

Translate this page into:
Identification and functional prediction of new triterpenoids from Alismatis Rhizoma using HPLC-HRMS and in-silico analysis
⁎Corresponding authors. 2262810416@qq.com (Xiao-rong Yan), yuanming@sicau.edu.cn (Ming Yuan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Alismatis Rhizoma (AR) is a crucial substance for discovering new triterpenoids to address hyperlipidemia and obesity. Currently, approximately 120 triterpenoids have been identified in AR, making the discovery of new triterpenoids increasingly challenging. Thus, based on the advantages of HPLC-HRMS, it was utilized to identify both reported and new triterpenoids, and then in-silico analysis was employed to predict the functions of these new triterpenoids. Twenty reported triterpenoids and four new triterpenoids (25-methoxy-16-oxo-11-anhydroalisol A, 25-methoxy-16-oxo-alisol A, 25-methoxy-16-oxo-alisol A 23-acetate, and 25-methoxy-16-oxo-alisol A 24-acetate) from AR were identified using HPLC-HRMS, and then KEGG analysis suggested four new triterpenoids might be activated the PPAR signaling pathway to relieve hyperlipemia. The results of PPI and molecular docking indicated PPARA might be the key targets for anti-hyperlipidemia of four new triterpenoids, of which 25-methoxy-16-oxo-11-anhydroalisol A has better binding activity with PPARA. Additionally, 25-methoxy-16-oxo-11-anhydroalisol A has more potential as a candidate drug because of its better absorption, distribution, metabolism, excretion, and lower toxicity. Hence, our findings proved there are new triterpenoids with anti-hyperlipidemia medicinal potential in AR, thereby guiding the direction of future works and reducing the consumption of time and financial resources.
Keywords
Alismatis Rhizoma
HPLC-HRMS
In-silico analysis
Molecular docking
ADMET prediction

- HPLC-HRMS
-
high performance liquid chromatography-high resolution mass spectrometry
- ADMET
-
absorption, distribution, metabolism, excretion, and toxicity
- AR
-
Alismatis Rhizoma. KEGG, Kyoto Encyclopedia of Genes and Genomes
- PPI
-
protein-protein interaction
- PPARA
-
peroxisome proliferator-activated receptor alpha
- EGFR
-
epidermal growth factor receptor
- EIC
-
extraction ion chromatogram
- OMe
-
methoxy
- OAc
-
acetoxy
Abbreviations
1 Introduction
In China, many plant products such as wolfberry, almond, and hawthorn have been recognized as raw materials for functional foods due to their long-standing edible and medicinal experience. Consequently, these plant products constitute good materials for discovering new bioactive compounds. Specifically, Alismatis Rhizoma (AR), which refers to the dried rhizome of Alisma plantago-aquatica Linn. and Alisma orientale (Sam.) Juzep, has been classified as raw material for functional foods for treating hyperlipidemia, obesity, and fatty liver in China (Feng et al., 2021; Wang et al., 2020; Yan et al., 2022). Remarkably, AR is the primary ingredient in approximately 220 functional foods that have received approval. Among these, 37 % are intended for the prevention of hypertension, and 34 % are designed to combat obesity (Fig. S1 and Table S1). AR is an essential material for discovering new bioactive compounds, and many scholars isolated and identified various triterpenoids for alleviating hyperlipidemia and obesity (Feng et al., 2021; Liu et al., 2020). Currently, approximately 120 triterpenoids have been identified in AR, making the discovery of new triterpenoids increasingly challenging (Wang et al., 2020).
The traditional research workflow for identifying new compounds is to first select suitable solvent and chromatographic column packing as mobile phase and separation medium respectively to isolate each compound monomer, then identify their structure and compare with the database to determine whether it is a new compound. However, the emergence of HPLC-HRMS has simplified this process (Aydoğan, 2020). HPLC-HRMS, which combines the high efficiency of HPLC in separating fractions and the high accuracy of HRMS in structural identification, is widely utilized for observing reported compounds and identifying new compounds (Atanasov et al., 2021). Notably, Jerman Klen et al. and Shi et al. used HPLC-HRMS to successfully identify three new phenolics in cruciferous vegetable-based dietary supplements and two new acylated glucosinolates in olive matrices, respectively (Jerman Klen et al., 2015; Shi et al., 2017). Moreover, HPLC-HRMS has also been employed in identifying new metabolites of microorganisms. For instance, Quinocinnolinomycins A-D, a new group of bioactive compounds, was discovered from bacteria using HPLC-HRMS (Kurita et al., 2015). Thus, HPLC-HRMS has great potential in the discovery of new bioactive compounds.
The cost of traditional new drug development strategies is as high as $1.8 billion because the attrition rate of candidate drugs is about 96 % (Paul et al., 2010). In-silico analysis tools have been developed and widely used for developing new drugs to reduce the attrition rate of candidate drugs. Several web tools, including SwissTargetPrediction, SuperPred, Similarity ensemble approach, and PharmMapper, have been created for the prediction of target proteins of bioactive compounds, enabling the exploration of their pharmacological activity (Daina et al., 2019; Gallo et al., 2022; Keiser et al., 2007; Wang et al., 2017). Furthermore, the molecular docking technique has been extensively utilized for the analysis of the affinity between bioactive compounds and target proteins (Pinzi and Rastelli, 2019). The potential development of bioactive compounds is heavily influenced by their ADMET properties. In response, ADMET predictive tools, such as PARDRIDGE, ADMETlab, and ProTox-II, have been created to quickly screen these compounds (Banerjee et al., 2018; Daina et al., 2017; Xiong et al., 2021). For example, Li et al. screened two dipeptidyl peptidase-IV inhibitory peptides by ADMET predictive tools and molecular docking (Li et al., 2023). Recently, in-silico analysis tools have emerged as powerful and cost-effective methods in the field of drug development due to their cost-time and cost-efficiency.
In this study, HPLC-HRMS was utilized to identify both reported and new triterpenoids, and the functions of these new triterpenoids were predicted with in-silico analysis. As a result, it proved that there are new triterpenoids with anti-hyperlipidemia medicinal potential in AR, thereby guiding future research endeavors and reducing the consumption of time and financial resources.
2 Materials and methods
2.1 Materials and chemicals
AR (A. plantago-aquatica L.) was collected from Yaan City, Sichuan Province, China in Jan. 2022. Acetonitrile was obtained from Merck Chemicals (Shanghai) Co., Ltd. Reference substances were purchased from Macklin Biochemical Co. (Shanghai, China).
2.2 Compounds analysis of the extract from AR
The dried AR (100 g) was extracted using a liquid to solid ratio of 30 mL/g at a temperature of 50 °C and ultrasonic power of 240 W for a duration of 3 h. After centrifugation at 4500 r/m for 20 min, the supernatant was collected and subjected to concentration and freeze-drying. Subsequently, the freeze-dried sample was dissolved in 90 % acetonitrile to obtain a concentration of 1 mg/mL for analysis with HPLC-HRMS (UltiMate 3000 HPLC and Q-Exactive Orbitrap mass spectrograph, Thermo Fisher Scientific Inc., USA).
HPLC-HRMS equipped with Waters ACQUITY BHE C18 (2.1 × 150 mm, 1.7 μm) column at 40 °C. The mobile phases were 0.1 % formic acid (A) and acetonitrile (B) at 0.3 mL/min with gradient elution as follows: 0–3 min, 90 % A; 3–7 min, 90 %-80 % A; 7–10, 80 %-50 % A; 10–15 min, 50 %-15 % A; 15–20 min, 15 %-0% A; 20–25 min; 0 %-90 % A. The mass spectrograph utilized an electrospray ionization (ESI) source in positive ion mode, with the detailed parameters specified in the published literature (Zhao et al., 2022).
2.3 Activity prediction of new compounds
To predict the targets of the new compounds, the SwissTargetPrediction, SuperPred, Similarity ensemble approach, and PharmMapper databases were consulted based on their structural formula, and then Metascape was applied for KEGG analysis (Daina et al., 2019; Gallo et al., 2022; Keiser et al., 2007; Wang et al., 2017; Zhou et al., 2019). According to the KEGG results, the relevant targets of hyperlipidemia were collected with OMIM, GeneCards, and Disgenet databases. After that, the crossover targets between hyperlipidemia and new compounds were screened, followed by conducting PPI analysis with STRING to identify key targets (Szklarczyk et al., 2021).
2.4 Molecular docking of the new compounds and key proteins
PPARA was identified as key targets through the PPI analysis results. To validate the binding activity between these key targets and the new compounds, molecular docking was performed. The structure of the new compounds was drawn and their energy was then minimized using the MM2 force field. The protein receptors for PPARA (6LX4) was obtained from the PDB database. For docking with 6LX4, a docking box with a center at x/y/z = 8.04/17.23/56.23 and size x/y/z = 11.25/15.50/12.75 was employed. The docking of ligands to the key targets and the computation of their binding energy were performed using Autodock Vina 1.1.2 (Trott and Olson, 2010).
2.5 Drug-likeness evaluation and ADMET prediction of the new compounds
The drug-likeness of new compounds was assessed using SwissADME for properties related to absorption, distribution, and metabolism, while predictions for excretion and toxicity properties were made with ADMETlab 2.0 and ProTox-II (Banerjee et al., 2018; Daina et al., 2017; Xiong et al., 2021).
3 Results and discussion
3.1 Identification of reported compounds
Based on the analysis of literature, triterpenoids were identified as the major natural products in AR(Liu et al., 2020; Wang et al., 2020). The molecular formula of these triterpenoids was summarized, and the response intensity of EIC was used as a criterion to filter out potential terpenoids with a threshold value set at 104 (Li et al., 2017; Liu et al., 2020; Shu et al., 2023; Song et al., 2013; Yang et al., 2020; Zhang et al., 2021). A total of 12 EICs met this criterion, and they contained 20 reported triterpenoids because of the existence of isomers (Fig. 1).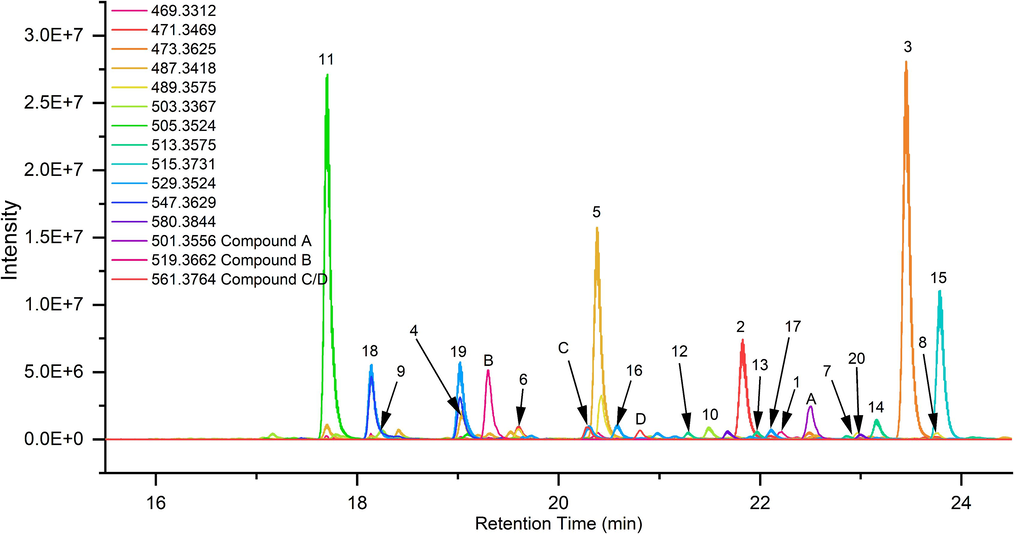
The EIC of reported triterpenes from AR.
The 20 triterpenoids were identified by MS/MS spectrometry combined with literature, and the results shown in Fig. 2, Table 1and Table S1 (Li et al., 2017; Song et al., 2013; Yang et al., 2020; Zhang et al., 2021; Zhang et al., 2022). Due to their strong response, peaks 11, 3, 5, and 15 were preferentially screened to summarize the law of mass fragmentation patterns for AR triterpenoids. Peak 11, 3, 5, and 15 were preferentially identified because of their intense response (Fig. 1). The molecular formula of peak 11 was C30H48O6 because its parent ion of 505.3504 [M + H]+ (Table 1). Consequently, it could potentially be one of the 16-oxo-alisol A, 16β-hydroperoxyalisol B, and alismanol M (Liu et al., 2020). Fortunately, 16β-hydroperoxyalisol B and alismanol M were exclusively found in Alisma orientale, while Alisma plantago-aquatica was found to have a high content of 16-oxo-alisol A, as reported by Zhang et al. (Zhang et al., 2022). Thus, it is likely that peak 11 corresponds to 16-oxo-alisol A. Subsequently, the structure of peak 11 was identified by analyzing its fragmentation process (Fig. 2a and Table 1). The loss of a series of H2O (18 Da) from the parent ion yielded some fragment ions at 487.3407 [M−H2O + H]+, 469.3295 [M−2H2O + H]+, 451.3197 [M−3H2O + H]+. Furthermore, the loss of C4H10O2 (90 Da), resulting from the rupture of the C23-C24 bond, led to the formation of fragment ions at 415.2828 [M−C4H10O2 + H]+ and 397.2723 [M−H2O−C4H10O2 + H]+. The fragment ion at 353.2465 [M−H2O−C4H10O2−C2H4O + H]+ was generated by the cleavage of the C20-C22 bond, resulting in the loss of C2H4O (44 Da) from 397.2723 [M−H2O−C4H10O2 + H] +. Ultimately, peak 11 was confirmed to be 16-oxo-alisol A based on comparison with reference substances and relevant literature (Song et al., 2013; Yang et al., 2020).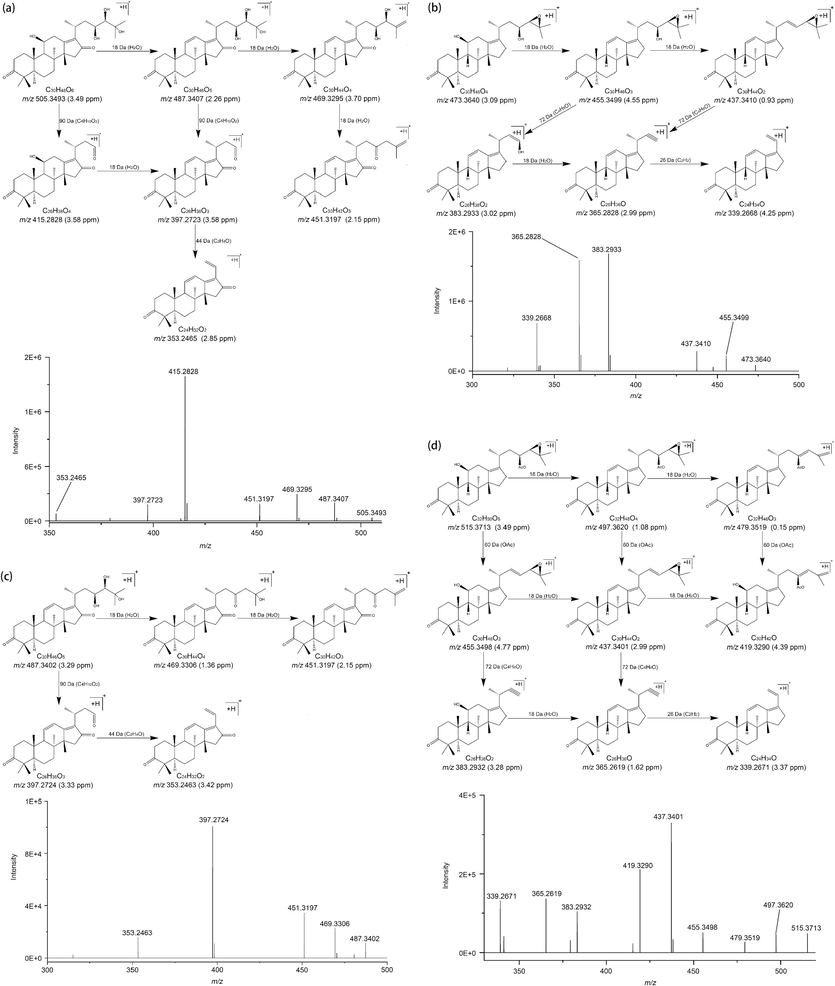
The fragmentation process of 16-oxo-alisol A (a), 16-oxo-11-anhydro-alisol A (b), Alisol B (c), and Alisol B 23-acetate (d).
No.
Identification
tR (min)
Selected ion
Molecular formula
Predicted
Measured
Error (ppm)
Fragment ion
1
Alisol L
22.22
M + H
C30H44O4
469.3312
469.3299
2.77
469.3320 (M + H; 1.63 ppm)
451.3190 (M−H2O + H; 3.70 ppm)
397.2722 (M−C4H8O + H; 3.83 ppm)
353.2461 (M−C4H8O−C2H4O + H; 3.98 ppm)
2
Alisol F
21.82
M−H2O + H
C30H48O5
471.3469
471.3452
3.61
381.2772 (M−H2O−C4H10O2 + H; 4.21 ppm)
339.2672 (M−H2O−C4H10O2−C2H2O + H; 3.07 ppm)
3
Alisol B
23.46
M + H
C30H48O4
473.3625
473.3610
3.17
473.3640 (M + H; 3.09 ppm)
455.3499 (M−H2O + H; 4.55 ppm)
437.3410 (M−2H2O + H; 0.93 ppm)
383.2933 (M−H2O−C4H8O + H; 3.02 ppm)
365.2828 (M−2H2O−C4H8O + H; 2.99 ppm)
339.2668 (M−2H2O−C4H8O−C2H2 + H; 4.25 ppm)
4
Alisol C
19.04
M + H
C30H46O5
487.3418
487.3399
3.90
487.3396 (M + H; 4.52 ppm)
469.3289 (M−H2O + H; 4.98 ppm)
451.3199 (M−2H2O + H; 1.71 ppm)
433.3095 (M−3H2O + H; 1.40 ppm)
415.2828 (M−C4H8O + H; 3.58 ppm)
397.2726 (M−H2O−C4H8O + H; 2.82 ppm)
353.2461 (M−H2O−C4H8O−C2H4O + H; 3.98 ppm)
5
16-oxo-11-anhydro-alisol A
20.38
M + H
C30H46O5
487.3418
487.3399
3.90
487.3402 (M + H; 3.29 ppm)
469.3306 (M−H2O + H; 1.36 ppm)
451.3197 (M−2H2O + H; 2.15 ppm)
397.2724 (M−C4H10O2 + H; 3.33 ppm)
353.2463 (M−C4H10O2−C2H4O + H; 3.42 ppm)
6
16-oxo-11-deoxy-alisol A
22.97
M + H
C30H48O5
489.3575
489.3554
4.29
489.3553 (M + H; 4.40 ppm)
471.3469 (M−H2O + H; 0.03 ppm)
453.3366 (M−2H2O + H; 0.61 ppm)
399.2884 (M−C4H10O2 + H; 2.43 ppm)
381.2771 (M−H2O−C4H10O2 + H; 4.48 ppm)
7
15, 16-dihydro-alisol A
19.60
M + H
C30H48O5
489.3575
489.3558
3.47
471.3460 (M−H2O + H; 1.03 ppm)
453.3337 (M−2H2O + H; 2.03 ppm)
435.3246 (M−3H2O + H; 2.66 ppm)
381.2780 (M−H2O−C4H10O2 + H; 2.12 ppm)
363.2669 (M−2H2O−C4H10O2 + H; 3.70 ppm)
351.2674 (M−H2O−C5H12O3 + H; 2.40 ppm)
337.2510 (M−H2O−C4H10O2−C2H4O + H; 4.72 ppm)
8
Neoalisol
23.76
M + H
C30H48O5
489.3575
489.3558
3.47
471.3457 (M−H2O + H; 2.52 ppm)
453.3354 (M−2H2O + H; 2.03 ppm)
435.3247 (M−3H2O + H; 2.43 ppm)
399.2878 (M−C4H10O2 + H; 3.94 ppm)
381.2781 (M−H2O−C4H10O2 + H; 1.85 ppm)
363.2672 (M−2H2O−C4H10O2 + H; 2.87 ppm)
9
Dehydro-16-oxo-alisol A
18.24
M + H
C30H46O6
503.3367
503.3348
3.91
485.3252 (M−H2O + H; 1.96 ppm)
467.3142 (M−2H2O + H; 2.97 ppm)
445.2925 (M−C3H8O + H; 2.81 ppm)
427.2843 (M−H2O−C3H8O + H; 0.90 ppm)
413.2679 (M−C4H10O2 + H; 1.78 ppm)
395.2568 (M−H2O−C4H10O2 + H; 3.22 ppm)
353.2452 (M−H2O−C4H10O2−C2H4O + H; 4.55 ppm)
10
20-hydroxy-alisol C
21.47
M + H
C30H46O6
503.3367
503.3348
3.74
485.3271 (M−H2O + H; 1.96 ppm)
467.3162 (M−2H2O + H; 1.31 ppm)
413.2667 (M−H2O−C4H8O + H; 4.68 ppm)
395.2557 (M−2H2O−C4H8O + H; 0.94 ppm)
387.2516 (M−C4H8O−C2H4O + H; 3.58 ppm)
385.2732 (M−H2O−C5H10O2+(H rearrangement) + H; 1.35 ppm)
369.2408 (M−H2O−C4H8O−C2H4O + H; 4.39 ppm)
367.2625 (M−2H2O−C5H10O2+(H rearrangement) + H; 1.79 ppm)
353.2458 (M−2H2O−C4H8O−C2H2O + H; 4.83 ppm)
11
16-oxo-alisol A
17.70
M + H
C30H48O6
505.3524
505.3504
3.96
505.3493 (M + H; 3.49 ppm)
487.3407 (M−H2O + H; 2.26 ppm)
469.3295 (M−2H2O + H; 3.70 ppm)
451.3197 (M−3H2O + H; 2.15 ppm)
415.2828 (M−C4H10O2 + H; 3.58 ppm)
397.2723 (M−C4H10O2−H2O + H; 3.58 ppm)
353.2465 (M−C4H10O2−H2O−C2H4O + H; 2.85 ppm)
12
16β-hydroxy-alisol B 23-acetate
21.28
M−H2O + H
C32H50O6
513.3575
513.3557
3.51
513.3554 (M−H2O + H; 4.00 ppm)
453.3345 (M−H2O−OAc + H; 4.02 ppm)
435.3246 (M−2H2O−OAc + H; 2.66 ppm)
399.2882 (M−OAc−C4H8O + H; 2.93 ppm)
13
13β,17β-epoxy-Alisol B 23-acetate
21.97
M−H2O + H
C32H50O6
513.3575
513.3557
3.51
513.3565 (M−H2O + H; 1.85 ppm)
453.3351 (M−H2O−OAc + H; 2.69 ppm)
435.3242 (M−2H2O−OAc + H; 3.58 ppm)
399.2885 (M−OAc−C4H8O + H; 2.18 ppm)
14
Alisol F 24-acetate
23.15
M−H2O + H
C32H50O6
513.3575
513.3557
3.51
381.2772 (M−H2O−C6H12O3 + H; 4.21 ppm)
363.2669 (M−2H2O−C6H12O3 + H; 3.70 ppm)
339.2672 (M−2H2O−C6H12O3−C2H2 + H; 3.07 ppm)
15
Alisol B 23-acetate
23.81
M + H
C32H50O5
515.3731
515.3712
3.65
515.3713 (M + H; 3.49 ppm)
497.3620 (M−H2O + H; 1.08 ppm)
479.3519 (M−2H2O + H; 0.15 ppm)
455.3498 (M−OAc + H; 4.77 ppm)
437.3401 (M−H2O−OAc + H; 2.99 ppm)
419.3290 (M−2H2O−OAc + H; 4.39 ppm)
383.2932 (M−OAc−C4H8O + H; 3.28 ppm)
365.2619 (M−H2O−OAc−C4H8O + H; 1.62 ppm)
339.2671 (M−H2O−OAc−C4H8O−C2H2 + H; 3.37 ppm)
16
Alisol C 23-acetate
22.11
M + H
C32H48O6
529.3524
529.3504
3.72
529.3510 (M + H; 0.88 ppm)
511.3377 (M−H2O + H; 2.35 ppm)
469.3295 (M−OAc + H; 0.29 ppm)
451.3196 (M−H2O−OAc + H; 1.70 ppm)
433.3087 (M−2H2O−OAc + H; 3.25 ppm)
415.2845 (M−C4H8O−Ac+(H rearrangement) + H; 2.13 ppm)
17
16-oxo-11-anhydro-alisol A 24-acetate
20.58
M + H
C32H48O6
529.3524
529.3505
3.53
529.3495 (M + H; 1.26 ppm)
511.3393 (M−H2O + H; 4.89 ppm)
469.3296 (M−OAc + H; 3.49 ppm)
451.3190 (M−H2O−OAc + H; 3.70 ppm)
433.3106 (M−2H2O−OAc + H; 1.14 ppm)
18
16-oxo-alisol A 23-acetate
18.14
M + H
C32H50O7
547.3629
547.3609
3.65
529.3510 (M−H2O + H; 2.58 ppm)
511.3403 (M−2H2O + H; 2.94 ppm)
469.3297 (M−H2O−OAc + H; 3.27 ppm)
451.3191 (M−2H2O−OAc + H; 3.48 ppm)
433.3091 (M−3H2O−OAc + H; 2.32 ppm)
415.2827 (M−C4H10O2−Ac+(H rearrangement) + H; 3.82 ppm)
397.2741 (M−C4H10O2−Ac+(H rearrangement)–H2O + H; 0.95 ppm)
353.2467 (M−C4H10O2−Ac+(H rearrangement)-C2H4O-H2O + H; 2.28 ppm)
19
16-oxo-alisol A 24-acetate
19.02
M + H
C32H50O7
547.3629
547.3611
3.29
529.3511 (M−H2O + H; 2.39 ppm)
511.3393 (M−2H2O + H; 4.89 ppm)
469.3299 (M−H2O−OAc + H; 2.85 ppm)
451.3194 (M−2H2O−OAc + H; 2.82 ppm)
433.3087 (M−3H2O−OAc + H; 3.25 ppm)
415.2828 (M−C4H10O2−Ac+(H rearrangement) + H; 3.58 ppm)
397.2728 (M−C4H10O2−Ac+(H rearrangement)–H2O + H; 2.32 ppm)
353.2466 (M−C4H10O2−Ac+(H rearrangement)-C2H4O-H2O + H; 2.57 ppm)
20
12-hydroxy-16-oxo-alisol A 24-acetate
22.99
M + NH4
C32H50O8
580.3844
580.3823
3.57
545.3469 (M−H2O + H; 0.70 ppm)
467.3135 (M−2H2O−OAc + H; 4.46 ppm)
431.2765 (M−C6H12O3 + H; 2.09 ppm)
413.2662 (M−C6H12O3−H2O + H; 0.81 ppm)
387.2520 (M−C6H12O3−H2O−C2H2 + H; 2.55 ppm)
Peak 3, with a parent ion at 473.3610 [M + H]+, suggests a molecular formula of C30H48O4 and may be identified as alisol B, 11-deoxy-13β, 17β-epoxyalisol B, or alisol G (Liu et al., 2020). However, alisol B was found to be abundant in our sample based on HPLC analysis, indicating that peak 3 is likely alisol B (Fig. S2). This is supported by the presence of fragment ions at 455.3499 [M−H2O + H]+ and 437.3410 [M−2H2O + H]+, which result from the loss of multiple H2O from the parent ion. Further fragmentation yielded fragment ions at 383.2933 [M−H2O−C4H8O + H]+ and 365.2828 [M−2H2O−C4H8O + H]+, corresponding to the loss of C4H8O, and the fragment ion at 339.2668 [M−2H2O−C4H8O−C2H2 + H] + was formed by the loss of C2H2. By comparing the obtained data with reference substances and literature, peak 3 was conclusively identified as alisol B (Zhang et al., 2021). This is illustrated in Fig. 2b and Table 1.
Peak 5, with a parent ion at 487.3399 [M + H]+ (Table 1), was identified as C30H46O5 by its molecular formula. This peak is likely to be either 16-oxo-11-anhydroalisol A or alisol C (Liu et al., 2020). The EIC of C30H46O5 has two peaks at 19.04 (peak 4) and 20.38 min (peak 5), respectively. Peak 5 might be 16-oxo-11-anhydroalisol A because the retention time of alisol C was shorter than that of 16-oxo-11-anhydroalisol A in the C18 column (Zhang et al., 2022). Fragment ions at 469.3306 [M−H2O + H]+ and 451.3197 [M−2H2O + H]+were generated by the loss of a series of H2O from the parent ion. Additionally, The fragment ions at 397.2724 [M−C4H10O2 + H]+ and 353.2463 [M−C4H10O2−C2H4O + H]+ was formed by losing C4H10O2 from parent ion and then losing C2H4O again (Table 1 and Fig. 2c). Consequently, through these observations, peak 5 was further identified as 16-oxo-11-anhydroalisol A (Zhang et al., 2022).
Peak 15 was identified as alisol B 23-acetate based on the molecular formula C32H50O5, calculated by the parent ion at 515.3712 [M + H]+ (Table 1). HPLC analysis of our sample indicated a high abundance of alisol B 23-acetate (Fig. S2), further supporting this identification. This conclusion is reinforced by the fragment ions at 497.3620 [M−H2O + H]+ and 479.3519 [M−2H2O + H]+, which were generated by the loss of a series of H2O (18 Da). Additional fragment ions at 455.3498 [M−OAc + H]+, 437.3401 [M−H2O−OAc + H]+, and 419.3290 [M−2H2O−OAc + H]+ were formed by the elimination of the acetoxy (OAc, 60 Da) group from the parent ion and 497.3620 [M−H2O + H]+, and 479.3519 [M−2H2O + H]+, respectively. Furthermore, the loss of C4H8O from 455.3498 [M−OAc + H]+ and 437.3401 [M−H2O−OAc + H]+ resulted in the formation of fragment ions at 383.2932 [M−OAc−C4H8O + H]+ and 365.2619 [M−H2O−OAc−C4H8O + H]+, respectively. Finally, the fragment ion at 339.2671 [M−H2O−OAc−C4H8O−C2H2 + H]+ was generated from 365.2619 [M−H2O−OAc−C4H8O + H]+ through the loss of C2H2 (Table 1 and Fig. 2d). All of these findings, along with the reference substance and existing literature, further confirm the identification of peak 15 as alisol B 23-acetate (Yang et al., 2020).
3.2 Identification of new compounds
The fragments m/z 339 or 353 are considered crucial for identifying AR triterpenoids by analzing the mass spectrum fragments of AR triterpenoids. Therefore, four new triterpenoid compounds were identified using this pattern, and named components A, B, C, and D, respectively. Compound A, with a molecular formula of C31H48O5, was identified through the analysis of its symmetric sharp single peak at 22.52 min in the EIC (Fig. 3a) and its corresponding parent ion of 501.3556 [M + H]+ (Table 2). The fragmentation pattern of compound A suggested that it belonged to the triterpenoid class, based on the loss of H2O (18 Da), C2H4O (44 Da), and C4H6 (54 Da) from its fragment ions, which conformed to the fragmentation rule of AR triterpenoids. As no triterpenoids with the molecular formula of C31H48O5 were previously reported in AR, the structure of compound A was inferred based on its fragment ions. The characteristic fragment ion at 353.2465, which is indicative of alisol C and 16-oxo-aliso types, was produced by the loss of C2H4O from 397.2724 (Zhang et al., 2021). The fragment ion at 397.2724, in turn, was obtained from 451.3191 by losing C4H6, where 451.3191 was formed through the loss of H2O from 469.3296. Notably, the difference value between 501.3588 and 469.3296 was approximately 32 Da, suggesting that 469.3296 might have resulted from the loss of the methoxy (OMe) group, which weighs 32 Da, from 501.3588. Additionally, the parent ion at 501.3588 lost a molecule of H2O to give rise to 483.3474. The characteristic of compound A has an OMe group and might locate at the C25 position because the methoxy group is mostly located at C25 of AR triterpenoid (Feng et al., 2021). Thus, compound A was identified as 25-methoxy-16-oxo-11-anhydroalisol A.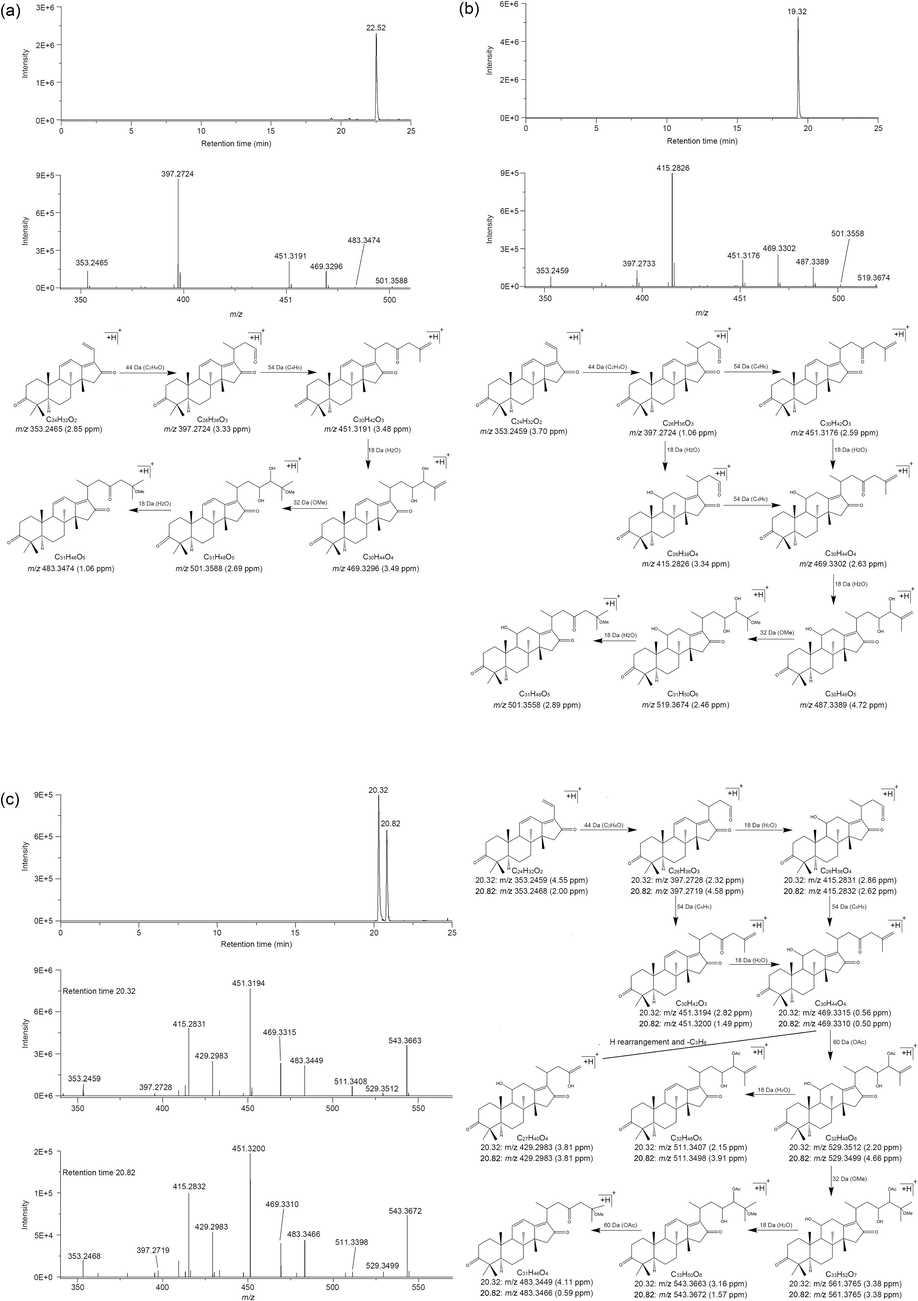
The EIC and fragmentation process of 25-methoxy-16-oxo-11-anhydroalisol A (a), 25-methoxy-16-oxo-alisol A (b), and 25-methoxy-16-oxo-alisol A 23 or 24-acetate (c).
Compound
Identification
tR (min)
Selected ion
Molecular formula
Predicted
Measured
Error (ppm)
Fragment ion
A
25-methoxy-16-oxo-11-anhydroalisol A
22.52
M + H
C31H48O5
501.3575
501.3556
3.65
501.3588 (M + H; 2.69 ppm)
483.3474 (M−H2O + H; 1.06 ppm)
469.3296 (M−OCH3 + H; 3.49 ppm)
451.3191 (M−OCH3−H2O + H; 3.48 ppm)
397.2724 (M−OCH3−H2O−C4H6 + H; 3.33 ppm)
353.2465 (M−OCH3−H2O−C4H6−C2H4O + H; 2.85 ppm)
B
25-methoxy-16-oxo-alisol A
19.31
M + H
C31H50O6
519.3680
519.3662
3.60
519.3674 (M + H; 2.46 ppm)
501.3558 (M−H2O + H; 2.89 ppm)
487.3389 (M−OCH3 + H; 4.72 ppm)
469.3302 (M−H2O−OCH3 + H; 2.63 ppm)
451.3176 (M−2H2O−OCH3 + H; 2.59 ppm)
415.2826 (M−H2O−OCH3−C4H6 + H; 3.34 ppm)
397.2733 (M−2H2O−OCH3−C4H6 + H; 1.06 ppm)
353.2459 (M−2H2O−OCH3−C4H6−C2H4O + H; 3.70 ppm)
C
25-methoxy-16-oxo-alisol A 24-acetate
20.31
M + H
C33H52O7
561.3786
561.3764
3.92
543.3663 (M−H2O + H; 3.16 ppm)
529.3512 (M−OCH3 + H; 2.20 ppm)
511.3407 (M−OCH3−H2O + H; 2.15 ppm)
483.3449 (M−H2O−OAc + H; 4.11 ppm)
469.3315 (M−OCH3−OAc + H; 0.56 ppm)
451.3194 (M−OCH3−OAc−H2O + H; 2.82 ppm)
429.2983 (M−OCH3−OAc−C3H6 + H; 3.81 ppm)
415.2831 (M−OCH3−OAc−C4H8 + H; 2.86 ppm)
397.2728 (M−OCH3−OAc−C4H8−H2O + H; 2.32 ppm)
353.2459 (M−OCH3−OAc−C4H8−H2O−C2H4O + H; 4.55 ppm)
D
25-methoxy-16-oxo-alisol A 23-acetate
20.82
M + H
C33H52O7
561.3786
561.3764
3.92
543.3672 (M−H2O + H; 1.50 ppm)
529.3499 (M−OCH3 + H; 4.66 ppm)
511.3398 (M−OCH3−H2O + H; 3.91 ppm)
483.3466 (M−H2O−OAc + H; 0.59 ppm)
469.3310 (M−OCH3−OAc + H; 0.50 ppm)
451.3200 (M−OCH3−OAc−H2O + H; 1.49 ppm)
429.2983 (M−OCH3−OAc−C3H6 + H; 3.81 ppm)
415.2832 (M−OCH3−C4H8O−Ac+(H rearrangement) + H; 2.62 ppm)
397.2719 (M−OCH3−H2O−C4H8O−Ac+(H rearrangement) + H; 4.58 ppm)
353.2468 (M−OCH3−OAc−C4H8−H2O−C2H4O; 2.00 ppm)
The parent ion at 519.3662 [M + H]+ of compound B suggested its molecular formula was C31H50O6 (Table 2). The fragment ions at 353.2459, 397.2733, 451.3176, and 469.3302 were consistent with compound A. In contrast, compound B exhibited additional fragment ions at 415.2826, 487.3389, 501.3558, and 519.3674. The fragment ion at 415.2826 was derived from 469.3302 through the loss of C4H6, where 469.3302 originated from the dehydration of 487.3389. Additionally, the fragment ion at 487.3389 was formed through the loss of the OMe group from 501.3588, as the difference value between them was 32 Da. Furthermore, the parent ion at 519.3674 lost a molecule of H2O to yield 501.3558, which could potentially be compound A (Fig. 3b). Overall, compound B was identified as 25-methoxy-16-oxo-alisol A.
Compounds C and D were isomers with the molecular formula of C33H52O7 because there was no difference in their parent and fragment ions (Table 2 and Fig. 3c). The fragment ions observed, such as 353, 397, 415, 451, and 469, were consistent with compound B. However, compounds C and D had additional fragment ions at 429, 483, 511, 529, and 543. The fragment ion at 429 was obtained from 469 by H rearrangement after losing C3H6. Similarly, the ion at 483 was obtained by removing the OAc group from 543 and then losing the OMe group, resulting in 451. Furthermore, the presence of the ion at 511 indicated the loss of H2O and subsequent loss of the OAc group, leading to 451. The ions at 529 and 543 were generated by the loss of the OMe group and H2O from the parent ion 561, respectively. These findings revealed that compounds C and D differed from compound B by just 42 Da, which corresponded to an acetyl group. Therefore, compounds C and D were likely formed by replacing one of the hydroxyl groups of compound B with an acetoxy group. Specifically, compounds C and D were identified as 25-methoxy-16-oxo-alisol A 23-acetate and 25-methoxy-16-oxo-alisol A 24-acetate, respectively, considering that acetoxy groups are typically located at the C23 and C24 positions of AR triterpenes (Liu et al., 2020). In summary, the 24 triterpenoids from AR were identified with HPLC-HRMS, including 20 reported triterpenoids and 4 new triterpenoids (Fig. 4).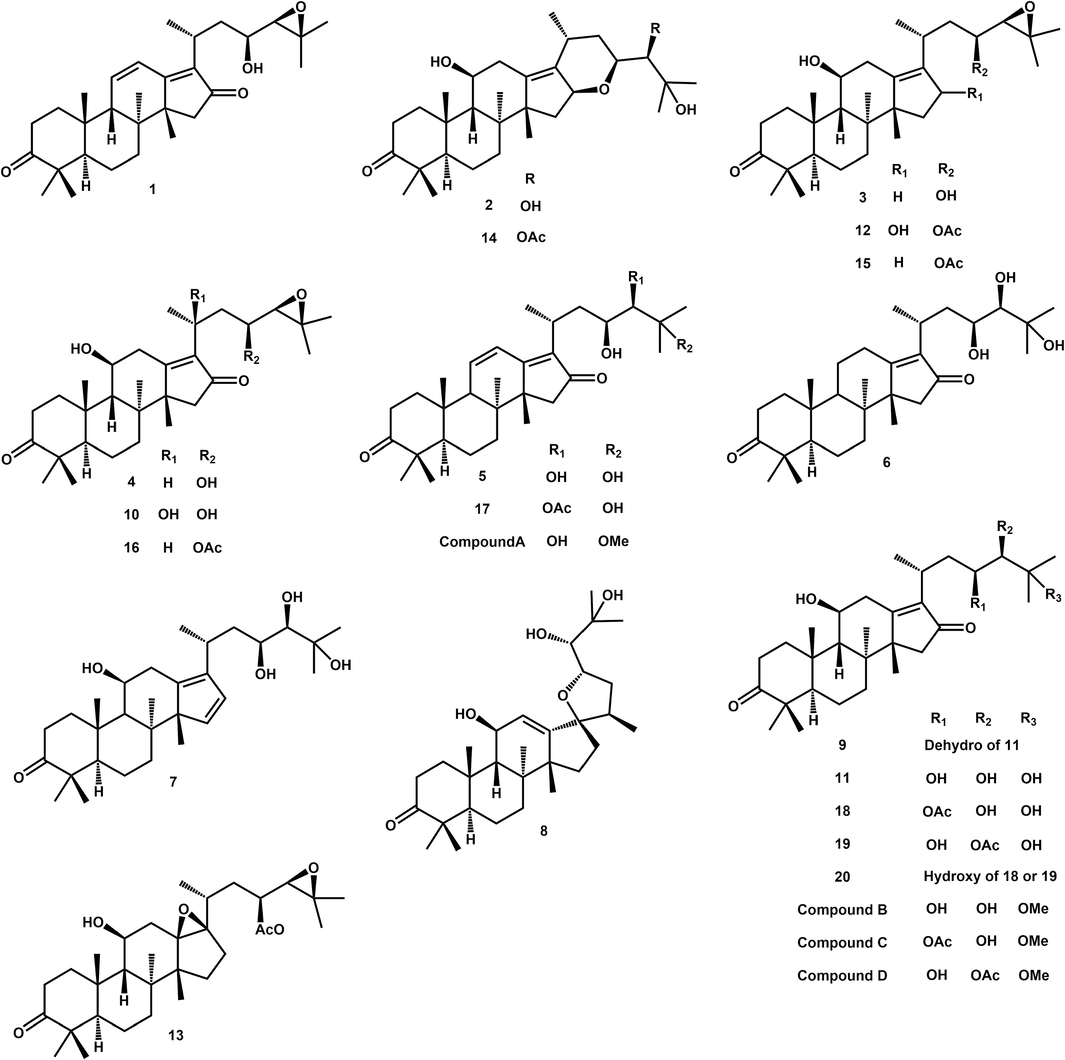
The structure of 20 reported triterpenes (a) and 4 new triterpenes (b).
3.3 Prediction of activity with new compounds
The targets of compounds A, B, C, and D were predicted using various databases such as SwissTargetPrediction, SuperPred, Similarity ensemble approach, etc., based on their structural formula. These compounds were found to have 184, 195, 162, and 429 targets, respectively. The predicted targets were then analyzed using Metascape for KEGG analysis, which revealed that they were significantly enriched in the PPAR signaling pathway (Fig. 5a-d). The PPAR signaling pathway is closely associated with hyperlipidemia as it regulates fatty acid β-oxidation, bile acid metabolism, and cholesterol metabolism (Bougarne et al., 2018). Additionally, Yan et al. indicated that AR triterpenoids can alleviate hyperlipidemia by activating the PPAR signaling pathway (Yan et al., 2022). Therefore, these new triterpenoids have the potential to alleviate hyperlipidemia by activating the PPAR signaling pathway.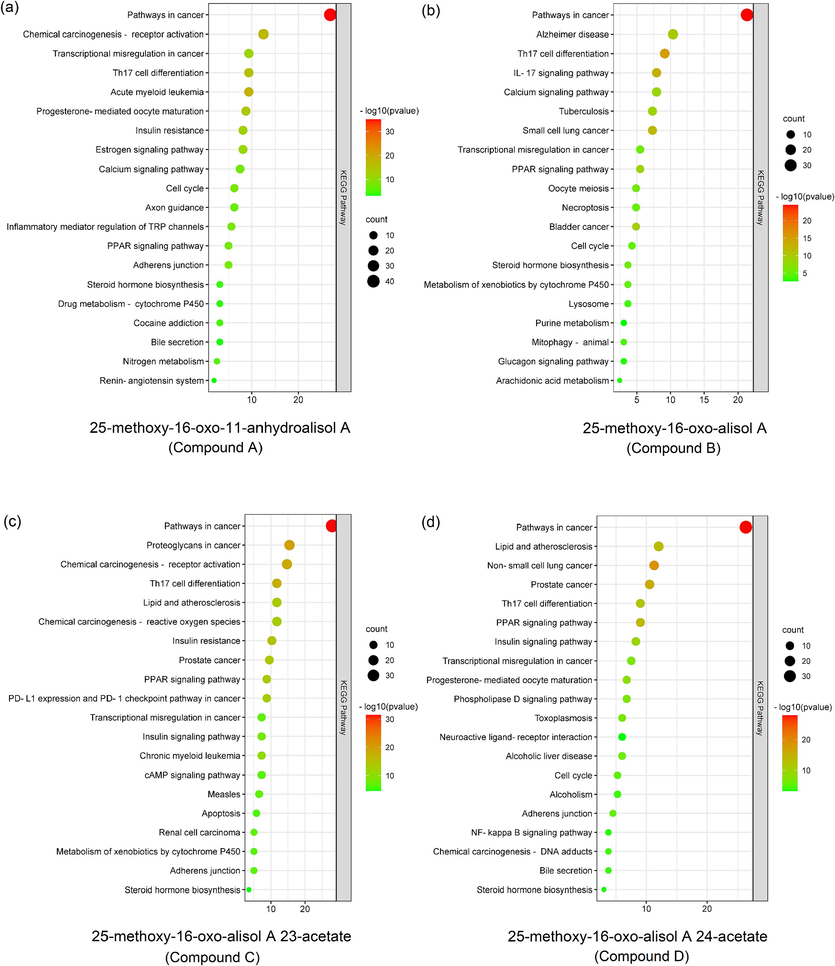
The KEGG analysis of targets from 25-methoxy-16-oxo-11-anhydroalisol A (a), 25-methoxy-16-oxo-alisol A (b), 25-methoxy-16-oxo-alisol A 23-acetate (c) and 25-methoxy-16-oxo-alisol A 24-acetate (d).
The PPI analysis was performed to screen the key targets. Afterwards, the targets of hyperlipidemia were collected and used to screen the crossover targets between new compounds and hyperlipidemia. As shown in Fig. 6a, 624 hyperlipidemia targets were collected, and 24, 31, 26, 24 crossover targets between compounds A-D and hyperlipidemia were filtered, respectively. Subsequently, the key targets were confirmed with a degree value greater than 10 in the PPI network. The PPARA and EGFR turned out to be potential key targets of compounds A-D in the treatment of hyperlipidemia (Fig. 6b and Table S2). PPARA regulates the expression of related genes for fatty acid β-oxidation, bile acid metabolism, and cholesterol metabolism and its agonist is a common clinical drug used in the treatment of hyperlipidemia (Duval et al., 2007). Additionally, EGFR is one of the target genes of PPARA. In normal circumstances, PPARA increases the expression of EGFR, but when PPARA binds to the ligand, its protein conformation changes and it cannot bind to the promoter of EGFR, leading to the downregulation of EGFR expression (Mahankali et al., 2015). The downregulation of EGFR is beneficial for hyperlipidemia because EGFR inhibitors reduce triglyceride and cholesterol levels in serum and liver tissues (Liang et al., 2018). Based on the PPI results, although both PPARA and EGFR may be potential key targets for compound A-D for treating hyperlipidemia, PPARA may be more important as literature suggested that AR triterpenoids can target the activation of PPARs (Li et al., 2016). Thus, PPARA was identified as target protein for compound A-D.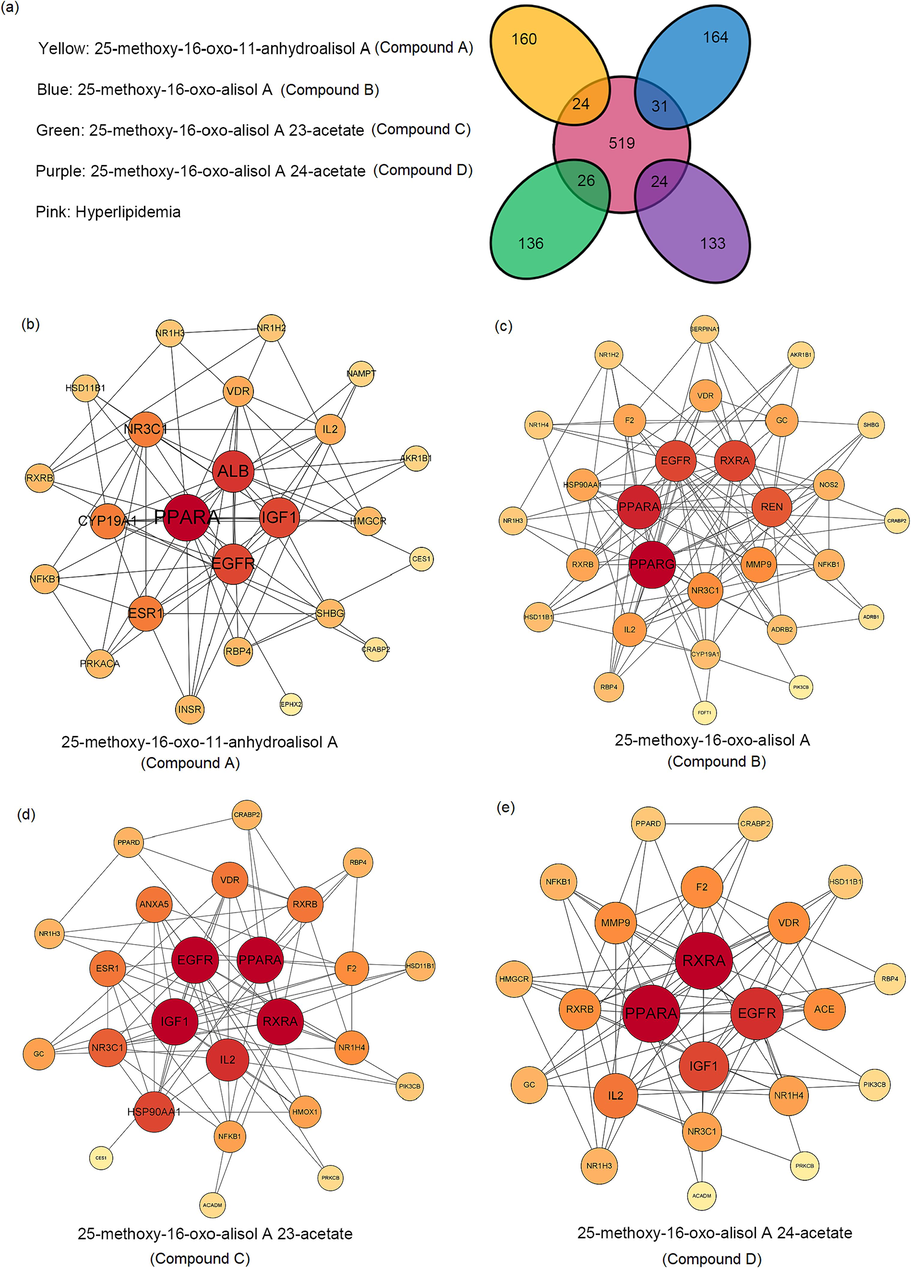
Prediction of anti-hyperlipidemic activity of four new triterpenoids. (a) Crossover targets of new compounds and hyperlipidemia. PPI analysis of crossover targets from 25-methoxy-16-oxo-11-anhydroalisol A (a), 25-methoxy-16-oxo-alisol A (b), 25-methoxy-16-oxo-alisol A 23-acetate (c) and 25-methoxy-16-oxo-alisol A 24-acetate (d) to hyperlipidemia.
Compounds A-D might bind with PPARA because their binding energy is less than −5 kJ/mol (Table 3) (An et al., 2021). The binding energy of compound A was less than that of the control group (fenofibric acid, PPARA agonist, −6.9 kJ/mol), suggesting that compound A has more potential as a PPARA agonist. The visualization results of molecular docking further supported this finding, as they showed that the binding energy was mainly influenced by hydrogen bonds rather than hydrophobic interaction (Fig. 7 and Table 3). This suggests that the intermolecular force of the hydrogen bond is much greater than that of the hydrophobic interaction. Specifically, compound A demonstrated strong binding affinity to PPARA through hydrogen bonds with Thr-279, Ala-333, and hydrophobic interactions with Ile-241, Leu-247, Glu-251, Leu-254, Lys-257, Ile-272, Cys-275, Val-332, Tyr-334, Ile-339, and Phe-421. In conclusion, compound A exhibits promising potential for the treatment of hyperlipemia by binding to PPARA.
Compound
binding energy (kJ/mol)
PPARA
Hydrogen-bond
Hydrophobic-interaction
25-methoxy-16-oxo-11-anhydroalisol A (Compound A)
−7.9
Thr-279, Ala-333
Ile-241, Leu-247, Glu-251, Leu-254, Lys-257, Ile-272, Cys-275, Val-332, Tyr-334, Ile-339, Phe-421
25-methoxy-16-oxo-alisol A (Compound B)
−6.9
Ala-333
Ile-241, Leu-247, Glu-251, Leu-254, Lys-257, Ile-272, Cys-275, Thr-279, Val-332, Tyr-334, Ile-339
25-methoxy-16-oxo-alisol A 23-acetate (Compound C)
−6.4
Ala-333
Ile-241, Leu-247, Ala-250, Leu-254, Lys-257, Ile-272, Cys-275, Cys-276, Thr-279, Val-332, Tyr-334, Ile-339, Phe-421
25-methoxy-16-oxo-alisol A 24-acetate (Compound D)
−6.7
Ala-333
Ile-241, Leu-247, Ala-250, Glu-251, Leu-254, Lys-257, Ile-272, Cys-275, Cys-276, Thr-279, Val-332, Tyr-334, Ile-339, Phe-421
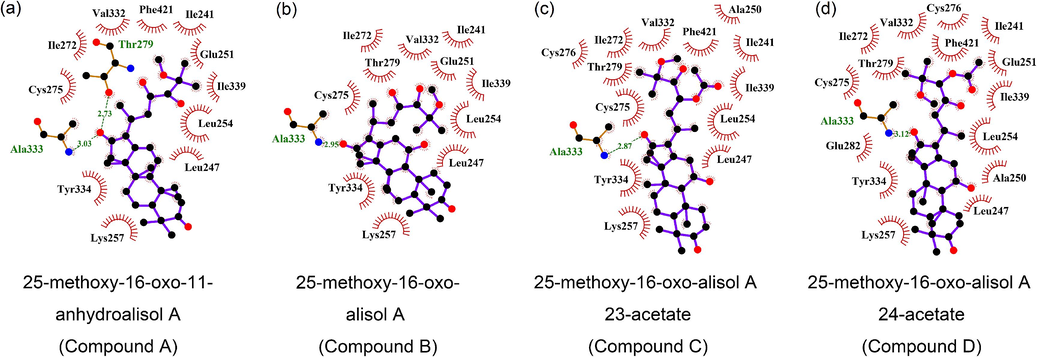
Molecular docking of 25-methoxy-16-oxo-11-anhydroalisol A (a), 25-methoxy-16-oxo-alisol A (b), 25-methoxy-16-oxo-alisol A 23-acetate (c), 25-methoxy-16-oxo-alisol A 24-acetate (d) to PPARA.
3.4 Drug-likeness and ADMET prediction of the new triterpenoids
Drug development is greatly facilitated by ADMET prediction, as it significantly reduces the workload. Therefore, evaluating drug-likeness is crucial, considering that drug-like characteristics are heavily influenced by their ADMET properties (Gleeson et al., 2011). Table 4 contained drug-like characteristics of the new compounds and common decision rules of drug-likeness (Egan et al., 2000; Ghose et al., 1999; Lipinski et al., 2001; Muegge et al., 2001; Veber et al., 2002). Due to the large molecular weight, molar refractivity, and atoms of the new triterpenoids, it does not conform to the Lipinski and Ghose rules (Table 4). However, it conformed to the Muege rule with high molecular weight requirements and the Egan and Veber rules without molecular weight requirements. Thus, the ADMET properties of the new compounds can be further predicted.
Compound
Formula
Molecular weight
LogP a
TPSA b
Rings
Corbon
Heteroatoms
Rotatable bonds
HBA c
HBD d
Molar Refractivity
Atoms
A
C31H48O5
500
3.98
83.83
4
31
5
6
5
2
144.53
84
B
C31H50O6
518
3.98
104.06
4
31
6
6
6
3
146.17
87
C
C33H52O7
560
4.72
110.13
4
33
7
8
7
2
155.91
92
D
C33H52O7
560
4.01
110.13
4
33
7
8
7
2
155.91
92
Muegge
200–600
−2–5
≤150
≤7
>4
>1
≤15
≤10
≤5
–
–
Lipinski
≤500
≤5
–
–
–
–
≤10
≤10
≤5
–
–
Ghose
160–480
−0.4–5.6
–
–
–
–
–
–
–
40–130
20–70
Egan
–
≤5.88
≤131.6
–
–
–
–
–
–
–
–
Veber
–
–
≤140
–
–
–
≤10
–
–
–
–
The ADMET properties of the new triterpenoids were evaluated and presented in Table 5. In absorption, the results showed that the new compounds demonstrated high HIA and exhibited moderate permeability across Caco-2 cells. Additionally, they displayed an ability to inhibit P-glycoprotein, indicating their potential to enter cells and maintain intracellular drug concentration. In terms of drug distribution within the human body, the evaluation of blood–brain barrier penetration and plasma protein binding became crucial (Chen et al., 2020; Pardridge et al., 1986). The new triterpenoids showed moderate blood–brain barrier penetration ability and weak plasma protein binding ability, suggesting their potential to enter the brain and intracellular compartments. Xu et al. also reported similar findings, confirming the ability of AR triterpenoids to cross the blood–brain barrier (Xu et al., 2017). Moreover, the involvement of cytochrome P450 in the metabolic process of these new triterpenoids is likely, as they seem to act as substrates and inhibitors of cytochrome P450. The new compounds demonstrated a moderate clearance rate and exhibited significant differences in their half-life during excretion. Specifically, components B, C, and D had a higher probability of having a half-life of more than 3 h compared to compound A. This finding is consistent with literature that indicates the half-life of AR triterpenoids is mostly greater than 3 h, although some may have a half-life of less than 3 h (Xu et al., 2017). In terms of toxicity, the new ingredients showed pronounced differences, with compounds A and D having a higher median lethal dose (LD50) compared to compounds C and D, suggesting that compounds A and D have a higher safety margin.
Classification
Index
Compound A
Compound B
Compound C
Compound D
Standard
Absorption
Human Intestinal Absorption (HIA, %)
94.638
91.729
95.448
95.448
Low (0–20); Middle (20–70); High (70–100)
Caco-2 cell permeability (nm/sec)
25.573
22.042
23.926
23.649
Low (<4); Middle (4–70); High (>70)
P-glycoprotein inhibitory activity
Inhibitor
Inhibitor
Inhibitor
Inhibitor
Distribution
Blood-brain barrier penetration ability
1.196
0.459
0.194
0.174
Low (<0.1); Middle (0.1–2); High (>2)
Plasma protein binding ability (%)
89.629
83.956
85.796
85.920
Weakly (<90); Strongly (>90);
Metabolism
CYP_2C19_inhibition
Non
Non
Non
Non
CYP_2C9_inhibition
Inhibitor
Inhibitor
Inhibitor
Inhibitor
CYP_2D6_inhibition
Non
Non
Non
Non
CYP_2D6_substrate
Non
Non
Non
Non
CYP_3A4_inhibition
Inhibitor
Inhibitor
Inhibitor
Inhibitor
CYP_3A4_substrate
Substrate
Substrate
Substrate
Substrate
Excretion
Clearance rate (mL/min/kg)
6.626
6.580
5.258
5.094
Low (<5);Middle (5–15); High (>15)
Half-life (probability of half-life > 3 h, %)
25.4
48.1
48.7
52.0
Toxicity
Median lethal dose (LD50, mg/kg)
4000
123
10
5000
Toxicity Class
Ⅴ
III
II
Ⅴ
4 Conclusion
The triterpenoids from AR were analyzed with HPLC-HRMS, and their mass fragmentation pattern was summarized to infer new triterpenoids. Twenty reported triterpenoids and four new triterpenoids (25-methoxy-16-oxo-11-anhydroalisol A, 25-methoxy-16-oxo-alisol A, 25-methoxy-16-oxo-alisol A 23-acetate, and 25-methoxy-16-oxo-alisol A 24-acetate) were observed from the AR extract. KEGG analysis suggested that the four new triterpenoids might activate the PPAR signaling pathway to relieve hyperlipemia. Additionally, the properties of absorption, distribution, and metabolism of the four new triterpenoids are consistent, but there are great differences in excretion and toxicity. The toxicity of 25-methoxy-16-oxo-11-anhydroalisol A and 25-methoxy-16-oxo-alisol A 24-acetate were far less than 25-methoxy-16-oxo-alisol A and 25-methoxy-16-oxo-alisol A 23-acetate. Moreover, 25-methoxy-16-oxo-11-anhydroalisol A had a better clearance rate and half-life compared with 25-methoxy-16-oxo-alisol A 24-acetate. Therefore, it has more potential as a candidate drug for anti-hyperlipidemia. Furthermore, the results of PPI analysis and molecular docking indicated that PPARA might be the key targets for anti-hyperlipidemia with the four new triterpenoids, with 25-methoxy-16-oxo-11-anhydroalisol A exhibiting better binding ability to PPARA than the other three new triterpenoids. In conclusion, HPLC-HRMS combined with in-silico analysis revealed the presence of new triterpenoids with anti-hyperlipidemia potential in AR, thereby guiding the direction of future endeavors and reducing the waste of time and financial resources.
5 Declaration of Generative AI and AI-assisted technologies in the writing process
During the preparation of this work, the authors used Acadwrite AI (http://acadwrite.cn/) to improve the readability of the article. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
CRediT authorship contribution statement
Tao Gao: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft. Sheng-lin Hu: Visualization, Writing – original draft. Rui Yan: Investigation, Methodology. Ling-zhi He: Formal analysis, Investigation. Nan Fang: Investigation. Zhong-hao Zhang: Investigation. Zhi-hao Duan: Investigation. Zi-zhong Tang: Resources. Yang-er Chen: Resources. Shu Yuan: Writing – review & editing. Lin Ye: Writing – review & editing. Xiao-rong Yan: Resources. Ming Yuan: Conceptualization, Resources, Supervision, Writing – review & editing.
Acknowledgments
This work was funded by the Transfer Payments Key Research and Development projects of Yaan (22ZDYFZF0009), Science and Technology Planning Project in 2022 of Dazhu County, Science and Technology Innovation Entrepreneurship Seedling Engineering Cultivation Project of Yaan.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Mechanisms of Rhizoma Coptidis against type 2 diabetes mellitus explored by network pharmacology combined with molecular docking and experimental validation. Sci. Rep.. 2021;11(1):20849.
- [CrossRef] [Google Scholar]
- Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov.. 2021;20(3):200-216.
- [CrossRef] [Google Scholar]
- Recent advances and applications in LC-HRMS for food and plant natural products: a critical review. Anal. Bioanal. Chem.. 2020;412(9):1973-1991.
- [CrossRef] [Google Scholar]
- ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucl. Acids Res.. 2018;46(W1):W257-W263.
- [CrossRef] [Google Scholar]
- Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev.. 2018;39(5):760-802.
- [CrossRef] [Google Scholar]
- Applicability of free drug hypothesis to drugs with good membrane permeability that are not efflux transporter substrates: a microdialysis study in rats. Pharmacol. Res. Perspect.. 2020;8(2):e00575.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7(1):42717.
- [CrossRef] [Google Scholar]
- SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucl. Acids Res.. 2019;47(W1):W357-W364.
- [CrossRef] [Google Scholar]
- Prediction of drug absorption using multivariate statistics. J. Med. Chem.. 2000;43(21):3867-3877.
- [CrossRef] [Google Scholar]
- Alisma genus: Phytochemical constituents, biosynthesis, and biological activities. Phytother. Res.. 2021;35(4):1872-1886.
- [CrossRef] [Google Scholar]
- SuperPred 3.0: drug classification and target prediction—a machine learning approach. Nucl. Acids Res.. 2022;50(W1):W726-W731.
- [CrossRef] [Google Scholar]
- A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem.. 1999;1(1):55-68.
- [CrossRef] [Google Scholar]
- Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov.. 2011;10(3):197-208.
- [CrossRef] [Google Scholar]
- Phenolic profiling of olives and olive oil process-derived matrices using UPLC-DAD-ESI-QTOF-HRMS analysis. J. Agric. Food Chem.. 2015;63(15):3859-3872.
- [CrossRef] [Google Scholar]
- Relating protein pharmacology by ligand chemistry. Nat. Biotechnol.. 2007;25(2):197-206.
- [CrossRef] [Google Scholar]
- Integration of high-content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc. Natl. Acad. Sci. U.S.A.. 2015;112(39):11999-12004.
- [CrossRef] [Google Scholar]
- The strategy for establishment of the multiple reaction monitoring based characteristic chemical profile of triterpenes in Alismatis rhizoma using two combined tandem mass spectrometers. J. Chromatogr. A. 2017;1524:121-134.
- [CrossRef] [Google Scholar]
- Development of a cell-based peroxisome proliferator-activated receptors (PPARs) screening model and its application for evaluation of triterpenoids isolate from Alismatis Rhizoma. China J. Chin. Mater. Med.. 2016;41(21):4015-4022.
- [CrossRef] [Google Scholar]
- Two novel dipeptidyl peptidase-IV (DPP-IV) inhibitory peptides identified from truffle (Tuber sinense) by peptidomics, in silico, and molecular docking analysis. J. Food Compost. Anal.. 2023;121:105384
- [CrossRef] [Google Scholar]
- Inhibition of EGFR attenuates fibrosis and stellate cell activation in diet-induced model of nonalcoholic fatty liver disease. BBA - Mol. Basis Dis. 2018;1864(1):133-142.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev.. 2001;46(1):3-26.
- [CrossRef] [Google Scholar]
- Advances in studies on chemical compositions of Alismatis Rhizoma and their biological activities. Chin. J. Chin. Mater. Med.. 2020;45(7):1578-1595.
- [CrossRef] [Google Scholar]
- Phosphatidic Acid (PA) can displace PPARα/LXRα binding to the EGFR promoter causing its transrepression in luminal cancer cells. Sci. Rep.. 2015;5(1):15379.
- [CrossRef] [Google Scholar]
- Simple selection criteria for drug-like chemical matter. J. Med. Chem.. 2001;44(12):1841-1846.
- [CrossRef] [Google Scholar]
- Blood-brain barrier: interface between internal medicine and the brain. Ann. Intern. Med.. 1986;105(1):82-95.
- [CrossRef] [Google Scholar]
- How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov.. 2010;9(3):203-214.
- [CrossRef] [Google Scholar]
- Molecular docking: shifting paradigms in drug discovery. Int. J. Mol. Sci.. 2019;20(18):4331.
- [CrossRef] [Google Scholar]
- Chemical profiling of glucosinolates in cruciferous vegetables-based dietary supplements using ultra-high performance liquid chromatography coupled to tandem high resolution mass spectrometry. J. Food Compost. Anal.. 2017;61:67-72.
- [CrossRef] [Google Scholar]
- Advanced data post-processing method for rapid identification and classification of the major triterpenoids of Alismatis rhizoma by ultra-performance liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry. Phytochem. Anal.. 2023;34(5):528-539.
- [CrossRef] [Google Scholar]
- Characterization of protostane triterpenoids in dried tuber of Alisma orientalis by Q-TOF mass spectrometry in both positive and negative modes. Asian J. Chem.. 2013;25(18):10296-10304.
- [Google Scholar]
- Correction to ‘The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets’. Nucl. Acids Res.. 2021;49(18):10800.
- [CrossRef] [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31(2):455-461.
- [CrossRef] [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem.. 2002;45(12):2615-2623.
- [CrossRef] [Google Scholar]
- PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database. Nucl. Acids Res.. 2017;45(W1):W356-W360.
- [CrossRef] [Google Scholar]
- Triterpenoids from Alisma Species: Phytochemistry, structure modification, and bioactivities. Front. Chem.. 2020;8:363.
- [CrossRef] [Google Scholar]
- ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucl. Acids Res.. 2021;49(W1):W5-W14.
- [CrossRef] [Google Scholar]
- Pharmacokinetics and tissue distribution of five major triterpenoids after oral administration of Rhizoma Alismatis extract to rats using ultra high-performance liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal.. 2017;146:314-323.
- [CrossRef] [Google Scholar]
- Network pharmacology combined with metabolomics and lipidomics to reveal the hypolipidemic mechanism of Alismatis rhizoma in hyperlipidemic mice. Food Funct.. 2022;13(8):4714-4733.
- [CrossRef] [Google Scholar]
- Establishing a rapid classification and identification method for the major triterpenoids of Alisma orientale. Phytochem. Anal.. 2020;31(3):384-394.
- [CrossRef] [Google Scholar]
- “Force iteration molecular designing” strategy for the systematic characterization and discovery of new protostane triterpenoids from Alisma Rhizoma by UHPLC/LTQ-Orbitrap-MS. Anal. Bioanal. Chem.. 2021;413(6):1749-1764.
- [CrossRef] [Google Scholar]
- Authentication of herbal medicines from multiple botanical origins with cross-validation mebabolomics, absolute quantification and support vector machine model, a case study of Rhizoma Alismatis. Arab. J. Chem.. 2022;15(10):104118
- [CrossRef] [Google Scholar]
- Chemical analysis of Chrysosplenium from different species by UPLC-Q exactive orbitrap HRMS and HPLC-DAD. J. Pharm. Biomed. Anal.. 2022;218:114861
- [CrossRef] [Google Scholar]
- Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun.. 2019;10(1):1523.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105793.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




