

Translate this page into:
Comprehensive review of liquid chromatography methods for fumonisin determination, a 2006–2022 update
⁎Corresponding authors. angelesrc@uaem.mx (M. Ángeles Ramírez-Cisneros), myolanda@uaem.mx (Maria Yolanda Rios)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Fumonisins are mycotoxins present worldwide. They are mainly found in corn and its derived foods; however, they also have an important presence in other grains, fruits, and vegetables. Their consumption in excessive amounts can affect animal and human health. The most abundant of these is fumonisin B1, associated with a range of toxicological effects in animals, including equine leukoencephalomalacia, porcine pulmonary edema, and rodent carcinogenicity. In humans this mycotoxin has been shown to increase rates of esophageal cancer. The International Agency for Research on Cancer has classified FB1 within the 2B group, considering it a possible human carcinogen. Thus, analytical methods that identify/quantify fumonisins become a necessity to ensure adequate control of food and crops. An analytic method needs to be sensitive, selective, and robust to provide reliable data that can aid in monitoring risk assessment, quality control, and research. Recently, colorimetric methods which use immunologic and molecular approaches based on dyes, enzymes and aptamers have gained attention; some of these using nanomaterials. However, these methods are still in development. Currently, chromatographic methods remain the most confident and robust analytic tool, especially for quantification purposes. There is a great deal of information reported in the literature regarding these methods; despite this, there has not been a compilation of the methods for fumonisin analysis to facilitate its consult since 2005. Being the most common method for fumonisin detection worldwide, the present review focuses on the compilation of liquid chromatography methods published between 2006 and 2022 organized by matrix, analytes, instrument, and method conditions, using diverse detectors including MS, fluorescence, and an evaporative light scattering detector. Additionally, These techniques have been applied to diverse matrices, namely food and beverages, including grains, milk, meat, beer, wine; as well as biological samples such as urine, plasma, serum, and tissues. Other aspects pertaining to legislation, extraction, cleanup (selective pressurized liquid extraction, strong anion-exchange, immunoaffinity chromatography, and QuEChERS), derivatization procedures, limit of detection and quantification of fumonisins are also included. This review had compiled and organized 88 chromatographic methods for fumonisins analysis, and the analysts can consult all the procedures with detail.
Keywords
Fumonisins
Fumonisin B1
Fumonisin analysis
Food analysis
Mycotoxins analysis method

1 Introduction
Despite the current improvement in processing, packing and labeling activities, food safety is still an important concern, not only for human consumption, but also for crop control, fresh food quality and safety. Fungi contamination of these and other products is a paramount problem, as it can cause diverse ailments to humans and animals, as well as compromise production yield of the different crops and livestock. Mycotoxins are small secondary metabolites (molecular weight -MW- ∼ 700) produced by microfungi; these are naturally occurring substances that are responsible for detrimental effects to the host, and are, for the most part, resistant to food processing (Bullerman and Bianchini, 2007, Turner et al., 2009). These compounds can be carcinogenic, nephrotoxic, hepatotoxic, neurotoxic, immunosuppressant, and can modify estrogen production (Jia et al., 2014). An important aspect pertaining to the consumption of mycotoxins is their ability to accumulate within an organism. Thus, different sources such as grains: wheat (Headley, 2022 in graphical abstract), oats, rice (Toro 2022 in graphical abstract), barley, and corn (Diogo 2011 in graphical abstract), fresh vegetables (Cumming 2022 in graphical abstract) and fruits (apples, raisins, and nuts) contribute to increase the amount of accumulated toxins in the host. This phenomenon continues in livestock whereby the ingestion of contaminated food sources increases the levels of toxins within their organisms, and are passed on to their derivatives (i.e. meat, milk, eggs, among others). As a result, human consumption of these products multiplies the chain of transmission, as crops and livestock (Embrenhar 2022 in graphical abstract) become saturated of mycotoxins from different sources; this is known as a carryover effect (Marasas 2001). Hence, contamination by mycotoxins has been recognized as a health problem, with special attention being put on aflatoxins, ochratoxins and fumonisins by their direct or accumulated toxicity (Requena et al., 2005).
Mycotoxins are generally characteristic to a specific genus. Some of the main genus producing mycotoxins are Aspergillus (aflatoxins and ochratoxins), Penicillium (patulin, ochratoxin A, citrinin, penicillic acid, cyclopiazonic acid, and penitrem), and Fusarium (trichothecenes and fumonisins) (Grajewski et al., 2012). Among aflatoxins, ochratoxins, and fumonisins, these last ones have been associated with important human diseases such as esophageal cancer (Marasas 2001), with an increased incidence of human immunodeficiency virus (HIV) infection (Williams 2010), liver and kidney disease, and growth impairment (Chen 2018). Some reviews have compiled the toxicity and mechanism of action of FB (Chen 2021, Stockmann-Juvala 2008). It has been estimated that mycotoxins are present in at least a quarter of the world’s agricultural products, and their stability at high temperatures guarantee their integrity even after passing through cooking and industrial procedures (Williams 2010). Despite these considerations, not all countries have legislation that regulate their concentration in food. The number of mycotoxins that are known to exert a toxic effect on human and animal health is constantly increasing, for this reason, generation and observance of legislation that ensures minimization of mycotoxins exposure is needed to ensure the quality of food (Bueno 2015). Diverse detection methods have been used to evaluate fumonisins, and some new methods have a promising future for easier and faster methodologies. Enzyme-linked immunosorbent assay (ELISA) methods based on antigens are specific and commercially available, however these have expiration date and need to be stored under refrigeration. Some enzymes have been proposed for colorimetric methods intended for more analytes, however these demonstrate low selectivity. Nanomaterials have arisen as a promising tool for mycotoxin detection, using immunoreactions or aptamers for detection. Despite this, for research purposes, characterization of nanomaterials is required, and instrumentation is expensive. Thus, this method may only prove favorable for future commercial applications if a high specificity, especially in real samples, can be achieved. These techniques have been recently reviewed (Majdinasab et al., 2021) and remain out of the scope of the present paper. In general, the most extensively used technique for mycotoxin determination is liquid chromatography associated with different detectors (Bueno 2015). This is because it has a well established and robust methodology that has been proven for all kinds of matrices. There is a considerable number of articles regarding fumonisin analysis (including reviews); however, there has been no compilation of this information available since 2006. This review aims to compile and organize the advances in the field from 2006 to 2022 in a single document including liquid chromatography coupled to mass spectrometry (LC-MS) and ultra-performance liquid chromatography (UPLC) methods currently used. Additionally, matrices, pretreatment procedures and instrument conditions are also reported, so that readers can easily find a method close to their needs in a single article.
2 Fusarium genus
Fusarium genus (syn Giberella) was first described by Link in 1803. It belongs to the Nectriaceae family and is widely spread in soil. Fusarium includes more than 150 species of filamentous fungi, classified into nine categories, and is considered one of the most mycotoxigenic genus. Fusarium phylogeny and morphology has been recently reviewed generating an online identification database (Crous 2021). It is of agricultural concern for its capacity to grow on plants, particularly crops, but also in fruits, contaminating food and feed (Tapia 2014, Grajewski et al., 2012). Approximately 20 species are considered pathogenic for their capacity to produce mycotoxins that affect plants, animals, and humans. F. verticillioides and F. proliferatum are the main producers of fumonisins (Gelderblom 1988); F. solani and F. oxysporum have been reported to cause minor health problems directly to humans, producing keratitis, endophthalmitis, onychomycosis, cutaneous and subcutaneous infections, sinusitis, arthritis and mycetoma. In immunocompromised patients, however, especially those with hematological disorders, they can cause severe disseminated infections that can reach mortalities of almost 100% (“Fungal Infections. Fusarium Solani” https://www.life-worldwide.org/fungal-diseases/fusarium-solani; “Fungal Infections. Fusarium Oxysporum,” https://www.life-worldwide.org/fungal-diseases/fusarium-oxysporum). Prolonged exposition to these fungi can also lead to chronic diseases such as cancer (Shier 2000). The distribution of Fusarium species has been studied mainly in commercial substrates, and particularly for certain geographical areas such as F. graminearum and F. culmorum in Europe (Pasquali 2016), F. oxysporum in Israel and Middle East (Maymon 2020), and F. oxysporum worldwide (Dita 2018).
3 Fumonisins
The first report regarding fumonisins was published in 1988 when they were first isolated by Gelderblom et al (Gelderblom 1988). The chemical structure of these mycotoxins was first proposed in the same year as a result of the collaboration between the Programme on Mycotoxins and Experimental Carcinogenesis (PROMEC) and the Council for Scientific and Industrial Research (CSIR) (Marasas 2001). Structurally, fumonisins are characterized by a long chain of polyhydroxy alkylamines containing two propane tricarboxylic acid moieties (tricarballylic acid, TCA) that are esterified to hydroxyl groups on adjacent carbon atoms. Currently twenty-eight different structures of fumonisins have been described (Agriopoulou 2020), which have been classified into four series: Series-A corresponds to amides, Series-B exhibits a free amine group and a terminal methyl, Series-C includes a terminal amine group, and Series-P incorporate an 3-hydroxypiridinium residue in their structures (Yazar 2008, Braun 2018).The fumonisins most frequently isolated from Fusarium are illustrated in Fig. 1.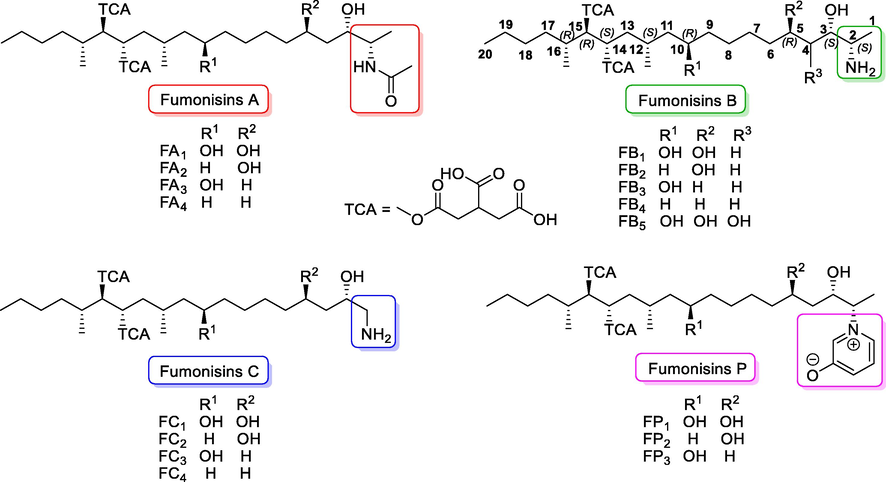
Selected chemical structures of fumonisins.
Within these groups of natural compounds, fumonisins B (FB1, FB2, FB3) are the most relevant because they have been found on various food products and crops (Arranz 2004). FB1 is the most abundant and toxic fumonisin of the group. Its chemical structure is a 2S-amino-12S,16R-dimethyl-3S,5R,10R,14S,15R-pentahydroxyeicosane, in which hydroxyl groups at C-14 and C-15 are substituted with a propane-1,2,3-tricarboxylic acid (TCA) residue. FB2 does not have the hydroxyl group at C-10. FB2 and FB3′s structural isomers, differ only in the location of an hydroxyl group (Fig. 1) (Bryła 2013). The FUM genes have been identified as the responsible for fumonisin biosynthesis (Alexander 2009).
3.1 Fumonisins in food
Fumonisins are present in a wide number of food products around the world. Cereals are the group with the highest documented concentration of these toxins (Kamle 2019). Maize, and maize-based products are particularly affected (Stępień 2011), with as much as an estimated 50% of products contaminated in varying degrees (Pagliuca 2005), depending mainly on agroclimatic and storage conditions (Bryła 2013). In particular, FB1 has been found in different types of food such as asparagus, garlic (Seefelder 2002), barley (Park 2002), beers (Kawashima 2007), dried figs (Heperkan 2012), and milk (Gazzotti 2009). Additionally, FB1 and FB2 have been reported in ‘black oats’ feed from Brazil, and forage grass in New Zealand. They have also been found in home-grown corn consumed in rural areas of Southern Africa, and in commercial corn-based human food products from retail outlets (Norhasima 2009).
Concentrations of FB1 and FB2 vary widely between products. They have been found in corn meal up to 2.98 µg FB1/g and 0.92 µg FB2/g, and in corn grits up to 2.55 µg FB1/g and 1.07 µg FB2/g, respectively. In contrast, Switzerland, the United States, and South Africa have reported very low concentrations of these toxins, being lower than 0.06 µg/g, in products such as corn breakfast cereal (Norhasima 2009). A meta-analysis including contamination of cereal-based foods revealed the highest concentration of fumonisins in corn-based products, followed by wheat-based products, other cereals, and barley-based foods. Regarding the occurrence, it was reported widely in other cereal-based foods, followed by corn-based foods, rice-based foods, and wheat-based foods (Farhadi 2021).
3.2 Stability
The integrity of fumonisins depend on a combination of conditions that include temperature, pH, humidity, biotic or abiotic conditions, matrix and, time in these conditions. Several studies on fumonisin stability were performed in the 90′s. It has been shown that FB1 is partially hydrolyzed at acidic or basic conditions, or at 100–125 °C, and completely degraded at 200 °C for 60 min in the absence of a matrix (Jackson 1996). Thus, the extent of FBs degradation, and their toxicity in food depend primarily on the cooking and processing conditions (Humpf 2004). FBs are known to be relatively heat stable and are minimally affected during food processing techniques such as baking, frying, broiling or extrusion cooking, where temperatures can reach 150–200 °C (Humpf 2004). In maize flour, at neutral and acidic conditions, FBs were reported stable at temperatures greater than 220 °C (25 min) (Bryła 2017). Selection and disposal of damaged grains, along with soaking and/or washing corn reduced the concentration of FBs by eliminating it from food material (Saunders 2001). Dry milling has been shown to maintain FB1 mostly intact (Kamle 2019), however, wet milling has been shown to produce products suitable for animal and human consumption (gluten, fiber, germ, and starch), as the water used in the process causes FB1 deterioration (Saunders 2001). Fumonisins can also interact with aminoacids, proteins or reducing sugars to form covalent bonds during heat processes. For instance, FB1 reacts with D-glucose, present in corn grits, during extrusion cooking at 160–180 °C and forms the reaction product N-(carboxymethyl) fumonisin B1 known as NCM (Seefelder 2002, Taylor 2012).
3.3 Toxicological effects
Fumonisin has been proven to induce growth and lipid disruption in plants, animals, and humans, especially FB1. Additionally, immunotoxicity, organ toxicity (liver, kidney, intestinal tract, heart, lungs, brain) and reproductive toxicity has been reported (Chen 2021). Structural similarity between sphingosine, sphinganine and fumonisin (e.g. FB1, Fig. 2) is cited as the key for their toxic effects, however oxidative stress, endoplasmic reticulum stress and altered tumor necrosis factor (TNF) signaling pathway, has also been recognized as a mechanisms of their toxicity (Chen 2021; Stockmann-Juvala 2008).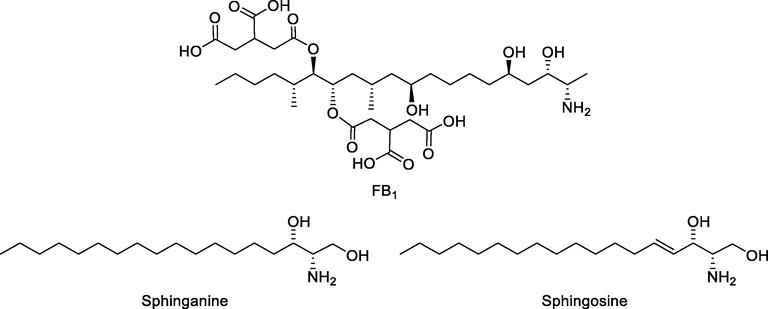
Chemical structures of sphinganine, sphingosine and FB1.
In banana plants, FB1 decreases the activity of certain enzymes such phenylalanine ammonia lyase (PAL), β-1,3-glucanase (GLU), and chitinase (CHI). It also enhances reactive oxygen species like malondialdehyde (MDA) and hydrogen peroxide, as well as transcription of genes associated to cell death (Xie 2021). In maize, FB1 competitively inhibits ceramide synthetase (CerS) disturbing lipid equilibrium and cell protection (Beccaccioli 2021).
In animals, the presence of FBs has been found to impair immune function, cause liver and kidney damage, decrease weight and increase mortality rate (Akande 2006). Fumonisins can cause an ample range of animal diseases, including leukoencephalomalacia (LEM) in horses (Lockett 2022 in graphical abstract) and rabbits, hemorrhage in rabbits, pulmonary edema in pigs, and liver cancer in rats. In addition, they are toxic to turkey poults and have been associated with diarrhea and reduced body weight in broiler chicks (Ghiasian 2009). Different species of fish are affected by FB1, in general, they induce weight and hematocrit reduction, as well as liver and kidney damage similar to other animal species (Oliveira 2020).
Fumonisins are associated with an increased risk of esophageal and liver cancer in humans (Liu 2017), and with a general increase of cancer incidence in regions where maize is the population’s dietary base (Martins 2012). The inhibition of CerS causes the accumulation of the sphingoid bases sphinganine (Sa) and sphingosine (So), and a decrease of complex sphingolipids (Cano-Sancho 2012). Currently, the interference with sphingolipid biosynthesis remains the main cause of toxicity in humans and animals (Soriano del Castillo 2007). Sphingolipids have recently been associated with control of cell growth and proliferation of cancer cells. Ceramide has an important role in limiting cancer progression by inducing cell death (Ogretmen and Avenue 2018). Thus, its inhibition by fumonisins can potentially enhance the development of cancer, which is why the International Agency for Research on Cancer (IARC) has classified FB1 as a probable carcinogenic to humans (group 2B) (Duarte-Vogel 2006). Exposure to fumonisins has also been shown to increase the risk of neural tube defects (NTD) in humans (Seyed Amir Ghiasian 2006). Furthermore, some studies have suggested a possible link between exposure to fumonisins and an increase in the mortality of infection by human immunodeficiency virus (HIV) in sub-Saharan Africa (Williams 2010). More recently, a preliminary study has demonstrated the presence of hydrolyzed FB1 (aminopentol) in the urine of women infected with human papillomavirus (HPV) and its absence in healthy women (Ramírez-Cisneros 2020).
Fusarium produces fumonisin to facilitate its entrance to the cell by producing lipid disruption in the host cell. As a corollary, cells affected by fumonisins become a target for other infection agents such as viruses. Additionally, this lipid disruption leads to alterations in cell metabolism that can lead to cancer and cell death.
Hydrolyzed fumonisins are structurally more similar to Sa and So, however their toxic effects are still unknown. Toxicodynamic studies, especially in humans are necessary to establish dose–response of fumonisins and their hydrolyzed forms.
3.4 Toxicokinetics
The bioavailability, distribution, and toxicokinetic studies in several animal species including laboratory rodents, primates, swine, ruminants, and poultry have shown that fumonisins are poorly absorbed and have a very low bioavailability. However, little amounts of fumonisins accumulate in tissues and organs (Shier 2000). The bioavailability for FB1 administered orally in non-human primates has been reported as < 5 % of the dose with Tmax = 1.02 h. Elimination half-live was found to be T1/2 = 3.15 h for plasma, T1/2 = 4.07 h for liver and T1/2 = 7.07 h for kidney. In contrast, when administered with feed, concentrations in the kidneys increase approximately 10-fold compared to liver concentrations; suggesting an increase in the rate of elimination (Voss 2017). Bioavailability studies have demonstrated that, of the total concentration of FBs (FB1+2+3) in the liver or kidney of rats, FB1 shows the highest concentration, finding FB2 and FB3 in very minor concentrations (Voss 2017). In contrast, FB1 is only detected in plasma and tissues at low levels, suggesting that its absorption is negligible.
Indeed, in cows and laying hens, systemic absorption of orally given FB1 is<1% (Bouhet 2007). Fumonisins were mostly excreted, almost unchanged, in feces and only a small percentage was excreted in urine. Nevertheless, urine is the most acceptable, and easiest, medium to investigate compared to feces (Van Der Westhuizen 2013).
Even though fumonisins have poor absorption, they have been demonstrated to be an important factor in the development of livestock and human diseases (Shier 2000). This poses the interesting question of why they have proven toxic effects despite their low bioavailability. Several investigations have tried to explain this phenomenon, including in vitro studies using Caco-2-cells to prove the absorption of FB1 in enterocytes. A study has established that the only form readily absorbed corresponds to the completely hydrolyzed form of FB1 (aminopentol). Another study using radiolabeled FB1, performed in nonhuman primates, demonstrated that after 24 h of administration, the intestinal epithelial cells contained 25% of the dose (Shephard 1992). Furthermore, recent data has indicated an interaction between FB1 and cholesterol and/or bile salts, which may lead to the incorporation of FB1 into mixed micelles. Thus, the metabolism of fumonisins could lead to an increased bioavailability (Bouhet 2007).
Some aspects of fumonisin toxicokinetics remain unknown, however, and pigs have been suggested as a model because of its similarity with fumonisin metabolism in humans (Schelstraete 2020).
4 Limits and legislation
Removal of mycotoxins from food products has proven to be a difficult process; therefore, maximum acceptable levels have been established for human consumption to ensure the safety of these products. Guidelines have been published in response to this need, that dictate the maximum concentration of these compounds that can be tolerated. There is a varied range of permissible amounts of mycotoxins in food according to different guidelines, encompassing ranges from 200 to 4000 μg/kg (Ponce-García 2018). Many organizations worldwide oversee strict regulations for mycotoxin control, and possible food contamination. Some of these are global organizations such as the Joint Expert Committee on Food Additives (JECFA); the scientific advisory board of the World Health Organization (WHO) and the Food and Agriculture Organization (FAO). Others are limited to geographical areas such as the European Food Safety Authority (EFSA) in the European Union, which gives counseling to European Commission; and the Food and Drug Administration (FDA) in the United States of America (Pereira 2014).
In 1997, fumonisins as a subgroup of mycotoxins, were subject to regulations in only one country (FAO, 1997). In 2005, the number of countries regulating fumonisins increased to six, and the limit for their presence in maize was established as a maximum of 3000 mg/kg (Panel 2015).
Currently, many countries have implemented several regulations to control the presence of fumonisins in food products by implementing prescribed acceptable and maximum limits (WHO-Department of Food Safety and Zoonoses 2018). The JECFA established a maximum tolerable daily intake (PMTDI) of 2 µg/kg b.w./day for FB1, FB2, and FB3 (alone or in combination). On the other hand, the European Union (EU Regulation 1126/2007) and the US, proposed acceptable upper limits of 4000 µg/kg for FB1 and FB2 (Agriopoulou 2020). These established safe limits are not homogenous as different countries change them mainly in relationship to food products. For example, the maximum permissible levels (MPL) for the combination of FB1 + FB2 is 4000 μg/kg for unprocessed maize; whereas for maize intended for direct human consumption is 1000 μg/kg; 800 μg/kg for maize-based breakfast cereals/snacks; and 1400/2000 μg/kg for maize milling fractions of particle size greater/<500 μm respectively. The Codex Alimentarius Commission on Food Contaminants recommends a limit of 5000 μg/kg for combined FB1 + FB2 + FB3 MPL for unprocessed corn grain and 2000 μg/kg MPL for processed maize-based products including flour (Bryła 2013) (WHO-Department of Food Safety and Zoonoses 2018). The main purpose of these legislations is to prevent the consumption of food that is potentially contaminated with mycotoxins, ensuring the protection of the inhabitants of developed countries (Alberts 2017). At present, there are limits established for raw maize (4000 µg/kg), as well as for maize flour and semolina (2000 µg/kg) (Alimentarius 2019). The European Commission has regulated acceptable levels of fumonisins with its most recent modification in 2010 indicating 2000 µg/kg for raw maize, 1000 µg/kg for maize products for coction, 400 µg/kg for direct ingest maize products and, 200 µg/kg for babies and kinder food (European Comission 2007). In contrast, countries with emerging economies lack similar regulations or have poor standards; this can lead to problems with overconsumption of food with high levels of mycotoxins, including fumonisins (Ponce-García 2018). To control and/or verify fumonisin presence in food and feed products, analytical methods are needed for a wide variety of matrices. These have been proven to affect fumonisin stability and thus, bioavailability (Tables 1-3). (ACN) Acetonitrile, (AcOH) Acetic acid, (AE) Appearance energy, (AmAc): ammonium acetate, (AmFo) Ammonium formate, (CaV) Capillary voltage, (CaT) Capillary temperature, (CGF) Cone gas flow, (CoG) Collision gas, (CUR) Curtain gas, (DG) Drying gas, (DGF) Desolvation gas flow, (DGT) Desolvation gas temperature, (EV) Extractor voltage, (FA) Formic acid, (Frag Vol) Fragmentor Voltage, (GF) Gas flow, (GT) Gas Temp, (LIT) linear ion tramp, (MeOH) Methanol, (MSPD) Matrix Solid Phase Dispertion, (NG) Nebulizer gas, (NR) Not reported, (PLE) Pressurize Liquid Extraction, (RT) Room temperature, (ST) Source temperature, (SV) Source voltage, (tan) analysis time, (tTot) total time including column conditioning. (ACN) Acetonitrile, (AcOH) Acetic acid, (AE) Appearance energy, (AmAc): ammonium acetate, (AmFo) Ammonium formate, (CaV) Capillary voltage, (CaT) Capillary temperature, (CGF) Cone gas flow, (CoG) Collision gas, (CUR) Curtain gas, (DG) Drying gas, (DGF) Desolvation gas flow, (DGT) Desolvation gas temperature, (EV) Extractor voltage, (FA) Formic acid, (Frag Vol) Fragmentor Voltage, (GF) Gas flow, (GT) Gas Temp, (LIT) linear ion tramp, (MeOH) Methanol, (MSPD) Matrix Solid Phase Dispertion, (NG) Nebulizer gas, (NR) Not reported, (PLE) Pressurize Liquid Extraction, (RT) Room temperature, (ST) Source temperature, (SV) Source voltage, (tan) analysis time, (tTot) total time including column conditioning. (ACN) Acetonitrile, (AcOH) Acetic acid, (AE) Appearance energy, (AmAc): ammonium acetate, (AmFo) Ammonium formate, (FA) Formic acid, (Hex) Hexane, (MeOH) Methanol, (MSPD) Matrix Solid Phase Dispertion, (NR) Not reported, (PBS) Phosphate Buffer Solution, (PLE) Pressurize Liquid Extraction, (RT) Room temperature, (tan) analysis time, (tTot) total time including column conditioning.
Ref
FBs
MatrixSample (g)
Sample treatment
Extraction procedureLC conditions
Column / Injection volume / Mobile Phase
Flow / Analysis TimeMS conditions, Limits
Mass Conditions / Limits
Maize and corn-based products
(Zitomer et al. 2008)
B1, B2, B3
Maize leaf0.01
Extraction: 1.-Add 2 mL ACN/H2O 1:1 (5% FA); 2.- Gently shaken for 3 h; 3.- Centrifugate to 15000 g; 4.- Filter; 5.- Dilute 1:10
Metachem Inertsil ODS-3, 150 x3 mm, 5 µm
Inj vol 20 µL, A) H2O/ACN/FA 97:2:1, B) H2O/ACN/FA 2:97:1.
70-50% B in 9 min, 50-100% B in 2 min, keep 10 min; initial conditions for 10 min
Flow: 0.20 mL/min, Time: tan=21 min, tTot=31 min
QTrap
CaT : 210°C
LOD: 0.01 µg/kg all FBs
(De Girolamo et al. 2014)
B1, B2, PHF (B1, B2), HF (B1, B2)
Maize based products20
Extraction: 1.- 100 mL MeOH/ACN/citrate-phosphate buffer 25:25:50; 2.- Shake 1h; 3.- Dilute 1:10 with MeOH/H2O 80:20 with 0.5% AcOH; 4.- Filter
Gemini C18, 150 x 2.0 mm, 5 µm at 40 °C
Inj vol 20 µL, A) H2O, B) MeOH, both with 0.5% AcOH
40-60% B in 30 min, 60 to 40% B in 1 min; initial conditions for 9 min
Flow: 0.2 mL/min, Time: tan=30 min, tTot=40 min
Orbitrap
CaV 45 V; SV 4 kV; RF Lens 75 V; ST 300 °C; CaT: 300 °C; SG 30 U; GF 10 skimmer V 18 V
LOD: 5 µg/kg, LOQ: 10 µg/kg all FBs
(Beltrán et al. 2009)
B1, B2
Maize, kernel, dry pasta, baby food2.5
Extraction: 1.- Add ACN/H2O 80:20 + 0.1% AcOH, 2.-shake 90 min, 3.-centrifuge to 4000 rpm, 10 min; 4.-dilute 1:2 with H2O, 5.-filter (0.22 mm nylon filter)
Acquity UPLC BEH C18, 50 x 2.1 mm, 1.7 µm at 40°C
Inj vol 20 µL, A) H2O, B) MeOH, both with 0.5 mM AmAc and 0.1% AcOH
10-90 % B in 4 min, initial conditions for 3 min
Flow: 0.3 mL/min, Time: tan=4 min, tTot=7 min
QQQ
CaV 3.5 kV; DGT 500°C; ST 120 °C; T 40 °C; DGF 1200 L/h, CoG 4 x 10-3 mbar
LOD: 1 µg/kg, LOQ: 3.5 µg/kg
(C. Dall’Asta et al. 2008)
B1, B2, B3
Maize, maize-based products25
Extraction LLE: 1.- Add 100 mL H2O/ACN/MeOH 50:25:25, 2.- blend (6000 rpm/5 min); 3.- take 4 mL; 4.- filter; 5.- dry N2; 6.- reconstitute 1mL in H2O/ACN 1:1; 7.- filter
XTerra C18, 250 × 2.1 mm, 5 µm at 30°C
Inj vol 10 µL, A) H2O, B) MeOH, both with 0.1% FA
0% B for 3 min, 0-45% B in 2 min, keep 5 min, 45-85% B in 15 min, keep for 10 min, initial conditions for 10 min
Flow: 0.2 mL/min, Time: tan=35 min, tTot=45 min
QQQ
CaV 3.2 kV; CV 30 V; EV 3 V; ST 120 °C; DGT 160 °C; CGF 70 L/h; DGF 650 L/h (N2 for both)
LOD: B1, B2 1 µg/kg, FB3 8 µg/kg
LOQ: B1, B2 5 µg/kg, FB3 12 µg/kg
(Arroyo-Manzanares et al. 2018)
B1, B2 and other toxins
Wheat, maize2
QuEChERS: 1.-Add 8 mL of H2O; 2.-shake 10 s; 3.-add 10 mL 5% FA in ACN; 4.-shake 2 min; 5.-add 4 g MgSO4 + 1 g NaCl; 6.-shake 1 min: 7.-vortex 2 min; 8.-centrifuge to 4500 rpm, 5 min, 4 °C; 9.-take 5 mL; 10-dry under N2 at 40 °C; 11.-reconstitute (0.2 mL MeOH/H2O 1:1); 12.-centrifuge to 14000 g, 5 min, 4 °C
ACQUITY HSS UPLC T3, 150 x 2.1 mm, 1.8 µm at 30 °C
Inj vol 10 µL, A) H2O, B) MeOH, both with 0.3% FA and 5 mM AmF
5% B, keep 0.5 min, 5-94% B in 19.5 min, keep 1 min, 94-5% B in 3 min; initial conditions for 4 min
Flow: 0.4 mL/min, Time: tan=21 min, tTot=28 min
QQQ
ST 150 °C; DGT 400 °C; NG 7 bar (N2); CGF 150 L/h; DGF 1000 L/h
LOD; 1.28 B1, 0.25 FB2, 0.27 B3 µg/kg LOQ: 4.24 B1, 0.82 FB2, 0.89 B3 µg/kg
(Chiara Dall’Asta, Galaverna, et al. 2009)
B1, B2, B3
Corn-based products5
Extraction: 1.- Add 50 mL H2O/MeOH 30:70; 2.- Blend to 6000 rpm, 10 min; 3.- Stir for 60 min; 4.- re-extract the solid (same way); 5.- Filter; 6.- Dry 4 mL; 7.- Dissolve in 2 mL MeOH
Xterra C18, 250 x 2.1 mm, 5 µm, at 30 °C
Inj vol 5 µL, A) H2O, B) MeOH, both with 0.2% FA
30% B for 2 min, 30-45% B in 3 min, 45-90% B in 20 min, keep for 10 min, 30% B in 1 min; initial conditions for 20 min
Flow: 0.2 mL/min, Time: tan=35 min, tTot=56 min
QQQ
CaV 3.2 kV; EV 3 V; ST 120 °C; DGT 160 °C; CGF 70 L/h; DGF 650 L/h (N2, both)
LOD: FB1 4 µg/kg, B2, FB3 8 µg/LOQ: B1 B2 5, B3 12 µg/kg
(Chiara Dall’Asta, Mangia, et al. 2009)
B1, B2, B3
Ground corn5
Extraction: 1.- Add 50 mL H2O/MeOH 30:70; 2.-Blend to 6000 rpm, 10 min; 3.- Stir for 50 min; 4.- Centrifuge to 3500 g, 15 min; 5.- Filter (2 mL)
Xterra C18, 250 x 2.1 mm, 5 µm at 30°C
Inj vol 10 µL, A) H2O, B) MeOH, both with 0.1% FA
30 % B for 2 min, 30-45% B in 3 min, 45-90% B in 20 min, keep for 10 min; initial conditions for 15 min
Flow: 0.2 mL/min, Time: tan=35 min, tTot=50 min
QQQ
CaV 4 kV; EV 2 V; ST 120°C; DGT 350 °C; CGF 50 L/h; DGF 600 L/h
LOD: 5 µg/Kg
(Chiara Dall’Asta, Mangia, et al. 2009)
B1, B2, B3
Corn-based products5
Extraction: 1.- Add 2 ml H2O/ACN/AcOH 20:79:1; 2.- Extract 90 min in rotatory shaker; 3.- Centrifuge 3000 rpm, 3 min; 4.- Take aliquot 350 µL and dilute 1:1 with extraction solvents
Gemini C18, 150 x 4.6 mm, 5μm at 25 °C
Inj vol 5 µL, A) H2O/ACN/AcOH 89:10:1, B) H2O/ACN/AcOH 2:97:1, both with 5 mM AmAc
0% B for 2 min, 0-100% B in 12 min, keep for 3 min; initial conditions for 4 min
Flow: 1.0 mL/min, Time tan=17 min, ttotal=21 min
QQQ
CaV 4.0 kV; EV 3 V; ST 550 °C; CUR 10 psi
LOD: 8 µg/kg
(G. B. de Oliveira et al. 2017)
B1, B2
Maize1
Extraction: 1.- Add 1 g Silica gel as dispersant; 2.- Mix in polypropylene cartridges, MSPD; 3.- Elute with 16 mL of 20 mM AmFo buffer:MeOH 9:1 (pH 7); 4.- Collect 2 mL fractions; 5.- Centrifuge to 4000 rpm, 10 min; 6.- Filter
Poroshell, C18, 100 x 3 mm, 2.7 µm, 40 °C
Inj vol 10 µL, A) Ultrapure H2O, B) ACN, both with 0.1% FA
20-90% B in 3 min, keep 0.4 min, 90-20 % B in 0.1 min; initial conditions for 6 min
Flow: 0.5 mL/min, Time: tan=3.4 min, tTot=9.5 min
QQQ
CaV 4.5 kV; EP 10 V; DGT 650 °C; NG 40 CUR 18 a.u,
LOD: B1 514, B2 176 µg/kg
LOQ: B1 594, B2 210 µg/kg
(D’Arco et al. 2008)
B1, B2, B3
Corn-based baby food3
Extraction: 1.- Add 100 µL of a 5 µg/mL Fbs solution (0.5 µg) and keep 15 min at RT; 2.-pack into 11 mL PLE pressure resistant stainless steel extraction cell; 3.-elute with 22 mL of MeOH 60% at 40°C and 34 atm, 2 min of preheating, 5 min of static time, 60 s of purge time; 4.-concentrate to 5 mL (40 °C and 80 mbar); 5.-transfer to a 15 mL conical tube; 6.-evaporate to dryness at 55°C with N2 ; 7.reconstitute 1 mL MeOH/H2O 50:50; 8.-filter
Luna C18, 150x4.6 mm, 5 µm (Temp NR)
Inj vol NR, A) H2O, B) MeOH, both with 0.5% FA
65% B for 3 min, 65-95% B in 4 min, keep 3 min, initial conditions in 10 min
Flow: 0.30 mL/min, Time: tan=10 min, tTot=20 min
QQQ
CaV 3.20 kV; CoV 50 V; EV 3 V; RF lens 0.2 V; ST 125 °C; DGT 300 °C; DGF 500 L/h; CGF gas 50 L/h
LOD: 0.7 B1 and B2, 1.5 µg/kg B3 LOQ: 2 B1 and B2, 5 µg/kg B3
(Chiara Dall’Asta, Mangia, et al. 2009)
B1, B2, B3
Raw corn100
Extraction: 1.- Add 50 mL KOH 2M; 2.-Centrifuge to 6000 rpm, 10 min; 3.- Stir (50 min); 4.- Add 50 mL ACN; 5.- Stir 10 min; 6.- Separate 20 mL and dry under N2; 7.- Redissolve in 50 mL KOH 2M; 8.- Centrifuge to 3500 rpm, 15 min; 9.- Dry under N2; 10.- Redissolve in H2O/MeOH 30:70
Hypersil C18, 150 x 2.1 mm, 5 μm at 25°C
Inj vol 10 µL, A) H2O, B) MeOH, both with 0.2% FA
20% B for 1 min, 20-100% B inwalnut 5 min, keep 3 min, initial conditions for 4 min
Flow: 0.6 mL/min, Time: tan=8 min, tTot=13 min
QQQ QTrap
CaV 4 kV; CoV 50 V; ST 425°C; DGT 350°C; CGF 50 L/h; DGF 600 L/h (N2, both)
LOD: <15 µg/kg
(Hu et al. 2019)
B1, B2
Raw maize1
Extraction: 1.- 10 mL ACN/H2O/AcOH 70:29:1; 2.- Shake 30 min; 3.- Centrifuge to 4500 rpm, 10 min; 4.- Filter supernatant; 5.- Take 1 mL; 6.- Add 10 µL, 1 µg/mL 13C-34 FB1 and 13C-34 FB2
Luna C18, 150 x 2 mm, 3 µm at 40°C
Inj vol 5 µL, A) H2O, B) MeOH, both with 2 mM AmAc
40-90% B in 6 min, keep 1 min, 90-100% B in 1 min, keep 1 min, 100-40% B in 2 min; initial conditions for 4 min
Flow: 0.2 mL/min, Time: tan=9 min, tTot=15 min
Qtrap
CaV 5.5 kV; EP 10 V; ST 600°C; CUR 40 psi; CoV 10 V; dwell time 100 ms
LOD: 7 B1; 6 B2 µg/kg
LOQ: 28 B1; 27 B2 µg/kg
(Bergmann, Hübner, and Humpf 2013)
B1
Maize10
Extraction: 1.- Add 20 mL ACN/H2O 70:30 with 1% FA; 2.-Vortex 30 s; 3.- Sonicate 10 min; 4.- Shake 15 min; 5.- Centrifugate to 8000 g, 15 min, 25 °C; 6.- Dilute 1:1 1% FA; 7.- Filter if necessary
Hyperclone C8 BDS, 150 x 2.0 mm, 3 µm at 40° C
Inj vol 20 µL, A) H2O, B) ACN, both with 1% FA
65% B for 4 min, 37.5% B for 0.5 min, 5% B for 2 min, keep for 0.5 min, initial conditions for 4 min
Flow: 0.30 mL/min, Time: tan=7 min, tTot=11 min
QTrap
CaV 5.5 kV; DG 350 °C; NG 35 psi; DG 45 psi; CUR (N2) 30 psi; CoG 5 x 10−5 Torr; QTrap CUR 20 psi
LOD: 53 µg/kg, LOQ: 188 µg/kg
(de Matos et al. 2021)
B1, B2, HB1, HB2
Corn products5
Extraction: 1.- Add ACN:H2O:FA 75.24:1; 2.- shake for 2 min; 3.- sonicate for 10 min; 4.- centrifuge at 3000 rpm for 7 min; 5.-take 0.05 mL of extract; 6.- dilute with 0.95 mL 0.05% of AF in MeOH:H2O 1.1; 7.- filter
ACQUITY BEH C18 100 x 2.1 mm, 1.7 μm at 35°C
Inj vol 5 μL, A) H2O (0.1% FA), B) MeOH
65-80% B in 3 min, hold for 1 min, 100% B in 1 min, initial condition for 2 min
Flow 0.3 mL/min, Time: tan= 5 min, tTot= 7 min
QQQ
CaV: 3kV; DGT: 400 °C; ST: 150 °C; CGF: 15 L/h; DGF:750 L/h
LOD: (B1: 0.43-1.98, FB2 0.19-1.37, HB1 0.72-1.39, HB2 0.36-0.70) μg/Kg
LOQ: (B1:1.43-6.59, FB2 0.60-4.60, HB12.40-4.60, HB2 1.20-2.30) μg/Kg
(Lin et al. 2011)
B1, B2
Corn5
Extraction: 1.- Add 25 mL MeOH/H2O 3:1; 2.-Ultrasonic bath for 10 min at RT, output powder 120 W; 3.- Centrifugate to 5000 g, 5 min; 4.- Filter (0.22 mm nylon filter)
Zorbax Eclipse XDB-C18, 150 x 2.1 mm, 3.5 µm at 30°C
Inj vol 10 µL, MeOH/H2O/FA 75:25:0.2
Flow: 0.20 mL/min, Time tan= tTot = 4 min
Q
CaV 3.5 kV; CoV 50 V; ST 120 °C; DGT 350°C; DGF 600 L/h
LOD: 3.5 B1, 2.5 µg/kg B2
LOQ: 11.7 B1, 8.3 µg/kg B2
(A. S. Silva et al. 2019)
B1, B2
Maize flour2
Extraction: 1.- Add 10 mL ACN 80%; 2.- Shake at 110 rpm, 1h; 3.- Centrifuge to 3000 rpm, 10 min; 4.- Remove supernatant; 5.- Re-extract the solid, same way; 6.- Centrifuge to 3000 rpm, 10 min; 7.- Dilute 1:1 with H2O; 8.- Filter
Zorbax Eclipse Plus C18, 2.1 x 50 mm, 1.8 µm at 30 °C
Inj vol 20 µL, A) 0.1% FA, B) ACN
10-70% B in 12 min, 70-90% B in 1 min, keep 1 min, 90-10% B in 1 min, initial conditions for 2 min
Flow: 0.5 mL/min, Time: tan=14 min, tTot=17 min
TOF
CaV 5.5 KV; ST 575 °C; CUR 30 psi; Gas 1 and Gas 2, 55 psi both; DP 100 V; Full scan 100-750 Da
LOD: 62.5 µg/kg,LOQ: 125 µg/kg all FBs
Other cereal and seeds
(Bartók et al. 2006)
B1, B2, B3, its analogs
Rice3
Extraction: 1.- Add 25 mL of ACN/H2O 75:25; 2.- Centrifuge to 13,500 rpm, 1 min; 3.- Shake 1 h; 4.- Centrifuge to 10,000 g, 10 min; 5.- Filter
Supelcosil ABZ Plus, 250 x 2.1 mm, 5 µm at 40 °C
Inj vol 1 µL, A) H2O, B) ACN, both with 0.1% FA
25-40 % B in 22 min, 40-100% B in 5 min, keep for 3 min.
Flow: 0.3 mL/min, Time: tan=27 min, tTot=30 min
QTrap
CaV 3.5 kV; EV 200 V; HED Voltage 7 kV; NG 40 psi; DGF 9 L/min; DGT 350 °C; trap drive 53.9; max accumulation time 300 ms; full scan 50-1100 m/z
LOD / LOQ: NR
(Soleimany, Jinap, and Abas 2012)
B1, B2
Cereals10
Extraction: 1.- Add 40 mL H2O/ACN/AcOH 20:79:1; 2.- Shake 60 min; 3.- Centrifuge the supernatant at 3000 rpm, 10 min; 4.- Dilute 1:1 in H2O/ACN/AcOH 79:20:1; 5.- Filter
Thermo Scientific C18, 150 x 4.6 mm, 3 µm at 30°C
Inj vol 20 µL; A) H2O, B) MeOH both with 0.1% AcOH
5% B for 8 min, 5-90% B in 14 min; 90-5% B in 3 min
Flow: 0.25 mL/min, Time: tan=22 min, tTot=25 min
QQQ
CaV 3 kV; ST 120°C; DGT 400 °C; spray gas N2
LOD: 20 ng/g, LOQ: 40 ng/g
(Rausch, Brockmeyer, and Schwerdtle 2020)
B1, B2, B3 and other toxins
Cereals1
QuEChERS: 1.- Add 2 mL H2O, 2.-mix 1 min, RT, 10 min; 3.- extract with 8 mL ACN/FA 75:5; 4.- Shake 15 min; 5.- add 4 g anhydrous MgSO4, 1 g NaCl, 1 g Na2HCit 1.5 H2O, Na3Cit 2 H2O, 6.- Mix 1 min; 7.- Shake 15 min; 7.- Centrifuge to 2140 g, 2 min; 8.- Filter; 9.- Take 500 µL, dry; 10.- Redissolved in 250 µL MeOH/H2O 20:80
Raptor Fluoro Phenyl 50 x 2.1 mm, 2.7 µm in series with
Raptor Biphenyl 50 x 2.1 mm, 2.7μm at 30 °C
Inj vol 10 µL, H2O, 0.3% FA, B) MeOH, both with 5 mM AmFo
20% B for 0.6 min, 20-40 % B in 0.4 min, 40-90% in 8 min, keep 1 min, initial conditions for 3.5 min
Flow: 0.4 mL/min
Time: tan=10 min, tTot=13.5 min
QQQ
CaV 4.5 kV; ST 500 °C; CUR 40 psi; ISG 1 60 psi; ISG 2 65 psi
LOQ: depending on the matrix, FBs 4-15 µg/kg
(Aurelien Desmarchelier et al. 2010)
B1, B2 and other mycotoxins
Cereals5
QuEChERS: 1.- Add 10 mL H2O + 10 mL 0.5% AcOH in ACN; 2.- Shake at 300 rpm, 5 min; 3.- Add 5 g MgSO4/NaCl 4:1, 4.- Shake; 5.- Centrifuge to 4000 g, 15 min, RT; 6.- Take 5 mL; 7.- Shake at 200 rpm, 5 min; 8.- Centrifuge to 4000 g, 1 min; 9.- Dry 1 mL at 40 °C (N2); 10.- Add 75 µL MeOH; 11.- Sonicate; 12.- Add 75 µL H2O, mix; 13.- Centrifuge to 8500 g, 10 min, RT; 14.- Dilute 60 µL with 140 µL H2O; 15.- Centrifugate to 8500 g, 10 min, RT
Zorbax Bonus-RP, 150 x 2.1 mm, 3.5 µm
Inj vol NR, A) H2O 0.15% FA, 10 mM AmFo, B) MeOH 0.05% FA
15% B 0.5 min, 15-100% B 8.5 min, keep for 6 min, 15% B in 1 min, initial conditions for 9.5 min
Flow: 0.25 mL/min, Time: tan=15 min, tTot=25.5 min
QTrap SRM
ST 550 °C; NG 50 psi; CUR 40 psi; TG 30 psi; CoG 1.2 x 10-4 psi
LOQ: 50 µg/kg all FBs
(Liao et al. 2013)
B1, B2 and other toxins
Finished grain, nut products1
Extraction: 1.-Add 5 mL H2O/ACN 15:85; 2.-shake to 1550 rpm, 30 min; 3.-centrifugate to 4500 rpm, 5 min; 4.-take 500 µL; 5.-add 20 µL of 13C-34 FB1 (25 µg/mL) + 480 µL 20 mM FA; 6.-vortex 15 s; 7.-filter
Ultra-Aqueous C18, 100 x 2.1 mm, 3 μm, at 40 °C
Inj vol 10 µL, A) H2O, B) MeOH, both with 0.1% FA+ 10 mM AmFo
10 % B for 1 min, 10-100% B in 6 min, keep for 3 min, initial conditions for 5 min
Flow: 0.5 mL/min, Time: tan=10 min, tTot=15 min
QTrap
Conditions NR
LOD: FBs 2.2-2.9 µg/kg, LOQ: FBs 7.3-9.6 µg/kg, depending on the matrix
(Bartók et al. 2010)
Isomers of B1
Rice1
Extraction: 1.-8 mL MeOH/H2O 75:25; 2.-homogenize 9,500 rpm, 4 min: 3.-centrifuge to 10,000 rpm, 10 min, 4.-filter
YMC-Pack J’sphere ODS H80, 250 x 2.1 mm, 4 µm, 40 °C
Inj vol 1 µL, A) H2O, B) ACN, both with 0.1% FA
24-40% B for 79 min, 40-100 % B for 15 min, keep for 10 min
Flow: 0.20 mL/min, Time: tan=79 min, tTot=104 min
TOF, full scan MS
CaV 3.5 kV; Fragmentor 170 V; skimmer 70 V; DGT 350 °C; DGF 10 mL/min; NG 20 psi; full scan 100-1700; acquisition rate 250 ms/spectrum
LOD/LOQ: NR
(Oueslati et al. 2012)
B1, B2
Cereals, derived products5
Extraction: 1.- Add 10 mL ACN/H2O 80:20; 2.-vortex 2 min, shake 60 rpm x 10 min; 3.-centrifuge to 5000 rpm, 5 min; 4.-filter 2 mL (0.20 μm, Millipore)
Acquity UPLC BEH C18, 100x2.1 mm, 1.7 µm at 30°C
Inj vol 5 µL, A) H2O with 5 mM AmFo, B) MeOH
25-75% B in 3 min, 75-100% B in 2 min, keep for 1.5 min, 100-25% B in 1 min; initial conditions for 1 min
Flow: 0.35 mL/min, Time: tan=6.5 min, tTot=8.5 min
QQQ
CaV 3.5 kV; CoV FB1 45 V, FB2 55 V; EV 3 V; ST 120 °C; DGT 350 °C; CGF 50 L/h; DGF 650 L/h
LOD: B1 and B2 1 µg/kg
LOQ: B1 and B2 5 µg/kg
(Rausch, Brockmeyer, and Schwerdtle 2021)
B1, B2, B3, HB1, HB2, HB3
Cereals2.5
Extraction: 1.- Add ACN:H2O:FA 79.20:1; shake for 15 min at RT; 3.- Add 20 µL of Deuterated internal standard; 4.- rotary agitation for 30 min; 5.- centrifuge at 1902 g, 6.- take an aliquot of supernatant, 7.- filter
First dimension: YMC-Pack Diol-NP C18 100 × 2.1 mm, 5 μm at 40 °C.
Vol. inj: 10 μL of sample, A) H2O, B) ACN:H2O 90:10 Both (0.1% FA, 10 mM AmFo)
100% B in 2.5 min, 100-90% B in 0.5 min, 90-20 % B in 0.8 min, hold for 3.8 min, 20-100% B in 0.20 min. initial condition for 17.20 min.
Second dimension: 2 columns connected in series Raptor FluoroPhenyl, 50 × 2.1 mm, 2.7 μm and Raptor Biphenyl 50 × 2.1 mm, 2.7 μm,
5% B for 1.2 min, 5-0% B in 0.10 min, hold for 7.15 min, 0-5% B in 0.05 min, 5-50% B in 1.1 min, 50-70% B in 4.4 min, 70-85% B in 2.5 min, 85-100% B in 3 min, hold for 2 min, 100-5% B in 0.10 min, initial condition for 4 min
Flow 0.2 mL/ min, 0.3 ml/min, Time: tan = 7.6 min tTot = 25 min, Time: tan = 15.50 min tTot = 25 min
QQQ
CaV: 4.5 kV; CUR: 40 psi; ST: 500 °C;
LOQ: (B1-3: 10, HB1-3: 100) μg/Kg
Other samples
(Škrbić, Živančev, and Godula 2014)
B1, B2 and other toxins
Crude extracts of nuts10
Extraction: 1.- Add 40 mL ACN/H2O/AcOH 79:20:1; 2.- Shake 1h; 3.- Filter; 4.- Take 20 mL; 5.- Add 20 mL hexane; 6.- Mix 2 min; 7.- Centrifuge to 5000 rpm, 5 min; 8.- Eliminate hexane phase. 9.- Filter aqueous phase
Hypersil GOLD C18, 50 x 2.1 mm, 1.9 µm at 25 °C
Inj vol 10 µL, A) H2O, B) MeOH, both with 1% AcOH and 5 mM AmAc
5 % B for 0.5 min, 5-95 % B in 2.5 min, keep 2 min, 95-5% B in 1.2 min, initial conditions for 1.8 min
Flow: 0.50 mL/min, Time: tan=6 min, tTot=8 min
QQQ
CaV 3.4 kV; ST 350 °C; SG 40 arbitrary units; aux gas 10 arbitrary units; CaT 270 °C
LOD: 0.24 B1, 0.05 B2 µg/kg
LOQ: 0.8 B1, 0.17 B2 µg/kg
(Yibadatihan, Jinap, and Mahyudin 2014)
B1, B2 and other toxins
Palm kernel cake5
Extraction: 1.- Add 20 ml H2O/ACN/FA 20:79:1; 2.- Shake 60 min; 3.- Centrifuge supernatant to 3000 rpm, 10 min; 4.-Dilute 1:4 with water; 5.-Filter
Symmetry C18, 150 x 2.0 mm, 3μm, 30 °C
Inj vol. 25 µL, A) H2O, 0.2% FA, B) MeOH
10% B for 8 min, 10-90 % B in 2 min, keep 7 min, from 90-10% B in 3 min, initial conditions for 5 min
Flow: 0.20 mL/min, Time: tan=17 min, tTot= 25 min
QQQ
CaV 3 kV; ST 120 °C; DGT 350 °C
LOD both: Std 5.6 µg/kg LOQ both: Std 18 µg/kg
LOD both: Samples 17.5 µg/kg LOQ both: samples 58 µg/kg
(Qian et al. 2018)
B1, B2 and other toxins
Feed2
QuEChERS: 1.- Add 1.5 g NaCl + 10 mL 3% AcOH in ACN/H2O 80:20; 2.-Vortex 1 min, 3.-Ultrasound 20 min; 4.-Add 2 g anh MgSO4; 5.-Vortex 1 min; 6.-Centrifuge to 8000 rpm, 5 min; 7.-Dry (N2, 40 °C); 8.-Dissolve in MeOH:H2O 1:1; 9.-Filter
ACQUITY UPLC HSS T3, 100 x 2.1 mm, 1.8 µm at 40°C
Inj vol. 5 µL, A) H2O, 0.1% FA, 1 mM AmAc; B) MeOH}
0-10% B in 1 min, 10-20% B in 2 min, 20-99% B in 8 min, keep 2.5 min; 99-10% B in 0.1 min; initial conditions for 5 min
Flow: 0.3 mL/min, Time: tan=13.5 min, tTot= 18.5 minQQQ
CaV 5.5 kV; ST 550°C; Auxiliary gas 40 psi
LOQ: 0.4 µg/kg for both B1 y B2
(Spanjer, Rensen, and Scholten 2008)
B1, B2, B3 and other toxins
Peanut, pistachio, wheat, maize, cornflakes, raisins, figs25
Extraction: 1.- Add 100 mL ACN/H2O 80:20, 2.- Shake 2h; 3.-Dilute 1:4 with H2O; 4.- Filter if necessary
(For raisins and figs use MeOH)
Alltima C18, 150 x 3.2 mm, 5 µm at 30 °C
Inj vol 20 µL, A) H2O, B) ACN, both with 0.1% FA10-70% B in 12 min (curve 1), keep 4 min, 70-90 % B in 1.5 min (curve 6), keep 2.5 min, 90-10 % B in 1 min (curve 1)
, initial conditions for 5 min
Flow: 0.3 mL/min, Time: tan=20 min, tTot=25 min
QQQ
CaV 2.5 kV; CoV 75 V; DGT 450°C; CGF 100 L/h (N2); DGF 600 L/h
LOQ: depending on the matrix, B1 5-100 µg/kg, B2 1-100 µg/kg
(Aurélien Desmarchelier et al. 2014)
B1, B2 and other toxins
Cereals, cocoa, oil, spices, infant formula, coffee, nuts25
Extraction: 1.- Add 50 mL H2O, 2.- Homogenize 1 min 10000 rpm, 3.-Take 5 g of sample (peanut, green cofee, cocoa, paprika) or 2 g (infant formula, sunflower oil), 4.-Add 100 µL of 13C-FB standard (FB1 and FB2 each 10 µg/mL), 5.-Add 10 mL H2O and 10 mL ACN, 0.5% AcOH, 6.- Add 5 g MgSO4:NaCl 4:1 Centrifuge 4000g, 15 min, 7.-Defat 5 mL ACN phase with 5 ml hexane. 8.- Take 1 mL of ACN phase, dry, 9.-Reconstitute in 150 µL H2O/MeOH 1:1, 10.-Centrifuge 8500 g, 10 min, 11.-Take 60 µL, add 140 µL H2O, 12.-Centrifuge 8500 g, 10 min
Zorbax Bonus-RP C18, 150 x 2.1 mm, 3.5 µm at 50 °C
Inj vol 20 µL, A) H2O, 0.15% FA, 10 mM AmFo, B) MeOH, 0.05% FA
15% B for 0.5 min, 15-100 % B in 6 min, keep for 4.5 min, 100-15% B in 0.5 min, initial conditions for 7.5 min
Flow: 0.35 mL/min, Time: tan=11 min, tTot= 19 min
QTrap, QQQ
ST 550 °C; CUR 40 psi, Nebulizer 50 psi; Turbo gas 30 psi
LOD/LOQ: NR
(Shar et al. 2020)
B1, B2 and other toxins
Feed, its ingredients5
Extraction: 1.- Add ACN/H2O/FA 79:20:1; 2.- Shake for 90 min to 180 rotations/s; 3.-Centrifuge to 4000 rpm, 2 min, 4.-Filter
Acquity C18, 100 x 2.1 mm, 1.8 µm, 40 °C
Inj vol 20 µL, A) H2O, 1% FA, B) MeOH/H2O/FA, 97:2:1, both with 10 mM AmFo.
0% B for 2 min, 0-50% B in 0.5 min, 50-100% B in 3.5 min, keep 1 min, initial conditions in 1 min, seal wash for 5 min
Flow: 0.5 mL/min, Time: tan=7 min, tTot= 8 min
sQ
CaV 2.79 kV; ST 150 °C; DGT 350 °C; CGF 50 L/h; CGF 600 L7h
LOD B1: 0.07 µg/kg,LOQ B1: 0.22 µg/kg
LOD B2: 0.03 µg/kg,LOQ B2: 0.08 µg/kg
(Frenich et al. 2009)
B1, B2
Maize, walnut, breakfast cereal, biscuit5
Extraction: 1.- Add 10 mL ACN/H2O 80:20 (for biscuit add 20 mL); 2.- Vortex 2 min; 3.- Shake to 60 rpm, 10 min; 4.- Centrifuge to 4500g, 5 min; 5.- Take and filter 2 mL
Acquity C18, 100 x 2.1 mm, 1.7 μm at 30°C
Inj vol 5 µL, A) H2O with AmFo 5 mM, B) MeOH
25-75% B in 3 min, 75-100% B in 2 min, keep for 1.5 min, 100- 25% B in 1 min; initial conditions for 1 min
Flow: 0.35 mL/min, Time: tan=6.5 min, tTot=8.5 min
sQ
CaV 3.5 kV; EV 3 V; ST 120°C; DGT 350°C; CGF 50 L/h; DGF 650 L/h (N2 for both)
LOD maize: B1 0.1 µg/kg, B2 0.2 µg/kg,LOQ maize: B1 0.5 µg/kg, B2 0.6 µg/kg; LOD breakfast cereal: B1 2.1 µg/kg, B2 0.7 µg/kg LOQ breakfast cereal: B1 6.2 µg/kg, B2 2.5 µg/kg
Beverages
(Rubert et al. 2011)
B1, B2, B3 and other toxins
Beer10 mL
Extraction: 1.- Sonicate 25 min, 2.-Condition SPE Oasis HLB cartridges with 5 mL ACN/MeOH 1:1; 3.- 5 mL H2O; 4.- 10 mL sample into cartridge; 5.-Wash with 5 mL H2O; 6.- Dry 30 min; 7.- Eluate with 4mL ACN:MeOH 1:1; 8.- Dry (N2, 35 °C), 9.- Reconstitute in 1 mL (ACN/MeOH 1:1); 10.-Filter
Gemini C18, 150 x 2.0 mm, 5 µm, at 35 °C
Inj vol 10 µL, A) H2O, 0.1% FA, B) MeOH, both with 5 mM AmFo
5-95% B in 10 min, 95-80% B in 5 min, initial conditions 5 min
Flow: 0.2 mL/min, Time: tan=10 min, tTot= 20 min
QQQ Orbitrap XL
CaV 30 V; SV 4 kV; Source Temp 275 °C; Capillary gas sheat 35 units; auxiliary gas 30 arbitrary units
LOD: 30-35 µg/L, LOQ: 90-105 µg/L all Fbs depending of the beer type
(Huang et al. 2018)
B1, B2 and other toxins
Liquorice2
QuEChERS: 1.-Add 100 µL of D-atrazine (60 µg/L), 15 mL acetate buffer pH 3.0, 10 mL 5% FA in ACN; 2.- Shake; 3.- Extract with ultrasonic (53 KHz, 5 min, 20°C); 4.- Add 4 g MgSO4 + 1 g NaCl + 0.5 g Na2HCit·1.5H2O, 1 g Na3Cit·2H2O; 5.- Shake to 1500 strokes/min, 5 min; 6.- Ice bath 10 min, 7.-Centrifuge to 18514 g, 10 min; 8.- Take 6.0 mL; 9.- Transfer supernatant into 15 mL centrifugation tube containing 900 mg MgSO4, 600 mg C18, 150 mg PSA, 150 mg Si; 10.- Shake 5 min, 11.- Centrifuge 10 min; 12.- Take 2 mL, reduce volume <0.5 mL with N2; 13.- Complete to 1 mL with H2O/MeOH 80:20; 14.-filter
Poroshell EC-C18, 150 x 3 mm, 2.7 µm at 20°C
Inj vol 5 µL, A) H2O, B) MeOH, 0.2% FA and 2 mM AmF
20% B for 2 min, 20-50% B in 2 min, 50-100% B in 7 min, keep 1 min, 100-20% B in 1 min, initial conditions for 2 min
Flow: 0.45 mL/min, Time: tan=12 min, tTot= 15 min
QQQ
CaV 5.5 kV; DP 150 eV; EP 10 eV; CUR 30 psi; GS1: 50 psi, turbo gas (gas 2) 50 psi, GT: 450°C
LOD: B1, B2, 0.05 µg/kg
LOQ: B1. B2, 0.125 µg/kg
(Tamura et al. 2012)
B1, B2, B3 and other toxins
Wine5 mL
Extraction: 1.-Add 25 mL AmAc 10 mM, mix, 2.-wash in Oasis HLB SPE Cartridge conditioned with 5 mL AmAc 10 mM, 3.-elute with 5 mL AmAc 10 mM/ACN 1:1, 4.-elute 5 mL ACN, mix, dry N2 40°C, 5.-dissolve in 1mL H2O, 6.-60 µL FA + 5 mL ACN, mix, 7.-apply to multistep #229 Ochra cartridge. 8.-Dry 4 mL of eluate with N2 40°C, 9.-dissolve in 500 µL AmAc 10 mM/ACN 85:15, 10.-filter
Acquity UPLC BEH C18, 100 x 2.1 mm, 1.7 µm at 40°C
Inj vol 5 µL, A) H2O; B) MeOH, with 2% AcOH, 0.1 mM AmAc
55-80% B in 5 min, initial conditions for 2 min
Flow: 0.3 mL/min, Time: tan=5 min, tTot= 7 min
QQQ
CaV 3 kV; ST 120°C; DGT 450 °C; CGF 50 L/h; DGF 800 L/h
LOD: 0.30 µg/L, LOQ: 1 µg/L all Fbs
(Miró-Abella et al. 2017)
B1, B2 and other toxins
Plant-based beverages10 mL
Extraction: 1.- Add 10 mL 1% FA in ACN in a 50 mL centrifuge tube, 2.- Shake 3 min; 3.- Add 4 g MgSO4 + 1 g NaCl; 4.- Shake vigorously 3 min; 5.- Centrifuge to 10000 rpm, 5 min, 20°C, 6.-dilute 1:1 with phase A 7.-filter
Cortecs UHPLC C18, 100 x 2.1 mm, 1.6 μm at 40°C
Inj vol 5 µL, A) H2O, B) MeOH, both with 0.1% AcOH, 5 mM AmAc
10-50% B in 4.5 min, 50-95% in 7.5 min, keep 2.5 min
Flow: 0.45 mL/min, Time: tan= 14.5 min, tTot= NR
QQQ
CaV 4 kV; DGF 18 L/min; DGT 160°C; nebulizer 35 psi; nozzle voltage 0.5 kV; Frag Vol 380 V
LOD: 0.80; LOQ: 2.68 µg/kg all Fbs
(B. Zhang et al. 2018)
B1 and other toxins
Grapes, wines5
Extraction: 1.- Add 5 mL distilled H2O, 10 mL 1% AcOH in ACN; 2.- Shake to 3000 rpm; 3.- Add 1 g NaCl + 4 g MgSO4, 4.- Centrifuge to 13000 rpm, 5 min, 10 °C; 5.- Transfer into 10 mL polypropylene tube containing 450 mg MgSO4; 6.- Shake 30 s; 7.-Centrifuge to 5000 rpm, 5 min, 10 °C
ZORBAX RRHD Eclipse Plus C18, 50 x 2.1 mm, 1.8 µm at 30°C,
Inj vol 2 µL, A) H2O, B) ACN, both with 0.1% FA
10-42% B in 2.4 min, 42-51% B in 3.6 min, 51-95% B in 0.2 min, 95-10% B for 0.8 min, initial conditions for 5 min
Flow: 0.3 mL/min, Time: tan=6.2 min, tTot= 12 min
QQQ
CaV 4 kV; DG temperature 350 °C; DG flow 10 L/min; Nebulizer 40 psi
LOD: 1 µg/L, LOQ: 3 µg/L
(Pizzutti et al. 2014)
B1, B2, B3 and other toxins
Wines5
Extraction: 1.- Add 5 mL H2O, 10 mL 1% AcOH in ACN, 25 µg/mL of: FB1 (ACN/H2O 1:2), FB2 (CAN/H2O 1:3), and FB3 (ACN); 2.-Mix to 300 rpm, 1 min; 3.- Add 3 g anh. MgSO4; 4.- Shake 1 min; 5.- Centrifuge 13000 rpm, 5 min, 6.-Take 3 mL of superior phase; 6.- Mix with 450 mg anh. MgSO4; 7.- Mix 10 s, centrifuge 4000 rpm, 4 min, 10 °C; 8.- Filter and dilute 1:1 with MeOH
Acquity UPLC BEH C18, 100 x 2.1 mm, 1.7 µm, 50 °C
Inj vol 5 µL A) H2O, B) ACN, both with 0.1% FA
10-70% B in 10 min, 90 % B for 2 min, initial conditions for 1 min
Flow: 0.4 mL/min, Time: tan=12 min, tTot= 13 min
QQQ
CaV 2 kV; ST 120 °C; DGT 400 °C; DGF 100 L/h; CGF 700 L/h
LOQ: 50 µg/kg all Fbs
(Pérez-Ortega et al. 2012)
B1 and other toxins
Wine4 mL
Extraction: Oasis HLB, Bond Elut Plexa 1.- SPE cartridges preconditioned with 4 mL MeOH, 2.- 4 mL H2O at 2 mL/min; 3.- Add sample into cartridge; 4.- Elute with MeOH/H2O 5:95; 5.- Dry in vacuum 1 min; 6.- Elute twice/4 mL MeOH, 1 mL/min; 6.- Evaporate (N2, 37°C); 7.-Reconstitute (1 mL MeOH:H2O 2:8); 8.- Filter
Zorbax Eclipse XDB-C18, 50 x 4.6 mm, 1.8 µm, temp NR
Inj vol 20 µL, A) H2O, 0.1% FA; B) ACN
10 % B for 2 min, 10-50% B in 3 min, 50-100% B in 10 min, keep 3 min
Flow: 0.5 mL/min, Time: tan=18 min, tTot= NR
TOF
CaV 4kV; NGP 40 psi; DGF 9 L/min; DGT 325 °C; Frag Vol 190 V; range 50 -1000
LOD: 0.8 µg/L, LOQ: 2.68 µg/L
Samples of animal origin
(Cao et al. 2018)
B1, B2
Urine, plasma200 µL urine
200 µL
plasma
Extraction: 1.- Add 50 µL β-glucuronidase + 20 µL SI (13C34-FB1 1 mg/mL); 2.-incubate 37 °C overnight; 3.-centrifuge to 10000 rpm, 5 min; 4.-take supernatant, add 730 µL H2O/ACN 90:10; 5.-filter1.- Add 50 µL β-glucuronidase + 20 µL SI
(13C34-FB1 1 mg/mL); 2.-incubate 27°C overnight; 3.-add 1mL ACN:AcOH 99:1; 4.-vortex 30 s; 5.-centrifuge to 5000 rpm, 10 min; 6.-dry at 45°C; 7.-reconstitute in 200 µL of H2O:ACN 9:1; 8.-mix 30 s; 9.-filter
Kinetex C18, 100 x 2.1 mm, 2.6 µm, 40°C
Inj vol 10 µL A) H2O, 0.2 mmol/L AcOH; B) MeOH
25% B for 1 min, 25-70% B in 2 min, 70-25% B in 0.5 min, initial conditions for 1.5 min
Flow: 0.2 mL/min, Time: tan=3 min, tTot= 5 min
QQQ, TISP
CUR 20 psi; CoG (CAD) 8 psi; GS1 20 psi; GS2 15 psi; GT 600℃; EP 10.0; CP 12.0
LOD B1: urine 0.12 µg/L, LOQ B1: urine 0.45 µg/L
LOD B1: plasma 0.19 µg/L, LOQ B1: plasma 0.39 µg/L
(Devreese et al. 2012)
B1 and other toxins
Pig plasma250 µL
Extraction: 1.- Add 12.5 µL 13C-34 FB1 (25 µg/mL in ACN) + 750 µL ACN (deproteinization); 2.-vortex 15 s; 3.-centrifuge to 8517 g, 10 min, 4°C; 4.-evaporate supernatant (N2, 45 °C); 5.-reconstitute with 200 µL H2O/MeOH 85:15; 6.-vortex 15 s, 7.-filter
Hypersil Gold C18, 50 x 2.1 mm, 1.9 μm at 45 °C
Inj vol 2.5-10 µL, A) H2O with 0.1% AcOH, B) MeOH
35 % B for 1.5 min, 90 % B in 0.5 min, keep 1.5 min, 90-35 % B in 0.2 min, initial conditions 2.3 min
Flow:0.30 mL/min, Time: tan=3.5 min, tTot= 6 min
QQQ
CaV 4 kV, ST 300 °C; Aux gas 18 au; ISGP 4 au; SGP 23 au; VT 300 °C;
LOD: 0.8 µg/L, LOQ: 1 µg/L
(Arroyo-Manzanares, García-Campaña, and Gámiz-Gracia 2013)
B1, B2
Milk thistle Silybum marianum
2
QuEChERS: 1.- Add 8 mL of 30 mM NaH2PO4 (pH 7.1); 2.-vortex 10 s; 3.-add 5 mL ACN with 5% FA; 4.- shake 2 min; 5.-sdd 4 g MgSO4 + 1 g NaCl + 1 g NaCit + 0.5 g Na2HCit 1.5 H2O; 6.- shake 1 min; 7.-centrifuge to 4500 rpm, 5min); 8.-take 1 mL; 9.- dry; 10.-reconstitute with 1 mL MeOH/H2O 1:1; 11.-filter
Zorbax Eclipse C18, 50 x 2.1 mm, 1.8 μm at 35 °C
Inj vol 5 µL, A) H2O, B) MeOH, both with 0.3% FA, 5 mM AmFo
5-50% B in 1 min, 50-72 % B for 2 min, 72-80 % B for 2 min, 80-90 %B for 2 min, 90-5% B in 0.2 min
Flow: 0.4 mL/min, Time: tan=7.2 min
QQQ
ST 500 °C; CUR 30 psi; ISV 5 kV; gas 1 and gas 2 50 psi
LOD: B1 3.9 µg/kg,13.7 µg/kg
LOQ: B1 13.5 µg/kg, B2 45.7 µg/kg
(S. Zhang et al. 2022)
B1, B2, B3
Broiler Chicken Feed and Excreta5
Extraction: 1.- Add 20 mL of ACN:H2O; 2.- shake for 30 min; 3.- ultrasonic for 30 min; 4.- take 50 μL; 5.- centrifuge at 8000 rpm for 15 min; 6.- add 950 μL of H2O and vortex; 7.- take 50 μL; 8.- add 10 μL of IS 13C-FBs; 9.- dilute with 850 μL of in MeOH:H2O 1:9 (0.2 % -FA)
CORTEX C18 10 x 4.6 mm, 5 μm at 40 °C
Vol. Inj NR, A) H2O B) MeOH both with 0.2 % -FA 10-90%B in 6 min; hold for 2 min; initial condition for 2 min
Flow 0.4 mL/min, Time: tan= 8 min tTot= 10 min
QQQ
CaV 2.5 kV; CoG: 0.15 mL/ min, DGT 500 °C; DGF: 800 L/h;
LOD: 50 μg/Kg all Fbs LOQ 160 μg/Kg all Fbs
(Weiying et al. 2022)
B1, B2
Milk1
Extraction: 1.- Add IS (13C34-FB1 117 (13C34-FB1), 13C34-fumonisin B2 (13C34-FB2) mixed internal standard (25 μg/mL); 2.- add 5 mL of ACN:H2O (2% FA); 3.- vortex for 10 min; 3.- Centrifuge at 3900 rpm for 3 min; 4.- evaporate to dryness at 40 °C under N2; 5.- redissolved in 5 mL of H2O; 6.- Add 6 mg of DSPME MIL-101 (Cr); 7.-ultrasonic for 10 min; 8.- centrifuge at 1200 rpm for 5 min; 9.- filter
Shimadzu C18 100 × 2.1mm, 1.8 μ m at 40 °C
Vol. Inj. 3 μL of sample, A) H2O (1% FA), B) CAN 5% B for 1 min
5 -90 %B in 3.5 min; hold for 2.5 min; initial condition in 0.1 min; hold for 1.9 min
Flow 0.4 mL/min, Time: tan= 8 min tTot= 10 min
Qtrap
CaV: 5.5 kV; CoG: 35 psi; CUR: 35 psi; GS2: 45 psi
LOD: 1.5 μg/Kg all Fbs LOQ 5 μg/Kg all Fbs
(Flores-Flores and González-Peñas 2018)
B1 B2, B3
Milk1 mL
Extraction LLE: 1.- Add 4 mL 2 % FA in ACN, 2.-shake 15 min; 3.-centrifuge 5000 rpm, 10 min, 4.-take 4 mL supernatant, 5.-add 60 mg NaOAc, 6.-shake 15 min, 7.- centrifuge 5000 rpm, 5 min, 8.-take 3.5 mL of ACN phase, dry at 65°C, 9.-reconstitute in 200 μL of mobile phase, 10.-filter
Ascentis Express C18, 150 x 2.1 mm, 2.7 μm, 45°C
Inj vol 20 µL, A) H2O, B) MeOH/H2O 95:5, both with 0.1% FA and 5 mM AmFo
5-28% B in 5 min, 28-45 in 5.5 min, 45-60% B in 0.5 min, 60-90% B in 5 min, keep for 1 min, initial conditions for 13 min
Flow: 0.4 mL/min, Time: 16 min
QQQ
CaV 4 kV; DGT (high-purity N2) 350°C; DGF 9 L/min; 275.8 Pa, dry gas 40 psi
LOD/LOQ: FB1 10 µg/L, FB2 2.5 µg/L, FB3 0.625 µg/L
(Song et al. 2013)
B1 and other toxins
Pig, human urine5 mL
Extraction: 1.- Add 10 mL MgSO4 (2 M) with EtOAc/FA 99:1, shake 15 min; 2.-centrifuge to 4000 g, 15 min; 3.-take aqueous phase, add 5 mL ACN/FA 99:1; 4.-repeat extraction; 5.-dry (N2, 60°C); 6.-reconstitute with 500 µL 1:1 A:B; 7.-filter; 8.-centrifuge to 10000 g, 5 min
Symmetry C18, 150 x 2.1 mm, 5 μm at RT
Inj vol 20 µL, A) H2O, B) MeOH, both with 0.3% FA, 5 mM AmFo
5% B for 1 min, 5-25% B in 4 min, 25-60%B in 2 min, 60-80% B in 8 min, 80-100 B in 1 min, keep 6 min, 100-5 % B in 3 min
Flow: 0.25 mL/min, Time: tan=22 min, tTot= 25 min
QQQ
CaV 3.2 kV; DGF 800 L/h; CGF 20 L/h; DGT 350 °C; ST 120 °C
LOD: 0.05 ng/mL, LOQ: 0.17 ng/mL
(K. Zhang et al. 2013)
B1, B2, B3 and other toxins
Milk based infant foods0.5
Extraction: 1.- Add 25 µL IS (13C34 FB1, 13C34 FB2, 13C34 FB3 500 ng/mL); 2.- Vortex 30 s; 3.- Add 5 mL ACN/H2O 1:1; 4.- Shake 10 min at 30-35 pulsations/min; 5.- Take an aliquot of 2 mL; 6.- Filter 2 mL; 7.-Centrifuge to 4500 rpm, 30 min
Phenomenex Kinetex XB-C18, 100 x 2.1 mm, 2.6 μm, 40°C
Inj vol 5 µL, A) H2O, B) MeOH, both with 0.1% FA, 10 mM AmFo
5-40% B lineal in 2 min, 40-100% exponential B in 7 min, keep 2.5 min, 100-5% B in 0.5 min, initial conditions for 3 min
Flow: 0.3 mL/min, Time: tan=11.5 min, tTot= 15 min
QQQ-IT
CaV 5.5 kV; CUR 30 psi; ST 450 °C; gas 1 and gas 2 60 psi
LOQ B1: 2 µg/kg all fbs
(Abia et al. 2013)
B1, B2 and other toxins
Urine1 mL
Extraction: 1.- Centrifuge to 5600 g, 3 min; 2.-take 100 µL 3.-add 900 µL H2O/ACN 9:1
Gemini 150 x 4.6 mm, 5 µm
Inj vol 5 µL, A) H2O, B) ACN, both with 0.1% AcOH
5 % B for 2 min, 5-30 % B in 8 min, 30-96 % B in 4 min, keep 1 min, initial conditions for 2.25 min
Flow: 0.6 mL/min, Time: tan=15 min, tTot= 17.25 min
QTrap
ST 650 °C, CUR 30 psi, SG 80 psi, DG 80 psi
LOD: B1 and B2 0.5 µg/L, LOQ: B1 and B2 1.7 µg/L
(Nualkaw et al. 2020)
B1, B2 and other toxins
Swine, Poultry, Dairy Feeds1
QuEChERS: 1.-Add 10 mL H2O 1% FA, 2.-soak 30
min; 4.-add 10 mL ACN: 5.-shake to 240 rpm, 30 min; 6.-add 1 g NaCl + 4 g MgSO4; 7.-shake 30 s: 8.-centrifuge to 10000 rpm, 5 min; 9.-take 2 mL; 10.-add 0.1 g silica C18 + 0.3 g MgSO4; 11.-mix; 12.-centrifugate 1 min; 13.-dry at 40 °C, 14.-reconstitute in 960 µL MeOH 20% + 40 µL (250 ng/mL 13C-34 FB1+50 ng/mL 13C-34 FB2); 15.-filter
Accucore C18, 100 x 2.1 mm, 2.6 µm, 25 °C
Inj vol 3 µL, A) deionized H2O, 0.1% FA, 5mM AmF; B) MeOH
0-20% B in 4 min, 20-40% B in 5.5 min; 40-100% B in 10.5 min, keep 2.5 min; initial conditions for 3 min
Flow: 0.4 mL/min, Time: tan=22.5 min, tTot= 25.5 min
Qtrap
Needle voltage 4.5 kV; CUR 30 psi; nebulizer (Gas1), turbo gas (Gas2) 55 psi; turbo gas temperature 500 °C
LOD: B1 15 µg/kg, B2 4.5 µg/kg; LOQ: B1 30 µg/kg, B2 9 ng/kg
(Osteresch et al. 2017)
B1 and other toxins
Blood or serum100 µL
Extraction LLE:
1.-Spott 4 times on filter paper; 2.-dry overnight at RT, 3.-Extract with 1 mL H2O/acetone/ACN 30:35:35 in 2 mL safe-lock tubes; 4.-Sonicate 30 min; 5.-Take 800 μL; 7.-Dry at 50°C under reduced pressure; 8.-Reconstitute with H2O/ACN/AcOH 95:5:0.1; 9.-Centrifuge to 22000 g, 10 min
Gravity SB C18, 100 x 2.0 mm, 3 µm at 45°C
Inj vol 30 μL, A) H2O, 0.1% AcOH, B) ACN, 2% AcOH
3-15% B in 3 min, 15-55% B in 1.5 min, keep for 1.5 min, 55-100% B in 2 min, keep 10 min, initial conditions 1.5 min
Flow: 0.75 (0-6), 0.85 (6.1-10), 0.75 (10.1-11.5)
mL/min, Time: tan=10 min, tTot= 11.5 min
QTrap
CaV 5.5 kV; ST 500 °C; DP 125 V; CUR 40 psi; GS1 45 psi; GS2 50 psi
LOD: 0.521 ng/L LOQ: 2.5 ng/mL
Ref
FBs
MatrixSample
(g)Sample treatment
Extraction procedureLC conditions
Column / Injection volume / Mobile Phase
Flow / Analysis TimeMS conditions
Mass Conditions / Limits
Maize and corn-based products
(Ren et al. 2011)
B1, B2, B3
Maize2.5
Extraction: 1.- Add 200 µL of IS (2.5 µg/mL 13C34- FB1, 1 µg/mL 13C34-FB2, 13C34-FB3); 2.- Add 10 mL ACN/H2O 1:1; 3.- Extract with ultrasonic 1h; 4.- Centrifuge to 15 000 rpm, 6 min; 5.- Adjust pH to 7-9 with NaOH; 6.- Take an aliquot of 3 mL; 7.-Dilute with MeOH/H2O (66.7:33.3)
Clean up: 1.- Load the dilute sample in MultiSep 211 FUM cartridge; 2.-Pass 8 mL of MeOH/H2O (66.7:33.3); 3.- Pass 10 mL of MeOH (1% AcOH), collect; 4.- Transfer 10 mL to a tube; 5.- Dry (N2, 50°C); 6.- Redissolve in 1 mL of MeOH:AmAc 10 mM/L (1:1); 7.- Shake 30s; 8.- Filter
BEH C18, 100 x 2.1 mm, 1.7 μm, 35°C
Inj vol 2 µL, A) H2O, 0.1% FA, B) ACN/MeOH 1:1
30-70% B in 2.3 min, 70% B for 1.7 min, 70-100% B in 0.2 min, keep for 0.6 min, 100-30% B in 0.2 min., re-equilibrate for 2 min
Flow: 0.3 mL/min, Time: tan=4.8min, tTot=7 min
QQQ
CaV 3.5 kV; CoV 45 V; ST: 120; CGF: 50L/h DGT 350°C; DGF 500 L/h
LOD:
(B1 0.45, FB2 0.50, B3 0.10) µg/kgLOQ:
(B1 1.50, FB2 1.65, B3 0.40) µg/kg
(L. Silva et al. 2009)
B1, B2 and other toxins
Corn-based products25
Extraction: 1.- Add 40 mL MeOH/H2O 80:20; 2.- Centrifuge to 2500 g, 15 min 3.- Extract the remaining solid with 30 mL MeOH/H2O 80:20; 4.- Filter
Clean up: 1.- Dilute 10 mL of filtrate with 40 mL of PBS; 2.- Take 20 mL 3.- Add to a FumoniTestTM immunoaffinity; 4.- Wash with 10 mL PBS; 5.- Eluted twice with 1.5 mL of MeOH; 6.- Evaporate (N2, 60 °C); 7.- Reconstitute in 50 μL MeOH/H2O (1:1)
Luna C18, 250 x 4.6 mm, 5 μm
Inj vol 10 µL, A) H2O, B) MeOH both with 0.5% FA
65% B for 4 min, 65-95% B in 4 min, keep 7 min
Flow: 0.50 mL/min, Time: tan=tTot=15 min
QQQ
CaV 4 kV, GT 350°C; DGF 13 L/min; NG 30 psi
LOD: 40 µg/kg, LOQ:110 µg/kg all Fbs
(Cavaliere et al. 2007)
B1, B2 and other toxins
Maize1
Extraction: 1.- 10 mL ACN/H2O 75:25; 2.- homogenize 15s; 3.- Transfer on cartridge (6 mL) with 100 mg of C18; 4.-Wash the extract with 7 mL of ACN/H2O 75:25, twice; 5.- Collect 25 mL; 6.- Take 5 mL; 7.- Dilute with 500 mL of H2O.
Clean up: 1.- Load sample dilute on SPE-Carbograph-4 (500 mg); 2.- Wash with 10 mL of H2O; 3.- Pass 0.3 mL of MeOH; 4.- Elute with 1 mL MeOH and 8 mL of DCM:MeOH 8:2 (50 mM of FA); 5.- Evaporate to 100 µL; 6.- Add IS (FB1, FB2 in MeOH/H2O 1:1 (1 mg/mL); 7.- Evaporate to 100 µL; 8.- Dilute with 100 µL of LC mobile phase
Alltima C18, 250 x 2.1 mm, 5 µm, 45 °C
Inj vol 20 µL, A) H2O, B) MeOH, both containing 25 mmol/L FA, adjusted to pH 3.8 with ammonia
60% B for 3 min, 60-90% B in 5 min, 100% for 10 min
Flow: 0.2 mL/min, Time: tan= tTot=18 min
QQQ
CoV 5.5 kV; CUR 35; GS1 35; GS2 40; GT 350 °C
LOD/LOQ: 10 mg/kb for FB1 and 5ng/kg FB2
(Lattanzio et al. 2007)
B1, B2 and other toxins
Maize10
Extraction: 1.- Add 50 mL de PBS; 2.- Shake 60 min; 3.- Centrifuged to 3000 g, 10 min; 4.- filtrate 35 mL of PBS (extract A); 5.- Add 35 mL of MeOH, to the remain solid, containing 15 mLPBS; 6.- extract again 7.- Shake 60 min; 7.- Centrifuge to 3000 g, 10 min; 8.- Dilute 10 mL of extract with 90 PBS (extract B); 9.- Filter
Clean up:
1.-Load 50 mL of extract B to the IAC; 2.- Wash with 20 mL of PBS; 3.- Add 5 mL of extract A; 4.- Wash with 10 mL of water; 5.- Eluate both extracts with 1.5 mL MeOH twice; 6.- Dry at 50 °C; 7.- Reconstitute with 200
L MeOH/H2O 4:6 (1 mM AmAc and 0.1% AcOH)
Gemini C18, 150 x 2 mm, 5 µm, 40°C
Inj vol 20 µL, A) H2O (0.5% AcOH, 1 mM AmAc)
B) MeOH (0.5% AcOH, 1 mM AmAc
20-40% B in 3 min, 40-63% B in 35 min, keep constant for 11 min, initial conditions for 10 min
Flow: 0.200 mL/min, Time: tan=49 min, tTot=59 min
QTrap
GT 350 °C; CUR 30 PSI; CoV: 4.5 kV: GS1: 10 psi, GS2 30 psi.
LOD: B1 1.1 µg/kg, B2 0.4 µg/kg
(Y. Wang et al. 2013)
B1 and other toxins
Maize10
Extraction: 1.- Add 50 mL of ACN/H2O/AcOH (79:20:1); 2.- Stir for 10 min; 3.- Filter; 4- Evaporate 10 mL to dry; 5.- Redissolve in 100 µL of MeOH; 6.- Vortex 1 min; 7.- Add 1.9 mL of H2O 8.- Vortex again for 1 min
Clean up: 1.- Active the Oasis HLB SPE cartridges with 2 mL of MeOH; 2.- Equilibrate with MeOH/H2O (05:95); 3.- Load sample; 4.- Wash with 2 mL MeOH/H2O (05:95); 5.- Elute with 2 mL of MeOH; 6.- Dry (N2, 50°C); 7.- Redissolve in 1 mL MeOH/H2O (2:8)
Shimadzu XR-ODS 75 x 3.0 mm, 2.2μm, 30°C
Inj vol 20 µL, A) H2O, B) MeOH both with 0.1% AcOH, 1 mM AmAc
50% B for 5 min, 50-10% B in 5 min, keep constant for 10 min, 10-50% B in 1 min, keep constant for 4 min
Flow: 0.30 mL/min, Time: tan=21 min, tTot=25 min
QTrap
GT 450°C; CUR 10 psi; GS1 50 psi; GS2 50 psi; SV 5.5 kV
LOD: 0.64 μg/kg, LOQ: 2.12 µg/kg
Other cereals and seeds
(Bryła, Renata, et al. 2013)
B1, B2, B3
Cereal products25
Extraction: 1.-Add 100 mL ACN/MeOH/H2O (25:25:50); 2.- Stir 30 min; 3.- Centrifuge to 10730 g, 10 min; 4.- Dilute the supernatant 1:1 with 10 mL deionized H2O
Clean up: 1.- Transfer 8 mL of dilute extract to a FumoZon cartridge; 2.- Preconditionate with 4 mL of MeOH and H2O; 3.- Wash with 6 mL ACN/H2O (25:75); 4.- Eluate with 4 mL of 2% FA in MeOH; 5.- Evaporate to dry; 6.- Redissolve in 1 mL of MeOH/H2O/AcOH (1:8.9:0.1)
Kinetex PFP, 100x2.1mm, 2.6μm
Inj vol 25 µL, A) MeOH:H2O:AcOH (20:79.9:0.1) B) MeOH:H2O:AcOH (79:19.9:0.1)
20% B for 4 min, 20-55% B in 6 min, keep constant for 15 min, 55-100% in 5 min, keep constant for 10 min, initial conditions for 20 min
Flow: 0.15 mL/min, Time: tan=40 min, tTot=60 min
IT
GF 45 a.u.; AGF 10 a.u.; CoV 4.5 kV; CaV 40 V; ST 260 °C
LOQ: 25 µg/kg all FBs
(Vaclavikova et al. 2013)
B1, B2, B3 and other toxins
Cereals, nuts5
Extraction:1.- Add 20 mL of ACN/H2O/AcOH (79.5:20:0.5) for 60 min; 2.- Centrifuge to 5000 rpm, 2 min; 3.- Dilute 2 mL of sample with 33 mL of PBS
Clean up: 1.- Load the aliquot on IAC; 2.- Wash with 10 mL of ultrapure H2O; 3.- Elute with 3 mL of MeOH, evaporate; 4.- Reconstitute in 0.5 mL of MeOH/H2O (0.5% AcOH) (1:1); 6.- Filter
Acquity UPLC HSS T3 RP 100 x 2.1 mm, 1.7μm, 40°C
Inj vol 10 µL, A) H2O, B) MeOH both with 5 mM AmAc
5-50% B in 1 min, 50-100% B in 6 min, keep 1 min, initial condition for 2 min.
Flow: 0.4 mL/min, Time: tan=8 min, tTot=10 min
QTrap
ST 450°C; CaV 4.5kV; CUR 20 a.u.; GS1 55 a.u, GS2: 55 a.u
LOD: 5 μg/kg, LOQ: 10 µg/kg all FBs
(Arroyo-Manzanares et al. 2014)
B1, B2 and other toxins
cereals, spelt, rice2
QuEChERS: 1.- Add 8 mL H2O into test tube; 2.- Shake for 10 s; 3.- Add 10 mL 5% FA in ACN; 4.- Shake 2 min; 5.- Add 4 g MgSO4, 1 g NaCl, 1 g sodium citrate, 0.5 g Na2HCit 1.5 H2O; 5.- Shake for 1 min; 6.- Centrifuge to 4500 rpm, 5 min; 7.- Transfer 2 mL of upper layer to a vial; 8.- Evaporate; 9.- Reconstitute with 1 mL of MeOH/H2O 50:50; 10.- Filter
Zorbax Eclipse Plus RRHD C18, 50 x 2.1 mm, 1.8μm, 35°C
Inj vol 5 µL, A) H2O, B) MeOH both with 0.3% FA, 5 mM AmFo
5% B for 1 min, 5-50% B in 1 min, 50-72% B in 2 min, 72-80% B in 2 min, 80-90% for 2 min, initial conditions in 0.2 min.
Flow: 0.4 mL/min, Time: tan=8 min, tTot=8.2 min
QQQ
GT: 500°C; CUR: 30 psi; CaV 5 kV; GS1 and GS2 50 psi
LOD: B1 0.20, B2 0.30 µg/kg
LOQ: B1 0.65, B2 1.01 µg/kg
(Cendoya et al. 2019)
B1, B2
wheat-based products25
Extraction: 1.- Add 50 mL of MeOH/H2O 3:1; 2.- Shake for 30 min; 3.-Filter
Clean up: 1.- Precondition with 5 mL of MeOH and 5 mL MeOH/H2O 3:1; 2.- Load 10 mL of filtrated; 3.- Wash with 8 mL of MeOH/H2O 3:1, 3 mL of MeOH; 4.- Elute with 14 mL of MeOH with 0.5% AcOH ; 5.- Dry (N2, 40°C)
XBridge™ C18, 150 x 2.1 mm, 3.5μm, 20°C
Inj vol 45 µL, A) H2O, B) MeOH both with 1% FA
9.5% B for 2 min, 9.5-50% B in 1 min, 50-97.5% B in 11 min, keep for 3 min, initial condition for 5 min.
Flow: 0.2 mL/min, Time: tan=17 min, tTot=22 min
QQQ
CaV 3.0 kV; ST: 150 °C; DGT 200 °C; DGF: 726 L/h; GF 109 L/h
LOD 0.01 µg/kg LOQ: 0.05 µg/kg all FBs
Products of animal origin
(Gazzotti et al. 2009)
B1
Bovine milk10
Extraction: 1.- Centrifuge to 6000 rpm, 15 min; 2.- Dilute 5 mL of sample 1:1 with H2O
Clean up: 1.- Load the dilute sample to Vicam FumoniTestTM
Immunoaffinity at 1 drop/s; 2.- Wash with 20 mL of PBS buffer at 5 mL/min; 3.- Elute with 1.5 mL of MeOH; 4.- Pass 1.5 mL of H2O, collect 3 mL; 5.- Evaporate 3 mL of eluate to 1 mL (40°C, N2)
XTerra MS C18, 150 x 2.15 mm, 5μm, 35°C
Inj vol 10 µL, A) H2O/ACN (90:10) with 0.3% FA, B) ACN (0.3% FA)
Elute isostatically with 75% A-25%B for 2 min, wash 80% B for 3 min
Flow: 0.30 mL/min, Time: tan=2 min, tTot=5 min
QQQ
CaV 3.25 kV; CoV 50 V; IST 140°C; DGT 400°C
LOD: 0.003 μg/kg, LOQ: 0.1 µg/kg
(Gazzotti et al. 2011)
B1 B2, HFB1 HFB2
Pig liver1
Extraction: 1.- Homogenize in 6 mL of MeOH/H2O 80:20; 2.- Stir for 20 min; 3.- Centrifuge to 3000 rpm, 5 min; 4.- Wash twice with 6 mL of hexane; 5.- Evaporate aqueous phase; 6.- Reconstitute with 2 mL of aqueous buffer with 2% of AcOH, 0.1% Et3N (pH 3.4)
Clean up: 1.- Condition the Oasis HLB SPE cartridges with 2 mL of MeOH and 2 mL of H2O; 2.- Load the sample; 3.- Wash twice: first 1 mL MeOH/H2O (05:95), then 1 mL MeOH/H2O/AcOH (05:94:01); 4.- Elute with 2 mL of MeOH; 5.- Evaporate to 200 μL; 6.- Reconstitute in 1mL of mobile phase of LC
XTerra MS C18, 150 x 2.15 mm, 5μm, 35°C
Inj vol 10 µL, A) ACN/H2O (90:10); B) ACN both with 0.3% FA
25% B for 4 min, 25-40% B in 4 min, keep for 4 min,
initial condition for 5 min
Flow: 0.30 mL/min, Time: tan=12 min, tTot= 17 min
QQQ
CaV 3.25 kV; ST 140 °C; GT 400 °C; GF 50 L/h; DGF 890 L/h
LOD: 0.05 μg/kg, LOQ: 10 µg/kg all FBs and analogues
(Sørensen, Mogensen, and Nielsen 2010)
B1, B2
Meat products0.7
Extraction: 1.- Add 140 μL of IS (13C-FB2 0.5 μg/mL), 4.5 mL of H2O, 2.5 mL of ACN, 6 mL of pentane; 2.- Shake for 1 h; 3.- Centrifuge to 8000 g, 10 min; 4- Discard upper phase; 5.- Transfer 3.5 mL of lower phase; 6.- Add 9 mL of acetone; 7.- Shake; 8.- Centrifuge to 8000 g, 10 min; 10.- Collect 100 mL upper phase; 11.- Evaporate to 1.5 mL (45°C), reconstitute in 0.25 mL of MeOH
Clean up: 1.- Load sample in Oasis (MAX) SPE cartridges; 2.-condition with 1 mL of MeOH followed by 1 mL of H2O, wash with 1 mL of 1% aqueous ammonia; 1 mL of MeOH/H2O/HCl 37% (40:59:1); 3.- elute with 2 mL of 2% AcOH in MeOH; 4.- evaporate (N2, 45°C), re-dissolve in 200 µL ACN/H2O (1:2).
Gemini C6-phenyl 50 x 2 mm, 3μm, 40°C
Inj vol 1 µL, A) H2O, B) ACN both with 20mM FA
20-55% B in 6 min, then 100% in 0.5 min, keep for 2.5 min.
Flow: 0.30 mL/min, Time: tTot=9 min
QQQ
IST 120°C; DGF 700L/h; DGT 350°C
LOD: B1 64 µg/kg, B2 6 µg/kg,LOQ: B1 and B2 150 µg/kg
(Liliana J.G. Silva et al. 2010)
B1, B2
Urine10 mL
Extraction: 1.- Filter, 2.- Dilute 1:1 with 10 mL of PBS, 3.- Mix for 3 min
Clean up: 1.- Load the sample in FumoniTestTM immunoaffinity column; 2.- Wash with 10 mL PBS; 3.- Elute with 5 mL of MeOH; 4.- Dry (N2, 60°C); 5.- Redissolve in 1 mL of MeOH/H2O (1:1)
Luna C18, 150 x 4.6mm, 5μm, 30°C
Inj vol 20 µL, A) H2O, B) MeOH both with 0.5% FA
65% B for 3 min, 65-75% B in 4 min, keep for 8 min, initial condition for 10 min
Flow: 0.50 mL/min, Time: tan=15 min, tTot=25 min
QQQ
CaV 3.20 kV; ST 125 °C; DGT 300°C; DG 500 L/h
LOD: 5 µg/L LOQ: 10 µg/L all FBs
(Šarkanj et al. 2018)
B1 and other toxins
Urine500 µL
Extraction: 1.- Centrifuge to 5600 g, 3 min; 2.- Incubate with 500 µL PBS (200 mM, pH 7.4) containing 3000 U of β-glucuronidase, 16 h, 37 °C
Clean up: 1.- Precondition with 1mL MeOH, 1mL H2O; 2.- Add sample to Oasis PRiME HLB; 3.- Wash twice with 500 µL H2O; 4.- Eluate with 200 µL ACN x 3; 5.- Evaporated (N2); 6.- Reconstitute with 470 µL of 10% ACN, 0.1% AcOH, add 30 µL IS (0.38 ng/mL 13C-FB1)
Acquity HSS T3, 100 x2.1 mm, 1.8μm, 35°C
Inj vol 10 µL, A) H2O, B) ACN, both with 0.1% AcOH
10% B for 2 min, 10-50% B in 13 min., 50-95% B in 5 min, hold 4 min, initial condition for 3 min.
Flow: 0.1 mL/min, Time: tan= 24 min, tTot= 27 min
Qtrap
ISV 4.50 kV; ST 550°C; CUR 30 psi; SG 80 psi; DG 80 psi
LOD: 0.001 µg/L, LOQ: 0.01 µg/L
Bevearages
(Nakagawa et al. 2020)
B1, B2, B3
Domestic wine5 mL
Extraction: 1.- Add 0.1 mL of IS (13C34-FB1 0.2 mg/L in acetonitrile: water (1:1); 2.- adjusted volume at 10 mL with wine; 3.- mix; 4.- add 8mL of PBS (1% PEG, 5% NaHCO3; 5.- mix;
Clean up:
1.- Equilibrate with 3 mL of PBS; 2.- Load sample in cartridge; 3.- Wash 6 mL (3 mL x 2 times) of H2O (0.5% NaHCO3) and 6 mL (3 mL x 2 times) of 10 mM AmAc; 4.- Elute with 3 mL of MeOH (2% AcOH); 5.- evaporate to dryness; 6.- reconstitute in 0.2 mL ACN:H2O 1:1.
ZORBAX Eclipse XDB-C18 250×3 mm, 5μm, 40°C
Inj vol 3–20 μL, A) H2O, B) ACN, both with 0.1% FA
10% B for 3 min, 10-90% B in 15 min, hold for 5 min, initial conditions for 10 min.
Flow: 0.3 mL/min, Time: tan= 20 min, tTot= 30 min
Qtrap
CaV 5 kV; CUR 10 psi; GS1 70 psi; SG 60 psi; ST 500°C
LOD: 1 μg/Kg all Fbs LOQ 2
(Romero-González et al. 2009)
B1, B2
Beer10 mL
Extraction: 1.- Sonicate for 20 min
Clean up: 1.- Precondition with 5 mL ACN/H2O (60:40) and 5 mL of H2O; 2.- Load sample in C18 cartridge; 3.- Wash with 5 mL of H2O; 4.- Elute 2 mL ACN/MeOH 60:40; 5.-Filter
Acquity UPLC BEH C18, 100 x 2.1 mm, 1.7μm, 30°C
Inj vol NR, A) H2O, B) MeOH both with 5 mM AmFo
25 to 100% B in 3.75 min, keep 1.25 min, 100 to 25% B in 0.5 min, initial condition for 1 min
Flow: 0.35 mL/min, Time: tan= 5.5 min, tTot= 6.5 min
QQQ
CaV 3.5 kV; ST 120; DGT 350°C; CGF 80 L/h; DGF 600 L/min
LOD: B1 0.07, B2 0.09 µg/kg; LOQ: B1 0.23, B2 0.30 µg/kg
(Tamzura, Uyama, and Mochizuki 2011)
B1, B2, B3, and other toxins
Beer-based drinks10 mL
Extraction: 1.- Sonicate for 15 min, 2.- Add 10 mL ACN, mix, 3.- Add the content of dSPE citrate extraction tube; 4.- Vortex for 20 s, 5.- Centrifuge to 2380 g, 5 min
Clean up:
1.- Precondition with 5 mL ACN; 2.- Load sample in InertSep C18, SPE; 3.- Elute with 5 mL ACN; 4.- Evaporate to dryness; 4.- Dissolved with 500 µL of 10 mM AmAc aqueous/ACN (85:15); 6.- Filter
Acquity UPLC BEH C18, 50 x 2.1 mm, 1.7μm, 40°C
Inj vol 5 µL, A) H2O, B) MeOH (2% AcOH, 0.1 mM AmAc)
55-80% B in 2 min
Flow: 0.50 mL/min, Time: tan= tTot=2 min
QQQ
CaV 3 kV; IST 120°C; DGT 450°C; CGF 50 L/h; DGF 800 L/h
LOQ: 5 µg/L all FBs
Other samples
(di Mavungu et al. 2009)
B1, B2, B3 and other toxins
Food supplements1
Extraction: 1.- Add 25 mL of AcOEt/FA 95:5 for 30 min ; 2.- Centrifuge; 3.- Evaporate 20 mL to dryness; 4.- Reconstitute in 5 mL of H2O/MeOH 1:1 and 10 mL Hex; 5.- Shake, 6.- Transfer aqueous fraction into a tube; 7.- Add H2O/MeOH 1:1 (2 x 5mL); 8.- Evaporate; 9.- Reconstitute in 400 µL H2O/MeOH 1:1; 10.- Centrifuge to 14000 g, 10 min; 12.- Take 250 µL, 13.- Filter; 14.- Dilute in 25 mL H2O
Clean up: SPE: 1.- Condition with 10 mL CH2Cl2/MeOH 8:2 with 50 mM FA, then 5 mL MeOH, 20 mL acidified H2O (10 mM HCl), finally 10 mL H2O; 2.- Add obtained solution to Oasis HLB SPE cartridge; 3.- Wash 10 mL H2O; 4.- Elute with 1 mL MeOH and 4 mL CH2Cl2/MeOH 8:2; 5.- Evaporate; 6. Reconstitute in 100 µL injection solvent; 7.- centrifuge 14000g for 10 min.
Symmetry C18, 150 x 2.1 mm, 5μm, RT
Inj vol 20 µL, A) H2O/MeOH/AcOH 94:5:1, B) MeOH/H2O/AcOH 97:2:1 both with 5mM AmAc
5-65 % B in 7 min, 65-75% B in 4 min, 75-100% B in 2 min, keep for 2 min, 100-60% B in 1 min, 60-40% B in 6 min, 40-5% B in 1 min, hold 2 min.
Flow: 0.3 mL/min, Time: tan= 16 min, tTot= 25 min
QQQ
CaV 3.2 kV; ST 150°C; DGT 350°C; CGF 20 L/h;
DGF500 L/h
LOD: B1 1, FB2 0.3, FB3 1 µg/kg
LOQ: B1 3, FB2 1, FB3 3 µg/kg
(Khayoon et al. 2010)
B1, B2
Food, feed10
Extraction: Add 40 mL ACN/H2O (1:1); 2.- Shake 5 min; 3.-Filter
Clean up: 1.- Take 1 mL of filtrate; 2.- Add 2.5 mL of 1% KCl; 2.- Precondition with 5 mL of MeOH, follow of 5 mL 1% KCl solution; 3.- Load in C18, SPE; 4.- wash with 3 ml 1% KCl, followed by 2 mL of ACN/1% KCl 1:9; 5.- Elute with 2 mL of MeOH/H2O (1:1)
Inertsil ODS, 350 x 2.1mm, 3μm, 40°C
Inj vol 20 µL, A) H2O, B) MeOH both with 0.2% FA
50-75% B in 4.0 min, 75-100% in 2.0 min, keep 6.5 min, B 100-50% in 3min
Flow: 12.5 min 0.20 mL/min, 3 min 0.3 mL/min, Time: tan=12.5, tTot=15.5 min
QQQ
CaV 4 kV; DGF 600 L/h; DGT 350°C
LOD: B1 10, B2 40 µg/kg
LOQ: B1 40, B2 130 µg/kg
(Jerome Jeyakumar, Zhang, and Thiruvengadam 2018)
B1, B2 and other toxins
Fungal cultures: Maize, AsparagusNR
Extraction: 1.- Add 25 mL AcOEt to cultures, shake to 8000 rpm; 2.-
After 2 h, mix with 5% acetone, isopropanol; 3.- Extracted with AcOEt 1:1; 4.- Collect upper layer, 6.- Evaporate; 7.- Reconstitute 10 mL isopropanol
Clean up SAX: 1.- Add 10 mL sample into cartridge; 2.- Eluate 3 mL MeOH followed by 5 mL of 1% KCl; 3.- Collect into a 5-mL tube; 4.- Dry
Supelco C18, 250 x 2.1 mm, 5μm
Inj vol 10 µL, A) H2O, 0.1% FA, B) CAN
15% B, 5 min, 15-100% B in 35 min, keep 10 min; 100-15 % B in 1 min, keep 9 min.
Flow: 0.2 mL/min, Time: tan=50 min, tTot=60 min
Qtrap
CaV 5 kV; ST 200°C; DGT 300°C, NGF 2μmL/min
LOD/LOQ: NR
(Facorro, Llompart, and Dagnac 2020)
B1, B2
Mixed Feed Rations2
QuEChERS: 1.- Add 10 mL of ACN/FA 90:10; 2.- Shake for 1 h, 25°C; 3.- Add 0.5 Na citrate sesquihydrate+1g NaCitrate+1g NaCl+4g MgSO4; 4.- Shake for 1 min; 5.- Centrifuge to 3398 g, 5 min
Clean up: 1.- Discard of supernatant; 2.- Load 1 mL in SPE Oasis PRiME HLB cartridge (3cc, 150 mg), collect; 3.- Transfer to a 2 mL dSPE tube; 4.- Add 150 mg MgSO4+50 mg PSA+30 mg C18 silica+30 mg Al-N; 4.- Centrifuge to 2360 g, 2 min; 5.- Take 500 µL, evaporate; 6.- Reconstitute with 350 µL of MeOH
Kinetex C18, 50 x 2.1 mm, 2.6 µm, 40°C
Inj vol 10 µL, a) H2O, B) MeOH, both buffered with 3 mM AmFo or AmAc.,
10%-100% B in 8 min, keep 7 min
Flow: 0.25mL/min, Time: tan=tTot=15 min
QTOF
CaV 5.5 kV; ST 550 °C, CUR 50 a.u.
LOQ: B1 2.9, B2 2.4 µg/L
(Jia et al. 2014)
B1, B2, B3 and other toxins
Dairy products15
Extraction: 1.- Add 10 mL MeOH/H2O (84:16) with 1% AcOH; 2.- Vortex 1 min, add 6 g MgSO4+1.45 g sodium acetate anhydrous; 3.- Shake for 1 min; 4.- Centrifuge to 4000 rpm, 5 min
Clean up: 1.- Add 8 mL of upper phase+1.2 g MgSO4+108 mg PSA+405 mg C18 silica to dSPE tube; 2.- Shake for 1 min; 3.- Centrifuge to 4000 rpm, 5 min; 4.- Transfer 200 µL; 5.- Add 300 µL of MeOH+500 µL 8 mM AmFo; 6.- Vortex 30 s; 7.- Filter 1 mL
Thermo Accucore C18, 100 x 2.1 mm, 2.6μm
Inj vol 5 µL, A) H2O, B) MeOH, both 0.1% FA, 4 mM AmFo
0% B for 1 min, 0-100% B in 6 min, keep 5 min, 100-0% B in 1 min, initial condition for 2 min
Flow: 0.30 mL/min, Time: tan=12 min, tTot=15 min
Q-Orbitrap
CaV 3kV; ST 320 °C; GT 350 °C; SG 18 L/min, Aux 3 L/min
LOD/LOQ: NR
(Monbaliu et al. 2009)
B1, B2, B3 and other toxins
Sweet pepper3
Extraction: 1.- Add 15 mL AcOEt, FA 1%); 2.- Shake 15 min; 3.- Centrifuge to 3300 g, 5 min; 4.- Filtrate; 5.- Repeat this process with 10 mL of the same mix solvent; 6.- Keep an aliquot (10 mL) for the SAX; 7.- Evaporate remaining part to 5 mL
Clean up: 1.- SPE: pass remaining through the NH2-SPE column; 2.- evaporate; 3.- Redissolve the evaporate in 3 mL of ACN/H2O (84:16); 4.- Pass through the SPE; 3.-SAX, evaporate aliquot to dry; 5.- Redissolve in 5 mL MeOH/H2O (75:25); 5.- Adjust pH at 5.8-6 with NaOH 0.25 M; 6.- Wash with 4 mL MeOH/H2O (75:25) and then 4 mL of MeOH; 7.- Elute with 4 mL MeOH, AcOH 1%; 8.- Evaporate; 9.- Redissolve in 100 µL H2O:MeOH:AcOH (57.2:41.8:1) and 5 mM of AmAc; 10.- Centrifuge to 14000 g, 15 min
Symmetry C18, 150 x 2.1 mm, 5 μm, 25 °C
Inj vol 20 μL, A) H2O/MeOH/AcOH (94:5:1). B) MeOH/H2O/AcOH (97:2:1) both with 5 mM AmAc
5-65% B in 7 min, 65-75% B in 4 min, 100% B for 2 min, initial conditions for 12 min
Flow: 0.30 mL/min, Time: tan=13 min, tTot=25 min
QQQ
CaV: 3.2 kV, ST: 150 °C, DGT: 350 °C
LOD: B1 13, FB2 6.5, B3 8.4 µg/kg
LOQ: B1 27, FB2 13, B3 17 µg/kg
(de Smet et al. 2009)
B1, B2, B3
Bell pepper, rice, corn flakes1
Extraction:1.- Add 8 mL of ACN/H2O 84:16; 2.- Shaker 30 min; 3.-Centrifuged to 2670 g, 20 min; 4.- Evaporate
Clean up: 1.- Condition with 2 mL MeOH; 2.- Wash with MeOH/H2O 75:25; 3.- Redissolve sample in 2 mL of MeOH/H2O (75:25); 4.- Adjust pH 5.8-6.5 with 0.1M NaOH; 5.- Eluate with 2 mL MeOH/FA 95:5; 6.- Evaporate; 7.- Redissolve in 100 µL of H2OACN 60:40 with 0.3% FA
Altima C18, 150 x 3.2 mm, 5μm
Inj vol 20 μL , H2O/ACN (60:40) with 0.3% FA
Isocratic condition
Flow: 0.3 mL/min, Time: 12 min
QQQ
CaV 3.6 eV; ST 140 °C; DGT 350 °C
LOD: B1 20, B2 7.5, B3 12.5 µg/kg
LOQ: B1 40, B2 15, B3 25 µg/kg
[Ref]
FBs
MatrixSample
(g)Sample treatment
LC conditions
Detector conditions, Limits
Maize and corn-based products
(Wall-Martínez et al. 2019)
B1, B2 and other toxins
Tortilla
25
Extraction:1.- Dry the tortilla at 60°C for 2.5 h; 2.- Milled and homogenize for 15 min at 30 rpm; 3.- Add 50 mL MeOH/H2O 80:20; 4.- Shake for 2 min; 5.- Centrifuge to 4000 rpm, 10 min; 6.- Take 10 mL of supernatant; 7.- Dilute adding 40 mL of PBS
Clean up IAC R-Biopharm: 1.- Precondition with 20 mL PBS (5.0 mL/min); 2.- Load 10 mL of sample diluted on the cartridge; 3.- Wash with 1.5 mL MeOH (0.5-1.0 mL/min) and 1.5 mL of H2O
Derivatization: Mix 100 μL of diluted extract with 100 μL OPA reagent (120 mg OPA, 3mL MeOH, 12 mL Na2B4O7· H2O 0.1 M, 179 µL 2-mercaptoehtanol) prior to injection.
Uptisphere type 5 ODB, ODS, 250 x 4.6 mm, 5μm, 40°C
Iny vol 10 µL, A) 99% H2O, B) ACN both with 1 % AcOH
41 % B 9 min, 61 % B for 7 min, keep 4 min, initial conditions for 5 min
Flow: 0.8 mL/min, Timea tan= 20 min, tTot=25 min
FDA
λex : 360 nm
λem: 450 nm
LOD: B1 0.13, B2 0.04 µg/kg
LOQ: B1 3.0, B2 2.7 µg/kg
(Caldas and Silva 2007)
B1, B2 and other toxins
Corn base food products
25 cornmeal, precooked corn flour, popcorn, sweet corn, corn flakes
12.5
corn snacks
Extraction: 1.- Add 100 mL MeOH/H2O (3:1) (cornmeal, PCF, popcorn, corn snacks), 50 mLMeOH/H2O (4:1) + 2.5 g NCl (sweet corn) 100 mL MeOH:0.4 M sodium tetraborate (3:1) (corn flakes), 2.- filter
Clean up SAX: 1.- Precondition with 5mL MeOH:H2O 1:1; 2.- Add 10 mL of filtrate on SAX column; 3.- Wash with 5mL MeOH:H2O 3:1; 3.- Elute 12 mL MeOH/AcOH (99:1) + 4 mL MeOH:AcOH (95:5) (cornmeal, PCF, popcorn, corn snacks), 12 mL MeOH/AcOH (99:1) + 8 mL MeOH:AcOH (95:5) (sweet corn, corn flakes); 4.- Dry at 40°C; 5.- Reconstitute in 500 μL
Derivatization: 1.- Add 480 μL of 0.05 M sodium borate buffer (pH 9.5), 170 μL sodium cyanide solution (0.013%) and 50 μL of 0.5 mg/mL naphthalene-2,3 dicarboxaldehyde (NDA) in MeOH; 2.- Vortex, 3.- Heat at 60 °C for 15 min, and cooled, 4.- Add 2.8 mL of 0.05 M phosphate buffer
C18, 150 cm x 4.6 mm, NR
Iny vol 10µL, A) H2O, B) ACN both 2.5% AcOH
55-80% B in 5 min, keep 8 min, initial conditions for 1min
Flow: 1 mL/min, Time: tan= 13 min tTot= 14 min
FDA
λex : 420 nm
λem: 500 nm
LOQ: 127-2040 µg/kg depending on the matrix
(Chiara Dall’Asta, Mangia, et al. 2009)
B1, B2, B3
Ground corn25
Extraction: 1.- Add 100 mL H2O/ACN 1:1; 2.- Shake for 1h; 3.- Filter; 4.- Adjust to pH 6-9 with 0.5 N NaOH; 4.-Take 3 mL, place into test tube; 5.- Add 8 mL MeOH/H2O 3:1
Clean up SPE MultiSep 211 Fum: 1.- Precondition with 5 mL MeOH, then 5 mL MeOH/H2O 3:1; 2.- Load sample 3.-Wash with 8 mL MeOH/H2O, then 3 mL MeOH; 4.-Elute 3:1 MeOH/AcOH 99:1; 5.- Dry at the eluate to 60oC; 6.- Reconstitute with 1 mL MeOH
Derivatization: 1.- Add 1 mL sodium borate buffer (0.05 M, pH 9.5), 0.5 mL NaCN reagent (13 mg NaCN in 100 mL water) and 0.5 mL NDA reagent (25 mg NDA in 100 mL MeOH). 2.- Heat for 20 min in a 60°C water bath and cooling for 4 min at 8°C, 3.- Dilute with 7 mL of phosphate buffer (0.05 M, pH 7.4)/ACN (2:3)
Brownlee C18, 100 x 4.6, 5μm, NR
Inj vol 80 µL, H2O/ACN/AcOH 52:47:1
Isocratic
Flow: 0.2 mL/min, Time: NR
FDA
λex: 420 nm
λem: 500 nm
LOD/LOQ: < 100 µg/kg
(L. J.G. Silva et al. 2007)
B1, B2
Maize base products25
Extraction: 1.- Add 40 mL MeOH/H2O 4:1; 2.- Centrifuge 2500 g, 15 min; 3.- Extract remaining solid twice with 30 mL MeOH/H2O 4:1; 4.- Filter
Clean up: FumoniTest TM IAC: 1.- Dilute 10 mL sample with 40 mL PBS; 2.- Filter; 3.- Load 20 mL; 3.- Wash 10 mL PBS; 5.- Elute 2 x 1.5 mL MeOH; 6.- Evaporated at 60°C
Derivatization: 1.- Reconstitute in 50 μL MeOH/H2O 1:1; 2.- Add 500 μL 0.05M sodium borate buffer (pH 9.5 adjusted with 1N NaOH), 500 μL NaCN reagent, and 150 μL NDA reagent (0.5 mg/mL in ACN); 2.-Heat 15 min at 60oC, cold to room temp
Nucleosil 120, C18, 250 x 4.6mm, 5μm, NR
Inj vol NR, ACN/H2O/AcOH 61:38:1
Isocratic
Flow: 1 mL/min, Time: tTot:14 min
FDA
λex: 420 nm
λem: 500 nm
LOD/LOQ: NR
(Muscarella et al. 2008)
B1
maize-based foods5
Extraction: 1.- Add 2x12.5mL of an ACN/MeOH/H2O 3:3:4; 2.-Sonicate 20 min; 3.-Centrifuge at 2112 g, 10 min; 4.- Dilute 3 mL with 12 mL PBS
Clean up IAC FUMONIPREP: 1.- Load 10 mL of dilute sample; 2.- Wash with 10mL of PBS; 3.- Elute with 4 mL of MeOH at 0.5 L/min; 4.- Evaporate; 5.- Dry, 40°C; 6.- Reconstitute in 0.5mL of MeOH/0.1M phosphate buffer at pH 3.15 3:2
Derivatization: OPA: NR
Eurospher C18, 150mm x 4.6mm, 3μm, 40 °C
Inj vol 100 µL, A) 0.1M phosphate buffer at pH 3.15, B) MeOH
60% B for 2 min; 60-65% B in 5 min, to 65-75% B 3 min; initial condition for 5 min
Flow: 0.8 mL/min, Time: tan= 10 min tTot= 15 min
FDA
λex: 343 nm
λem: 445 nm
LOD B1 4, B2 5 µg/L
LOQ: B1 13, B2 16 µg/L **
(Liu et al. 2017)
B1, B2
Maize10
Extraction: 1.- Hydrate for 12 h with 10 mL ultrapure H2O; 2.- Add 30 mL ACN, 3.- Shake 120 rpm, 1 h; 4.-Filter
Clean up SAX: 1.- Precondition with 5 mL MeOH followed by 5 mL ACN /H2O 3:1 at a flow rate 1 mL/min; 2.- Load 8 mL of sample; 3.- Wash with 5 mL MeOH; 4.- Elute with 10 mL AcOH/MeOH 1:99, 5.-Dry, 6.- Reconstitute in 2 mL ACN/H2O 1:1
Derivatization: OPA post column in HPLC pump 1 (1% NaOH) 0.8 mL/min, pump 2 (3g OPA, 9 mL MeOH, potassium borate, 9 mL 2-mercaptoethanol) 0.6 mL/min
Agilent C18, 250 x 4.6 mm, 5μm, 40°C
Inj vol 50 µL, A) 0.05 M citric acid buffer (pH = 4), B) MeOH
55-65% B in 10 min, 65-70% B in 12 min, keep 3 min, 70-55% B in 3 min
Flow: 1 mL/min, Time: tan= 25 min tTot= 28 min
FDA
λex: 335 nm
λem: 440 nm
LOD: B1 6, B2 7 µg/kg
LOQ: B1 20, B2 23 µg/kg
(Sokolovic 2022)
B1, B2
Maize20
Extraction: 1.- Add 100 mL of MeOH:H2O 7:3; 2.- shake for 3 min; 3.- filter; 4.- dilute 1:20 with deionized H2O
Derivatization: OPA: NR
Zorbax Eclipse C18 125 x 4 mm, 5 μL
Vol. Inj: NR, A) 20 % H2O (1% Na3PO4) B) 80% MeOH
isocratic elution
Flow: 1 mL/min,Time: tTot= 30 min
FDA
λex: 335 nm
λem: 440 nm
LOD: 223 µg/kg for all FBs
(Gnonlonfin et al. 2008)
B1 and other toxins
Chips10
Extraction: 1.- Add 50 mL of MeOH/H2O 75:25; 2.- Mix for 1 min; 3.- Filter
Clean up SAX: 1.- Precondition with 5 mL H2O, 5 mL MeOH, and 5 mL MeOH/H2O 75:25 at a flow rate of 1 mL/min; 2.- Apply 10 mL of sample; 3.- Wash 8 mL with 5 mL MeOH/H2O 75:25 and 8 mL MeOH; 4.- Elute 14 mL MeOH/AcOH 99:1 at a flow 1 mL/min; 5.- Evaporate; 6.- Reconstitute in 1 mL of MeOH, 7.-Evaporate, 8.-Reconsitute in 200 µL MeOH
Derivatization OPA: 1.- Mix 50 µL sample with 200 μL OPA (40 mg OPA in 1 ml of methanol
followed by addition of 5 ml of 0.L M sodium borate solution and 50 μL of 2-mercaptoethanol.)
Supercosil C18, 150 x 4 mm, 3μm, 30°C
Inj vol 10 µL, MeOH/0.1 M sodium dihydrogen phosphate 80:20 adjust pH 3.35 with phosphoric acid
Isocratic
Flow: 1 mL/min
Time: NR
FDA
λex : 335
λem: 440
LOD: 0.025 µg/kg
(Tardieu et al. 2008)
B1
Animal tissues,
liver, kidney1
Extraction: 1.- Homogenize in 2 mL of distilled H2O (Liver, 500 rpm; breast muscle 3000rpm, 20 s) with a teflon Potter, 2.- Precipitate proteins with 2 mL of MeCN/MeOH 1:1 and 25mg of NaCl; 3.-Stir to 300 rpm, 120 min; 4.- Centrifuge 3000 g, 15 min; 5.-Take 3 mL of supernatant; 6.-Add 4 mL Hex; 7.- Centrifuge 3000 g, 15 min; 8.- Take 2 mL aqueous phase; 9.- Dilute with 8 mL PBS
Clean up: IAC FUMONIPREPC: 1.- Pass the sample through cartridge; 2.- Wash 10 mL of PBS (pH 7.4); 3.-Elute 1.5 mL of MeOH, 1.5mL of H2O; 4.- Evaporate, 40°C; 5.-Reconstitute with 200 μL ACN/H2O 1:1
Derivatization OPA:1.- Add to 50 μL of sample: 50 μL of OPA, 50 μL of 0.1M borate buffer at pH 8.3, and 50 μL of H2O
Prontosil C18, 250 x 4.6mm, 5μm
Inj vol 20 µL, MeOH/NaH2PO4 0.1M pH 3.35, 75:25
Isocratic
Flow: 1 mL/min
Time: tTot 15 min
FDA
λex : 335
λem: 440
LOD: 10 µg/kg
LOQ: 13 µg/kg
(Kawashima, Vieira, and Valente Soares 2007)
B1 and other toxins
Beer>50 mL
NR
Clean up: SAX: 1.- Adjust pH 5.8-6.5 with 1N NaOH; 2.- Filter; 3.- Precondition with 10 mL of MeOH, 10 mL MeOH/H2O 3:1; 4.- Load 50 mL; 5.- Apply into SAX; 6.- Wash with 10 mL MeOH/H2O 3:1 and 6 mL MeOH; 7.- Elute with 20 mL MeOH/AcOH 95:5; 8.-Dry with N2 at 60°C
Derivatization: OPA: 1.- Reconstitute in 500 μL ACN/H2O 1:1; 2.- Take 100 μL; 3.- Add 200 μL OPA reagent (40 mg O-ftaldialdehyde in 1 mL ethanol diluted with 0.1 M borate buffer and 50 μL 2-mercaptoethanol; 4. Ultrasonic bath at 5-15 °C, 30 sec
Spherisorb ODS-2, 250 x 4.6 mm 2, 5μm
Inj vol 20 µL, ACN/H2O/AcOH 54:46:1 Isocratic
Flow: 1 mL/min, Time: tTot 19 min
FDA
λex : 335
λem: 440
LOD/LOQ: NR
(Jerome Jeyakumar, Zhang, and Thiruvengadam 2018)
B1, B2 and other toxins
SugarcaneNR
>50 mL
Extraction: 1.- Add 25 mL AcOEt to the culture; 2.- Shake to 8000 rpm; 3.- Filtrate after 2h; 4.- Mix with 5% acetone, isopropanol; 5.-Extract liquid phase with AcOEt in a 1:1 ratio; 6.- Collect upper phase, 7- Evaporate; 8.- Reconstitute in isopropanol. All x 3
Clean up SAX: 1.- Load 10 mL; 2.- Wash with 3 mL MeOH followed by 5 mL of 1% KCl; 3.- Collect into 5 mL tube; 4.- Evaporate
Derivatization OPA: 1. Pre-column derivatization [50 mg of OPA in 1.25 mL of methanol + 50 μL of 2-mercaptoethanol + 11.2 mL of 0.1 M sodium borate buffer (pH 9.5)]; 2.- Mix 100 μL of sample with 25 μL of OPA; 3.-incubate 2 min to rt.
C18, 250 x 4.6 mm, 5μm
Inj vol 20 µL, Isocratic, MeOH/0.1 M sodium dihydrogen phosphate buffer pH 3.3; 75:25
Flow: 0.3 mL/min, Time: 16 min
FDA
λex: 335 nm
λem: 440 nm
LOD/LOQ:NR
(Piacentini et al. 2017)
B1 and other toxins
Beer>50 mL
25
Clean up: 1.- Adjust pH 5.8-6.5 with 1N NaOH; 2.- Filter; 3.- Precondition with 10 mL of MeOH, 10 mL MeOH/H2O 3:1; 4.- Load 50 mL; 5.- Apply into SAX; 6.- Wash with 10 mL MeOH/H2O 3:1 and 6 mL MeOH; 7.- Elute with 5 mL MeOH/AcOH 95:5; 8.-Dry with N2 at 60°C, 7.- Reconstitute in 300 μL ACN/H2O 1:1, 8.-Filter
Derivatization OPA: 1.-Take 500 μL; 2.-Add 200 μL OPA reagent (40 mg O-ftaldialdehyde in 1 mL ethanol diluted with 5 mL 0.1 M borate buffer and 50 μL 2-mercaptoethanol.
Luna C18, 150 x 4.60 mm, 5 µm, Temp NR
Inj vol 20 µL, ACN/H2O/AcOH 520:480:5
Isocratic
Flow: 1 mL/min, Time: 15 min
FDA
λex: 335 nm
λem:440 nm
LOD: 2 µg/L, LOQ: 6.3 µg/L
(Smith et al. 2017)
B1, B2
Feed25
10
Extraction: 1.- Add 50 mL MeOH/ACN/H2O 1:1:2; 2.-Vortex for 30 s; 3.-Shake 20 min, 3.-Filter, 4.-Dilute 1:5 with 0.01 M PBS
Clean up SPE: 1.- Take 5 mL aliquot; 2.-apply into SPE column; elute rate approximately 1-2 drops/s, 3.-Wash with 10 mL 0.01 M PBS, 4.-Removed solvent (vacuum, 5 min), 5.-Elute with 1.5 mL MeOH then 1.5 mL H2O
Derivatization Fmoc: 1.- Take 500 µL, 2- Add boric acid (1 M, pH 7.5, 125 µL), control pH during derivatization; 3.- Add Fmoc (125 µL, 0.12 g Fmoc, 40 mL ACN, 2.88 g citric acid, 1.10 g tetramethylammonium chloride in 1 L distilled and deionized water), mix and wait 10 min, 4.- vortex; 5.-Add 1 mL anhydrous pentane, 6.- vortex and allowed to separate; 7.- discard the organic (top) layer; 8.- transfer aqueous (bottom) layer to a amber autosampler vial for HPLC-FLD analysis.
Acclaim 120 C18, 4.6 x 150 mm; 3 µ, 35°C
10 µL of sample, A) citrate buffer (pH 4.7): ACN (70:30)], 20% B [citrate buffer (pH 4.7): ACN (30:70)
20-95% B in 20 min post-injection, keep 5 min, 95-20 % B in 1 min, initial conditions for 4 min
Flow: 1 mL/min, Time: tan25 min, tTot: 30 min
FDA
λex: 263 nm
λem: 313 nm
LOQ: B1 7.55, B2 8.5 µg/L
(J. Wang, Zhou, and Wang 2008)
B1
corn products10
Extraction: 1.- Add ACN/H2O 1:1; 2.- Shake over night; 3.- Filter; 4.-Take 10 mL; 5.- Place on the ice for 15 min; 6.- Centrifuge to 7000 rpm, 10 min at 4 °C
Clean up: 1.- Preconditione with 2 mL of MeOH, 2.- Transfer 50 mL of sample; 3.- Apply to centrifugal tube with 300 mg of amberlite XAD-4; 4.- Stir for 5 h; 5.- Wash with 40 mL with deionized H2O; 6.- Elute with 3 mL MeOH; 7.- Collect 8.- Dry 65 °C, 9.- Reconstitute
Alltima C18, 250 x 4.6 mm, 5μm
20 µL of sample, A) H2O/TFA, B) ACN/FA
0-20% B from 0 to 5 min, 20-40% B from 5 to 10 min, 40-80% B from 10 to 15 min, 80% B from 15 to 20 min, 80-0% B from 20 to 25 min
Flow: 1 mL/min, Time: 25 min
ELSD
45°C of drift tube temperature, 2.0 L/min N2 gas flow, gain value of 1 in the impactor-on mode
LOD/LOQ: 3000 µg/L
5 Analytical methods
There are a lot of reported methods for fumonisin analysis. These have been mainly developed to analyze their presence in grains and grain-based products as there is a high concern for their presence in these types of matrices. However, other matrices such as fruits, vegetables, animal tissues, cereals and beverages should also be considered, as their carry over and cumulative effects ensure their presence in these types of food products. Moreover, analysis in human matrices is of special importance to completely establish toxicokinetics, as well as to elucidate the mechanisms by which fumonisins relate to some diseases.
This review compiles and organizes 88 analytical methods for fumonisins between 2006 and 2022, including liquid chromatography coupled with MS detectors (single quadrupole -sQ-, triple quadrupole -QQQ- and time of flight -TOF-, with or without ion tramp), fluorescence and light scattering. The workflow for fumonisin determination includes 1) extraction, sometimes followed by 2) clean up or derivatization, and finally 3) separation and detection (Fig. 3), being the first and third steps the fundamental ones. The detailed methodology used depends on the matrix analyzed, as well as the instrumentation available (Ridgway 2012). Matrices included in this work were classified as maize and corn-based products (34 methods), other cereal and seeds (11 methods), beverages (12 methods), products of animal origin (17 methods) and other samples (14 methods). Instrumentation used and conditions are detailed. Table 1 includes methods describing extraction and separation/detection using chromatography coupled to mass detectors without clean-up procedure; Table 2 shows those methods that include a clean-up stage after extraction, followed by separation/detection using chromatography coupled to mass detectors; Table 3 refers to methods describing extraction and separation/detection using chromatography coupled to fluorescence or light scattering detectors.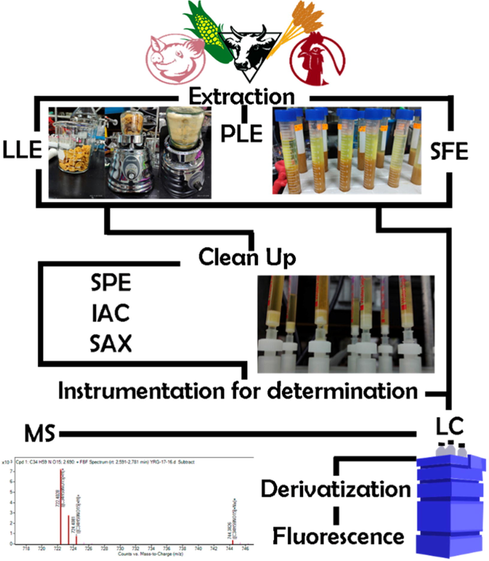
Workflow for fumonisin chromatographic analysis.
5.1 Extraction
Extraction is needed to obtain the enriched extract with the desired analytes, and to enhance sensitivity of the method, diminishing interferences with other components of the sample. Organic solvents, such as chloroform and hexane, which are commonly used in other mycotoxin extraction, are not recommended for FBs determination (Patel et al., 2011); this is due to the structure of FBs, which includes multiple hydroxy, amine and carbonyl groups that make polar solvents necessary for its extraction (Scott 1993). Therefore, a mixture of water and acetonitrile (ACN) or methanol (MeOH) is the most used solvent. However, some matrices are aqueous rendering these mixtures useless as the matrices are miscible with these solvents.
FBs’ ability to conjugate with proteins and sugars, allows it to be extracted with organic acids, the most commonly used are acetic acid (AcOH), formic acid (FA) and trifluoracetic acid (TFA); some authors have even used strong acids such as hydrochloric or sulphuric acid in the extraction of FBs (Zöllner and Mayer-Helm, 2006). To enhance the solubility of fumonisins in organic solvents, pressure has sometimes been used during extraction. Reported methods include liquid–liquid extraction (LLE) (Lucci et al., 2015), pressurized liquid extraction (PLE) (D’Arco 2008) and supercritical fluid extraction (Selim et al., 1996) (Tables 1-3). Matrix and analysis method defines the extraction method to be used and/or the extraction yield (Damiani et al., 2019.
Reported methods use an aqueous:organic proportion ranging from 10 to 85 % of organic solvent, however, typically more than 50% of MeOH or ACN and, from 0.1 to 3% of acid is used. Some mixes of ACN:MeOH:H2O were used keeping the mentioned range for aqueous, and some used 100 % ethyl acetate (Monbaliu et al., 2009) for extraction. Immunoaffinity extraction was also reported for urine samples. Usually, suspension of sample into extraction solvents, was followed by shaking, for periods of time ranging from seconds up to 3 h, filtering or centrifugation, from 3,000 to 10,000 rpm for 2 to 15 min (Tables 1-3).
In 2012 Pietri and collaborators observed problems during the extraction step which resulted in unexpected low recoveries in maize flour samples due to the interactions between fumonisins and matrix components (Damiani et al., 2019).
5.1.1 Liquid-liquid extraction (LLE)
LLE is the most commonly used technique which, depending on the composition of the food matrix, uses a mixture of acidified solvents (Lucci et al., 2015). Examples include: methanol–water (Paepens et al., 2005), acetonitrile–water (Zitomer et al., 2008 or methanol–acetonitrile–water and a non-polar phase (Bryła et al., 2013. It is based on the distribution of toxin in immiscible phases (aqueous and organic phase). The non-polar contaminants (lipids and cholesterol) are removed with non-polar organic solvents such as hexane and cyclohexane, while polar toxin compounds are extracted in the aqueous phase. This method is useful for both liquid and solid samples, the latter are homogenized and remain suspended in a polar solvent. In both cases, centrifugation is carried out, after which drying is performed under a nitrogen atmosphere, and, finally, reconstitution is done in a mixture of the chosen solvent. LLE is suitable for several toxins at small-scale preparations, however, its main disadvantage is that it is time consuming and there can be loss of sample during handling (Nawaz 2017).
5.1.2 Pressurized liquid extraction (PLE)
PLE, also known as Accelerated Solvent Extraction (ASE), uses temperatures around 100–180 °C and 1500–2000 psi of pressure to modify the conditions of the solvent and the sample, and facilitate the extraction of analytes (Kou 2003). The sample is initially dispersed with an inert material and further loaded into an extraction cell where the solvent is pumped in. Then, the extraction cell is heated to the desired temperature (above 200 °C) for 5 to 9 min, and pressurized (D’Arco 2008).
5.1.3 Supercritical fluid extraction (SFE)
Supercritical fluids are helpful in the extraction of analytes from a matrix. Their unique properties (low density and viscosity) make them superior to conventional extraction solvents, facilitating the extraction of compounds in samples. The most used fluid is CO2, however, analytes with polar characteristics do not adequately dissolve. To increase its efficiency towards polar analytes, modifiers such as methanol, ethanol or acetone are added. Limitations of this technique include high cost and the need for sophisticated equipment (Selim et al., 1996, Nawaz et al., 2017).
5.2 Clean-up
A great number of methods have included a clean-up step after extraction (Table 2). The aim is to eliminate major impurities like organic acids, polar pigments, sugars, among others. The most used are QuEChERS, solid-phase extraction (SPE) with reverse phase, strong anion exchange (SAX) cartridges, and immunoaffinity columns (IAC) (Damiani et al., 2019, Marschik et al., 2013). It has been shown that solvent temperature used in this process can deeply influence the recovery of fumonisins (Lawrence et al., 2000).
5.2.1 QuEChERS
QuEChERS is a technique initially developed by Anastassiades and collaborators in 2003 (Anastassiades 2003). They coined the acronym QuEChERS which stands for Quick, Easy, Cheap, Effective, Rugged and Safe. It involves micro-scale extraction with acetonitrile, followed by a cleanup based on a dispersive solid-phase extraction (d-SPE) (Wilkowska and Biziuk, 2011). In the extraction step, magnesium sulphate is used to reduce water in the sample, along with sodium chloride, while in the cleanup step, a primary secondary amine (PSA) or C18 is usually used as sorbent to retain co-extracted compounds such as sugar and fatty acids (Ridgway 2012, Zhang et al., 2012). Other salts such as magnesium chloride, sodium nitrate, sodium sulfate and lithium chloride have been used to eliminate water, finding magnesium sulfate as the most effective for separation of both phases, eliminating water from the organic phase. QuEChERS has become the most popular pre-treatment for some matrices like as corn, wheat, oats, rice, and other cereals, as it boasts several advantages such as the decrease in volume of solvent, materials, time, as well as a reduction in cost of analysis.
5.2.2 Solid phase extraction (SPE)
SPE is a variation of traditional chromatography, and thus, is based on the same principle, the use of a mobile and a stationary phase. Separation is performed according to affinity using small disposable cartridges packed with silica gel or bonded phases which are in the stationary phase. The sample is first dissolved and loaded into a cartridge, after which it is rinsed to remove most of the contaminants and is subsequently extracted from the cartridge with a polarity compatible solvent. All this is done under reduced pressure. The SPE cartridges contain different binding phases, for example silica gel, C18 (octadecylsilane), floredil, phenyl, aminopropyl, ion exchange (anionic and cationic) or SAX, immunosorbents, and molecular imprinting polymers. These last two are affinity materials which provide them with a high binding capacity for small molecules making them excellent candidates for cleanup in terms of specificity, however, they have a high cost, and are not compatible with organic solvents, limiting their use to aqueous systems. This is a disadvantage compared to more common binding phases such as SAX or C18 (Turner et al., 2009).
Regarding fumonisin analysis, C18 is the most used stationary phase for SPE due to its easy acquisition, low costs, and the possibility of extraction of hydrolyzed forms. The second most used phase are SAX resins, whose efficiency is based on the interaction with fumonisin carbonyl groups, making them not appropriate for hydrolyzed forms (Zöllner and Mayer-Helm, 2006). Its elution has been reported with MeOH acidified with 0.05% AcOH achieving a pH < 7. When ion exchange resins are used for this purpose, it is necessary that the analyzed mycotoxin be in its ionic form and in an aqueous solvent. For this reason, pH regulation of the medium is an important factor. This methodology has been used for the extraction of fumonisins and moniliformin. SAX columns consist of resins with weakly basic functional groups, such as NH2, NHCH3 or N(CH3)2, or with quaternary ammonium strongly basic groups (N(CH3)OH) in which OH is replaceable by mycotoxin. Several types exist in both, anionic and cationic phases. SAX is the favored material for mycotoxin extraction (Turner et al., 2009).
IAC uses antibodies, present in the stationary phase, that bind selectively to mycotoxins present in the extract. This poses an important advantage, as there is a specific interaction between the antibody and the analyte, resulting in a greater speed of interaction. After antibody binding, mycotoxins are recovered by elution with a miscible solvent or by antibody denaturation. Disadvantages of this process include the necessity of combination with other techniques such as LLE or SPE for complex samples; and the requirement for the extract to be in aqueous solution containing little or no organic solvents, as their presence, even in low concentrations, can denature antibodies (Pereira et al., 2014). Recently, a rapid and sensitive method for determination of seven mycotoxins (including FB1) using immunomagnetic (monoclonal antibodies conjugated with CNBr) solid-phase extraction (IMPSE) coupled to UPLC-MS/MS has been developed for peanut, maize, and wheat matrices (Wang et al., 2022).
5.3 Derivatization
The main objective of derivatization is to change the chemical and physical properties of compounds by modifying their chemical structure (Qi et al., 2014). Thus, derivatization reagents react with target compounds containing various functional groups, including carbonyl (O’Brien-Coker et al., 2001 hydroxyl (Barry et al., 2003), carboxyl (Santa et al., 2009), amine (Vanhoenacker et al., 2009, and thiol (Vichi et al., 2013.
This strategy has been of utmost importance in the development of new methodology for the detection of fumonisins, as these compounds are not capable of developing fluorescence or absorbance in UV–VIS light, due to their lack of a suitable chromophore or fluorophore group for detection. Derivatization with fluorescent derivatives including 9-fluorenylmethylchloroformate (FMOC-CL), 4-flouro-7-nitro-benzofurazan (NBD-F), o-phthaldialdehyde (OPA), naphthalene-2,3-dicarboxaldehyde (NDA) and dansyl chloride (DnS-Cl) (Ndube et al., 2011, Ndube 2009, Silva et al., 2009) allows for fumonisin detection with HPLC coupled to fluorescence or UV-Vanhoenacker et al., 2009), albeit with a low sensitivity. Despite these limitations, UV detection is still used although the methods are not new (Cardinael et al., 2015). Out of the fluorescent derivatives, OPA is the most used due to its low detection limits (50 ng/g), followed by NBD-F which is detected at 100 ng/g. NDA has an even lower detection limit than OPA, however, its use is generally avoided as potassium cyanide is required during derivatization, representing a high health risk (Table 3).
5.4 Instrumentation for determination
Once the sample is obtained, extracted and, in some cases purified or cleaned-up, different instrumentation can be used for fumonisin analysis; being HPLC and UPLC the most frequently employed. Chromatographic column is used in a reverse phase, most commonly with C18 as a stationary phase; nevertheless, diphenyl, amide and C8 may also be used. The chromatographer can be coupled with a fluorescence (Table 3) or ESI source with mass spectrometry detectors (Tables 1-2). For these last ones, QQQ is the most widely used analyzer, although sQ and TOF analyzers had also been utilized. These are all used in positive mode and all acquisition modes are reported, full scan, single reaction mode (SRM) or multiple reaction mode (MRM). Some analyzers use an Ion Trap array.
5.4.1 Separation
Fumonisins have a higher molecular weight (around 721.83 g/mol) compared to other mycotoxins such as Ocratoxin A (403.81 g/mol), Zaralenone (318.36 g/mol) or Patulin (154.12 g/mol). Because of their high polarity, reverse phase LC is an excellent option for its separation. Previous extraction methods involve an aqueous phase, which is included as mobile phase (Tables 1-3). Different proportions of solvents are used for the composition of the mobile phase, MeOH:H2O is preferred, followed by ACN:H2O, especially when derivatization is used to provide better sensibility (Velázquez et al., 2000). There is a clear tendency of using a greater proportion of organic solvents in these mixtures with gradients reaching 100 % organic concentration, as well as the addition of FA or AcOH, and in some cases ammonium salts. This is done to enhance the ionization process necessary for mass detection, to control pH, and to increase the efficiency of separation.
Temperature used for these analyses vary between 10 and 45 °C for MeOH as mobile phase and 30–50 °C for ACN; flows from 0.1 to 1 mL/min are reported. Column dimension is another important aspect to consider when analyzing fumonisins. According to the literature compiled in the present article, there is a great variation between column dimensions, ranging from 50 to 250 mm in length, diameters going from 2.0 to 4.6 mm, and particle size ranging from 1.6 to 5 µm. The most used, however, oscillate between 100 and 150 × 2 mm, with a particle size of 4.6 µm.
A recent work by Sultan et. al. evaluated the efficacy of 5 columns with different dimensions and particle sizes. FB1 and FB2 were analyzed using liquid–liquid extraction, followed by a cleaning procedure using SPE, and fluorescence detection, using OPA as a fluorophore group. They conclude that the use of reverse phase SPE, followed by derivatization with OPA is an effective method for the determination of fumonisins, which agrees with the information gathered by this review. In that work the comparison between columns Nucleosil Cronus (150 mm × 4.6 mm, 5 μm) and Poroshell (75 mm × 4.6 m, 2.7 μm) yielded similar results regarding time and solvent use. However, it is of note that the use of columns with porous particles or those with a solid nucleus affect separation. Similarly, both the diameter of the column, and particle size used are also important parameters to determine in fumonisin analysis (Sultan et al., 2022).
5.4.2 Detection
Although many methods for fumonisin detection exist, such those based in fluorescence, the methods based on MS are the most sensitive. Among the methods included in this work, the lowest FB1 LOD for fluorescence detector was 0.025 versus 0.0005 µg/Kg obtained with MS QTrap detector. Besides, MS detectors offer a great advantage as they do not require derivatization (Tables 1-2).
FDA methods depend on the presence of a chromophore or fluorophore that allows for the correct detection of the analytes, as has been mentioned previously, various derivatizing agents exist (see section 4.3), despite this disadvantage, these methos are still commonly used due to their low costs, and their applicability to a great number of matrices including beer, maize, and biological fluids, among others. Fumonisins can be detected at the following longitudes: λex: 420 nm, λem: 500 nm.
On the other hand, mass spectrometry for the detection of fumonisins is carried out with an ESI interphase, and IT, orbitrap, QQQ and TOF analyzers used in positive mode. In this analysis, the ion [M + H] + has been found to be the most abundant, with or without a high grade of fragmentation. Additionally, in negative mode, the formation of doubly charged molecular ions has been reported. The positive mode is used more frequently, although some authors have reported that it favors the formation of adducts that may present a problem with sensitivity. Despite this, the positive mode is still the most used mode as the [M + H] + ion is three times more abundant than [M - H] -.
According to the present review, various mass analyzers such as sQ, QQQ, and TOF have been used, some of them with an ion trap (IT). IT methods are theoretically more sensitive, yet, not all ion trap (IT), Trap or QTrap methods reported here have been the most sensitive, with some QQQ or even sQ methods being able to detect lower concentrations (Tables 1 and 2).
Lower limits for FB1, FB2, and FB3 have been reported by different authors, including Šarkanj et al for urine analysis (0.001 µg/L FB1 and FB2) (Šarkanj 2018), Zitomer et al regarding maize tissues analysis (0.01 µg/kg for FB1 and FB2) (Zitomer et al., 2008), Huang et al for liquorice (0.05 µg/L FB1 and FB2) (Huang et al., 2018). Among the different fumonisins analyzed, the most reported is FB1, with [M + H] 722.2 m/z being the most abundant ion. Additionally, the 334.3 and 352.3 m/z product ions can also be obtained by using a collision energy of 38–56 and 38–40 eV respectively (Table 4). (CE) Collision Energy, (CoV) Cone Voltage, (DP) Declustering potential, all in V.
FB1,
Transitions (m/z)
CE
DP
CoV
722.2 → 704.3
31
70–76
50
722.2 → 352.3
38–40
70–76
50–60
722.2 → 334.3
38–56
70–76
50–65
FB2/FB3
706.2 → 354.4
37
68–75
50
706.2 → 336.5
40–47
68–75
50–55
706.2 → 318.4
40–55
68–75
50–65
A light scattering method has also been reported. Even though its reported LOD and LOQ are high, these limits approach those that are permissible. Thus, it may prove useful in screening, as detection by this method correlates with level above the permissible limits (Ramalho et al., 2022, Mirón-Mérida et al., 2021).
5.4.3 Non chromatographic methods for fumonisins detection
Aside from conventional chromatographic methods, there is a wide variety of methods for fumonisin determination. These can be classified into two groups: immunological and molecular (Table 5) (Deepa and Sreenivasa, 2019).
Immunological methods
Molecular methods
Enzyme Linked Immuno Sorbent Assay (ELISA)
Internal transcriber spacer (ITS)
Dipstick
Intergenic spacers (IGS)
Biosensor
Polyketide synthase
Immunoaffinity
FUM genes
Colloidal gold immune assay
Microarray
Polymerase chain reaction (PCR)
The immunological methods are based on the interaction between the mycotoxin and a specific antibody. These antibodies act by recognizing specific chemical groups; as such, they can recognize structural analogs. To facilitate antibody detection, a marker is added which can be radioactive, chromogenic or fluorogenic in nature. The most popular, commercial, immunological method for fumonisin detection is enzyme-linked immunosorbent assay (ELISA) (Pereira et al., 2014). This method has been used to determine fumonisin concentration in corn and other cereals (Wang et al., 2006fresh and dehydrated commercial garlic (Tonti 2017); during industrial cornflakes processing (Castells et al., 2008); and maize and gluten meal (Coronel et al., 2016). Techniques such as time-resolved immunochromatographic assays, enzyme-linked aptamer assays, chemiluminescence immunoassays, fluorescence immunoassays, fluorescence resonance energy transfer immunoassays, and metal-enhanced fluorescence assays have been implemented in the detection of mycotoxins (Majdinasab et al., 2021, Chauhan et al., 2016).
Although molecular methods do not directly determine the presence of fumonisins, they are nonetheless important as they allow rapid detection of fumonisin-producing species. These DNA-based identification methods are fast, sensitive, and reliable (Deepa and Sreenivasa, 2017) because they are independent of the morphology and cultivability of the fungi. Of these, PCR is the most frequently used technology for detection of mycotoxin-producing Fusarium species (Gong et al., 2015). Today, aptamer-based methods are having a great impact in the detection of mycotoxins. Due to their exceptional affinity and specificity, they can be comparable to antibodies, with certain advantages such as easy nucleobase and chemical modification, and exponential self-amplification (Mirón-Mérida et al., 2021).
Also, these methods take advantages of nanomaterials to improve LOD, cost, analysis time, reduce instrument use for final users and overall, pretreatment and manipulation of samples. However, at a research level, nanomaterials need to be characterized, requiring instrumentation that is not common. Many of these technologies are still under development, with a large amount of research proposing them for fumonisin determination. Much of this information has been compiled over the years in various review papers (Majdinasab et al., 2021, Deepa and Sreenivasa, 2019, Gong et al., 2015, Mirón-Mérida et al., 2021). Until these methodologies achieve the robustness of chromatographic techniques, especially for absolute quantification, the latter techniques remain the techniques of choice.
6 Remarks
Fumonisins are mycotoxins widely distributed in food products, mainly due to the contamination of cereals (such as bread, bread, pasta, boxed cereals, flour, among others) by species of the Fusarium genus. Additionally, their presence in livestock feed, along with the eventual accumulation of these mycotoxins within their tissues, increases the transmission chain.
Fumonisins have a high capacity to withstand the processes used in the food industry. They have been found to be thermically stable, at a neutral pH, in temperatures ranging from 100 to 125 °C, only observing small degrees of degradation in alkaline or acidic mediums at temperatures above 175 °C. The analysis of the stability of its hydrolyzed forms in corn-based products indicates that their decomposition begins at temperatures above 250 °C, with the loss of the TCA groups. Even so, their decomposition does not exceed 20% of total fumonisins. The conjugation of fumonisins with sugars, proteins and even metals also occur in food products that are rich in these chemical entities. Currently there are no specific methodologies for analysis, detection, and quantification of hydrolyzed or conjugated forms of fumonisins for all the interest matrices, representing a niche of opportunity from an analytical and application point of view in the food industry. The basis of food in Mexico is corn; therefore, its population may be exposed to the consumption of Fusarium mycotoxins. Currently, there is a lack of legislation regulating the consumption of these compounds. The creation of new legislation is important to achieve adequate control and management of mycotoxin levels in food to ensure adequate food health in this regard.
Supporting material can be consulted at https://www.sciencedirect.com/journal/arabian-journal-of-chemistry and provides Tables 1-3 as excel file to facilitate the user experience by allowing the reader to sort by matrix, detector, LOD, LOQ, analysis time, etc.
7 Conclusion
Cheap, easy, and fast analytical methods for fumonisin detection are important worldwide, especially for countries where the content of these toxins is not regulated. Implementing regulation aids in the control of food products and contributes to food safety. Molecular, immunologic, and chromatographic methods can be used for fumonisin analysis. Molecular methods present the disadvantage of being only qualitative but widely used to identify fumonisin producer species. While immunologic and chromatographic methods can be utilized for both, qualitative and quantitative analysis. Immunologic methods are highly specific and useful for free fumonisins; however, these are not recommended for conjugated fumonisins. Immunological or molecular assays are still in development and could be a reasonable screening approach with final quantification being carried out by robust chromatographic methods, although some ELISA methods are commercially available. There are a wide variety of chromatographic methods. These are used for all kind of studies and applied to all kinds of samples because they can be coupled to different detectors. Chromatographic analysis of FBs can be qualitative or quantitative, another advantage is that they can analyze different FBs at the same time. Among the chromatographic methods, different sensibilities can be reached thus, although ELDS presents LOD very close to the maximum permissible levels it is still a viable option as a screening method. The use of mass spectrometry analyzers provides a high sensibility and is appropriate when analyzing samples of different origin. UPLC and HPLC methods are reported, as well as different analyzers. Very low limits of detection can be achieved. Sample pretreatment can be sufficient by extraction with an organic solvent or mixtures of ACN, MeOH and water, sometimes using weak acids. Similarly, these mixtures are employed in a gradient elution with 0.1–0.3% of acid, however clean-up is suggested for these mixtures. Chromatographic methods have a greater versatility regarding the combination of columns and detectors that are available, which is part of the reason these methods are still employed generating more sensitive, shorter, and reliable results. The information of the chromatographic methods for fumonisin analysis developed in the last 16 years has been included in this review. This paper will facilitate to the reader to consider the methodological aspects of a method to analytical success. Thus, the readers will be able to combine and adapt these aspects between methods to their own necessity.
Availability of data and material
Not applicable.
Code availability
Not applicable.
For the present review, ethics approval, consent to participate and consent for publication are not applicable.
Funding
No funding was received for conducting this study. Y.D.O.-A. scholarship CVU-811460.
Author contributions
Y.D.O.A. compiled all the methods parameters and wrote the first version of the manuscript, E.S.R. reviewed the medical and biological implications of fumonisins, M.Á.R.C. reviewed all methods parameters, M.Y.R. reviewed the general redaction of the complete manuscript. M.Á.R.C and M.Y.R. guided the complete work. All authors participated in the redaction of the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Bio-monitoring of mycotoxin exposure in Cameroon using a urinary multi-biomarker approach. Food Chem. Toxicol. 2013;62:927-934.
- [CrossRef] [Google Scholar]
- Advances in occurrence, importance, and mycotoxin control strategies: Prevention and detoxification in foods. Foods. 2020;9(2)
- [CrossRef] [Google Scholar]
- Nutritional and health implications of mycotoxins in animal feeds: A review. Pak J Nut. 2006;5(5):398-403.
- [CrossRef] [Google Scholar]
- Technological and community-based methods to reduce mycotoxin exposure. Food Control. 2017;73(May):101-109.
- [CrossRef] [Google Scholar]
- Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Reviews. 2009;28(2–3):198-215.
- [CrossRef] [Google Scholar]
- Norma general para los contaminantes y las toxinas presentes en los alimentos y piensos CXS 193–1995. FAO: WHO; 2019. p. :1-76.
- Fast and easy multiresidue method employing acetonitrile extraction/partitioning and. J AOAC Int. 2003;86:412-431.
- [CrossRef] [Google Scholar]
- Review: HPLC determination of fumonisin mycotoxins. Crit Rev Food Sci Nutr. 2004;44(3):195-203.
- [CrossRef] [Google Scholar]
- Multiclass mycotoxin analysis in Silybum marianum by ultra high performance liquid chromatography-tandem mass spectrometry using a procedure based on QuEChERS and dispersive liquid-liquid microextraction. J. Chromatogr. A. 2013;1282:11-19.
- [CrossRef] [Google Scholar]
- Simple methodology for the determination of mycotoxins in pseudocereals, spelt and rice. Food Control. 2014;36(1):94-101.
- [CrossRef] [Google Scholar]
- In-house validation of a rapid and efficient procedure for simultaneous determination of ergot alkaloids and other mycotoxins in wheat and maize. Anal. Bioanal. Chem. 2018;410(22):5567-5581.
- [CrossRef] [Google Scholar]
- Use of S-pentafluorophenyl tris(2,4,6-trimethoxyphenyl)phosphonium acetate bromide and (4-hydrazino-4-oxobutyl) [tris(2,4,6-trimethoxyphenyl)phosphonium bromide for the derivatization of alcohols, aldehydes and ketones for detection by liquid chromatograp. Rapid Commun. Mass Spectrom. 2003;17(5):484-497.
- [CrossRef] [Google Scholar]
- Detection of new fumonisin mycotoxins and fumonisin-like compounds by reversed-phase high-performance liquid chromatography/electrospray ionization ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 2006;20(16):2447-2462.
- [Google Scholar]
- Detection and characterization of twenty-eight isomers of fumonisin B1 (FB1) mycotoxin in a solid rice culture infected with Fusarium verticillioides by reversed-phase high-performance liquid chromatography/electrospray ionization time-of-flight and ion t. Rapid Commun. Mass Spectrom. 2010;24(1):35-42.
- [Google Scholar]
- The effect of Fusarium verticillioides fumonisins on fatty acids, sphingolipids, and oxylipins in maize germlings. Int J Mol Sci. 2021;22(5):1-17.
- [CrossRef] [Google Scholar]
- Determination of mycotoxins in different food commodities by ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2009;23(12):1801-1809.
- [Google Scholar]
- Stable isotope dilution analysis of small molecules with carboxylic acid functions using 18O labeling for HPLC-ESI-MS/MS: Analysis of fumonisin B1. J. Agric. Food Chem. 2013;61(33):7904-7908.
- [CrossRef] [Google Scholar]
- The intestine as a possible target for fumonisin toxicity. Molr Nut Food Res. 2007;51(8):925-931.
- [CrossRef] [Google Scholar]
- Exposure, Occurrence, and Chemistry of Fumonisins and their Cryptic Derivatives. Compr. Rev. Food Sci. Food Saf. 2018;17(3):769-791.
- [CrossRef] [Google Scholar]
- Bryła, M., Renata, J., Roszko, M., and Szymczyk, K., 2013a. Application of molecularly imprinted polymers to determine B 1 , B 2 , and B 3 fumonisins in cereal products., 578–584. doi:10.1002/jssc.201200753.
- Fumonisins in plant-origin food and fodder - a review. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2013;30(9):1626-1640.
- [CrossRef] [Google Scholar]
- Effects of pH and temperature on the stability of fumonisins in Maize products. Toxins. 2017;9(3):1-16.
- [CrossRef] [Google Scholar]
- Determination of Mycotoxins in Food: A Review of Bioanalytical to Analytical Methods. Appl Spect Rev. 2015;50(9):728-774.
- [CrossRef] [Google Scholar]
- Stability of mycotoxins during food processing. Int J Food Microbiol. 2007;119(1–2):140-146.
- [CrossRef] [Google Scholar]
- Mycotoxins in corn-based food products consumed in Brazil: An exposure assessment for fumonisins. J. Agric. Food Chem. 2007;55(19):7974-7980.
- [CrossRef] [Google Scholar]
- Occurrence of fumonisins in Catalonia (Spain) and an exposure assessment of specific population groups. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2012;29(5):799-808.
- [CrossRef] [Google Scholar]
- Quantitative determination of carcinogenic mycotoxins in human and animal biological matrices and animal-derived foods using multi-mycotoxin and analyte-specific high performance liquid chromatography-tandem mass spectrometric methods. J. Chromatogr. B: Analyt Technol Biomed Life Sci. 2018;1073:191-200.
- [CrossRef] [Google Scholar]
- Distribution of fumonisins and aflatoxins in corn fractions during industrial cornflake processing. Int. J. Food Microbiol. 2008;123(1–2):81-87.
- [CrossRef] [Google Scholar]
- Mycotoxins produced by Fusarium genus in maize: determination by screening and confirmatory methods based on liquid chromatography tandem mass spectrometry. Food Chem. 2007;105(2):700-710.
- [CrossRef] [Google Scholar]
- Fumonisin occurrence in wheat-based products from Argentina. Food Addit Contam: Part B Surveillance. 2019;12(1):31-37.
- [CrossRef] [Google Scholar]
- Recent advances in mycotoxins detection. Biosens Bioelectron. 2016;81:532-545.
- [CrossRef] [Google Scholar]
- Exposure to aflatoxin and fumonisin in children at risk for growth impairment in rural Tanzania. Environ Int. 2018;115(March):29-37.
- [CrossRef] [Google Scholar]
- Research progress on fumonisin b1 contamination and toxicity: A review. Molecules. 2021;26(17):1-21.
- [CrossRef] [Google Scholar]
- Fumonisins in maize and gluten meal analysed in Argentinean wet milling industrial plants by ELISA compared with HPLC-FLD method. Food Control. 2016;67:285-291.
- [CrossRef] [Google Scholar]
- Fusarium: more than a node or a foot-shaped basal cell. Studies in Mycology. 2021;98:100116
- [CrossRef] [Google Scholar]
- Cumming, R., 2022. iNaturalist Mexico. https://www.inaturalist.org/observations/134863617. Last access February 7th, 2022.
- Analysis of fumonisins B1, B2 and B3 in corn-based baby food by pressurized liquid extraction and liquid chromatography/tandem mass spectrometry. J. Chromatogr. A. 2008;1209(1–2):188-194.
- [CrossRef] [Google Scholar]
- Dall’Asta, C., Galaverna, G., Aureli, G., Dossena, A., and Marchelli, R., 2008. A LC/MS/MS method for the simultaneous quantification of free and masked fumonisins in maize and maize-based products. World Mycotoxin J, 1(3), 237–246. doi:10.3920/wmj2008.x040.
- Dall’Asta, C., Mangia, M., Berthiller, F., et al. 2009b. Difficulties in fumonisin determination: The issue of hidden fumonisins. Anal. Bioanal. Chem, 395(5), 1335–1345. doi:10.1007/s00216-009-2933-3.
- Dall’Asta, C., Galaverna, G., Mangia, M., Sforza, S., Dossena, A., and Marchelli, R., 2009a. Free and bound fumonisins in gluten-free food products. Mol Nut Food Res, 53(4), 492–499. doi:10.1002/mnfr.200800088.
- Damiani, T., Righetti, L., Suman, M., Galaverna, G., and Dall’Asta, C., 2019. Analytical issue related to fumonisins: A matter of sample comminution? Food Control, 95, 1–5. doi:10.1016/j.foodcont.2018.07.029.
- Use of liquid chromatography-high-resolution mass spectrometry for isolation and characterization of hydrolyzed fumonisins and relevant analysis in maize-based products. J Mass Spectrom. 2014;49(4):297-305.
- [CrossRef] [Google Scholar]
- de Matos, N.A.V., de Moraes, M.H.P., Sartori, A.V., and do Couto J.S., 2021. Optimization and Validation of an Analytical Method for the Determination of Free and Hidden Fumonisins in Corn and Corn Products by UHPLC-MS/MS. Food Anal Methods, 14(8), 1611–1624. doi:10.1007/s12161-021-01984-8.
- de Oliveira, G.B., de Castro Gomes Vieira, C.M., Orlando, R.M., and Faria, A.F., 2017. Simultaneous determination of fumonisins B1 and B2 in different types of maize by matrix solid phase dispersion and HPLC-MS/MS. Food Chem, 233, 11–19. doi:10.1016/j.foodchem.2017.04.091.
- Molecularly imprinted solid-phase extraction of fumonisin B analogues in bell pepper, rice and corn flakes. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2009;26(6):874-884.
- [CrossRef] [Google Scholar]
- Fumonisins: A Review on its Global Occurrence, Epidemiology, Toxicity and Detection. J. Vet. Med. Res. 2017;4(6):1093.
- [Google Scholar]
- Molecular methods and key genes targeted for the detection of fumonisin producing Fusarium verticillioides – An updated review. Food Biosci. 2019;32(October):100473
- [CrossRef] [Google Scholar]
- Development and comparison of two multiresidue methods for the analysis of 17 mycotoxins in cereals by liquid chromatography electrospray ionization tandem mass spectrometry. J. Agric. Food Chem. 2010;58(13):7510-7519.
- [CrossRef] [Google Scholar]
- Combining the quick, easy, cheap, effective, rugged and safe approach and clean-up by immunoaffinity column for the analysis of 15 mycotoxins by isotope dilution liquid chromatography tandem mass spectrometry. J. Chromatogr. A. 2014;1337:75-84.
- [CrossRef] [Google Scholar]
- Quantitative determination of several toxicological important mycotoxins in pig plasma using multi-mycotoxin and analyte-specific high performance liquid chromatography-tandem mass spectrometric methods. J. Chromatogr. A. 2012;1257:74-80.
- [CrossRef] [Google Scholar]
- LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2009;26(6):885-895.
- [CrossRef] [Google Scholar]
- Diogo, L., 2011. iNaturalist Mexico. https://www.inaturalist.org/observations/36434139. Last access February 7th, 2022.
- Fusarium wilt of banana: Current knowledge on epidemiology and research needs toward sustainable disease management. Front Plant Sci. 2018;871(October):1-21.
- [CrossRef] [Google Scholar]
- Embrenhar, 2022. iNaturalist Mexico. https://www.inaturalist.org/observations/136754985. Last access February 7th, 2022.
- Reglamento (CE) 1126/2007 de 28 de septiembre de 2007 que modifica el Reglamento (CE) no 1881/2006 por el que se fija el contenido máximo de determinados contaminantes en los productos alimenticios por lo que se refiere a las toxinas de Fusarium en el maí. Diario Oficial de La Unión Europea. 2007;L 255:14-17.
- [Google Scholar]
- Combined (d)SPE-QuEChERS Extraction of Mycotoxins in Mixed Feed Rations and Analysis by High Performance Liquid Chromatography-High-Resolution Mass Spectrometry. Toxins. 2020;12(3):206-225.
- [CrossRef] [Google Scholar]
- Prevalence and concentration of fumonisins in cereal-based foods: a global systematic review and meta-analysis study. Environ Sci Pollution Res. 2021;28(17):20998-21008.
- [CrossRef] [Google Scholar]
- Short communication: Analysis of mycotoxins in Spanish milk. J Dairy Sci. 2018;101(1):113-117.
- [CrossRef] [Google Scholar]
- Simple and high-throughput method for the multimycotoxin analysis in cereals and related foods by ultra-high performance liquid chromatography/tandem mass spectrometry. Food Chem. 2009;117(4):705-712.
- [CrossRef] [Google Scholar]
- “Fungal Infections. Fusarium Oxysporum.” n.d. http://www.life-worldwide.org/fungal-diseases/fusarium-oxysporum.
- “Fungal Infections. Fusarium Solani.” n.d. Accessed August 26, 2021. http://www.life-worldwide.org/fungal-diseases/fusarium-solani.
- Determination of fumonisin B1 in bovine milk by LC-MS/MS. Food Control. 2009;20(12):1171-1174.
- [CrossRef] [Google Scholar]
- Analysis of fumonisins B1, B2 and their hydrolysed metabolites in pig liver by LC-MS/MS. Food Chem. 2011;125(4):1379-1384.
- [CrossRef] [Google Scholar]
- Fumonisins–novel mycotoxins with cancer-promoting activity produced by Fusarium moniliforme. App Environ Microbiol. 1988;54(7):1806-1811.
- [Google Scholar]
- Incidence of Fusarium verticillioides and levels of fumonisins in corn from main production areas in Iran. J. Agric. Food Chem. 2006;54(16):6118-6122.
- [CrossRef] [Google Scholar]
- Occurrence of fumonisins in maize imported into Iran during 2001–2002. Mycotoxin Rese. 2009;25(1):25-28.
- [CrossRef] [Google Scholar]
- Mycoflora and natural occurrence of aflatoxins and fumonisin B1 in cassava and yam chips from Benin, West Africa. Int. J. Food Microbiol. 2008;122(1–2):140-147.
- [CrossRef] [Google Scholar]
- Molecular strategies for detection and quantification of mycotoxin-producing Fusarium species: A review. J Sci Food Agricult. 2015;95(9):1767-1776.
- [CrossRef] [Google Scholar]
- Occurrence of mycotoxins in Polish animal feed in years 2006–2009. Journal of Animal Physiology and Animal Nutrition. 2012;96(5):870-877.
- [CrossRef] [Google Scholar]
- Headley, J., 2022. iNaturalist Mexico. https://www.inaturalist.org/observations/143416143. Last access February 7th, 2022.
- Mycoflora and natural occurrence of aflatoxin, cyclopiazonic acid, fumonisin and ochratoxin A in dried figs. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2012;29(2):277-286.
- [CrossRef] [Google Scholar]
- Free and hidden fumonisins in raw maize and maize-based products from China. Food Addit Contam: Part B Surveillance. 2019;12(2):90-96.
- [CrossRef] [Google Scholar]
- Optimized QuEChERS method combined with UHPLC-MS/MS for the simultaneous determination of 15 mycotoxins in liquorice. Journal of AOAC International. 2018;101(3):633-642.
- [CrossRef] [Google Scholar]
- Effects of thermal food processing on the chemical structure and toxicity of fumonisin mycotoxins. Mol Nut Food Res. 2004;48(4):255-269.
- [CrossRef] [Google Scholar]
- Effects of time, temperature, and pH on the stability of fumonisin B1 in an aqueous model system. J. Agric. Food Chem. 1996;44(3):906-912.
- [CrossRef] [Google Scholar]
- Determination of mycotoxins by HPLC, LC-ESI-MS/MS, and MALDI-TOF MS in Fusarium species-infected sugarcane. Microbial Pathogenesis. 2018;123:98-110.
- [CrossRef] [Google Scholar]
- Multi-mycotoxin analysis in dairy products by liquid chromatography coupled to quadrupole orbitrap mass spectrometry. J. Chromatogr. A. 2014;1345:107-114.
- [CrossRef] [Google Scholar]
- Fumonisins: Impact on agriculture, food, and human health and their management strategies. Toxins. 2019;11(6):1-23.
- [CrossRef] [Google Scholar]
- Fumonisin B1 and ochratoxin A in beers made in Brazil. Ciencia E Tecnologia de Alimentos. 2007;27(2):317-323.
- [CrossRef] [Google Scholar]
- A reversed phase high performance liquid chromatography method for the determination of fumonisins B1 and B2 in food and feed using monolithic column and positive confirmation by liquid chromatography/tandem mass spectrometry. Anal Chimica Acta. 2010;679(1–2):91-97.
- [CrossRef] [Google Scholar]
- Extraction of Semivolatile Organic Compounds from Solid Matrices. Sample Preparation Techniques in Analytical Chem. 2003;139–182
- [CrossRef] [Google Scholar]
- Simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in maize by liquid chromatography/tandem mass spectrometry after multitoxin immunoaffinity cleanup. Rapid Commun. Mass Spectrom. 2007;21(20):3253-3261.
- [Google Scholar]
- Effect of temperature and solvent composition on extraction of fumonisins B1 and B2 from corn products. J AOAC Int. 2000;83(3):604-611.
- [Google Scholar]
- Multi-mycotoxin analysis of finished grain and nut products using high-performance liquid chromatography-triple-quadrupole mass spectrometry. J. Agric. Food Chem. 2013;61(20):4771-4782.
- [CrossRef] [Google Scholar]
- Anthracene and Anthraquinone Derivatives from the Stem Bark of Juglans mandshurica Maxim. Helv. Chim. Acta. 2011;94:1488.
- [Google Scholar]
- Natural occurrence of fumonisins B1 and B2 in maize from eight provinces of China in 2014. Food Addit Contam: Part B Surveillance. 2017;10(2):113-117.
- [CrossRef] [Google Scholar]
- Lockett, D., 2022. iNaturalist Mexico. https://www.inaturalist.org/observations/147363346. Last access February 7th, 2022.
- Lucci, P., Castro-ríos, R., and Núñez, O., 2015. State-of-the-art in the analysis of fumosinins by liquid chromatography-mass spectrometry in: Evans C.M. (Eds.) Fumonisins: Natural Occurrence, Management Practices and Health Concerns, 116p, ISBN: 978-1-63482-789-8.
- Advances in colorimetric strategies for mycotoxins detection: Toward rapid industrial monitoring. Toxins. 2021;13(1):13-47.
- [CrossRef] [Google Scholar]
- Discovery and occurrence of the fumonisins: A historical perspective. Environ Health Persp. 2001;109(suppl. 2):239-243.
- [CrossRef] [Google Scholar]
- Extracting fumonisins from maize: Efficiency of different extraction solvents in multi-mycotoxin analytics. Mycotoxin Res. 2013;29(2):119-129.
- [CrossRef] [Google Scholar]
- Occurrence of fumonisins in feed for swine and horses. Revista Iberoamericana de Micologia. 2012;29(3):175-177.
- [CrossRef] [Google Scholar]
- The origin and current situation of Fusarium oxysporum f. sp. cubense tropical race 4 in Israel and the Middle East. Scientific Rep. 2020;10(1):1-11.
- [CrossRef] [Google Scholar]
- Determination of mycotoxins in plant-based beverages using QuEChERS and liquid chromatography–tandem mass spectrometry. Food Chem. 2017;229:366-372.
- [CrossRef] [Google Scholar]
- Aptamer-based detection of fumonisin B1: A critical review. Anal Chimica Acta. 2021;1160
- [CrossRef] [Google Scholar]
- Development of a multi-mycotoxin liquid chromatography/tandem mass spectrometry method for sweet pepper analysis. Rapid Commun. Mass Spectrom. 2009;23(1):3-11.
- [Google Scholar]
- Development of a new analytical method for the determination of fumonisins B1 and B2 in food products based on high performance liquid chromatography and fluorimetric detection with post-column derivatization. J. Chromatogr. A. 2008;1203(1):88-93.
- [CrossRef] [Google Scholar]
- Detection and Determination of Fumonisins B1, B2, and B3 Contaminating Japanese Domestic Wine by Liquid Chromatography Coupled to Tandem Mass Spectrometry (LC–MS/MS) Curr Microbiol. 2020;77(10):3057-3064.
- [CrossRef] [Google Scholar]
- Determination of Mycotoxins in Food. In: Wong Y.C., Lewis R.J., eds. Analysis of Food Toxins and Toxicants. Vol 2 Volume Set, 139. John Wiley & Sons, Ltd; 2017. p. :137-168. Online ISBN:9781118992685
- [Google Scholar]
- Determination of fumonisins in maize by HPLC with ultraviolet detection of o-phthaldialdehyde derivatives. Mycotoxin Res. 2009;25(4):225-228.
- [CrossRef] [Google Scholar]
- HPLC determination of fumonisin mycotoxins in maize: A comparative study of naphthalene-2,3-dicarboxaldehyde and o-phthaldialdehyde derivatization reagents for fluorescence and diode array detection. J. Chromatogr. B: Analyt Technol Biomed. Life Sci. 2011;879(23):2239-2243.
- [CrossRef] [Google Scholar]
- Simultaneous determination of multiple mycotoxins in swine, poultry and dairy feeds using ultra high performance liquid chromatography-tandem mass spectrometry. Toxins. 2020;12(4):1-18.
- [CrossRef] [Google Scholar]
- Aldehyde analysis by high performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2001;15(12):920-928.
- [CrossRef] [Google Scholar]
- Sphingolipid metabolism in cancer signalling and therapy. Nature Reviews Cancer. 2018;18(1):33-50.
- [CrossRef] [Google Scholar]
- Occurrence of mycotoxins in fish feed and its effects: A review. Toxins. 2020;12(3)
- [CrossRef] [Google Scholar]
- Multi-mycotoxin analysis using dried blood spots and dried serum spots. Anal. Bioanal. Chem. 2017;409(13):3369-3382.
- [CrossRef] [Google Scholar]
- Multi-mycotoxin determination in cereals and derived products marketed in Tunisia using ultra-high performance liquid chromatography coupled to triple quadrupole mass spectrometry. Food Chem. Toxicol. 2012;50(7):2376-2381.
- [CrossRef] [Google Scholar]
- Development of a liquid chromatography / tandem mass spectrometry method for the quantification of fumonisin B1, B2 and B3 in cornflakes. Rapid Commun. Mass Spectrom.. 2005;19:2021-2029.
- [CrossRef] [Google Scholar]
- Simple method for the simultaneous isolation and determination of fumonisin B1 and its metabolite aminopentol-1 in swine liver by liquid chromatography-fluorescence detection. J. Chromatogr. B: Analyt Technol Biomed. Life Sci. 2005;819(1):97-103.
- [CrossRef] [Google Scholar]
- Opinion of the Scientific Panel on Contaminants in Food Chain on a request from the Commission related to fumonisins as undesirable substances in animal feed. EFSA J. 2015;235:1-32.
- [Google Scholar]
- Natural co-occurrence of aflatoxin B1, fumonisin B1 and ochratoxin A in barley and corn foods from Korea. Food Addit Contam. 2002;19(11):1073-1080.
- [CrossRef] [Google Scholar]
- A European database of Fusarium graminearum and F. culmorum trichothecene genotypes. Front Microbiol. 2016;7(apr):1-11.
- [CrossRef] [Google Scholar]
- Patel, S., Hazel, C., and Shah, I., 2011. Report Number : c03061 investigation of the loss of parent fumonisin mycotoxins during food processing. (March), 1–96. https://old.food.gov.uk/sites/default/files/C03061.pdf (accessed 8 Feb 2023).
- Mycotoxins in cereals and related foodstuffs: A review on occurrence and recent methods of analysis. Trends Food Sci. Technol. 2014;36(2):96-136.
- [CrossRef] [Google Scholar]
- Generic sample treatment method for simultaneous determination of multiclass pesticides and mycotoxins in wines by liquid chromatography-mass spectrometry. J. Chromatogr. A. 2012;1249:32-40.
- [CrossRef] [Google Scholar]
- Mycotoxin analysis of industrial beers from Brazil: The influence of fumonisin B1 and deoxynivalenol in beer quality. Food Chem. 2017;218:64-69.
- [CrossRef] [Google Scholar]
- Development, optimization and validation of a multimethod for the determination of 36 mycotoxins in wines by liquid chromatography-tandem mass spectrometry. Talanta. 2014;129:352-363.
- [CrossRef] [Google Scholar]
- Fumonisins and their analogues in contaminated corn and its processed foods – A review. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2018;35(11):2183-2203.
- [CrossRef] [Google Scholar]
- Derivatization for liquid chromatography-mass spectrometry. TrAC - Trends Anal Chem. 2014;59:121-132.
- [CrossRef] [Google Scholar]
- Detection of 13 mycotoxins in feed using modified QuEChERS with dispersive magnetic materials and UHPLC–MS/MS. J Separation Sci. 2018;41(3):756-764.
- [CrossRef] [Google Scholar]
- Ramalho, R.R.F., Pereira, I., da S. Lima, G., et al., 2022. Fumonisin B1 analysis in maize by Molecularly Imprinted Polymer Paper Spray Ionization Mass Spectrometry (MIP-PSI-MS). J Food Compost Anal, 107, 104362. doi:https://doi.org/10.1016/j.jfca.2021.104362.
- Ramírez-Cisneros, M.Á., Rivera, V., Rios, M.Y., Vargas-De-León, C., Dávila, S., and Peralta, R., 2020. Hydrolyzed FB1 (aminopentol) detection on urine of HPV positive Mexican women: A preliminary study. 08 March 2021, PREPRINT (Version 2) available at Research Square (https://doi.org/10.21203/rs.3.rs-68390/v2).
- Development and Validation of a QuEChERS-Based Liquid Chromatography Tandem Mass Spectrometry Multi-Method for the Determination of 38 Native and Modified Mycotoxins in Cereals. J. Agric. Food Chem. 2020;68(16):4657-4669.
- [CrossRef] [Google Scholar]
- Development and validation of a liquid chromatography tandem mass spectrometry multi-method for the determination of 41 free and modified mycotoxins in beer. Food Chem. 2021;338:127801
- [CrossRef] [Google Scholar]
- Simultaneous determination of fumonisins B1, B2 and B3 contaminants in maize by ultra high-performance liquid chromatography tandem mass spectrometry. Analytica Chimica Acta. 2011;692(1–2):138-145.
- [CrossRef] [Google Scholar]
- Application of conventional solid-phase extraction for multimycotoxin analysis in beers by ultrahigh-performance liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2009;57(20):9385-9392.
- [CrossRef] [Google Scholar]
- Application of hybrid linear ion trap-high resolution mass spectrometry to the analysis of mycotoxins in beer. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2011;28(10):1438-1446.
- [CrossRef] [Google Scholar]
- Synthesis of benzofurazan derivatization reagents for short chain carboxylic acids in liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI-MS/MS) Biomed Chromatog. 2009;23(4):443-446.
- [CrossRef] [Google Scholar]
- Ultra-sensitive, stable isotope assisted quantification of multiple urinary mycotoxin exposure biomarkers. Anal Chim Acta. 2018;1019:84-92.
- [CrossRef] [Google Scholar]
- Control of fumonisin: Effects of processing. Environ. Health Perspect. 2001;109(suppl. 2):333-336.
- [CrossRef] [Google Scholar]
- Comparative toxicokinetics of Fusarium mycotoxins in pigs and humans. Food Chem. Toxicol. 2020;137(October 2019):111140-111153.
- [CrossRef] [Google Scholar]
- Analysis of Fumonisin B1 in Fusarium proliferatum-infected asparagus spears and garlic bulbs from Germany by liquid chromatography-electrospray ionization mass spectrometry. J. Agric. Food Chem. 2002;50(10):2778-2781.
- [CrossRef] [Google Scholar]
- Supercritical Fluid Extraction of Fumonisin B1 from Grain Dust. J. Agric. Food Chem. 1996;44(10):3224-3229.
- [CrossRef] [Google Scholar]
- Natural co-occurrence of Fusarium toxins in poultry feed and its ingredients. Journal Fur Verbraucherschutz Und Lebensmittelsicherheit. 2020;15(4):341-350.
- [CrossRef] [Google Scholar]
- Fate of a single dose of the 14C-labelled mycotoxin, fumonisin B1, in rats. Toxicon. 1992;30(7):768-770.
- [CrossRef] [Google Scholar]
- The fumonisin paradox: a review of research on oral bioavailability of fumonisin B1, a mycotoxin produced by Fusarium moniliforme. J Toxicol: Toxin Reviews. 2000;19(2):161-187.
- [Google Scholar]
- UHPLC-ToF-MS method for determination of multi-mycotoxins in maize: Development and validation. Curr Res Food Sci. 2019;1:1-7.
- [CrossRef] [Google Scholar]
- Analysis of fumonisins in corn-based food by liquid chromatography with fluorescence and mass spectrometry detectors. Food Chem. 2009;112(4):1031-1037.
- [CrossRef] [Google Scholar]
- Occurrence of fumonisins B1 and B2 in Portuguese maize and maize-based foods intended for human consumption. Food Addit Contam. 2007;24(4):381-390.
- [CrossRef] [Google Scholar]
- Fumonisins determination in urine by LC-MS-MS. Anal. Bioanal. Chem. 2010;396(2):809-816.
- [CrossRef] [Google Scholar]
- Multimycotoxin analysis of crude extracts of nuts with ultra-high performance liquid chromatography/tandem mass spectrometry. J Food Compost Anal. 2014;34(2):171-177.
- [Google Scholar]
- Quantitation of fumonisin B1 and B2 in feed using FMOC pre-column derivatization with HPLC and fluorescence detection. Food Chem. 2017;234:174-179.
- [CrossRef] [Google Scholar]
- Sokolovic, M., 2022. HPLC and ELISA Methods for Detection and Quantification of Fumonisins in Naturally Contaminated Maize., Research Square, PREPRINT available at Research Square (https:// doi: 10.21203/rs.3.rs-1908427/v1).
- Determination of mycotoxins in cereals by liquid chromatography tandem mass spectrometry. Food Chem. 2012;130(4):1055-1060.
- [CrossRef] [Google Scholar]
- Development and application of salting-out assisted liquid/liquid extraction for multi-mycotoxin biomarkers analysis in pig urine with high performance liquid chromatography/tandem mass spectrometry. J. Chromatogr. A. 2013;1292:111-120.
- [CrossRef] [Google Scholar]
- Simultaneous determination of ochratoxin A, mycophenolic acid and fumonisin B2 in meat products. Anal. Bioanal. Chem. 2010;398(3):1535-1542.
- [CrossRef] [Google Scholar]
- Soriano del Castillo, J.M., 2007. Micotoxinas en alimentos, Ediciones Díaz de Santos. ISBN: 978-84-7978-808-7.
- LC–MS/MS multi-method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2008;25(4):472-489.
- [CrossRef] [Google Scholar]
- Genetic and phenotypic variation of Fusarium proliferatum isolates from different host species. J App Gen. 2011;487–496
- [CrossRef] [Google Scholar]
- A review of the toxic effects and mechanisms of action of fumonisin B 1. Hum Exp Toxicol. 2008;27(11):799-809.
- [CrossRef] [Google Scholar]
- Efficacy of different c18 hplc analytical columns in the analysis of fumonisins b1 and b2 in different matrices. Biointerface Res. Appl. Chem. 2022;12(2):1721-1734.
- [CrossRef] [Google Scholar]
- A method for multiple mycotoxin analysis in wines by solid phase extraction and multifunctional cartridge purification, and ultra-high-performance liquid chromatography coupled to tandem mass spectrometry. Toxins. 2012;4(6):476-486.
- [CrossRef] [Google Scholar]
- Development of a multi-mycotoxin analysis in beer-based drinks by a modified quechers method and ultra-high-performance liquid chromatography coupled with tandem mass spectrometry. Anal Sci. 2011;27(6):629-635.
- [CrossRef] [Google Scholar]
- Determination of Fumonisin B1 in animal tissues with immunoaffinity purification. J. Chromatogr. B: Analyt Technol Biomed Life Sci. 2008;870(1):140-144.
- [CrossRef] [Google Scholar]
- Taylor, P., and Scott, P.M., 2012. Food Additives & Contaminants: Part A - Recent research on fumonisins : a review., (May 2015), 37–41.
- Detection of Fumonisins in Fresh and Dehydrated Commercial Garlic. J. Agric. Food Chem. 2017;65(32):7000-7005.
- [CrossRef] [Google Scholar]
- Toro, S., 2022. iNaturalist Mexico. https://www.inaturalist.org/observations/134863617. Last access February 7th, 2022.
- Analytical methods for determination of mycotoxins: A review. Anal Chim Acta. 2009;632(2):168-180.
- [CrossRef] [Google Scholar]
- Application of single immunoaffinity clean-up for simultaneous determination of regulated mycotoxins in cereals and nuts. Talanta. 2013;117:345-351.
- [CrossRef] [Google Scholar]
- Fumonisin biomarkers in maize eaters and implications for human disease. World Mycotoxin. 2013;6(3):223-232.
- [CrossRef] [Google Scholar]
- Determination of arylamines and aminopyridines in pharmaceutical products using in-situ derivatization and liquid chromatography–mass spectrometry. J. Chromatogr. A. 2009;1216(16):3563-3570.
- [CrossRef] [Google Scholar]
- Effect of solvents on the fumonisins analysis by high-performance liquid chromatography with AccQ.Fluor as the derivatizing reagent. J. Chromatogr. A. 2000;870(1–2):469-472.
- [CrossRef] [Google Scholar]
- Determination of volatile thiols in lipid matrix by simultaneous derivatization/extraction and liquid chromatography–high resolution mass spectrometric analysis. Application to virgin olive oil. J. Chromatogr. A. 2013;1318:180-188.
- [CrossRef] [Google Scholar]
- Fumonisins. Reprod Dev Toxicol. 2017;1:925-943.
- [CrossRef]
- Risk assessment of exposure to mycotoxins (aflatoxins and fumonisins) through corn tortilla intake in Veracruz City (Mexico) Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2019;36(6):929-939.
- [CrossRef] [Google Scholar]
- The health and toxic adverse effects of Fusarium fungal mycotoxin, fumonisins, on human population. Am. J. Infect. Dis. 2009;5(4):280-288.
- [CrossRef] [Google Scholar]
- Rapid determination of fumonisin B1 in food samples by enzyme-linked immunosorbent assay and colloidal gold immunoassay. J. Agric. Food Chem. 2006;54(7):2491-2495.
- [CrossRef] [Google Scholar]
- Development and application of a method for the analysis of 9 mycotoxins in maize by HPLC-MS/MS. J. Food Sci.. 2013;78(11):1752-1756.
- [CrossRef] [Google Scholar]
- Simultaneous determination of aflatoxins, fumonisin B1, T-2 and cyclopiazonic acid in agri-products by immunomagnetic solid-phase extraction coupled with UHPLC-MS/MS. Food Chem. 2022;378:132020
- [CrossRef] [Google Scholar]
- Analysis of mycotoxin fumonisins in corn products by high-performance liquid chromatography coupled with evaporative light scattering detection. Food Chem. 2008;107(2):970-976.
- [CrossRef] [Google Scholar]
- Weiying, W., Weiwei, Z., Shunli, H., et al. 2022. Determination of fumonisins in edible vegetable oil by MIL-101(Cr) based dispersive solid phase microextraction combined with high performance liquid chromatography tandem mass spectrometry., 11, 133–143.
- WHO-Department of Food Safety and Zoonoses (2018) Fumonisins. Retrieved February 28, 2021, from https://www.who.int/foodsafety/FSDigest_Fumonisins_EN.pdf.
- Determination of pesticide residues in food matrices using the QuEChERS methodology. Food Chem. 2011;125(3):803-812.
- [CrossRef] [Google Scholar]
- HIV and hepatocellular and esophageal carcinomas related to consumption of mycotoxin-prone foods in sub-Saharan Africa. Am. J. Clin. Nutr. 2010;92(1):154-160.
- [Google Scholar]
- Fumonisin B1 induced aggressiveness and infection mechanism of Fusarium proliferatum on banana fruit. Environ. Pollut. 2021;288:117793
- [CrossRef] [Google Scholar]
- Fumonisins, trichothecenes and zearalenone in cereals. Int J Mol Sci. 2008;9(11):2062-2090.
- [CrossRef] [Google Scholar]
- Simultaneous determination of multi-mycotoxins in palm kernel cake (PKC) using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Food Addit. Contam. - Chem. Anal. Control Expo. Risk Assess. 2014;31(12):2071-2079.
- [CrossRef] [Google Scholar]
- Simultaneous analysis of 20 mycotoxins in grapes and wines from Hexi Corridor region (China): Based on a QuEChERS–UHPLC–MS/MS method. Molecules. 2018;23(8)
- [CrossRef] [Google Scholar]
- A review of sample preparation methods for the pesticide residue analysis in foods. Cent Eur J Chem. 2012;10(3):900-925.
- [CrossRef] [Google Scholar]
- Determination of mycotoxins in milk-based products and infant formula using stable isotope dilution assay and liquid chromatography tandem mass spectrometry. J. Agric. Food Chem. 2013;61(26):6265-6273.
- [CrossRef] [Google Scholar]
- LC-MS/MS Analysis of Fumonisin B1, B2, B3, and Their Hydrolyzed Metabolites in Broiler Chicken Feed and Excreta. Toxins. 2022;14(2):1-10.
- [CrossRef] [Google Scholar]
- A single extraction method for the analysis by liquid chromatography/tandem mass spectrometry of fumonisins and biomarkers of disrupted sphingolipid metabolism in tissues of maize seedlings. Anal. Bioanal. Chem. 2008;391(6):2257-2263.
- [CrossRef] [Google Scholar]
- Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry. J. Chromatogr. A. 2006;1136(2):123-169.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104716.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2
Table 1. LC-MS methods for FBs without clean up.




