

Translate this page into:
Anticancer activity, spectroscopic and molecular docking of some new synthesized sugar hydrazones, Arylidene and α-Aminophosphonate derivatives
⁎Corresponding author at: Department of Chemistry, Turabah University College, Turabah, Taif University, 21995, Saudi Arabia. s.alosaimi@tu.edu.sa (Saad H. Alotaibi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
In this work, we design and then synthesis of new derivatives of nucleosides, oxadiazoline derivatives containing acetylated sugars and α-aminophosphonate derivatives. The synthesized compounds have been elucidated by different spectroscopic analysis, such as, elemental analysis, 13C NMR, infrared (IR) and 1H NMR. The compounds previously synthesized were purified and then tested against breast cancer cells (MCF-7). The results showed that the compounds 5d, 6b, 7a and 9h showed moderate to very high activity against breast cancer, with incidence rates of 78.45%, 84.60%, 93.45% and 95.78%, respectively. The reference ratio of 5-fluorouracil was inhibitory of 96.02%. The binding potential of synthesized compounds against thymidylate synthase (TS) and Cathepsin B (CB) has been investigated and the compounds 7a and 9h exhibits highly binding with thymidylate synthase (TS) and Cathepsin B (CB).
Keywords
4-Hydroxybenzaldehyde
Nucleosides
Arylidene derivatives
Molecular docking
Anticancer agents

1 Introduction
It is noticeable in recent times that a large number of researchers in the field of organic and medical chemistry have tended to prepare new derivatives of nucleosides that contain monosaccharaides in their composition as well as contain heterocyclic rings known for their biological activity (Ayoup et al., 2016). A study has been conducted on sugars hydrazones derived from the compounds oxadiazole and oxadiazoline against cancer cells, and the results showed the effectiveness of these compounds compared to 5-fluorouracil (Amer et al., 2018; Alotaibi, 2020). It is known that heterocycles containing one or more nitrogen atoms have multiple biological activity, so thienyl and thiazolopyrimidine derivatives associated with monosaccharaides were synthesized and showed high results as anti-cancer (Basiony et al., 2020; Dwivedi et al., 2020). Scientists working in the field of discovering new drugs seek to synthesize simple compounds with high biological activity. Among these compounds are Schiff bases known for their high effectiveness as anti-cancer (Ejidike and Ajibade, 2016), as well as found that they work as antioxidants (Kumar et al., 2017), anti-viral (Seley-Radtke, 2020) and antibacterial (Amer et al., 2017; Mesbah et al., 2018; Alotaibi and Amer, 2020). Schiff's bases associated with hydrazone have been linked to copper (II), zinc (II), and cobalt (II) ions to form complexes that have been found to have effective anticancer effects (Fekri et al., 2019; Abd-Elzaher et al., 2016). In this work, we synthesized new nucleosides and shiff bases derived from 4-Hydroxybenzaldehyde and the synthesized compounds were tested against brest cancer cells.
2 Material and methods
2.1 Chemistry
Melting points were determined with a Kofler block apparatus and are uncorrected. The nuclear magnetic resonance analysis for hydrogen 1HNMR was measured on spectrometer (400 MHz) using CDCl3 as a solvent and TMS (δ) as the internal standard in King Saud University, Riyadh, Saudi Arabia. The IR spectra were recorded on a by ic50 model FTIR (Thermo) using KBr disks in University College of Turbah, Taif University, Turbah, Taif, Saudi Arabia. TLC using aluminum silica gel plates 60 F245 monitored the progress of the reactions. The anticancer activity of the synthesized compounds was carried out in Al-Azhar University, Cairo, Egypt.
2.1.1 Ethoxycarbonylmethyl parahydroxybenzaldhyde (2)
In a dry acetone (250 ml), a mix of p-hydroxybenzaldhyde 1 (10 mmole) and ethylchloroacetate (10 mmole) was boiled in the presence of condenser for 12 h in the presence of anhydrous K2CO3 (10 mmole). After we make sure that the reaction has ended, the mixture is filtered, and the filtrate is evaporated under pressure to give the yellow oil in 85% yield. Rf = 0.50 (3% Ethylacetate in Diethyl ether). 1H NMR (DMSO‑d6): δ = 1.22 (t, 3H, J = 8.1 Hz, CH3CH2), 4.11 (q, 2H, J = 8.1 Hz, CH3CH2), 4.63 (s, 2H, CH2), 6.93 (d, 1H, J = 5.5 Hz, H-2), 7.56 (d, 1H, J = 5.5 Hz, H-3), 10.49 (s, 1H, CHO). Anal. Calc. for C11H12O4: C, 63.45; H, 5.81; Found C, 63.49; H, 5.85.
2.1.2 2-(4-formylphenoxy)acetohydrazide (3)
A mixture of 2 (10 mmol) and hydrazine hydrate (30 mmole) in ethanol (30 ml) was refluxed for 5 h. The separated product is recrystallized by ethanol to yield a white powder in 90% yield, m.p. 203–205 °C, Rf = 0.31 (3% Ethylacetate in Diethyl ether). 1H NMR (DMSO‑d6): δ = 4.35 (brs, 2H, NH2), 4.63 (s, 2H, CH2), 6.92 (d, 2H, J = 5.5 Hz, Ar-H), 7.49 (d, 2H, J = 5.5 Hz, Ar-H), 9.33 (brs, 1H, NH), 10.42 (s, 1H, CHO). Anal. Calc. for C9H10N2O3: C, 55.67; H, 5.19; N, 14.43. Found C, 55.72; H, 5.23; N, 14.38.
2.1.3 General procedure for the preparation of nucleosides derived from 2-(4-formylphenoxy) acetohydrazide 5a-d
To a solution of hydrazide 1 (10 mmole) in absolute ethanol (40 ml), appropriate sugar derivatives (D-(+)-mannose, D-(+)-galactose, D-(+)-glucose and L-(+)-arabinose) (10 mmole) respectively and acetic acid few drops were added. The mixture was heated for 8–12 h in the presence of a condenser. After the reaction finished the solvent was removed by evaporation and the formed precipitate was filtered off, washed by absolute ethanol, dried to give the nucleosides 5a-d in 80–85% yields respectively.
2.1.3.1 2-(4-formylphenoxy)-N'--2,3,4,5,6-pentahydroxyhexylidene)acetohydrazide (5a)
White powder, 85% yield, m.p. = 252–254 °C, Rf = 0.68 (5% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3310 (OH), 3050 (Ar-H), 1787 (CO of aldehyde), 1640 (CO of amide); 1H NMR (400 MHz, DMSO‑d6) δ: 2.75 (1H, dd, J = 5.2 Hz, OH), 3.55 (3H, dd, J = 5.2 Hz, 3xOH), 3.63 (1H, t, J = 5.2 Hz, OH), 3.59 (1H, d, J = 2.50 Hz, H-1), 3.35–3.80 (6H, m, H-2, H-3, H-4, H-5, H-6), 4.60 (2H, s, CH2), 7.43 (1H, s, CH), 7.17 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.85 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.00 (H, brs, NH), 9.85 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 64.42 (CH2OH of sugar), 69.21 (CH2), 60.75, 70.62, 71.73, 72.33 (CH-Sugar), 114.82, 129.21, 132.33, 163.92 (Ar-CH), 153.52 (CH⚌N), 171.12 (CONH), 191.06 (CHO); Anal. Calcd for C15H20N2O8: C, 50.56; H, 5.66; N, 7.86. Found C, 50.51; H, 5.72; N, 7.90.
2.1.3.2 D-(+)-Galactose-2-(4-formylphenoxy) acetohydrazide (5b)
White powder, 82% yield, m.p. = 283–285 °C, Rf = 0.65 (5% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3310 (OH), 3050 (Ar-H), 1787 (CO of aldehyde), 1640 (CO of amide); 1H NMR (400 MHz, DMSO‑d6) δ: 2.75 (1H, dd, J = 5.2 Hz, OH), 3.57 (3H, dd, J = 5.2 Hz, 3xOH), 3.65 (1H, t, J = 5.2 Hz, OH), 3.58 (1H, d, J = 2.50 Hz, H-1), 3.37–3.82 (6H, m, H-2, H-3, H-4, H-5, H-6), 4.63 (2H, s, CH2), 7.40 (1H, s, CH), 7.18 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.83 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.00 (H, brs, NH), 9.85 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 64.45 (CH2OH of sugar), 69.24 (CH2), 60.65, 70.67, 71.75, 72.33 (CH-Sugar), 114.81, 129.21, 132.33, 163.92 (Ar-CH), 153.52 (CH⚌N), 171.12 (CONH), 191.06 (CHO); Anal. Calcd for C15H20N2O8: C, 50.56; H, 5.66; N, 7.86. Found C, 50.61; H, 5.71; N, 7.91.
2.1.3.3 D-(+)-Glucose-2-(4-formylphenoxy) acetohydrazide (5c)
White powder, 80% yield, m.p. = 225–227 °C, Rf = 0.55 (5% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3310 (OH), 3050 (Ar-H), 1787 (CO of aldehyde), 1640 (CO of amide); 1H NMR (400 MHz, DMSO‑d6) δ: 2.75 (1H, dd, J = 5.2 Hz, OH), 3.57 (3H, dd, J = 5.2 Hz, 3xOH), 3.65 (1H, t, J = 5.2 Hz, OH), 3.58 (1H, d, J = 2.50 Hz, H-1), 3.37–3.82 (6H, m, H-2, H-3, H-4, H-5, H-6), 4.63 (2H, s, CH2), 7.40 (1H, s, CH), 7.18 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.83 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.00 (H, brs, NH), 9.85 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 64.45 (CH2OH of sugar), 69.24 (CH2), 60.65, 70.67, 71.75, 72.33 (CH-Sugar), 114.81, 129.21, 132.33, 163.92 (Ar-CH), 153.52 (CH⚌N), 171.12 (CONH), 191.06 (CHO); Anal. Calcd for C15H20N2O8: C, 50.56; H, 5.66; N, 7.86. Found C, 50.60; H, 5.70; N, 7.91.
2.1.3.4 L-(+)-Arabinose-2-(4-formylphenoxy) acetohydrazide (5d)
White powder, 84% yield, m.p. = 298–300 °C, Rf = 0.78 (5% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3310 (OH), 3050 (Ar-H), 1787 (CO of aldehyde), 1640 (CO of amide); 1H NMR (400 MHz, DMSO‑d6) δ: 2.75 (1H, dd, J = 5.2 Hz, OH), 3.57 (2H, dd, J = 5.2 Hz, 2xOH), 3.65 (1H, t, J = 5.2 Hz, OH), 3.58 (1H, d, J = 2.50 Hz, H-1), 3.37–3.82 (4H, m, H-2, H-3, H-4, H-5), 4.63 (2H, s, CH2), 7.40 (1H, s, CH), 7.18 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.83 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.00 (H, brs, NH), 9.85 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 64.15 (CH2OH of sugar), 69.09 (CH2), 60.55, 72.15, 74.13 (CH-Sugar), 114.81, 129.21, 132.33, 163.92 (Ar-CH), 153.52 (CH⚌N), 171.12 (CONH), 191.06 (CHO); Anal. Calcd for C14H18N2O7: C, 51.53; H, 5.56; N, 8.59. Found C, 51.48; H, 5.60; N, 8.54.
2.1.4 General procedure for the synthesis of acetylated nucleosides 6a-d
A solution of nucleoside derivatives 5a-d (10 mmole) in 20 ml of anhydrous pyridine, then the acetic anhydride (60 mmole) was added and The mixture was stirred at room temperature overnight (TLC). Then the product was poured over 80 gm of ice to give precipitates. The resultant precipitates were filtered, washed with water and dried to give the acetylated nucleosides 6a-d in 83=91% yields.
2.1.4.1 2,3,4,5,6-Penta-O-acetyl-D-(+)-Mannose-2-(4-formylphenoxy)acetohydrazone (6a)
Pale brown powder, 87% yield, m.p. = 198–200 °C, Rf = 0.55 (7% ethyl acetate in CHCl3). IR sp-ectra (KBr) (v, cm−1):, 3370 (NH), 3050 (Ar-H), 1735 (COCH3), 1787 (CO of aldehyde), 1640 (CO of amide), 1375 (CH3); 1H NMR (400 MHz, DMSO‑d6) δ: 2.11. 2.14. 2.15, 2.17, 2.20 (15H, 5 s, 5xCOCH3), 4.53 (1H, d, J = 2.50 Hz, H-1), 4.45–5.14 (5H, m, H-2, H-3, H-4, H-5, H-6), 4.63 (2H, s, CH2), 7.48 (1H, s, CH), 7.19 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.86 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.08 (H, brs, NH), 9.91 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.73 (2CH3 of 2COCH3), 21.08 (3CH3 of 3COCH3), 69.18 (CH2), 58.67, 61.65, 67.54, 70.22, 71.37 (CH-Sugar), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 153.43 (CH⚌N), 170.31 (CO of acetyl groups of sugar), 171.03 (CONH), 191.12 (CHO); Anal. Calcd for C25H30N2O13: C, 53.00; H, 5.34; N, 4.49. Found C, 52.99; H, 5.29; N, 4.45.
2.1.4.2 2,3,4,5,6-Penta-O-acetyl-D-(+)-Galactose-2-(4-formylphenoxy)acetohydrazone (6b)
Pale brown powder, 85% yield, m.p. = 234–236 °C, Rf = 0.45 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3050 (Ar-H), 1735 (COCH3), 1787 (CO of aldehyde), 1640 (CO of amide), 1375 (CH3); 1H NMR (400 MHz, DMSO‑d6) δ: 2.11. 2.14. 2.15, 2.17, 2.20 (15H, 5 s, 5xCOCH3), 4.53 (1H, d, J = 2.50 Hz, H-1), 4.45–5.14 (5H, m, H-2, H-3, H-4, H-5, H-6), 4.63 (2H, s, CH2), 7.48 (1H, s, CH), 7.19 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.86 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.08 (H, brs, NH), 9.91 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.73 (2CH3 of 2COCH3), 21.08 (3CH3 of 3COCH3), 69.18 (CH2), 58.67, 61.65, 67.54, 70.22, 71.37 (CH-Sugar), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 153.43 (CH⚌N), 170.31 (CO of acetyl groups of sugar), 171.03 (CONH), 191.12 (CHO); Anal. Calcd for C25H30N2O13: C, 53.00; H, 5.34; N, 4.49. Found C, 52.99; H, 5.29; N, 4.45.
2.1.4.3 2,3,4,5,6-Penta-O-acetyl-D-(+)-Glucose-2-(4-formylphenoxy)acetohydrazone (6c)
Pale brown powder, 83% yield, m.p. = 291–293 °C, Rf = 0.50 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3050 (Ar-H), 1735 (COCH3), 1787 (CO of aldehyde), 1640 (CO of amide), 1375 (CH3); 1H NMR (400 MHz, DMSO‑d6) δ: 2.11. 2.14. 2.15, 2.17, 2.20 (15H, 5 s, 5xCOCH3), 4.53 (1H, d, J = 2.50 Hz, H-1), 4.45–5.14 (5H, m, H-2, H-3, H-4, H-5, H-6), 4.63 (2H, s, CH2), 7.48 (1H, s, CH), 7.19 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.86 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.08 (H, brs, NH), 9.91 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.73 (2CH3 of 2COCH3), 21.08 (3CH3 of 3COCH3), 69.18 (CH2), 58.67, 61.65, 67.54, 70.22, 71.37 (CH-Sugar), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 153.43 (CH⚌N), 170.31 (CO of acetyl groups of sugar), 171.03 (CONH), 191.12 (CHO); Anal. Calcd for C25H30N2O13: C, 53.00; H, 5.34; N, 4.49. Found C, 52.99; H, 5.29; N, 4.45.
2.1.4.4 2,3,4,5,6-Penta-O-acetyl-D-(+)-Arabinose-2-(4-formylphenoxy)acetohydrazone (6d)
Pale brown powder, 91% yield, m.p. = 258–260 °C, Rf = 0.60 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3370 (NH), 3050 (Ar-H), 1735 (COCH3), 1787 (CO of aldehyde), 1640 (CO of amide), 1375 (CH3); 1H NMR (400 MHz, DMSO‑d6) δ: 2.08. 2.13. 2.16, 2.18, (12H, 4 s, 4xCOCH3), 4.53 (1H, d, J = 2.50 Hz, H-1), 4.21–5.09 (4H, m, H-2, H-3, H-4, H-5), 4.63 (2H, s, CH2), 7.48 (1H, s, CH), 7.20 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.90 (2H, dd, J = 6.2 Hz, CH-aromatic), 8.12 (H, brs, NH), 9.78 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.75 (2CH3 of 2COCH3), 21.11 (2CH3 of 2COCH3), 58.34, 61.30, 67.05, 68.47 (CH-Sugar), 69.20 (CH2), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 153.48 (CH⚌N), 170.22 (CO of acetyl groups of sugar), 171.07 (CONH), 191.08 (CHO); Anal. Calcd for C22H26N2O11: C, 53.44; H, 5.30; N, 5.67. Found C, 53.49; H, 5.36; N, 5.70.
2.1.5 General procedure for the synthesis of Oxadiazoline derivatives 7a-d
A mixture of acetylated nucleoside derivatives 6a-d (10 mmole) was dissolved in acetic anhydride (60 mmole), then the reaction mixture was heated in the presence of condenser at 80–90 °C for 1–2 h (TLC). The resultant mixture was poured into 80 gm of ice, and then chloroform was added to the aqueous solution in separating funnel. We then obtained the separated organic layer dissolved in the solvent and then evaporated the solvent to give the corresponding oxadiazoline derivatives 7a-d in 70–75% yields.
2.1.5.1 3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Mannose-2-(4-formylphenoxy)methyl – 1,3,4- Oxadiazoline (7a)
Brown gum, 75% yield, Rf = 0.63 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3050 (Ar-H), 1738 (COCH3), 1785 (CO of aldehyde), 1375 (CH3), 1450 (CH2); 1H NMR (400 MHz, DMSO‑d6) δ: 2.05 (3H, s, COCH3 of Oxadiazoline), 2.10, 2.13, 2.15, 2.18, 2.22 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.24–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.25 (1H, d, J = 2.50 Hz, H-1), 5.94 (1H, s, CH of Oxadiazoline), 7.20 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.88 (2H, dd, J = 6.2 Hz, CH-aromatic), 9.87 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.71 (CH3 of COCH3), 21.04 (4CH3 of 4COCH3), 23.38 (CH3 of COCH3 of Oxadiazoline), 69.99 (CH2), 61.60, 62.17, 67.78, 68.75, 75.82 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 158.25 (CH⚌N of Oxadiazoline), 168.73 (CO of N-acetyl of Oxadiazoline), 170.20 (CO of acetyl groups of sugar), 190.43 (CHO); Anal. Calcd for C27H32N2O14: C, 53.29; H, 5.30; N, 4.60. Found C, 53.33; H, 5.36; N, 4.65.
2.1.5.2 3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Galactose-2-(4-formylphenoxy)methyl – 1,3,4- Oxadiazoline (7b)
Brown gum, 73% yield, Rf = 0.67 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3050 (Ar-H), 1738 (COCH3), 1785 (CO of aldehyde), 1375 (CH3), 1450 (CH2); 1H NMR (400 MHz, DMSO‑d6) δ: 2.05 (3H, s, COCH3 of Oxadiazoline), 2.10, 2.13, 2.15, 2.18, 2.22 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.24–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.25 (1H, d, J = 2.50 Hz, H-1), 5.94 (1H, s, CH of Oxadiazoline), 7.20 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.88 (2H, dd, J = 6.2 Hz, CH-aromatic), 9.87 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.71 (CH3 of COCH3), 21.04 (4CH3 of 4COCH3), 23.38 (CH3 of COCH3 of Oxadiazoline), 69.99 (CH2), 61.60, 62.17, 67.78, 68.75, 75.82 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 158.25 (CH⚌N of Oxadiazoline), 168.73 (CO of N-acetyl of Oxadiazoline), 170.20 (CO of acetyl groups of sugar), 190.43 (CHO); Anal. Calcd for C27H32N2O14: C, 53.29; H, 5.30; N, 4.60. Found C, 53.33; H, 5.36; N, 4.65.
2.1.5.3 3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Glucose-2-(4-formylphenoxy)methyl – 1,3,4- Oxadiazoline (7c)
Brown gum, 70% yield, Rf = 0.60 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3050 (Ar-H), 1738 (COCH3), 1785 (CO of aldehyde), 1375 (CH3), 1450 (CH2); 1H NMR (400 MHz, DMSO‑d6) δ: 2.05 (3H, s, COCH3 of Oxadiazoline), 2.10, 2.13, 2.15, 2.18, 2.22 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.24–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.25 (1H, d, J = 2.50 Hz, H-1), 5.94 (1H, s, CH of Oxadiazoline), 7.20 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.88 (2H, dd, J = 6.2 Hz, CH-aromatic), 9.87 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.71 (CH3 of COCH3), 21.04 (4CH3 of 4COCH3), 23.38 (CH3 of COCH3 of Oxadiazoline), 69.99 (CH2), 61.60, 62.17, 67.78, 68.75, 75.82 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 158.25 (CH⚌N of Oxadiazoline), 168.73 (CO of N-acetyl of Oxadiazoline), 170.20 (CO of acetyl groups of sugar), 190.43 (CHO); Anal. Calcd for C27H32N2O14: C, 53.29; H, 5.30; N, 4.60. Found C, 53.33; H, 5.36; N, 4.65.
2.1.5.4 3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Arabinose-2-(4-formylphenoxy)methyl – 1,3,4- Oxadiazoline (7d)
Brown gum, 74% yield, Rf = 0.65 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3050 (Ar-H), 1738 (COCH3), 1785 (CO of aldehyde), 1375 (CH3), 1450 (CH2); 1H NMR (400 MHz, DMSO‑d6) δ: 2.06 (3H, s, COCH3 of Oxadiazoline), 2.10, 2.13, 2.15, 2.18, 2.20 (12H, 5 s, 4xCOCH3), 4.60 (2H, s, CH2), 4.21–5.15 (4H, m, H-2, H-3, H-4, H-5), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.99 (1H, s, CH of Oxadiazoline), 7.18 (2H, dd, J = 6.00 Hz, CH-aromatic), 7.87 (2H, dd, J = 6.2 Hz, CH-aromatic), 9.87 (1H, s, CHO); 13C NMR (100 MHz, CDCl3): δ: 20.71 (CH3 of COCH3), 21.04 (3CH3 of 3COCH3), 23.25 (CH3 of COCH3 of Oxadiazoline), 70.00 (CH2), 61.62, 64.65, 68.73, 75.63 (CH-Sugar), 76.79 (CH-N-Ac of Oxadiazoline), 114.79, 129.23, 132.33, 163.90 (Ar-CH), 158.30 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.23 (CO of acetyl groups of sugar), 191.12 (CHO); Anal. Calcd for C24H28N2O12: C, 53.73; H, 5.26; N, 5.22. Found C, 53.77; H, 5.31; N, 5.36.
2.1.6 Preparation of α-aminophosphonates 9a-l
Equivalent amounts of Oxadiazoline derivatives 7a-d (10 mmole) triphenylphosphite (10 mmole) and different aromatic amines (1-Napthyl amine (10 mmole), 2-Nitro aniline (10 mmole) and 4-Methyl aniline (10 mmole) respectively were dissolved in CH3CN, the addition of few drops of perchloric acid occurred, later the stirring was occurred at 25 °C overnight. The solvent evaporated and then the residues of a-aminophosphonate derivatives were treated with diethyl ether to afford the corresponding α-Aminophosphonates 9a-l in 85–92% yields.
2.1.6.1 5-((4-((diphenoxyphosphoryl)(naphthalen-1-ylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Mannose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9a)
Pale yellow powder, 85% yield, m.p. = 278–280 °C, Rf = 0.75 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (21H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C49H50N3O16P: C, 60.80; H, 5.21; N, 4.34. Found C, 60.86; H, 5.25; N, 4.40.
2.1.6.2 5-((4-((diphenoxyphosphoryl)((2-nitrophenyl)amino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Mannose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9b)
Pale yellow powder, 90% yield, m.p. = > 300 °C, Rf = 0.80 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1570, 1380 (NO2), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C45H48N4O18P: C, 56.13; H, 4.92; N, 5.82. Found C, 56.08; H, 5.00; N, 5.78.
2.1.6.3 5-((4-((diphenoxyphosphoryl)(p- tolylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Mannose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9c)
Pale yellow powder, 87% yield, m.p. = > 300 °C, Rf = 0.70 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 2.35 (3H, s, CH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 21.41 (CH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C46H50N3O16P: C, 59.29; H, 5.41; N, 4.51. Found C, 59.33; H, 5.37; N, 4.55.
2.1.6.4 5-((4-((diphenoxyphosphoryl)(naphthalen-1-ylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Galactose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9d)
Pale yellow powder, 92% yield, m.p. = 267–269 °C, Rf = 0.72 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (21H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C49H50N3O16P: C, 60.80; H, 5.21; N, 4.34. Found C, 60.86; H, 5.26; N, 4.40.
2.1.6.5 5-((4-((diphenoxyphosphoryl)((2-nitrophenyl)amino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Galactose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9e)
Pale brown powder, 88% yield, m.p. = 290–292 °C, Rf = 0.65 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1570, 1380 (NO2), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 147.48, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); 31P NMR (162 MHz, CDCl3): d = 17.23 (O = P-CH-); Anal. Calcd for C45H48N4O18P: C, 56.13; H, 4.92; N, 5.82. Found C, 56.19; H, 5.00; N, 5.89.
2.1.6.6 5-((4-((diphenoxyphosphoryl)(p- tolylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Galactose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9f)
White powder, 88% yield, m.p. = > 300 °C, Rf = 0.55 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 2.35 (3H, s, CH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 21.41 (CH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C46H50N3O16P: C, 59.29; H, 5.41; N, 4.51. Found C, 59.34; H, 5.37; N, 4.58.
2.1.6.7 5-((4-((diphenoxyphosphoryl)(naphthalen-1-ylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Glucose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9g)
Brown gum, 85% yield, Rf = 0.73 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (21H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C49H50N3O16P: C, 60.80; H, 5.21; N, 4.34. Found C, 60.86; H, 5.25; N, 4.40.
2.1.6.8 5-((4-((diphenoxyphosphoryl)((2-nitrophenyl)amino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Glucose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9h)
Brown gum, 87% yield, Rf = 0.72 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1570, 1380 (NO2), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C45H48N4O18P: C, 56.13; H, 4.92; N, 5.82. Found C, 56.18; H, 4.86; N, 5.76.
2.1.6.9 5-((4-((diphenoxyphosphoryl)(p- tolylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Glucose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9i)
Brown powder, 88% yield, m.p. = > 300 °C, Rf = 0.66 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.11, 2.14, 2.17, 2.21 (15H, 5 s, 5xCOCH3), 2.35 (3H, s, CH3), 4.60 (2H, s, CH2), 4.22–5.12 (5H, m, H-2, H-3, H-4, H-5, H-6), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (4CH3 of 4COCH3), 21.41 (CH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C46H50N3O16P: C, 59.29; H, 5.41; N, 4.51. Found C, 59.35; H, 5.36; N, 4.57.
2.1.6.10 5-((4-((diphenoxyphosphoryl)(naphthalen-1-ylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Arabinose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9j)
White powder, 92% yield, m.p. = 212–214 °C, Rf = 0.63 (7% methanol in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.08, 2.13, 2.17, 2.21 (12H, 4 s, 4xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (4H, m, H-2, H-3, H-4, H-5), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (21H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (3CH3 of 3COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C46H46N3O14P: C, 61.67; H, 5.18; N, 4.69. Found C, 61.73; H, 5.25; N, 4.74.
2.1.6.11 5-((4-((diphenoxyphosphoryl)((2-nitrophenyl)amino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Arabinose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9k)
White powder, 89% yield, m.p. = 277–279 °C, Rf = 0.56 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1570, 1380 (NO2), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.11, 2.14, 2.17, 2.21 (12H, 4 s, 4xCOCH3), 4.60 (2H, s, CH2), 4.22–5.12 (4H, m, H-2, H-3, H-4, H-5), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (3CH3 of 3COCH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 61.67, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C42H43N4O16P: C, 56.63; H, 4.87; N, 6.29. Found C, 56.68; H, 4.94; N, 6.35.
2.1.6.12 5-((4-((diphenoxyphosphoryl)(p- tolylamino)methyl)phenoxy)methyl)-3-Acetyl-2-(2,3,4,5,6-Penta-O-acetyl-D-(+)-Arabinose-2-phenoxy)methyl – 1,3,4- Oxadiazoline (9l)
Brown gum, 88% yield, Rf = 0.57 (7% ethyl acetate in CHCl3). IR spectra (KBr) (v, cm−1):, 3350 (NH), 3050 (Ar-H), 1738 (COCH3), 1375 (CH3), 1450 (CH2); 1HNMR (400 MHz, DMSO‑d6) δ: 2.03 (3H, s, COCH3 of Oxadiazoline), 2.11, 2.14, 2.17, 2.21 (12H, 4 s, 4xCOCH3), 2.35 (3H, s, CH3), 4.60 (2H, s, CH2), 4.22–5.12 (4H, m, H-2, H-3, H-4, H-5), 5.27 (1H, d, J = 2.50 Hz, H-1), 5.65 (1H, t, J = 5.5 Hz, CH-P-), 5.90 (1H, s, CH of Oxadiazoline), 6.85–8.10 (18H, m, Ar-H), 8.98 (1H, brs, NH); 13C NMR (100 MHz, CDCl3): δ: 20.73 (CH3 of COCH3), 21.08 (3CH3 of 3COCH3), 21.41 (CH3), 23.41 (CH3 of COCH3 of Oxadiazoline), 69.65 (CH-P-), 72.31 (CH2) 62.27, 67.72, 68.43, 75.76 (CH-Sugar), 76.48 (CH-N-Ac of Oxadiazoline), 109.34, 114.09, 119.12, 120.33, 121.23, 124.65, 124.76, 125.06, 126.11, 127.66, 127.92, 128.4, 128.6, 10.1, 134.34, 14748, 150.42, 156.18 (Ar-CH), 158.27 (CH⚌N of Oxadiazoline), 168.55 (CO of N-acetyl of Oxadiazoline), 170.25 (CO of acetyl groups of sugar); Anal. Calcd for C43H46N3O14P: C, 60.07; H, 5.39; N, 4.89. Found C, 60.00; H, 5.43; N, 4.94.
2.2 Anticancer activity
2.2.1 Cell line propagation
At the start of pretreatment, cell propagation was established via Dulbecco modified Eagle Medium (DMEM), consisting of 1% L-glutamine, 50 μg/mL gentamicin, and 10% heat inactivated fetal bovine serum and HEPES buffer. Cells were placed in an incubation at 37 °C. Hence, they were promoted twice a week. (Mosmann, 1983). Cytotoxicity was determined via a viability assay in which a 96-well plate was used to prepare cells to accommodate a concentration of 1x 104 per well in 100 μl growth medium. After 24 h, different concentrations of the medium were applied and then added using a multichannel pipette, the serial double dilution of the tested chemical compounds was assigned to the confluent cell. Monolayers were distributed in 96 well plates. Adjustment of microtiter plate incubation for 48 h with moisture at 37 °C and 5% CO2 was performed. The test sample was concentrated with three wells. Then, each control cell was incubated without test containment as well as with the addition of dimethylsulphoxide and without. The maximum inactive concentration of dimethylsulphoxide was 0.1%. After the cell incubation at 37 °C, and with a 24-hour incubation setting, different concentrations of samples were produced. A colorimetric technique was used to determine the cell productivity. After incubation, 1% violet crystalline solution was placed into the remaining cell media in each well for 30 min. The remaining stains were washed by distilled water. 30% glacial acetic acid was added in the wells followed by absorption measurements at 490 nm followed by a spectral background correction across the well without spots. By the absence of the compounds tested. Samples were compared with cellular controls as the experiment was conducted in triplicate; the efficacy of cytotoxicity was then calculated. Hence, a microplate reader was used to detect the optical density of the samples. The viable cells were mapped together with the percentage survival by the following equation: [(ODt/ODc)] × 100%, where ODc is the average optical density of untreated cells and ODt is the average optical density of all wells treated with samples tested; A graph of live cells and drug concentration was made to understand the degree of survival of tumor cells alive after treatment (Gomha et al., 2015). Graphic plots of the dose–response-curve for all the concentration help to determine the estimation of the IC50 of intact cells (GraphPad Prism software; San Diego, CA USA).
2.3 Docking studies
Into TS binding site (PDB ID: 1JU6, resolution: 2.89 Å) and CB (PDB ID: 2DCD, resolution: 2.50 Å). The co-crystallized ligands (LYA and 78A) were used as reference molecules against TS and CB, respectively. The binding free energies (ΔG) were reported in (Table 6) against thymidylate synthase (TS) and Cathepsin B (CB). The crystal structures of the target proteins; thymidylate synthase (TS) (PDB ID: JU6, resolution: 2.89 Å) and Cathepsin B (CB) (PDB ID: 2DCD, resolution: 2.50 Å) were downloaded from Protein Data Bank (http://www.pdb.org). Molecular Operating Environment (MOE) was used for the docking analysis (Nasser et al., 2020). In these studies, the free energies and binding modes of the examined molecules against target proteins were determined. At first, the water molecules were removed from the crystal structures of target proteins, retaining only one chain which essential for binding. The co-crystallized ligands (LYA and 78A for TS and CB, respectively) were used as reference ligands. Then, the protein structures were protonated and the hydrogen atoms were hided. Next, the energy was minimized and the binding pockets of each protein was defined (Li et al., 2020; El-Gamal et al., 2018). The structures of the examined compounds and the co-crystallized ligands were drawn using ChemBioDraw Ultra 14.0 and saved as SDF formats. Then, the saved files were opened using MOE software and 3D structures were protonated. Next, the energy of the molecules was minimized. Validation processes were performed for each target receptor by running the docking process for only the co-crystallized ligand. Low RMSD values between docked and crystal conformations indicate valid performance (Ibrahim, 2017; Elmetwally et al., 2019). The docking procedures were carried out utilizing a default protocol. In each case, 20 docked structures were generated using genetic algorithm searches. The output from of MOE software was further analyzed and visualized using Discovery Studio 4.0 software (Nasser et al., 2020; Elmetwally et al., 2019; Mahdy, 2020; El-Zahabi et al., 2019; El-Naggar, 2020).
2.4 In silico toxicity
The toxicity parameters of the synthesized compounds were calculated using Discovery studio 4.0. Sorafenib was used as a reference drug. At first, the CHARMM force field was applied then the compounds were prepared and minimized according to the preparation of small molecule protocol. Then different parameters were calculated from the toxicity prediction (extensible) protocol.
3 Results and discussion
3.1 Chemistry
4-Hydroxybenzaldehyde (1) was reacted with ethylchloroacetate and potassium carbonate in dry acetone under reflux to give Ethoxycarbonylmethyl parahydroxybenzaldhyde (2) in 85% yield. Ethoxycarbonylmethyl parahydroxybenzaldhyde (2) was hydrazinolysis by hydrazine hydrate in absolute ethanol to give 2-(4-formylphenoxy) acetohydrazide (3) (Scheme 1). The elucidation of compounds 2 by 1HNMR spectra appears that triplet peak at 1.22 for CH3CH2, quartet at 4.11 for CH3CH2 and 10.49 for CHO group; on the other hand, the elucidation of 3 by 1HNMR spectra appears that broad peak at 4.35 for NH2 group, broad peak at 9.33 for NH and singlet at 10.42 for CHO group.
Hydrazide 3 was reacted with sugar moieties (D-(+)-Mannose, D-(+)-Galactose, D-(+)-Glucose and D-(+)-arabinose respectively) 4a-d in absolute ethanol and in the presence of acetic acid as catalyst under reflux to give the corresponding nucleosides 5a-d in 80–85% yields which were acetylated by acetic anhydride in pyridine at room temperature to give the acetylated nucleosides 6a-d in yields, on the other hand nucleosides 5a-d were reacted with acetic anhydride at 80–90 °C to give oxadiazoline derivatives 7a-d in yields (Scheme 2). The elucidation of nucleosides 5a-d by IR spectra showed that the disappearance of NH2 and appearance of 1640 of CONH, 1778 for CHO, 3310 for OH and 3370 for NH; 1HNMR spectra appears that the disappearance of NH2 and appearance of peaks around 3.35–3.80 for ((CH) groups of sugar moieties), singlet peaks around 2.75–3.63 for (OH) groups of sugar moieties), doublet of doublet 7.17 and 7.85 for CH-aromatic, broad peak around 8.00 for (NH group) and singlet peak around 9.85 for CHO group; 13CNMR spectra appears that peaks around 60.75 to 72.53 for (CH of sugar moieties), 64.42 for hydroxyl groups of sugar moieties, the peaks of (CH– aromatics) appears around 114.82 to 163.92, peak around 153.52 for (CH⚌N), peak around 171.12 for (CONH) and peak around 191.06 for formyl group. The elucidation of nucleosides 6a-d by IR spectra showed that the disappearance of hydroxyl group and appearance of 3370 for NH, 1735 for (COCH3) and 1640 of CONH, 1778 for CHO; 1HNMR spectra appears that the disappearance of hydroxyl groups of sugar moieties and appearance of singlet peaks around 2.08 to 2.20 for (COCH3 groups of sugar moieties), singlet peaks around 4.45–5.14 for ((CH) groups of sugar moieties), doublet of doublet 7.19 and 7.86 for CH-aromatic, singlet peak around 7.84 for (CH⚌N), broad peak around 8.08 for (NH group) and singlet peak around 9.91 for CHO group; 13CNMR spectra appears that peaks around 20.73 to 21.08 for (CH3 of acetyl groups of sugar moieties), peaks around 58.67 to 71.37 for (CH of sugar moieties), peaks of (CH– aromatics) appears around 114.79 to 163.90, peak around 153.43 for (CH⚌N), peak around 170.31 for (CO of acetyl groups of sugar moieties), peak around 171.03 for (CONH) and peak around 191.12 for formyl group. The elucidation of nucleosides 7a-d by IR spectra showed that the appearance of 1785 for CHO and 1735 for (COCH3); 1HNMR spectra appears that the appearance of singlet peak around 2.05 for acetyl group of oxadiazoline, singlet peaks around 2.10 to 2.22 for (acetyl groups of sugar moieties), singlet peaks around 4.24–5.25 for ((CH) groups of sugar moieties), doublet of doublet 7.20 and 7.88 for CH-aromatic, singlet peak around 5.94 for (CH⚌N of oxadiazoline), and singlet peak around 9.87 for CHO group; 13CNMR spectra appears that peaks around 20.71 to 21.04 for (CH3 of acetyl groups of sugar moieties), singlet peak around 23.38 for acetyl group of oxadiazoline ring, peaks around 61.60 to 75.82 for (CH of sugar moieties), peak around 76.48 for (CH-N-Ac of oxadiazoline), peaks of (CH– aromatics) appears around 114.79 to 163.90, peak around 158.25 for (CH⚌N), peak around 168.73 for (CO of N-acetyl of oxadiazoline), peak around 170.20 for (CO of acetyl groups of sugar moieties), peak around 171.03 for (CONH) and peak around 190.43 for formyl group.
Oxadiazoline derivatives 7a-d were reacted with primary amine derivatives 8a-l and triphenylphosphite in acetonitrile and in the presence of perchloric acid as catalyst at room temperature to give α-aminophosphonate derivatives 9a-l in 85–92% yields (Scheme 3). The elucidation of nucleosides 9a-l by IR spectra showed that the disappearance of formyl group and appearance of peak around 3350 for (NH), peak around 3050 for (Ar-H) and peak around 1738 for (COCH3); 1HNMR spectra appears that the appearance of singlet peak around 2.03 for acetyl group of oxadiazoline, singlet peaks around 2.08 to 2.21 for (acetyl groups of sugar moieties), singlet peaks around 4.22–5.27 for ((CH) groups of sugar moieties), peak around 5.65 for (CH-P-), peak around 5.90 for (CH⚌N of oxadiazoline), multiplet peaks around 6.85 to 8.00 for (CH-aromatic) and broad peak around 8.98 for NH group; 13CNMR spectra appears that peaks around 20.73 to 21.08 for (CH3 of acetyl groups of sugar moieties), singlet peak around 23.41 for acetyl group of oxadiazoline ring, peaks around 61.67 to 75.76 for (CH of sugar moieties), peak around 69.65 for (CH-P-) peak around 76.48 for (CH-N-Ac of oxadiazoline), peaks of (CH– aromatics) appears around 109.34 to 156.18, peak around 158.27 for (CH⚌N of oxadiazoline), peak around 168.55 for (CO of N-acetyl of oxadiazoline) and peak around 170.25 for (CO of acetyl groups of sugar moieties).
3.2 Anticancer activity
In this work, the compounds synthesized as anti-breast cancer compounds were tested. These compounds were nucleosides, acetylated nucleosides, and oxadiazoline derivatives, alpha-aminophosphonates 6d, 4d, 6b and 4c against the line cells of MCF-7. The well-known drug as the anticancer 5-fluorouracil (5-FU) was used as a reference, which is an important analog of Pyrimidines. Table 5 and Fig. 5. The results showed that the compound 9h containing phosphonate group had very high inhibitory activity against MCF-7cell line, with 95.78% inhibition against breast cancer cells; Compared to the inhibition by the standard 5-FU of 96.02% (Table 4). The results also showed that the compound 7a containing the sugar acetylated and oxadiazoline ring (Table 3) had high activity against the MCF-7 cell line, with 93.45% inhibition. On the other hand, the results showed that the two compounds 5d and 6b had moderate activity against breast cancer (Tables 1, 2), with 78.45% and 84.60% inhibition, respectively.
Sample Concentration (µg/ml)
Inhibition
Viability (%)
SD (±)
500
78.45
21.55
0.73
250
63.56
36.44
2.01
125
42.14
57.86
1.27
62.5
20.50
79.50
0.50
31.25
8.23
91.77
0.38
15.6
1.78
98.22
0.21
7.8
0.55
99.45
0.14
3.9
0
100
–
0
0
100
–
Sample Concentration (µg/ml)
Viability (%)
Inhibition
SD (±)
500
15.40
84.60
0.25
250
29.80
70.20
1.11
125
41.85
58.15
1.95
62.5
63.76
36.24
2.08
31.25
81.75
18.43
0.65
15.6
92.36
7.64
0.26
7.8
97.45
2.55
0.28
3.9
99.80
0.20
0.05
0
100
0
–
Sample Concentration (µg/ml)
Viability (%)
Inhibition
SD (±)
500
6.55
93.45
0.28
250
15.57
84.43
0.55
125
24.44
75.56
0.85
62.5
36.28
63.72
2.55
31.25
47.42
52.58
2.25
15.6
74.35
25.65
1.76
7.8
92.90
7.10
0.80
3.9
99.33
0.77
0.14
0
100
0
–
Sample Concentration (µg/ml)
Viability (%)
Inhibition
SD (±)
500
4.22
95.78
0.15
250
11.24
88.76
0.31
125
21.60
78.40
0.42
62.5
31.53
68.47
1.00
31.25
39.55
60.45
2.30
15.6
71.24
28.76
2.00
7.8
89.76
10.24
0.34
3.9
98.50
1.50
0.18
0
100
0
–
3.3 Docking study
In this work, the binding potential of twenty-four compounds against thymidylate synthase (TS) and Cathepsin B (CB) has been investigated. This investigational work was performed to get further insight into the binding modes into TS binding site (PDB ID: 1JU6, resolution: 2.89 Å) and CB (PDB ID: 2DCD, resolution: 2.50 Å) (Nasser et al., 2020). The co-crystallized ligands (LYA and 78A) were used as reference molecules against TS and CB, respectively. The binding free energies (ΔG) were reported in (Table 6). To validate the docking process, the co-crystallized ligands (LYA and 78A) were re-docked against the isolated pockets of the target proteins. It was found that, the RMSD values between the re-docked conformer and the co-crystallized conformers of LYA and 78A were 1.34 and 1.51, respectively, which confirms the validity of the used docking protocols (Fig. 1).
Sample Concentration (µg/ml)
Viability (%)
Inhibition
SD (±)
500
3.98
96.02
0.16
250
8.12
91.88
0.24
125
14.91
95.09
0.33
62.5
27.84
72.16
0.18
31.25
39.58
60.42
0.62
15.6
46.79
53.21
2.31
7.8
62.43
37.57
1.69
3.9
78.15
21.58
0.41
0
100
0
–
Comp.
Thymidylate Synthase
Cathepsin-B
1
5a
−22.66
−14.78
2
5b
−22.27
−16.64
3
5c
−22.65
−15.08
4
5d
−21.62
−15.36
5
6a
−31.57
−18.66
6
6b
−30.17
−17.57
7
6c
−30.05
−18.27
8
6d
−28.29
−18.10
9
7a
−33.54
−19.39
10
7b
−30.91
−15.96
11
7c
−32.07
−17.25
12
7d
−29.58
−17.62
13
9a
−35.53
−18.68
14
9b
−35.31
−17.20
15
9c
−37.25
−19.89
16
9d
−34.22
−17.07
17
9e
−36.31
−16.18
18
9f
−35.74
−16.90
19
9g
−37.23
−18.88
20
9h
−38.22
−20.02
21
9i
−39.47
−18.17
22
9j
−33.78
−18.74
23
9k
−33.31
−18.68
24
9l
−32.63
−17.15
25
co-crystallized ligand (LYA)
−23.30
–
25
co-crystallized ligand(78A)
–
−18.59
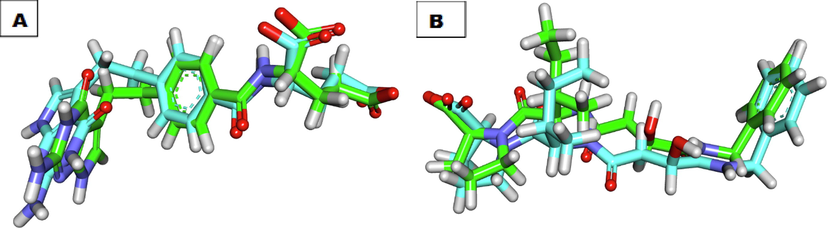
Superimposition of the co-crystallized pose (green) and the docking pose (maroon) of the same ligands, A) LYA, B) 78A.
The binding mode of the co-crystallized ligand (LYA) against TS showed an affinity value of −23.30 Kcal/mole. It formed four hydrogen bonds and six hydrophobic interactions. The 2-oxopyrrolidin-3-yl moiety occupied the first pocket of TS forming two hydrogen bonds with Arg78 and Lys77. Furthermore, it formed three hydrophobic interactions with Lys308, Phe225, and Ile108. The central phenyl ring occupied the spacer region of TS forming three hydrophobic interactions with Ile108, Phe225, and Leu225. Besides, the glutamic acid moiety occupied the 2nd pocket of the receptor and was incorporated in two hydrogen-bonding interactions with Val313 and Cys195 (Fig. 2). Compound 7a exhibited a binding mode like that of the co-crystallized ligands against TS with an affinity value of −33.54 Kcal/mole. It formed four hydrogen bonds. The benzaldehyde moiety occupied the first pocket of TS forming a hydrogen bond with Met309. Additionally, the 3-acetyl-2, 3-dihydro-1, 3, 4-oxadiazole moiety occupied the spacer region of the TS forming a hydrogen bond with Trp109. The pentane-1, 2, 3, 4, 5-pentayl pentaacetate occupied the 2nd pocket of the receptor forming two hydrogen bonds with Ala312 and Arg50 (Fig. 3). Compound 9i exhibited a binding mode like that of the co-crystallized ligands against TS with an affinity value of −39.47 Kcal/mole. It formed four hydrogen bonds and ten hydrophobic interactions. The diphenyl (((2-nitrophenyl) amino)(phenyl)methyl)phosphonate moiety occupied the first pocket of TS forming two hydrogen bonds with Lys309. In addition, it formed seven hydrophobic interactions with Lys308, Leu221, Lys77, and Lys107. The 3-acetyl-2, 3-dihydro-1, 3, 4-oxadiazole moiety occupied the spacer region of TS forming Three hydrophobic interactions with Leu221, Ile108, and Phe225. The 3-(acetoxymethyl) pentane-1, 2, 4, 5-tetrayl tetraacetate occupied the 2nd pocket of the receptor. It formed one hydrogen bond with Ser216 (Fig. 4).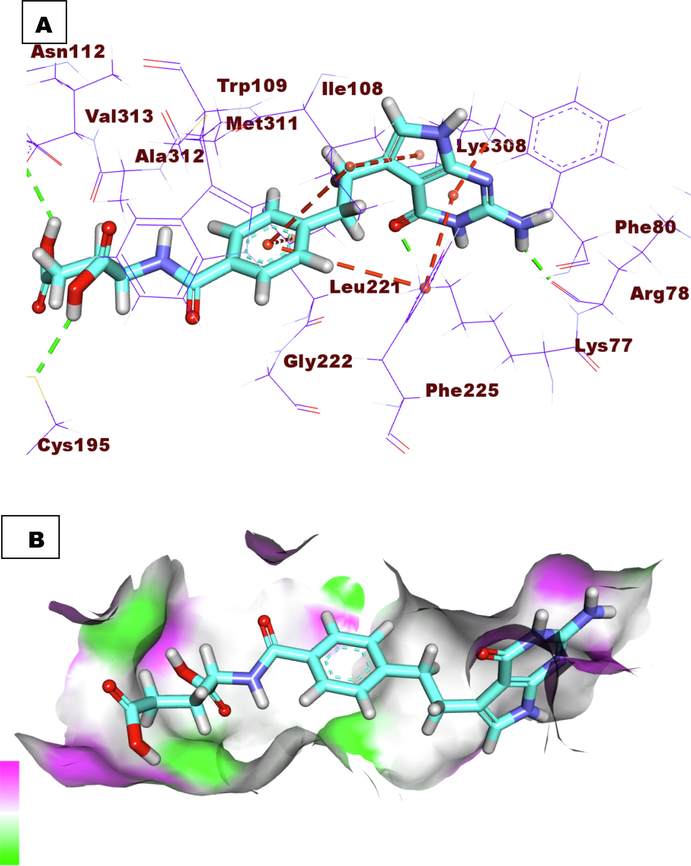
A) Co-crystallized ligand (LYA) docked into the active site of TS. B) Mapping surface showing co-crystallized ligand (LYA) occupying the active pocket of TS.
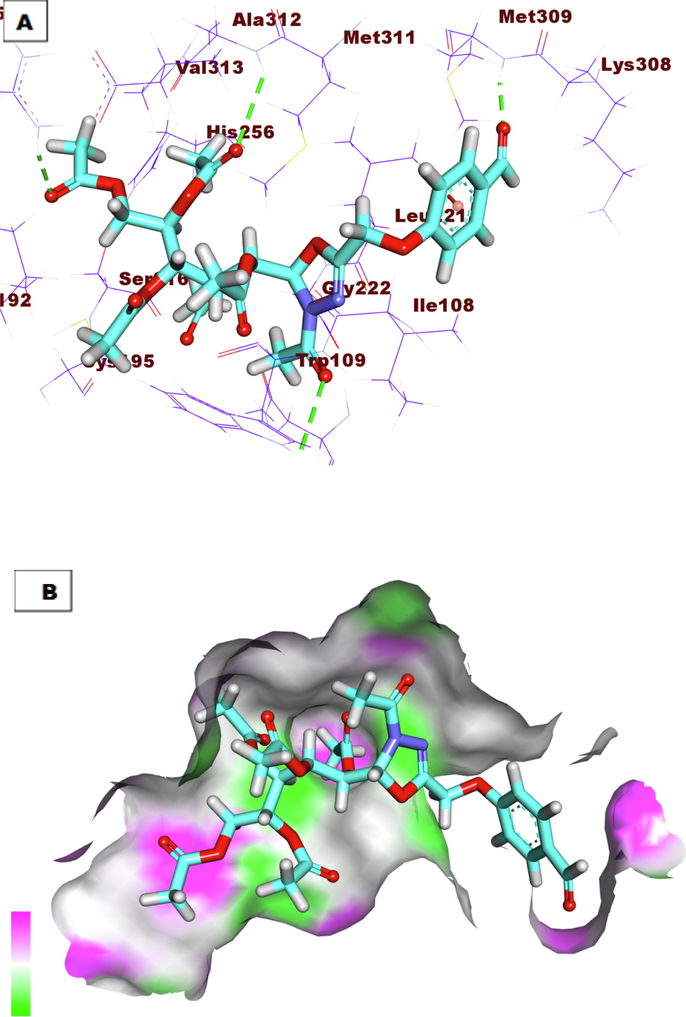
A) Compound 7a docked into the active site of TS. B) Mapping surface showing compound 7a occupying the active pocket of TS.
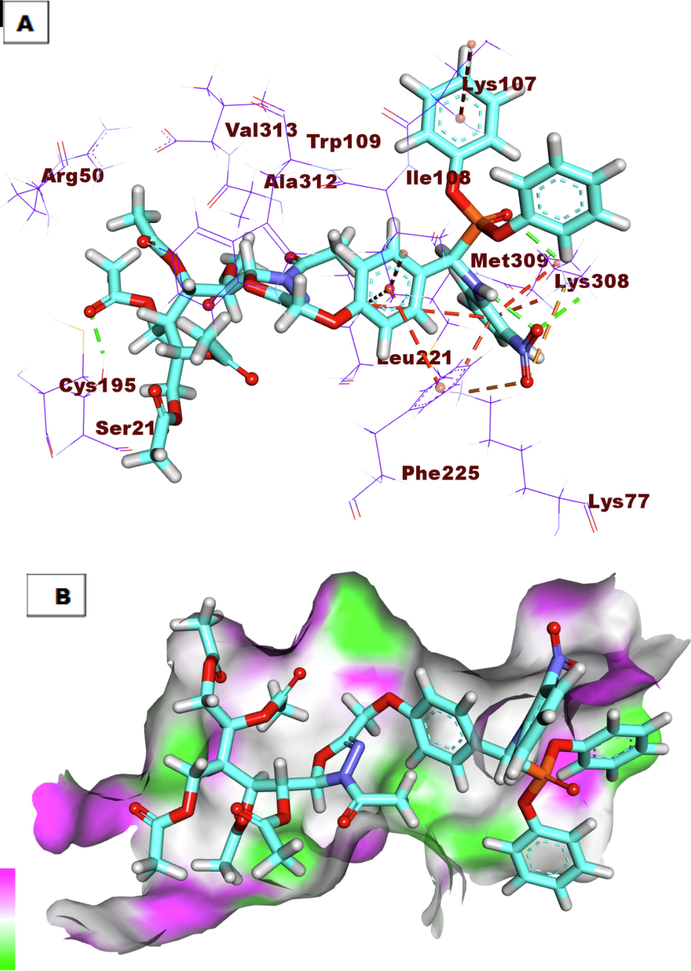
A) Compound 9i docked into the active site of TS. B) Mapping surface showing compound 9i occupying the active pocket of TS.
3.3.1 Docking against Cathepsin-B (CB)
The proposed binding mode of the co-crystallized ligand (78A) showed an affinity value of −18.59 kcal/mole. The benzylcarbamoyl was involved in hydrogen bonding interaction forming three bonds with Gly74, Cys29, and Gly198. In addition, the L-isoleucyl moiety formed two hydrogen bonds with Trp221 and Gln23. In addition, it formed two hydrophobic interactions with Val176 and Trp221. Moreover, the L-proline moiety formed three hydrophobic interactions with Cys119, His110, and Trp221 (Fig. 5). Compound 7a exhibited a binding mode like that of the co-crystallized ligands against CB with an affinity value of −19.39 Kcal/mole. The pentane-1,2,3,4,5-pentayl pentaacetate formed four hydrogen bonds with the key amino acid residues in the active site including Gln23, Trp221, His110, and His111(Fig. 6). Compound 9h exhibited a binding mode like that of the co-crystallized ligands against CB with an affinity value of −20.02 Kcal/mole. In detail, the diphenyl (((2-nitrophenyl) amino)(phenyl) methyl)phosphonate moiety formed a hydrogen bond with Gly74. Additionally, it formed one hydrophobic interaction with Phe75, and one electrostatic interaction with Gln245. The 3-acetyl-2, 3-dihydro-1, 3, 4-oxadiazole moiety formed one hydrogen bond with Gln23. Finally, the 3-(acetoxymethyl) pentane-1, 2, 4, 5-tetrayl tetraacetate formed one hydrogen bond with His111 (Fig. 7).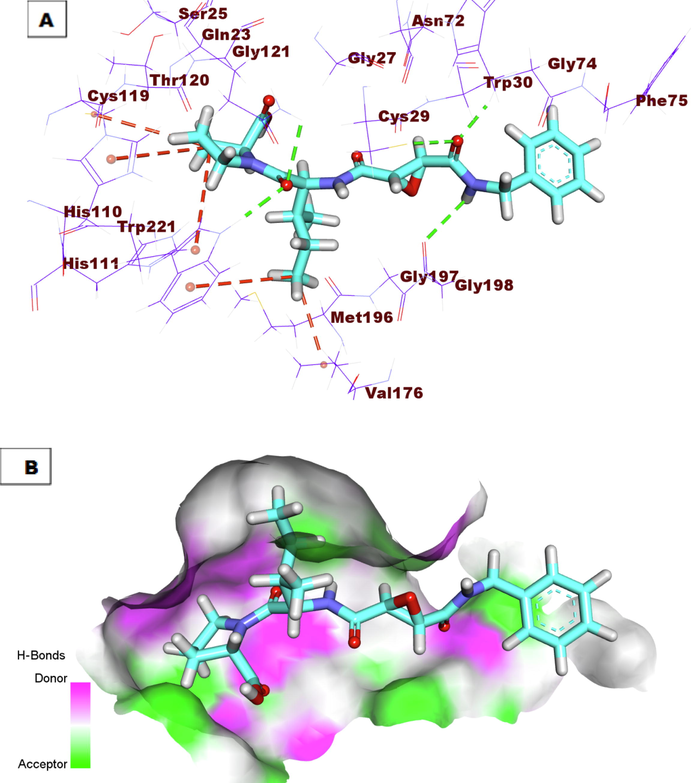
A) Co-crystallized ligand (78A) docked into the active site of Cathepsin B. B) Mapping surface showing co-crystallized ligand (78A) occupying the active pocket of Cathepsin B.
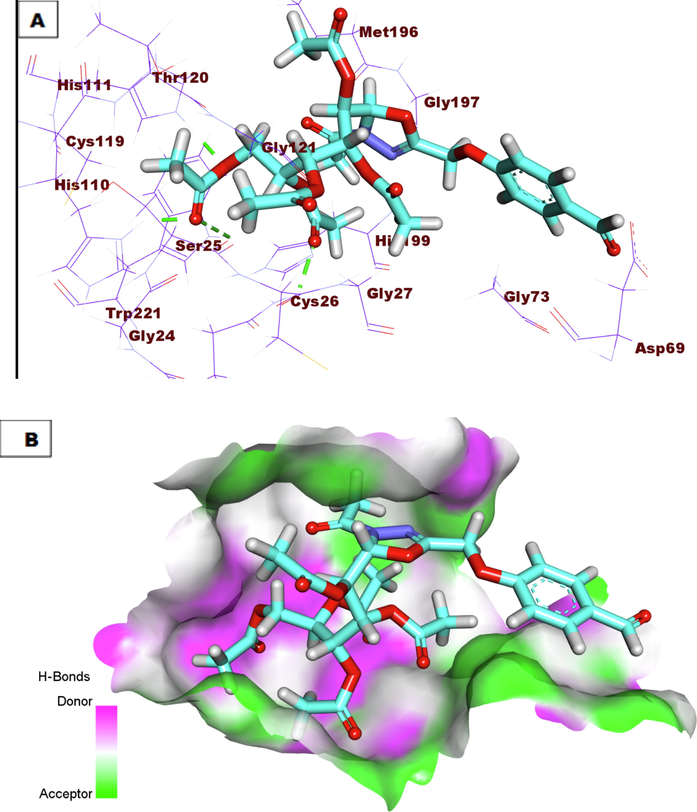
A) Compound 7a docked into the active site of Cathepsin B. B) Mapping surface showing compound 7a occupying the active pocket of Cathepsin B.
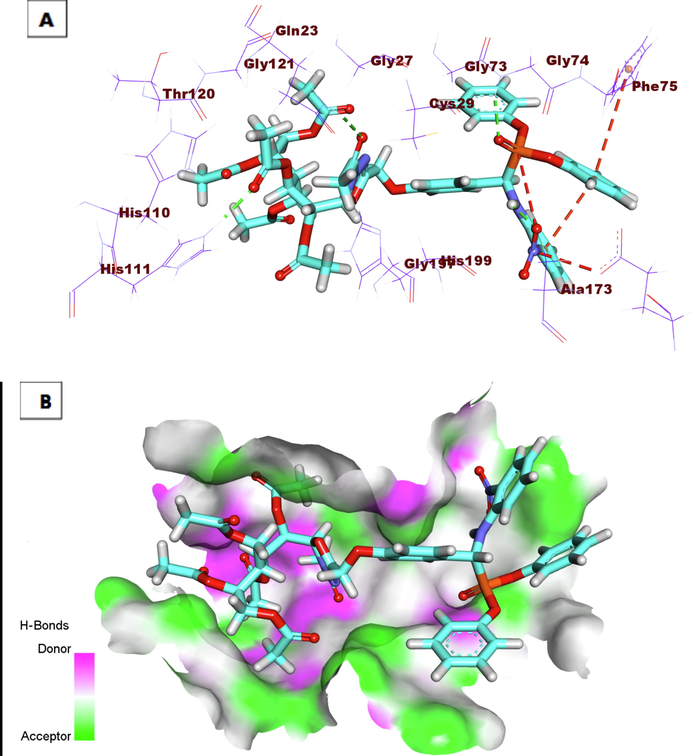
A) Compound 9h docked into the active site of Cathepsin B. B) Mapping surface showing compound 9h occupying the active pocket of Cathepsin B.
3.4 In silico toxicity studies
The toxicity profile of the synthesized compounds was determined according to the validated and constructed models in Discovery studio software (Xia et al., 2004). These models include i) FDA rodent carcinogenicity, which computes the probability of a submitted chemical structure being a carcinogen. ii) Carcinogenic potency TD50 which predicts the tumorigenic dose rate 50 (TD50) of a chemical in a rodent chronic exposure toxicity test of carcinogenic potency (Venkatapathy et al., 2011). iii) Rat maximum tolerated dose, which predicts the rat maximum tolerated dose (MTD) of a chemical (Shlyakhter et al., 1992). iv) Developmental toxicity potential which predicts whether a particular compound is likely to be toxic in a developmental toxicity potential assessment (Louisse et al., 2015). v) Rat oral LD50 which predicts the rat oral acute median lethal dose (LD50) in the toxicity test of a chemical (Diaza et al., 2015). vi) Rat chronic LOAEL which predicts the rat chronic lowest observed adverse effect level (LOAEL) value of a chemical (Pizzo and Benfenati, 2016; Venkatapathy et al., 2004). vii) Ocular irritancy predicts whether a particular compound is likely to be an ocular irritant and how severe the irritation is in the Draize test (Wilhelmus, 2001). viii) Skin irritancy predicts whether a particular compound is likely to be a skin irritant and how severe it is in a rabbit skin irritancy test [13]. As shown in Table 7, most compounds showed in silico low toxicity profile against the tested models. All compounds were predicted to be non-carcinogenic against the FDA rodent carcinogenicity model. Moreover, all compounds showed carcinogenic potency TD50 more than the reference compounds; raltitrexed (carcinogenic potency TD50 = 2.437 mg/kg body weight/day) except compound 7j. The tested compound had carcinogenic potency TD50 values ranging from 2.581 to 327.59 mg/kg body weight/day). Besides, the rat maximum tolerated doses of compounds 4a-d were estimated to be between 0.620 and 0.888 g/kg body weight, which were higher than that of raltitrexed (rat maximum tolerated dose = 0.188 g/kg body weight). The other derivatives were predicted to have less rat maximum tolerated doses. Moreover, all compounds with an exception of 4a-d were predicted to be non-toxic against the developmental toxicity potential model. For the rat oral LD50 model, the tested compounds showed oral LD50 values ranging from 0.389 to 6.385 mg/kg body weight/day. Such values less than that of raltitrexed (oral LD50 = 8.753 mg/kg body weight/day). Moreover, all the tested compounds were predicted to be irritant against the ocular irritancy model except compounds 9a, 9d, 9g, and 9j. Finally, all compounds were predicted to be non-irritant against the skin irritancy model except compounds 7a-d.
No.
FDA rodent carcinogenicity(Mouse)
Carcinogenic Potency TD50(Rat)a
Rat Maximum Tolerated Dose (Feed)b
Developmental Toxicity Potential
Rat Oral LD50 b
Chronic LOAEL b
Ocular Irritancy
Skin Irritancy
5a
Non-Carcinogen
55.062
0.888
Toxic
0.323
0.893
Irritant
Non-Irritant
5b
Non-Carcinogen
55.062
0.888
Toxic
0.323
0.893
Irritant
Non-Irritant
5c
Non-Carcinogen
55.062
0.888
Toxic
0.323
0.893
Irritant
Non-Irritant
5d
Non-Carcinogen
53.248
0.620
Toxic
0.237
0.606
Irritant
Non-Irritant
6a
Non-Carcinogen
327.059
0.048
Non-Toxic
0.129
1.448
Irritant
Non-Irritant
6b
Non-Carcinogen
0.048
327.059
327.059
0.129
1.448
Irritant
Non-Irritant
6c
Non-Carcinogen
0.048
327.059
0.048
0.129
1.448
Irritant
Non-Irritant
6d
Non-Carcinogen
0.059
261.443
Non-Toxic
0.120
1.357
Irritant
Non-Irritant
7a
Non-Carcinogen
0.023
64.843
Toxic
0.027
6.335
Irritant
Irritant
7b
Non-Carcinogen
0.023
64.843
Toxic
0.027
6.335
Irritant
Irritant
7c
Non-Carcinogen
0.023
64.843
Toxic
0.027
6.335
Irritant
Irritant
7d
Non-Carcinogen
0.028
52.365
Toxic
0.025
5.996
Irritant
Irritant
9a
Non-Carcinogen
0.016
2.581
Non-Toxic
0.003
0.420
Non-Irritant
Non-Irritant
9b
Non-Carcinogen
0.013
4.359
Non-Toxic
0.027
6.335
Irritant
Non-Irritant
9c
Non-Carcinogen
0.016
2.581
Non-Toxic
0.027
6.335
Irritant
Non-Irritant
9d
Non-Carcinogen
0.013
4.359
Non-Toxic
0.027
6.335
Non-Irritant
Non-Irritant
9e
Non-Carcinogen
0.015
7.344
Non-Toxic
0.005
0.606
Irritant
Non-Irritant
9f
Non-Carcinogen
0.016
3.150
Non-Toxic
0.004
0.928
Irritant
Non-Irritant
9g
Non-Carcinogen
0.013
5.319
Non-Toxic
0.005
0.606
Non-Irritant
Non-Irritant
9h
Non-Carcinogen
0.013
5.319
Non-Toxic
0.005
0.606
Irritant
Non-Irritant
9i
Non-Carcinogen
0.021
2.189
Non-Toxic
0.005
0.606
Irritant
Non-Irritant
9j
Non-Carcinogen
0.017
3.694
Non-Toxic
0.003
0.417
Non-Irritant
Non-Irritant
9k
Non-Carcinogen
0.019
6.207
Non-Toxic
0.005
0.601
Irritant
Non-Irritant
9l
Non-Carcinogen
0.133
2.437
Non-Toxic
0.004
0.919
Irritant
Non-Irritant
Raltitrexed
Non-Carcinogen
0.021
2.189
Non-Toxic
0.175
8.753
Irritant
Non-Irritant
4 Structure-Activity relationships (SAR)
The activity of synthesized compound determined by the activity of functional groups in the composition. The theoretical study showed that compounds 9h and 7a have high activity of binding with TS and CB respectively, when we make comparison with biological study we found that compounds 9h and 7a have high activity against breast cancer. These high activities refers to the presence of phosphonate group and acetylated sugar in compound 9h while refers to the presence of oxadiazoline ring and acetylated sugar in compound 7a.
5 Conclusions
In our study we were designed a new compounds as nucleosides, acetylated nucleosides, oxadiadiazoline and α-aminophosphonate derivatives and then these compounds were purified and elucidated by different spectroscopic analysis. The synthesized compounds were tested against breast cancer cells (MCF-7). The binding potential of synthesized compounds against thymidylate synthase (TS) and Cathepsin B (CB) has been investigated and the compounds 7a and 9h exhibits highly binding with thymidylate synthase (TS) and Cathepsin B (CB).
Acknowledgements
We acknowledge Taif University for Researchers Supporting Project number (TURSP- 2020/83), Taif University, Taif, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis, anticancer activity and moleculardocking study of Schiff base complexescontaining thiazole moiety. Beni-suef Univ. J. Basic Appl. Sci.. 2016;5:85-96.
- [Google Scholar]
- Synthesis, characterization, anticancer activity, and molecular docking of some new sugar hydrazone and arylidene derivatives. Arabian J. Chem.. 2020;13:4771-4784.
- [Google Scholar]
- Synthesis, spectroscopic and molecular docking studies on new schiff bases, nucleosides and a-aminophosphonate derivatives as antibacterial agents. Saudi J. Biolog. Sci.. 2020;27:3481-3488.
- [Google Scholar]
- Synthesis of Some new Nucleosides Derived from 2-Mercapto Benzimidazole With Expected Biological Activity. Orient. J. Chem.. 2017;33:2303-2310.
- [Google Scholar]
- Synthesis of some new 1, 3, 4-oxadiazole derivatives bearing sugars and α-aminophosphonate derived from 4-nitrophenol as anticancer agents. National J. Physiol., Pharmacy Pharmacol.. 2018;8:1275-1286.
- [Google Scholar]
- Synthesis, Docking, and Evaluation of Antimicrobial Activity of a New Series of Acyclo C-Nucleosides of 1, 2, 4-Triazolo [4, 3-a] quinoxaline Derivatives. J. Heterocyclic Chem.. 2016;53:153-163.
- [Google Scholar]
- Synthesis and Cytotoxic Activity of New Thiazolopyrimidine Sugar Hydrazones and Their Derived Acyclic Nucleoside Analogues. Molecules. 2020;25:2-16.
- [Google Scholar]
- Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res.. 2015;26:1-27.
- [Google Scholar]
- Anti-proliferative potential of triphenyl substituted pyrimidines against MDA-MB-231, HCT-116 and HT-29 cancer cell lines. Bioorg. Med. Chem. Lett.. 2020;30:127468.
- [Google Scholar]
- Synthesis, Characterization, Anticancer, and Antioxidant Studies of Ru(III) Complexes of Monobasic Tridentate Schiff Bases. Bioinorg. Chem. Appl.. 2016;2016:1-11.
- [Google Scholar]
- Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J. Mol. Struct.. 2018;1166:15-33.
- [Google Scholar]
- Design, synthesis and anticancer evaluation of thieno [2, 3-d] pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg. Chem.. 2019;88:102944.
- [Google Scholar]
- Design, eco-friendly synthesis, molecular modeling and anticancer evaluation of thiazol-5 (4 H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv.. 2020;10:2791-2811.
- [Google Scholar]
- Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorg. Chem.. 2019;91:103115.
- [Google Scholar]
- Synthesis, characterization, anticancer and antibacterial evaluation of Schiff base ligands derived from hydrazone and their transition metal complexes. Inorg. Chim. Acta. 2019;484:245-254.
- [Google Scholar]
- Synthesisand Anticancer Activities of Thiazoles, 1,3-Thiazines, and Thiazo-lidine Using Chitosan-Grafted-Poly(vinylpyridine) as Basic Cata-lyst. Heterocycles. 2015;91:1227-1243.
- [Google Scholar]
- Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4 (3H)-one derivatives as potential PPARγ and SUR agonists. Bioorg. Med. Chem.. 2017;25:4723-4744.
- [Google Scholar]
- Synthesis, characterization and antioxidant activities of Schiff bases are of cholesterol. J. Saudi Chem. Soc.. 2017;21:5322-5328.
- [Google Scholar]
- Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem.. 2020;2020:1-19.
- [Google Scholar]
- Prediction of in vivo developmental toxicity of all-trans-retinoic acid based on in vitro toxicity data and in silico physiologically based kinetic modeling. Arch. Toxicol.. 2015;89:1135-1148.
- [Google Scholar]
- Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4 (3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg. Chem.. 2020;94:103422.
- [Google Scholar]
- Synthesis, characterization, spectroscopic studies and antimicrobial activity of three new Schiff bases derived from Heterocyclic moiety. 2018;1151:41-48.
- Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55-63.
- [Google Scholar]
- Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org. Biomol. Chem.. 2020;18:7608-7634.
- [Google Scholar]
- In silico models for repeated-dose toxicity (RDT): prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. Silico Methods Predicting Drug Toxicity. 2016:163-176.
- [Google Scholar]
- Seley-Radtke, K., 2020. Discovery, Design, Synthesis, and Application of Nucleoside/Nucleotides. In: Multidisciplinary Digital Publishing Institute. Molecules. vol. 25, pp. 526–1528.
- Monte Carlo Simulation of Rodent Carcinogenicity Bioassays. Risk Anal.. 1992;12:73-82.
- [Google Scholar]
- Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Comput. Sci.. 2004;44:1623-1629.
- [Google Scholar]
- Structure-Activity Relationships for Carcinogenic Potential, General. Appl. Syst. Toxicol. 2011
- [Google Scholar]
- Classification of kinase inhibitors using a Bayesian model. J. Med. Chem.. 2004;47:4463-4470.
- [Google Scholar]




