

Translate this page into:
A novel class of 1,4-disubstituted 1,2,3-triazoles: Regioselective synthesis, antimicrobial activity and molecular docking studies
⁎Corresponding authors at: Malladi Drugs & Pharmaceuitcals Ltd., R&D Centre, Chennai 600124, Tamil Nadu, India (K.C. Rao); Department of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia (N. Arumugam). kcrao2009@gmail.com (Chennakesava Rao Kella), anatarajan@ksu.edu.sa (Natarajan Arumugam)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A Novel class of 1,4-disubstituted 1,2,3-triazoles have been synthesized in good to excellent yields via Cu(I) accelerated azide-alkyne click chemistry reaction strategy. The newly synthesized compounds were assessed for their in vitro antimicrobial activity against five Gram-positive, seven Gram-negative bacteria and three fungi. Most of the synthesized compounds displayed significant activity against the tested Gram-positive and Gram-negative bacteria. Molecular docking study revealed that all docked compounds are bound efficiently with the active site of Topoisomerase IV (4EMV) receptor with the observed the free energy of binding from −7.79 to −9.44 kcal/mol. Interestingly, compound 13a forms four hydrogen bonds and displayed high binding energy (−9.44 kcal/mol) with the Topoisomerase IV (4EMV) receptor which correlated with their in vitro antimicrobial assays.
Keywords
1,4-Disusbstituted-1,2,3-triazoles
Click chemistry
Antimicrobial activity
Docking study

1 Introduction
The increasing emergence of drug resistance, intractable pathogenic microorganisms and newly rising pathogens have become a serious health threat for humanity worldwide. These circumstances stimulate an essential need to develop novel class of antimicrobial agents (Duraipandiyan et al., 2009), particularly, structurally diverse chiral small molecule with unique mechanism of action from currently available clinical antimicrobial drugs (Satish et al., 2020). In this context, the construction of chiral hybrid heterocycles is essential to explore new pharmaceutical agent and agrochemicals. Among the heterocyclic pharmacophores, 1,2,3-triazoles occupy protuberant place (Leiling et al., 2019) in drug discovery owing to their multifarious pharmaceutical activities. For instance, anti-fungal (Yi et al., 2020), anti-bacterial (Adnan et al., 2017), anti-HIV (Lin et al., 2020; Giffin et al., 2008), anti-tubercular (Patpi et al., 2012; Pravin et al., 2020; Abdul Aziz et al., 2017), anti-inflammatory (Hong-Jian et al., 2017) and antiproliferative activities (Zhi et al., 2019; Hupe et al., 1991). In addition, triazole comprising amino acids have been established for peptide drug conjugates, glycopeptides, peptide fluorescence labelling (Wei et al., 2018) and inhibitors of glycogen synthase kinase (Imran et al., 2016), antagonists of GABA receptors (Alessandro et al., 2018; Bascal et al., 1996). Tazobactam, Cephalosporin and Cefatrizine are clinically used efficient drug candidates for antibacterial infections, these drugs possesses 1,2,3-triazole as an active moiety.
In recent years 1,2,3-triazoles gains special attention in the drug discovery because their unique structural features in view of stability to metabolic degradation and are capable of hydrogen bonding, which is a favorable aspect for binding the bio-molecular targets and can also improve the solubility (Era et al., 2020; Anlian et al., 2019). In addition, these structural motifs were synthesized easily through 1,3-dipolar cycloaddition Click chemistry approach (Leiling et al., 2020; Filip et al., 2020; Jie-Ping et al., 2016). The unique structural features of 1,2,3-triazole heterocycles including their biological importance prompted us towards their design, synthesis and antimicrobial activity of pure 1,4-disubsituted-1,2,3-triazole employing copper catalyzed azide-alkyne click chemistry reaction (Cu-AAC). Further, computational molecular docking study was also carried out to examine the binding interface template of the synthesized compounds to the amino acid residues combining active site of the respective receptor has been described in the manuscript.
2 Materials and methods
2.1 General experimental procedure to synthesize chloramine 2a and 2b
A 250 mL reaction flask was charged with (1R,2S)-phenylpropanolamine 1a (150 g, 1.0 mol), chloroform (700 mL) and stirred for 10 min. to get a clear solution. Thionyl chloride (180 g, 1.5 mol) was very slowly added to the above solution at 30 to 50 °C for 4 h and continued the heating at 50 °C for 2 h. The excess SOCl2 and chloroform were distilled off completely when the reaction was completed. Acetone (200 mL) was added to the solid mass and continued the distillation to remove traces of thionyl chloride. Later the solid mass was stirred with acetone (300 mL) at ambient temperature, filtered, washed with acetone (100 mL) and dried under vacuum at 60 °C to get the HCl salt of (1S,2S)-chloramine 2a. The same procedure was repeated with (1S,2R)-phenylpropanolamine 1b instead of 1a to obtain the HCl salt of (1R,2R)-chloramine 2b. Analytical data of 2a and 2b is presented below.
Compound 2a: Yield 182 g (89%); mp 182–183 °C (lit 178–179 °C); SOR + 93.05° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3438, 2938, 2880, 2771, 1605, 1579, 1450, 1456, 1373, 1198, 713 and 700; 1H NMR (400 MHz) δH: 0.99 (3H, d, J = 6.8 Hz), 4.42 (1H, m), 5.09 (1H, d, J = 3.9 Hz), 7.34–7.98 (5H, m) and 8.45 (3H, bs); 13C NMR (100 MHz) δC: 17.87, 50.51, 66.40, 128.38, 128.80, 128.83 and 139.14; ESI-MS: m/z 170 and 172 in the ratio of 3:1.
Compound 2b: Yield 19 2 g (94%), mp 179–180 °C [lit 168–170 °C; SOR −92.68° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3440, 2939, 2880, 2772, 1603, 1578, 1499, 1456, 1373, 1198, 712 and 619;1H NMR (400 MHz, DMSO‑d6) δH: 1.01 (2H, J = 6.8 Hz), 4.44 (1H, m), 5.12 (1H, d, J = 6.9 Hz)), 7.36 – 8.00 (5H, m) and 8.34 (3H, bs); 13C NMR (100 MHz) δC: 17.90, 50.50.55, 66.42, 128.32, 128.82, 128.8 and 139.15; ESI-MS: m/z 170 and 172 in the ratio of 3:1.
2.2 General experimental procedure to synthesize N-Boc-chloramine 5a and 5b
Triethylamine (126 g, 1.24 mol) was slowly added at 0–5 °C to the HCl salt of (1S,2S)-chloramine 2a (125 g, 0.61 mol) and chloroform (500 mL) for 2 h followed by addition of Boc anhydride (135 g, 0.62 mol) at the same temperature for 30 min for 6 h. After completion of the reaction (TLC), the solvent was removed and EtOAc (500 mL) was charged. Stirred for 10 min and separated the white crystalline HCl salt of triethylamine by filtration. The clear filtrate was collected and EtOAC was removed using a rota evaporator. The obtained solid was stirred with n-heptane (170 mL), filtered and dried at 60 °C for overnight to get N-Boc-(1S,2S)-chloramine 5a (155 g). N-Boc-(1R,2R)-chloramine 5b was obtained by following the same procedure with (1R,2R)-chloramine 2b in place of 2a. Analytical data of 5a and 5b is presented below.
Compound 5a: White crystalline solid; Yield 155 g (95%); mp 94–96 °C; SOR +72.22° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3385, 3030, 2982, 2970, 2930, 1687, 1514, 1450, 1388, 1338, 1248, 1167, 1055, 748 and 706; 1H NMR (400 MHz) δH: 1.20 (d, 3H, J = 6.7 Hz), 1.39 (9H, s), 4.18 (1H, bs), 4.63 (1H, m), 4.98 (1H, d, J = 4.3 Hz) and 7.26–7.38 (5H, m); 13C NMR (100 MHz) δC: 18.28, 28.32, 51.85, 66.52, 79.62, 127.74, 128.34, 138.30 and 154.94. MS-ESI: m/z 270 and 272 in the ratio of 3:1.
Compound 5b: White solid; Yield 152 g (93%); mp 95–96 °C; SOR −69.13° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3385, 3031, 2982, 2970, 2930, 1688, 1514, 1450, 1388, 1338, 1248, 1167, 1055, 748 and 706; 1H NMR (400 MHz) δH: 1.21 (d, 3H, J = 6.7 Hz), 1.39 (s, 3H), 4.18 (1H, bs), 4.62 (1H, m), 4.97 (1H, d, J = 4.3 Hz) and 7.26–7.38 (5H, m); 13C NMR (100 MHz) δC: 18.29, 28.32, 51.86, 66.52, 79.61, 127.74, 128.36, 138.32 and 154.95. MS-ESI: m/z 270 and 272 in the ratio of 3:1.
2.3 General experimental procedure for synthesis of N-Boc-azidamine 6a and 6b
(1S,2S)-N-Boc-chloramine 5a (150 g, 0.56 mol), sodium azide (37 g, 0.57 mol) and dimethylsulfoxide (600 mL) were mixed and stirred at 25–30 °C for 10 h. About 2.0 L of water was added, the precipitate was filtered, washed with water and dried under vacuum over-night to get the (1R,2S)-N-Boc-azidamine 6a as a white solid. The same experiment was repeated with (1R,2R)-N-Boc-chloramine 5b instead of 5a to obtain the (1S,2R)-N-Boc-azidamine 6b. Analytical data N-Bocazidamines 6a and 6b is presented below.
Compound 6a: Yield 132 g, (86%), mp 80–81 °C; SOR −19.36° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3375, 3032, 2980, 2104, 1684, 1528, 1454, 1321, 1263, 1165, 758, 743 and 704; 1H NMR (400 MHz) δH: 0.99 (3H, d, J = 6.6 Hz), 1.46 (9H, s), 3.92 (1H, m), 4.68 (m, 1H), 4.90 (m, 1H), 7.26–7.40 (m, 5H, ArH); 13C NMR (100 MHz) δC: 14.4, 28.4, 51.3, 69.4, 126.9, 128.0, 128.7, 137.1, 155.0; MS-ESI: m/z at 277 [M+H]+; Anal. calcd for C14H20N4O2: C, 60.85; H, 7.30; N, 20.28. Found C, 61.12; H, 7.36; N, 20.31.
Compound 6b: Yield 121 g (79%); mp 82–85 °C; SOR +20.42° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3375, 3032, 2980, 2104, 1684, 1526, 1454, 1321, 1163, 743 and 704; 1H NMR (400 MHz) δH: 0.98 (3H, d, J = 6.6 Hz), 1.45 (9H, s), 3.91 (1H, m), 4.68(1H, m), 4.89 (1H, m), 7.25–7.39 (5H, m); 13C NMR (100 MHz) δC: 14.4, 28.4, 51.3, 69.5, 126.9, 128.0, 128.7, 137.1, 155.0; MS-ESI: m/z at 277 [M+H] +; Anal. calcd for C14H20N4O2: C, 60.85; H, 7.30; N, 20.28. Found C, 61.12; H, 7.36; N, 20.31.
2.4 General experimental procedure for the synthesis of triazole derivatives 9a-13a and 9b-13b
Terminal alkyne (8a-e, 36 mmol), (1R,2S)-N-Boc-azidamine (6a or 6b, 10 g, 36 mmol), N,N-diisopropylethylamine (5.6 g, 43 mmol) and copper(I) iodide (0.68 g, 10 mol%) were mixed with 100 mL of solvent mixture (methanol/water/tetrahydrofuran in equal volume) and stirred at 40–45 °C for 12–16 h to form the N-Boc protected triazole 7. Progress of the reaction monitored by TLC, the obtained residue was filtered to remove the copper salts and the filtrate was concentrated in the rota evaporator to obtain a oily mass. The residue was diluted with H2O (100 mL) and extracted with CH2Cl2 (100 mL). The organic layer was washed well with water followed by drying over anhyd. MgSO4. The dried organic layer was added with CF3COOH (10 mL) at 20–25 °C and stirred for 2–4 h. The progress of reaction monitored by TLC (10% methanol in methylenedichloride). The reaction mass was reduced completely under vacuum at 50 °C using a rota evaporator. The residue was diluted with 100 mL of water and basified with sodium hydroxide to pH above 9.0. The obtained solid was filtered and recrystallized in isopropanol to get the novel 1,4-disubstituted 1,2,3-triazoles 9–13 as a white to off-white solids in 87 to 95% yield.
Compound 9a: Prepared from N-Boc-azidamine 6a and terminal alkyne 8a. Yield 67%; mp 250–251 °C; SOR −35.5° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3375, 3122, 3030, 2926, 2852, 1606, 1514, 1493, 1454, 1227, 1165, 752 and 700; 1H NMR (400 MHz) δH: 1.25 (3H, d, J = 6.4 Hz), 1.60 (2H, m), 1.63 (4H, m), 1.83 (4H, m), 4.92 (1H, m), 5.61 (1H, d, J = 9.4 Hz), 7.28–8.15 (6H, m); 13C NMR (100 MHz) δC: 18.5, 22.2, 23.3, 25.7, 38.2, 38.3, 47.3, 68.2, 68.4, 121.2, 128.4, 128.6, 128.7, 138.0, 156.1, 168.7; MS-ESI: m/z at 283 [M+H] + as dehydrated one; Anal. calcd for C17H24N4O: C, 67.97; H, 8.05; N, 18.65. Found C, 68.18; H, 8.10; N, 18.72.
Compound 9b: Prepared from N-Boc-azidamine 6b terminal alkyne 8a.Yield 60%; mp 250–252 °C; SOR +30.1° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3371, 3297, 3030, 2936, 2855, 1493, 1452, 1379, 1157, 766 and 702; 1H NMR (400 MHz) δH: 1.20 (3H, d, J = 6.4 Hz), 1.58 (2H, m), 1.60 (4H, m), 1.68 (2H, m), 1.70 (m, 2H), 4.38 (1H, m), 5.73 (m, 1H), 7.40–7.61 (6H, m), 8.41 (2H, brs); 13C NMR (100 MHz) δc: 16.2, 22.3, 22.4, 25.9, 26.2, 49.3, 66.5, 74.5, 120.3, 124.5, 128.7, 129.5, 135.5, 148.9; MS-ESI: m/z at 283 [M+]+ as dehydrated one; Anal. calcd for C17H24N4O: C, 67.97; H, 8.05; N, 18.65. Found C, 68.15; H, 8.09; N, 18.61.
Compound 10a: Prepared from N-Boc-azidamine 6a terminal alkyne 8b. Yield 87%; mp 86–87 °C, SOR: −22.8° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3366, 3280, 3061, 2966, 2924, 1551, 1456, 1375, 1138, 754 and 709; 1H NMR (400 MHz) δH: 0.89 (3H, t, J = 7.3 Hz), 1.08 (3H, d, J = 5.9 Hz), 1.34 (2H, m), 1.62 (2H, m); 2.70 (2H, t, J = 7.7 Hz), 4.0 (1H, bs), 5.16 (1H, d, J = 7.6 Hz), 7.26–7.47 (6H, m); 13C NMR (100 MHz) δC: 13.79, 22.33, 25.37, 31.47, 50.02, 71.91, 77.23, 120.49, 128.26, 128.75, 128.93, 136.60 and 148.16; MS-ESI: m/z at 259 [M+H]+; Anal. calcd for C15H22N4: C, 69.73; H, 8.58; N, 21.69. Found C, 69.54; H, 8.54; N, 21.80
Compound 10b: Prepared from N-Boc-azidamine 6b terminal alkyne 8b. Yield 94%; mp 84–86 °C; SOR: +23.4° (c = 1.0%, methanol, 25 °C); IR (cm−1) 3368, 3281, 3060, 2966, 2924, 1550, 1458, 1375, 1138, 754 and 707; 1H NMR (400 MHz) δH: 0.89 (3H, t, J = 7.3 Hz), 1.09 (3H, d, J = 6.0 Hz), 1.34 (2H, m), 1.62 (2H, m); 2.67 (2H, t, J = 7.7 Hz), 4.02 (1H, bs), 5.18 (1H, d, J = 7.6 Hz), 7.28–7.48 (6H, m); 13C NMR (100 MHz) δC: 13.82, 22.36, 25.38, 31.48, 50.01, 72.01, 77.22, 120.51, 128.30, 128.86, 128.96, 136.65 and 148.20; MS-ESI: m/z at 259 [M+H]+; Anal. calcd for C15H22N4: C, 69.73; H, 8.58; N, 21.69. Found C, 69.42; H, 8.63; N, 21.74.
Compound 11a: Prepared from N-Boc-azidamine 6a terminal alkyne 8c. Yield 87%; mp 117–118 °C; SOR: −10.1° (c = 1.0%, methanol, 25 °C); IR (cm−1); 3364, 3285, 3071, 2965, 1715, 1589, 1493, 1456, 1279, 1194, 1105, 1053, 864, 767, 735 and 710; 1H NMR (400 MHz) δH: 1.07 (d, 3H, J = 6.3 Hz), 2.37 (3H, s), 4.02 (1H, m), 5.19 (1H, d, J = 7.8 Hz), 5.43 (2H, s), and 7.26–7.83 (10H, m); 13C NMR (100 MHz) δC: 20.8, 21.2, 50.1, 58.0, 72.2, 123.9, 126.9, 128.3, 129.0, 129.7, 130.3, 133.9, 136.1, 138.2, 142.7, 166.6; MS-ESI: m/z at 351 [M+H]+; Anal. calcd for C20H22N4O2: C, 68.55; H, 6.33; N, 15.99. Found C, 68.69; H, 6.30; N, 16.00
Compound 11b: Prepared from N-Boc-azidamine 6b terminal alkyne 8c. Yield 76%; mp 118–120 °C; SOR: +9.7° (c = 1.0%, methanol, 25 °C); IR (cm−1); 3364, 3069, 2965, 1715, 1684, 1528, 1456, 1279, 1192, 1105, 865, 739 and 708; 1H NMR (400 MHz) δH: 1.13 (d, 3H, J = 6.3 Hz), 2.42 (3H, s), 4.08 (1H, m), 5.25 (1H, d, J = 7.8 Hz), 5.48 (2H, s), 7.31–7.89 (10H, m, ArH); 13C NMR (100 MHz) δC: 19.9, 20.5, 49.4, 57.3, 71.5, 123.2, 126.2, 127.6, 128.2, 128.3, 129.5, 133.2, 135.3, 137.4, 141.9, 165.9; MS-ESI: m/z at 351 [M+H]+; Anal. calcd for C20H22N4O2: C, 68.55; H, 6.33; N, 15.99. Found C, 68.61; H, 6.39; N, 16.06.
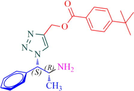
Compound 12a: Prepared from N-Boc-azidamine 6a terminal alkyne 8d. Yield 95%; mp 110–113 °C; SOR: −12.4° (c = 1.0%, methanol, 25 °C); IR (cm−1); 3372, 3136, 2967, 2905, 1711, 1609, 1454, 1275, 1115, 1097, 852, 779 and 704; 1H NMR (400 MHz) δH: 1.07 (3H, d, J = 6.7 Hz), 1.32 (9H, s), 4.00 (1H, m), 5.18 (1H, d, J = 4.3 Hz), 5.22 (2H, s), 7.27–7.96 (10H, m) and 8.77 (2H, bs); 13C NMR (100 MHz) δC: 20.77, 31.09, 35.09, 50.09, 57.94, 72.25, 123.88, 125.36, 126.97, 128.30, 128.95, 129.03, 129.64, 136.13, 142.81, 156.92 and 166.48; MS-ESI: m/z at 393 [M+H]+; Anal. calcd for C23H28N4O2: C, 70.38; H, 7.19; N, 14.27. Found C, 70.51; H, 7.22; N, 14.30.
Compound 12b: Prepared from N-Boc-azidamine 6b terminal alkyne 8d Yield 90%; mp 114–115 °C; SOR: +12.9° (c = 1.0%, methanol, 25 °C); IR (cm−1); 3372, 3136, 2967, 2905, 1711, 1609, 1454, 1276, 1115, 1098, 1049, 852, 779 and 704; 1H NMR (400 MHz) δH: 1.07 (d, 3H, J = 6.7 Hz), 1.32 (s, 9H), 4.04 (1H, m), 5.18 (1H, d, J = 4.3 Hz), 5.43 (2H, s), 7.27–7.96 (10H, m) 13C NMR (100 MHz) δC: 20.8, 31.0, 35.1, 50.1, 57.9, 72.3, 123.9, 125.4, 126.9, 128.3, 128.9, 129.0, 129.6, 136.1, 142.8, 156.9 and 166.5; MS-ESI: m/z at 393 [M+H]+; Anal. calcd for C23H28N4O2: C, 70.38; H, 7.19; N, 14.27. Found C, 70.48; H, 7.25; N, 14.32.
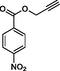
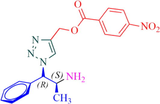
Compound 13a: Prepared from N-Boc-azidamine 6a terminal alkyne 8e. Yield 95%; mp 193–195 °C, SOR: −20.6° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3383, 3134, 3076, 2980, 2932, 1686, 1528, 1450, 1365, 1277, 1163, 1061, 856, 768, 750 and 708; 1H NMR (400 MHz) δH: 1.10 (d, 3H, J = 6.4 Hz), 3.85 (1H, m), 5.02 (1H, d, J = 7.8 Hz), 5.12 (2H, s) and 7.18–7.88 (10H, m) and 8.78 (2H, bs); 13C NMR (100 MHz) δH: 19.12, 50.08, 54.88, 70.42, 124.45, 125.78, 127.56, 129.46, 130.17, 131.44, 132.36, 134.94, 138.78, 141.42 and 167.86; MS-ESI: m/z at 382 [M+H]+; Anal. calcd for C19H19N5O4: C, 59.84; H, 5.02; N, 18.36. Found C, 59.96; H, 5.08; N, 18.42.
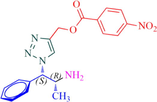
Compound 13b: Prepared from N-Boc-azidamine 6b terminal alkyne 8e. Yield 89%; mp 198–200 °C; SOR: +22.8° (c = 1.0%, methanol, 25 °C); IR (cm−1): 3390, 3130, 3075, 2980, 2930, 1695, 1530, 1450, 1365, 1277, 1162, 1060, 858, 768, 750 and 710; 1H NMR (400 MHz) δH: 1.11 (d, 3H, J = 6.4 Hz), 3.88 (1H, m), 5.03 (1H, d, J = 7.8 Hz), 5.10 (2H, s) and 7.16–7.87 (10H, m) and 8.65 (2H, bs); 13C NMR (100 MHz) δH: 19.18, 50.12, 54.87, 70.46, 124.48, 125.77, 127.55, 129.48, 130.16, 131.42, 132.38, 134.95, 138.82, 141.40 and 167.78; MS-ESI: m/z at 382 [M+H]+; Anal. calcd for C19H19N5O4: C, 59.84; H, 5.02; N, 18.36. Found C, 60.03; H, 5.06; N, 18.45.
3 Results and discussion
3.1 Chemistry
Our synthetic work commenced with readily available chiral pure precursor d- and l- isomers of phenylpropanolamine (1a and 1b) which was subjected to sequence of synthetic transformations as described in Scheme 1. Thus, phenylpropanolamine 1 was converted into its chloro derivative 2 by treatment with thionyl chloride. Nguyenet al., carried out the chlorination using PCl5 with 78% yield. Angelina et al. and Raul et al. reported the chlorination of phenylpropanolamine 1 using thionyl chloride with 74% and 72% yield, respectively. Interestingly, we obtained 2a in 89% and 2b in 94% yield when the reaction was carried out at 50 °C against the literature method (0–30 °C). Noticed inversion configuration at benzylic carbon which was confirmed with their specific optical rotations which are having opposite sign from their precursors. The obtained values of 2a and 2b are equal in value and opposite in sign which indicates the formation of pseudo isomers 2a with (1S,2S)- and 2b with (1R,2R)-configuration (Table 1). The obtained specific optical rotation (SOR) values of 2a and 2b are matches with values reported in the literature (Nguyen et al., 2000) and the structure of 2b was confirmed by XRD study (Angelina et al., 1998). The chloramines 2a and 2b were found to be enantiomers (pseudo) since they were prepared from their corresponding enantiomers 1a and 1b respectively.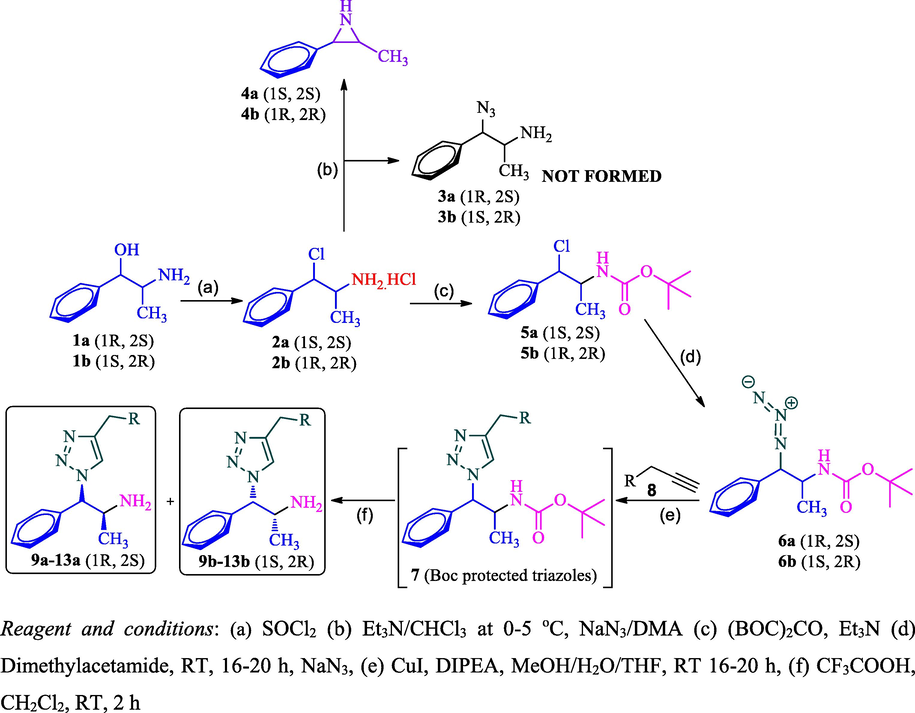
Synthesis of novel 1,4-disubstituted 1,2,3-triazoles 9–13.
Our next task was to convert the pseudo-chloramine 2 to azidamine 3. The direct conversion of the chloro group to azide was attempted by azidation with sodium azide. But the reaction led to the formation of aziridine 4 which was confirmed by mass spectral analysis. The plausible reason for the formation of aziridine 4 was due to the lone pair of electrons on the nitrogen of the amine group of 2 attacks on β-carbon where chlorine is attached which leads to the formation of aziridien 4 by eliminating chlorine atom as described in Scheme 2. The formation of aziridine 4 was confirmed through LCMS analysis and shown molecular ion [M+H]+ at m/z 149 instead of 177 of expected azide 3 and it further confirms the formation of aziridine 4 as evidenced by literature (Nguyen et al., 2000). Hence, a two-step sequence involving the protection of the amine group and subsequent azidation was adopted to obtain requisite azide 6. The Boc protected amine 5 restricts the formation of azidridine 4 due to the non-availability of nitrogen lone pair due to keto-enol tautomerism as shown in Scheme 2. The Boc protected amino compound 5 was successfully executed in the subsequent azidation reaction without any difficulties. Thus, compound 5a and 5b were treated with sodium azide at room temperature to furnish the respective N-Boc-azidamines 6a and 6b respectively in excellent yield. Here observed one more inversion of configuration at benzylic carbon which was confirmed with their specific optical rotations (6a and 6b) which are having opposite sign from their precursors 5a and 5b (Table 2). Hence overall retention of configuration at N-Boc-azidamine is expected with respect to phenylpropanolamine 1 (Scheme 1).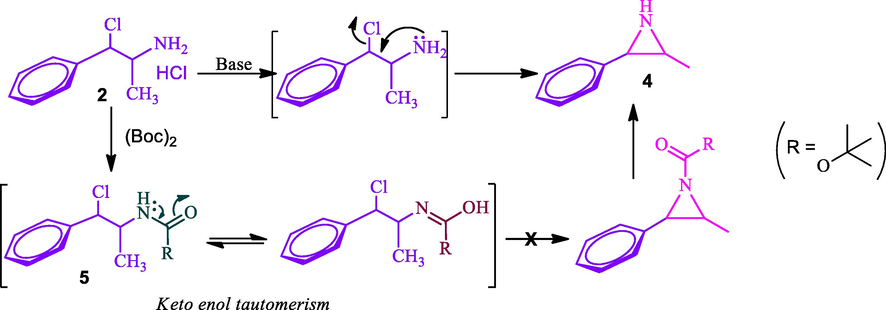
Azidation of chloramines 2 with and without the protection of amine group.
Having synthesized the N-Boc-azidamine 6 in excellent yield, one-pot click chemistry reaction was performed for the regioselective synthesis of 1,4-disubstituted-1,2,3-triazoles (Scheme 1). Thus, the N-Boc-azidamine 6 was treated with various terminal alkynes 8 to get N-Boc protected triazoles 7 as in situ and immediate hydrolysis in presence of trifluoroacetic acid to obtain the desired 1,4-disubstituted-1,2,3-triazoles 9–13 (Scheme 2) in good to excellent yields. The absolute configuration at the benzylic carbon of compounds 9–13 was expected to be retained with respect to starting material phenylpropanolamine 1. Initially, optimization of the click chemistry reaction was investigated under various condition including different catalyst and temperature as described in Table 3. Among them, the reaction with MeOH/H2O/THF (1:1:1) in the presence of CuI at 35–40 °C afforded excellent yield with less reaction time (Table 3, entry 35). The structure of compounds 9–13 (Table 4) was elucidated by 1H, 13C and mass spectroscopic analysis as illustrated for a representative example 11a. In the 1H NMR spectrum of 11a, a singlet at δ 2.37 ppm ascribable to methyl hydrogens of the phenyl ring. A doublet at δ 1.07 ppm assignable to the ester methylene hydrogens. The multiplet at δ 4.02 ppm belongs to methine hydrogen adjacent to the methyl group. A doublet at δ 5.19 ppm was assigned to methine hydrogen adjacent to the triazole ring. In the 13C NMR spectrum, the ester carbonyl carbon displayed at 166.6 ppm. Finally, the structure of N-acetyl derivative of compound 9b and its absolute stereo configuration have been unambiguously ascertained by X-ray diffraction analysis, which is shown in Fig. 1 (CCDC No.1443803). The molecular structure of 9b was stabilized by intermolecular O-H….N hydrogen bonding interaction. The detailed crystal data has been provided in supplementary file. S1: n-BuOH/water (1:1); S2: n-BuOH; S3: THF; S4: Methanol/water (1:1); S5:Methanol/water/THF (1:1:1); S6:MeOH; R1: No reaction; R2: Reaction proceeded but not completed; R3: Reaction completed; aTLC, mobile phase 10% methanol in dichlormetrhane, bYield of 7a after hydrolysis; DIPEA: N,N-Diisopropylethylamine; TEA:Triethylamine; NBA:n-Butyl alcohol.
Entry
Cat.
mol%
Base
mol%
Solvent
Temp (°C)
Time (h)
TLCa
Yieldb (%)
1
CuCl
10
DIPEA
200
S1
40–45
48
R1
–
2
CuCl
20
DIPEA
200
S3
40–45
48
R1
–
3
CuCl
50
DIPEA
200
S4
40–45
48
R1
–
4
CuCl
100
DIPEA
200
S5
40–45
48
R1
–
5
CuCl
100
DIPEA
200
S1
70–80
48
R1
–
6
CuCl
100
TEA
200
S1
70–80
48
R1
–
7
CuCl
100
DIPEA
300
S1
100–110
48
R1
–
8
CuCl
100
DIPEA
200
S2
70–80
48
R1
–
9
CuCl
100
DIPEA
200
S5
70–80
48
R1
–
10
CuBr
10
DIPEA
200
S1
40–45
48
R1
–
11
CuBr
50
DIPEA
200
S1
40–45
48
R2
–
12
CuBr
100
DIPEA
200
S2
40–45
48
R2
–
13
CuBr
100
DIPEA
200
S1
70–80
48
R2
–
14
CuBr
100
TEA
200
S5
70–80
48
R1
15
CuBr
100
DIPEA
200
S2
70–80
48
R2
16
CuBr
100
DIPEA
200
S3
70–80
48
R2
17
CuBr
100
DIPEA
200
S4
70–80
48
R2
18
CuBr
100
DIPEA
200
S5
70–80
48
R2
19
CuI
5
DIPEA
200
S1
70–80
48
R2
–
20
CuI
5
DIPEA
200
S2
70–80
48
R2
–
21
CuI
5
DIPEA
200
S3
70–80
48
R2
–
22
CuI
5
DIPEA
200
S4
70–80
48
R2
–
23
CuI
5
DIPEA
200
S5
70–80
48
R2
–
24
CuI
5
DIPEA
200
S6
60–65
48
R2
–
25
CuI
10
DIPEA
200
S1
70–80
24
R3
72
26
CuI
10
DIPEA
200
S2
70–80
48
R2
–
27
CuI
10
DIPEA
200
S3
70–80
48
R2
–
28
CuI
10
DIPEA
200
S4
70–80
24
R3
75
29
CuI
10
DIPEA
200
S5
70–80
12
R3
79
30
CuI
10
DIPEA
200
S6
60–65
48
R2
–
31
CuI
20
DIPEA
200
S1
70–80
20
R3
81
32
CuI
20
DIPEA
200
S4
70–80
12
R3
72
33
CuI
20
DIPEA
200
S5
70–80
12
R3
88
34
CuI
20
TEA
200
S1
70–80
48
R1
–
35
CuI
20
TEA
200
S4
70–80
48
R1
–
36
CuI
20
TEA
200
S5
70–80
48
R1
–
37
CuI
20
NBA
200
S5
70–80
48
R1
–
38
CuI
10
DIPEA
200
S5
20–25
24
R3
82
39
CuI
10
DIPEA
200
S5
35–40
12
R3
95
40
CuI
10
DIPEA
200
S5
55–60
12
R3
83
41
CuSO4
20
DIPEA
200
S5
70–80
48
R2
–
42
CuSO4
50
TEA
200
S5
70–80
48
R2
–
Entry
Terminal alkyne
1,2,3-Triazole
1,2,3-Triazole
SOR
Yield
1
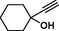 8a
8a
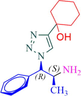 9a
9a
 9b
9b
−35.5°(9a)/ +30.1° (9b)
67% (9a)/ 60% (9b)
2
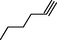 8b
8b
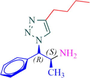 10a
10a
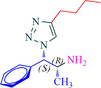 10b
10b
−22.8°(10a)/ +23.4°(10b)
87% (10a)/ 94% (10b)
3
 8c
8c
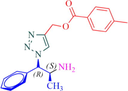 11a
11a
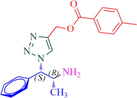 11b
11b
−10.1°(11a)/ +9.7°(11b)
87% (11a)/ 76% (11b)
4
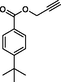 8d
8d
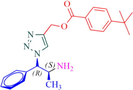 12a
12a
 12b
12b
−12.4(12a)/ +12.9(12a)
95% (12a)/ 90% (12a)
5
 8e
8e
 13a
13a
 13b
13b
−20.6°(13a)/ +22.8°(13b)
95% (13a)/ 89% (13a)
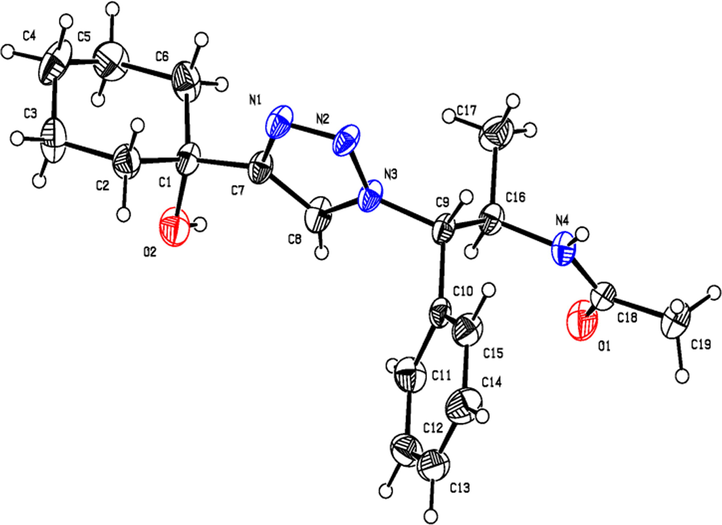
ORTEP diagram of N-acetyl derivative of 9b.
The feasible pathway for the formation of 1,2,3-triazole derivatives 9–13 is shown in Scheme 3. Initially, phenylpropanolamine 1 was reacted with thionyl chloride to form corresponding alkyl hydrochloride salt 2 with inversion of configuration at benzylic carbon. Compound 2 reacts with BOC anhydride in the presence of Et3N to afford N-Boc product 5 in good yield. The N-Boc protected compound 5 was involved SN2 reaction with sodium azide furnished azide 6 with one more inversion of configuration at benzylic carbon (overall retention of configuration). Simultaneously, the copper (I) generated in situ π-complex with terminal alkyne 8 in the presence of a base, the terminal hydrogen of the alkyne is deprotonated and consequently initiation of the reaction to form copper (I) acetylide 14 and then the azide 6 coordinates to copper acetylide to form copper complex 15 followed by the formation of regioselective 5-cuprated triazole 16. Finally, acid hydrolysis furnishes the desired triazole product 9–13 along with the regeneration of the active catalyst.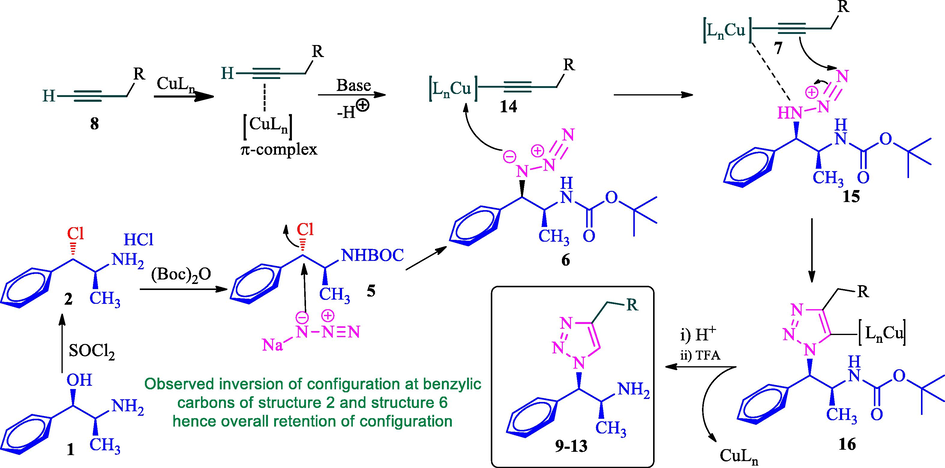
The feasible mechanism for the formation of 1,2,3-triazoles.
3.2 Antimicrobial activity
All the newly synthesized compounds 9a-13a and 9b-13b were assessed for their antimicrobial activities against five Gram-positive and seven Gram-negative bacteria and three fungi using in vitro disk diffusion method (Balachandran et al. 2012). The bacterial strains were obtained from Institute of Microbial Technology (IMTECH), Chandigarh, India-160 036 and fungal strains were obtained from the Department of Microbiology, Christian Medical College, Vellore, Tamil Nadu, India. As shown in Table 5, compounds 9a, 9b, 10a, 10b, 11a, 11b, 13a and 13b exhibited promising antibacterial activity against tested Gram-positive and Gram-negative bacteria at 1 mg/disk when compared with standard antimicrobial drug streptomycin (for bacteria). However, compounds 9a-13a and 9b-13b showed moderate activity against fungi. The minimum inhibitory concentration (MIC) of active compounds such as 9a, 9b, 10a, 10b, 11a, 11b, 13a and 13b were performed according to the standard reference methods (Balachandran et al., 2012; CLSI, 2008; NCCLS/CLSI, 2002) for Gram-positive and Gram-negative bacteria. The MIC values of all active compounds 9a, 9b, 10a, 10b, 11a, 11b, 13a and 13b showed potential activity against Gram-positive and Gram-negative bacteria with a range of 31.25 to 62.5 µg/mL are given in Table 6. Particularly, compounds 9a, 9b, 10a, 11a and 11b were showed significant MIC values against tested Gram-positive and Gram-negative bacteria when compared to control. At the same time antimicrobial activity of the synthesized compounds was correlated against their substitutions, it was understood that aliphatic chain/ring substitution attached to triazole ring (9a, 9b and 10a) and substitutions in aryl ring such as p-nitro (13a and 13b), p-methyl (11b) and p-tert-butyl (12a) enhanced the activity to the maximum. Only moderate activities were observed for the triazoles 10b, 11a and 12b of other isomers displayed moderate to good activity. NA- no activity, C-Streptomycin (standard antibacterial agent) C-Ketoconazole (standard antifungal agent) NA-no activity, C-Streptomycin (standard antibacterial agent).
S. No
Name of the microorganism
9a
9b
10a
10b
11a
11b
12a
12b
13a
13b
C
Gram positive Bacteria
1
B. subtilis
20
22
18
15
25
16
15
22
22
16
22
2
M. luteus
26
25
22
15
17
25
16
17
14
13
26
3
S. aureus
19
23
20
17
15
20
NA
NA
14
12
14
4
S. epidermidis
24
26
22
19
20
23
14
15
19
15
25
5
S. aureus MRSA
20
22
21
18
NA
18
NA
NA
18
12
30
Gram negative Bacteria
1
E. aerogenes
22
20
19
18
18
22
NA
NA
15
14
22
2
S. typhimurium
22
24
20
18
17
15
NA
NA
20
11
24
3
K. pneumoniae
20
22
19
NA
15
22
NA
NA
NA
NA
20
4
P. vulgaris
20
22
19
17
18
14
NA
NA
17
12
30
5
S. paratyphi-B
22
20
19
17
18
17
NA
16
19
12
18
6
S. flexneri
22
21
18
NA
17
20
15
NA
20
15
30
7
P. aeruginosa
24
16
19
17
25
19
19
16
14
23
30
Fungi
1
C. albicans
14
12
NA
NA
14
12
NA
NA
12
12
28
2
M. pachydermatis
13
10
10
NA
NA
NA
NA
NA
14
14
26
3
A. flavus
15
NA
NA
NA
17
NA
16
19
NA
NA
24
S. No.
Name of the microorganism
9a
9b
10a
10b
11a
11b
13a
13b
C
Gram positive Bacteria
1
B. subtilis
125
500
250
250
62.5
125
500
500
25
2
M. luteus
31.25
31.25
31.25
125
62.5
31.25
250
500
6.25
3
S. aureus
62.5
31.25
62.5
125
125
62.5
250
500
6.25
4
S. epidermidis
31.25
31.25
31.25
62.5
62.5
31.25
62.5
250
25
5
S. aureus MRSA
62.5
31.25
31.25
125
NA
125
62.5
500
6.25
Gram negative Bacteria
1
E. aerogenes
31.25
62.5
62.5
125
62.5
31.25
250
250
25
2
S. typhimurium
31.25
31.25
62.5
125
125
250
62.5
500
6.25
3
K. pneumoniae
62.5
31.25
62.5
NA
250
31.25
NA
NA
25
4
P. vulgaris
62.5
31.25
62.5
125
62.5
250
62.5
500
6.25
5
S. paratyphi-B
31.25
62.5
62.5
125
62.5
125
62.5
500
30
6
S. flexneri
31.25
31.25
125
NA
125
62.5
62.5
250
6.25
7
P. aeruginosa
125
125
125
500
250
125
250
250
25
3.3 Molecular docking studies
Docking simulation of the compounds 9–13 was performed on Topoisomerase IV (4EMV) (Takei et al., 2002) using AutoDock4 (Morris et al., 2009). The binding free energy was estimated and the docking scores are summarized in Table 7. Molecular docking results of all novel triazoles 9–13 with 4EMV receptor show that all the docked compounds bind efficiently with the receptor and exhibits free energy of binding value from −7.79 to −9.44 kcal/mol. Interestingly, among all the compounds docked, compound 13a exhibited very high binding with 4EMV receptor and forms four hydrogen bonds resulting in a binding energy of −9.44 kcal/mol.
The compound 13a, NH2 interacts with the ASP-78 and forms two hydrogen bonds with bond lengths of 1.8 and 2.3 Å. Also, NO2 interacts with the ARG-140 and forms two hydrogen bonds with bond lengths of 1.9 and 2.0 Å. In addition to the hydrogen bonds, C⚌O forms a polar interaction with the GLU-55 and triazole nitrogen forms a polar interaction with the ASN-51. Binding interaction of 13a with the active site residues of the 4EMV receptor is shown in Fig. 4. Docking diagram of method validation using crystallized and docked ligand with 4EMV receptor and docking mode of all synthesized triazoles in the active site of the receptor are shown in Figs. 2 and 3. From the docking results, compound 13a displayed higher binding energy (−9.44 kcal/mol). These docking results strongly correlate with in vitro antimicrobial activity.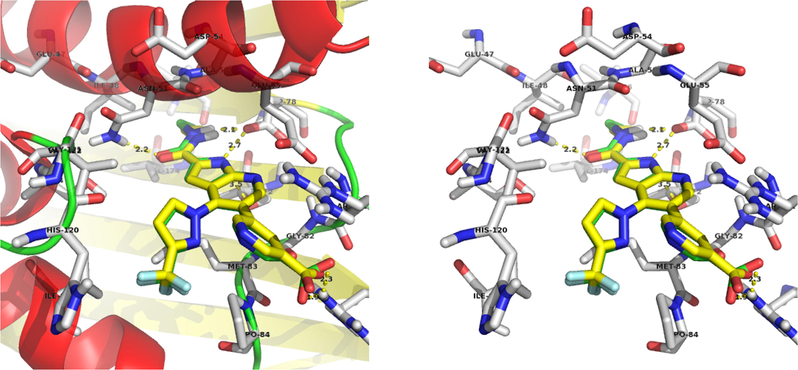
Method validation using crystallised and docked ligand with 4EMV receptor.
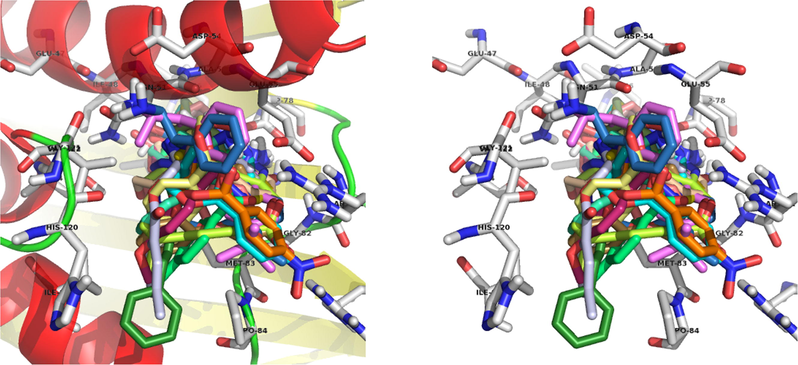
Docking mode of all synthesized triazoles in the active site.
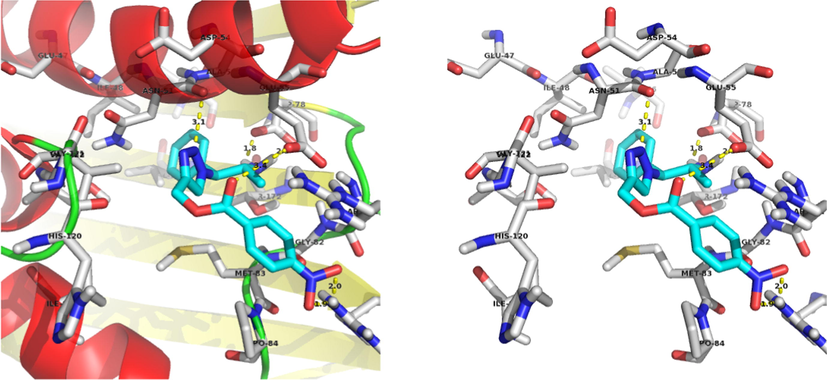
Docking mode of the most binding energy compound 13a.
4 Conclusion
A series of hitherto unexplored novel class of 1,2,3-triazoles were synthesized in good to excellent yields employing copper supported azide-alkyne 1,3-dipolar cycloaddition reaction. Most of the synthesized triazole compounds were displayed potent antibacterial activities against tested Gram-positive and Gram-negative bacteria. Particularly the compounds with aliphatic groups present in ester moiety showed significant antimicrobial activities when compared with others. Furthermore, all the synthesized 1,2,3-triazoles were also investigated for their docking simulation with Topoisomerase IV (4EMV) receptor and the result disclose that all compounds were binding efficiently with the receptor with binding free energy from −7.79 to −9.44 kcal/mol. Interestingly, compound 13a forms four hydrogen bonds with high binding energy (−9.44 kcal/mol) which strongly correlated to the in vitro findings due to presence of nitro function.
Acknowledgement
The project was supported by Researchers Supporting Project number (RSP-2020/231), King Saud University, Riyadh, Saudi Arabia. The authors C.R.K. and E.K. thanks to Malladi Drugs & Pharmaceuticals Ltd., R&D Centre, Chennai-600124, TN, India.
References
- Synthesis and biological evaluation of novel 1,2,3-triazole derivatives as anti-tubercular agents. Bioorg. Med. Chem. Lett.. 2017;27:3698-3703.
- [Google Scholar]
- Synthesis and antibacterial activity of 1-N-(β-d-glucopyranosyl)-4-((1-substituted-1H-1,2,3-triazol-4-yl)ethoxymethyl)-1,2,3-triazoles Arabian. J. Chem.. 2017;10:S3508-S3514.
- [Google Scholar]
- 4-Hydroxy-1,2,3-triazole moiety as bioisostere of the carboxylic acid function: a novel scaffold to probe the orthosteric γ-aminobutyric acid receptor binding site. Eur. J. Med. Chem.. 2018;158:311-321.
- [Google Scholar]
- Chlorination reactions of ephedrines revisited. Stereochemistry and functional groups effect on the reaction mechanisms. Tetrahedron Asymmetry. 1998;9:1661-1671.
- [Google Scholar]
- Multi-component syntheses of diverse 5-fluoroalkyl-1,2,3-triazoles facilitated by air oxidation and copper catalysis. Green Chem.. 2019;21:3407-3412.
- [Google Scholar]
- Antimicrobial and antimycobacterial activities of methyl caffeate isolated from Solanum torvum Swartz. Fruit. Indian J. Microbiol.. 2012;52:676-681.
- [Google Scholar]
- Novel azole derivatives are antagonists at the inhibitory GABA receptor on the somatic muscle cells of the parasitic nematode Ascaris suum. J. Parasitolo.. 1996;112:253-259.
- [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI), 2008. Reference method for Broth dilution antifungal susceptibility testing of filamentous fungi; approved standard second edition. CLSI document M38-A2 (ISBN 1-56238-668-9). Clinical and Laboratory Standards Institute, Wayne.
- Methods for antimicrobial susceptibility testing of aerobic bacteria approved standard M07–A8 (ninth ed.). Wayne: National Committee for Clinical Laboratory Standards; 2008.
- Antibacterial and antifungal activity of Flindersine isolated from the traditional medicinal plant, Toddaliaasiatica (L.) Lam. J. Ethnopharmacol.. 2009;123:494-498.
- [Google Scholar]
- Synthesis and biological evaluation of a library of AGE-related amino acid triazole crosslinkers. Eur. J. Org. Chem. 2020:5368-5379.
- [Google Scholar]
- Metal-free 1,2,3-triazole synthesis in deep eutectic solvents. Synlett. 2020;31:605-609.
- [Google Scholar]
- A copper(I)-catalyzed 1,2,3-triazole azide-alkyne click compound is a potent inhibitor of a multidrug-resistant HIV-1 protease variant. J. Med. Chem.. 2008;51:6263-6270.
- [Google Scholar]
- Design, synthesis, anti-inflammatory activity, and molecular docking studies of perimidine derivatives containing triazole. Bioorg. Med. Chem. Lett.. 2017;27:4409-4414.
- [Google Scholar]
- The inhibition of receptor-mediated and voltage-dependent calcium entry by the antiproliferative L-651,582. J. Biol. Chem.. 1991;266:10136-10142.
- [Google Scholar]
- Synthesis of pyrimidin-4-one-1,2,3-triazole conjugates as glycogen synthase kinase-3β inhibitors with anti-depressant activity. Bioorg. Chem.. 2016;68:41-55.
- [Google Scholar]
- Base-promoted synthesis of N-substituted 1,2,3-triazoles via enaminone-azide cycloaddition involving regitz diazo transfer. Org. Lett.. 2016;18:6034-6037.
- [Google Scholar]
- In-water synthesis of 5-thiolated 1,2,3-triazoles from β-thioenaminones by diazo transfer reaction. J. Org. Chem.. 2019;84:14179-14186.
- [Google Scholar]
- Transition metal-free annulation of enamines and tosyl azide toward N-heterocycle fused and 5-amino-1,2,3-triazoles. Eur. J. Org. Chem. 2020:5606-5609.
- [Google Scholar]
- Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur. J. Med. Chem.. 2020;190:112085
- [Google Scholar]
- AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [Google Scholar]
- M27–A2 reference method for broth dilution antifungal susceptibility testing of yeasts: proposed standard. Wayne: National Committee for Clinical Laboratory Standards; 2002.
- Synthesis of chiral cis-1,2,3-trisubstituted aziridines. Tetrahedron. 2000;56:7299-7304.
- [Google Scholar]
- Design, synthesis, and structure-activity correlations of novel dibenzo[b, d]furan, dibenzo[b, d]thiophene, and N-methylcarbazole clubbed 1,2,3-triazoles as potent inhibitors of Mycobacterium tuberculosis. J. Med. Chem.. 2012;55:3911-3922.
- [Google Scholar]
- Novel isoniazid embedded triazole derivatives: Synthesis, antitubercular and antimicrobial activity evaluation. Bioorg. Med. Chem. Lett.. 2020;30:127434
- [Google Scholar]
- Recent advances in DNA gyrase-targeted antimicrobial agents. Eur. J. Med. Chem.. 2020;199:112326
- [Google Scholar]
- Contribution of the C-8-methoxy group of gatifloxacin to inhibition of type II topoisomerases of Staphylococcus aureus. Antimicrob. Agents. Chemother.. 2002;46:3337-3338.
- [Google Scholar]
- BODIPY peptide labeling by late-stage C(sp3)−H activation. Angew. Chem. Int. Ed.. 2018;57:10554-10558.
- [Google Scholar]
- Synthesis of 1, 2, 4-triazole benzoyl arylamine derivatives and their high antifungal activities. Eur. J. Med. Chem.. 2020;200:112463
- [Google Scholar]
- Zhi, X., Shi-Jia, Z., Yi, L., 2019. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 183, 111700.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.10.026.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




