

Translate this page into:
A facile lyophilisation-based sample preparation approach for the determination of selected wastewater-borne antiretroviral drugs and metabolites by SFC-MS/MS
⁎Corresponding author. mosekiemang@ub.ac.bw (Tlou Mosekiemang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The presence of pharmaceuticals in aquatic ecosystems is at the centre-stage of research after the realization that wastewater treatment processes are generally ineffective in the removal of these entities in wastewater given the high likelihood of effluent reuse after disposal. In addition, the continued efforts to scout for new and emerging aquatic contaminants has until recently elicited proliferation of numerous analytical methods for the determination of various (un)known contaminants, since emphasis is now placed in the development of environmentally benign approaches. Herein, we propose and discuss a novel and relatively eco-friendly analytical method based on lyophilization for sample preparation and SFC-MS/MS for the determination of eight (8) antiretroviral drugs (ARVDs) and three (3) metabolites in wastewater samples. This method proved useful in the improvement for the recoveries of lamivudine (3TC) and emtricitabine (FTC) by up to 99% compared to as low as 23% with solid phase extraction (SPE) method. Indeed, previous literature reports has reported poor recoveries for the polar ARVDs, especially on reversed phase (RP) SPE. In contrast, lyophilization promoted matrix effects as evidenced by ion suppression of up to 50% experienced on late eluting compounds. Despite this, lyophilization-SFC-MS/MS method was successfully validated for the quantification of all target analytes, partial exceptions were for ritonavir metabolite (RTVM) which could not be quantified using lyophilization possibly due to lyophilization-induced losses. Generally, the obtained data has proved that lyophilization is an alternative to SPE and SFC is a suitable alternative to LC.
Keywords
Antiretroviral drugs
Lyophilization
Metabolites
SFC-MS/MS, Wastewater

1 Introduction
Human-excreted remnants of intact and bio-transformed pharmaceutical residues which are mostly resistant to wastewater treatment processes are continuously discharged into rivers and streams along with (partially)treated effluent (Mosekiemang et al., 2019; Nannou et al., 2020; Zhang et al., 2021). The implication is that despite the accomplished improvements in sewer treatment technologies, they are still ineffective to completely remove residual pharmaceutical contaminants (Muriuki et al., 2020; Nannou et al., 2020). Therefore, the perpetual release of these pharmaceutical pollutants into aquatic environments is a worrisome phenomenon due to the possible ecosystem alterations that may arise due to their presence in the environment and worse-still their likelihood to enter the human food chain (Akenga et al., 2021). The latter aspect may be true considering that nowadays effluent is considered an alternative resource to both surface- and groundwater although for purposes excluding human consumption (Yalin et al., 2023). Until now, residues of antiretroviral drugs (ARVDs) continue to be detected in raw- and treated domestic wastewaters at substantial concentrations (Abafe et al., 2018; Fekadu et al., 2019; Madikizela et al., 2020; Mlunguza et al., 2020; Mosekiemang, 2021; Mosekiemang et al., 2019; Muriuki et al., 2020; Nannou et al., 2020; Prasse et al., 2010; Wood et al., 2015). The first occurrence of ARVDs in environmental waters was reported in 2010 (Prasse et al., 2010) and from then on reported in most African countries (K’oreje et al., 2016; Madikizela et al., 2020; Mosekiemang et al., 2019; Nannou et al., 2020; Wood et al., 2015), until more recently when the first ARVDs metabolites were detected in South African domestic wastewater (Mosekiemang et al., 2021) and in the River Thames in the UK (Richardson et al., 2021). Even so, there are no known human health cases that resulted from such exposure to ARVDs-impacted water thus far, but significant toxicological effects have been reported on other living species (Cid et al., 2021). For instance, the interactive- and/or combined toxic effects of wastewater-borne ARVDs such as efavirenz, lamivudine, tenofovir etc., has been demonstrated on phytoplankton (Kitamura et al., 2023). Undoubtedly, these represent a category of emerging and environmentally persistent contaminants that necessitate monitoring especially in regions experiencing high consumption per capita for these drugs (Abafe et al., 2018; Mlunguza et al., 2020; Mosekiemang et al., 2019; Muriuki et al., 2020; Peng et al., 2014; Prasse et al., 2010; Wood et al., 2015).
Regular environmental monitoring for the presence of ARVDs and their metabolites in aquatic environment require extremely accurate and highly reliable analytical methods. A typical analytical technique used for this purpose is the liquid chromatography-tandem mass spectrometry (LC-MS/MS) due to its excellent selectivity and sensitivity conferred by the tandem-MS configuration and/or the multiple reaction monitoring (MRM) scanning mode (Chen et al., 2016; Hermes et al., 2018; Nannou et al., 2019). In this mode, the mass-to-charge ratios (m/z) for the precursor ions and their respective fragment ions (i.e., transition ion pairs/triplets) are mass-selected and exclusively monitored throughout the successive stages of the MS instrument (i.e., in a tandem-in-space MS fashion). This method is designed to exclusively monitor targeted transition ions during the MS cycle time (tcycle), in this way conferring excellent sensitivity and detectability especially for trace level ions contained in complex matrix (Chen et al., 2016; Hermes et al., 2018).
A comparable alternative and yet a low-cost chromatographic mode for the (non)targeted determination of several wastewater-borne polar pharmaceutical pollutants is the supercritical fluid chromatography coupled to tandem MS (SFC-MS/MS) due to the several analytical benefits it offers relative to the liquid chromatography (LC) (Si-hung et al., 2023; Si-hung and Bamba, 2022). In fact, the SFC-MS/MS method is now evolving rapidly in other fields except for environmental applications (Si-hung and Bamba, 2022). Some of the advantages of SFC-MS/MS over LC-MS/MS include the lower viscosity and diffusivity attributes of the gaseous CO2-based mobile phase which enables faster chromatographic separation (Desfontaine et al., 2015; Desfontaine and Guillarme, 2015; Dispas et al., 2016; Romand et al., 2016; Svan and Hedeland, 2018; Tarafder, 2018). Especially noteworthy is that the achievable high flow rates obtainable by modern SFC instruments are fostered by the advent of finer particle size columns (i.e., sub 2 µm-particle sizes) which allow for faster run-times without loss of chromatographic efficiency (Grand-Guillaume Perrenoud et al., 2014; Nováková et al., 2014; C. West and Lesellier, 2006). In addition, the gaseous CO2 and small amount of organic solvent mobile phase used in SFC creates a perfect match with the electrospray ionization (ESI-MS) and facilitates analyte ionization and improved detection by the MS detector (Bieber et al., 2023). Despite these attributes, this technique (SFC-MS/MS) has found limited application in the determination of ARVDs in wastewater (Si-hung and Bamba, 2022; Svan and Hedeland, 2018; Wang et al., 2022).
Instrumental assaying is normally preceded by a lengthy and often challenging process based on solid phase extraction (SPE) that entails analyte extraction-, purification- and enrichment steps which are not as straightforward to accomplish due to the multiple steps involved in the process (Backe and Field, 2012; Kostopoulou and Nikolaou, 2008; Nannou et al., 2019). The analyte enrichment step during the SPE process may over-amplify concentrations of native analytes (i.e., target analytes already present in the sample) to levels beyond the instrument handling capacity (Backe and Field, 2012; Simarro-gimeno et al., 2023). This often leads to the contamination of the flow path resulting in carry over effects and possible saturation of the detector resulting in truncated peaks. Regarding ARVDs, the extreme polarity range displayed by these compounds may present a challenge due to non-retention of polar classes especially on RP-SPE (Mosekiemang et al., 2019; Simarro-gimeno et al., 2023; Wood et al., 2015). For instance, the nucleoside reverse transcriptase inhibitors (NRTIs) class contain pyrimidine bases and a ribose sugar in their molecular structures (Fig. 1) and are predominantly polar (Andrade et al., 2011; Mosekiemang et al., 2021). Consequently, poor and irreproducible recoveries due to non-retention on RP-SPE have been reported for emtricitabine, lamivudine tenofovir etc. (Andrade et al., 2011; Brewer and Lunte, 2015; Mosekiemang, 2021). Notwithstanding this, poor recoveries and ion source-induced analyte losses are often circumvented by incorporating an appropriate isotopically labelled internal standards (ILIS) at the early stages of sample preparation to circumvent the possibility of analyte losses. Against this background, it is clear that a more universal sample preparation technique is required especially when the aim is to develop a method targeting compounds with diverse physico-chemical properties (i.e., multiclass ARVDs) (Mosekiemang et al., 2019; Wood et al., 2015). In the present study, we attempted to evade the limitations of SPE by employing lyophilization as a novel and more universal sample processing technique to improve the poor recoveries previously experienced for polar analytes (Hu et al., 2014; Narayan et al., 2011; Ramirez et al., 2014; Zhang et al., 2021). Lyophilization is a desiccation technique in which frozen samples are sublimated under vacuum to force a solid-to-gas phase transition. The dried residues are then re-constituted in a smaller volume relative to the initial volume resulting in elevation of concentrations of target analytes to detectable levels. It is a promising technique due to the several benefits it offers that includes its low operation costs, high sample throughput and the fact that it is predominantly solventless and therefore eco-friendly (Hirsch et al., 1998; Ramirez et al., 2014). However, there are potential drawbacks associated with this technique that include potential loss of volatile analytes and possible promotion of matrix effects because it lacks the sample cleaning aspect (de Voogt et al., 2000; Ramirez et al., 2014; Zhang et al., 2021). In the present study, a rather novel approach that combines lyophilisation for sample preparation followed by analysis using SFC-MS/MS was evaluated. The objective for this was to evaluate the analyte-enrichment capability of lyophilization using SPE as a reference for the processing of polar ARVDs previously known for non-retention, poor- and irreproducible recoveries on RP-SPE (Boulard et al., 2018; Mosekiemang, 2021; Mosekiemang et al., 2019; Nannou et al., 2020, 2019; Wood et al., 2015). The resulting data were compared to UHPLC-MS/MS employed as a reference.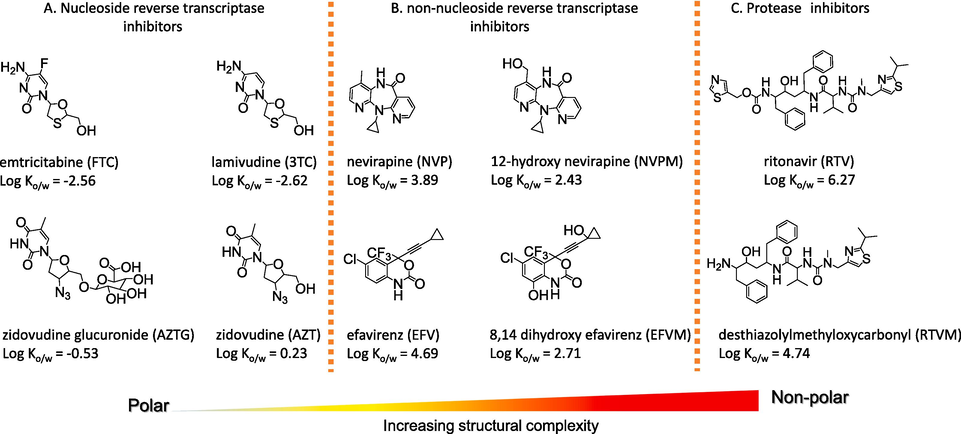
Typical physico-chemical properties and molecular structures of investigated compounds. The compounds are grouped according to pharmaceutical classes: (A) nucleoside reverse transcriptase inhibitors (FTC, 3TC, AZTG), (B) non-nucleoside reverse transcriptase inhibitors (NVP, NVPM, EFV, EFVM) and (C) protease inhibitors (RTV, RTVM). The hypothetic polarity diagram (i.e., polarity scale) underscores the diversity in the physico-chemical properties of the target compounds.
2 Experimental
2.1 Reagents and chemicals
HPLC-grade chemicals, reagents and standards were used in all experiments. Analytical standards (i.e., >98 % purity) for the target compounds (Fig. 1) were procured either from ClearSynth™ (Mumbai, India) as it is the case with efavirenz, emtricitabine, lamivudine, nevirapine, ritonavir, zidovudine, or Toronto Research Chemicals (Toronto, Canada) for 8,14-dihydroxy efavirenz, deuterated nevirapine, desthiazolylmethyloxycarbonyl ritonavir and zidovudine glucuronide (Mosekiemang, 2021; Mosekiemang et al., 2019; Venter and Onselen, 2023). Acetonitrile (ACN), methanol (MeOH), ammonium hydroxide (NH4OH) and formic acid (HCOOH) were all procured from Romil Ltd. (Waterbeach, Cambridge, GB).
2.2 Instrumentation and stationary phase evaluation
A Waters Acquity UPC2 SFC system (Acquity Ultraperformance Convergence Chromatography™) was used in this study. It comprised of a binary solvent delivery pump, temperature-regulated column compartment kept at 60 °C, sample manager module and an automatic back pressure regulator (ABPR) set at 1700 psi. The mobile phases (CO2 and MeOH) were set to be delivered at a flow rate of 1.30 mL min−1. A 5 µL sample volume was injected into the column housed in a temperature-controlled (60 °C) column compartment. Several Acquity™ UPC2 stationary phases (HSS C18 SB, Torus-1 AA, Torus-2 PIC, 2-EP and BEH) were evaluated for their suitability in the separation of target analytes (Fig. 1). All columns (100 mm × 3.0 mm, 1.7 µm dp) were made of sub-2 µm fully porous silica particles except for the HSS C18 SB (100 mm × 3.0 mm, 1.8 µm dp). The best results were obtained out of the BEH column and thereafter used for the rest of the study. The mobile phases’ gradient was set as follows: 98 % CO2 (0–0.2 min), 98–77.7 % CO2 (0.2–5 min), 77.7–40 % CO2 (5–7 min), 40 % CO2 (7–7.2 min), 40– 98 % CO2 (7.20–9 min), and 98 % CO2 until re-equilibration.
2.3 Mass spectrometric operating conditions
A Xevo TQ-S QqQ MS (Waters®) was hyphenated to an UPC2 system equipped with an ancillary pump (Waters 515™) designed to deliver an additional modifier (0.1 % HCOOH in MeOH at 0.3 mL min−1) to the mobile phase prior to the ESI in a configuration known as ‘pre-BPR splitter with sheath pump interface’ (Akbal and Hopfgartner, 2020; Gros et al., 2021; Mosekiemang, 2021; Perrenoud et al., 2014). The temperatures for the ion source and desolvation gas were set at 150 °C and 350 °C, respectively. The N2 cone gas- and desolvation gas flow rates were set at 150 and 900 L h−1, respectively. This included the capillary- and cone voltages which were set at 3.8 kV and 20 V, respectively.
The MS detector was operated in the MRM scanning mode to monitor two transition ion pairs (i.e., precursor- and two fragment ions) and/or two identification points per cycle time (tcycle). The MRMs were optimized using one ionization mode at a time (i.e., ESI– followed by ESI+ and vice versa). In the final method, all analytes were ionised in the positive mode (ESI+). Briefly, individual standard (52 ng/mL) for each analyte was infused post-column directly into the ESI at the following series of cone voltages (CV): 15, 20, 25 and 30 V to determine the best possible CV that produced the most intense precursor ion, and the obtained conditions were transferred to set-up the daughter ion scan mode. Then in this mode, the precursor ions were subjected to a series of collision energies (CE) in the range 15, 20, 25 and 30 eV to determine the optimum CE yielding the most intense fragment ions. Other parameters such as the dwell- (tdwell) and cycle times (tcycle) were software-set (MassLynx™ version 4.1) at ∼ 10 data points/peak. These were deemed adequate points/peak not to require implementation of a peak smoothing algorithm during peak integration considering the importance of peak purity and/or peak symmetry in quantitative chromatography. In that way all quantifier (Q) ions and their corresponding qualifier (q) ions were successfully assigned. Peak identity was confirmed if the ratio of peak intensities (q/Q or Q/q) was constant over the calibration range and independent from matrix and vice-versa. Similarly, the retention time shifts (ΔtR) between the Q- and q ions was monitored and expected to be negligible and independent of concentration and matrix effects (Kruve et al., 2015a, 2015b). Optimised MRM experimental conditions are summarized in Table S1 ‘supplementary information (SI)’ section. Even under these conditions, AZTG and EFVM could not be detected possibly due to the acidified ion source chamber owing to the flow injection of 0.1 % HCOOH in MeOH modifier solvent.
2.4 Preparation of calibration standard solutions
Calibration standards in the range of 0.610– 312 ng mL−1 were prepared by an appropriate dilution of a 20-µg mL−1 cocktail standard with neat MeOH. That is, from each concentration level, a 2000 µL aliquot of the cocktail standard was drawn to which an additional 900 µL of ILIS (100 ng mL−1) was added and diluted to volume (3 mL) with MeOH. In that way, a ten (10)-point equidistant calibration level system (0.407–208 ng mL−1), with each calibration point containing a uniform concentration of ILIS (100 ng mL−1 NVP-D3) was produced. This resulted in a constant volume fraction of a cocktail standard to a diluent throughout the experiment.
2.5 Sample collection and preparation
Wastewater samples were collected as previously described (Mosekiemang, 2021; Mosekiemang et al., 2019). Samples were vacuum filtered using a Whatman® filter paper (150 mm diameter and 1 µm pore size) and subsequently the sample pH was adjusted to ∼ 7 by a dropwise addition of 5 % NH4OH or 5 % HCOOH for increment or reduction of pH, accordingly. Prior to lyophilization- and SPE processing, method blanks were prepared by addition of a 300 μL ILIS (NVP-D3) and diluting to 50 mL with pH-adjusted wastewater matrix. The spike and recovery samples (i.e., fortified samples) were prepared in the same way except for an additional 75 µL spike of cocktail standards at appropriate concentrations to give the final spiking concentrations at low- (0.03 ng mL−1), mid- (0.3 ng L-1) and high (3 ng mL−1) levels prior to processing by SPE or lyophilization. Samples were split into two batches for processing by each method as shown in Fig. 2.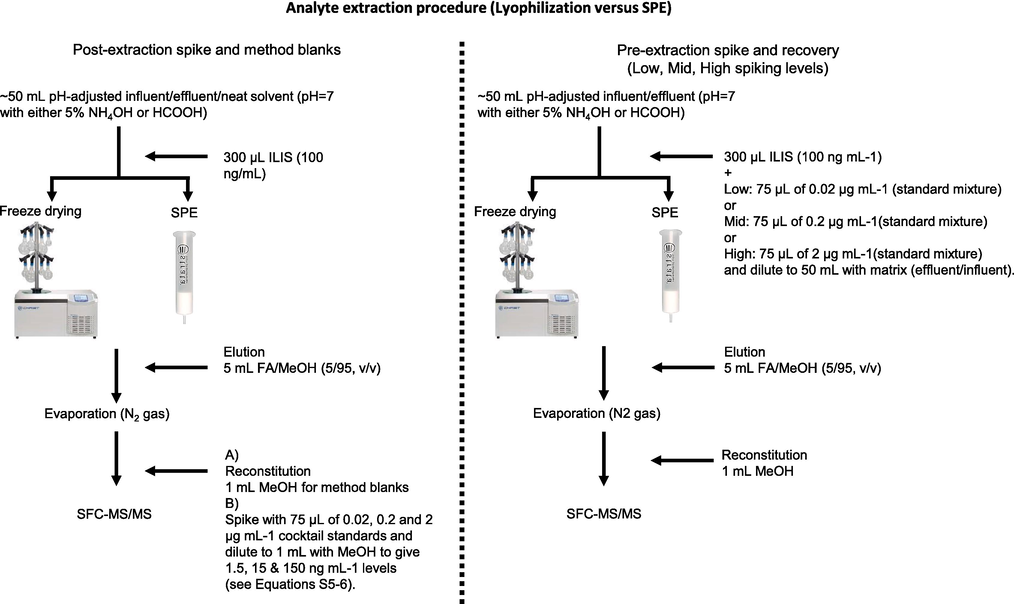
An illustrative experimental design for the comparison of lyophilization and SPE. Preparation process for method blanks and fortified samples intended for the spike and recovery experiments is also shown.
2.5.1 Lyophilisation
In this instance, all pH-adjusted- and ILIS-amended samples as described in the ‘Sample collection and preparation’ section above were each aliquoted into appropriate 100 mL lyophilization containers. Afterwards, the samples were frozen in a liquid N2 bath and then mounted on a Beta 1–8 LD plus lyophilizer (Christ, Germany) and processed overnight at −90 °C and ∼ 0.09 mbar vacuum. The resulting desiccants were then re-constituted in ∼ 5 mL acidified methanol (HCOOH/MeOH, 5/95, v/v) to ensure complete dissolution of residues. The resultant eluates were quantitatively transferred to appropriate polytop vials and subsequently dried using N2 gas. The workflow is illustrated in Fig. 2.
2.5.2 Solid phase extraction
This batch of samples as described in the ‘Sample preparation and processing’ section was processed by SPE. Here, a Resprep QR-24 port SPE manifold (Restek, Bellefonte, USA) was used to process Strata-SDBL cartridges (200 mg/6 mL, Phenomenex, Torrance, USA) (Mosekiemang, 2021). These SPE cartridges were successively conditioned and equilibrated with 5 mL ACN, MeOH and pH-adjusted water (pH 7), respectively. Thereafter, ∼50 mL pH-adjusted- and ILIS-amended samples were percolated into the SPE cartridges at ∼ 1 mL min−1 prior to washing/rinsing with pH-adjusted water. The loaded SPE cartridges were then vacuum dried for ∼ 10 min and eluted with 5 mL acidified methanol (HCOOH/MeOH, 5/95, v/v) and finally dried using N2 gas.
2.5.3 Resuspension of dried residues
The desiccants and/or residues described in Sections 2.5.1–2 above were reconstituted in the same way except that the ILIS-amended blanks were spiked with appropriate concentrations of cocktail standards prior to reconstitution to serve as post-extraction fortified samples. In short, an additional 75 µL aliquot of cocktail standards (0.02 µg mL−1) was added to the non-spiked ILIS-amended residue blanks prior to reconstitution to 1 mL with MeOH to serve as the post-extraction low spiking level (1.5 ng mL−1). In the same way, additional 75 µL aliquots of 0.2 and 2 µg mL−1 were added to the respective non-spiked ILIS-amended residue blanks and reconstituting to 1 mL with MeOH to yield the post-extraction mid- (15 ng mL−1) and high spiking levels (150 ng mL−1), respectively.
Similarly, residues for the pre-extraction fortified samples were resuspended in 1 mL MeOH. This meant a fifty-fold (50 × ) enrichment factor (EF), considering a reduction of 50 mL (initial sample volume) to 1 mL (reconstitution volume). In the same way, the initial spiking concentrations at low- (0.03 ng mL−1), mid- (0.3 ng L-1) and high (3 ng mL−1) levels as described Section 2.5 were now enriched 50 × to 1.5 ng mL−1 at low-, 15 ng mL−1 at mid- and 150 ng mL−1 at high spiking levels, respectively. Finally, all reconstituted pre- and post-extraction fortified samples now contained equal concentrations of target analytes across the spiking levels thus facilitating the implementation of Equations S5-6, (SI) for the evaluation of recoveries and matrix effects across the spiking levels.
2.6 Method validation
The method fitness for purpose was assessed in terms of the traditional validation approaches to ensure accuracy and reliability of the quantitative results. At the start, a response function was established, this is the relationship between the instrument response (peak area/intensity) as a function of concentration. Then, the calibration data was tested for statistical normality and the best regression model for data-fitting was determined. This task entailed performing several statistical tests for linearity such as inspection of the spread of residuals to establish (hetero)homoscedasticity, test for outliers using the Cochran’s test and the analysis of variance lack-of-fit (ANOVALOF) (Kruve et al., 2015b, 2015a; Van Loco et al., 2002).
For most analytes, linearity range was up to 208 ng mL−1 while NVP and its metabolite (NVPM) displayed narrow/reduced linear ranges of up to 104 ng mL−1, possibly due to over-saturation of the detector. Other parameters such as the detection- and quantification limits (LODs and LOQs) were determined from the calibration data according to Equations S1–S4, (SI), while recovery and matrix effects were determined according to Equations S5–S5. Errors (%RSD) related to repeatability and reproducibility were determined from the spike and recovery data (Evard et al., 2016; Hewavitharana et al., 2018; Kruve et al., 2015a, 2015b).
3 Results and discussion
3.1 Method performance and column selection
A Water Acquity UHPLC system coupled to a Xevo TQ-S MS system was previously fine-tuned for the detection of the same target analytes (Mosekiemang, 2021; Mosekiemang et al., 2019). In this study, re-optimisation was performed, this time the same MS detector was coupled to a Waters Acquity UPC2 system using CO2 and MeOH as mobile phases to which an additional make-up flow of 1 % HCOOH in MeOH modifier was added in a post column fashion (Guillarme et al., 2018; Perrenoud et al., 2014; Tarafder, 2018). The optimised experimental conditions such as the transition ions (MRMs), Q/q ion ratios, collision energies etc., are shown in Table S1, (SI). AZTG and EFVM, previously detected in the negative ionisation mode (Mosekiemang et al., 2019), were not detected this time around, possibly due to the infusion of acidified organic modifier to the ESI chamber. The non-detection of these compounds under acidified ionisation chamber conditions could be ascribed to the ‘wrong-way-around’ phenomenon as recently observed for the same compounds (Venter and Onselen, 2023). Briefly, the ‘wrong-way-round’ ionisation implies that contrary to expectations, protonated ions can be produced under basic conditions and vice versa where deprotonated ions are formed under acidic conditions. This often leads to the formation of weak precursor ions characterised by low peak intensities that often falls below the threshold detection limits leading to non-detection (i.e., false negative detection).
Column screening was performed to identify the SFC-phase that provided sufficient chromatographic separation for the target analytes, considering that SFC inherently produces poor peak shapes, and that peak symmetry is a crucial attribute in quantitative chromatography (Grand-Guillaume Perrenoud et al., 2012a; Wahab et al., 2017). Five SFC column phases were evaluated using peak asymmetry factors (As) as a criterion for selection (Ashraf-Khorassani and Taylor, 2010; Desfontaine et al., 2016; Grand-Guillaume Perrenoud et al., 2012a; Wahab et al., 2017). Interpretation of results for these experiments considered the extensive findings of previous work on SFC column classification especially by computational models such as linear solvation energy relationships (LSER) and other relevant data related to ARVDs on the same stationary phases (Khater et al., 2013; C West and Lesellier, 2006a; C. West and Lesellier, 2006).
The elution order of ARVDs on all columns was in reverse to what was previously observed on reversed phase (Desfontaine et al., 2016; Mosekiemang et al., 2019) and generally resembled that of normal phase, where nonpolar compounds (EFV, NVP, log KO/W≥2) showed markedly lower retention compared to their polar counterparts. Early eluting compounds, particularly EFV, NVP and NVPM displayed gaussian peaks compared to late eluting compounds (i.e., FTC, RTVM and 3TC) on all columns. Surprisingly, the RTVM peak displayed a tailing phenomenon which was worse off on the BEH 2EP and Torus 2 PIC but phenomenally improved on the Torus 1AA, BEH and HSS C18 SB. In contrast, RTV (i.e., a structural analogue of RTVM) was sufficiently symmetrical on all columns. At this stage, the reason for these variations in peak asymmetry factors for these structurally similar compounds is unexplainable but could be partially ascribed to an interplay of silanol and π-π interactions (Ashraf-Khorassani and Taylor, 2010; Desfontaine et al., 2016; Grand-Guillaume Perrenoud et al., 2012a). AZT was characterized by noisy peaks on all columns except for the BEH.
Also noteworthy is the fact that it took significantly high proportions of the organic mobile phase to elute the highly retained compounds, especially for RTVM. But, under these conditions (i.e., 60 % MeOH), the gaseous CO2 is no more supercritical but subcritical, and the chromatography can be more accurately termed as ‘enhanced fluidity’, and better-still with extended scope of applicability to separate even the extremely polar compounds (Bennett et al., 2019; Cui and Olesik, 1991).
The column chemistries assessed in this study comprised of a polar unbonded (BEH), polar bonded (BEH-2EP, 1-AA, 2-PIC) and a nonpolar straight chain non-end capped phase (HSS C18 SB) with some polar character (Akbal and Hopfgartner, 2020; Farrell, 2017; Grand-Guillaume Perrenoud et al., 2014; Nováková et al., 2014). The key thing to note here is that the pH of the CO2/MeOH mobile phase is predominantly acidic at (sub-)supercritical conditions (i.e., pH∼4) (Desfontaine et al., 2016) and at this pH the compounds are expected to be protonated because of their high pKa’s (∼10–14). For instance, 3TC, FTC, NVP, NVPM, RTV and RTVM are predominately protonated under acidic conditions. Therefore, similar elution orders were observed on the 2-EP, 1-AA, 2-PIC (Fig. 3). For these phases, analyte-stationery phase-interaction proceeds by a way of hydrogen bonding and dipole–dipole interactions while the effects of secondary ionic interactions are greatly suppressed by the electrostatic forces resulting from the protonation of basic bonded ligands (Desfontaine et al., 2016). This could possibly be the reason for the gaussian peak shapes observed on the 2-EP, 1-AA and 2-PIC especially for the early eluting analytes (e.g., EFV, NVP, AZT and NVPM). Amongst these phases, late eluting analytes (e.g., FTC, RTVM and 3TC) were to a greater extent highly retained on the 1-AA, a phenomenon ascribed to the reduced electrostatic repulsion forces due to the high pKa of 1-aminianthracene and possibly the contribution of additional analyte-stationery phase interactions (i.e., π-π interactions) (Grand-Guillaume Perrenoud et al., 2014; Wahab et al., 2017).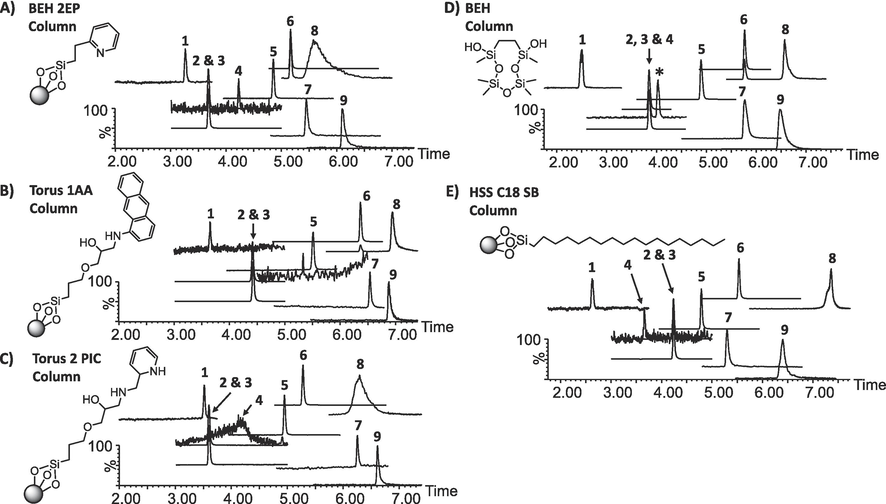
Typical total ion chromatograms (TICs) of target analytes separated on the five (5) stationary phases evaluated. Each TIC indicates the column name and the corresponding ligand chemistry. Target analytes are arranged according to elution order 1–9, corresponding to EFV, NVP, NVP-D3, AZT, NVPM, RTV, FTC, RTVM and 3TC, respectively. Compounds abbreviations were already described in Fig. 1.
The BEH phase provided sufficiently symmetrical peaks, a phenomenon possibly attributed to the uniform distribution of silanol functional groups and that the ethylene bridge contained the BEH silica material is known to provide good peaks compared to erstwhile silica raw material (C West and Lesellier, 2006b). Also noteworthy is that AZT produced a symmetrical peak only on this column compared to the noisy peaks observed on other columns (Fig. 3).
The HSS C18 SB phase also provided overall symmetrical peaks for most compounds except for slight peak-tailing tendencies observed for the late eluting compounds (FTC and 3TC) and peak-fronting with slight shouldering for RTVM. The retention mechanisms for this phase are predominantly governed by the hydrophobic interactions of the octadecyl functionality (C18 straight chain backbone). Therefore, the observed tailing observed for the late eluting compounds may be an indication of the presence of acidic residual silanols which may have facilitated additional interaction with basic analytes (Ashraf-Khorassani and Taylor, 2010; Hirose et al., 2019; C West and Lesellier, 2006b).
The data discussed above are conclusive regarding the elution order observed for the test analytes with late eluting analytes (FTC, 3TC and RTVM) suffering slight tailing effects due to the onset of secondary ionic interactions. In contrast, early- and mid-eluting peaks were generally symmetrical, but AZT displayed moderately noisy peaks on all columns except for the BEH which was eventually selected for the present study. Comparatively, better peak shapes were obtained in our previous study using RP-LC (Mosekiemang et al., 2019) and also by Venter and van Onselen (Venter and Onselen, 2023), although not unexpected considering that SFC inherently suffers poor peak shapes which may be improved by addition of suitable additives including small amount of water (Grand-Guillaume Perrenoud et al., 2014, 2012a, 2012b; Wahab et al., 2017).
Considering the above, the best column for the present study was considered the BEH column because all compounds were sufficiently separated with good peak symmetries. Partial co-elution was observed for NVP/AZT and RTV/FTC, but these pairs were subsequently separated based on their unique m/z ratios of the transition ions. This may be true because as stated earlier, the MRM detection mode was optimised for the best sensitivities for all analytes, which in a way demonstrated the excellent selectivity offered by the MRM mode to detect and distinguish between closely eluting analytes (Chen et al., 2016; Hermes et al., 2018). As an example, the retention time-aligned peaks for the respective Q- and q transition ions of AZT/NVP peaks are shown in Fig. 4. As can be seen, there is a tR-difference between the Q- and q ions for the partially coeluting peaks (i.e., AZT, tR 3.97 min. eluted earlier than NVP, tR 3.98 min.).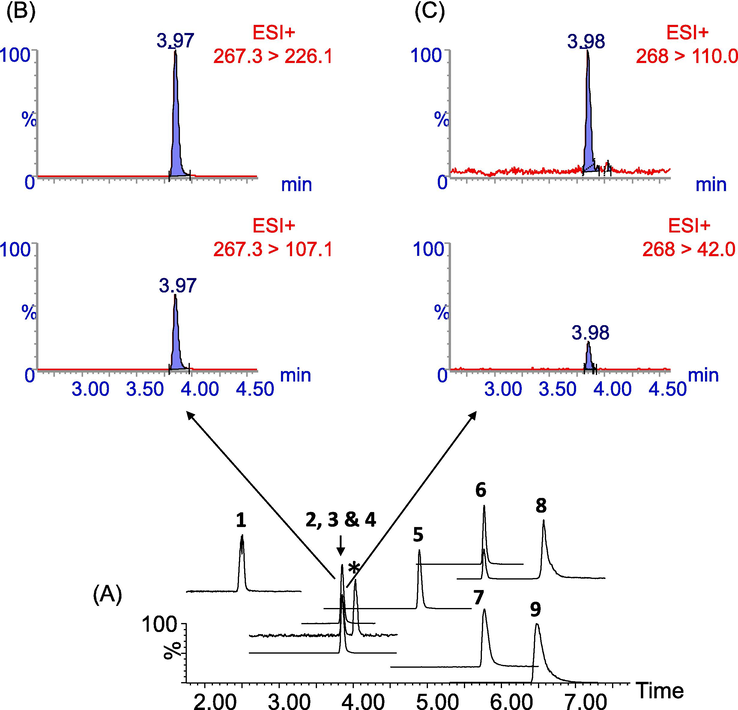
A typical total ion chromatograph (TIC) obtained using the Acquity UPC2 BEH column for the separation of target analytes (A). Peak annotations 1–9 represents elution orders for target analytes as already described in Fig. 3. Peaks 2 and 4 are partially co-eluting but successfully separated by the differences in their tR. Integrated peaks show that AZT eluted at tR 3.97 min (B) while NVP eluted at tR 3.98 min (C). The peak denoted by an asterisk is possibly baseline noise or an unknown entity.
3.2 Evaluation of lyophilisation and SPE
Previous studies reported poor SPE-recoveries for the polar NRTI ARVDs such as abacavir, indinavir and 3TC (Aminot et al., 2015), which was also confirmed by our previous study for other wastewater-borne NRTI compounds (i.e., AZTG, 3TC, AZT and FTC) (Mosekiemang et al., 2019). This prompted the present work to probe lyophilisation process for analyte extraction and enrichment, as this was considered a solventless alternative procedure to RP-SPE and most importantly to prevent analyte losses associated with poor recoveries for polar analytes (Hu et al., 2014; Narayan et al., 2011; Ramirez et al., 2014; Zhang et al., 2021). To achieve this, an ILIS was added before the analyte extraction process to make up for sample handling variations and both methods were assessed in terms of the ILIS- and non-ILIS-corrected recoveries (Hewavitharana et al., 2018). Recovery data was evaluated at low- (0.03 ng mL−1), mid- (0.3 ng mL−1) and high spiking levels (3 ng mL−1). Relevant data are presented in Table S4–S6, (SI).
The relative recoveries (ILIS-corrected) were adequate and in the range 65–99 % for effluent samples and 68–100 % for influent samples processed by lyophilisation across the spiking levels. Surprisingly, RTVM could not be quantified in all fortified samples due losses ascribed to irreversible adsorption to the system’s tubing or vacuum-induced losses during lyophilization (de Voogt et al., 2000). Regarding SPE, recoveries for the influent and effluent samples were only good for the apolar analytes of the nNRTI and PI pharmaceutical classes (EFV, NVP, NVPM, RTV and RTVM) and poor for the polar NRTI class (FTC and 3TC). This observation is in line with findings of previous studies (Aminot et al., 2015; Mosekiemang et al., 2019). Generally, the obtained results were conclusive that lyophilisation offered improved recoveries for all pharmaceutical classes compared to SPE which suffered losses for the polar analytes particularly for FTC and 3TC. This is a clear indication of the suitability of this form of sample preparation to process analytes with diverse physico-chemical properties.
For completeness, non-ILIS-corrected recoveries were also considered. The results obtained in this way are informative in highlighting the true analyte extraction efficiency of a sample preparation method (i.e., not compensated for analyte losses due to sample handling variations). A few notable differences were observed between non-ILIS- and ILIS-corrected recoveries implying that minimal analyte losses may have been incurred during sample preparation except for the known poor recoveries of the polar analytes due to non-retention on SPE. Problems associated with analyte losses due to sample handling can be easily overcome by incorporation of a suitable ILIS, but this may not always work as expected due to ILIS-analyte structural dissimilarities (Boix et al., 2016; Ibáñez et al., 2021; Simarro-gimeno et al., 2023). For instance, the results clearly show poor ILIS- and non-ILIS-corrected recoveries for 3TC and FTC by SPE in all samples considered. The recoveries in the range 23–42 % and 25–41 % were obtained for FTC and 3TC, respectively in ILIS-corrected effluent samples while almost similar results in the range 22–40 % as well as 18–31 % were obtained for the respective compounds in non-ILIS effluent samples. The same trend was manifested in influent samples where recoveries for the ILIS-corrected samples were measured in the range 23–66 % and 20–52 % in contrast to slightly worse recoveries in the range 13–34 % and 11–22 % for FTC and 3TC, respectively in non-ILIS-corrected influent samples. The poor recoveries particularly for the ILIS-corrected samples could be ascribed to the ILIS-analyte structural mismatches that may have resulted in the partial compensation for sample handling- and ionization-induced analyte losses. Table 1 provides a detailed summary for these data. To this end, lyophilisation can be viewed as an improvement to (large volume)direct injection (LVDI) (Backe and Field, 2012; Mosekiemang et al., 2019; Simarro-gimeno et al., 2023) in the context of improving instrument sensitivity (i.e., high LODs), considering that unlike LVDI it allows for preconcentration of large sample volumes and thus a substantial EF (50 × ) needed to considerably lower achievable method detection limits. Lyophilisation SPE Lyophilisation SPE Lyophilisation SPE Lyophilisation SPE Superscripts denote measurements determined at:
Matrix
Recovery (%)
Analytes
EFV
NVP
AZT
NVPM
RTV
FTC
3TC
RTVM
Effluent
ILIS-corrected
83.4–95.8
72.7–74.0a
82.0b
75.5–76.4a
63.9–84.4
64.5–75.6
74.1–98.9
n.q.d
86.2–96.0
98.1–104a
114b
99.6–101a
78.4–88.5
22.8–41.7
25.0–41.2
56.3–58.7
Non-ILIS-corrected
50.2–118
76.2–90.2a
85.8b
80.8–86.4a
75.6–86.3
58.7–88.0
66.1–82.4c
n.q.
49.5–98.3
89.8–101a
89.6b
89.5–102a
73.4–88.1
22.2–40.4
18.1–30.8
33.1–57.8
Influent
ILIS-corrected
100–104
94.5–94.8a
99.0b
87.8–97.7a
84.1–89.9
67.8–103
99.3–102
n.q.
63.2–100
99.2–103a
103b
100–111a
87.8–95.6
23.1–66.3
19.5–51.7
78.1–91.1
Non-ILIS-corrected
64.3–110
86.8–88.5a
96.8b
86.5–92.3a
70.5–90.1
67.5–81.8
92.5–100
n.q.
71.9–98.7
89.5–103a
110b
83.7–105a
76.1–99.3
12.7–33.9
10.5–21.6
73.9–80.5
3.3 Method validation and performance
The first parameter of paramount importance in quantitative studies is the determination of accuracy for the proposed method and this is determined in several ways that includes identification of the best regression model that fits the calibration data, identification, and elimination of outliers in the data, inspection of residuals and calculation of the coefficient of determination (R2) for the regression plots. All these data are presented in Table 2. As can be seen, all plots turned up R2 values above 0.9996 with significantly low residual errors (<10 %) in all the calibration plots. Follow up Cochran’s tests for outliers for the individual plots were also in agreement with the assumption of homoscedasticity in all the plots of residuals. Homoscedasticity is confirmed when the experimental Cochran’s C value (CCalc.) is less than the critical Cochran’s value (CTab.) (Van Loco et al., 2002). In addition, the ANOVALOF test confirmed the ordinary least square regression model as the best model to fit the obtained calibration data, in turn confirming the assumption of linearity over the calibration ranges for the individual calibration plots. A worked-out example for these chemometric tests is illustrated in Table S3. For all analytes linearity was confirmed for up to 208 ng mL-1calibration level, except for NVP and NVPM which displayed reduced linearities for up to 104 ng mL−1 calibration level due to peak truncation and/or detector saturation beyond 104 ng mL−1.
Analytes
tR
(min)Range
(ng/mL)Statistical test for linearity
MDL
(ng/mL)MQL
(ng/mL)Asb
Precision (% RSD)c
R2
aCochran’s C
aANOVALOF
Treated wastewater
Raw wastewater
Intra-day
Inter-day
Intra-day
Inter-day
EFV
2.76
LOD–208
0.9997
0.6021 [0.6161]
2.2 × 10-29 [3.2592]
0.0306
0.104
1.01
3 / 1 / 3
6 / 4 / 4
3 / 3 / 7
5 / 6 / 7
NVP
3.97
LOD–104
0.9993
0.6554 [0.6838]
2.9 × 10-30 [3.7083]
0.0378
0.126
1.03
3 / 7 / 5
10 / 7 / 5
3 / 1 / 3
11 / 2 / 3
AZT
3.98
LOD–208
0.9993
0.5000 [0.6838]
0.00 [3.7083]
0.114
0.368
1.06
− / − / 3
− / − / 2
− / − / 4
− / − / 4
NVPM
5.04
LOD–104
0.9992
0.5783 [0.6838]
0.00 [3.7083]
0.0396
0.132
1.04
3 / 5 / 5
6 / 4 / 5
5 / 1 / 5
5 / 15 / 6
RTV
5.85
LOD–208
0.9969
0.2415 [0.5157]
2.2 × 10-33 [2.7413]
0.0343
0.126
1.06
3 / 8 / 6
7 / 12 / 14
9 / 4 / 7
9 / 7 / 7
FTC
5.87
LOD–208
0.9972
0.0498 [0.5157]
1.2 × 10-05 [2.7413]
0.0318
0.106
1.16
6 / 7 / 6
8 / 13 / 5
9 / 2 / 2
7 / 6 / 9
3TC
6.55
LOD–208
0.9990
0.0498 [0.5157]
1.2 × 10-05 [2.7413]
0.0307
0.103
1.22
3 / 8 / 1
2 / 3 / 5
8 / 6 / 6
5 / 5 / 7
RTVM
6.64
LOD–208
0.9999
0.7047 [0.6161]
4.7 × 10-28 [3.2592]
0.0326
0.109
1.21
− / − / 9
− / − / 6
− / − / 5
− / − / 5
The method detection limits (MDLs) were calculated from the LOD data considering the EF (50 × ). As such, all MDLs were measured from 30 to 37 ng L-1 except for 113 ng L-1obtained for AZT. The method quantification limits (MQLs) were determined the same way using the LOQ data and taking into account the EF (50 × ). The MQLs ranged from 103 to132 ng L-1, a typical range at which in-sewer compounds are normally detected. The method precision was measured in terms of intra- (repeatability) and inter-day precision (reproducibility). As expected, the method proved to be extremely repeatable since all repeatability tests yielded %RSD values less than 9 %. In contrast, reproducibility values were not that excellent as evidenced by %RSD values of ∼ 15 %. Nonetheless, all the precision data were within acceptable levels.
Matrix effects (MEs). Despite the complementary assembly and/or hyphenation of SFC to ESI, the configuration may be susceptible to matrix effect-like challenges due to analyte precipitation resulting from the gaseous CO2 decompression at the interface junction where SFC connects to the ion source (i.e., ESI) (Akbal and Hopfgartner, 2020; Perrenoud et al., 2014). In addition, interferents displaying similar chromatographic behaviour as target analytes are also notorious for introducing matrix effects such as the ion source-induced ion enhancement or suppression. Given the importance matrix effects in quantitative chromatography (Stahnke et al., 2012; Svan et al., 2018), Equation S6 (SI) was employed to evaluate the extent of ion suppression or enhancement for the present method. The results obtained herein (Table 3) show that ion suppression was largely encountered in influent samples regardless of the sample preparation method and in late eluting compounds as evidenced by the strong retention observed for FTC (tR 5.87 min.) and 3TC (tR 6.55 min.). The ion suppression phenomenon observed here could be attributed to several factors, the main one being that raw wastewater is naturally dirtier than treated wastewater and may contain more interferents. In addition, matrix effects seemed to have been tR-dependent and varied at the different chromatographic regions as evidenced by ion suppression especially for the late eluting compounds. The ion suppression was worse in lyophilised samples compared to SPE-processed. This may be a true observation considering that the mobile phase gradient went up to 60 % MeOH at the 7–7.2 min. region to facilitate elution of highly retained compounds and to flush out dirt from the column. Therefore, it is logical to suspect severe matrix effects for compounds eluting at this chromatographic region (See Section 2.1 for mobile phase gradient description). Also, noteworthy is that RTVM could not be detected and quantified in all fortified samples processed by lyophilisation possibly due to losses related to volatility during lyophilization (de Voogt et al., 2000). Apart from the ion suppression effects observed for FTC, 3TC and the non-detection of RTVM, lyophilization technique has proved to be a suitable and promising sample preparation method and compares adequately with SPE. Lyophilisation SPE Lyophilisation SPE Lyophilisation SPE Lyophilisation SPE Lyophilisation SPE Lyophilisation SPE
Matrix
Spiking level
Analytes
EFV
NVP
AZT
NVPM
RTV
FTC
3TC
RTVM
Effluent
Low spiking level
(0.03 ng mL−1)
111 (±5)
125 (±6)
n.d.a
113 (±6)
127 (±5)
98.8 (±5)
5.1 (±6)
n.q.b
75.4 (±4)
103 (±4)
n.d.
89.4 (±3)
102 (±4)
97.2 (±3)
104 (±1)
111 (±8)
Mid spiking level
(0.3 ng mL−1)
108 (±6)
128 (±6)
n.d.
127 (±6)
128 (±8)
111 (±2)
4.2 (±8)
n.q.
110 (±10)
126 (±8)
n.d.
100 (±3)
114 (±5)
93.1 (±5)
99.7 (±3)
118 (±2)
High spiking level
(3 ng mL−1)
103 (±6)
120 (±3)
117 (±3)
120 (±2)
104 (±5)
115 (±7)
4.8 (±7)
n.q.
101.0 (±6)
105 (±1)
95.0 (±5)
101 (±7)
103 (±4)
107 (±3)
93.1 (±8)
111 (±5)
Influent
Low spiking level
(0.03 ng mL−1)
85.8 (±5)
113 (±4)
n.d.
82.9 (±6)
84.5 (±4)
56.8 (±7)
59.3 (±7)
n.q.
97.3 (±6)
108 (±9)
n.d.
113 (±6)
96.2 (±6)
78.7 (±2)
92.9 (±3)
136 (±8)
Mid spiking level
(0.3 ng mL−1)
103 (±3)
102 (±8)
n.d.
64.8 (±7)
103 (±8)
95.5 (±1)
95.7 (±1)
n.q.
100 (±7)
136.8 (±6)
n.d.
101 (±8)
100 (±9)
94.3 (±7)
94.8 (±5)
158 (±9)
Low spiking level
(3 ng mL−1)
n.q.
100 (±4)
115 (±5)
68.1 (±4)
89.2 (±5)
89.1 (±3)
2.7 (±8)
n.q.
158 (±3)
103 (±2)
98.1 (±4)
95.4 (±3)
93.5 (±9)
93.5 (±3)
99.8 (±7)
158 (±7)
3.4 Quantitative data: SFC-MS/MS versus UHPLC-MS/MS
The quantitative data obtained by lyophilization-SFC-MS/MS method was compared to that of direct injection-UHPLC-MS/MS as described in our previous study (Mosekiemang et al., 2019). As already discussed, AZT was not detected in the present study while RTVM was only detected in SPE-processed samples and could not be detected in lyophilized samples, possibly to due losses associated with lyophilization (de Voogt et al., 2000) or severe ion suppression as already hypothesized elsewhere. Despite these disparities, there was an excellent agreement in the overall quantitative data obtained by both methods, as it is established by the statistical mean differences test of < 3.5 % for both methods and also verified by an insignificant paired t-test (p > 0.05). All data pertaining to such comparison are presented in Table 4. Values in parenthesis denotes the standard deviations (±SD) for six measurements (n = 6).
Instrument
Concentrations in ng mL−1
EFV
NVP
AZT
NVPM
RTV
3TC
FTC
RTVM
UHPLC
19.2 (±3)
1.43 (±2)
n.d.
4.30 (±8)
20.0 (±3)
48.7 (±7)
352(±9)
n.d.
SFC
18.9 (±2)
1.42 (±7)
n.d.
4.15 (±1)
19.4 (±9)
49.7 (±3)
343 (±1)
n.d.
% Difference
1.6
1.0
–
3.5
3.2
−2.0
2.7
–
p-value
(paired t-test)0.77
0.83
–
0.19
0.07
0.24
0.17
–
3.5 Comparative method performance between SFC and UHPLC
On evaluation of the present method and in comparison to the preciously published one based on direct injection-UHPLC-MS/MS for the same suite of analytes, several accomplishments and shortcomings were noted. Regarding achievements, LODs, LOQs, and recoveries were markedly improved especially for FTC and 3TC in lyophilized samples (Backe and Field, 2012; Hermes et al., 2018; Mosekiemang et al., 2019; Richardson et al., 2021; Simarro-gimeno et al., 2023). This was not unexpected because the calculations for MDLs, MQLs and recoveries took into consideration the EF (50 × ) according to Eqs. S1–6. Conversely, the high MDLs and MQLs observed for the direct injection UHPLC method was because of the small sample volume used.
RTVM could not be satisfactorily validated by UHPLC due to poor reproducibility. In contrast, the same compound was accurately validated using SFC-MS/MS but could not be detected in real samples possibly because it was absent in the considered samples. Similarly, it could not be detected in fortified lyophilized samples due to losses associated with lyophilization but was successfully quantified in the same samples processed by SPE.
4 Conclusions
An eco-friendly lyophilization-SFC-MS/MS method was successfully developed for the quantitative determination of six (6) wastewater-borne antiretroviral drugs and two (2) metabolites. Incorporation of two (2) extra metabolites (AZT and EFVM) proved futile and could not be processed further. Our finding indicates that sample preconcentration by lyophilization not only improves recoveries for the polar analytes (i.e., FTC and 3TC) but also improves the overall method’s sensitivity in terms of providing low detection limits. Like SPE, lyophilization utilizes large volumes of sample (typically more than 50 mL) in so doing it confers an enrichment factor which when incorporated in the calculation of MDL and MQL sufficiently lowers the detection limit. This contrasts with direct injection and/or large volume direct injection which is known to suffer elevated detection limits due to miniaturized sample volume. However, lyophilization is susceptible to matrix effects compared to SPE because it is essentially a desiccation method that does not provide any form of sample clean-up. Our findings indicated a severe ion suppression phenomenon of up to 95 % for late eluting compounds such as FTC and 3TC. Overall, there were no notable differences between sample preparation by lyophilization and SPE. In addition, there was a strong similarity in the data obtained by SPE- and lyophilization-SFC-MS/MS as evidenced by negligible mean differences and an insignificant paired t-test between the two data sets. While there is no universal method for the determination of multiclass ARVDS, lyophilization-SFC-MS/MS has so far provided promising results in terms of providing acceptable recoveries for 3TC and FTC which previously could not be retained using SPE-based sample preparation methods. Overall, lyophilization has proved to be an alternative to SPE as much as SFC has proved to be an to LC.
CRediT authorship contribution statement
Tlou Mosekiemang: Writing – review & editing, Writing – original draft, Investigation, Formal analysis, Data curation, Conceptualization. Sithandile Ngxangxa: Writing – review & editing, Methodology, Conceptualization. Matlhogonolo Kelepile: Writing – review & editing.
Acknowledgements
TRECCAfrica II initiative is gratefully acknowledged for awarding a PhD fellowship to TM. The Central Analytical Facility at Stellenbosch University is gratefully acknowledged for providing the state-of-the-art analytical instrumentation used in this study. The University of Botswana is acknowledged for offering a study leave to TM.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- LC-MS/MS determination of antiretroviral drugs in influents and effluents from wastewater treatment plants in KwaZulu-Natal, South Africa. Chemosphere. 2018;200:660-670.
- [CrossRef] [Google Scholar]
- Hyphenation of packed column supercritical fluid chromatography with mass spectrometry: where are we and what are the remaining challenges? Anal. Bioanal. Chem.. 2020;412:6667-6677.
- [CrossRef] [Google Scholar]
- Uptake, accumulation and impact of antiretroviral and antiviral pharmaceutical compounds in lettuce. Sci. Total Environ.. 2021;766:144499
- [CrossRef] [Google Scholar]
- Development and application of a multi-residue method for the determination of 53 pharmaceuticals in water, sediment, and suspended solids using liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem.. 2015;407:8585-8604.
- [CrossRef] [Google Scholar]
- Twenty-six years of HIV science: An overview of anti-HIV drugs metabolism. Brazilian J. Pharm. Sci.. 2011;47:209-230.
- [CrossRef] [Google Scholar]
- Subcritical fluid chromatography of water soluble nucleobases on various polar stationary phases facilitated with alcohol-modified CO2 and water as the polar additive. J. Sep. Sci.. 2010;33:1682-1691.
- [CrossRef] [Google Scholar]
- Is SPE necessary for environmental analysis? A quantitative comparison of matrix effects from large-volume injection and solid-phase extraction based methods. Environ. Sci. Technol.. 2012;46:6750-6758.
- [CrossRef] [Google Scholar]
- Enhanced fluidity liquid chromatography : A guide to scaling up from analytical to preparative separations. J. Chromatogr. A. 2019;1595:190-198.
- [CrossRef] [Google Scholar]
- Electrospray Ionization Efficiency Predictions and Analytical Standard Free Quantification for SFC / ESI / HRMS. 2023
- [CrossRef]
- Analytical methodologies based on LC-MS/MS for monitoring selected emerging compounds in liquid and solid phases of the sewage sludge. MethodsX. 2016;3:333-342.
- [CrossRef] [Google Scholar]
- Utilization of large volume zwitterionic hydrophilic interaction liquid chromatography for the analysis of polar pharmaceuticals in aqueous environmental samples: Benefits and limitations. J. Chromatogr. A. 2018;1535:27-43.
- [CrossRef] [Google Scholar]
- Analysis of nucleosides in municipal wastewater by large-volume liquid chromatography tandem mass spectrometry. Anal. Methods. 2015;7:5504-5510.
- [CrossRef] [Google Scholar]
- Tracking the signals of living archaea: A multiple reaction monitoring (MRM) method for detection of trace amounts of intact polar lipids from the natural environment. Org. Geochem.. 2016;97:1-4.
- [CrossRef] [Google Scholar]
- Toxicity of Antiretrovirals on the Sea Urchin Echinometra lucunter and Its Predicted Environmental Concentration in Seawater from Santos Bay (Brazilian Coastal Zone) Resources. 2021;10:1-12.
- [CrossRef] [Google Scholar]
- High-Performance Liquid Chromatography Using Mobile Phases with Enhanced Fluidity. Anal. Chem.. 1991;63:1812-1819.
- [CrossRef] [Google Scholar]
- Freeze-drying brings about errors in polychlorinated biphenyl recovery calculations. Trends Anal. Chem.. 2000;19:292-299.
- [Google Scholar]
- SFC – MS versus RPLC – MS for drug analysis in biological samples. Bioanalysis. 2015;7:1193-1195.
- [Google Scholar]
- Supercritical fluid chromatography in pharmaceutical analysis. J. Pharm. Biomed. Anal.. 2015;113:56-71.
- [CrossRef] [Google Scholar]
- Evaluation of innovative stationary phase ligand chemistries and analytical conditions for the analysis of basic drugs by supercritical fluid chromatography. J. Chromatogr. A. 2016;1438:244-253.
- [CrossRef] [Google Scholar]
- Screening study of SFC critical method parameters for the determination of pharmaceutical compounds. J. Pharm. Biomed. Anal.. 2016;125:339-354.
- [CrossRef] [Google Scholar]
- Tutorial on estimating the limit of detection using LC-MS analysis, part I: Theoretical review. Anal. Chim. Acta. 2016;942:23-39.
- [CrossRef] [Google Scholar]
- W.P. Farrell Practical Approaches to Column Selection for Supercritical Fluid 2017 Elsevier Inc. Supercritical Fluid Chromatography 10.1016/B978-0-12-809207-1.00003-3.
- Pharmaceuticals in freshwater aquatic environments: A comparison of the African and European challenge. Sci. Total Environ.. 2019;654:324-337.
- [CrossRef] [Google Scholar]
- Analysis of basic compounds by supercritical fluid chromatography: Attempts to improve peak shape and maintain mass spectrometry compatibility. J. Chromatogr. A. 2012;1262:205-213.
- [CrossRef] [Google Scholar]
- Comparison of ultra-high performance supercritical fluid chromatography and ultra-high performance liquid chromatography for the analysis of pharmaceutical compounds. J. Chromatogr. A. 2012;1266:158-167.
- [CrossRef] [Google Scholar]
- On-line supercritical fl uid extraction-supercritical fl uid chromatography (SFE-SFC) at a glance : A coupling story. Trends Anal. Chem.. 2021;144:116433
- [CrossRef] [Google Scholar]
- What are the current solutions for interfacing supercritical fl uid chromatography and mass spectrometry ? J. Chromatogr. B. 2018;1083:160-170.
- [CrossRef] [Google Scholar]
- Quantification of more than 150 micropollutants including transformation products in aqueous samples by liquid chromatography-tandem mass spectrometry using scheduled multiple reaction monitoring. J. Chromatogr. A. 2018;1531:64-73.
- [CrossRef] [Google Scholar]
- Standard addition with internal standardisation as an alternative to using stable isotope labelled internal standards to correct for matrix effects — Comparison and validation using liquid chromatography- tandem mass spectrometric assay of vitamin D. J. Chromatogr. A. 2018;1553:101-107.
- [CrossRef] [Google Scholar]
- Comparison of Retention Behavior between Chromatography with Various Stationary Phases. Molecules. 2019;24:2425-2438.
- [Google Scholar]
- Determination of antibiotics in different water compartments via liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. A. 1998;815:213-223.
- [CrossRef] [Google Scholar]
- Determination of 26 veterinary antibiotics residues in water matrices by lyophilization in combination with LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci.. 2014;949–950:79-86.
- [CrossRef] [Google Scholar]
- Occurrence of pharmaceutical metabolites and transformation products in the aquatic environment of the Mediterranean area. Trends Environ. Anal. Chem.. 2021;29:e00118.
- [Google Scholar]
- Occurrence patterns of pharmaceutical residues in wastewater, surface water and groundwater of Nairobi and Kisumu city, Kenya. Chemosphere. 2016;149:238-244.
- [CrossRef] [Google Scholar]
- Characterization of five chemistries and three particle sizes of stationary phases used in supercritical fluid chromatography. J. Chromatogr. A. 2013;1319:148-159.
- [CrossRef] [Google Scholar]
- The phytoremediation capacity of Lemna minor prevents deleterious effects of anti-HIV drugs to nontarget organisms. Environ. Pollut.. 2023;329:121672
- [Google Scholar]
- Analytical problems and the need for sample preparation in the determination of pharmaceuticals and their metabolites in aqueous environmental matrices. Trends Anal. Chem.. 2008;27:1023-1035.
- [CrossRef] [Google Scholar]
- Tutorial review on validation of liquid chromatography-mass spectrometry methods: Part II. Anal. Chim. Acta. 2015;870:8-28.
- [CrossRef] [Google Scholar]
- Tutorial review on validation of liquid chromatography-mass spectrometry methods: Part I. Anal. Chim. Acta. 2015;870:29-44.
- [CrossRef] [Google Scholar]
- Analysis, occurrence and removal of pharmaceuticals in African water resources: A current status. J. Environ. Manage.. 2020;253:109741
- [CrossRef] [Google Scholar]
- Determination of selected antiretroviral drugs in wastewater, surface water and aquatic plants using hollow fibre liquid phase microextraction and liquid chromatography - tandem mass spectrometry. J. Hazard. Mater.. 2020;382:121067
- [CrossRef] [Google Scholar]
- Novel analytical methods for the determination of antiretroviral drugs and their metabolites in environmental water samples. Stellenbosch University; 2021. PhD Thesis
- Simultaneous quantification of commonly prescribed antiretroviral drugs and their selected metabolites in aqueous environmental samples by direct injection and solid phase extraction liquid chromatography - Tandem mass spectrometry. Chemosphere. 2019;220:983-992.
- [CrossRef] [Google Scholar]
- Ultra-high pressure liquid chromatography coupled to travelling wave ion mobility-time of flight mass spectrometry for the screening of pharmaceutical metabolites in wastewater samples: Application to antiretrovirals. J. Chromatogr. A. 2021;1660:462650
- [CrossRef] [Google Scholar]
- Mass loading, distribution, and removal of antibiotics and antiretroviral drugs in selected wastewater treatment plants in Kenya. Sci. Total Environ.. 2020;743:140655
- [CrossRef] [Google Scholar]
- Analytical strategies for the determination of antiviral drugs in the aquatic environment. Trends Environ. Anal. Chem.. 2019;24:e00071.
- [Google Scholar]
- Antiviral drugs in aquatic environment and wastewater treatment plants: A review on occurrence, fate, removal and ecotoxicity. Sci. Total Environ. 2020
- [CrossRef] [Google Scholar]
- Quantification of organophosphate insecticides in drinking water in urban areas using lyophilization and high-performance liquid chromatography – electrospray ionization-mass spectrometry techniques. Int. J. Mass Spectrom.. 2011;300:12-20.
- [CrossRef] [Google Scholar]
- Modern analytical supercritical fluid chromatography using columns packed with sub-2μm particles: A tutorial. Anal. Chim. Acta. 2014;824:18-35.
- [CrossRef] [Google Scholar]
- Profile and behavior of antiviral drugs in aquatic environments of the Pearl River Delta. China. Sci. Total Environ.. 2014;466–467:755-761.
- [CrossRef] [Google Scholar]
- Coupling state-of-the-art supercritical fluid chromatography and mass spectrometry : From hyphenation interface optimization to high-sensitivity analysis of pharmaceutical compounds. J. Chromatogr. A. 2014;1339:174-184.
- [CrossRef] [Google Scholar]
- Antiviral drugs in wastewater and surface waters: A new pharmaceutical class of environmental relevance? Environ. Sci. Technol.. 2010;44:1728-1735.
- [CrossRef] [Google Scholar]
- A simple method for routine monitoring of glyphosate and its main metabolite in surface waters using lyophilization and LC-FLD + MS/MS. Case study: Canals with influence on Biscayne National Park. Sci. Total Environ.. 2014;496:389-401.
- [CrossRef] [Google Scholar]
- Rapid direct analysis of river water and machine learning assisted suspect screening of emerging contaminants in passive sampler extracts. Anal. Methods. 2021
- [CrossRef] [Google Scholar]
- Separation of substrates and closely related glucuronide metabolites using various chromatographic modes. J. Chromatogr. A. 2016;1435:54-65.
- [CrossRef] [Google Scholar]
- Trends in Analytical Chemistry Current state and future perspectives of supercritical fl uid chromatography. Trends Anal. Chem.. 2022;149:116550
- [CrossRef] [Google Scholar]
- Analytica Chimica Acta First proof-of-concept of UC / HILIC for extending the versatility of the current art of supercritical fluid separation. Anal. Chim. Acta. 2023;1240:340741
- [CrossRef] [Google Scholar]
- Evaluation of direct sample injection as a fast, no-sample handling, approach for the LC-MS / MS monitoring of pharmaceuticals in different water matrices. Microchem. J.. 2023;193:108985
- [CrossRef] [Google Scholar]
- The influence of electrospray ion source design on matrix effects. J. Mass Spectrom.. 2012;47:875-884.
- [CrossRef] [Google Scholar]
- The differences in matrix effect between supercritical fl uid chromatography and reversed phase liquid chromatography coupled to ESI / MS. Anal. Chim. Acta. 2018;1000:163-171.
- [CrossRef] [Google Scholar]
- The differences in matrix effect between supercritical fluid chromatography and reversed phase liquid chromatography coupled to ESI/MS. Anal. Chim. Acta. 2018;1000:163-171.
- [CrossRef] [Google Scholar]
- Designs and methods for interfacing SFC with MS. J. Chromatogr. B. 2018;1091:1-13.
- [CrossRef] [Google Scholar]
- Linearity of calibration curves: Use and misuse of the correlation coefficient. Accredit. Qual. Assur.. 2002;7:281-285.
- [CrossRef] [Google Scholar]
- Evaluating the “ wrong - way - round ” electrospray ionization of antiretroviral drugs for improved detection sensitivity. Anal. Bioanal. Chem.. 2023;1187–1193
- [CrossRef] [Google Scholar]
- Total peak shape analysis: detection and quantitation of concurrent fronting, tailing, and their effect on asymmetry measurements. J. Chromatogr. A. 2017;1509:163-170.
- [CrossRef] [Google Scholar]
- The key role of hydrophobicity in the determination of pharmaceuticals by liquid chromatography – electrospray ionization - mass spectrometry under the interference of natural organic matter. Environ. Sci. Pollut. Res.. 2022;83071–83080
- [CrossRef] [Google Scholar]
- Characterisation of stationary phases in subcritical fluid chromatography with the solvation parameter model IV. Aromatic Stationary Phases. J. Chromatogr. A. 2006;1115:233-245.
- [CrossRef] [Google Scholar]
- Characterization of stationary phases in subcritical fluid chromatography by the solvation parameter model I. Alkylsiloxane-Bonded Stationary Phases. 2006;1110:181-190.
- [CrossRef] [Google Scholar]
- The occurrence of anti-retroviral compounds used for HIV treatment in South African surface water. Environ. Pollut.. 2015;199:235-243.
- [CrossRef] [Google Scholar]
- Mitigating risks and maximizing sustainability of treated wastewater reuse for irrigation. Water Res. X. 2023;21:100203
- [CrossRef] [Google Scholar]
- Determination of 38 pharmaceuticals and personal care products in water by lyophilization combined with liquid chromatography-tandem mass spectrometry. Anal. Methods. 2021;13
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105924.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




