

Bandgap engineering and optoelectronic properties of all-inorganic lead-free Pd-based double perovskites
⁎Corresponding authors. rjsa@mju.edu.cn (Rongjian Sa), liudiwen1987@163.com (Diwen Liu)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The various physical properties of lead-free double perovskites A2PdX6 (A = K, Rb, Cs; X = Cl, Br, I) are revealed for the first time. The calculated structural parameters of these Pd-based compounds are consistent with the experimental data. It is likely to possess a tetragonal structure for K2PdI6 at room temperature. Cs2PdBr6 is dynamically stable when the pressure is in the range of 0–6.95 GPa. The mechanical properties are analysed and all the compounds are mechanically stable. The band gap trend of the A2PdX6 compound is identified when the A-site cation and halide anion are varied. Three A2PdBr6 compounds exhibit suitable band gaps for photovoltaic applications. An optimum band gap can be achieved for Cs2PdBr6 when the moderate pressure is applied. In addition, the electron shows better mobility than that of the hole for three A2PdBr6 compounds. The optical absorption coefficient of the A2PdBr6 compound is improved when the A-site cation changes from Cs to Rb to K. Applying pressure is beneficial to enhance light absorption capacity of Cs2PdBr6. The findings of this work can provide guidance for the design of potential A2PdBr6 compounds for photovoltaic applications.
Keywords
Pd-based double perovskite
Mechanical properties
Suitable band gap
Optical coefficient

1 Introduction
New energy materials have been developed to meet the needs of various photoelectric applications, such as solar cells (Chirilă et al., 2011; Nie et al., 2015) and light-emitting diodes (Ajia et al., 2018; Muhammed et al., 2017). It is reported for the first time that lead halide perovskites are considered as light absorbers in 2009 (Kojima et al., 2009). Lead-based hybrid perovskites have been successfully used for solar cells during the past decade. The latest power conversion efficiencies (PCEs) of these materials has reached 25.2% according to a recent report (Jeong et al., 2021). Many unique optoelectronic properties are found for these lead-based perovskites, such as the adjustable direct band gap, small electron and hole effective masses, high absorption coefficient, and long carrier diffusion length (Frohna et al., 2018; Giorgi et al., 2013; Stranks et al., 2013). However, two major challenges still remain for practical applications: the toxicity of lead and the poor stability. These two issues have received considerable attention in recent years. The basic chemical formula of halide perovskite is ABX3, where A is inorganic or organic monovalent cation, B is divalent metal cation, and X is halogen anion, such as CsBX3 (B = Pb, Sn; X = I, Br, Cl) (Khera et al., 2021). Lead-free double perovskites (A2B1+B3+X6 and A2B4+X6) have been proposed as novel candidates for optoelectronic applications. For example, the A2B1+B3+X6 structure is considered as cubic phase by replacing two B-site cations with one B1+ and one B3+ cation (e.g., Cs2AgBiBr6 (Filip et al., 2016), Cs2AgSbCl6 (Karmakar et al., 2018), and Cs2AgInCl6 (Lee et al., 2019)). The electronic properties of these compounds are greatly affected from such modification. Unfortunately, these compounds usually show large indirect band gaps (over 2.0 eV), which cannot achieve effective optical absorption and limit the performance. Recently, the predicted band gaps for Rb2AgTlCl6 and Cs2AgTlCl6 are 0.78 eV and 0.74 eV, which make them suitable for the optoelectronics applications (Murtaza et al., 2021).
The A2BX6 perovskite can be considered as a derivation from ABX3 when half of the metal cations are removed with leaving a vacancy. The result implies that the B-site should be a tetravalent metal cation to maintain the charge neutrality for the whole structure. Most of the A2BX6 perovskites possess cubic structure (space group: Fm3m) and good stability at room temperature. The optoelectronic and transport properties of these A2BX6 compounds have been studied experimentally and theoretically in recent years. The most typical case is that Cs2SnI6 shows excellent stability at ambient temperature and has an ideal direct band gap with ∼1.3 eV (Bounos et al., 2018; Lee et al., 2014; Yang, X. et al., 2020). The Cs2TiIxBr6−x (x = 0–6) compounds exhibit tunable wide band gaps (1.0–1.8 eV) (Ju et al., 2018). In particular, Cs2TiI2Br4 shows an optimum direct band gap value (1.38 eV) for single-junction solar cells (Ju et al., 2018). In addition, Te-based double perovskites A2TeI6 (A = CH3NH3, CH(NH2)2) have suitable band gaps (1.4–1.5 eV) and good stability in ambient conditions (Ju et al., 2019). The solid-solution Cs2Sn1−xTexI6 (x = 0, 0.25, 0.50, 0.75, 1) have been synthesized experimentally and the structure-property relationship is also elucidated (Maughan et al., 2016). Novel lead-free double perovskite Cs2PtI6 exhibits an optical band gap of 1.37 eV and excellent stability (Yang, S. et al., 2020). Through the investigation of the electronic and phonon transport properties of Cs2PtI6, it has the potential to be a thermoelectric material (Sajjad et al., 2020). In addition, Cs2PtI6 can be used as an electrocatalyst for the hydrogen evolution reaction owing to its extraordinary stability under ambient conditions (Hamdan and Chandiran, 2020). A heterojunction of BiVO4/Cs2PtI6 is made and used as a photoanode for photoelectrochemical (PEC) water oxidation (Jayaraman et al., 2021). The photovoltaic properties of Cs2PtI6 is assessed and the PCE of 13.88% is achieved (Schwartz et al., 2020). The optoelectronic and thermoelectric properties of Pt-based double perovskites have been widely explored by means of density functional theory (DFT) (Mahmood, Q. et al., 2021; Sa et al., 2021; Suzuki and Tsuyama, 2021; Zhao et al., 2021).
Another new type of lead-free Cs2PdBr6 is reported for the first time in 2017 and it has a narrow band gap of 1.6–1.69 eV, which is a potential compound for optoelectronic applications (Sakai et al., 2017; Zhou et al., 2018). The structure, electronic, optical, and transport properties of several Pd-based compounds have been demonstrated by different research groups through the DFT investigations in recent years (Bhamu et al., 2018; Du et al., 2020; Faizan et al., 2021a). Although K2PdCl6 and K2PdBr6 have been synthesized (Ketblaar and van Walsem, 1938), the theoretical investigations are lacking so far. In this work, the DFT method is employed to explore the various physical properties of lead-free A2PdX6 with the chemical composition variation. The electronic properties of all-inorganic Pd-based compounds with varying the A-site cation and halogen anion are calculated, and the results are further analyzed and discussed in detail.
Our calculated results yield the following conclusions. Firstly, a tetragonal structure is likely to be adopted for K2PdI6 at room temperature. Cs2PdBr6 is dynamically stable at all pressures from 0 to 6.95 GPa. Secondly, the band gap is found to decrease when the halide anion varies from Cl to Br to I. It leads to an increase in band gap when the ionic radius of the A-site cation increases from K to Rb to Cs. Three A2PdBr6 compounds are predicted to be the promising materials for perovskite solar cells. In addition, when the pressure is applied, the band gap of Cs2PdBr6 can be adjusted to an ideal value on appropriate conditions. Finally, the band gaps of these Pd-based compounds are closely related with their bond lengths (Pd−X).
2 Computational details
In this study, the DFT calculations were performed for cubic structure Pd-based double perovskites using the projector augmented wave (PAW) (Blöchl, 1994) method as implemented in Vienna ab initio simulation package (VASP) (Kresse and Furthmüller, 1996). The generalized gradient approximation (GGA) of Perdew-Burke-Ernzerhof (PBE) was used (Perdew et al., 1996). The tetrahedron method with Blochl corrections was employed for structural optimization. The plane wave cutoff of 500 eV was adopted. The convergence criteria of energies and all forces were set to be 10−5 eV and 0.01 eV/Å, respectively. The Brillouin zone was sampled with 6 × 6 × 6 k-mesh. The band gaps of Pd-based double perovskites were significantly underestimated by the standard DFT method, so the Heyd-Scuseria-Ernzerhof (HSE06) hybrid functional with the default parameters (Heyd et al., 2003) was used for the electronic and optical properties. The crystal structure of A2PdX6 was visualized by the VESTA tool (Momma and Izumi, 2011).
3 Results and discussion
3.1 Structural properties
In this work, nine lead-free double perovskites A2PdX6 (A = K, Rb, Cs; X = Cl, Br, I) with cubic structure (space group: Fm3m) are simulated. The crystal structure of A2PdX6 is depicted in Fig. 1. The calculated lattice constants and available experimental data are displayed in Fig. 2. The lattice constants of most compounds are in good agreement with the reported experimental values (Engel, 1935; Ketblaar and van Walsem, 1938; Schüpp et al., 2000). It is acceptable that the maximum deviation is less than 5% between the theoretical and experimental data. It is found that the lattice constants are not sensitive with the A-site cation, while the lattice constants are greatly increased from Cl to Br and I. In addition, the bond length Pd–X will vary with the change of chemical composition. The relationship between the bond length and band gap will be discussed in the following text. To the best of our knowledge, no experimental lattice constants have been reported for K2PdI6 so far. It is reported that K2PtI6 adopts a tetragonal structure at room temperature, while Rb2PtI6 and Cs2PtI6 have cubic structures (Thiele et al., 1983). In addition, the energy difference between cubic and tetragonal structures is calculated for K2PtI6 in the previous theoretical work (Cai et al., 2017). It is found that the tetragonal structure has a lower total energy than that of cubic structure. The results are consistent with the fact that the tetragonal structure of K2PtI6 is only experimentally observed. Based on this point, the energy variation between cubic and tetragonal structures is also calculated for K2PdI6 in the present study. We find that the total energy of cubic structure is higher by 129 meV/f.u. than that of tetragonal structure. Therefore, it is presumed to be a tetragonal structure for K2PdI6 at room temperature.

- Crystal structure of double perovskite A2PdX6.
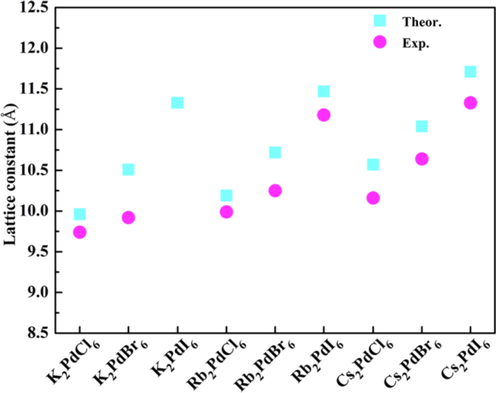
- Theoretical and experimental lattice constants of different double perovskites A2PdX6.
The experimental band gap of Cs2PdBr6 is between 1.6 and 1.69 eV (Sakai et al., 2017; Zhou et al., 2018). It is expected to tune the band gap of Cs2PdBr6 by applying the pressure. In our previous theoretical works (Jing et al., 2019; Liu et al., 2021a; Liu et al., 2019; Liu et al., 2021b; Sa et al., 2020), an optimum band gap can be obtained for a material with large optical band gap when the pressure is used. The stability is an important factor for practical applications in solar cells. Therefore, the dynamical stability of Cs2PdBr6 under different pressures is also studied. The computed phonon spectra of Cs2PdBr6 is presented in Fig. 3. The positive frequency of all phonon modes confirms that Cs2PdBr6 is dynamically stable at different pressures from 0 to 6.95 GPa.

- Phonon spectrum of Cs2PdBr6 under pressure from 0 to 6.95 GPa.
It is well known that there are three independent elastic constants (C11, C12, and C44) for the cubic double perovskites. The mechanical stability of the cubic structure should satisfy the following relations (Born and Huang, 1955):
It is proved from Table 1 that nine double perovskites are mechanically stable. In addition, the elastic constants of each compound can be used to obtain other mechanical parameters when the Voigt–Ruess–Hill method is employed (Hill, 1952). The brittle or ductile nature of a material has been judged by two empirical criteria Pugh’s ratio (B/G) and Poisson ratio (v) (Huang et al., 2011; Pugh, 1954). The critical values are 1.75 and 0.26 for Pugh’s ratio and Poisson ratio, respectively. When the value of B/G (or v) is larger (lower) than 1.75 (or 0.26), the material is ductile (brittle). The results suggest that K2PdBr6 is a brittle material while the rest of the compounds are ductile materials. Moreover, the order of the B/G and v values is A2PdBr6 < A2PdCl6 < A2PdI6.
| C11 | C12 | C44 | B | G | E | B/G | v | |
|---|---|---|---|---|---|---|---|---|
| K2PdCl6 | 28.56 | 18.00 | 5.75 | 21.52 | 5.56 | 15.35 | 3.87 | 0.38 |
| K2PdBr6 | 11.92 | 5.04 | 5.84 | 7.33 | 4.72 | 11.67 | 1.55 | 0.23 |
| K2PdI6 | 18.56 | 15.64 | 4.70 | 16.65 | 2.98 | 8.45 | 5.58 | 0.41 |
| Rb2PdCl6 | 25.82 | 15.72 | 6.27 | 19.08 | 5.75 | 15.67 | 3.32 | 0.36 |
| Rb2PdBr6 | 10.67 | 4.17 | 3.38 | 6.34 | 3.33 | 8.49 | 1.90 | 0.28 |
| Rb2PdI6 | 16.95 | 13.74 | 4.65 | 14.81 | 3.04 | 8.53 | 4.88 | 0.40 |
| Cs2PdCl6 | 19.40 | 9.54 | 4.54 | 12.83 | 4.70 | 12.55 | 2.73 | 0.34 |
| Cs2PdBr6 | 10.80 | 4.76 | 3.87 | 6.77 | 3.50 | 8.96 | 1.93 | 0.28 |
| Cs2PdI6 | 18.46 | 11.07 | 3.81 | 12.53 | 3.06 | 8.48 | 4.10 | 0.39 |
3.2 Electronic properties
The elastic and optoelectronic properties of double perovskites have been investigated for optoelectronic devices (Khan et al., 2021; Saeed et al., 2022; Saeed et al., 2021). The band structure usually plays an important role in understanding the physical properties of a material such as the carrier mobility and absorption spectra. The electronic band structures of nine lead-free Pd-based compounds are investigated and plotted in Fig. 4(a)-(i). It is found that the valence band maximum (VBM) and conduction band minimum (CBM) of all band structures are at the same point Γ (0, 0, 0), indicating they are direct band gap semiconductors. The electronic properties of A2PdX6 (A = K, Rb, Cs; X = Cl, Br, I) have been studied experimentally and theoretically in recent years (Bhamu et al., 2018; Cai et al., 2017; Du et al., 2020; Faizan et al., 2021a; Faizan et al., 2021b; Mahmood, Qasim et al., 2021; Sakai et al., 2017; Wang et al., 2020; Xu and Liu, 2020; Zhou et al., 2018). It is reported from the experiments that lead-free Cs2PdBr6 has an optical band gap of 1.6–1.69 eV (Sakai et al., 2017; Zhou et al., 2018). In this work, the calculated band gap of Cs2PdBr6 is 1.72 eV, which matches the experimental values (Sakai et al., 2017; Zhou et al., 2018). The results suggest that the band gap calculated by the HSE06 method is reliable for Pd-based double perovskites. Moreover, our calculated results are consistent well with the previous theoretical data (Cai et al., 2017; Wang et al., 2020).

- Electronic band structures of (a) K2PdCl6, (b) Rb2PdCl6, (c) Cs2PdCl6, (d) K2PdBr6, (e) Rb2PdBr6, (f) Cs2PdBr6, (g) K2PdI6, (h) Rb2PdI6, and (i) Cs2PdI6.
The band gap values of all the compounds are presented in Fig. 5(a). It can be clearly seen that the band gap is decreased for each compound when the Cl atoms are replaced with the Br or I atoms. Moreover, the band gap is slightly increased when the ionic radii of the A-site cation increases. We find that the band gap is either too large or small for A2PdCl6 and A2PdI6, which are not suitable for photovoltaic applications. The band gaps of three A2PdBr6 is between 1.30 and 1.80 eV, so they are the potential materials for perovskite solar cells. Furthermore, the band structures of K2PdX6 (X = Cl, Br, I) are revealed for the first time in the present study. According to the latest theoretical prediction (Zelai et al., 2022), the band gap (1.60 eV) for Ga2PdBr6 is suitable for optoelectronic in the visible region.
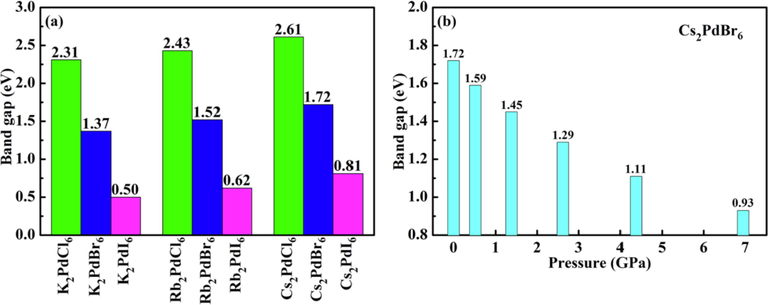
- The band gap trend for (a) A2PdX6 and (b) Cs2PdBr6 under pressure.
We find that Cs2PdBr6 and CsPbI3 have similar band gap values (∼1.7 eV) (Eperon et al., 2015), but they are not suitable for single-junction solar cells. According to the Shockley–Queisser theory (Shockley and Queisser, 1961), the maximum theoretical efficiency will be achieved when the band gap is between 1.3 and 1.4 eV. For instance, it is predicted that the best photovoltaic performance of CsPbI3 may be realized at 1.40 GPa because it has an optimum band gap (1.34 eV) at this condition (Jing et al., 2019). Therefore, an ideal band gap can be obtained for Cs2PdBr6 when the pressure is used. The band gap trend of Cs2PdBr6 under different pressures is presented in Fig. 5(b). The band gap of Cs2PdBr6 decreases when the pressure increases. Cs2PdBr6 shows an ideal band gap value in the range of 1–3 GPa according to our calculated results.
The effective masses of electron and hole (Γ → R) of three A2PdBr6 compounds are shown in Fig. 6. The electron’s effective mass is much smaller than that of the hole for each compound, indicating that the electron mobility is better than the hole mobility. In addition, the effective masses of electron and hole are slightly decreased from Cs to K. The effective mass of the electron fluctuates between 0.5 and 0.8 m0, while the effective mass of the hole varies from 1.80 to 2.43 m0.
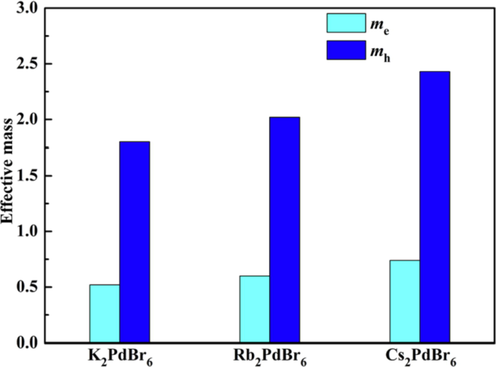
- The effective masses of electron and hole for the A2PdBr6 compound.
Fig. 7 shows the relationship between the band gap and bond length Pd–X for Pd-based double perovskites. It is observed from Fig. 7(a) that the impact of the A-site cation on the bond length is quite small, so the band gap variation is limited. For example, the band gap change is less than 0.4 eV between K2PdX6 and Cs2PdX6. The bond length Pd–X is greatly increased from Cl to I, which leads to the lower band gap. When the pressure is applied for Cs2PdBr6, the bond length Pd–Br becomes smaller. The results show that the longer the bond length, the larger the band gap, as shown in Fig. 7(b). The band gap of perovskite material can be effectively regulated by changing the halogen composition and applying the pressure.

- The relationship between the band gap and bond length Pd–X of (a) A2PdX6 and (b) Cs2PdBr6 under pressure.
The total and partial density of states of three A2PdBr6 double perovskites are plotted in Fig. 8. The VBM of each compound is mainly contributed from the Br-4p orbitals, while the CBM is made of the mixing contribution of the Pd-4d and Br-4p orbitals.

- The calculated density of states of (a) K2PdBr6, (b) Rb2PdBr6, and (c) Cs2PdBr6.
3.3 Optical properties
The optical properties of these A2PdBr6 compounds are calculated by using the HSE06 method. The expression of dielectric function ε(ω) is given as follows:
The real part ε1(ω) of dielectric function can be obtained by using the Kramers-Kronig relation (Rehman et al., 2018). The calculated optical absorption coefficients of A2PdBr6 double perovskites are shown in Fig. 9. The light absorption curves of the three compounds are similar in Fig. 9(a). The light absorption capacity of K2PdBr6 is slightly stronger than those of Rb2PdBr6 and Cs2PdBr6. The maximum absorption coefficients (∼4.0 × 105 cm−1) of each compound are found at 300 and 450 nm, and the lower absorption coefficient is located between 300 and 400 nm. It can be seen from Fig. 9(b) that the absorption coefficient of Cs2PdBr6 is redshifted when the pressure is applied. The absorption coefficient of Cs2PdBr6 is greatly improved when the pressure gradually increases. In general, three A2PdBr6 compounds show high absorption coefficients in the visible light region, which make them suitable for solar cell applications.

- Optical absorption spectrum of (a) A2PdBr6 and (b) Cs2PdBr6 under pressure.
4 Conclusion
In summary, the comprehensive investigation of the structural, mechanical, electronic, and optical properties of all-inorganic A2PdX6 double perovskites has been performed by using the DFT approach. The optimized lattice constants of these Pd-based compounds are consistent with the available experimental data. It is likely to have a tetragonal structure for K2PdI6 at room temperature. The dynamical stability of Cs2PdBr6 is proved at different pressures. The elastic constants are calculated and all the compounds are mechanically stable. K2PdBr6 is only a brittle material among the studied systems. The HSE06 method is employed to accurately calculate the electronic and optical properties of the studied Pd-based compounds. The band gaps of these Pd-based double perovskites can be tuned by varying the A-site cationand X ions. The band gap values of three A2PdBr6 are suitable for applicaions in perovskite solar cells. Moreover, the electron displays better mobility than that of the hole for three A2PdBr6 compounds. The band gap is closely related with the structural parameter, such as the bond length (Pd−X). The light absorption capacity of A2PdBr6 is gradually increased from Cs to Rb to K. The optical absorption coefficient of Cs2PdBr6 is improved when the pressure is used. The present study is meaningful for exploring potentail lead-free double perovskites for solar cell applications.
Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (No. 2020J01858).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Generated Carrier Dynamics in V-Pit-Enhanced InGaN/GaN Light-Emitting Diode. ACS Photonics. 2018;5:820-826.
- [Google Scholar]
- Revealing optoelectronic and transport properties of potential perovskites Cs2PdX6 (X = Cl, Br): A probe from density functional theory (DFT) Sol. Energy. 2018;162:336-343.
- [Google Scholar]
- Defect Perovskites under Pressure: Structural Evolution of Cs2SnX6 (X = Cl, Br, I) J. Phys. Chem. C. 2018;122:24004-24013.
- [Google Scholar]
- Computational Study of Halide Perovskite-Derived A2BX6 Inorganic Compounds: Chemical Trends in Electronic Structure and Structural Stability. Chem. Mater.. 2017;29:7740-7749.
- [Google Scholar]
- Highly efficient Cu(In, Ga)Se2 solar cells grown on flexible polymer films. Nat. Mater.. 2011;10:857-861.
- [Google Scholar]
- Theoretical investigations of structural, electronic, and physical properties of Rb2BX6 (B = Ti, Se, Pd; X = F, Cl, Br, I) double perovskites. J. Appl. Phys.. 2020;128:235110.
- [Google Scholar]
- Die Kristallstrukturen einiger Hexachlorokomplexsalze. Z. Kristallogr.-Cryst. Mater.. 1935;90:341-373.
- [Google Scholar]
- Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A. 2015;3:19688-19695.
- [Google Scholar]
- Electronic and optical properties of vacancy ordered double perovskites A2BX6 (A = Rb, Cs; B = Sn, Pd, Pt; and X = Cl, Br, I): a first principles study. Sci. Rep.. 2021;11:6965.
- [Google Scholar]
- Faizan, M., Khan, S.H., Khachai, H., Seddik, T., Omran, S.B., Khenata, R., Xie, J., Al-Anazy, M.m., 2021b. Electronic, optical, and thermoelectric properties of perovskite variants A2BX6: Insight and design via first-principles calculations. Int. J. Energy Res. 45, 4495-4507.
- Band Gaps of the Lead-Free Halide Double Perovskites Cs2BiAgCl6 and Cs2BiAgBr 6 from Theory and Experiment. J. Phys. Chem. Lett.. 2016;7:2579-2585.
- [Google Scholar]
- Inversion symmetry and bulk Rashba effect in methylammonium lead iodide perovskite single crystals. Nat. Commun.. 2018;9:1829.
- [Google Scholar]
- Small Photocarrier Effective Masses Featuring Ambipolar Transport in Methylammonium Lead Iodide Perovskite: A Density Functional Analysis. J. Phys. Chem. Lett.. 2013;4:4213-4216.
- [Google Scholar]
- Cs2PtI6 Halide Perovskite is Stable to Air, Moisture, and Extreme pH: Application to Photoelectrochemical Solar Water Oxidation. Angew. Chem. Int. Ed.. 2020;59:16033-16038.
- [Google Scholar]
- Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys.. 2003;118:8207-8215.
- [Google Scholar]
- The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A. 1952;65:349-354.
- [Google Scholar]
- First-principles calculations of mechanical and thermodynamic properties of YAlO3. Comput. Mater. Sci.. 2011;50:3056-3062.
- [Google Scholar]
- BiVO4/Cs2PtI6 Vacancy-Ordered Halide Perovskite Heterojunction for Panchromatic Light Harvesting and Enhanced Charge Separation in Photoelectrochemical Water Oxidation. ACS Appl. Mater. Interfaces. 2021;13:16267-16278.
- [Google Scholar]
- Pseudo-halide anion engineering for α-FAPbI3 perovskite solar cells. Nature. 2021;592:381-385.
- [Google Scholar]
- Tuning electronic and optical properties of CsPbI3 by applying strain: A first-principles theoretical study. Chem. Phys. Lett.. 2019;732:136642.
- [Google Scholar]
- Tellurium-Based Double Perovskites A2TeX6 with Tunable Band Gap and Long Carrier Diffusion Length for Optoelectronic Applications. ACS Energy Lett.. 2019;4:228-234.
- [Google Scholar]
- Earth-Abundant Nontoxic Titanium(IV)-based Vacancy-Ordered Double Perovskite Halides with Tunable 1.0 to 1.8 eV Bandgaps for Photovoltaic Applications. ACS Energy Lett.. 2018;3:297-304.
- [Google Scholar]
- Cu(II)-Doped Cs2SbAgCl6 Double Perovskite: A Lead-Free, Low-Bandgap Material. Chem. Mater.. 2018;30:8280-8290.
- [Google Scholar]
- Die Krystallstruktur des Ammonium-, Kalium-, Rubidium- und Cäsiumpalladiumhexa-chlorids und -Bromids. Recl. Trav. Chim. Pays-Bas. 1938;57:964-966.
- [Google Scholar]
- Khan, I., Shahab, Haq, I.U., Ali, A., Ali, Z., Ahmad, I., 2021. Elastic and Optoelectronic Properties of Cs2NaMCl6 (M = In, Tl, Sb, Bi). J. Electron. Mater. 50, 456-466.
- Theoretical Investigation of CsBX3 (B = Pb, Sn; X = I, Br, Cl) Using Tran-Blaha Modified Becke-Johnson Approximation for Flexible Photoresponsive Memristors. Adv. Theory Simul.. 2021;4:2100011.
- [Google Scholar]
- Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc.. 2009;131:6050-6051.
- [Google Scholar]
- Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci.. 1996;6:15-50.
- [Google Scholar]
- Air-Stable Molecular Semiconducting Iodosalts for Solar Cell Applications: Cs2SnI6 as a Hole Conductor. J. Am. Chem. Soc.. 2014;136:15379-15385.
- [Google Scholar]
- Colloidal Synthesis of Lead-Free Silver-Indium Double-Perovskite Cs2AgInCl6 Nanocrystals and Their Doping with Lanthanide Ions. J. Phys. Chem. C. 2019;123:2665-2672.
- [Google Scholar]
- First-principles study of the stability, electronic and optical properties of CdTe under hydrostatic pressure. Chem. Phys. Lett.. 2021;764:138272.
- [Google Scholar]
- Pressure-induced effects in the inorganic halide perovskite CsGeI3. RSC Adv.. 2019;9:3279-3284.
- [Google Scholar]
- Pressure-induced band gap tuning in Cs2TiBr 6: A theoretical study. J. Solid State Chem.. 2021;300:122244.
- [Google Scholar]
- Optoelectronic and thermoelectric properties of double perovskite Rb2PtX6 (X = Cl, Br) for energy harvesting: First-principles investigations. J. Phys. Chem. Solids. 2021;148:109665.
- [Google Scholar]
- First principle study of lead-free double perovskites halides Rb2Pd(Cl/Br)6 for solar cells and renewable energy devices: A quantum DFT. Int. J. Energy Res.. 2021;45:14995-15004.
- [Google Scholar]
- Defect Tolerance to Intolerance in the Vacancy-Ordered Double Perovskite Semiconductors Cs2SnI6 and Cs2TeI6. J. Am. Chem. Soc.. 2016;138:8453-8464.
- [Google Scholar]
- VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst.. 2011;44:1272-1276.
- [Google Scholar]
- High-Efficiency InGaN/GaN Quantum Well-Based Vertical Light-Emitting Diodes Fabricated on β-Ga2O3 Substrate. ACS Appl. Mater. Interfaces. 2017;9:34057-34063.
- [Google Scholar]
- Lead Free Double Perovsites Halides X2AgTlCl6 (X = Rb, Cs) for solar cells and renewable energy applications. J. Solid State Chem.. 2021;297:121988.
- [Google Scholar]
- High-efficiency solution-processed perovskite solar cells with millimeter-scale grains. Science. 2015;347:522-525.
- [Google Scholar]
- Wave-Number-Dependent Dielectric Function of Semiconductors. Phys. Rev.. 1962;128:2093-2097.
- [Google Scholar]
- Generalized Gradient Approximation Made Simple. Phys. Rev. Lett.. 1996;77:3865-3868.
- [Google Scholar]
- XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag.. 1954;45:823-843.
- [Google Scholar]
- Exploring novel phase of tin sulfide for photon/energy harvesting materials. Sol. Energy. 2018;169:648-657.
- [Google Scholar]
- Exploring electronic and optical properties of Ge-based perovskites under strain: Insights from the first-principles calculations. Spectrochim. Acta A. 2020;229:118013.
- [Google Scholar]
- Exploring the electronic and optical properties of vacancy-ordered double perovskites Cs2PtX6 (X = Cl, Br, I) J. Solid State Chem.. 2021;304:122602.
- [Google Scholar]
- First-principles prediction of the ground-state crystal structure of double-perovskite halides Cs2AgCrX6 (X = Cl, Br, and I) J. Phys. Chem. Solids. 2022;160:110302.
- [Google Scholar]
- Optoelectronic and elastic properties of metal halides double perovskites Cs2InBiX6 (X = F, Cl, Br, I) Chin. Opt. Lett.. 2021;19:030004.
- [Google Scholar]
- Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3. Phys. Rev. B. 2000;62:8828-8834.
- [Google Scholar]
- Ultralow Lattice Thermal Conductivity in Double Perovskite Cs2PtI6: A Promising Thermoelectric Material. ACS Appl. Energy Mater.. 2020;3:11293-11299.
- [Google Scholar]
- Solution-Processed Cesium Hexabromopalladate(IV), Cs2PdBr 6, for Optoelectronic Applications. J. Am. Chem. Soc.. 2017;139:6030-6033.
- [Google Scholar]
- Zwei neue Iodopalladate mit gleicher Summenformel: Rb2PdI4 · I2 – ein neuer Strukturtyp mit eingelagerten I2-Molekülen – und Rb2PdI6. Z. Anorg. Allg. Chem.. 2000;626:202-207.
- [Google Scholar]
- Air Stable, High-Efficiency, Pt-Based Halide Perovskite Solar Cells with Long Carrier Lifetimes. Phys. Status Solidi RRL. 2020;14:2000182.
- [Google Scholar]
- Detailed Balance Limit of Efficiency of p-n Junction Solar Cells. J. Appl. Phys.. 1961;32:510-519.
- [Google Scholar]
- Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science. 2013;342:341-344.
- [Google Scholar]
- Structural, electronic, and optical properties of Pt-based vacancy-ordered double perovskites A2PtX6 (A = K, Rb, Cs; X = Cl, Br, I) in tetragonal P4/mnc polymorph. Opt. Mater.. 2021;119:111323.
- [Google Scholar]
- Über hexaiodoplatinate (IV) M2PtI6 (M = K, Rb, Cs, NH4, Tl) Darstel-lungsverfahren, Eigenschaften und Kristallstrukturen/On hexaiodoplatinates (IV) M2PtI6 (M = K, Rb, Cs, NH4, Tl)-preparation, properties and structural data. Z. Naturforsch. B. 1983;38:905-910.
- [Google Scholar]
- Electronic Structure and Optical Properties of Vacancy-Ordered Double Perovskites Cs2PdBrxCl6-x by First-Principles Calculation. J. Phys. Chem. C. 2020;124:13310-13315.
- [Google Scholar]
- Photovoltaic properties of all-inorganic lead-free perovskite Cs2PdBr 6: A first-principles study. AIP Adv.. 2020;10:115203.
- [Google Scholar]
- Novel Lead-Free Material Cs2PtI6 with Narrow Bandgap and Ultra-Stability for Its Photovoltaic Application. ACS Appl. Mater. Interfaces. 2020;12:44700-44709.
- [Google Scholar]
- Composition effects on structure and optical properties in double perovskite derivatives semiconductors Cs2SnI6-xBrx (x = 0–6) APL Mater.. 2020;8:021102.
- [Google Scholar]
- First-principles study of lead-free double perovskites Ga2PdX6 (X = Cl, Br, and I) for solar cells and renewable energy. J. Mater. Res. Technol.. 2022;16:631-639.
- [Google Scholar]
- First-principles study on the structural, electronic and optical properties of vacancy-ordered double perovskites Cs2PtI6 and Rb2PtI6. Opt. Mater.. 2021;114:110952.
- [Google Scholar]
- All-Inorganic Lead-Free Cs2PdX6 (X = Br, I) Perovskite Nanocrystals with Single Unit Cell Thickness and High Stability. ACS Energy Lett.. 2018;3:2613-2619.
- [Google Scholar]




