

Translate this page into:
Biology-oriented drug synthesis of nitrofurazone derivatives: Their α-glucosidase inhibitory activity and molecular docking studies
⁎Corresponding authors. khalid.khan@iccs.edu (Khalid Mohammed Khan), yeong@umt.edu.my (Yeong Yik Sung)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In the present study, twenty (20) structural variants of nitrofurazone were synthesized based on BIODS (Biology-oriented drug synthesis) approach. The structure elucidation of the synthetic molecules (1–20) was carried out using different spectroscopic techniques, and their α-glucosidase inhibitory activity was also determined. The synthetic molecules 1–20 exhibited good α-glucosidase inhibition than the parent, nitrofurazone. Four compounds 2, 4, 6, and 7 showed potential inhibition against α-glucosidase with IC50 values ranging between 0.63 ± 0.25–1.29 ± 0.46 µM as compared to the standard acarbose (IC50 = 2.05 ± 0.41 µM). Nevertheless, compounds 15 (IC50 = 0.74 ± 0.12 µM), and 19 (IC50 = 0.54 ± 0.3 µM) also displayed good α-glucosidase inhibition and compound 19 was the most active compound of the series. Kinetic study of the active compounds 7 and 19 was also carried out to confirm the mode of inhibition. The binding interactions of the most active compounds within the active site of enzyme were determined by molecular docking. Moreover, molecular dynamic simulation of compound 19 was also performed in order to determine the stability of the overall complex (α-glucosidase + c19) in an explicit watery environment. The synthetic molecules were predicted as non-cytotoxic, however, seven compounds 1, 3, 4, 9, 10, 11, and 12 were predicted as carcinogenic.
Keywords
Nitrofurazone
Diabetes
In silico studies
α-glucosidase inhibition

1 Introduction
α-Glucosidase belongs to the family of hydrolase enzymes and contains calcium as a metal atom. The primary purpose of this enzyme is to degrade glycogen into glucose in lysosomes. However, it is responsible for the increased blood glucose level in type-2 diabetes mellitus. Therefore, it is quite necessary to target and inhibit the excess activity of this enzyme to control diabetes. The compounds capable of inhibiting the intestinal α-glucosidase enzyme usually slow down the digestion and absorption of carbohydrates. Thus, meal-related hyperglycemia can be controlled through these inhibitors. Moreover, the inhibition of α-glucosidase has a profound effect on the glycan structure which consequently affects the maturation, transport, secretion, and function of glycoproteins (Nashiru et al., 2001; Park et al., 2008; Bosenberg and Van Zyl, 2008; Abbasi et al., 2017; Davies and Henrissat, 1995; Van de Laar, 2008; Azam et al., 2012). Several α-glucosidase inhibitors have been reported in the literature, such as acarbose, nojirimycin, 1-deoxynojirimycin, miglitol, and voglibose (Bosenberg and Van Zyl, 2008; Khan et al., 2014; Liu et al., 2006). The α-glucosidase inhibitors can effectively treat diabetes and other clinical conditions that are carbohydrate mediated, such as cancer, obesity, hepatitis, and hyperlipoproteinemia (de Melo et al., 2006; Fischer et al., 1995). According to the World Health Organization, the number of type 2 diabetic cases will reach 360 million by 2030. The goal of diabetes treatment is to control blood glucose levels, particularly after a meal, obesity, cholesterol, and triglyceride levels, as well as to predict the progression of complications (Yoshikawa et al., 2010).
Biology-oriented drug synthesis is focused on the chemical transformation and modification of commercially available drugs to create the libraries of compounds that can serve as lead molecules and might be evaluated for their diversified biological properties (Khan et al., 2018). During past years, our group has targeted and synthesized several libraries of compounds based on commercial drugs under the umbrella of biology-oriented syntheses. The synthetic molecules were found to possess diversified biological activities i.e. flurbiprofen derivatives were reported as novel α-amylase inhibitors (Yoshikawa et al., 2010); S-naproxen derivatives were found to have potential urease inhibitory activity (Mohiuddin et al., 2019); metronidazole derivatives showed good β-glucuronidase and α-amylase inhibitory activities (Muhammad et al., 2017), piroxicam derivatives were also reported for their antinociceptive activity (Ullah et al., 2016), and atenolol derivatives were also reported as urease inhibitors (Wahid et al., 2020). In continuation of our research on biology-oriented drug synthesis (BIODS), we had targeted another commercially available drug nitrofurazone to synthesize a library of compounds and evaluated their α-glucosidase inhibitory activity.
Nitrofurazone is available commercially with the trade name of furacin (Ullah et al., 2016) and is the first 5-NO2-furan medicine (Brito et al., 2013) approved as an antichagasic drug regardless of its toxicity. The causative agent of this disease is Trypanosoma cruzi, commonly found in Latin America and was responsible for almost 25 million deaths in 2008. Nitrofurazone has shown antitrypanosomal activity by inhibiting a specific parasite detoxifying enzyme trypanothione reductase. It is also known to inhibit the parasite protease i.e., cruzipain. The mechanistic action of nitrofurazone is still not fully understood, however, it has a wide range of activities against different organisms including parasites and bacteria (Chin Chung et al., 2011; Blumenstiel et al., 1999; Paulino et al., 2005). Nitrofurazone also possesses a wide spectrum of antimicrobial properties and is effective against both Gram-positive and Gram-negative bacterial strains. Nitrofurazones are found to be effective against trypanosomes (Chamberlain, 1976). Nitrofurazone and its analogs are found to be active against Staphylococcus spp. ATTC and Bacillus spp. ATTC (Popiołek and Biernasiuk, 2016).
In our previous work, we demonstrated the synthesis and antidiabetic activities of different classes of compounds namely indoles, oxadiazoles, hydrazides, etc. (Taha et al., 2017; Khan et al., 2018; Gollapallia et al., 2018; Abbasi et al., 2017). Fig. 1 represents the classes of compounds that were found to have α-glucosidase and α-amylase inhibitory activities, though the parent moieties of these compounds are different, these compounds have few structural similarities i.e. presence of amide linkage. Keeping that in mind, we have modified the nitrofurazone molecule by treating it with different substituted N-alkyl/aryl derivatives to afford synthetic molecules 1–20 and screened them for α-glucosidase inhibitory activity (Figure-1 in Supp. inf.). Compounds 1–20 were synthesized using eight alkyl bromides and twelve benzyl chlorides. Out of the synthesized compounds, compounds 1–12, and 16–20 are newly synthesized while compounds 13–19, and 17 are already reported in the literature (Tocher, 1997; Brondani et al., 2007; Bartel et al., 2009). Thus, this study reports the synthesis of nitrofurazone derivatives 1–20 and the in vitro α-glucosidase inhibitory activities along with the in silico study to confirm the binding interactions of compounds within the enzyme pocket.
2 Results and discussion
2.1 Chemistry
Twenty compounds 1–20 were derived from nitrofurazone via one-step synthetic transformation. Nitrofurazone was allowed to react with substituted alkyl/aryl halides in dimethylformamide (DMF) while potassium carbonate (K2CO3) was used as a catalyst. The temperature of the reaction mixture was maintained at 100 ˚C upon refluxing for 12 h and its progress was monitored through TLC. After the disappearance of starting material on TLC, potassium carbonate was filtered off. The product was then extracted with dichloromethane (DCM) (Scheme 1). Structure elucidation of the synthetic molecules was carried out by EI-MS, HR-EIMS, 1H-, and 13C NMR spectroscopy.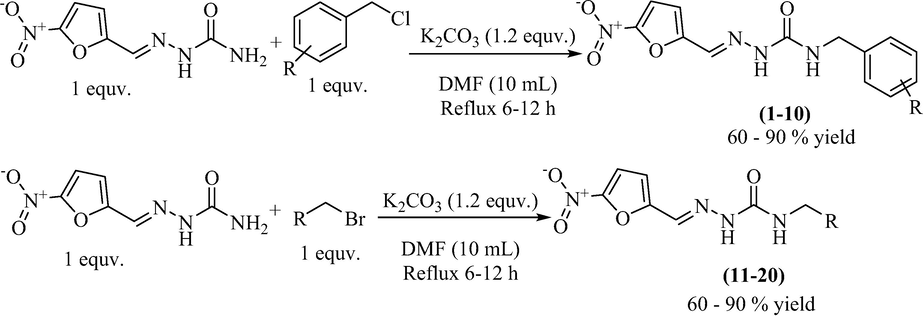
Synthesis of nitrofurazone derivatives (1–20).
2.2 In vitro α-glucosidase inhibitory activity
All twenty derivatives of nitrofurazone along with the parent molecule nitrofurazone were examined for their activity towards α-glucosidase enzyme inhibition, while acarbose was used as a standard. Almost all compounds showed higher α-glucosidase inhibition than the parent compound, nitrofurazone (IC50 = 37.5 ± 2.87 µM), nonetheless, compounds 2, 4, 6, 7, 9, 12, 15, and 19 showed good inhibitory potential within the range of 0.63 ± 0.25–1.63 ± 0.28 µM as compared to the standard acarbose (IC50 = 2.05 ± 0.41 µM) (Table 1). The remaining compounds exhibited weak activity against the α-glucosidase enzyme. The general structural features of nitrofurazone derivatives 1–20 are represented in Figure-2 in Supp. inf. SEMa (Mean ± Standard error of the mean); Standardb (Inhibitor for α-glucosidase activity).
C. No.
R
IC50 ± SEM (µM)
C. No.
R
IC50 ± SEM (µM)
1
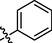
17.2 ± 3.21
11
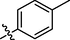
34.3 ± 3.38
2
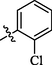
1.29 ± 0.46
12
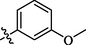
1.24 ± 0.38
3
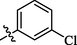
28.1 ± 1.34
13

12.7 ± 5.38
4
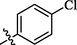
0.63 ± 0.25
14
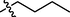
20.2 ± 1.21
5
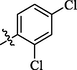
12.4 ± 1.54
15

0.74 ± 0.12
6
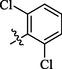
0.63 ± 0.25
16

28.4 ± 2.11
7
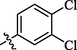
0.65 ± 0.22
17

30.9 ± 3.21
8
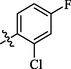
14.2 ± 0.93
18

24.0 ± 2.12
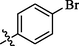
9
1.63 ± 0.28
19

0.54 ± 0.30
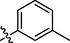
10
37.1 ± 2.09
20

27.3 ± 1.56
Nitrofurazone
37.5 ± 2.87
Standardb = Acarbose
2.05 ± 0.41
2.3 Structure-activity relationship (SAR) for α-glucosidase enzyme inhibition
Limited structure–activity relationship (SAR) suggested that the position and nature of different substituted groups on the benzyl part of nitrofurazone derivatives have a variable influence on the α-glucosidase inhibition. The synthetic analogs were separated into two categories based on the substitution of the nitrogen atom of nitrofurazone. Category ‘A’ comprises of the aryl/benzyl part substituted analogs 1–12, however, category ‘B’ is composed of alkyl substitutions 13–20.
2.4 Category A
The parent compound nitrofurazone (IC50 = 37.5 ± 2.87 µM) exhibited weak inhibitory activity against α-glucosidase enzyme in comparison with the standard acarbose (IC50 = 2.05 ± 0.41 µM) and all synthetic derivatives. Compound 1 (IC50 = 17.2 ± 3.21 µM) with unsubstituted aryl ring displayed good inhibitory activity as compared to nitrofurazone, however, it was less active than acarbose (Figure-3 in Supp. inf.).
The halogen-substituted analogs also displayed good to moderate inhibitory activities. Amongst them, compounds 2 (IC50 = 1.29 ± 0.46 µM), 3 (IC50 = 28.1 ± 1.34 µM), and 4 (IC50 = 0.63 ± 0.25 µM) with ortho, meta, and para-substituted chloro groups, respectively, exhibited good inhibitory activities as compared to their parent molecule, however, compound 3 was less active than the standard as well as compounds 2 and 4 (Figure-4 in Supp. inf.).
Compounds 5, 6, and 7 with dichloro substitution displayed good inhibitory potential. Among them, compounds 6 (IC50 = 0.63 ± 0.25 µM) and 7 (IC50 = 0.65 ± 0.22 µM) exhibited identical activities, however, compound 5 (IC50 = 12.4 ± 1.54 µM) was found to be less active. Furthermore, the mono chloro substituted analogs 2 and 4 showed good inhibitory potential among all chloro substituted molecules, suggesting that the ortho and para-substituted chloro atoms have good interactions within the enzyme pocket. Nevertheless, the activity pattern is a bit different in the case of dichloro substituted compounds, amongst them compounds 6 with chloro groups at ortho positions and 7 with chloro groups at meta and para positions were found to be more active as compared to compound 5 having two chloro groups at ortho para positions. This difference in the activity of compounds might be due to the presence of two chloro groups on the aryl part which might interact differently with the active site of the enzyme (Figure-5 in Supp. inf.).
Compound 9 (IC50 = 1.63 ± 0.28 µM) with para-bromo substitution also displayed good inhibition, however, compound 8 (IC50 = 14.2 ± 0.93 µM) with chloro and fluoro groups was less active in comparison with other molecules of similar type (Figure-6 in Supp. inf.).
Among methyl and methoxy substituted compounds 10 (IC50 = 37.1 ± 2.09 µM), 11 (IC50 = 34.3 ± 3.38 µM), and 12 (IC50 = 1.24 ± 0.38 µM), compound 12 with methoxy group at meta position displayed potential inhibition against the α-glucosidase enzyme, might be due to the inductive effect of methoxy group present at meta position (Figure-7 in Supp. inf.).
2.5 Category B
This category comprises of total eight compounds 13–20. Among them, compounds 15 (IC50 = 0.74 ± 0.12 µM) and 19 (IC50 = 0.54 ± 0.30 µM) displayed good inhibitory activities as compared to the standard acarbose. Compound 19 was discovered to be the most active of the series, which suggests that the octyl chain attached to the nitrofurazone has achieved a configuration that fits into the enzyme's active site. However, compound 15 with pentyl chain also displayed good inhibition but was less active than compound 19 of nitrofurazone derivatives (Figure-8 in Supp. inf.).
2.6 Enzyme kinetics study for α-glucosidase inhibition
Enzyme kinetics studies were performed to find out the mode of enzyme inhibition. The most potent compounds 7 and 19 among the tested ones were selected for kinetic studies. Both the compounds were used at a concentration of 0, 0.3, 0.6, and 0.9 µM. Compound 7 showed competitive inhibition while compound 19 revealed uncompetitive inhibition against α-glucosidase (Figure-9 in Supp.inf.).
2.7 Molecular docking study
A molecular docking study was performed to further understand the binding mode of all synthesized compounds against the α-glucosidase enzyme. The lead compound nitrofurazone results (Figure-10 in Supp. inf.) were compared with all the top conformations from each compound and were found in a well-accommodated pattern in the active site of the enzyme, except compound 20, which hold a much bulky group, this compound was found much bigger than the radius of the active site, which unable to accommodate well, also adopted π-stacking interactions with some local residues. While other active compounds in the series showed numerous key interactions with active site residues that might play important role in the enhancement of enzymatic activity. The details of the protein–ligand interaction profile have been enlisted in Table 2. Moreover, the docking results of the standard lead compound revealed key interactions with the active site residues, comparatively, it has been observed that the docking results of our enlisted compounds are far better than the standard compound.
C. No.
Interaction details
Docking score (S)
Ligands
Receptor
Distance
E cal/mol
residue
NFZ
O
OH
H-accepter
2.63
−2.3
TYR344
−8.2098
1
N1
OD2
H-donor
4.14
−1.8
ASP349
−9.0991
N5
6-ring
π-H
4.34
−0.8
TYR71
2
C15
OD2
H-donor
2.84
−0.1
ASP349
−10.8071
N2
ND2
H-accepter
3.49
−0.1
ASN347
O4
CE1
H-accepter
2.44
−0.1
HIS348
O13
CA
H-accepter
3.18
-O.5
PHE300
3
C15
OD2
H-donor
3.12
−0.5
ASP349
−9.0012
4
CL22
OE2
H-donor
4.05
−0.3
GLU304
−9.2390
O14
CB
H-accepter
2.91
−0.1
PHE177
CL22
CB
H-accepter
3.30
−0.1
PHE300
6-ring
CD
π-H
4.23
−0.5
ARG312
5
N1
OE4
H-donor
2.89
−9.4
GLU276
−10.5777
O4
ND2
H-accepter
3.23
−2.2
ASN347
6
N5
ODI
H-donor
2.16
−3.1
ASN347
−11.7045
O14
NH1
H-donor
2.71
−0.1
ARG439
O4
ND2
H-accepter
1.79
−4.1
ASN347
Cl23
CD
H-accepter
2.84
−3.8
ARG439
7
C15
OD1
H-donor
3.03
−1.2
ASP214
−11.5945
O4
NH2
H-accepter
2.93
−0.3
ARG212
O4
NE2
H-accepter
2.53
−3.6
HIS348
O13
CB
H-accepter
3.36
−0.1
PHE300
8
N5
OD1
H-donor
3.19
−1.4
ASN347
−9.1643
O4
ND2
H-accepter
2.55
−2.5
ASN347
9
C6
OD1
H-donor
3.25
−0.3
ASN347
−10.6506
O4
NE2
H-accepter
3.31
−2.6
HIS348
O14
OH
H-accepter
3.23
−1.6
TYR344
6-ring
CE1
π-H
3.85
−0.3
PHE158
10
5-ring
CD
π-H
3.75
−0.6
ARG312
−8.2309
11
O4
ND2
H-accepter
2.52
−2.8
ASN347
−8.9435
12
N5
ODI
H-donor
3.25
−1.1
ASN347
−11.5231
C15
ODI
H-donor
3.31
−0.0
ASN347
O4
NH2
H-accepter
3.23
−2.1
ARG212
O4
CZ
H-accepter
3.85
−0.1
PHE300
13
N1
O
H-donor
2.73
−10.9
ASP408
−9.9815
C15
OD2
H-donor
3.11
−0.1
PHE157
C16
OD2
H-accepter
3.34
−0.1
ARG312
C21
O
H-accepter
2.74
−4.2
ARG312
14
O14
CE1
H-accepter
2.72
−0.6
HIS111
−9.5983
5-ring
6-ring
π-π
3.84
−0.0
PHE177
15
N1
O
H-donor
3.40
−0.2
ASP349
−10.9647
N5
O
H-donor
3.23
−1.4
ASP349
C17
OE1
H-donor
3.95
−0.1
GLN350
O7
CZ
H-accepter
3.99
−0.1
PHE158
16
O4
NE2
H-accepter
3.60
−1.0
HIS348
−9.1089
5-ring
6-ring
π-π
4.00
−0.0
PHE300
17
5-ring
6-ring
π-π
3.93
−0.0
PHE177
−8.8934
18
C6
OE1
H-donor
3.06
−2.2
GLU276
−9.0012
5-ring
ND2
π-H
4.90
−0.5
ASN347
19
N1
O
H-donor
3.70
−3.3
PHE157
−12.0905
N1
CD1
H-acceptor
3.81
−0.1
PHE157
O14
CD2
H-acceptor
3.23
−0.1
LEU218
O4
N
H-acceptor
1.9
−4.4
ARG312
C22
OD1
H-donor
3.0
−0.1
ASN347
C21
O
H-donor
3.6
−0.1
ASP349
C15
6-ring
H-π
4.41
−0.1
TYR313
C20
6-ring
H-π
4.42
−0.2
PHE300
C22
6-ring
H-π
3.98
−0.7
PHE300
5-ring
CD2
π-H
4.38
−0.1
PHE300
20
O31
OH
H-accepter
2.87
−0.6
TYR344
−9.1220
5-ring
6-ring
π-π
2.86
−0.0
PHE300
We have enlisted the synthetic compounds into two categories based on the substituted groups, category-A bearing the benzene (aryl) substitution, while Category-B holds the alkyl substitutions. Among both the categories, we have noticed several potent compounds compared among the categories. Also, we have observed that changing the position of the substituted group ultimately affects the enzymatic activity. The magnitude of activation of the alkyl group is more moderate than the compounds that hold the deactivated group, even though they are di-substituted, i.e., compound 5–7, and 8 which holds two deactivated groups at different positions of the benzene ring. The magnitude of deactivation of these halogen groups is much weaker than the alkyl group, which could donate most of their electronic density to the π-system, while the deactivation groups, which could withdraw most of the electronic density from the π-system, resulting in the partial positive charge on the ring, and hence remain unstable and this way the enzymatic activity remains less than the compound that holds the activated group and resulting donating electronic density, hence the access of the π-system are richer and enhance the enzymatic activity overall. Figure-11 (Supp. inf.) displays the mode of interaction of the most active compounds.
We also noticed that the compounds substituted with the alkyl groups showed less activity on a gradual pattern, i.e., compounds 13–14, 16–18, and 20, while compound 19 which has 9 carbon showed the highest potential. These results delineate that the compound holding the alkyl group by the pattern (=>6 < 9) showed the highest potential than other compounds. The increase in the carbon quantity does matter, due to the active site of the enzyme, which couldn’t accommodate this high-profile compound, i.e., compound 20 (20 carbons). The highest potential of ranked first compound might be due to the well-accommodated pattern of binding in the active site of the enzyme. In general, the molecular docking results explain that the compounds that hold the donating groups showed the best potential than the compounds that hold withdrawing groups. Also, we have noticed that compounds that hold longer carbon chain at their substituted position (compound 20) were not well accommodated in the active site, resulting in less activity than the compound that holds a similar group i.e., alkyl but with less no. of carbons (compound 19).
2.8 Inspecting the stability of α-glucosidase in presence of potent compound
Molecular docking results revealed a fit-well binding pattern of compound 19 with the adaptation of several key interactions with active site residues. Furthermore, we have performed molecular dynamics (MD) simulation, to show the stability of the overall complex (α-glucosidase + c19) in an explicit watery environment. Generally, it was observed that the compound showed an inverted V-like illustration in the active site of the enzyme, where one end of the compound (the alkyl group) of the compound remains stick with the key residues and the other side stick with other residues and the overall active site of the compound represent a cave-like structure. For the ease of clarification and support of dynamics results, we further analyzed the dynamics trajectory.
The deviation of backbone atoms was examined by root-mean-square-deviation (RMSD). The RMSD of the system relative to the original structures illustrates that 50 ns simulation time is adequate to attain equilibration at temperature 310 K (Fig. 12A). The low RMSD curve supports the high stability of the conformation and vice versa. Initially, the RMSD curve gradually goes upward and oscillates after 37 ns simulation time, hence we have extracted that conformation, and were compared it with the rest of the conformations extracted from the later simulation time. Interestingly, we have found both the conformations the same, but solely found differences in the 6-AF conformation, where the alkyl group over the benzene ring rotates by 10˚. The RMSD curve indicates the stabilized behaviors of the overall complex, wherein most of the simulation time the c19 resides tightly with the bonded residues in the active site. This tight binding to the active site indicates a crucial role of c19 in protein stability, which rescues the protein from activation. To clarify further the specificity of this compound bonding in the active site dynamically. We have analyzed the root-mean-square-fluctuation (RMSF). The results indicate a high fluctuation in the active site where this compound bonded with several key residues, and some local fluctuation was observed in the terminal region of the enzyme, where a bunch of single loops fluctuated most of the simulation time (Fig. 12B). These results delineate that this compound binding stabilized the overall conformation of the protein and might rescue them from activation.
2.9 Cytotoxicity and carcinogenicity prediction
All the twenty compounds were predicted as non-cytotoxic and the probability of the cytotoxicity is described in Table 3. Furthermore, the carcinogenicity was also evaluated by the ProtoxII server. Out of 20 compounds, a total of seven compounds 1, 3, 4, 9, 10, 11, and 12 were predicted as carcinogenic with the probability scores of carcinogenicity 0.88, 0.65, 0.65, 0.69, 0.89, 0.89, and 0.62, respectively. All the remaining compounds were predicted as non-carcinogenic.
C. No.
Carcinogenicity
Cytotoxicity
Probability of carcinogenicity
Probability of cytotoxicity
1
Active
Inactive
0.88
0.91
2
Inactive
Inactive
0.50
0.85
3
Active
Inactive
0.65
0.86
4
Active
Inactive
0.65
0.86
5
Inactive
Inactive
0.50
0.85
6
Inactive
Inactive
0.50
0.85
7
Inactive
Inactive
0.50
0.85
8
Inactive
Inactive
0.56
0.83
9
Active
Inactive
0.69
0.60
10
Active
Inactive
0.89
0.91
11
Active
Inactive
0.89
0.91
12
Active
Inactive
0.62
0.89
13
Inactive
Inactive
0.55
0.79
14
Inactive
Inactive
0.54
0.81
15
Inactive
Inactive
0.65
0.81
16
Inactive
Inactive
0.52
0.8
17
Inactive
Inactive
0.58
0.78
18
Inactive
Inactive
0.58
0.78
19
Inactive
Inactive
0.58
0.78
20
Inactive
Inactive
0.58
0.78
2.10 Conclusion
In conclusion, twenty (1–20) different nitrofurazone derivatives were synthesized by using alkyl/benzyl halides. Out of twenty derivatives, fourteen were new, while six have been reported previously. All synthetic compounds showed higher α-glucosidase inhibition than nitrofurazone. The synthetic molecules were divided into two categories based on their substitution, among benzyl derivatives, the chloro substituted derivatives 2, 4, 6, and 7 showed potential inhibition against the α-glucosidase enzyme, which might be associated with the position and number of chloro groups helping these molecules to attain the best position to fit in the active pocket of the enzyme. Nevertheless, the alkyl-substituted compounds 15 and 19 also displayed good α-glucosidase inhibition, and compound 19 was the most active compound of the series. The binding conformation of most active compounds was determined by a molecular docking study. The docking studies revealed that structural features of active compounds play a vital role in the binding interactions of compounds with the enzyme. The kinetic studies of most active compounds 7 and 19 showed that these compounds exhibit a competitive and uncompetitive mode of inhibition, respectively. The synthetic molecules were also examined for cytotoxicity and carcinogenicity. All the compounds were predicted as non-cytotoxic, however, few compounds 1, 3, 4, 9, 10, 11, and 12 were predicted as carcinogenic. The current study has identified new and potential α-glucosidase inhibitors which may act as lead compounds for drug discovery and research. However, further studies must be carried out to evaluate their cytotoxic effect thus confirming them as potential lead molecules or drug candidates.
3 Experimental
3.1 Materials and method
Dried glasswares were used for all the reactions, chemical reagents were purchased either from E-Merck or Sigma-Aldrich (USA). Evaporation of organic solvent was carried out by rotatory evaporator (Büchi rotavapor 210) with a water bath temperature of 35 °C. Nitrofurazone was provided by Nabi Qasim Pharmaceutical Industries Pvt. TLC (thin layer chromatography) was performed on Merck Kieselgel 60 F254 silica gel sheets. Visualizations of spots were carried out with UV light (254 and 366 nm) and iodine vapors. Melting points (m.p.) were measured by Büchi melting point M.560 apparatus in open glass capillary tubes and are uncorrected. EI-MS and HR-EIMS spectra were recorded on JEOL JMS 600H-1 and JEOL JMS-HX-110, respectively. Bruker Avance spectrophotometer 400/100 MHz was used for 1H NMR and 13C NMR, respectively. Chemical shifts (δ) are in ppm and coupling constants (J) are in Hz.
3.2 General synthetic procedure for compounds 1–20
In a 100 mL round-bottomed flask, nitrofurazone (2 mmol) was dissolved in DMF (10 mL), K2CO3 (1.2 equivalent) was also added and the reaction mixture was stirred for 10 min, then alkyl/ substituted benzyl halides (2 mmol) were added and it was refluxed for 6–12 h. The reaction progress was monitored through TLC (Hex: EtOAc, 1:1). When the reaction was completed, the reaction mixture was filtered to separate K2CO3 and the filtrate was poured into distilled water. The product was extracted by using dichloromethane. Anhydrous sodium sulfate (Na2SO4) was used to remove water molecules from organic, further, it was evaporated in vacuo and washed with ethyl acetate to afford the pure product (1–20) (Khan et al., 2018).
3.3 α-Glucosidase inhibition assay
α-Glucosidase inhibition assay was performed following the literature procedure (Solangi et al., 2020) Briefly, the solutions of α-glucosidase (Saccharomyces cerevisiae 2.5 U Ml-1 purchased from Sigma-Aldrich, USA) and the substrate, p-nitrophenyl α-D-glucopyranoside (p-NPG) were prepared in 0.07 M phosphate buffer (pH 6.8). The buffer (70 μL) was pre-incubated along with 10 μL of the enzyme and 10 μL of the test compound at 37 °C for 5 min. The 96 well plates were further incubated for 30 min at the same temperature after the addition of 10 μL of p-NPG (10 mM) to each well. 80 μL Na2CO3 solution (0.2 M) was added to quench the reaction. The test compounds were not added in negative control wells, however, 10 μL of buffer was additionally added to them. Acarbose was used as the positive control. The absorbance was measured at a wavelength of 405 nm using a microplate reader (Bio-Tek ELx 800™, Winooski, USA) which determined the activity of test compounds against α-glucosidase. The following formula was used for the calculation of percent inhibition.
Dose-response curves of potential inhibits (≥50%) were obtained and IC50 was determined with the help of the GraphPad Prism 5.0 Software Inc., San Diego, California, USA.
3.4 Molecular docking (MD) protocol
Molecular docking studies were carried out to explore the inhibition mechanism of nitrofurazone derivatives against the α-glucosidase enzyme. The α-glucosidase model used in this study was previously reported by our group (Chemical Computing Group (CCG) Inc. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2019). The chemical structure of nitrofurazone derivatives was built by Molecular Operating Environment (MOE) version 2019.0137 builder module and minimized by MMFF94x forcefield. MOE-dock Program was used to explore the binding modes of each molecule. The stability of molecules was ensured by analyzing hydrogen bonding and hydrophobic interactions of molecules with the respective target through MOE-Protein Ligand Interaction Fingerprint (PLIF) Program.
3.5 Molecular dynamic simulation
First, the generalized Amber force field (GAFF) (Wang et al., 2004) was used to parameterize c19. Single-point energy calculations utilizing Schrodinger's quantum chemistry module, Jaguar, employing the Hartree-Fock level of theory and the 6–311 g** basis set established the partial atomic charges. The Antechamber module (Wang et al., 2006) was used to assign the GAFF atom types, and the AMBER LEaP module was used to generate the parameter file (Delano, 2010). AMBER version 2018 (Case et al., 2018) was used to perform all-atom MD simulations and essential dynamics analyses. Hydrogen atoms were included in the homology model of the enzyme using the LEaP module. Counter ions were then introduced to keep the system neutral. The system was solvated in the TIP3P water model truncated octahedral box with a cut-off of 10.0 Å buffer. All MD simulations were accelerated using the CUDA version of PMEMD. The detail molecular dynamics simulation methodology has been described in detail in our previous study (Rehman et al., 2020; Rehman et al., 2019; Rehman et al., 2019; Rehman et al., 2021).
3.6 Kinetics of α-glucosidase inhibition
To find out inhibition mode (competitive, uncompetitive, non-competitive, or mixed) of the most potent compounds against α-glucosidase, the enzyme kinetics study was performed according to the described protocol by Elahabaadi et al. with slight modification (Elahabaadi et al., 2021). Compounds 7 and 19 having IC50 0.65 ± 0.22 and 0.54 ± 0.30 µM, respectively, were selected for this study. These compounds were tested with concentrations of 0, 0.3, 0.6, and 0.9 µM against enzyme at various concentrations of p-nitrophenyl α-D-glucopyranoside (4–20 mM). The reaction mixture was incubated at 37 °C for 30 min and the reading was taken at a wavelength of 405 nm using a microplate reader (Bio-Tek ELx 800™, Winooski, USA). Lineweaver–Burk's plots were obtained with the help of GraphPad prism 5.0 Software Inc., San Diego, California, USA.
3.7 Carcinogenicity and cytotoxicity prediction
Carcinogens are chemicals that can cause tumors or develop the occurrence of tumors (Zhang et al., 2014). The ability to predict cytotoxicity is critical for screening chemical compounds that might cause the undesired as well as the desired cell harm, the latter being particularly relevant in the case of tumor cells (Banerjee et al., 2018). The ProTox-II online server was used to predict the carcinogenicity of the compounds. The ProTox-II is a simple and self-explanatory user interface server. The name of the compound or the SMILES string of the compound is required as input to estimate the probable toxicities of the compound. The chemical editor (https://www.chemdoodle.com/) also allows the user to draw the structure of the compounds. The ProTox-II server was used to predict the carcinogenicity and cytotoxicity of each compound (Kroes et al., 2004).
3.8 N-(Benzyl)-2-((5-nitrofuran-4-yl)methylene)hydrazine Carboxamide (1)
Compound 1 was obtained as yellow amorphous powder with 66% yield. Rf value = 0.46 (Hex:EtOAc, 1:1); Melting point; 167–168 °C; 1H NMR: (400 MHz, DMSO‑d6): δH 7.74 (d, 1H, J2, 3 = 4.0 Hz, H-2), 7.52 (s, 1H, H-5), 7.40 (d, 1H, J3,2 = 4.0 Hz, H-3), 7.36 (d, 2H, J2′,6′ = 7.2 Hz, H-2ʹ, H-6ʹ), 7.26 (m, 4H, H-3ʹ, H-4ʹ, H-5ʹ, NH-1), 3.16 (s, 1H, NH-2), 5.18 (s, 2H, CH2). EI-MS: m/z (rel. abund. %) 288 (M+, 18), 245 (56), 202 (20), 154 (33), 149 (85), 106 (43), 91 (1 0 0), 79 (23), 51 (12). HREI-MS: m/z Calcd. for C13H12N4O4 288.0859, Found 288.0849.
3.9 N-(2ʹ-Chlorobenzyl)-2-((5-nitrofuran-2-yl) methylene)hydrazinecarboxamide (2)
Compound 2 was obtained as pale-yellow amorphous powder with 62% yield. Rf value = 0.34 (Hex:EtOAc, 1:1); Melting point; 198–199 °C; 1H NMR: (400 MHz, MeOD-d): δH 7.80 (s, 1H, H-5), 7.51 (d, 1H, J2,3 = 4.0 Hz, H-2), 7.39 (t, 1H, J5′(4′/6′) = 7.7 Hz, H-5ʹ), 7.29 (d, 1H, J3′, 6′ = 1.6 Hz, H-3ʹ), 7.26 (t, 1H, J4′(3′/5′) = 1.6 Hz, H-4ʹ), 7.24 (d, 1H, J6′,3′ =1.6 Hz, H-6ʹ), 7.04 (d, 1H, J3,2 = 4.0 Hz, H-3), 4.56 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 154.9 (C = O), 152.8 (C-1), 151.3 (C-4), 137.0 (C-1ʹ), 131.5 (C-3ʹ), 129.0 (C-6ʹ), 128.3 (C-2ʹ), 128.1 (C-5ʹ), 127.1 (C-5), 115.1 (C-2), 112.0 (C-3), 40.46 (CH2). EI-MS: m/z (rel. abund. %) 323 (M+, 1), 305 (9), 287 (98), 276 (2), 255 (3), 181 (5), 167 (8), 155 (1 0 0), 140 (25), 132 (26), 125 (89), 106 (29), 89 (19), 79 (23), 51 (20). HREI-MS: m/z Calcd. for C13H11ClN4O4 322.0469, Found 322.0455.
3.10 N-(3ʹ-Chlorobenzyl)-2-((5-nitrofuran-2-yl) methylene)hydrazinecarboxamide (3)
Compound 3 was obtained as pale-yellow amorphous powder with 64% yield. Rf value = 0.47 (Hex:EtOAc, 1:1); Melting point; 229–230 °C; 1H NMR:(400 MHz, MeOD-d): δH (s, 1H, H-5), 7.51 (d, 1H, J2, 3 = 4.0 Hz, H-2), 7.34 (s, 1H, NH-1), 7.29 (s, 1H, H-2ʹ), 7.27 (d, 1H, J4′,5′ = 5.2 Hz, H-4ʹ), 7.24 (d, 1H, J6′,4′ = 2.0 Hz, H-6ʹ), 7.22 (d, 1H, J5′,4′ = 7.6 Hz, H-5ʹ), 4.44 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 154.9 (C = O), 152.8 (C-1), 143.0 (C-1ʹ), 132.8 (C-3ʹ), 130.1 (C-5), 127.9 (C-2ʹ), 126.8 (C-6ʹ), 126.5 (C-4ʹ), 125.7 (C-5ʹ), 115.1 (C-2), 112.6 (C-3), 42.0 (s, 2H, CH2). EI-MS: m/z (rel. abund. %) 322 (M+, 9), 183 (3), 169 (93), 155 (1 0 0), 140 (34), 132 (70), 125 (81), 111 (9), 106 (30), 96 (4), 89 (13), 77 (15), 51 (10). HR-EIMS: m/z Calcd. for C13H11ClN4O4 322.0469, Found 322.0467.
3.11 N-(4ʹ-Chlorobenzyl)-2-((5-nitrofuran-2-yl)methylene)hydrazine Carboxamide (4)
Compound 4 was obtained as pale-yellow amorphous powder with 66% yield. Rf value = 0.46 (Hex:EtOAc, 1:1); Melting point; 269–270 °C; 1H NMR: (400 MHz, MeOD-d): δH 7.86 (s, 1H, NH-1), 7.50 (s, 1H, H-5), 7.48 (d, 2H, J2′,6′ = 3′,5′ = 8.4 Hz, H-3ʹ, H-5ʹ), 7.46 (d, 1H, J2,3 = 3.6 Hz, H-2), 7.17 (d, 2H, J2′,6′ = 3′,5′ = 8.4 Hz, H-2ʹ, H-6ʹ), 7.01 (d, 1H, J3,2 = 4.0 Hz, H-3), 5.19 (s, 1H, NH-2), 4.55 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 154.9 (C = O), 152.9 (C-1), 151.3 (C-4), 139.4 (C-1ʹ), 131.1 (C-4ʹ), 128.9 (C-3ʹ, C-5ʹ), 128.1 (C-2ʹ, C-6ʹ), 127.8 (C-5), 115.1 (C-2), 112.5 (C-3), 41.9 (CH2). EI-MS: m/z (rel. abund. %) 322 (M+, 5), 305 (34), 181 (4), 166 (20), 155 (77), 140 (8), 79 (8), 51 (8), 44 (9). HREI-MS: m/z Calcd. for C13H11ClN4O4 322.0469, Found 322.0466.
3.12 N-(2ʹ,4ʹ-Dichlorobenzyl)-2-((5-nitrofuran-2-yl) methylene)hydrazine carboxamide (5)
Compound 5 was obtained as pale-yellow amorphous powder with 72% yield. Rf value = 0.44 (Hex:EtOAc, 1:1); Melting point; 236–237 °C; 1H NMR: (400 MHz, DMSO‑d6): δH 7.80 (s, 1H, H-5), 7.76 (d, 1H, J3′,5′ = 4.0 Hz, H-3ʹ), 7.69 (d, 1H, J5′,3′ = 2.0 Hz, H-5′), 7.43 (d, 2H, J6′,5′ = 5.0 Hz, H-6ʹ), 7.21 (s, 2H, NH-1, NH-2), 6.81(d, 1H, J3,2 = 8.4 Hz, H-3), 5.15 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 154.9 (C = O), 152.7 (C-1), 136.4 (C-4), 132.4 (C-4), 132.4 (C-1′), 131.9 (C-2′), 129.5 (C-4), 128.4 (C-3′), 128.2 (C-5′), 127.7 (C-6′), 127.3 (C-5), 115.1 (C-2), 112.8 (C-3), 40.1 (CH2). EI-MS: m/z (rel. abund. %) 357 (M+, 1), 341 (16), 321 (32), 201 (25), 159 (1 0 0), 155 (94), 140 (63), 79 (35), 51 (41), 43 (8). HR-EIMS: m/z Calcd. for C13H10Cl2N4O4 356.0079, Found 356.0098.
3.13 N-(2ʹ,6ʹ-Dichlorobenzyl)-2-((5-nitrofuran-2-yl)methylene)hydrazine Carboxamide (6)
Compound 6 was obtained as yellow amorphous powder with 61% yield. Rf value = 0.45 (Hex:EtOAc, 1:1); Melting point; 240–241 °C; 1H NMR: (400 MHz, MeOD-d): δH 7.80(s, 1H, H-5), 7.51 (d, 1H, J2,3 = 4.0 Hz, H-2), 7.39 (t, 2H, J4′(3′/5′) = 7.2 Hz, H-4ʹ, NH-1), 7.28 (m, 2H, H-3ʹ, H-5ʹ), 7.04 (d, 1H, J3,2 = 4.0 Hz, H-3), 5.46 (s, 1H, NH-2), 4.56 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 154.0 (C = O), 152.5 (C-1), 151.3 (C-4), 135.3 (C-1′), 133.8 (C-2′), 130.1 (C-5), 128.7 (C-3′, C-5′), 128.1 (C-4′), 115.0 (C-2), 113.0 (C-3), 39.40 (CH2). EI-MS: m/z (rel. abund. %) 357 (M+, 1), 341 (2), 321 (32), 201 (25), 174 (26), 166 (71), 159 (1 0 0), 155 (94), 140 (63), 123 (22), 111 (19), 79 (35), 70 (19), 51 (1), 43 (8). HREI-MS: m/z Calcd. for C13H10Cl2N4O4 356.0079, Found 356.00160.
3.14 (3ʹ,4ʹ-Dichlorobenzyl)-2-((5-nitrofuran-2-yl)methylene)hydrazine carboxamide (7)
Compound 7 was obtained as yellow amorphous powder with 69% yield. Rf value = 0.46 (Hex:EtOAc, 1:1); Melting point; 236–237 °C; 1H NMR: (400 MHz, MeOD-d): δH 8.30 (s, 1H, H-3ʹ), 7.54 (s, 1H, H-5), 7.43 (dd, 3H, J3′,5′ = 8.8 Hz, H-3ʹ, H-5ʹ, NH-1), 7.34 (d, 1H, J2,3 = 3.6 Hz, H-2), 7.19 (dd, 1H, J2′,6′ = 8.4 Hz, H-6ʹ), 6.79 (d, 1H, J3,2 = 4.0 Hz, H-3), 5.27 (s, 1H, NH-2), 4.50 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 154.9 (C = O), 152.7 (C-1), 141.7 (C-1ʹ), 130.4 (C-5), 129.0 (C-3ʹ), 129.0 (C-4ʹ), 128.08 (C-2ʹ, C-5ʹ), 127.4 (C-6ʹ), 115.1 (C-2), 112.7 (C-3), 41.6 (CH2). EI-MS: m/z (rel. abund. %) 357 (M+, 1), 339 (4), 201 (34), 174 (25), 166 (1 0 0), 159 (76), 140 (27), 123 (24), 109 (11), 89 (10), 75 (14), 51 (14), 44 (7), HREI-MS: m/z Calcd. for C13H10Cl2N4O4 356.0079, Found 356.0098.
3.15 N-(2ʹ-Chloro-4ʹ-flourobenzyl)-2-((5-nitrofuran-2-yl) methylene) hydrazine carboxamide (8)
Compound 8 was obtained as brown amorphous powder with 77% yield. Rf value = 0.46 (Hex:EtOAc, 1:1); Melting point; 198–199 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.31 (d, 1H, J2, 3 = 3.6 Hz, H-5), 7.18 (dd, 1H, J5′,3′ = 2.4 Hz, J5′,6′ = 8.4 Hz, H-5ʹ), 7.11 (s, 1H, H-5), 6.98 (dd, 1H, J6′,3′ = 2.4 Hz, J6′,5′ = 8.4 Hz, H-6ʹ), 6.95 (dd, 1H, J3′,5′ = 2.4 Hz, J3′,6′ = 7.6 Hz, H-3ʹ), 6.72 (d, 1H, J3,2 = 4.0 Hz, H-3), 4.55 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δH 163.6 (C-4′), 160.2 (C=O), 155.9 (C-1), 151.7 (C-4), 149.6 (C-1ʹ), 133.1 (C-2ʹ), 128.4 (C-6ʹ), 125.4 (C-5), 117.6 (C-3ʹ), 115.0 (C-5ʹ), 14.7 (C-2), 113.1 (C-3), 42.3 (CH2). EI-MS: m/z (rel. abund. %) 340 (M+, 30), 305 (26), 297 (80), 254 (26), 201 (52), 167 (28), 158 (33), 143 (1 0 0). HREI-MS: m/z Calcd. for C13H10ClFN4O4 340.0375, Found 340.03806.
3.16 N-(4ʹ-Bromobenzyl)-2-((5-nitrofuran-2-yl)methylene)hydrazine Carboxamide (9)
Compound 9 was obtained as brown amorphous powder with 63% yield. Rf value = 0.47 (Hex:EtOAc, 1:1); Melting point; 235–236 °C;1H NMR: (400 MHz, MeOD-d): δH 7.50 (d, 2H, J2′,6′ = 3′,5′ = 8.4 Hz, H-3ʹ, H-5ʹ), 7.48 (s, 1H, NH-1), 7.46 (d, 1H, J2,3 = 3.6 Hz, H-2), 7.17 (d, 2H, J3′,5′ = 2′,6′ = 8.4 Hz, H-2ʹ, H-6ʹ), 7.01 (d, 1H, J3,2 = 4.0 Hz, H-3), 5.19 (s, 1H, NH-2), 4.55 (s, 2H, CH2). 13C NMR: (75 MHz, DMSO‑d6): δC 155.8 (C = O), 153.4 (C-1), 151.4 (C-4), 134.8 (C-1ʹ), 131.5 (C-3ʹ, C-5ʹ), 128.9 (C-2ʹ, C-6ʹ), 125.2 (C-4ʹ), 120.2 (C-5), 115.2 (C-2), 111.2 (C-3), 43.1 (CH2). EI-MS: m/z (rel. abund. %) 368 (M+, 4), 323 (11), 291 (2), 282 (7), 229 (53), 184 (27), 169 (1 0 0), 154 (10), 132 (4), 90 (23), 79 (8), 63 (4), 51 (7), 44 (2). HREI-MS: m/z Calcd. for C13H11BrN4O4 365.9964, Found 365.9971.
3.17 N-(3-Methylbenzyl)-2-((5-nitrofuran-2-yl)methylene)hydrazine Carboxamide (10)
Compound 10 was obtained as yellow amorphous powder with 71% yield. Rf value = 0.47 (Hex:EtOAc, 1:1); Melting point ; 189–190 °C; 1H NMR: (400 MHz, DMSO‑d6): δH 7.74 (d, 1H, J2, 3 = 4.0 Hz, H-2), 7.49 (s, 1H, H-5), 7.40 (d, 1H, J3,2 = 4.0 Hz, H-3), 7.22 (t, 2H, J5′(4′/6′) = 7.6 Hz, H-5ʹ, NH-1), 7.06 (d, 1H, J2′,4′ =7.6 Hz, H-2ʹ), 7.01 (d, 2H, J4′,6′ = 11.2, H-4ʹ, H-6ʹ), 6.51 (s, 1H, NH-2), 5.14 (s, 2H, CH2), 2.26 (s, 3H, aromatic CH3). 13C NMR: (75 MHz, DMSO‑d6): δC 156.2 (C = O), 153.8 (C-1), 138.2 (C-4), 135.5 (C-1ʹ), 128.9 (C-3ʹ), 128.2 (C-2ʹ), 127.3 (C-5ʹ), 125.3 (C-4ʹ), 123.9 (C-5) 115.6 (C-2), 111.6 (C-3), 44.0 (CH2), 21.3 (aromatic CH3). EI-MS: m/z (rel. abund. %) 302 (M+, 2), 259 (32), 240 (2), 225 (4), 200 (3), 163 (17), 138 (3), 120 (13), 57 (6), 43 (12). HREI-MS: m/z Calcd. for C14H14N4O4 302.1015, Found 302.1006.
3.18 N-(4-Methylbenzyl)-2-((5-nitrofuran-2-yl)methylene)hydrazine carboxamide (11)
Compound 11 was obtained as yellow amorphous powder with 86% yield. Rf value = 0.47 (Hex:EtOAc, 1:1); Melting point; 185–186 °C; 1H NMR: (500 MHz, MeOD-d): δH 8.54 (s, 1H, H-5), 7.44 (d, 1H, J2,3 = 4.0 Hz, H-2), 7.22 (d, 2H, J2′,6′=3′,5′ = 8.4 Hz, H-2ʹ, H-6ʹ), 7.48 (s, 1H, NH-1), 7.17 (d, 2H, J3′,5′=2′,6′ = 8.4 Hz, H-3ʹ, H-5ʹ), 6.22 (d, 1H, J3,2 = 4.0 Hz, H-3), 5.19 (s, 1H, NH-2), 4.41 (s, 2H, CH2), 2.31 (s, 3H, aromatic CH3). 13C NMR: (75 MHz, DMSO‑d6): δC 158.5 (C = O), 154.0 (C-1), 151.3 (C-4), 135.4 (C-1ʹ), 129.0 (C-3ʹ, C-5ʹ), 127.7 (C-2ʹ, C-6ʹ), 127.8 (C-5), 125.1 (C4ʹ), 118.3 (C-2), 116.3 (C-3), 50.4 (CH2), 20.6 (aromatic CH3). EI-MS: m/z (rel. abund. %) 302.2 (M+, 2), 259 (32), 240 (2), 225 (4), 200 (3), 163 (17), 138 (3), 120 (13), 57 (6), 43 (12). HREI-MS: m/z Calcd. for C14H14N4O4 302.1015, Found 302.1008.
3.19 N-(3ʹ-Methoxybenzyl)-2-((5-nitrofuran-2-yl) methylene)hydrazine carboxamide (12)
Compound 12 was obtained as yellow amorphous powder with 74% yield. Rf value = 0.44 (Hex:EtOAc, 1:1); Melting point; 199–200 °C; 1H NMR: (400 MHz, MeOD-d): δH 7.80 (s, 1H, H-5), 7.75 (d, 1H, J2,3 = 3,2 = 4.0 Hz, H-2), 7.51 (s, 1H, H-2′), 7.40 (d, 1H, J 3,2 = 2,3 = 4.0 Hz, H-3), 7.27 (t, J5′(4′/6′) = 8.0 Hz, H-5′), 7.11 (s, 1H, NH-1), 6.82 (s, 1H, NH-2), 6.80 (t, 1H, J6′(5′/2′) = 8.0 Hz, H-6ʹ), 6.72 (d, 1H, J4′,5′ = 7.6 Hz, H-4ʹ), 5.14 (s, 2H, CH2), 3.71 (s, 3H, aromatic OCH3). 13C NMR: (75 MHz, DMSO‑d6): δC 159.5 (C=O), 155.9 (C-1), 153.4 (C-4), 136.9 (C-1ʹ), 129.8 (C-5), 125.2 (C-5ʹ), 118.5 (C-6ʹ), 115.2 (C-2), 112.6 (C-4ʹ), 112.3 (C-3), 111.4 (C-2ʹ), 54.9 (aromatic OCH3), 43.6 (CH2), EI-MS: m/z (rel. abund. %) 318 (M+, 4), 275 (11), 179 (52), 154 (4), 136 (33), 121 (1 0 0), 91 (18), 77 (5), 51 (4). HREI-MS: m/z Calcd. for C14H14N4O5 318.0964, Found 318.0955.
3.20 2-((5-Nitrofuran-2-yl)methylene)-N-ethyl hydrazine carboxamide (13)
Compound 13 was obtained as yellow amorphous powder with 69% yield. Rf value = 0.48 (Hex:EtOAc, 1:1); Melting point; 155–156 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.42 (s, 1H, H-5), 7.36 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 4.02 (q, 2H, J = 7.2 Hz, CH2), 1.19 (t, 3H, J = 6.8 Hz, CH3). EI-MS: m/z (rel. abund. %) 226 (M+, 30), 210 (2), 183 (92), 168 (1 0 0), 154 (35), 138 (6), 121 (30), 109 (15), 93 (26), 79 (71), 67 (13), 51 (50), 42 (15).
3.21 2-((5-Nitrofuran-2-yl)methylene)-N-propyl hydrazine carboxamide (14)
Compound 14 was obtained as yellow amorphous powder with 83% yield. Rf value = 0.47 (Hex:EtOAc, 1:1); Melting point; 184–185 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.39 (s, 1H, H-5), 7.36 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 3.91 (t, 2H, J = 7.6 Hz, H-1ʹa,b), 1.62 (m, 2H, H-2ʹa, b), 0.97 (t, 3H, J = 7.3 Hz, CH3). EI-MS: m/z (rel. abund. %) 240 (M+, 13), 197 (26), 168 (1 0 0), 154 (11), 128 (6), 121 (9), 93 (7), 79 (9), 63 (7), 51 (5), 43 (7), 4 (5).
3.22 2-((5-Nitrofuran-2-yl) methylene)-N-butyl hydrazine carboxamide (15)
Compound 15 was obtained as yellow amorphous powder with 79% yield. Rf value = 0.44 (Hex:EtOAc, 1:1); Melting point; 184–185 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.39 (s, 1H, H-5), 7.36 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 3.91 (t, 2H, J = 7.6 Hz, H-1ʹa, b), 1.62 (m, 4H, H-2ʹa,b, H-3ʹa,b), 0.97 (t, 3H, J = 7.3 Hz, CH3). EI-MS: m/z (rel. abund. %) 269 (M+, 13), 197 (26), 168 (100), 154 (11), 128 (6), 121 (9), 93 (7), 79 (9), 63 (7), 51 (5), 43 (7), 4 (5).
3.23 2-((5-Nitrofuran-2-yl)methylene)-N-but-3-enyl hydrazine carboxamide (16)
Compound 16 was obtained as yellow crystalline form with 83% yield. Rf value = 0.46 (Hex:EtOAc, 1:1); Melting point; 135–136 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.41(s, 1H, H-5), 7.37 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.78 (d, 1H, J3,2 = 4.0 Hz, H-3), 5.82 (m, 1H, H-3ʹ), 5.13 (m, 2H, H-2ʹa,b), 4.02 (t, J = 7.2 Hz, H-1ʹa,b), 2.35 (dd, J = 7.2 Hz, 14.4 Hz, H4ʹa,b). 13C NMR: (75 MHz, DMSO‑d6): δC 155.5 (C = O), 154.2 (C-1), 151.4 (C-4), 135.0 (C-5), 124.6 (C-3′), 116.8 (C-1ʹ), 115.4 (C-2), 111.2 (C-3), 39.4 (C-1ʹ), 29.3 (C-4ʹ). EI-MS: m/z (rel. abund. %) 252 (M+, 2), 208 (3), 206 (17), 168 (100), 149 (14), 121 (5), 93 (5), 55 (5). HREI-MS: m/z Calcd. for C10H12N4O4 252.0859, Found 252.0861.
3.24 2-((5-Nitrofuran-2-yl)methylene)-N-pentyl hydrazine carboxamide (17)
Compound 17 was obtained as yellow amorphous powder with 81% yield. Rf value = 0.45 (Hex:EtOAc, 1:1); Melting point; 199–200 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.39 (s, 1H, H-5), 7.37 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 3.93 (t, 2H, J = 7.6 Hz, H-1ʹa,b), 1.35 (m, 6H, H-2ʹa,b, H-3ʹa,b, H-4ʹa,b), 0.89 (t, 3H, J = 6.8 Hz, CH3). 13C NMR: (75 MHz, DMSO‑d6): δC 155.6 (C = O), 154.3 (C-1), 151.0 (C-4), 124.3 (C-5), 115.5 (C-2), 111.1 (C-3), 40.0 (C-1′), 24.4 (C-3ʹ), 21.9 (C-4ʹ), 13.9 (C-5ʹ). EI-MS: m/z (rel. abund. %) 268 (M+, 56), 251 (8), 225 (16), 208 (28), 191 (8), 182 (12), 168 (1 0 0), 154 (46), 141 (9), 121 (38), 93 (20), 79 (36), 70 (48), 51 (20). HR-EIMS: m/z Calcd. for C11H16N4O4 268.1172, Found 268.11840.
3.25 2-((5-Nitrofuran-2-yl) methylene)-N-hexyl hydrazine carboxamide (18)
Compound 18 was obtained as yellow crystals with 79% yield. Rf value = 0.44 (Hex:EtOAc, 1:1); Melting point; 200–201 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.39 (s, 1H, H-5), 7.37 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 3.93 (t, 2H, J = 7.6 Hz, H-1ʹa,b), 1.31 (m, 8H, H-2ʹa,b, H-3ʹa,b, H-4ʹa,b, H-5ʹa,b), 0.89 (t, 3H, J = 6.8 Hz, CH3). EI-MS: m/z (rel. abund. %) 282 (3), 265 (4), 239 (36), 222 (14), 168 (1 0 0), 154 (25), 121 (42), 70 (39), 55 (20).
3.26 2-((5-Nitrofuran-2-yl) methylene)-N-octyl hydrazine carboxamide (19)
Compound 19 was obtained as yellow amorphous powder with 91% yield. Rf value = 0.47 (Hex:EtOAc, 1:1); Melting point; 66–67 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.38 (s, 1H, H-5), 7.36 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 3.92 (t, 2H, J = 7.6 Hz, H-1ʹa,b), 1.31 (m, 12H, H-2ʹa,b, H-3ʹa,b, H-4ʹa,b, H-5ʹa,b, H-6ʹa,b), 0.87 (t, 3H, J = 6.8 Hz, CH3). 13C NMR: (75 MHz, DMSO‑d6): δC 155.5 (C = O), 154.2 (C-1), 151.0 (C-4), 124.3 (C-5), 115.4 (C-2), 111.1 (C-3), 39.9 (C-1′), 24.4 (C-3′), 21.9 (C-4′), 13.9 (C-5′). EI-MS: m/z (rel. abund. %) 310 (M+, 19), 293 (9), 267 (56), 250 (23), 168 (1 0 0), 154 (31), 138 (26), 121 (58), 93 (30), 79 (35), 70 (58), 55 (21), 41 (37). HR-EIMS: m/z Calcd. for C14H22N4O4 310.641, Found 310.6380.
3.27 2-((5-Nitrofuran-2-yl) methylene)-N-octadecyl hydrazine carboxamide (20)
Compound 20 was obtained as yellow amorphous powder with 85% yield. Rf value = 0.59 (Hex:EtOAc, 1:1); Melting point; 85–86 °C; 1H NMR: (400 MHz, CDCl3-d): δH 7.38 (s, 1H, H-5), 7.36 (d, 1H, J2,3 = 4.0 Hz, H-2), 6.77 (d, 1H, J3,2 = 4.0 Hz, H-3), 3.93 (t, 2H, J = 7.6 Hz, H-1ʹa,b), 1.31 (m, 32H, H-2ʹa,b, H-3ʹa,b, H-4ʹa,b, H-5ʹa,b, H-6ʹa,b, H-7ʹa,b, H-8ʹa,b, H-9ʹa,b, H-10ʹa,b, H-11ʹa,b, H-12ʹa,b, H-13ʹa,b, H-14ʹa,b, H-15ʹa,b, H-16ʹa,b, H-17ʹa,b), 0.87 (t, 3H, J = 6.8 Hz, CH3). 13C NMR: (75 MHz, DMSO‑d6): δC 156.0 (C = O), 154.2 (C-1), 151.3 (C-4), 124.8 (C-5), 115.6 (C-2), 111.7 (C-3), 48.8 (C-1ʹ), 40.3 (C-2ʹ), 31.5 (C-3ʹ), 29.1 (C-4ʹ, C-5ʹ, C-6ʹ, C-7ʹ, C-8ʹ, C-9ʹ, C-10ʹ, C-11ʹ, C-12ʹ, C-13ʹ), 28.9 (C-14ʹ), 26.3 (C-15ʹ), 24.89 (C-16), 22.3 (C-17ʹ), 14.1 (CH3). EI-MS: m/z (rel. abund. %) 450 (M+, 1), 407 (11), 362 (11), 335 (9), 266 (6), 240 (10), 181 (12), 168 (1 0 0), 123 (19), 70 (23), 55 (29), 43 (49). HR-EIMS: m/z Calcd. for C24H42N4O4 450.3206, Found 450.32495.
Acknowledgement
We thankfully acknowledge the financial support of Sindh Higher Education Commission (SHEC), Pakistan vide letter No. NO.DD/SHEC/1-14/2014, Project code SHEC/SRSP/Med-3/15/2021-21.
References
- Novel α-glucosidase from extreme thermophile Thermus caldophilus GK24. BMB Reports. 2001;34(4):347-354.
- [Google Scholar]
- Discovery of novel α-glucosidase inhibitors based on the virtual screening with the homology-modeled protein structure. Bioorg. Med. Chem.. 2008;16(1):284-292.
- [Google Scholar]
- The mechanism of action of oral antidiabetic drugs: a review of recent literature. J. Endocrinol. Metabol. Diabet. South Africa. 2008;13(3):80-88.
- [Google Scholar]
- Abbasi, M. A., Shah, S. A. H., Aziz-ur-Rehman, Siddiqui, S. Z., Hussain, G., Khan, K. M., Ashraf, M., Ejaz, S. A. (2017). Synthesis of (E)-N'-[1-(2,4-dihydroxyphenyl)ethylidene]substituted hydrazides as possible α-glucosidase and butyryl cholinesterase inhibitors, Journal of the Chemical Society of Pakistan, 39, 248-253.
- Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vascular Health and Risk Management. 2008;4(6):1189.
- [Google Scholar]
- Structure and dynamics of alpha-glucosidase through molecular dynamics simulation studies. J. Mol. Liq.. 2012;174:58-62.
- [Google Scholar]
- Synthesis and molecular docking studies of potent α-glucosidase inhibitors based on biscoumarin skeleton. Eur. J. Med. Chem.. 2014;81:245-252.
- [Google Scholar]
- Synthesis and pharmacological activities of xanthone derivatives as α-glucosidase inhibitors. Bioorg. Med. Chem.. 2006;14(16):5683-5690.
- [Google Scholar]
- α-and β-Glucosidase inhibitors: chemical structure and biological activity. Tetrahedron. 2006;62(44):10277-10302.
- [Google Scholar]
- The α-glucosidase inhibitor N-butyldeoxynojirimycin inhibits human immunodeficiency virus entry at the level of post-CD4 binding. J. Virol.. 1995;69(9):5791-5797.
- [Google Scholar]
- Inhibitory effect of CuSO4 on α-glucosidase activity in ddY mice. Metallomics. 2010;2(1):67-73.
- [Google Scholar]
- Flurbiprofen derivatives as novel α-amylase inhibitors: biology-oriented drug synthesis (BIODS), in vitro, and in silico evaluation. Bioorg. Chem.. 2018;81:157-167.
- [Google Scholar]
- Biology-oriented drug synthesis (BIODS), in vitro urease inhibitory activity, and in silico study of S-naproxen derivatives. Bioorg. Chem.. 2019;83:29-46.
- [Google Scholar]
- Biology-oriented drug synthesis (BIODS) of 2-(2-methyl-5-nitro-1H-imidazol-1-yl) ethyl aryl ether derivatives, in vitro α-amylase inhibitory activity and in silico studies. Bioorg. Chem.. 2017;74:1-9.
- [Google Scholar]
- Piroxicam sulfonates biology-oriented drug synthesis (BIODS), characterization and anti-nociceptive screening. Med. Chem. Res.. 2016;25(7):1468-1475.
- [Google Scholar]
- Atenolol thiourea hybrid as potent urease inhibitors: design, biology-oriented drug synthesis, inhibitory activity screening, and molecular docking studies. Bioorg. Chem.. 2020;94:103359
- [Google Scholar]
- Nitrofurazone and its nitroheterocyclic analogues: a study of the electrochemical behavior in aqueous medium. J. Braz. Chem. Soc.. 2013;24(12):1964-1973.
- [Google Scholar]
- A prodrug approach to improve the physico-chemical properties and decrease the genotoxicity of nitro compounds. Curr. Pharm. Des.. 2011;17(32):3515-3526.
- [Google Scholar]
- Nitrofuran drugs as common subversive substrates of Trypanosoma cruzi lipoamide dehydrogenase and trypanothione reductase. Biochem. Pharmacol.. 1999;58(11):1791-1799.
- [Google Scholar]
- The chemotherapy of Chagas' disease: an overview. Mini Rev. Med. Chem.. 2005;5(5):499-519.
- [Google Scholar]
- Chemotherapeutic properties of prominent nitrofurans. J. Antimicrob. Chemother.. 1976;2:325-336.
- [Google Scholar]
- Synthesis and investigation of antimicrobial activities of nitrofurazone analogues containing hydrazide-hydrazone moiety. Saudi Pharm. J.. 2016;25
- [CrossRef] [Google Scholar]
- Biology-oriented drug synthesis (BIODS) of 2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl aryl ether derivatives, in vitro α-amylase inhibitory activity and in silico studies. Bioorg. Chem.. 2017;74:1-9.
- [Google Scholar]
- Flurbiprofen derivatives as novel α-amylase inhibitors: biology-oriented drug synthesis (BIODS), in vitro, and in silico evaluation. Bioorg. Chem.. 2018;81:157-167.
- [Google Scholar]
- Synthesis of Bis-indolylmethane sulfonohydrazides derivatives as potent α-Glucosidase inhibitors. Bioorg. Chem.. 2018;80:112-120.
- [Google Scholar]
- Abbasi, M.A., Shah, S.A.H., Aziz-ur-Rehman, Siddiqui, S.Z., Hussain, G., Khan, K.M., Ashraf, M., Ejaz, S.A. (2017). Synthesis of (E)-N'-[1-(2,4-dihydroxyphenyl)ethylidene]substituted hydrazides as possible α-glucosidase and butyrylcholinesterase inhibitors. Journal of Chemical Society of Pakistan, 39, 248-253.
- Reductive activation of nitroheterocyclic compounds. Gen. Pharmacol.. 1997;28(4):485-487.
- [Google Scholar]
- A new and efficient N-alkylation procedure for semicarbazides/semicarbazones derivatives. Tetrahedron Lett.. 2007;48(22):3919-3923.
- [Google Scholar]
- Nitroreductive metabolic activation of some carcinogenic nitro heterocyclic food contaminants in rat mammary tissue cellular fractions. Food Chem. Toxicol.. 2009;47(1):140-144.
- [Google Scholar]
- Solangi, M., Kanwal, Khan, K. M., Saleem, F., Hameed, S., Iqbal, J., Shafique, Z., Qureshi, U., Zaheer Ul-Haq, Taha, M., Perveen, S. (2020). Indole acrylonitriles as potential anti-hyperglycemic agents: Synthesis, α-glucosidase inhibitory activity and molecular docking studies. Bioorganic & Medicinal Chemistry, 28, 115605-115615.
- Chemical Computing Group (CCG) Inc. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2019.
- Wang, J., Wolf, R. M., Caldwell, J.W., Kollman P.A., Case, D. A., 2004. Development and testing of a general amber force field. 25(9), 1157-1174.
- Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model.. 2006;25(2):247-260.
- [Google Scholar]
- Delano, W.L., 2010. The PyMOL molecular graphics system, Version 1.3 r1, Schrödinger, LLC, New York, pp. 1-10.
- AMBER 2018. San Francisco, California: University of California; 2018.
- Decoding allosteric communication pathways in protein lysine acetyltransferase. Int. J. Biol. Macromol.. 2020;149:70-80.
- [Google Scholar]
- Gain-of-function SHP2 E76Q mutant rescuing autoinhibition mechanism associated with juvenile myelomonocytic leukemia. J. Chem. Inf. Model.. 2019;59(7):3229-3239.
- [Google Scholar]
- Exploring the pyrazinamide drug resistance mechanism of clinical mutants T370P and W403G in ribosomal protein S1 of Mycobacterium tuberculosis. J. Chem. Inf. Model.. 2019;59(4):1584-1597.
- [Google Scholar]
- Design, synthesis, and molecular docking of novel hybrids of coumarin-dithiocarbamate α-glucosidase inhibitors targeting type 2 diabetes mellitus. Polycyclic Aromat. Compd. 2021:1-11.
- [Google Scholar]
- Commentary emerging approaches in predictive toxicology. Environ. Mol. Mutagen.. 2014;55:679-688.
- [Google Scholar]
- ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.. 2018;46(W1):W257-W263.
- [Google Scholar]
- Structure-based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Food Chem. Toxicol.. 2004;42:65-83.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103806.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2
Supplementary data 3
Supplementary data 3




