2615-2627.
- [CrossRef] [Google Scholar]
- Synthesis, evaluation, molecular dynamics simulation and targets identification of novel pyrazole-containing imide derivatives. J. Biomol. Struct. Dyn.. 2021;39:2176-2188.
- [Google Scholar]
- ATPase mechanism of Eg5 in the absence of microtubules: insight into microtubule activation and allosteric inhibition by monastrol. Biochemistry. 2005;44:16633-16648.
- [Google Scholar]
- A mini-review on Biginelli adducts with notable pharmacological properties. J. Adv. Res.. 2015;6:363-373.
- [Google Scholar]
- Synthesis and photophysical study of new fluorescent proton transfer dihydropyrimidinone hybrids as potential candidates for molecular probes. New J. Chem.. 2017;41:15305-15311.
- [Google Scholar]
- Interaction of the mitotic inhibitor monastrol with human kinesin Eg5. Biochemistry. 2003;42:338-349.
- [Google Scholar]
- Design, synthesis, and molecular docking study of new monastrol analogues as kinesin spindle protein inhibitors. Arch. Pharm.. 2020;353(8):e2000060.
- [CrossRef] [Google Scholar]
- Apoptosis: a review of programmed cell death. Toxicol. Pathol.. 2007;35:495-516.
- [CrossRef] [Google Scholar]
- Advances in the discovery of kinesin spindle protein (Eg5) inhibitors as antitumor agents. Eur. J. Med. Chem.. 2013;62:614-631.
- [Google Scholar]
- Design, synthesis and biological evaluation of novel o-aminobenzamide derivatives as potential anti-gastric cancer agents in vitro and in vivo. Eur. J. Med. Chem.. 2022;227:113888
- [CrossRef] [Google Scholar]
- Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet.. 2020;21:49.
- [CrossRef] [Google Scholar]
- Synthesis and anticancer evaluation of novel indole based Arylsulfonylhydrazides against human breast cancer cells. ACS Omega. 2022;7:42036-42043.
- [CrossRef] [Google Scholar]
- Dihydropyrimidine-2-thiones as Eg5 inhibitors and L-type calcium channel blockers: potential antitumour dual agents. MedChemComm. 2019;10:1589-1598.
- [CrossRef] [Google Scholar]
- Nonsense-mediated mRNA decay: Degradation of defective transcripts is only part of the story. Annu. Rev. Genet.. 2015;49:339-366.
- [CrossRef] [Google Scholar]
- Targeted anti-mitotic therapies: can we improve on tubulin agents? Nature reviews. Cancer. 2007;7:107-117.
- [CrossRef] [Google Scholar]
- Discovery of the Biginelli hybrids as novel caspase-9 activators in apoptotic machines: Lipophilicity, molecular docking study, influence on angiogenesis gene and miR-21 expression levels. Bioorg. Chem.. 2019;86:569-582.
- [CrossRef] [Google Scholar]
- The well-accepted notion that gene amplification contributes to increased expression still remains, after all these years, a reasonable but unproven assumption. J. Carcinogen.. 2016;15
- [Google Scholar]
- Solvothermal synthesis of multiple dihydropyrimidinones at a time as inhibitors of Eg5. Molecules (Basel, Switzerland). 2021;26
- [CrossRef] [Google Scholar]
- Structural basis for inhibition of Eg5 by dihydropyrimidines: Stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J. Med. Chem.. 2010;53:5676-5683.
- [CrossRef] [Google Scholar]
- Rational design, synthesis, biological evaluation and molecular docking studies of chromone-pyrimidine derivatives as potent anti-cancer agents. J. Mol. Struct.. 2021;1239:130502
- [CrossRef] [Google Scholar]
- Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem.. 2017;132:108-134.
- [Google Scholar]
- Synthesis, biological assessment, and computational investigations of nifedipine and monastrol analogues as anti-leishmanial major and anti-microbial agents. Mol. Divers. 2022
- [CrossRef] [Google Scholar]
- Cytotoxic and apoptotic effects of novel pyrrolo[2,3-d]pyrimidine derivatives containing urea moieties on cancer cell lines. Anticancer Agents Med Chem.. 2018;18:1303-1312.
- [CrossRef] [Google Scholar]
- New chemical tools for investigating human mitotic kinesin Eg5. Bioorg. Med. Chem.. 2007;15:6474-6488.
- [CrossRef] [Google Scholar]
- Novel Biginelli dihydropyrimidines with potential anticancer activity: a parallel synthesis and CoMSIA study. Eur. J. Med. Chem.. 2009;44:4192-4198.
- [Google Scholar]
- Autophagy and multidrug resistance in cancer. Chin. J. Cancer. 2017;36:52.
- [CrossRef] [Google Scholar]
- Synthesis, characterization, and anticancer activities evaluation of compounds derived from 3, 4-dihydropyrimidin-2 (1 H)-one. Molecules. 2019;24:891.
- [Google Scholar]
- Isolation, synthesis and bioactivity evaluation of isoquinoline alkaloids from Corydalis hendersonii Hemsl. against gastric cancer in vitro and in vivo. Bioorg. Med. Chem.. 2022;60:116705
- [CrossRef] [Google Scholar]
- Pyrimidine: a review on anticancer activity with key emphasis on SAR. Future J. Pharma. Sci.. 2021;7:123.
- [Google Scholar]
- Evidence that monastrol is an allosteric inhibitor of the mitotic kinesin Eg5. Chem. Biol.. 2002;9:989-996.
- [Google Scholar]
- Small-molecule and mutational analysis of allosteric Eg5 inhibition by monastrol. BMC Chem. Biol.. 2006;6:1-9.
- [Google Scholar]
- Mechanisms of apoptotic phosphatidylserine exposure. Cell Res.. 2013;23:1247-1248.
- [CrossRef] [Google Scholar]
- Pyrrole as an important scaffold of anticancer drugs: recent advances. J. Pharm. Pharm. Sci.. 2022;25:24-40.
- [Google Scholar]
- Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol.. 2013;5:a008656
- [Google Scholar]
- Synthesis, characterization, and biological evaluation of tetrahydropyrimidines: Dual-activity and mechanism of action. Pharmaceutics. 2022;14:2254.
- [Google Scholar]
- Synthesis, characterization, and biological evaluation of tetrahydropyrimidines: Dual-activity and mechanism of action. Pharmaceutics. 2022;14
- [CrossRef] [Google Scholar]
- Design and synthesis of tetrahydropyrimidinone(thione)-triazole hybrid scaffolds and evaluation of their biological activities. Phosphorus Sulfur Silicon Relat. Elem.. 2021;196:1109-1116.
- [CrossRef] [Google Scholar]
- Synthetic chemistry of pyrimidines and fused pyrimidines: A review. Synth. Commun.. 2016;46:645-672.
- [Google Scholar]
- Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy. Future Med. Chem.. 2016;8:463-489.
- [CrossRef] [Google Scholar]
- Advances in the discovery of DHPMs as Eg5 inhibitors for the management of breast cancer and glioblastoma: A review. Results Chem. 2022:100718.
- [Google Scholar]
- Nyamaa, B., H. K. Kim, Y. Jeong, et al., 2014. Kinesin Spindle Protein Inhibition in Translational Research. Journal of Lipid and Atherosclerosis. 3, 63. doi: 10.12997/jla.2014.3.2.63.
- Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol.. 2013;5:a008672
- [Google Scholar]
- Repurposing of anticancer phytochemicals for identifying potential fusion inhibitor for SARS-CoV-2 using molecular docking and molecular dynamics (MD) simulations. J. Biomol. Struct. Dyn.. 2022;40:7744-7761.
- [CrossRef] [Google Scholar]
- Structure-activity relationships and molecular docking of novel dihydropyrimidine-based mitotic Eg5 inhibitors. ChemMedChem. 2010;5:1760-1769.
- [CrossRef] [Google Scholar]
- Synthesis, structure–activity relationship of novel substituted 4H-chromen-1, 2, 3, 4-tetrahydropyrimidine-5-carboxylates as potential anti-mycobacterial and anticancer agents. Bioorg. Med. Chem. Lett.. 2011;21:2855-2859.
- [Google Scholar]
- Cytotoxic and antimicrobial activities of some novel heterocycles employing 6-(1, 3-diphenyl-1 H-pyrazol-4-yl)-4-oxo-2-thioxo-1, 2, 3, 4-tetrahydropyrimidine-5-carbonitrile. Heterocycl. Commun.. 2019;25:107-115.
- [Google Scholar]
- Design, synthesis and evaluation of cytotoxic, antimicrobial, and anti-HIV-1 activities of new 1,2,3,4-tetrahydropyrimidine derivatives. Res. Pharma. Sci.. 2019;14:155-166.
- [CrossRef] [Google Scholar]
- Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol.. 2004;5:897-907.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation and molecular docking study of dihydropyrimidine derivatives as potential anticancer agents. J. Heterocycl. Chem.. 2020;57:1023-1033.
- [Google Scholar]
- Inhibitors of mitotic kinesins: Next-generation antimitotics. ChemMedChem. 2006;1:293-298.
- [CrossRef] [Google Scholar]
- Cysteine cathepsins as therapeutic targets in immune regulation and immune disorders. Biomedicines. 2023;11:476.
- [Google Scholar]
- Sepehri, S., H. P. Sanchez and A. Fassihi, 2015. Hantzsch-Type dihydropyridines and Biginelli-type tetra-hydropyrimidines: a review of their chemotherapeutic activities. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques. 18, 1-52. 10.18433/j3q01v.
- Hantzsch-type dihydropyridines and biginelli-type tetra-hydropyrimidines: a review of their chemotherapeutic activities. J. Pharm. Pharm. Sci.. 2015;18:1-52.
- [Google Scholar]
- Synthesis, biological evaluation, and molecular docking studies of novel 4-[4-Arylpyridin-1(4H)-yl]benzoic acid derivatives as anti-HIV-1 agents. Chem. Biodivers.. 2017;14
- [CrossRef] [Google Scholar]
- Design, synthesis, and anti-HIV-1 evaluation of a novel series of 1,2,3,4-tetrahydropyrimidine-5-carboxylic acid derivatives. Chem. Biodivers.. 2018;15:1700502.
- [CrossRef] [Google Scholar]
- Synthesis, structure–activity relationship and biological evaluation of 2,4,5-trisubstituted pyrimidine CDK inhibitors as potential anti-tumour agents. Eur. J. Med. Chem.. 2013;70:447-455.
- [CrossRef] [Google Scholar]
- Design, synthesis, and bioactivity of dihydropyrimidine derivatives as kinesin spindle protein inhibitors. Bioorg. Med. Chem.. 2019;27:115126
- [CrossRef] [Google Scholar]
- Novel pyrimidine derivatives as potential anticancer agents: Synthesis, biological evaluation and molecular docking study. Int. J. Mol. Sci.. 2021;22:3825.
- [Google Scholar]
- Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci.. 2008;105:12815-12819.
- [CrossRef] [Google Scholar]
- Pyrimidinone-peptoid hybrid molecules with distinct effects on molecular chaperone function and cell proliferation. Bioorg. Med. Chem.. 2008;16:3291-3301.
- [CrossRef] [Google Scholar]
- Discovery of novel 2, 4-disubstituted pyrimidines as Aurora kinase inhibitors. Bioorg. Med. Chem. Lett.. 2020;30:126885
- [Google Scholar]
- Anticancer activity of new substituted pyrimidines, their thioglycosides and thiazolopyrimidine derivatives. J. Appl. Pharma. Sci.. 2017;7:021-032.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105448.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




