

Translate this page into:
A first-principle study of the stability and electronic properties of halide inorganic double perovskite Cs2PbX6 (X = Cl,I) for solar cell application
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The problem of lead toxicity in perovskites materials that are currently performing with the most efficiency can be partially solved by choosing double perovskites compounds Cs2PbX6 (X = Cl,I), which have considerably reduced lead contents. These materials are slightly more stable, and substituting Cl and I with Br in small percentages further improves their mechanical stability and electronic properties. In this study, the properties of these promising materials were investigated in their pure and mixed forms.
Keywords
Cs2PbCl6
Simulation and modeling
Solar energy materials
Perovskite
Quantum ESPRESSO

1 Introduction
Harvesting energy from the sun is the most promising method for meeting the electricity needs of the future; it is sustainable, renewable, and much more environmentally friendly. A large and diverse collection of materials is used for this purpose, with many factors influencing material selection including cost, efficiency, and safety. Lead-based perovskite compounds have attracted the attention of many researchers, given their quick development, low cost, and promising results. Over the last decade, efficiencies in lead-based perovskites have improved dramatically and now exceed 25%, with a potential for continued improvement (https://www.nrel.gov). The great success in increasing efficiency has shifted the research focus toward other issues of concern with these materials, namely, their stability (Wang et al., 2016; Emami et al., 2015) and toxicity (Zhang et al., 2018; Babayigit et al., 2018). At room temperature, the stable phase of most of these compounds are light-inactive yellow phases and the light-active black phases turn into them under ambient conditions. Lead toxicity is the second major concern; solar cells are most effective when installed in open locations, which maximizes their exposure to sun light. However, such open locations also expose them to humidity and, given the high water solubility of lead, may allow lead to infiltrate our natural resources. Researchers have proposed many remedies to mitigate lead toxicity, including replacing lead with other elements such as tin, germanium, silver, and even bismuth (Zhang et al., 2018; Kour et al., 2019; Savory et al., 2016). But even with some very promising results, the efficiencies of these materials are much lower than that of lead compounds. In addition, some elements such as tin are water soluble, which degrade their stability. Toxicity is usually looked at from the limited viewpoint of the compound but when analyzed in the context of other technologies currently used for energy production processes, contamination from the lead contents of solar cells is moderate. Billen and colleagues introduced a metric called “toxicity potential payback time” to provide context for the relative scale of the toxicity problem; they found that lead levels in lead halide perovskites are up to four times lower than levels in current US electricity mixes (Billen et al., 2019). Based on this finding, a plausible path is to prioritize efficiency over other factors, at least for the current time, and attempt to decrease lead content by utilizing other compounds that contain less percentages of it.
For this study, double perovskite compounds with much lower lead contents, Cs2PbCl6 and Cs2PbI6 were chosen as alternatives to the perovskite CsPbX3. These compounds are known to be slightly more stable due to the 4 + lead oxidation state, and we found that replacing a small portion of the halogen atoms with bromine further stabilizes the material (Ruess et al., 2016). The structural and electronic properties of the pure and mixed states of these compounds were investigated and compared with those of conventional perovskite. In Section 2, the computational calculations are detailed, and in Section 3, the results of the calculations are presented and analyzed.
2 Computational details
Calculations were performed using density functional theory (DFT) and were implemented in the pseudopotential-based package Quantum ESPRESSO (Giannozzi et al., 2009; Giannozzi et al., 2017). The exchange-correlation functional PBSol was used, which is a version of the Perdew, Buke, and Erhzarhof (PBE) functional that has been optimized for solids (Perdew et al., 1996; Perdew et al., 2008). Supercells of 72 atoms ( of the basic unitcell) were studied using -shifted k-mesh and a plane-wave cutoff of 540 eV. These values were converged and optimized previously for this and similar compounds, and all structures and their unitcells were relaxed until the accuracy of energy fell below eV/atom and the force on atoms fell below 1 meV/A. Subsequently, the calculation of the density of states (DOS) using the same k-mesh and band structure along the desired path were evaluated in reciprocal space.
3 Results
3.1 Structural properties
A double perovskite (sometimes called defect or alternative perovskite) is a cubic
structure that can be derived from ABX3 by removing alternating halves of the B sites, which makes the BX6 octahedra isolated, rather than sharing corner (Brik and Kityk, 2011), as shown in Fig. 1. The calculations show that Cs2PbX6 has a smaller volume per atom than CsPbX3 (Table 1) and smaller B-X bond lengths (2), a manifestation of increased stability. Replacing some chlorine and iodine atoms with bromine in small amounts is known to increase the stability of some perovskite compounds (Ruess et al., 2016). The supercells each contained 72 atoms (formula multiplicity factor = 8), and up to three atoms of chlorine and iodine were replaced with bromine (2–6%).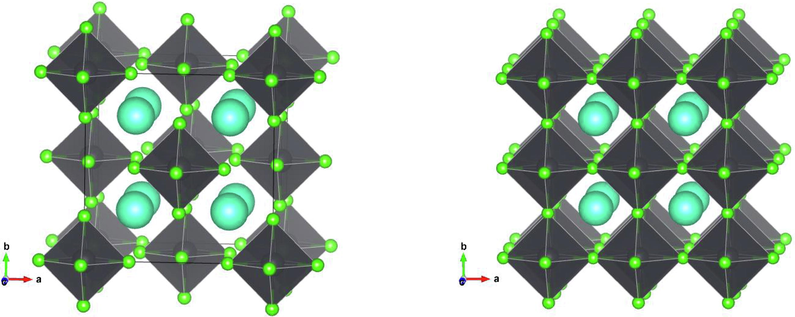
Crystal structure for Cs2PbCl6 (left) and CsPbCl3 (right). Cyan circles are Cs atoms, green are halogen atoms, and lead atoms are enclosed inside the octahedra.
X
Cs2PbX6
CsPbX3
Cl
10.23
11.21 (Moller, 1958)
(10.415) (Engel, 1933)
Br
10.69
11.74 (Moller, 1958)
I
11.48
12.58 (Trots and Myagkota, 2008)
Tables 1 and 2 summarize the main structural properties of pure compounds. The lattice parameter for Cs2PbCl6 is within 2% of the experimental values and it increases proportionally with atomic sizes. As expected due to the difference in atom sizes, when Br was used to replace Cl atoms, the volume increased, and when it was used to replace I atoms the volume decreased, as shown in Fig. 2. Interestingly, however, atom separation in all replaced structures tended to decrease compared to the separation in pure states, indicating increased phase stability (Figs. 3 and 4).
Cs2PbX6
CsPbX3
Bond
length (A)
length (A)
X = Cl
Pb-Cl
2.54
2.80
Cl-Cl
3.59
3.96
Cs-Cl
3.62
3.96
Pb-Cs
4.43
4.85
X = Br
Pb-Br
2.72
2.78
Br-Br
3.72
3.93
Cs-Br
3.78
3.93
Pb-Cs
4.63
4.81
X = I
Pb-I
3.00
3.06
I-I
3.88
4.33
Cs-I
4.06
4.33
Pb-Cs
4.97
5.36
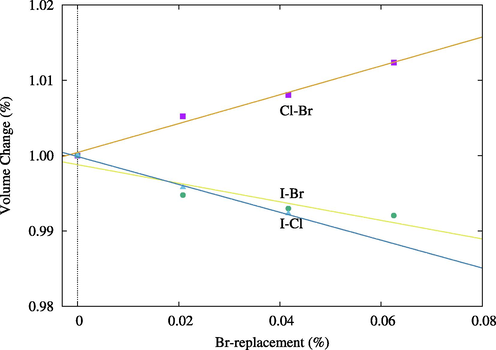
Volume percentage change when Cl and I atoms are replaced by Br atoms. Blue triangle and line are for the change when replacing I atoms with Cl.
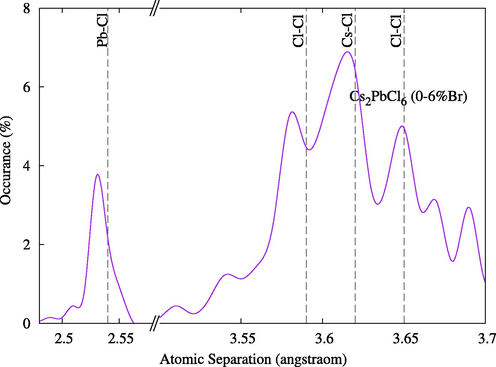
Optimized atom separations for compounds Cs2Pb(Cl1-xBrx)6. Indicated vertical lines are for pure state Cs2PbCl6.
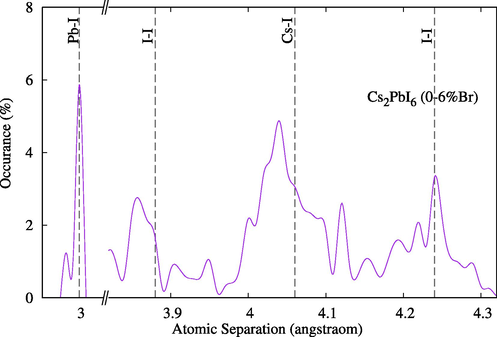
Optimized atom separations for compounds Cs2Pb(I1-xBrx)6. Indicated vertical lines are for pure state Cs2PbI6.
3.2 Electronic properties
The experimentally known stable phase among the family is Cs2PbCl6. It is a direct-bandgap material with a bandgap of 1.2 eV located at the
point (Fig. 5). Other studies reported bandgap values of 1.351 eV (Saal et al., 2013; Kirklin et al., 2015),1.36 eV (Klintenberg et al., 2002) and 1.6 eV (Jain et al., 2013). DFT is known to underestimate the values of bandgaps due to the uncertainty in determining the excited states energies. The DFT estimates of bandgaps can be improved using hybrid functionals, but these methods require more extensive resources than were available for the current study, particularly given the number of compounds investigated and the breadth of the searches for optimized structures involved. Because our purpose was a qualitative investigation of the effect of Br replacement, we concluded the possible underestimation of the bandgap would not present a significant issue. This conclosion is supporte by what Wang and collaborators found when they analyzed the effect of using PBE vs. modified Becke-Jonson (mBJ) functionals and showed that the difference in their estimates of band gaps is noticeable but qualitatively similar (Wang et al., 2015), and by the fact that PBE functional is known to exhibits a good compromise between accuracy and demand (Hernández-Haro et al., 2019).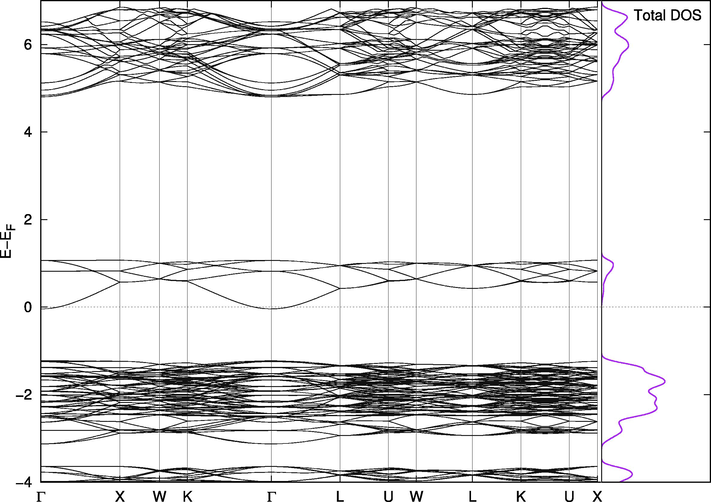
Band Structure for Cs2PbCl6.
It is instructive to comment on the nature of the band structures of pure compounds because it affects the band structures of the mixed phases (Figs. 5–7). In Cs2PbCl6, the bandgap is 1.2 eV with the Fermi level at the lowest part of the conduction band, which is not a very dense band. As mentioned above, the actual bandgap is expected to be larger than the calculated bandgap, which makes the material very suitable for the absorption of sunlight in a specific range of wavelengths.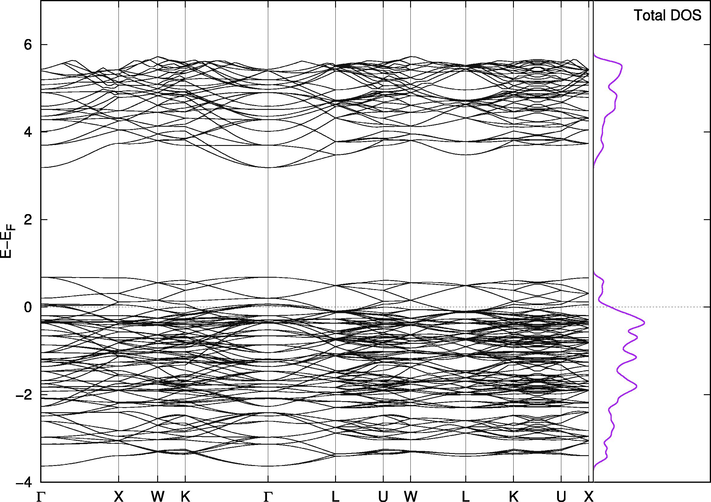
Band Structure for Cs2PbI6.
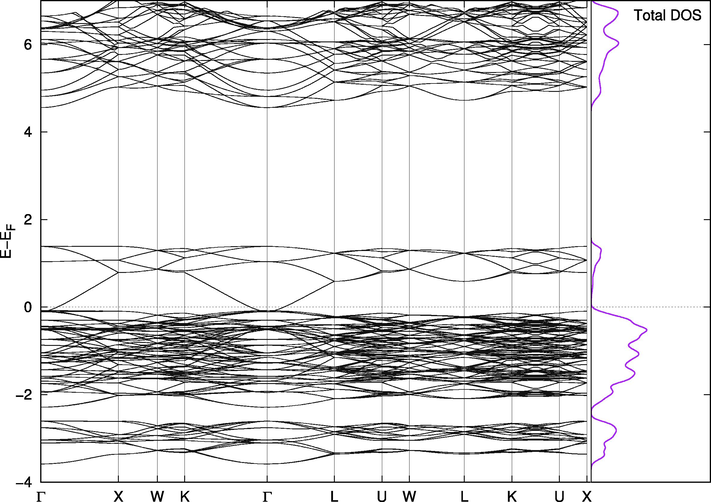
Band Structure for Cs2PbBr6.
On the other hand, Cs2PbI6 and Cs2PbBr6 are not naturally suitable for this purpose. The valence band in Cs2PbBr6 (Fig. 8) has a portion at the top above the Fermi level making the compound almost metallic. However, this portion is separated from the band by less than 0.1 eV when we included the spin orbit coupling effect and Wang found it to be 0.5 eV with PBE and 1.26 eV using mBJ (Wang et al., 2015). The states in this small band play vital roles in the mixed phases. The replacement effect on the band structure of Cs2PbCl6 is interesting; a single Br-Cl replacement (2%) introduces two very close, but separate, bands above the valence band of the pure Cl phase. They are separated from each other and from the band below them by amounts that are one order of magnitude larger than thermal fluctuations. In this configuration, the material is suitable for use in intermediate-band solar cells, which are thought to be better or comparable in efficiency to tandem cells (Green, 2001; Luque et al., 2012). The existence of intermediate bands allows for absorption of a wider range of wavelengths, and the newly introduced bands in the mixed phase are most likely due to the upper part of the valence band in pure Br compounds mentioned above. Their widths and locations can be controlled through adjusting the concentration of Br atoms in the compounds, as shown in Figs. 8–11. Replacing iodine atoms with Br or Cl did not introduce similar bands in useful locations in Cs2PbI6 (Fig. 12).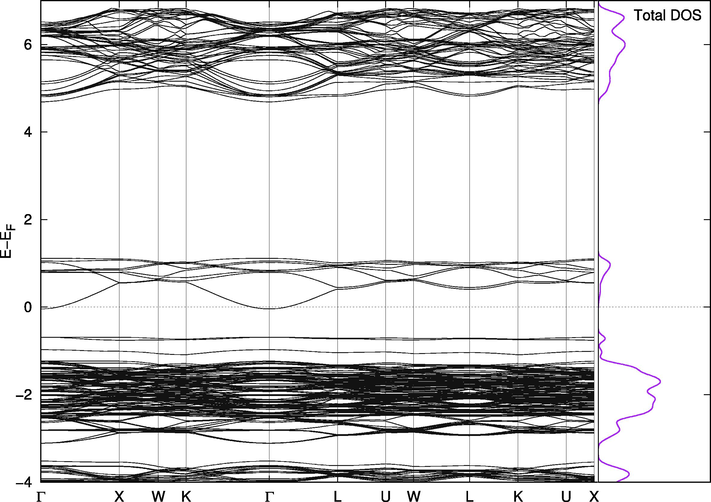
Band Structure for Cs2Pb(Cl98%Br2%)6.
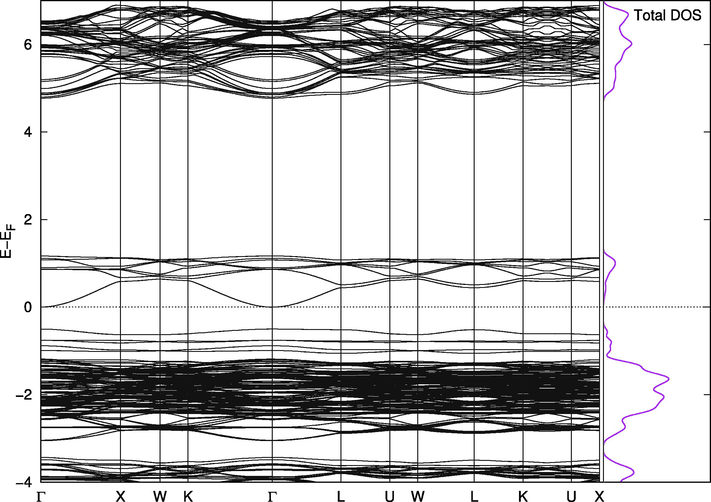
Band Structure for Cs2Pb(Cl96%Br4%)6.
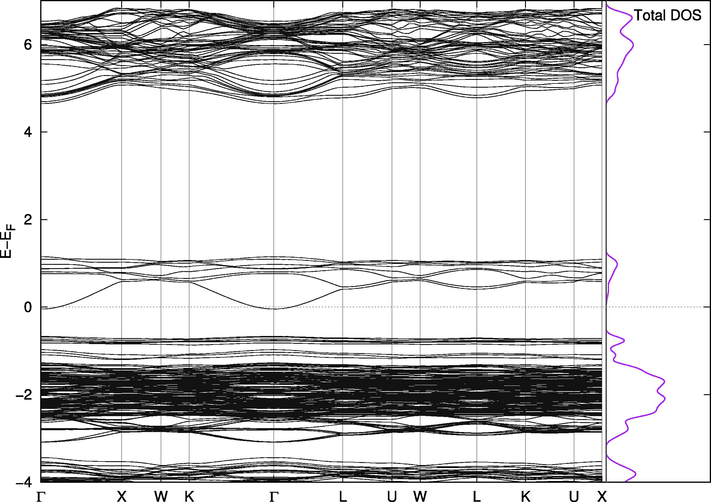
Band Structure for Cs2Pb(Cl94%Br6%)6.
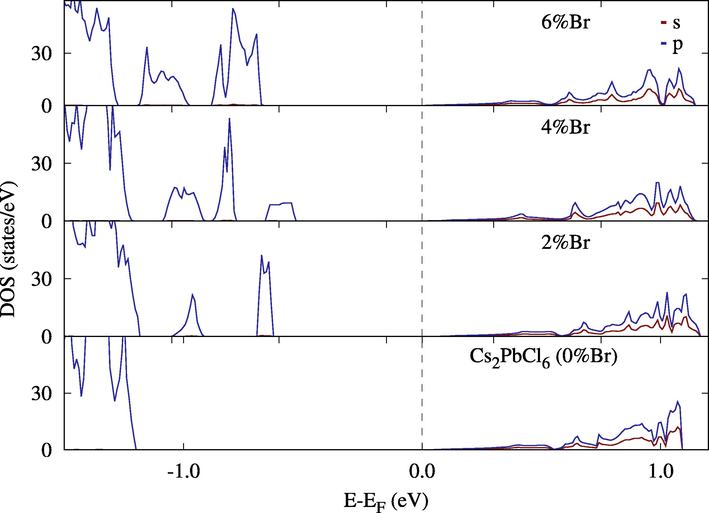
Density of States (DOS) for Cs2Pb(Cl(1−x)B4x)6. The peaks at the lowest percentage of Br are center at 2.46 eV and 3.28 eV above Fermi level.
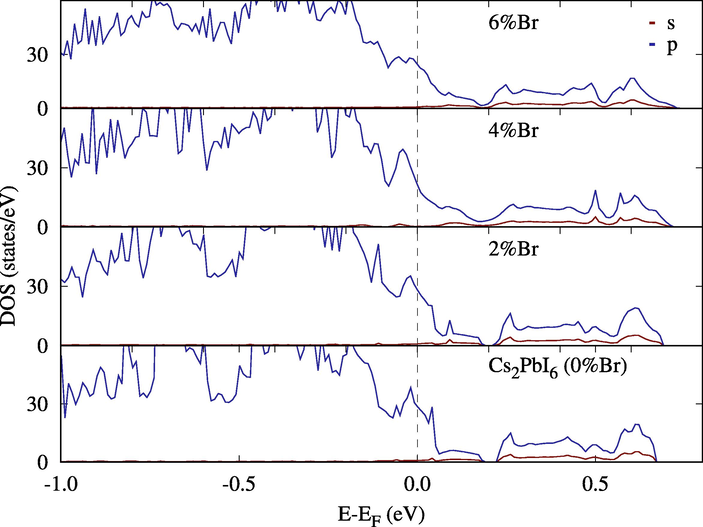
Density of States (DOS) for Cs2Pb(I1−xB4x)6. Most of the changes Br atoms introduced are within already existing bands.
This effect is clearer when looking at the density of states. In Fig. 11, a single replacement introduced two intermediate bands separated by at least 0.15 eV from the lower band and from each other and 0.7 eV from the conduction band. Since the separation can be controlled by the percentage of Br added to the compound; Lower concentrations can isolate these peaks to an optimal band separation for intermediate solar cells (two sub-bandgaps of 0.71 eV and 1.24 eV (Luque et al., 2012)). Replacing I with Br altered states around the Fermi level, which is already in the middle of the band. That limited the effect of I-Br mixing to the mechanical stability of Cs2PbI6 (Fig. 12).
4 Conclusion
Structural and electronic properties of double perovskite compounds Cs2PbX6 were studied as emerging solar cell materials. These compounds are more stable than the perovskite compound CsPbX3 and contain less lead. Moreover, this study showed that traces of Br can stabilize Cs2PbI6 and Cs2PbCl6 and enhance the latter’s light absorption by introducing intermediate bands within the gap around the Fermi level.
Acknowledgments
The calculations in this work were performed on AZIZ, the supercomputer at King Abdulaziz University’s High Performance Computing Center (http://hpc.kau.edu.sa). Compound figures were drawn using VESTA (Momma and Izumi, 2011) and gnuplot was used to produce the graphs.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Environment versus sustainable energy: The case of lead halide perovskite-based solar cells. MRS Energy Sustain. Rev. J.. 2018;1–15
- [CrossRef] [Google Scholar]
- Comparative evaluation of lead emissions and toxicity potential in the life cycle of lead halide perovskite photovoltaics. Energy. 2019;166:1089-1096.
- [CrossRef] [Google Scholar]
- Modeling of lattice constant and their relations with ionic radii and electronegativity of constituting ions of A2XY6 cubic crystals (A=K, Cs, Rb, Tl; X=tetravalent cation, Y=F, Cl, Br, I) J. Phys. Chem. Solids. 2011;72:1256-1260.
- [CrossRef] [Google Scholar]
- Recent progress in long-term stability of perovskite solar cells. U. Porto J. Eng.. 2015;1(2):52-62.
- [Google Scholar]
- The crystal structures of K2PtCl6 type compounds. Naturwissenschaften. 1933;21:704.
- [CrossRef] [Google Scholar]
- Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys.: Condens. Matter. 2009;21:395502.
- [Google Scholar]
- QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter. 2017;29:465901.
- [Google Scholar]
- Multiple band and impurity photovoltaic solar cells: general theory and comparison to tandem cells. Prog. Photovolt: Res. Appl. 2001;9:137-144.
- [Google Scholar]
- DFT prediction of band gap in organic-inorganic metal halide perovskites: An exchange-correlation functional benchmark study. Chem. Phys.. 2019;516:225-231.
- [CrossRef] [Google Scholar]
- The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater.. 2013;1(1):011002.
- [CrossRef] [Google Scholar]
- The Open Quantum Materials Database (OQMD): assessing the accuracy of DFT formation energies. npj Comput. Mater.. 2015;1:15010.
- [CrossRef] [Google Scholar]
- Klintenberg, M., Derenzo, S.E., Weber, M.J., 2002, A systematic search for new scintillators using electronic structure calculations, Tech. Connect Briefs, 2, 427–430. Technical Proceedings of the 2002 International Conference on Computational Nanoscience and Nanotechnology.
- Potential substitutes for replacement of lead in perovskite solar cells: a review. Glob. Challanges. 2019;3:1900050.
- [CrossRef] [Google Scholar]
- Understanding intermediate-band solar cells. Nat. Photon. 2012;6:146-152.
- [CrossRef] [Google Scholar]
- Crystal structure and photoconductivity of caesium plumbohalides. Nature. 1958;182:1436.
- [CrossRef] [Google Scholar]
- VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr.. 2011;44:1272-1276.
- [Google Scholar]
- Generalized gradient approximation made simple. Phys. Rev. Lett.. 1996;77:3865-3868.
- [CrossRef] [Google Scholar]
- Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett.. 2008;100:13.
- [CrossRef] [Google Scholar]
- Stabilization of organic-inorganic perovskite layers by partial substitution of iodide by bromide in methylammonium lead iodide. ChemPhysChem. 2016;17:1505-1511.
- [CrossRef] [Google Scholar]
- Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD) JOM. 2013;65:1501-1509.
- [CrossRef] [Google Scholar]
- Can Pb-free halide double perovskites support high-efficiency solar cells? ACS Energy Lett.. 2016;1(5):949-955.
- [CrossRef] [Google Scholar]
- High-temperature structural evolution of caesium and rubidium triiodoplumbates. J. Phys. Chem. Solids. 2008;69:2520-2526.
- [CrossRef] [Google Scholar]
- Electronic Structure and Optical Properties of Cs2AX’2X4 (A=Ge, Sn, Pb;X’, X=Cl, Br, I) AIP Adv.. 2015;5:127224.
- [CrossRef] [Google Scholar]
- Stability of perovskite solar cells. Sol. Energy Mater. Sol. Cells. 2016;147:255-275.
- [CrossRef] [Google Scholar]
- Perovskite solar cells: must lead be replaced - and can it be done? Sci. Technol. Adv. Mater.. 2018;19:225-442.
- [CrossRef] [Google Scholar]




