

Translate this page into:
Bio-approach synthesis of nanosilver impregnation on calcium hydroxyapatite by biological activated ammonia from urinary waste
⁎Corresponding authors at: Research Chair in Laser Diagnosis of Cancer, Department of Physics and Astronomy, College of Science, King Saud University, P.O. Box -2455, Riyadh 11451, Saudi Arabia. malsahi@ksu.edu.sa (Mohamad S. AlSalhi), rajasekargood@tvu.edu.in (Aruliah Rajasekar)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A novel biological approach is attempted to convert the human urinary waste into a well-designed bionanomaterial. In the present study, biological activated ammonia gas (NH3(g)) mediated synthesis of hydroxyapatite material (B-HAp) and then impregnation of silver nanoparticles (AgNPs) on the B-HAp material surface was performed by photoreduction method and was followed by an evaluation of its antibacterial activity and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. X-ray diffraction spectroscopy (XRD) and Field emission-scanning electron microscopy (FE-SEM) were engaged to analyze the synthesized materials. Analytical studies revealed the morphology of the crystalline B-HAp synthesized by biologically activated NH3(g) as spherical shaped with AgNPs impregnated on over it. Atomic Absorption Spectrometers (AAS) estimated 2–7 ppm of Ag+ ion were released from the 100 ppm of Ag concentration was impregnated with B-HAp material (B-HAp-Ag-10). It was also found to be an excellent performance of antibacterial activity against Pseudomonas sp, E.coli and S. aureus. The cell- material interaction study of the thus synthesised B-HAp-Ag-10 was found to exhibit a minimal cytotoxicity level when incorporated in MG63 osteosarcoma cell lines, thus confirming the prospective biological application of our material in the biomedical field.
Keywords
Hydroxyapatite
Nano-silver
Urine
Biologically activated ammonia
Antibacterial

1 Introduction
Depletion of natural resources and exceeding amounts of wastes have created a growing focus worldwide on developing methods to achieve the 3 'R's concept of reducing, reuse and recycle that can fulfil the need of energy and daily requirements (Kannan and Ronan, 2017). Excretes from human being, urine was estimated 1–2 L /day as a waste product. Human urine consists of urea, proteins, glucose, and minerals like phosphorus, potassium, sulphur, magnesium, sodium, and calcium (Kirchmann and Pettersson, 1995). Urea, a product of human protein degradation metabolism entered into the bloodstream and excreted in the urine (Bouatra et al., 2013). In general, urine consists of 20–30% of urea that was biotransformed into ammonia by ureolytic bacteria under environmental conditions (Phillips et al., 2013). This ammonia can be potentially utilized for biomaterial synthesis, which has been attempted in the present study.
Hydroxyapatite material (HAp) has a good bioactive property, such as adhesion, proliferation, and new bone formation in osteoblasts (Samavedi et al., 2013). The standard chemical-based approaches reported so far for the synthesis of HAp material using Ca(NO3)2·4H2O, (NH4)2HPO4 precursors and ammonium hydroxide solution (Wang et al., 2012; Wei et al., 2012; Bertran et al., 2014; Kramer et al., 2014; Ye et al., 2014; Akram et al., 2015; Brundavanam et al., 2015; Shi et al., 2015; Othman et al., 2016; Anwar et al., 2016; Mary et al., 2018). Iafisco et al. (2010) reported that synthesis of nanocrystals of carbonate–hydroxyapatite as plate like morphology with the NH4HCO3 solution as pH adjustors (Iafisco et al., 2010). Crystal formation of calcium phosphate in controlled manner using L-alanine, L-aspartic acid, and L-arginine by the vapour diffusion method (Gómez-Morales et al., 2011). The decomposition of NH4HCO3 solution to produces vapours of NH3 and CO2 which act as pH adjustors in aqueous solution of Ca(CH3COO)2 and (NH4)2HPO4 to form needle‐like morphology of carbonate–hydroxyapatite (Iafisco et al., 2011). Previous reports on the use of ureolytic and calcium precipitating bacteria for the synthesis of biomaterials like calcite, struvite, and HAp at microscale have been reported in the literature (Yong et al., 2004; Subramanian et al., 2012; Anusha Thampi et al., 2015). The ureolytic bacterial strains play a significant role in the conversion of urea into ammonia by urease enzyme (Phillips et al., 2013). Thus synthesised biogenic ammonia was released from the bacterial surface. It promoted the building-up of calcium-based precipitates around the cell matrix, which leads to the development of irregular 2D and 3D particle morphology (Chen et al., 2009; Prywer and Torzewska, 2009). The synthesized HAp material was reported to find its application in overcoming the failures in bone implants that are commonly associated with bacterial infections (Arciola et al., 2018). This was also found to avert the production of antibiotic-resistant bacteria due to the extended use of typical antibiotic treatment against the disease, causing microbes which precede the reduction of antibacterial efficiency (Arciola et al., 2018). In recent research attempts, the synthesis of HAp material incorporated with AgNPs at optimum concentration was implied for selective toxicity against bacterial species without causing any adverse impacts on human cell lines has been reported (Shi et al., 2015; Stanić et al., 2011; Vukomanovi et al., 2015). It owes this speciality due to the sustained release of Ag+ ion from the implant coated surface, thus hindering the bacterial infection (Stanić et al., 2011; Vukomanovi et al., 2015). In the present study, biological activated NH3(g) mediated synthesis of hydroxyapatite material (B-HAp) and then impregnation of AgNPs on the B-HAp material surface was performed by photoreduction method. The synthesized materials were characterized by various analytical techniques such as UV–Visible spectroscopy, Fourier-transform infrared spectroscopy (FT-IR), XRD and FE-SEM. In order to explore its biomedical potential, the synthesized material was evaluated for antibacterial activity against gram-negative (E. coli & Pseudomonas. sp) and gram-positive (S. aureus) bacterial species by standard methods. These materials were further tested for cell viability studies using MG63 osteosarcoma cell lines.
2 Materials and method
2.1 Materials
Calcium nitrate tetrahydrate (Ca(NO3)2·4H2O) (≥99%), Ammonium phosphate dibasic ((NH4)2HPO4) (≥99%), Urea (≥98%), Calcium chloride (CaCl2) anhydrous (≥99%), Magnesium chloride (MgCl2) anhydrous (≥99%), Potassium chloride (KCl) (≥98%), Sodium chloride (NaCl2) (≥99%), Sodium sulfate (Na2SO4) (≥98%), Ammonium chloride (NH4Cl) (≥99%), Dipotassium phosphate (≥99%), Creatinine (≥99%), Fetal bovine serum (FBS) (≥98%), dimethyl sulfoxide (DMSO) (≥99.9%), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Phosphate buffer solution (PBS), Silver nitrate (AgNO3) (≥99.0%), Nitric acid (HNO3), 70%, (≥99%), Phenol crystal (≥99%), Sodium nitroprusside dihydrate (≥99%), Glucose (≥99%), was attained from Sigma Aldrich India. Ammonia solution (NH3) 25% (MERCK, India). Dulbecco’s modified eagle medium (DMEM), urea base broth, tryptone, nutrient broth and mueller hinton agar were purchased from Hi-Media, India.
2.2 Selection of ureolytic bacteria for ammonia production
The ureolytic bacterial strains of S. ureilytica (HM475278) and B. subtilis (HM475276) were used in the present study for the production of biologically activated ammonia from synthetic urine and utilized for the synthesis of B-HAp material. These bacterial species were reported to assist for the efficient production of ammonia from synthetic urine in our previous report (Dhandapani et al., 2014). The following substrate medium, viz., urea base broth, synthetic urine and fresh urine sample were used for the production of biologically activated ammonia. i) The urea base broth medium was prepared and sterilized by autoclave method and it cool. After that, 0.66 M of the urea content was prepared in a 100 ml of sterile distilled water and then filtered by membrane filter technique (pore size is about 0.2 µm).The filtrate solution was added to sterilized urea broth (900 ml) for the production of ammonia. ii) The composition of synthetic urine (M) is as follows: Calcium chloride – 0.0058, Magnesium chloride- 0.0068, Potassium chloride- 0.0214, Sodium chloride- 0.0787, Sodium sulfate – 0.0161, Ammonium chloride- 0.0065, Creatinine- 0.0097, Dipotassium phosphate- 0.0160, Tryptone-1% w/v and 0.66 M of urea solution was prepared and sterilized protocol as described by urea broth media preparation. iii) Fresh urine sample: urine samples were collected from young male adult (25–28 years) at Vellore, Tamil Nadu, India. After urine collection, the sample was immediately frozen and stored at −20 °C. Directly before usage for the production of ammonia, urine samples were thawed to room temperature and then filtered by membrane filter technique (pore size is about 0.2 µm) to estimate the urea, protein and glucose content in each sample were analyzed by commercial available kit method. The above medium of urea base broth, synthetic urine medium and urine sample were inoculated with ureolytic bacterial cultures. It kept at room temperature under mild shaking conditions for five days, where the change in concentration of ammonia content was estimated by the indophenol blue method (Dhandapani et al., 2014). Sample was collected from enriched ureolytic culture at different time interval and centrifuged (10,000 rpm for 10 min). After that supernatant solution was collected and filtered by cellulose acetate membrane (pore size of 0.45 µm). The filtrate was tested for occurrence of ammonia concentration by indophenols method. It consisted of the 1.0 ml of tested sample, 0.5 ml of phenol-sodium nitroprusside solution (phenol, 0.53 (M) ; sodium nitroprusside, 0.0008 (M)), 0.5 ml of sodium hypochlorite solution (0.1 M), and 2 ml of distilled water and further incubation at 30 min at 37 °C and finally ammonia concentrations was measured at 625 nm by UV–visible spectroscopy. The pH of the ureolytic bacterial culture was also monitored at regular time intervals. Based on the results, the synthetic urine medium was used for further studies. Furthermore, after five days of old ureolytic culture was centrifuged for 10 min 10,000 rpm to obtained precipitates, then washed with distilled water at five times. After that, precipitate was dry with vacuum oven at 60 °C for 12 h. This precipitate sample noted as USC and then further analysis of organic functional groups by FT-IR spectroscopy, the nature of crystal phase by XRD and particle morphology by SEM.
2.3 Effect of urea source for the production of ammonia
Synthetic urine medium was prepared at various concentrations of urea such as 0.166, 0.333, 0.499 M and inoculated with ureolytic bacterial culture. This medium was kept in room temperature for five days. To observe the effect of urea concentration on the generation of biologically activated ammonia as alternative monitoring the pH of the medium at regular time intervals by employing pH meter (Eutech pH 700).
2.4 Biological activated ammonia gas mediated synthesis of HAp material
HAp material was prepared from base substances Ca(NO3)2·4H2O, (NH4)2HPO4 and biological activated NH3 (g) act as pH adjusters. Firstly, 160 ml of distilled water was taken in a clean 250 ml beaker. After that, 0.062 M of Ca(NO3)2·4H2O substance was added to a beaker under mild string condition for 10 min and 0.037 M of (NH4)2HPO4 was also added to beaker. This mixture of medium was transferred in to vessel. Meanwhile, the biological activated NH3 (g) was generated from the enrichment of ureolytic bacterial culture and purged with oxygen gas (O2(g)) to evolved NH3(g) as slowly pass through the HAp precursors solution. The solution pH was monitored (Eutech pH 700) with respect to time intervals. The HAp precursor’s solution of the pH was raise upto 8.5 by control of O2(g) purging on/off method and then maintained for 10 min. After that, this medium was transferred into beaker and heated to 80 °C under the stirring condition for 6 h. The precipitated material was separated by centrifugation for 30 min at 6000 rpm and followed by washing with distilled water for five times to remove excess ammonium, calcium and phosphate ions. The obtained precipitate sample was dried in a hot air oven. This precipitate sample noted as B-HAp. Similar method was conducted to synthesis of HAp material by using chemical ammonia solution as pH adjusters to obtain precipitate sample noted as C-HAp, in order to make the comparison between the particle morphology of the biologically activated NH3 (g) mediated synthesis of sample and chemical ammonia solution mediated synthesis of sample.
2.5 AgNPs impregnated with B-HAp material surface by photoreduction method
The deposition of AgNPs on the B-HAp material surface by photoreduction was performed as follows; 1 g of B-HAp material was dispersed in 100 ml of distilled water in a 250 ml of quartz beaker and kept in sonication for 30 min. The silver-ammonium complex was prepared 100 ppm by mixing the biologically activated ammonia solution (0.028 M) and AgNO3 (0.005 M). This medium was supplemented with glucose (0.1 mM). The mixture medium was kept in the dark condition for 10 min under the stirring condition and then transferred to a solar simulator sample desk (Class AAA solar simulator Science Tech-Canada). During illumination, the interior medium was analyzed by UV–Vis spectroscopy (Evolution 201, UV–Visible spectrophotometer-Thermo Scientific) at every 30 sec time intervals. The particles were collected by centrifugation for 20 min at 10,000 rpm and rinsed with distilled water for five times to remove the residual silver-ammonium substance. This precipitated sample was dried with vacuum oven at 50 °C for 12 h. Similar method was conducted for the 50 ppm of concentration of AgNPs was impragated with B-HAp material noted as B-HAp-Ag-5. For comparison of antibacterial activity and cytotoxicity level of B-HAp, the B-HAp-Ag-5 and B-HAp-Ag-10 samples were used respectively.
2.6 Characterization of materials
The crystal phase of the precipitation samples was analyzed by Bruker D8 Advance diffractometer in with Cu Kα radiation and detected using a Bruker Lynx Eye detector. XRD spectra were recorded in the range 10-90° 2θ at a step size of 0.02°. FT-IR spectra were recorded for the precipitation of the samples from the two systems (USC, & B-HAp). For FT-IR characterization, each sample was directly mixed placed with KBr pellet, and the spectra were recorded in the transmittance mode in the region of 400–4000 cm−1 with a resolution of 4 cm−1 for 64 scans (Bruker Optik GmbH-Tensor 27, FT-IR spectrometer, Germany). SEM-Tescan VEGA3 SBU was utilized to study the surface morphology of the particle. AgNPs deposited on the B-HAp material surface was observed by FE-SEM-Carl Zeiss, Supra 40 VP. Cumulative release of Ag+ ion from that AgNPs impregnated with B-HAp material at different time intervals sample were collected and centrifugation at 10,000 rpm for 20 min. The supernatant solution was collected and added with 1 ml of HNO3 solution (0.1 N) for 1 hr at room temperature. After acid digestion process, the sample was analysed by Atomic absorption spectroscopy (AAS), Thermo scientific model iCE 3000 series.
2.7 Evaluation of antibacterial activity
E. coli (MTCC-4296), S. aureus (MTCC-3196) and Pseudomonas. sp (MTCC-424) was cultured in nutrient broth in separate vials and incubated at 37 °C for 24 h. Synthesized materials were evaluated for antibacterial activity against the above tested bacterial strains by agar diffusion and wet interfacial method. In the agar diffusion method, the following powder material of B-HAp, B-HAp-Ag-5, and B-HAp-Ag-10 was weighed to 0.25 g and compactly packed into 10 mm disc. Completely dried sample discs were placed aseptically on the Mueller Hinton agar plates which were spread with fresh bacterial cultures of E.coli, S. aureus, and Pseudomonas. sp separately. The plates were kept in a bacterial incubator for 24 h to attain the bacterial growth inhibition zone. Based on the agar diffusion study, E.coli was selected for further detailed antibacterial studies. The wet interfacial method for the direct contact of E. coli cells on the material surface was done to study the survival of bacterial cell colonies at a short period. The samples (B-HAp, B-HAp-Ag-5, & B-HAp-Ag-10) were coated over a clean glass slide by doctor blade method and kept in a hot air oven for overnight at 180 °C. Then, this material surface was sterilized by UV-light exposure for 5 min. Subsequently, 100 µl of the E. coli cell (x104 CFU/ml) was dropped into each coated sample surface and incubated at 37 °C for 30 min in an aseptic condition. Later, the incubated sample was laid on the surface of the Mueller Hinton agar plate for 2 min and immediately removed from the agar plate. This plate was kept in a bacterial incubator at 37 °C for 24 h to observe the survival of E. coli cell colonies.
2.8 MTT assay
The human MG-63 osteosarcoma cell lines were procured from National Centre for cell science (NCCS)-Pune and India and maintained in DMEM with 10% FCS and 1% gentamicin. Cultures were kept at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. 80% of the confluent cells were detached from the culture flask through trypsinisation process, centrifuged to separate the media and re-suspended in fresh medium. After enrichment, the MG-63 osteosarcoma cell lines were used to assess the cell viability of the as-prepared B-HAp, B-HAp-Ag-5 and B-HAp-Ag-10 sample. MTT(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay is based on the metabolic ability of the viable cells to reduce soluble MTT by mitochondrial enzyme into an insoluble color formazan product, which was measured by spectrophotometrically. Brief, cells were incubated with 250 µl PBS (phosphate buffer solution) containing MTT (0.5 mg / ml) in a CO2 incubator at 37 °C . After 3 h of incubation, the PBS was removed and the formazan product formed was solubilised in 150 µl of DMSO. The absorbance was measured at 570 nm using BioRadmicroplate reader which was performed as per earlier report (Rajendran and Pattanayak, 2014).
2.9 Statistical analysis
Data were investigated statistically through the SPSS version 18 statistical software. Data were illustrated as mean ± SD of triplicate tests. The one way ANOVA test subsequently DMRT study was executed to examine the variations among the groups and the p-value less than 0.05 noted as significant.
3 Results
Fig. 1a represents the variation of pH in each medium (urea broth, synthetic urine & urinary sample) were inoculated with ureolytic bacterial species at regular time intervals. On observing the variation of pH, it showed 9.8, 10.2 and 8.3 in urea broth, synthetic urine and urinary sample systems were respectively. Whereas, the control system showed no significant change in any of the medium. The production of biologically activated ammonia in each medium was estimated concerning time intervals, as shown in Fig. 1b. The production of ammonia content was gradually increased with respect to time intervals and corresponds to pH of the medium also increased. It was found that the production of biologically activated ammonia (ppm) in the following order, urine sample (3,200) < urea broth (14,500) < synthetic urine (21,000). Based on the Fig. 1b to reveals that higher production of ammonia was observed in synthetic urine when compare to urea broth and urine sample. In the present study, our ureolytic bacterial strain as favourable for the production of ammonia in synthetic urine system.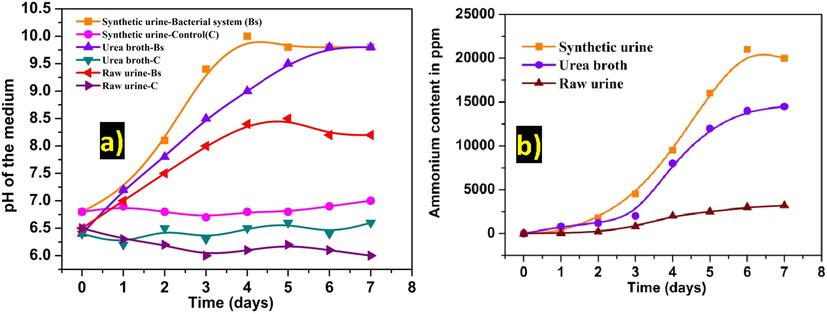
(a) pH and (b) ammonia concentration in the medium at a regular time interval with and without bacterial inoculation.
3.1 Effect of urea content for the production of ammonia
Production of ammonia directly depends on the concentration of urea present in the sample. In this regard, the presence of urea, protein and glucose content in various real urine samples of different aged adults were estimated and tabulated in Table 1. It can be seen that the urea content varied from 20 to 28 g/l. Therefore, various concentrations of urea (0.166, 0.333, 0.499 M) added to synthetic urine was examined for ammonia production in the presence of mixed ureolytic cultures with regular monitoring of pH of the medium. Fig. S1 represented the maximum increase of pH corresponded to the increased level of ammonia production with increasing content of urea in the synthetic urine sample. Whereas, in the control system (without urea), no significant change in pH value was observed. This study revealed that the production of ammonia certainly depends upon the presence of urea content.
Urine Sample
Age (year)
PH
Urea (g/L)
Protein (mg/L)
Glucose (mg/L)
1
24
6.2 ± 0.5
21 ± 0.2
0.1 ± 0.0
0.1 ± 0.0
2
26
6.4 ± 0.3
25 ± 0.9
–
0.1 ± 0.0
3
28
6.5 ± 0.1
28 ± 0.6
0.1 ± 0.0
0.1 ± 0.0
4
28
6.4 ± 0.1
20 ± 0.2
0.1 ± 0.0
0.2 ± 0.0
5
30
6.4 ± 0.3
22 ± 0.5
0.1 ± 0.0
0.2 ± 0.0
3.2 Biological activated ammonia gas mediated synthesis of HAp material
For the synthesis of B-HAp, the solution pH was gradually increased up to 8.5 and maintained by a switch on/off of O2(g).The pH of 8.5 was reached within 120 sec, and this was maintained up to 10 min (Fig. S2). White colour precipitation was observed after the treatment. There was no precipitate formed when only O2(g) purging (without biologically activated ammonia gas) through the control system.
3.3 AgNPs impregnated with B-HAp material surface by photoreduction method
Fig. 2 depicts the UV–Visible spectra of the photoreduction of Ag+ ion on the B-HAp material. During photoreduction, a small hump was observed at a maximum absorption wavelength of 424 nm after 1 min of light exposure. Later, the absorption peak shifted to 439 nm with a gradual increase in absorbance intensity after 2 min of light exposure. On further light exposure up to 3 min, it showed the shifting of maximum absorption peaks to 444 nm with stabilized absorbance intensity. The similar absorption band shift was observed in both concentrations (Fig. 2a-b). No absorption peak was found in the B-HAp. The solid mode UV–Visible absorption spectra reveal that the B-HAp-Ag-10 material exhibited higher absorption intensity at 430 nm compared to B-HAp-Ag-5, due to the varying amount of silver content (Fig. 2c).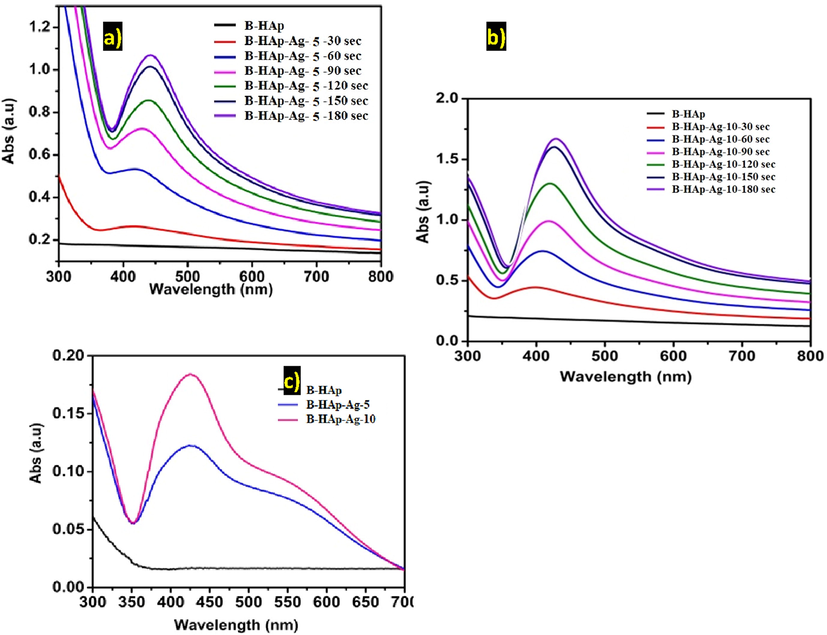
The UV–visible spectra of (a-b) photoreduction of Ag+ ion on B-HAp material under solution suspension and (c) comparative plotted from the precipitate sample (B-HAp-Ag-5 and B-HAp-Ag-10), after 5 min light exposure.
3.4 XRD analysis
Fig. 3 shows the X-ray diffraction patterns of the precipitates of USC, B-HAp, B-HAp-Ag-5, B-HAp-Ag-10, and C-HAp sample were respectively. All the diffraction peaks are identified as CaCO3, Ca (HPO4), HAp and Ag material with reference data of standard CaCO3 JCPDS.No 24-0030, Ca (HPO4) JCPDS.No 89-5969, HAp JCPDS.No 09-0432 and Ag JCPDS.No 87-0719 were respectively. In USC precipitate sample to obtain the high intensity of the polycrystalline phases of CaCO3 and Ca(HPO4) by ureolytic bacterial activity as resultants of precipitation of Ca2+ and PO42- ions at alkaline condition due to the production of ammonium and carbonate ions from synthetic urine medium, after five days over the incubation time periods (Fig. 3). Though the peaks were almost similar in B-HAp and C-HAp material, the B-HAp peaks were of high-intensity corresponding to the HAp JCPDS.No 09-0432. However, XRD pattern with an absence of CaCO3 crystal phases in B-HAp sample (Fig. 3(b-d)) due to lack of ammonium carbonate and ammonium bicarbonate. In the present study, biologically activated ammonia gas alone was used for the synthesis of B-HAp material. Based on the XRD result to confirmed as our synthesized product as HAp material. To improve the antibacterial behaviour of B-HAp material surface was deposited AgNPs by photochemical reduction method. The XRD patterns of B-HAp-Ag-5 and B- HAp-Ag-10 to exhibits low intensity of diffraction peak at 38.5, corresponding to silver crystal phase plane (1 1 1) in JCPDs No. 87-0719 (Fig. 3c-d).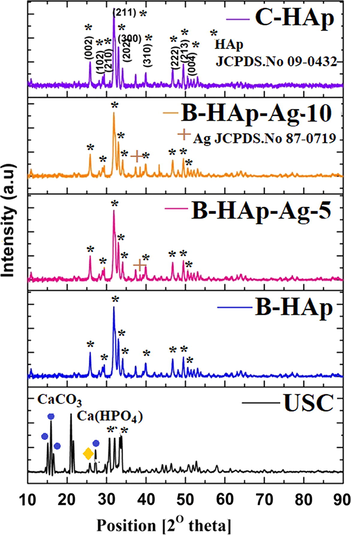
XRD patterns of (a) USC, (b) B-HAp, (c) B-HAp-Ag-5, (d) B-HAp-Ag-10, and (e) C-HAp sample.
3.5 FT-IR analysis
FT-IR spectra of the precipitated samples of USC and B-HAp are given in Fig. S3. For USC precipitate, presence of IR dual absorption bands at 568 and 609 cm−1 was due to the presence of asymmetric and symmetric deformation modes of the [PO4] group, (O-P-O) and a strong absorption band at 1043 cm−1 corresponds to the asymmetrical stretching mode of [PO4] group (P-O). The band at 1442 cm−1 can be assigned to the asymmetrical stretching mode of the [CO3] group, (C-O) and a weak absorption band at 874 cm−1 corresponded to a substitution of the ‘B-type’ [PO4] group with [CO3] group in the HAp crystal lattice (Brundavanam et al., 2015; Sadat-Shojai et al., 2013; Stanić et al., 2011). A band at 3487 cm−1 can be assigned to the OH– group of H2O absorbed in the sample (Fig. S3). The obtained results of FT-IR spectra clearly indicated the presence of calcium phosphate and carbonate groups along with HAp in the USC precipitate. Since, the [CO3] functional group was suppressed (Fig. S3) in B-HAp precipitated sample, the use of biologically activated ammonia gas seems to show a good possibility to synthesise HAp material without CaCO3 impurities.
3.6 Particle morphology
SEM depict the USC precipitates that have been obtained from the ureolytic bacteria inoculated with a synthetic urine culture at the end of 5th day and presented in Fig. 4(a-b). The calcite particle shows little honeycombed structures with microvoid space in the middle of their structure and flake-like structure. This void may be due to the occupancy of the ureolytic bacteria. On the other hand, C-HAp shows sharp needle-shaped clustered structures (Fig. 4(c-d)). B-HAp material displayed spherical shaped and particle size distribution ranging from 200 to 450 nm (Fig. 5a-b). However, the morphology of the B-HAp-Ag-5 and B-HAp-Ag-5 material was observed to be slightly increased distribution of particle size ranging from 230 to 490 nm by photoreduction process (Fig. 5c-f). Enlarged view of the figures shows the presence of impregnated AgNPs like a dot at the centre of HAp material surface, distributed uniformly with the particle size ranging from 10 to 20 nm (Fig. 5f).
SEM images of (a-b) USC precipitate and (c-d) C-HAp sample.

FE-SEM images of (a-b) B-HAp, (c-d) B-HAp-Ag-5, and (e-f) B-HAp-Ag-10 sample.
3.7 Cumulative release of Ag+ ion study
AAS measurement for cumulative release of Ag+ ion from AgNPs that were photo impregnated onto B-HAp material at two different concentrations, B-HAp-Ag-5, B-HAp-Ag-10 under solution immersed condition in phosphate buffer (pH 7.2) at various time intervals and presented in Fig. 6. In a typical initial stage as a pellet form sample of B-HAp-Ag-5 and B-HAp-Ag-10, the corresponding concentration of Ag+ ion was noted as 12 ± 1.3 and 29 ± 2.2 ppm respectively. According to the Fig. 6 shown, the mid stage of the cumulative release of Ag+ ion was found to be 1.2 ± 0.4 and 2.5 ± 0.7 ppm to attribute the B-HAp-Ag-5 and B-HAp-Ag-10 were respectively. The cumulative release of Ag+ ion gradually increased with increasing time intervals at a maximum of 3 ± 0.4 ppm to the corresponding sample of B-HAp-Ag-5 and 7 ± 0.9 ppm of B-HAp-Ag-10. Furthermore, both the samples released more Ag+ ion upon incubation up to 12–24 h. The reason for the release of Ag+ ion from B- HAp-Ag-10 material pellet was may be due to internal core compact or porous nature of the material, in which Ag+ ion was entrapped.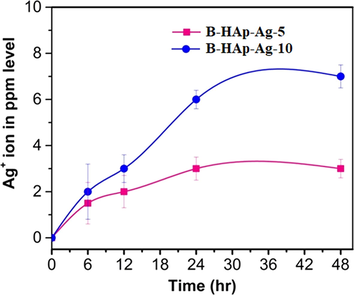
The cumulative Ag+ ion release from B-HAp-Ag-5 and B-HAp-Ag-10 material on time intervals in phosphate buffer saline at pH 7.2.
3.8 Antibacterial activity
On performing the antibacterial studies, the inhibition zone (mm) exhibited by B-HAp-Ag-5 and B-HAp-Ag-10 against E. coli, S. aureus, and Pseudomonas. sp, are shown in Table 2. The antibacterial activity for B-HAp-Ag-5 against E. coli was found to be 18.0 ± 2.1 mm with a significant increase in antibacterial activity on increasing the Ag loading to B-HAp-Ag-10 sample (26 ± 1.9). However, the B-HAp disc showed no bacterial inhibition zone. The antibacterial activity was found in the following order: E. coli > S. aureus > Pseudomonas. sp. The rationlise of this work was to prove the effectiveness of the B-HAp-Ag-5 and B-HAp-Ag-10 coatings towards the prevention of bacterial cell colonization or biofilm formation onto the implant base material surfaces (Fig. 7a). Low concentration of E. coli cells (x104 CFU/ml) was used for this study. On inducing the direct contact between the E. coli cell and the material surface, no colony growth was observed with both the samples (B-HAp-Ag-5 & B-HAp-Ag-10) coated over the glass slide after 30 min (Fig. 7b). On the contrary, the bare HAp material exhibited E. coli colony growth on the agar plate.
S. No
Tested disc
Bacterial inhibition zone (mm)
E. coli
S. aureus
Pseudomonas. sp
1
B-HAp
–
–
–
2
B-HAp-Ag-5
18 ± 2.1
14 ± 0.4
14 ± 1.4
3
B-HAp-Ag-10
26 ± 1.9
20 ± 0.5
21 ± 0.9

Schematic represent on (a) wet interface antibacterial activity of HAp-Ag material tested in E. coli cells and (b) experimental result observation.
3.9 MTT assay
To validate our results further, interaction studies of the B-HAp, B-HAp-Ag-5 and B-HAp-Ag-10 sample with cell lines is much needed. Therefore, we have chosen MG-63 osteosarcoma cell lines for this study. The B-HAp, B-HAp-Ag-5 and B-HAp-Ag-10 sample were evaluated concerning cell viability of MG63 osteosarcoma cell lines by MTT assay (Fig. 8). The cell viability was found to be 96 ± 3.4, 90 ± 2.5 and 84 ± 3.0% of the corresponding material in B-HAp, B-HAp-Ag-5 and B-HAp-Ag-10 were respectively. There is less distinction with cell viability of B-HAp-Ag-5 and B-HAp-Ag-10 sample. The untreated cell viabilty was found to be as 98 ± 2.9. The present result was indicated that our synthesized material have good bioactivity when compared to control value (untreated cell line) and then possible osteoblast growth on the material surface.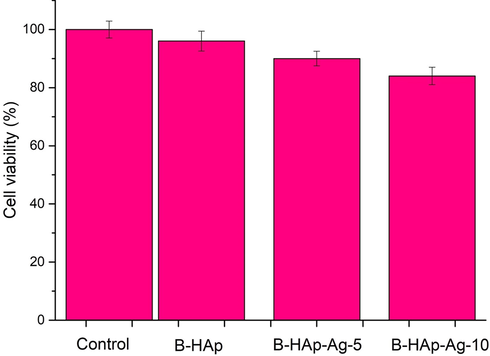
Cell viability study in MG63 osteosarcoma cell lines for (a) B-HAp, (b) B-HAp-Ag-5 and (c) B-HAp-Ag-10 by MTT assay.
4 Discussion
The ureolytic bacterial species are converted ammonia and carbonic acids from synthetic urine by urease and ammonia production as described by our previous report (Dhandapani et al., 2020) which was utilized for the synthesis of ZnO nanomaterial. In the present study, we demonstrated the production of biologically activated ammonia from the various medium such as urea broth, synthetic urine and fresh urinary sample by employing mixed ureolytic bacterial species of S. ureilytica, and B. subtilis. Among the urea substrate medium, the synthetic urine was suitable for the higher production of ammonia content when compared to another medium (Fig. 1). Low ammonia concentration in a fresh urine sample can be attributed to the suppressed bacterial growth may be due to the presence of uric acids and other excreted biochemicals substance. Further study needs much more that bacterial survival and proliferation on the urine waste water. It is possible overcome for a practical application by indigenous bacterial strain from toilet dropping area and urease enzyme activity. The pH range was maintained for the synthesis of B-HAp by biologically activated ammonia gas (our present study), when compare to particle morphologies of chemical ammonia solution mediated synthesis of HAp by various method were tabulated and presented in the Table 3. Therefore, it can be concluded that the biologically activated ammonia gas can be identified as for the synthesis of spherical shaped B-HAp materials for biomedical application.
Precursor
pH adjusters
pH range
Method
Morphology size
Ref.
Ca(OH)2, H3PO4
NH4OH
9
Wet chemical precipitation
NPs
(Wang et al., 2012)
Ca(NO3)2·4H2O, (NH4)2HPO4
NH3
10.5
Hydrothermal
NRs
(Wei et al., 2012)
DNA, Ca(NO3)2, (NH4)2HPO4
NH4OH
10
Precipitation
HAp-DNA nanocrystals
(Bertran et al., 2014)
Ca(NO3)2·4H2O, (NH4)2HPO4,
NH4OH
10–12
Precipitation
NRs
(Kramer et al., 2014)
Ca(NO3)2·4H2O, (NH4)2HPO4
NH4OH
8
Precipitation/ Hydrothermal
Porous 3D
(Ye et al., 2014)
Ca(NO3)2·4H2O, (NH4)2HPO4
NH4OH
10
Continuous microwave flow
Mesoporous HAp
(Akram et al., 2015)
Ca(OH)2, H3PO4
NH3
10
Ultrasound precipitation
NPs
(Brundavanam et al., 2015)
Ca(NO3)2·4H2O, (NH4)2HPO4
NH4OH
10
Precipitation
Microcube nested bundles
(Shi et al., 2015)
Ca(NO3)2·4H2O, (NH4)2HPO4 , AgNO3
NH3·H2O
9
Chemical precipitation
Nanocrystals
(Othman et al., 2016)
Ca(NO3)2·4H2O, (NH4)2HPO4, Urea
NH4OH
11
Co-precipitation
Nanocrytals
(Anwar et al., 2016)
Ca(NO3)2·4H2O, (NH4)2HPO4, Ca(OH)2, NaHCO3
NH4OH
8.5–11
Chemical precipitation
NPs
(Mary et al., 2018)
Ca(NO3)2·4H2O, (NH4)2HPO4
Biological activated ammonia gas
8.5
Bio-chemical precipitation
Spherical
Present work
Further, HAp material was subjected to nanosilver deposition at two different concentrations viz., B-HAp-Ag-5 and B-HAp-Ag-10 by photoreduction method. The silver ammonium complexes as cationic species are physically adsorbed on the B-HAp material surface by the influence of electrostatic interactions. The photochemical reaction can reduce this complex, and the electron transfer occurs from the glucose molecules to produce AgNPs around the material surface (Panáček et al., 2006; Montazer et al., 2012). In addition, we did not observe any of the Ag mirror grown on the quartz beaker inner wall during the photoreduction process, thus signifying our study from earlier reports regarding the effect of light intensity, photoreactor medium, ageing of AgNPs with reaction time and peak shifts, crystal size, and morphology (Panáček et al., 2006; Montazer et al., 2012; Alfuraydi et al., 2019). The present XRD result was in good in accordance with previous reports regarding silver doped HAp material preparation by various methods (Shi et al., 2015; Stanić et al., 2011; Vukomanovi et al., 2015). This characterization is yet another supporting study that shows the use of biologically activated ammonia gas in the place of chemical ammonia to prepare B-HAp material with uniform coated AgNPs.
The antibacterial activity results showed the combat nature of released Ag+ ion from B-HAp-Ag-5 and B-HAp-Ag-10 disc towards bacterial cells (E. coli, S. aureus, and Pseudomonas. sp) on the agar diffusion plate (Table 3). The AAS results confirmed the cumulative release of Ag+ ion concentration from the two samples of B-HAp-Ag-5 and B-HAp-Ag-10 at 24 hr was 3 ± 0.4 and 7 ± 0.9 ppm, respectively. Therefore, we demonstrated that bacterial inhibit zones well depend upon the release of Ag+ ion concentrations from B-HAp-Ag-5 and B-HAp-Ag-10 sample disc. The previous investigators have also proposed several mechanisms on antibacterial activity by AgNPs (AlSalhi et al., 2016; Devanesan et al., 2018; Vukomanovi et al., 2015). The wet interfacial study revealed that might have acted as the driving force for Ag+ ion release from the B-HAp-Ag-5 and B-HAp-Ag-10 coated glass slide, thus controlling the E.coli proliferation on the material surface. Though exposed for short time duration, it was believed to be enough for causing a microbial infection on implant metals due to the direct contact between the metal and bacterial species. From our results, it was evident that the B-HAp-Ag-5 and B-HAp-Ag-10 coating material have found to be effective in destroying the infection-causing microbes that are commonly associated with implant materials. In addition, AgNPs loaded with HAp material to providing the increase surface roughness and improved interaction with osteoblast growth, when compare to bare material (Tian et al., 2016). The HAp material surface of osteoblast adhesion/proliferation (i.e. osteoconduction) and their role in stimulating new bone formation as described in earlier reports (Samavedi et al., 2013; AlSalhi et al., 2019). In addition, the obtained spherical shape of B-HAp material with uniform deposition of AgNPs was analysis by MTT assay. The present result was indicated that our synthesized material has good cell viability for MG63 osteosarcoma cell lines. The AAS results discussed earlier in this study too supported the fact of trace amounts of Ag+ ion release from the coated material (B-HAp-Ag-5 and B-HAp-Ag-10), which aided in retaining the cell viability. Besides, the antibacterial activity exhibited by our material was also found to prevent bacterial infection. Thus, the inclusion of AgNPs in B-HAp material at an optimised concentration renders antibacterial activity, without affecting the bone cells, which paves the way to use our material in implant procedures.
5 Conclusions
The present study demonstrated the production of biologically activated ammonia from the broth of urea, synthetic urine and fresh urinary sample under optimum conditions. The biologically activated ammonia gas was employed to synthesise spherical shaped B-HAp material, which was uniformly coated with AgNPs by photoreduction method. The synthesis and deposition were confirmed by UV–Visible absorption spectra, XRD pattern and FE-SEM images. The two different concentrations of B-HAp-Ag-5 and B-HAp-Ag-10 have been used in demonstrating bacterial growth inhibition studies with three different infection-causing clinical species. Antibacterial reviews were found in the following order, E. coli > S. aureus > Pseudomonas. sp. The wet interfacial studies too supported the complete growth retardation of E.coli cells by B-HAp-Ag-5 and B-HAp-Ag-10 coating on the implanting material surfaces within 30 min of direct contact. Further, MTT assay of MG-63 osteosarcoma cell lines also revealed the cell viability owing to the release of low concentration of Ag+ ions from B-HAp-Ag-10 material without affecting the bone cell growth. Thus, we conclude that the biologically activated ammonia gas mediated synthesis of B-HAp material with AgNPs coating will undoubtedly serve as an excellent antibacterial agent against the common clinical pathogens associated with implant procedures.
Acknowledgements
The authors are grateful to the Deanship of Scientific Research, King Saud University for funding through Vice Deanship of Scientific Research Chairs. Dr Dhandapani, P., thank to Dr D.S.Kothari postdoctoral Fellowship (File.No.F.4-2/2006 (BSR) BL/17-18/0343).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Continuous microwave flow synthesis of mesoporous hydroxyapatite. Sci. Eng. C. 2015;56:356-362.
- [Google Scholar]
- Eco-friendly green synthesis of silver nanoparticles from the sesame oil cake and its potential anticancer and antimicrobial activities. J. Photochem. Photobiol. B. 2019;192:83-89.
- [Google Scholar]
- Green synthesis of silver nanoparticles using Pimpinella anisum seeds: antimicrobial activity and cytotoxicity on human neonatal skin stromal cells and colon cancer cells. Int. J. Nanomedicine. 2016;11:4439-4449.
- [Google Scholar]
- M.S. AlSalhi, S. Devanesan, P. Shanmugam, Y.O. Kim, J.T. Kwon, H.J. Kiim, Synthesis and biocompatible role of hierarchical structured carbon nanoplates incorporated α-Fe2O3 nanocomposites for biomedical applications with respect to cancer treatment, Saudi J. Biol. Sci, Online Nov (2019).
- Enhancement of bioactivity of titanium carbonitride nanocomposite thin films on steels with biosynthesized hydroxyapatite. Int. J. Nanomed.. 2015;10:107-118.
- [Google Scholar]
- Low temperature synthesis and characterization of carbonated hydroxyapatite nanocrystals. J. Mol. Struct. 2016;1117:283-286.
- [Google Scholar]
- Implant infections: adhesion, biofilm formation and immune evasion. Nat. Rev. microbial.. 2018;16:397.
- [Google Scholar]
- Mineralization of DNA into nanoparticles of hydroxyapatite. Dalton Trans.. 2014;43:317-327.
- [Google Scholar]
- Synthesis of a hydroxyapatite nanopowder via ultrasound irradiation from calcium hydroxide powders for potential biomedical applications. Nanosci. Nanoeng.. 2015;3:1-7.
- [Google Scholar]
- Bacteria-mediated synthesis of metal carbonate minerals with unusual morphologies and structures. Cryst. Growth Des.. 2009;9:743-754.
- [Google Scholar]
- Antimicrobial and cytotoxicity effects of synthesized silver nanoparticles from punica granatum peel extract. Nanoscale Res. Lett.. 2018;13:315.
- [Google Scholar]
- Bio-approach: Ureolytic bacteria mediated synthesis of ZnO nanocrystals on cotton fabric and evaluation of their antibacterial properties. Carbohydr. Polym.. 2014;103:448-455.
- [Google Scholar]
- Ureolytic bacteria mediated synthesis of hairy ZnO nanostructure as photocatalyst for decolorization of dyes. Mater. Chem. Phys. 2020122619
- [Google Scholar]
- Amino acidic control of calcium phosphate precipitation by using the vapor diffusion method in microdroplets. Cryst. Growth Des.. 2011;11:4802-4809.
- [Google Scholar]
- Biomimetic carbonate–hydroxyapatite nanocrystals prepared by vapor diffusion. Adv. Eng. Mater.. 2010;7:218-223.
- [Google Scholar]
- Formation of calcium phosphates by vapour diffusion in highly concentrated ionic micro-droplets. Cryst. Res. Technol.. 2011;8:841-846.
- [Google Scholar]
- Conversion of biowastes to biomaterial: An innovative waste management approach. Waste Manage.. 2017;67:67-72.
- [Google Scholar]
- Human urine - Chemical composition and fertilizer use efficiency. Fert. Res.. 1995;40:149-154.
- [Google Scholar]
- Control of hydroxyapatite nanoparticle morphology using wet synthesis techniques: Reactant addition rate effects. Mater. Lett.. 2014;131:145-147.
- [Google Scholar]
- Surfactant-free solvothermal synthesis of hydroxyapatite nested bundles for the effective photodegradation of cationic dyes. J. Phys. Chem. Solids. 2018;116:180-186.
- [Google Scholar]
- In situ synthesis of nano silver on cotton using Tollens’ reagent. Carbohydr. Polym.. 2012;87:1706-1712.
- [Google Scholar]
- Effect of calcium precursors and pH on the precipitation of carbonated hydroxyapatite. Procedia Chem.. 2016;19:539-545.
- [Google Scholar]
- Silver colloid nanoparticles: synthesis, characterization, and their antibacterial activity. J. Phys. Chem. B. 2006;24:16248-16253.
- [Google Scholar]
- Engineered applications of ureolytic biomineralization: a review. Biofouling.. 2013;29:715-733.
- [Google Scholar]
- Bacterially induced struvite growth from synthetic urine: experimental and theoretical characterization of crystal morphology. Cryst. Growth Des.. 2009;9:3538-3543.
- [Google Scholar]
- Silver incorporated antibacterial, cell compatible and bioactive titania layer on Ti metal for biomedical applications. RSC Adv. 2014;4:61444-61455.
- [Google Scholar]
- Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013;9:7591-7621.
- [Google Scholar]
- Calcium phosphate ceramics in bone tissue engineering: a review of properties and their influence on cell behavior. Acta Biomater.. 2013;9:8037-8045.
- [Google Scholar]
- Ultra-trace silver-doped hydroxyapatite with non-cytotoxicity and effective antibacterial activity. Mater. Sci. Eng. C. 2015;55:497-505.
- [Google Scholar]
- Synthesis of antimicrobial monophase silver-doped hydroxyapatite nanopowders for bone tissue engineering. Appl. Surf. Sci. 2011;257:4510-4518.
- [Google Scholar]
- Biosynthesis of calcium hydroxylapatite coating on sputtered Ti/TiN nano multilayers and their corrosion behavior in simulated body solution. J. Biomater. Appl.. 2012;26:687-705.
- [Google Scholar]
- Silver nanoparticle-loaded hydroxyapatite coating: structure, antibacterial properties, and capacity for osteogenic induction in vitro. RSC Adv.. 2016;11:8549-8562.
- [Google Scholar]
- Is Nano-Silver Safe within Bioactive Hydroxyapatite. Composites? ACS Biomater. Sci. Eng. 2015;1:935-946.
- [Google Scholar]
- A facile method to in situ formation of hydroxyapatite single crystal architecture for enhanced osteoblast adhesion. J. Mater. Chem.. 2012;22:19081-19087.
- [Google Scholar]
- Carbonated hydroxyapatite coatings with aligned crystal domains. Cryst. Growth Des.. 2012;12:3474-3480.
- [Google Scholar]
- Fabrication and characterization of porous 3D whisker-covered calcium phosphate scaffolds. Mater. Lett.. 2014;128:179-182.
- [Google Scholar]
- Synthesis of nanophase hydroxyapatite by a Serratia sp. from waste-water containing inorganic phosphate. Biotechnol. Lett.. 2004;26:1723-1730.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.04.024.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




