

Translate this page into:
Rapid identification of natural acetylcholinesterase inhibitors from Glycosmis parviflora stem utilizing dereplication, in vitro and in silico approach
⁎Corresponding authors at: School of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei 11031, Taiwan (S.-J.Hsu, C.-K.Lee). juliashu1101@gmail.com (Su-Jung Hsu), juliahsu@tmu.edu.tw (Su-Jung Hsu), cklee@tmu.edu.tw (Ching-Kuo Lee)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Dereplication approach to rapidly identify AChE-interacting compounds by comparing UPLC-MS/MS profile screening. The potential AChE inhibitors of eleven were isolated and identified from Glycosmis parviflora stems, including a new alkaloid. AChE inhibitors of Glycosmis parviflora stem interact with the peripheral anionic site of Acetylcholinesterase. Four of these potent AChE inhibitors exhibited mixed-type inhibition by binding to either the allosteric site or the substrate-binding site of AChE. ADMET predictions indicate favorable blood–brain barrier permeability for three compounds, and relatively lower permeability for one compounds.
Abstract
Acetylcholinesterase (AChE) inhibition is a significant strategy for preventing Alzheimer's disease (AD) and neurodegenerative diseases. In this study, a dereplication system was utilized to rapidly identify and characterize acetylcholinesterase-interacting compounds by comparing UPLC-MS/MS profile screening approach and molecular docking analysis, derived from the extracts and fractions of the stem of Glycosmis parviflora (Sims) Little. Eleven potential AChE inhibitors were isolated and identified from the ethyl acetate extract of G. parviflora, including an undescribed alkaloid (9), namely glybomine D, eight known alkaloids (1−8), a flavonoid (10), and a phytosterol (11). The inhibitory potential of these compounds against AChE was assessed, with O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), skimmianine (4) and arborine (2), regarded as effective inhibitors, yielding IC50 values of 39.81 μM, 41.53 μM, 49.40 μM, and 59.92 μM, respectively. Notably, O-methylglycosolone exhibited the highest potency. Four of these potent AChE inhibitors exhibited mixed-type inhibition. However, O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), and arborine (2) were first reported modulating with acetylcholinesterase activity. Furthermore, molecular docking revealed O-methylglycosolone (6) superior binding affinity (–23.749 kcal/mol) compared to other compounds, mainly by interacting with the peripheral anionic site of AChE, which forms hydrogen bonds and hydrophobic forces may play an important role, interaction with amino acid residues such as Tyr341, Tyr72, Ser293, and Arg296 in the active cavity, which is crucial for effective and selective inhibition of AChE activity. ADMET predictions suggest that arborine (2), skimmianine (4), and O-methylglycosolone (6) demonstrate favorable permeability across the blood–brain barrier, while 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1) exhibits comparatively reduced permeability. These findings highlight the potential of these compounds as natural AChE inhibitors for treating neurodegenerative diseases.
Keywords
Glycosmis parviflora
Alzheimer's disease
Acetylcholinesterase inhibitor
Dereplication
Molecular docking

- AD
-
Alzheimer's disease
- AChE
-
Acetylcholinesterase
- BuChE
-
Butyrylcholinesterase
- ChE
-
Cholinesterase
- EtOAc
-
Ethyl acetate
- BuOH
-
n-butanol
- MeOH
-
Methanol
Abbreviations
1 Introduction
Alzheimer's disease (AD) is the leading cause of dementia among the elderly, affecting over 45 million people, and is projected to triple by 2050 due to increased life expectancy (Prince et al., 2015). Cognitive decline and neuronal loss are hallmarks of AD (Selkoe, 2002). Its pathology is explained by two hypotheses, the cholinergic and amyloid hypotheses (Ali et al., 2016). According to the cholinergic hypothesis, AD is caused by a lack of acetylcholine, a neurotransmitter essential for memory and cognition. It is proposed that cholinergic neuron loss in the brain contributes to the cognitive deficits seen in AD (Mukherjee et al., 2007; Schliebs and Arendt, 2011). Acetylcholinesterase (AChE) quickly hydrolyzes acetylcholine, butyrylcholine delivered into the synaptic cleft, and blocking this catabolic process results in increased neurotransmitter levels, which may partially recover the cholinergic deficit found in AD (Beard et al., 1995). Butyrylcholinesterase (BuChE) is primarily expressed in glial cells and the white matter of the human brain. As indicated by its name, this enzyme chiefly facilitates the breakdown of butyrylcholine. Importantly, in AD, a significant elevation is observed in the BuChE levels (Heo et al., 2020). Acetylcholinesterase and butyrylcholinesterase are constituents of the cholinesterase enzyme family. The distinction between these enzymes lies in their respective distributions; AChE predominantly occurs in the bloodstream and neural synapses, whereas BuChE is primarily present in hepatic tissues and other organs such as the intestine and heart (Durmaz et al., 2022). As cholinergic markers have been observed to exhibit a significant reduction in postmortem brain samples from patients diagnosed with AD (Chear et al., 2016).
AD treatments currently focus on symptomatic relief through cholinesterase inhibitors and NMDA receptor antagonists (Melnikova, 2007). The inhibition of cholinesterase (ChE) is one of the critical strategies for treating Alzheimer’s disease (Saleem et al., 2023). Cholinesterase inhibitors enhance cholinergic activity by inhibiting enzymes (AChE and/or BuChE); this treatment improves behavioral symptoms in Alzheimer's disease AD patients (Mushtaq et al., 2014). Available cholinesterase inhibitors have had the disadvantages of unexpected side effects and short validity periods (Tuzimski and Petruczynik, 2022).
Physostigmine, a plant alkaloid and the first acetylcholinesterase inhibitor used in AD treatment was discontinued clinically due to its peripheral side effects despite its ability to cross the blood–brain barrier (Thal et al., 1983; Mushtaq et al., 2014). Currently, there are four drugs such as donepezil, galantamine, rivastigmine, and memantine, available on the market for the treatment of Alzheimer's disease (Boada-Rovira et al., 2004; Sharma, 2019). However, the effectiveness of these drugs is limited, and they exhibit various dose-related side effects, such as nausea, vomiting, and muscle cramps (Weinstock, 1999). Therefore, the discovery of safe, effective, and potent inhibitors with minimal toxicity has become a critical focus in medical research to address the urgent need for new treatments against AD.
Previously, the dereplication approach was employed, which compared the sample-treated enzymes and sample-untreated enzymes (Hsu et al., 2021) by using the UPLC/MS-MS profile. Subsequently, effectively identified the active compounds that inhibited xanthine oxidase. This same methodology was applied in this research to find natural constituents with significant therapeutically targeting acetylcholinesterase, which is crucial for treating various neurological disorders. Additionally, LC/MS spectrum may reveal the presence of the secondary metabolite compounds (Saleem et al., 2020).
Medicinal plants have been used for centuries for therapeutic purposes worldwide. Their derivatives underpin both ancient and modern medicine and are frequently discovered in commercial medications. According to scholarly literature, around one-fourth of prescription drugs are thought to be derived from plants or herbs (Saleem et al., 2022).
The genus Glycosmis consists of evergreen, globous shrubs within the Rutaceae family, renowned for their orange berries (Teja et al., 2021). Species Glycosmis parviflora, which has a long history in traditional medicine, the root and stem bark have been used in folk medicine to cure inflammation of the skin, scabies, boils, and wounds on the skin (Liu et al., 2014). Despite its potential therapeutic benefits, an evaluation of cholinesterase activity within the stem of G. parviflora remains insufficiently represented in existing literature. Addressing this gap constitutes a significant objective for the current study. Of notable significance is the examination of natural alkaloids, distinguished by their intricate nitrogen-containing structures, which may influence the active site of cholinesterase, thereby positioning them as pivotal sources for investigating anti-cholinesterase inhibitors (Konrath et al., 2013; Saleem et al., 2023). According to scientific literature, the chemical composition of G. parviflora, including alkaloids like carbalexine A-C and sulfur-containing amide derivatives, has been documented (Hofer and Greger, 2000; Yasir et al., 2019).
A previous study revealed AChE inhibition effects from an isolated alkaloid within the Glycosmis genus (Cardoso-Lopes et al., 2010; Yang et al., 2012). However, despite this promising finding, an investigation has yet to delve into the AChE activity of the stem parts of G. parviflora. Inspired by this unexplored potential, this study aims to discover AChE-inhibiting components within G. parviflora stem sourced from Vietnam using a rapid dereplication system, which integrated enzyme-interacting dereplication of UPLC-MS/MS profiling. Furthermore, there is an intention to shed light on their plausible action mechanisms through molecular docking analysis, in silico ADMET (Absorption, distribution, metabolism, excretion, and toxicity), and prediction of compounds to blood–brain barrier permeability, thus providing a deeper understanding of their therapeutic potential.
2 Materials and methods
2.1 Plant materials
The fresh stems of Glycosmis parviflora (Sims) Little were collected in March 2021 from Daklak, Vietnam. The plant voucher (VGP202105) was authenticated by Dr. Thanh-Hoa Vo (Vietnam National University Ho Chi Minh City, Vietnam), deposited at the School of Medicine, Vietnam National University Ho Chi Minh City, Vietnam, and the Department of Pharmacognosy, College of Pharmacy, Taipei Medical University, Taipei, Taiwan.
2.2 Extraction
The dried stems of G. parviflora (2.5 kg) were extracted thrice with ethanol 95 % at 50 °C. The crude extract (120.0 g) was obtained by evaporating the filtrate under vacuum. The crude extract was suspended in the distilled water and partitioned with ethyl acetate (EtOAc) and n-butanol (BuOH) to obtain an EtOAc residue (42.0 g), a BuOH residue (49.0 g), and an aqueous residue (21.0 g), respectively. The anti-AChE activity of these layers was investigated.
2.3 Cholinesterase inhibitory activity assay
The cholinesterase inhibitory activity analysis was performed as described by Ellman et al. (Ellman et al., 1961) with slight modifications. AChE from electric eel and BuChE from equine serum was provided by Sigma Aldrich (CAS No. 9000-81-1, 9001-08-5, respectively). In brief, assays were performed using the concentration at 0.03 U/mL of AChE (Enzyme was dissolved in Tris buffer solution pH 8.0) in the presence of 1.5 mM 5,5′-Dithiobis (2-nitrobenzoic acid),1.5 mM acetylthiocholine iodide, and inhibitors in 200 µL reaction mixtures, and continuously monitored for 10 mins at 405 nm, 37 °C. 5,5′-Dithiobis (2-nitrobenzoic acid) was used for color development, caused by a reaction between it and thiocholine (a product of the hydrolysis by AChE and BChE). BChE activity was assayed using the same method, but butyrylthiocholine iodide, and Tris buffer solution pH 7.4 were used instead of acetylthiocholine iodide, and Tris buffer solution pH 8.0, respectively. Physostigmine (Santa Cruz, Texas, USA, CAS No. 57-47-6) at the dose of 2.75 µg/mL was used as a positive control. The control group was assigned 100 %, and the compound (or the fraction)-treated groups were calculated relative to the control. Percentages of inhibitory activity were calculated with the following formula: where in:
ASE = absorbance of sample experiment (with ChE).
ASC = absorbance of sample control (without ChE).
AEC = absorbance of experimental control (with ChE).
ABC = absorbance of blank control (without ChE).
2.4 Preparation of acetylcholinesterase-substracted EtOAc layer extract for UPLC-MS/MS
The previously outlined method (Hsu et al., 2021) for removing acetylcholinesterase binding components was adhered to. The blank solution was made up of 50 µL of EtOAc extract of the stem of G. parviflora (20.000 ppm in dimethyl sulfoxide), 325 µL of phosphate buffer, and 150 µL of tris buffer. In parallel, instead of utilizing 150 µL of Tris buffer, 50 µL of Tris buffer and 100 µL of AChE were added to the test solution. Both solutions underwent stirring and were incubated for 40 min in a 37 °C water bath. Regarding that, 475 µL of acetonitrile was added to each solution to terminate the enzyme reaction. Both test solutions were filtered through 0.22 µm with a final concentration of 1000 ppm. UPLC-MS/MS was implemented to analyze the membrane working solutions.
2.5 Acetylcholinesterase dereplication of UPLC/MS/MS metabolome profiles analysis
The sample was analyzed using a UPLC system (ACQUITY UPLC; Waters, USA) coupled with a linear ion trap-orbitrap mass spectrometer (Q Exactive Plus Hybrid Quadrupole Orbitrap Mass (Thermo Fisher Scientific Inc., USA). Separation was achieved using an AcclaimTM RSLC PA2 column (2.2 μm, 120 Å, 2.1 mm x 150) (Thermo Fisher Scientific Inc., Waltham, MA, USA) at a flow rate of 0.3 mL/min with a gradient program of mobile phase A (dd water) and B (MeOH). The gradient program included a 5–30 % phase B from 0-3 min, 30–60 % phase B from 3 − 7 min, and 60–100 % phase B from 7 − 11 min. The column was washed with 100 % phase B for 2 min and re-equilibrated for 3 mins. The mass spectrometer was operated in both positive and negative ion modes using the Xcalibur software (version 2.0, Thermo Fisher Scientific, Bremen, Germany) with high-resolution full scan cycles (m/z 100–1500) and dd MS2. The sample was injected at a volume of 2 µL.
For finding the disparity in metabolome profiles between the EtOAc extract (Blank) and the AChE protein-subtracted extract (Test), data containing retention time, m/z, and peak intensity from UPLC–ESI–MS analyses were exported as text files. Subsequent processing involved the utilization of Xcalibur software and Microsoft Excel 2010 for manual pre-processing. Comparative analysis was conducted to discern differences in composition profiles between the Blank and Test extracts. The intensity ratio for each peak in the Test extract chromatogram, relative to the Blank extract, was calculated using the provided equation. Peaks exhibiting an intensity change exceeding 10 % were considered significantly altered due to AChE protein subtraction, designating them as potential AChE-interacting compounds (Hsu et al., 2021). Identification of chemical formulas and structures corresponding to these peaks was achieved by interrogation the ChemSpider and Reaxys databases.
2.6 Isolation and identification
The bioactive EtOAC residue (42.0 g) was fractionated into twelve fractions (Fr.1 ∼ 12) using the MPLC system (Isolera ONE, Uppsala, Sweden) with a silica gel column (Biotage SNAP, KP-Sil, 340.0 g), the gradient mixture of n-hexane–ethyl acetate (70:30, 60:40, 50:50, 25:75, 0:100, v/v) and the mixture of acetone–methanol (MeOH) (1:1, v/v). The semi-preparative HPLC method was conducted on six fractions with Luna silica gel column (250 × 10 nm, 5 µm Silica (2) 100 Å). The mixture of n-hexane – EtOAc (7:3, v/v) was eluted to obtain compounds 6 (5.6 mg), compound 8 (6.7 mg), and 9 (4.6 mg) from fraction Fr.2. Four pure compounds were yielded from Fr.4 by using the similar mobile phase, including compounds 3 (13.0 mg), 4 (13.8 mg), 5 (5.9 mg) and 7 (6.1 mg). When using the mixture of n-hexane – EtOAc (6:4, v/v), compound 1 (32.4 mg) was purified from Fr.5, and from Fr.7 and Fr.8 obtained compound 10 (46.0 mg) when using the mobile phase of n-hexane – EtOAc (5:5, v/v); Eluted by 100 % EtOAc for Fr.12, compounds 11 (17.6 mg) and 2 (25.6 mg) were yielded, respectively as show in Table S1. The extraction and isolation procedure had been repeated to obtain the required amount of each compound for chemical and biological experiments.
The structures of isolated compounds were elucidated by ESI-MS, 1D, and 2D NMR data and compared with the literature references. All NMR spectra were recorded on Bruker spectrometers (Rheinstetten, Germany) at 300 MHz, 500 MHz or 600 MHz with methanol‑d4, dimethyl sulfoxide-d6, acetone‑d6, or chloroform-d as solvent and tetrametylsilane as reference. The UV–Vis spectra were recorded with a Hitachi L-7455 Photo Diode Array Detector (Japan). ESI-MS data was obtained on a Q Exactive Plus Hybrid Quadrupole Orbitrap Mass (Thermo Fisher Scientific Inc., USA).
2.7 Kinetic studies of acetylcholinesterase inhibition
Kinetic studies were performed with the same test conditions, using seven concentrations of substrate acetylthiocholine iodide (0.1, 0.125, 0.167, 0.25, 0.5, 1, and 2 mM) and two concentrations of inhibitor at around their respective IC50 values. The initial rates were determined based on the absorbance increase at 405 nm observed between 0.5 and 5 min. The Lineweaver-Burk plots were plotted as Eq: Where v, [S], and Km values represent the rate of the enzymatic reaction, the concentration of substrate, and the Michaelis-Menten constant, respectively.
2.8 Molecular docking
The CDOCKER algorithm in Discovery Studio 2021 (Accelrys Software, Inc., San Diego, CA, USA) was used to assess the probable molecular binding mode between four potential AChE inhibitors, physostigmine as a positive control, and the acetylcholinesterase enzyme. The Protein Data Bank (https://www.rcsb.org/pdb/) was used to obtain the crystal structure of acetylcholinesterase (PDB ID: 1C2B). The compounds' 2D structures were created in ChemBioDraw Ultra 13.3 and converted to standard 3D formats in DS 2021. The transformed structures were then subjected to energy reduction in the CHARMm force field using the conjugate gradient approach (convergence criterion of 0.001 kcal/mol) (Brooks et al., 1983). Following that, the energy-minimized structures were implemented in the molecular docking experiments.
The apo-crystal structure of the acetylcholinesterase receptor (PDB code: 1C2B) was created by eliminating water molecules and adding hydrogen atoms. The available module represented missing loop sections in the prepared protein. Subsequently, the protein structure was subjected to ionization and protonation computations, followed by a final round of energy minimization optimized for molecular docking. The docking analysis binding site was determined by referring to the PDB site records and then updated using the binding site module. Upon this, the molecular docking analysis focused on the discovered binding site sphere within the 1C2B structure (coordinates: x = 40.1361, y = 84.2962, z = 29.1651; radius = 18.9). Then, using the CDOCKER program included in DS 2021, the energy-minimized structures of the four selected candidate compounds, positive control, were docked into the binding site of 1C2B. Each compound, 10 docking poses were generated and ranked based on CDOCKER energy which was computed by the CHARMm energy (−CDOCKER_ENERGY, interaction energy plus ligand strain) and the binding energy (−CDOCKER_INTERACTION_ENERGY). The pose with the highest CDOCKER energy score was chosen as the best match for each chemical. Furthermore, the ligand-binding free energy was calculated for each complex using the Generalized Born with Molecular Volume (GBMV) approach (Lee et al., 2003).
2.9 ADMET profiles using in silico method
In silico ADMET (Absorption, distribution, metabolism, excretion, and toxicity) profiles of each bioactive compound were forecasted using admetSAR 2.0 (https://lmmd.ecust.edu.cn/admetsar2/) to anticipate human intestinal absorption, Caco-2 permeability, blood–brain barrier permeability, P-gp inhibition, CYP3A4 substrate activity, inhibition of CYP3A4, CYP2C19, CYP2D6, and CYP1A2, hepatotoxicity, respiratory toxicity, reproductive toxicity, nephrotoxicity, plasma protein binding, and acute oral toxicity (Khanal et al., 2019; Dwivedi et al., 2021).
2.10 Statistical analysis
All data were repeated three times (n = 3) and denoted as means ± standard deviation (SD). Data was analyzed using GraphPad Prism edition 9.5.1 (GraphPad Software Inc, California, USA). The inhibitory activities of the crude extract and layers were compared using one-way ANOVA and Dunnett's Test (p-values < 0.05 and < 0.01, respectively).
3 Results
3.1 Inhibitory against cholinesterase activity of the extracts and layers from G. parviflora
To trail the activity efficacy of the extract of G. parviflora stem in preventing AD, it was fractionated into three layers: the ethyl acetate layer (EtOAc layer), the butanol layer (BuOH layer), and the aqueous layer. The findings demonstrated that the ethyl acetate layer extracted from the stem of G. parviflora displayed the highest AChE inhibition, reaching 58.79 ± 4.31 % at the concentration of 125 µg/mL, surpassing both the aqueous and n-butanol layer (Fig. 1). In comparison, the anti-acetylcholinesterase activity displayed significantly greater potency towards butyrylcholinesterase inhibition when utilizing identical extraction concentrations and layers. The BuChE inhibitory effect demonstrated a value of 26.26 ± 4.00 % for the ethyl acetate layer, representing more half the magnitude of the activity against AChE (Fig. S1). Based on these findings, it is inferred that the compounds present in the EtOAc layer possess the potential to serve as AChE inhibitors.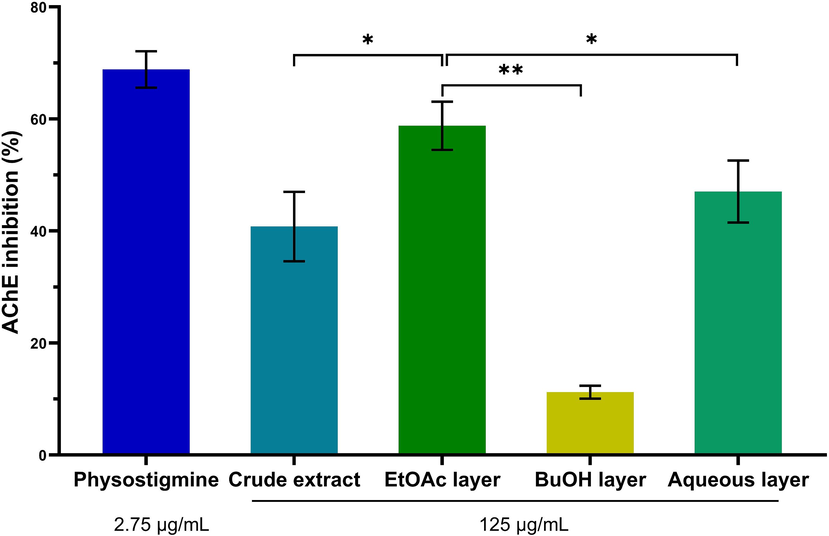
Comparative on acetylcholinesterase (AChE) inhibition between crude extract, and fractionated layers in G. parviflora stems. The crude extracts and fraction layers at 125 µg/mL, Physostigmine as a positive control at 2.75 µg/mL. The data were presented as mean ± S.D. (n = 3). EtOAc stands for ethyl acetate, and BuOH stands for n-butanol. * p-value < 0.05, ** p-value < 0.01 in comparison with the ethyl acetate layer.
3.2 Fast screening potential acetylcholinesterase-inhibiting constituents by enzyme-interacting dereplication of UPLC-MS/MS profiling
The dereplication approach was employed to rapidly screen the compounds that inhibit AChE from the EtOAc layer of the stem of G. parviflora; comparing enzyme-treated and untreated samples (Hsu et al., 2021). This process involved analyzing UPLC-MS/MS chromatographic profiles to efficiently isolate and identify active compounds inhibiting AChE. The study showed the differences in signal intensities change between AChE-treated and untreated extracts at specific peaks a–f; the representative BPI chromatograms, as depicted in Fig. 2, and Table S2, are provided herein. These differences suggested that the components associated with these peaks which were caused by the AChE protein subtraction could be potential AChE inhibitor candidates.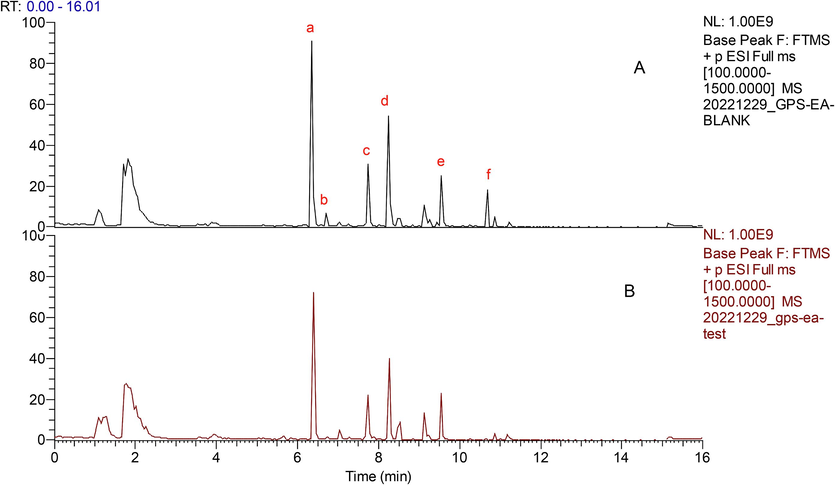
The base peak chromatograms (BPI) of untreated (A) and acetylcholinesterase −treated samples (B) of the ethyl acetate extraction from G. parviflora stems in positive mode.
3.3 Isolation and identification of the structure of the potential acetylcholinesterase inhibitors
According to the above, by this dereplication approach, potential targets were quickly narrowed down to eleven compounds from the six significant peaks. Next, merging using MPLC and semi-preparative HPLC isolated eleven compounds from the EtOAc layer of the stem of G. parviflora. As a result, eight pure alkaloids (1–9), a flavonoid (10), and a phytosterol (11) were successively isolated. Compound 9 is demonstrated as an undescribed alkaloid.
Compound 9 generated a colorless oil; [α] 28D − 27.6 (c 0.1, MeOH). The UV spectrum (MeOH) revealed three strong absorption peaks at the values of λmax 219, 261, 334 nm and one medium at λmax 294 nm, respectively (Fig. S9). The chemical formula of C20H23NO2 was determined by the positive HR ESI-MS [M + H]+ at m/z 310.1803 (calcd for a C20H24NO2 310.1807) (Fig. S2). On the 1H NMR spectrum, the signal of a para-tetrasubstituted benzene ring and an ortho-tetrasubstituted benzene ring was determined from two aromatic protons at δH [6.99 (d, 8.6, H-7); 7.16 (d, 8.6, H-7)]. Additionally, two methoxy signals were detected at δH 3.91 (s, 3H), 3.87 (s, 3H), one methyl group attached to an aromatic ring at δH 2.35 (s, 3H), one proton of NH group at δH 7.77 (s, 1H) and signals of prenyl group at δH 3.93 (m, 2H, H-1′), 5.33 (tqq, 6.5, 1.5, 1.5, H-2′), 1.93 (brs, 3H, H-4′) were also exhibited on the proton NMR analysis (Fig. S3). The structure of compound 9 was found using 13C NMR, DEPT, and HSQC data to yield five CH3, one CH2, five CH, and nine quaternary C groups (Fig. S4−6). The positions of two methoxy groups and a methyl group were established using HMBC data correlation between δH 3.91/ δC-2, δH 3.87/ δC-6 and δH 2.35/ δC-2,C-3,C-4, respectively (Fig. S7−8). Furthermore, the HMBC spectrum crosspeak of H-1′/C-5,C-6 δH-1ʹ/ δC-5,C-6 was used to identify the link of the prenyl group to the aromatic ring at the C-5 location. Other important HMBC and COSY signals are shown in Fig. 3, and 1D NMR data is clearly displayed in Table 1. Moreover, the 1D NMR spectroscopic data of compound 9 was quite comparable to that of compound 8 (Ito et al., 2004), except for substituting the –OH group to –OCH3 at the C-2 position. Based on the data presented above, the structure of the undescribed carbazole alkaloid 9 was found to be 2,6-dimethoxy-3-methyl-5-(3-methylbut-2-en-1-yl)-9H-carbazole, namely glybomine D (Fig. 4). 1H and data were measured at 500 MHz. 13C NMR data were measured at 125 MHz.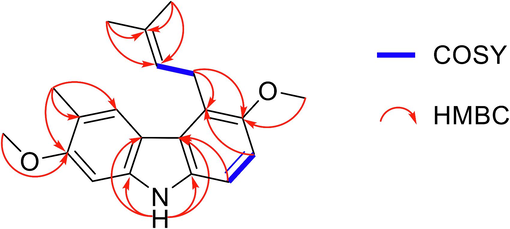
Key HMBC and COSY of compound 9.
Position
δH (J in Hz)
δC
1
6.83 (s)
92.4
2
157.2
2-OCH3
3.91 (s, 3H)
55.6
3
118.8
3-CH3
2.35 (s, 3H)
17
4
7.83 (s)
124.3
4a
116.6
4b
123.1
5
124.4
6
151.1
6-OCH3
3.87 (s, 3H)
58.12
7
6.99 (d, 8.6)
110.9
8
7.16 (d, 8.6)
107.8
8a
135.1
9a
140.6
1′
3.93 (m, 2H)
25.8
2′
5.33 (tqq, 6.5, 1.5, 1.5)
122.8
3′
132.3
4′
1. 93 (brs, 3H)
18.4
5′
1.69 (d, 1.5, 3H)
25.9
NH
7.77 (brs)
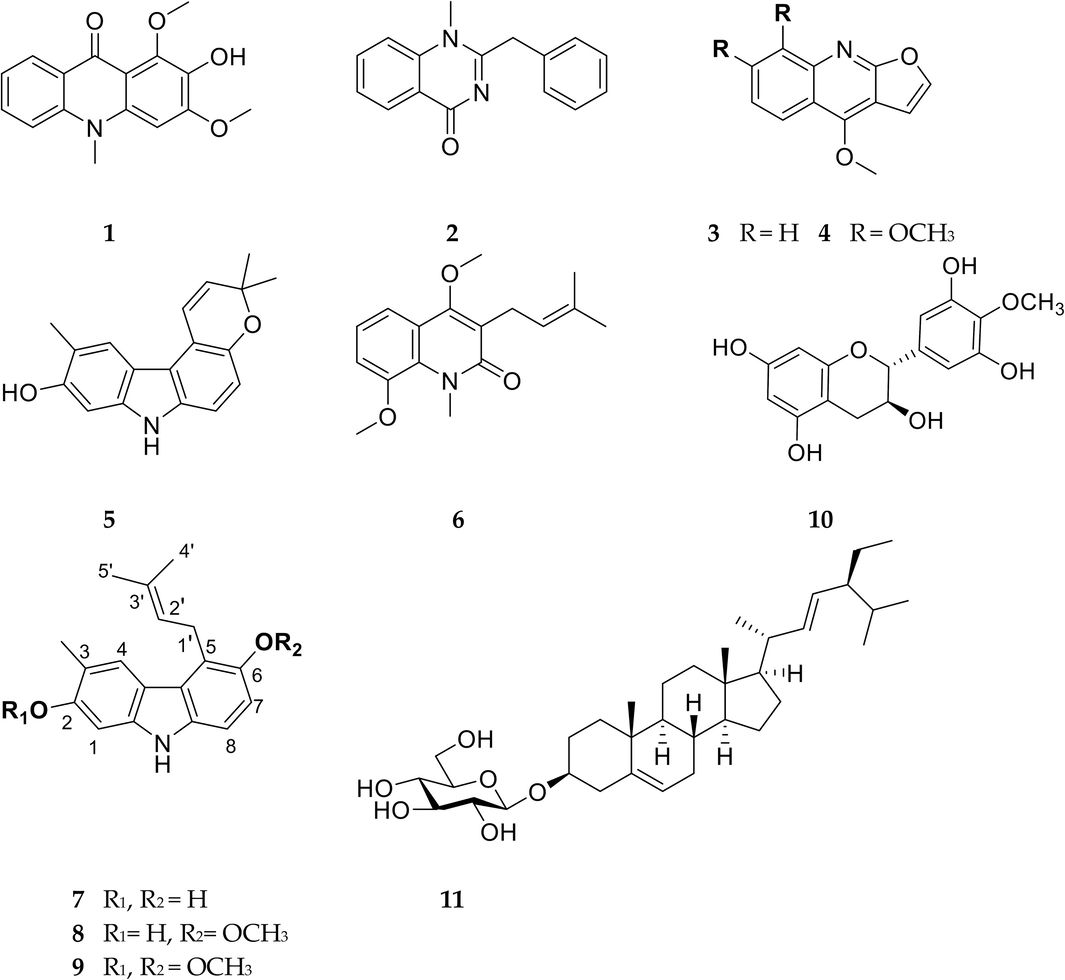
The structures of isolated compounds 1–11 from G. parviflora stems.
The other compounds were identified as 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), arborine (2), dictamnine (3), skimmianine (4), glycoborinine (5), O-methylglycosolone (6), glybomine C (7), glybomine B (8), 4′-O-Methyl-gallocatechin (10), stigmast-5,22-dien-3-O-β-D-glucopyranoside (11) (Fig. 4) by comparison with spectroscopic (1D and 2D-NMR) and LC-MS data from the literature.
Compound 1: C16H15NO4; HR-ESI-MS [M + H]+ m/z 286.1069; for 1H NMR (300 MHz, CDCl3): 8.49 (dd, 8.1, 1.8, H-8), 7.74 (ddd, 8.7, 7.0, 1.7, H-6), 7.52 (d, 8.7, H-5), 7.32 (ddd, 7.9, 7.0, 0.9, H-7), 6.31 (s, H-4), 4.03 (s, 3-OMe), 3.94 (s, 1-OMe), 3.87 (s, N-Me) (Bunalema et al., 2017) (Fig. S10).
Compound 2: C16H14N2O; HR-ESI-MS [M + H]+ m/z 251.1173; for 1H NMR (300 MHz, CDCl3): 8.38 (dd, 7.9, 1.7, 0.5, H-4), 7.72 (ddd, 8.7, 7.2, 1.7, H-1), 7.46 (d, 8.1, 7.2, 1.0, H-2, H-3), 7.25–7.38 (m, H-8, 9, 10, 11, 12, 13, 14), 4.25 (s, H-6), 3.61 (s, N-Me) (Murugan et al., 2020) (Fig. S11).
Compound 3: C12H9NO2; HR-ESI-MS [M + H]+ m/z 200.0706; for 1H NMR (500 MHz, CDCl3): 8.28 (dd, 8.5, 1.5, H-5), 8.01 (dd, 8.4, 1.0, H-8), 7.69 (ddd, 8.4, 6.7, 1.5, H-7), 7.63 (d, 2.8, H-2′), 7.45 (ddd, 8.2, 6.7, 1.2, H-6), 7.09 (d, 2.8, H-3′), 4.46 (s, 4-OMe); 13C NMR (125 MHz, CDCl3): 164.0 (C-2), 157.0 (C-4), 145.8 (C-10), 143.7 (C-2′), 129.8 (C-7), 127.9 (C-8), 123.9 (C-6), 122.5 (C-5), 118.9 (C-9), 104.9 (C-3′), 103.6 (C-3), 59.2 (C-4) (de Oliveira et al., 2016) (Fig. S12−13).
Compound 4: C14H13NO4; HR-ESI-MS [M + H]+ m/z 260.0911; for 1H NMR (300 MHz, CDCl3): 8.02 (d, 9.4, H-5), 7.58 (d, 2.8, H-2′), 7.23 (d, 9.4, H-6), 7.05 (d, 2.8, H-3′), 4.44 (s, 4-OMe), 4.10 (s, 7-OMe), 4.03 (s, 8-OMe) (Cardoso-Lopes et al., 2010) (Fig. S14).
Compound 5: C18H17NO2; HR-ESI-MS [M + H]+ m/z 280.1332; for 1H NMR (500 MHz, CDCl3): 7.83 (s, H-4), 7.21 (d, 9.8, H-1′), 7.10 (d, 8.5, H-8), 6.83 (d, 8.4, H-7), 6.80 (s, H-1), 5.80 (d, 9.8, H-2′), 2.40 (s, 3-Me), 1.48 (s, Me-4′, Me-5′); 13C NMR (125 MHz, CDCl3): 153.0 (C-2), 146.7 (C-6), 140.7 (C-9a), 134.9 (C-8a), 131.3 (C-2′), 124.0 (C-4), 120.4 (C-1′), 119.1 (C-4b), 117.5 (C-3), 116.3 (C-4a), 115.1 (C-5), 114.1 (C-7), 110.2 (C-8), 96.6 (C-1), 75.2 (C-3′), 27.4 (C-4, 5′), 16.4 (Me-3) (Chakravarty et al., 1999) (Fig. S15−16).
Compound 6: C17H21NO3; HR-ESI-MS [M + H]+ m/z 288.1595; for 1H NMR (500 MHz, CDCl3): 7.43 (dd, 8.0, 1.4, H-5), 7.16 (t, 8.0, H-6), 7.04 (dd, 8.0, 1.4, H-7), 5.26 (tq, 5.5, 1.5, H-2′), 3.95 (s, N-Me), 33.89 (s, 8-OMe), 3.87 (s, 4-OMe), 3.39 (d, 6.9, H2-1′), 1.80 (s, 4′ −Me), 1.68 (s, 5′ −Me); 13C NMR (125 MHz, CDCl3): 165.0 (C-2), 160.0 (C-4), 148.8 (C-8), 132.5 (C-3′), 130.6 (C-8a), 122.7 (C-3), 122.4 (C-6), 121.5 (C-2′), 120.2 (C-4a), 116.0 (C-5), 113.5 (C-7), 8-OMe), 56.7 (4-OMe), 35.5 (N-Me), 25.73 (5′-Me), 24.4 (C-1′), 18.0 (4′-Me) (Chakravarty et al., 1999) (Fig. S17−18).
Compound 7: C18H19NO2; HR-ESI-MS [M + H]+ m/z 282.1485; for 1H NMR (300 MHz, (CD3)2CO): 7.81 (s, H-4), 7.07 (d, 8.5, H-8), 6.85 (d, 8.4, H-7), 6.89 (s, H-1), 5.34 (m, H-2′), 3.94 (d, 6.4, H2-1′), 2.34 (s, 3-Me), 1.94 (s, 4′ −Me), 1.69 (s, 5′-Me) (Ito et al., 2004) (Fig. S19).
Compound 8: C19H21NO2; HR-ESI-MS [M + H]+ m/z 294.1496; for 1H NMR (600 MHz, CDCl3: 7.80 (s, H-4), 7.09 (d, 8.6, H-8), 6.97 (d, 8.6, H-7), 6.72 (s, H-1), 5.31 (m, H-2′), 3.91 (t, 5.7, H2-1′), 3.86 (s, MeO-6), 2.38 (s, Me-3), 1.92 (d, 1.4, Me-3′), 1.69 (d, 1.6, 5′ −Me); 13C NMR (150 MHz, CDCl3): 152.8 (C-2), 151.0 (C-6), 140.6 (C-9a), 135.2 (C-8a), 132.2 (C-3′), 124.5 (C-4), 122.8 (C-4b), 122.5, (C-2′) 117.5 (C-4a), 115.7 (C-3), 107.7 (C-8), 96.23 (C-1), 58.0 (6-OMe), 25.7 (C-1′), 25.6 (C-5′), 18.2 (C-4′), 16.3 (Me-3) (Brutting et al., 2016) (Fig. S20−21).
Compound 10: C16H16O7; HR-ESI-MS [M + H]+ m/z 319.0809; for 1H NMR (300 MHz, (CD3OD): 6.41 (s, H-2′, 6′), 5.93(d, 2.3, H-6), 5.88 (d, 2.3, H-8), 4.56 (d, 7.1, H-2), 3.96 (m, H-3), 3.79 (s, 4′-OMe), 2.82 (dd, 16.2, 5.3, H-4b), 2.51 (dd, 16.2, 7.7, H-4a) (Garcia et al., 1993) (Fig. S22).
Compound 11: C35H58O6; HR-ESI-MS [M + Na]+ m/z 597.4724; for 1H NMR (600 MHz (CD3)2SO): 5.31 (br d, 4.8, H-6), 5.16 (dd, 15.2, 8.7, H-22), 5.02 (dd, 15.2, 8.7, H-23), 4.85 (m, OH-2′, 3′, 4′), 4.42 (s, OH-6′), 4.22 (d, 7.8H-1′), 3.64 (m, H-6′), 3.46 (td, 11.2, 5.6, H-3), 3.12 (t, 8.9, H-3′), 3.07 (m, H-4′), 3.01 (m, H-5′), 2.89 (td, 8.4, 3.6, H-2′), 2.37 (m, H-1), 2.12 (t, 12.5, H-1), 1.93 (m, H2-16), 1.80 (m, H-4), 1.63 (m, H-8), 1.50 (m, H-7), 1.39 (ddd, 11.8, 8.0, 5.0, H-20), 1.23 (m, H-17), 1.15 (m, H-2), 0.99 (d, 6.6, H-19), 0.95 (s, H-9, 24), 0.89 (t, 6.3, H-29), 0.82 (s, H-26, 27), 0.65 (s, H-18).; 13C NMR (150 MHz, (CD3)2SO): 140.5 (C-5), 138.0 (C-22), 128.9 (C-23), 121.2 (C-6), 100.8 (C-1′), 77.0 (C-3), 76.8 (C-3′), 76.8 (C-5′), 73.5 (C-4′), 70.2 (C-2′), 61.1 (C-6′), 56.3 (C-14), 55.4 (C-17), 49.6 (C-9), 41.9 (C-12), 41.8 (C-13), 38.3 (C-1), 36.9 (C-4), 36.3 (C-10), 35.5 (C-20), 33.4 (C-2), 31.5 (C-24, 25), 31.4 (C-8), 31.4 (C-7), 29.3 (C-16), 25.5 (C-15), 23.9 (C-28), 22.6 (C-11), 19.1 (C-26), 19.0 (C-27), 18.9 (C-19), 18.7 (C-21), 11.8 (C-18), 11.7 (C-29) (Ridhay et al., 2012) (Fig. S23−24).
3.4 Assessment of acetylcholinesterase inhibitory activities of the eleven potential compounds
To further validate the inhibitory activities of potential AChE inhibitors identified from the EtOAc layer of the stem of G. parviflora, the inhibitory abilities of these eleven potential AChE inhibitors were measured through biochemical assays. This study utilized an acetylcholinesterase inhibition assay with physostigmine as the positive control to evaluate the effectiveness of purified compounds from the stem of G. parviflora EtOAc extracts in preventing AD. The results show that among eleven compounds, 6, 1, 4, and 2 showed lower IC50 values of 39.81 ± 1.76 µM, 41.53 ± 8.84 µM, 49.40 ± 7.10 µM, and 59.92 ± 12.71 µM, respectively, indicating that these four compounds exhibited more notable inhibitory activity towards AChE. As shown in Table 2, the remaining compounds seemed to have slight inhibition effects (IC50 > 100 µM). In particular, compound 6 showed the greatest inhibition, indicating its potential as a lead compound for acetylcholinesterase inhibitors. Results were expressed as mean ± SD (n = 3). Physostigmine was used as a positive control, IC50 = 0.75 ± 0.14 µM. IC50: the concentration required for fifty percent inhibition.
Compound
IC50 (µM)
1
41.53 ± 8.84
2
59.92 ± 12.71
3
159.72 ± 25.77
4
49.40 ± 7.10
5
162.20 ± 21.94
6
39.81 ± 1.76
7
337.79 ± 78.50
8
569.61 ± 129.83
9
251.00 ± 37.68
10
141.19 ± 26.07
11
149.13 ± 25.64
3.5 Evaluation of acetylcholinesterase kinetics for four highly potential compounds
Acetylcholinesterase inhibitors have been reported to exhibit competitive, noncompetitive, and mixed-type inhibitory (Heo et al., 2020; Zhang et al., 2023). In the present study, Lineweaver–Burk double reciprocal plots revealed that compounds 6, 1, 4, and 2 were mixed-type AChE inhibitors with a mode of action similar to physostigmine (Fig. 5) (Robaire and Kato, 1975). Subsequently, the interaction of the active compounds with the AChE enzyme was determined through molecular docking. The results suggest that compounds 6, 1, 4, and 2 inhibited the enzyme by binding with the free enzyme and the enzyme-substrate complex (Tabrez and Damanhouri, 2019).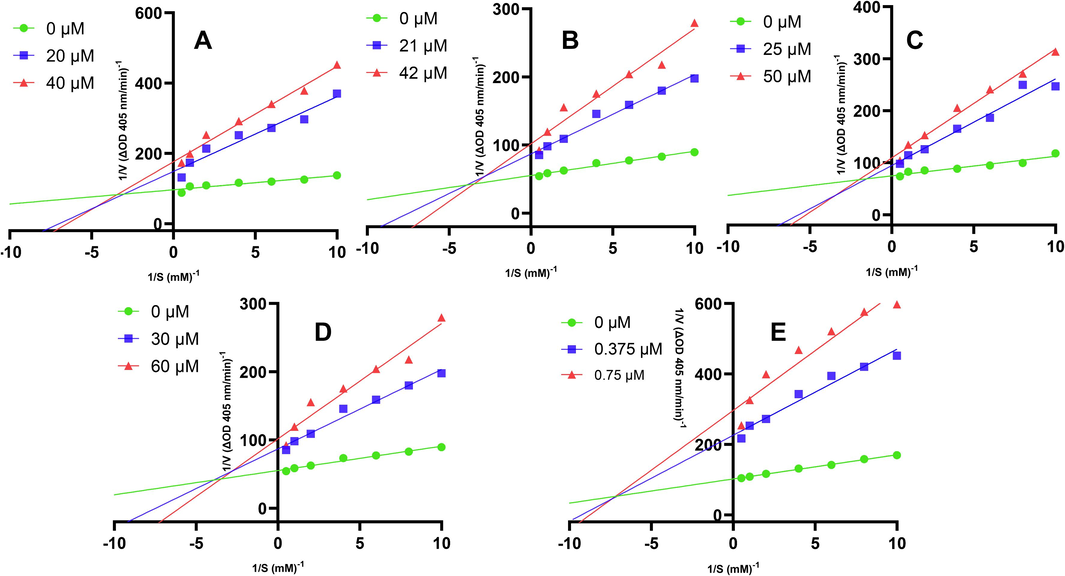
Lineweaver–Burk plots for the inhibitions of AChE by (A) O-methylglycosolone (6), (B) 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), (C) skimmianine (4), (D) arborine (2) and (E) Physostigmine; Substrates were employed at seven diferent concentrations (0.1–2.0 mM). Experiments were carried out at two inhibitor concentrations at around their respective IC50 values. Initial reaction rates are expressed as increases in absorbance per min. Each point is the average value from three independent experiment.
3.6 Mechanism of four AChE inhibitors in the binding potency of acetylcholinesterase
A molecular docking system was used to further understand the binding affinity and mechanism of the four inhibitors (compounds 6, 1, 4, and 2) inhibitory on AChE. For comparison, physostigmine was employed as a reference standard. The higher negative value of CDOCKER energy points out higher binding affinity and, consequently, higher interaction potency with acetylcholinesterase protein. As a result, the positive control, namely physostigmine and four of the potential compounds from the stem of G. parviflora including O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), skimmianine (4), and arborine (2) were reported to be as –23.749, −20.213, −13.006, −11.815, and −11.387 kcal/mol, respectively (Table 3), had negative values of CDOCKER energy, which were considered as potential AChE inhibitors. O-methylglycosolone displayed the highest docking score among the four potential compounds, with a ΔG (binding free energy) as high as −20.213 Kcal/mol, indicating its highest binding interaction. For the AChE complex, Physostigmine formed hydrogen bonds with the side chains Gln291, Ile294, Phe295, and Ser293 of the complex, and the phenyl ring formed pi-pi T-shaped with Tyr286, pi-alkyl interactions with key residues Leu286, Trp341 and Phe338 in CAS and PAS region of the complex. O-methylglycosolone (6) interaction with the AChE complex formed a hydrogen bond with Tyr341, Tyr72, Ser293, and Arg296, the phenyl ring formed pi-pi T-shaped with Tyr341, pi-alkyl interactions with Leu76, Trp286 and Phe297. Compounds 1 and 4 have similar CDOCKER binding energy that might form H-bonding with the side chains Tyr72 and Ser293, and the phenyl ring formed pi-pi T-shaped with Trp 286 of the complex. Arborine (2) only formed hydrogen bonds with the side chains Tyr341 of the complex, which might cause low binding affinity (Fig. 6, Fig. S25). The results show that hydrogen bonds and hydrophobic forces may play an important role, especially the inhibitor interaction with amino acid residues such as Tyr341, Tyr72, Ser293, and Arg296 in the active cavity, which is crucial for effective and selective inhibition of AChE activity. The docking conformation image of each compound in the selected pose is shown in Fig. 6. In addition, the CDOCKER interaction energies and binding free energies (ΔG) of the four compounds had the same trend as the IC50 values in the in vitro method, supporting those the subsequent molecular docking predictions were reliable. *Positive control, −: no interaction.
Compound
CDOCKER interaction energy (Kcal/mol)
△G (binding free energy)
(Kcal/mol)
Interaction residue
Hydrogen bond
pi-pi T-shaped
pi-alkyl
O-methylglycosolone (6)
−28.6111
−20.213
Tyr341, Tyr72,
Ser293, Arg296
Tyr341
Leu76, Trp286, Phe297
1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1)
−26.7249
−13.006
Tyr341, Tyr72, Ser293
Trp286
Leu76
Skimmianine (4)
−26.2951
−11.815
Ser293, Tyr72, Asp74
Trp286
Leu76, Typ341
Arborine (2)
−25.5012
−11.387
Tyr341
−
−
Physostigmine *
–33.8218
–23.749
Gln291, Ile294, Phe295, Ser293
Trp286
Leu289, Tyr341, Phe338
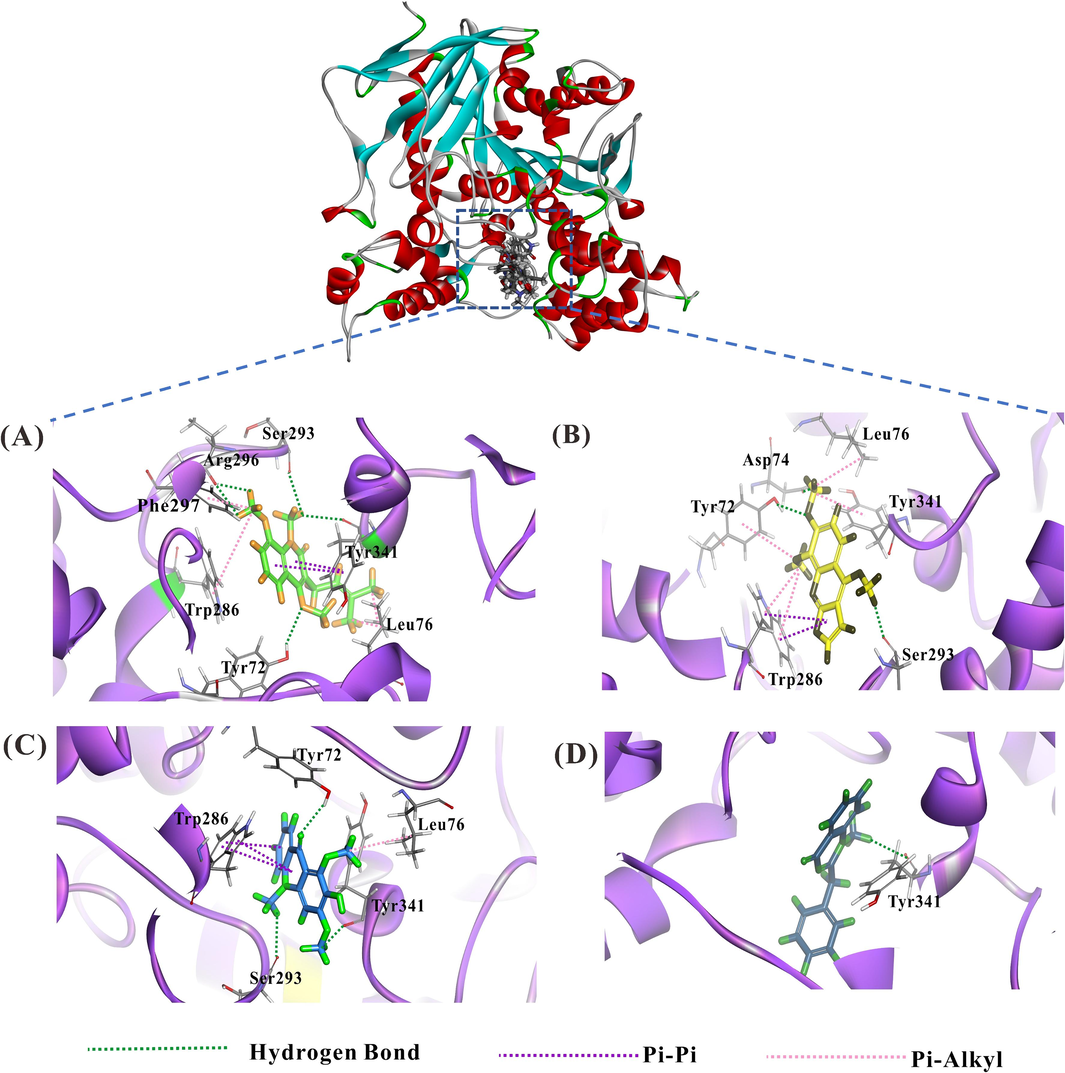
The binding modes between sites of acetylcholinesterase (PDB code: 1C2B) and AChE-inhibiting components were calculated by molecular docking. View of docking conformations for the four inhibitors binding to the active sites of active sites of acetylcholinesterase (1C2B), (A): O-methylglycosolone (6); (B): skimmianine (4); (C): 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1) and (D): arborine (2). Key residues are depicted as line models against a background, with hydrogen bonds denoted by green dashed lines. Pi-pi interactions are illustrated with purple dashed lines, while pi-alkyl interactions are indicated by pink dashed lines.
3.7 Prediction of pharmacokinetic properties
The pharmacokinetic characteristics of each bioactive compound are depicted in a heat map, illustrating human intestinal absorption (HIA), Caco-2 permeability (Caco-2p), blood–brain barrier (BBB) permeability, P-glycoprotein (P-gp) inhibition, CYP3A4, CYP2C19, CYP1A2 inhibition/substrate activity, hepatotoxicity, respiratory toxicity, reproductive toxicity, nephrotoxicity, plasma protein binding, and acute oral toxicity. The ADMET profile of each phytoconstituent is visualized as a heat map (Fig. 7). According to the ADMET prediction results, all compounds exhibit a high probability of human intestinal absorption (>90 %), which is equivalent to the result of the physostigmine-positive control. Among the compounds, 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1) has the lowest probability of blood–brain barrier penetration (42.5 %), while arborine (2) demonstrates the highest probability (97.5 %), followed by O-methylglycosolone (6) (75 %), and skimmianine (4) (65 %). Regarding pharmacokinetic distribution, all compounds are predicted to have a high probability of plasma protein binding (>70 %). Metabolism prediction suggests that the compounds primarily undergo metabolism via CYP3A4 substrate/inhibition, except for O-methylglycosolone (6), metabolized through CYP2C19 inhibition (probability = 72.9 %). O-methylglycosolone (6) is predicted to have the lowest probability of hepatoxicity (50.34 %) compared to other compounds.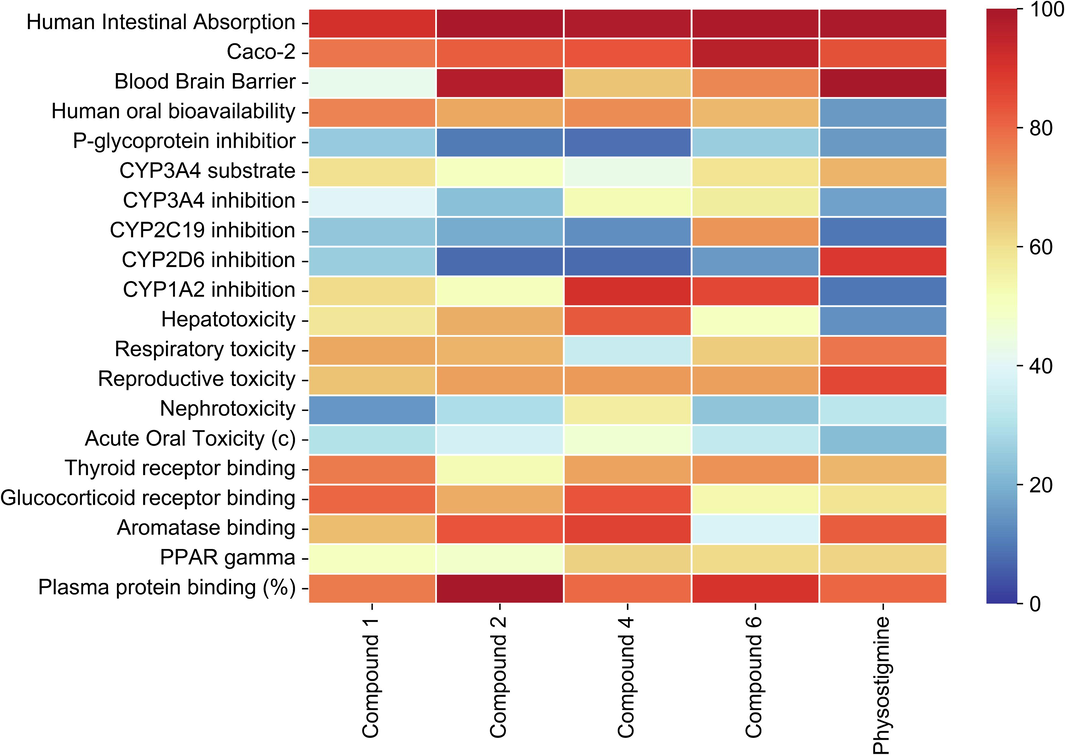
ADMET profile of bio-actives via heat map presenting 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), arborine (2), skimmianine (4), O-methylglycosolone (6), physostigmine (positive control).
4 Discussion
Acetylcholinesterase (AChE) has been demonstrated to be a potent drug target for treating neurodegenerative diseases, i.e., Alzheimer’s, Parkinson’s, and dementia (Milicevic and Sinko, 2022). In prior research, a dereplication methodology was devised to compare chromatographic profiles between enzyme-treated and non-treated extracts and then further isolate and identify the active compounds that inhibited the target enzyme (Hsu et al., 2021). Therefore, applying this approach has successfully isolated and identified of the eleven active compounds that are potential AChE inhibitors from the EtOAc layer of the stem of G. parviflora, confirming this system’s feasibility. The hypothesis was that by comparing the LC/MS fingerprinting profiles of the AChE-treated extract with the non-treated extract, the peaks that disappeared in the treated extract might have an affinity to the acetylcholinesterase, curtailing enzymatic activity for the virtual substrate, acetylthiocholine iodide, and were treated as potential enzyme-inhibiting components.
This study shows that the retention time, m/z, and intensity of peaks changed due to the AChE protein-subtracted extract. Peaks a, b, c, d, e, and f (Fig. 2) were identified as potential AChE-interacting components. Signal quality was notably enhanced in positive ion mode, resulting in a clearer chromatogram using UPLC-ESI-MS. This phenomenon may be attributed to the presence of alkaloid groups in the major components of the ethyl acetate fractions, which are easier to ionize in the positive mode, thus leading to increased signal intensity. Peaks a, d, e, and f were isolated and identified as compounds 2, 4, 1, and 6 displaying [M + H]+ ions at m/z 251.1178, 260.0916, 286.1071, and 288.1594 respectively, which corresponded to in the fractions including fraction Fr.12 (m/z 251.1178), Fr.4 (m/z 260.0916), Fr.5 (m/z 286.1071) and Fr.2 (m/z 288.1594). Peak b showed a [M + H]+ ion at m/z 274. 2739, and peak c showed a [M + H]+ ion at m/z 190.0864 was not isolated. Inferentially, the amount of these components from the fractions might be too low for isolation, causing the AChE-inhibiting potential compounds not to be obtained.
This study isolated and identified eleven potential AChE inhibitors (Table 2), including an undescribed alkaloid, which is compound 9, eight known alkaloids (1 − 8), a flavonoid (10), and a phytosterol (11). Nine of these compounds belong to the alkaloid category. A previous study by (Konrath et al., 2013) reported that natural alkaloids not only hold high potential value in pharmacological activity, including antibacterial, antifungal, antiviral, mutagenic, cytotoxic, antiplatelet, and enzyme-inhibitory effects, but also serve as important sources of AChE inhibitors (Konrath et al., 2013; Szewczyk and Peczek, 2023).
Further investigation confirmed the inhibitory activity of eleven compounds against AChE. The discovery of the compound glybomine D (9) as an undescribed alkaloid, while showing only a mild modulation of acetylcholinesterase with an IC50 of 251 µM, presents a lower value compared to the known compounds glybomine C (7) and glybomine B (8). Nevertheless, it still holds potential for further testing against other biological activities. In addition, O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), skimmianine (4), and arborine (2) exhibited inhibitory effects on acetylcholinesterase activity. Notably, O-methylglycosolone (6) was the most potent, with an IC50 value of 39.81 ± 1.76 μM. Our findings align with previous studies, indicating skimmianine (4) as a modulate AChE activity (Cardoso-Lopes et al., 2010; Yang et al., 2012), suggesting that with the alkaloid structure of quinoline, the methoxy group at the C-7 explained for more potent effects (Yang et al., 2012). Previous literature has shown skimmianine exhibits promise for Alzheimer's disease treatment by effectively inhibiting AChE and suppressing the production of nitric oxide (NO), thereby influencing inflammation within nervous tissue (Adamska-Szewczyk et al., 2016). Additionally, the neuroprotective effects of skimmianine may be attributed to its ability to inhibit neuroinflammation in LPS-activated microglia by targeting the NF-κB activation pathway (Ogunrinade et al., 2023). However, there have been no previous reports on the inhibitory activity of the other three constituents (O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1) and arborine (2)) against AChE, worthying for further research.
To understand the four potency acetylcholinesterase inhibitors catalytic action possible mechanisms using molecular docking. Previously, Cheung et al. (2012) reported the binding site of AChE has been extensively examined and that the binding site forms a deep and narrow channel gorge comprised of several distinct domains, namely the catalytic, anionic, acyl, oxyanion, and peripheral anionic (Cheung et al., 2012). The catalytic anionic site (CAS) located at the bottom of the binding site and the peripheral anionic site (PAS) situated at the entrance and within the channel cavity are particularly crucial. The PAS not only plays a pivotal role in the initial recognition of the positively charged substrate through the involvement of key amino acid residue Tyr72, Asp74, Tyr124, Trp286, and Tyr341 but also exhibits the ability to relate to modulate the activity of the catalytic surface (Richard et al., 1970).
From the molecular docking result, it was revealed that among the potential compounds from the stem of G. parviflora, O-methylglycosolone (6); 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1); and skimmianine (4) were found to interact with the PAS site of acetylcholinesterase which forming hydrogen bonds with amino acid Tyr72, Asp74, Tyr341 and Ser 293, with Try286 forming pi-pi T-shaped, as well as Leu76 and Try341 pi-alkyl bonds. These interactions help stabilize the complex-ligand and contribute to the inhibition of AChE. In contrast, arborine (2) showed weak inhibition due to the lack of amino residues with pi-pi and pi-alkyl, especially hydrogen bond residues, which are crucial for the catalytic residues of the PAS site at the acetylcholinesterase protein, resulting in the low binding affinity (−11,387 kcal/mol) and the value of IC50 (59.92 ± 12.71 µM) compared to the other potential compounds.
In addition, it has been reported that phenyl rings are also determinants of the ligand-enzyme complex stability because of their aromatic-pi stacking interactions with the key amino acid residues of the peripheral anionic site (PAS) and catalytic anionic site (CAS) (Uddin et al., 2020). In this study, methoxyl groups of O-methylglycosolone (6) and the amino acid Tyr341, Tyr72, Ser293, and Arg 296 to form hydrogen bonds at the active side of AChE protein, the phenyl ring also formed pi-pi T-shaped interactions with Tyr341, pi-alkyl interactions with key residues Leu76, Trp286 and Phe297 of CAS and PAS region as well.
It can be seen that this compound has the substrate possibility trapped by PAS, which is guided by the other domains to reach CAS. The selective binding to AChE is determined by the acyl subsite consisting of the crucial residues Leu76, Try286, and Phe297, consistent with results from a previous study (Fisher et al., 2012). These findings explained O-methylglycosolone (6) exhibited the best binding affinity (−20.213 kcal/mol) and the lowest IC50 value (39.81 ± 1.76 µM) among all potential AChE inhibitory compounds. The results from the in vitro AChE inhibition assay were trend consistent with the CDOCKER interaction binding energy and binding free energy (△G), confirming that O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), skimmianine (4) and arborine (2) were potent AChE inhibitor in the ethyl acetate layer of the extract from the stem of G. parviflora.
It is said that the blood–brain barrier is a formidable obstacle to the entry of most drugs into the brain. This property is attributed to tight junctions resembling epithelial barriers within the endothelium of brain capillaries. In Alzheimer's disease, there's a need for drugs capable of passing this barrier, which is not merely a physical separation between blood and brain but functions as a selective filter, allowing only specific molecules to penetrate the brain. Some small molecule drugs may traverse the BBB through lipid-mediated diffusion, provided they meet certain criteria, such as having a molecular weight below 400 Da and forming fewer than eight hydrogen bonds (Pardridge, 2012; Jakki et al., 2018). O-methylglycosolone (6), 1,3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1), skimmianine (4) and arborine (2) have been demonstrated to possess potent AChE inhibition activity through in vitro screening and have been further validated via molecular docking. Remarkably, ADMET predictions suggest that arborine (2), skimmianine (4), and O-methylglycosolone (6) exhibit good blood–brain barrier permeability, whereas 3-dimethoxy-2-hydroxy-10-methyl-9(10H)-acridinone (1) demonstrates lower permeability. Therefore, these compounds could be promising candidates for AD treatment and warrant further investigation in future studies.
A significant advantage is offered by the approach through the implementation of dereplication treatment. It enables the rapid identification of potentially valuable compounds with an affinity for target AChE by comparing the UPLC metabolomic profiles of treated and untreated materials. The utilization of dereplication markedly reduces the number of compounds requiring further MS/MS analysis, thereby streamlining the process and minimizing both cost and labor.
The ongoing challenge of deficient therapeutic options for neurological disorders, such as Alzheimer's disease, underscores the urgent need for researchers to expedite the discovery of novel therapeutic agents. In the current study, a dereplication strategy was aimed to be elucidated for identifying lead compounds in plant materials, yielding promising results that highlight the underexplored potential of this methodology in investigating natural acetylcholinesterase inhibitors for therapeutic purposes.
5 Conclusion
The prevalence of Alzheimer's disease is continuously rising, creating an urgent need for new AChE inhibitors. This study indicates that the stem of G. parviflora represents a promising natural source of AChE inhibitors and may be utilized in the prevention or treatment of related drug development. Through enzyme inhibitory activity and molecular docking analysis demonstrated that four isolated alkaloids out of eleven compounds inhibited AChE. In addition, molecular docking analysis declares those inhibitors' binding affinity mainly by interacting with the peripheral anionic site of AChE, which forms hydrogen bonds and hydrophobic forces. In silico ADMET studies, these compounds have a good blood–brain barrier permeability in the pharmacokinetic profile. This approach not only proves the effectiveness but also finds new applicable compounds. These findings indicate that the alkaloids derived from the stem of G. parviflora possess the potential to be employed as a natural therapy for the treatment of Alzheimer's disease. Further research can be conducted using neuronal cells and animal models.
Funding
This research was supported by the Ministry of Science and Technology of the Republic of China (Taiwan) [grant number MOST [110-2320-B-038 -039 -MY3].
CRediT authorship contribution statement
Truc-Ly Thi Duong: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. Ta-Wei Liu: Formal analysis, Methodology, Software. Quoc-Dung Tran Huynh: Formal analysis, Investigation, Methodology, Software, Writing – original draft. Dang-Khoa Nguyen: Formal analysis, Methodology, Software, Writing – original draft. Yun-Han Wang: Methodology. Man-Hsiu Chu: Methodology. Thanh-Hoa Vo: Resources. Su-Jung Hsu: Conceptualization, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. Ching-Kuo Lee: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing.
Acknowledgments
The authors would like to acknowledge the NMR data acquisition 333 system Core Facility at the Taipei Medical University and UHPLC-Obitrap-MS system Core Facility at National Taiwan University.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Ching-Kuo Lee reports financial support was provided by National Science and Technology Council. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Furochinoline alkaloids in plants from Rutaceae family—A review. Curr. Issues Pharm. Med. Sci.. 2016;29:33-38.
- [CrossRef] [Google Scholar]
- Anti-Alzheimer's disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med.. 2016;9:103-111.
- [CrossRef] [Google Scholar]
- The prevalence of dementia is changing over time in Rochester, Minnesota. Neurology. 1995;45:75-79.
- [CrossRef] [Google Scholar]
- Efficacy and safety of donepezil in patients with Alzheimer's disease: results of a global, multinational, clinical experience study. Drugs Aging. 2004;21:43-53.
- [CrossRef] [Google Scholar]
- CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem.. 1983;4:187-217.
- [CrossRef] [Google Scholar]
- Synthesis of Glycoborine, Glybomine A and B, the Phytoalexin Carbalexin A and the beta-Adrenoreceptor Antagonists Carazolol and Carvedilol. Chemistry. 2016;22:16897-16911.
- [CrossRef] [Google Scholar]
- Potential of Zanthoxylum leprieurii as a source of active compounds against drug resistant Mycobacterium tuberculosis. BMC Complement. Altern. Med.. 2017;17:89.
- [CrossRef] [Google Scholar]
- Alkaloids from stems of Esenbeckia leiocarpa Engl. (Rutaceae) as potential treatment for Alzheimer disease. Molecules. 2010;15:9205-9213.
- [CrossRef] [Google Scholar]
- Carbazole alkaloids from roots of Glycosmis arborea. Phytochemistry. 1999;50:1263-1266.
- [CrossRef] [Google Scholar]
- Cholinesterase inhibitory activity and chemical constituents of Stenochlaena palustris fronds at two different stages of maturity. J. Food Drug Anal.. 2016;24:358-366.
- [CrossRef] [Google Scholar]
- Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem.. 2012;55:10282-10286.
- [CrossRef] [Google Scholar]
- Two New Labdane-type Diterpenoids and others Compounds from Conchocarpus cyrtanthus (Rutaceae) Rev. Virtual Quim.. 2016;8:87-96.
- [CrossRef] [Google Scholar]
- Synthesis, anticholinesterase activity, molecular docking, and molecular dynamic simulation studies of 1,3,4-oxadiazole derivatives. Arch. Pharm. (Weinheim). 2022;355:e2200294.
- [Google Scholar]
- Exploring the therapeutic mechanisms of Cassia glauca in diabetes mellitus through network pharmacology, molecular docking and molecular dynamics. RSC Adv.. 2021;11:39362-39375.
- [CrossRef] [Google Scholar]
- A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol.. 1961;7:88-95.
- [CrossRef] [Google Scholar]
- Chapter 13 - Acetylcholine. In: Brady S.T., Siegel G.J., Albers R.W., eds. Basic Neurochemistry (eighth Edition). New York: Academic Press; 2012. p. :258-282.
- [Google Scholar]
- Acetylcholinesterase and butyrylcholinesterase inhibitory activities of khellactone coumarin derivatives isolated from Peucedanum japonicum Thurnberg. Sci. Rep.. 2020;10:21695.
- [CrossRef] [Google Scholar]
- Sulfur-containing amides from Glycosmis species (Rutaceae) Fortschr. Chem. Org. Naturst.. 2000;80:187-223.
- [CrossRef] [Google Scholar]
- Fast dereplication of xanthine oxidase-inhibiting compounds in alfalfa using comparative metabolomics. Food Res. Int.. 2021;141:110170
- [CrossRef] [Google Scholar]
- Chemical constituents of Glycosmis arborea: three new carbazole alkaloids and their biological activity. J. Nat. Prod.. 2004;67:1488-1491.
- [CrossRef] [Google Scholar]
- The Blood Brain Barrier and its Role in Alzheimer's Therapy: An Overview. Curr. Drug Targets. 2018;19:155-169.
- [CrossRef] [Google Scholar]
- Network pharmacology-based assessment to elucidate the molecular mechanism of anti-diabetic action of Tinospora cordifolia. Clin. Phytosci.. 2019;5:35.
- [CrossRef] [Google Scholar]
- Alkaloids as a source of potential anticholinesterase inhibitors for the treatment of Alzheimer's disease. J. Pharm. Pharmacol.. 2013;65:1701-1725.
- [CrossRef] [Google Scholar]
- New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem.. 2003;24:1348-1356.
- [CrossRef] [Google Scholar]
- Gas chromatography-mass spectrometric analysis of essential oil of aerial parts of Glycosmis parviflora (Sims) Little (Rutaceae) Trop. J. Pharm. Res.. 2014;13:275-280.
- [CrossRef] [Google Scholar]
- Therapies for Alzheimer's disease. Nat. Rev. Drug Discov.. 2007;6:341-342.
- [CrossRef] [Google Scholar]
- Use of connectivity index and simple topological parameters for estimating the inhibition potency of acetylcholinesterase. Saudi Pharm. J.. 2022;30:369-376.
- [CrossRef] [Google Scholar]
- Acetylcholinesterase inhibitors from plants. Phytomedicine. 2007;14:289-300.
- [CrossRef] [Google Scholar]
- Glycosmis pentaphylla (Rutaceae): A Natural Candidate for the Isolation of Potential Bioactive Arborine and Skimmianine Compounds for Controlling Multidrug-Resistant Staphylococcus aureus. Front. Public Health. 2020;8:176.
- [CrossRef] [Google Scholar]
- Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer's disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets. 2014;13:1432-1439.
- [CrossRef] [Google Scholar]
- Neuroprotection by Skimmianine in Lipopolysaccharide-Activated BV-2 Microglia. Molecules. 2023;28
- [CrossRef] [Google Scholar]
- Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab.. 2012;32:1959-1972.
- [CrossRef] [Google Scholar]
- Prince, M., A. Wimo, M. Guerchet, et al., 2015. The global impact of dementia: an analysis of prevalence, incidence, cost and trends. World Alzheimer Report. 2015.
- On the Question: Is Acetylcholinesterase an Allosteric Protein. Mol. Pharmacol.. 1970;6:108.
- [Google Scholar]
- A STIGMASTEROL GLYCOSIDE FROM THE ROOT WOOD OF Melochia Umbellata (Houtt) Stapf var. degrabrata K. Indones. J. Chem.. 2012;12:100-103.
- [CrossRef] [Google Scholar]
- Effects of edrophonium, eserine, decamethonium, d-tubocurarine, and gallamine on the kinetics of membrane-bound and solubilized eel acetylcholinesterase. Mol. Pharmacol.. 1975;11:722-734.
- [Google Scholar]
- UHPLC-MS phytochemical profiling, biological propensities and in-silico studies of Alhagi maurorum roots: a medicinal herb with multifunctional properties. Drug Dev. Ind. Pharm.. 2020;46:861-868.
- [CrossRef] [Google Scholar]
- Buxus papillosa CK Schneid.: A comprehensive review on traditional uses, botany, phytochemistry, pharmacology, and toxicology. Biocatal. Agric. Biotechnol.. 2022;46:102547
- [Google Scholar]
- Nutritional and medicinal plants as potential sources of enzyme inhibitors toward the bioactive functional foods: an updated review. Crit. Rev. Food Sci. Nutr.. 2023;1–24
- [CrossRef] [Google Scholar]
- The cholinergic system in aging and neuronal degeneration. Behav. Brain Res.. 2011;221:555-563.
- [CrossRef] [Google Scholar]
- Cholinesterase inhibitors as Alzheimer's therapeutics (Review) Mol. Med. Rep.. 2019;20:1479-1487.
- [CrossRef] [Google Scholar]
- Furoquinoline Alkaloids: Insights into Chemistry, Occurrence, and Biological Properties. Int. J. Mol. Sci.. 2023;24
- [CrossRef] [Google Scholar]
- Computational and Kinetic Studies of Acetylcholine Esterase Inhibition by Phenserine. Curr. Pharm. Des.. 2019;25:2108-2112.
- [CrossRef] [Google Scholar]
- Traditional uses, phytochemistry, pharmacology, toxicology and formulation aspects of Glycosmis species: a systematic review. Phytochemistry. 2021;190:112865
- [CrossRef] [Google Scholar]
- Oral physostigmine and lecithin improve memory in Alzheimer disease. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc.. 1983;13:491-496.
- [Google Scholar]
- Determination of Anti-Alzheimer's Disease Activity of Selected Plant Ingredients. Molecules. 2022;27
- [CrossRef] [Google Scholar]
- Evaluation of Cholinesterase Inhibitory Potential of Different Genotypes of Ziziphus nummularia, Their HPLC-UV, and Molecular Docking Analysis. Molecules. 2020;25
- [CrossRef] [Google Scholar]
- Selectivity of cholinesterase inhibition: clinical implications for the treatment of Alzheimer’s disease. CNS Drugs. 1999;12:307-323.
- [Google Scholar]
- Skimmianine, a furoquinoline alkaloid from Zanthoxylum nitidum as a potential acetylcholinesterase inhibitor. Med. Chem. Res.. 2012;21:722-725.
- [CrossRef] [Google Scholar]
- The genus Glycosmis [Rutaceae]: a comprehensive review on its phytochemical and pharmacological perspectives. J. Nat. Prod.. 2019;9:98-124.
- [Google Scholar]
- Sulfur-fluoride exchange (SuFEx)-enabled lead discovery of AChE inhibitors by fragment linking strategies. Eur. J. Med. Chem.. 2023;257:115502
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105811.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




