

Translate this page into:
Design and synthesis of some novel pyridothienopyrimidine derivatives and their biological evaluation as antimicrobial and anticancer agents targeting EGFR enzyme
⁎Corresponding authors. e.mohi.2010@live.com (Eman M. Mohi El-Deen), manal.hasan52@live.com (Manal M. Anwar)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
A new series of pyridothienopyrimidine derivatives was designed and evaluated as antimicrobial and anticancer agents. The target compounds were synthesized starting with 3-aminothieno[2,3-b]pyridine-2-carboxamide derivative 1 which underwent cyclocondensation reaction with aromatic aldehydes to give the key intermediates 2a,b. By further treatment of 2a,b with various reagents, the target 2,4-disubstituted-pyrido[3′,2′:4,5]thieno[2,3-d]pyrimidines 3a,b–11a,b were obtained. To evaluate the antimicrobial activity of the new compounds, they were tested against five bacterial and five fungal strains. Compounds 6c, 8b, 9a and 9b revealed the most significant antimicrobial activity against the tested microorganisms with MIC values range (4–16 μg/mL). Also, compounds 2a,b–11a,b were screened for their in vitro cytotoxic activity against HepG-2 and MCF-7 cancer cell lines compared with doxorubicin and cisplatin as references drugs. Moreover, compounds (2b, 4a, 6a, 7b, 7c and 9a) which exhibited the most potent anticancer activity, were further subjected to EGFRWT enzyme inhibition assay utilizing erlotinib as a standard drug. The compounds 6a, 7b, 7c and 9a which showed the most promising suppression effects were also evaluated as inhibitors against the mutant forms EGFRL858R and EGFRT790M. The 4-aminopyrazolone analogue 9a showed superior anticancer activity against both HepG-2 and MCF-7 cell lines (IC50 = 1.27, 10.80 μM, respectively) and more potent enzymatic inhibition activity against EGFRWT and its mutant forms EGFRL858R and EGFRT790M than that obtained by erlotinib (IC50 = 0.021, 0.053, 0.081 µM, respectively, IC50erlotinib; 0.027, 0.069, 0.550 µM, respectively). Finally, the molecular docking study showed good binding patterns of the most active compounds with the prospective target EGFRWT.
Keywords
Pyridothienopyrimidines
Antimicrobial activity
Anticancer activity
EGFR inhibitors

1 Introduction
Cancer disease represents a serious threat to human health due to its massive and complicated etiology (Wu et al., 2018). Liver cancer is a global health issue representing the sixth most common cancer type, and the third leading reason for cancer-related death. Furthermore, 75%–85% of liver cancer cases include hepatocellular carcinoma (HCC) (Sung et al., 2021). Although the spread of HCC, few drugs are available for its clinical treatment, especially in the advanced stages, thereby many efforts should be made in the development of new drugs for this serious cancer type (Luo et al., 2021). On the other hand, breast cancer (BC) is the world’s most prevalent cancer resulting in about 14% of the cancer-related deaths among women (Sung et al., 2021, Presti and Quaquarini, 2019). Despite the recent discovery of several promising novel therapies which exhibited considerable therapeutic success, drug resistance still remains one of the most important challenges in BC treatment (Lainetti et al., 2020).
Moreover, many recent studies exhibited that epidermal growth factor receptor (EGFR) was generally altered in many types of cancers. Thus, the inhibition of EGFR's kinase activity became a primary target for developing many cancer therapeutics (Thomas and Weihua, 2019). In clinical use, there are many small molecules acting as EGFR inhibitors such as: Osimertinib (AZD9291), olmutinib, PF06747775, WZ4002, Rociletinib, Nazartinib and others belong to the 3rd generation of EGFR kinase inhibitors (Liang et al., 2021; Phan et al., 2019). However, various side effects have been detected with their uses in the clinical application (Shah and Shah, 2019; Wang and Cang, 2016). Also, several mechanisms of resistance have been described to EGFR-TKIs, such as secondary and tertiary mutations in the EGFR geneT790M and C797S, respectively (Morgillo et al., 2016; Jänne et al., 2015). Accordingly, many efforts are still required to reach new EGFR inhibitors which can overcome resistance mechanisms and have high selectivity to minimize the adverse effects.
In addition, the immunosuppression, which usually results due to the anticancer drug regimen and the destruction of the mucosal barrier due to utilizing invasive devices in cancer patients making them at risk of different infectious diseases that need long-term prophylactic antibiotics (Sharma et al., 2021). Thus, there is an increasing urgency in designing novel anticancer agents having dual anticancer and antimicrobial activities to protect cancer patients against microbial infections (Felício et al., 2017).
Thieno[2,3-b]pyridine nucleus plays a vital role in the field of drug discovery, producing a wide spectrum of biological activities such as; anticancer (Abdelaziz et al., 2018; Arabshahi et al., 2015; Eurtivong et al., 2017; Al-Trawneh et al., 2021), antimicrobial (Ghattas et al., 2015; Mohi El-Deen et al., 2019; Mansour, 2020), antiviral (Amorim et al., 2017; Schnute et al., 2007), anti-inflammatory (Liu et al., 2013; Madhusudana et al., 2012), and antiangiogenic (Rizka et al., 2019) activities. In addition, the strategies of different new studies have been directed to the synthesis of compounds bearing thieno[2,3-b]pyridine fused with pyrimidine ring giving rise to the pyrido[3′,2′:4,5]thieno[3,2-d] pyrimidine scaffold as significant anticancer (Aziz et al., 2015a; Sirakanyan et al., 2019) and antimicrobial candidates (Mohi El-Deen et al., 2020; Ge Zayda et al., 2020; Sanad and Mekky, 2021; Fayed et al., 2014; El-Essawy et al., 2016) (Fig. 1).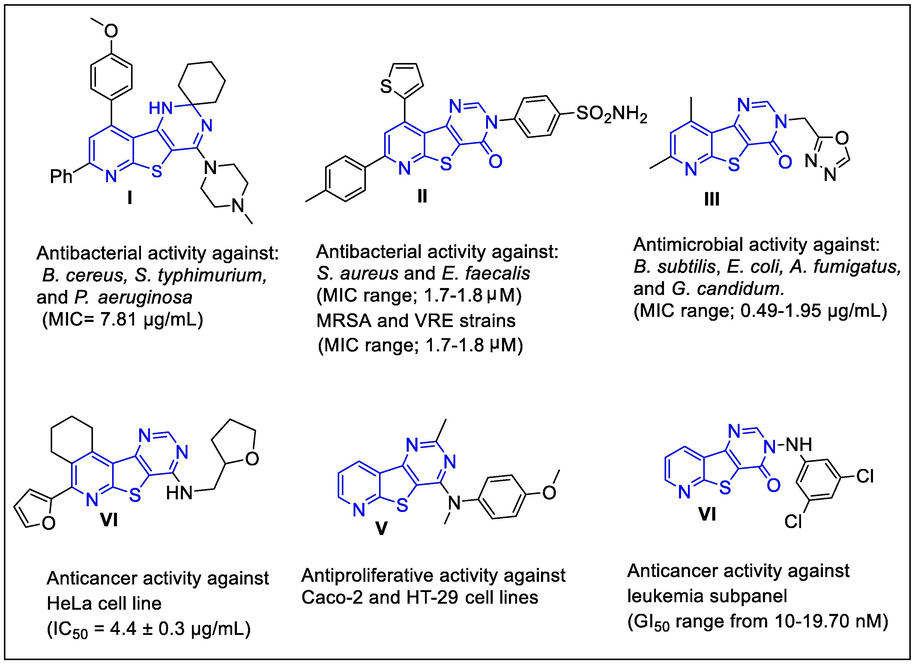
Some previously synthesized pyridothienopyrimidine derivatives as antimicrobial agents (I-III) and as anticancer agents (IV-VI).
Moreover, many biological studies revealed that the pyridothienopyrimidine-based derivatives produce their anticancer activities via protein kinase suppression effects such as; Pim-1 kinase, casein kinase 1 (CK1), dual-specificity protein kinase (CLK1), vascular endothelial growth factor receptor (VEGFR-2), and EGFR kinase (El-Nassan et al., 2018; Loidreau et al., 2012; Aziz et al., 2015b).
It has been reported that there are four essential pharmacophoric features shared by the reported and clinically used EGFR-TKIs: (i) a flat heterocyclic-aromatic ring system (ii) a terminal hydrophobic head (iii) NH-linker (iv) a hydrophobic tail (Elmetwally et al., 2019). Chia-Hsien Wu et al., have synthesized compound VII utilizing a knowledge-based design procedure for ATP-competitive inhibitors binding with the active site of the EGFR-TK. This compound showed high efficacy for suppressing the targeted enzyme, EGFR-TK (Wu et al., 2010). Furthermore, Milik et al. have reported the thieno[2,3-d]pyrimidine compound VIII as a potent anticancer agent targeting EGFR and HER2-TK (Milik et al., 2018) (Fig. 2).![The proposed hypothetic model of the new pyridothieno[3,2-d]pyrimidine target derivatives as EGFR TKIs.](/content/184/2022/15/4/img/10.1016_j.arabjc.2022.103751-fig3.png)
The proposed hypothetic model of the new pyridothieno[3,2-d]pyrimidine target derivatives as EGFR TKIs.
In view of the above issues, this work deals with the design and synthesis of a new series of pyridothienopyrimidine compounds (2a,b–11a,b) as antimicrobial and as anticancer agents bearing the reported essential pharmacophoric features of EGFR-TKIs. As the thieno[3,2-d]pyrimidine sector of the pyridotheinopyrimidine represents the central hetero-aromatic ring since the nitrogen atoms of the pyrimidine ring serve as hydrogen-bond acceptors resulting in promising EGFR-TK inhibitory potency. While, the substituent at position-4 was the linker (spacer) area, where the length of the linker in addition to the number of its hydrogen-bond acceptor and/or hydrogen-bond donor groups was modified. The different linkers were NH as compounds 5a,b, 6a–c, NH-N = C as 7a–c as well as a direct attachment with pyrazoline-N as compounds 8a,b–11a,b was also carried out in order to study its positive or negative impact on the target enzyme inhibition activity. The hydrophobic tail was based on the pyridine nucleus to occupy the front hydrophobic region of the ATP binding site. Also, the hydrophobic head is an alkyl or aryl group (Elmetwally et al., 2019, Nasser et. Al., 2020) (Fig. 2).
Next, all the target compounds were evaluated as antimicrobial agents against a panel of gram-positive, gram-negative bacterial, yeast and fungal strains. Also, the target compounds were assessed as anticancer agents against human liver carcinoma cell line (HepG2) and human breast cancer cells (MCF-7). The safety of the most potent anticancer candidates (2b, 4a, 6a, 7b, 7c and 9a) was evaluated against the normal WISH cell line. In addition, these compounds were further evaluated for their ability to suppress EGFR kinase. Also, molecular docking study was conducted to find out the plausible binding modes of the most promising derivatives in the active pocket of EGFR.
2 Results and discussion
2.1 Chemistry
The new set of pyridothienopyrimidine derivatives (2a,b–11a,b) was prepared utilizing the synthetic pathways outlined in Schemes 1, 2. The molecular structures of all the new compounds were confirmed via spectral data (IR, 1H NMR, 13C NMR and Mass) and elemental microanalyses. The bicyclic compound 3-amino-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide (1) (Youssefyeh, et al., 1984) was refluxed with aldehydes (4-methoxybenzaldehyde or furan-2-carbaldehyde) in glacial acetic acid to give, via cyclocondensation reaction, the tricyclic compounds 2,3-dihydropyridothienopyrimidin-4(1H)-ones 2a,b. Upon treatment of 2a,b with benzoquinone as an oxidizing agent, they underwent dehydrogenation to give the pyridothienopyrimidin-4(3H)-ones 3a,b. Subsequently, the latter derivatives 3a,b were treated with POC13/PCl5 mixture to yield the corresponding 4-chloro derivatives 4a,b (Scheme 1).![Synthesis of new pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine-based compounds 2a,b–6a-c.](/content/184/2022/15/4/img/10.1016_j.arabjc.2022.103751-fig4.png)
Synthesis of new pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine-based compounds 2a,b–6a-c.
![Synthesis of new pyrido[3′,2′:4,5] thieno[3,2-d]pyrimidine-based compound 7a-c–11a,b.](/content/184/2022/15/4/img/10.1016_j.arabjc.2022.103751-fig5.png)
Synthesis of new pyrido[3′,2′:4,5] thieno[3,2-d]pyrimidine-based compound 7a-c–11a,b.
The tricyclic structures of 2a,b were confirmed by 1H NMR spectra of 2a and 2b, they showed the signals of the aromatic protons of the appropriate aldehyde at δ 6.31–7.61 ppm alongside the two singlets signals corresponding to the two pyrimidinone NH groups at δ (7.02, 8.32 ppm) and δ (7.16, 8.35 ppm), respectively. Also, 1H NMR spectra of 2a and 2b revealed a singlet signal at δ 5.81 ppm and at δ 5.90 ppm corresponding to the CH of the pyrimidinone ring. The vanishing of one signal of the two singlets signals corresponding to the two pyrimidinone NH groups with the CH-pyrimidinone signal in the 1H NMR spectra of 3a,b confirmed the dehydrogenation reaction of 2a,b. The signal of the other NH group appeared as a downfield singlet at δ 12.83 and 13.0 ppm in the spectra of 3a and 3b, respectively, which indicated the presence of NH adjacent to the C = O group and confirmed the 1,2-dehydrogenation of 2a,b instead of the 2,3-dehydrogenation. Also, the 13C NMR spectra of 2a,b and 3a,b gave additional support to their structures. In the 13C NMR spectra of 2a,b the signals of CH-pyrimidinone appeared at δ 61.41–66.13 ppm and shifted downfield with the aromatic carbons in the 13C NMR spectra of 3a,b. Moreover, the IR and 1H NMR spectra of the 4-chloro analogues 4a,b confirmed their molecular structures by the absence of C = O and NH bands and the disappearance of the signal of the NH group, respectively.
In addition, compounds 4a,b acted as facile intermediates for nucleophilic substitution reactions with hydrazine hydrate and various amino acids, namely: glycine, valine, and alanine to afford the corresponding target compounds 5a,b and 6a-c, respectively (Scheme 1). IR spectra of compounds 5a,b exhibited characteristic absorption bands at the range 3380–3288 cm−1 confirming the existence of NH, NH2. Also, 1H NMR spectra of 5a and 5b showed a D2O exchangeable signal at δ 4.94 ppm assignable to the NH2 group beside the signal corresponding to the NH group at δ 9.02, 9.25 ppm, respectively. Moreover, IR spectra of the amino acid derivatives 6a–c exhibited broad absorption bands at 3400–3393 cm−1 due to OH groups, and at 1676–1670 cm−1 related to C = O groups. Also,1H NMR spectra of 6a-c showed two D2O exchangeable signals at the ranges δ 6.69–7.59 ppm and δ 12.23–12.83 ppm representing NH and OH protons alongside the parent protons which appeared at their expected regions. An additional singlet appeared by the glycine derivative 6a at δ 4.11 ppm due to NH-CH2 protons, while valine derivative 6b revealed douplet-multiplet signals at δ 0.99 and 1.90 ppm assignable for the protons of (CH3)2 and –CH of the isopropyl group, respectively. Also, 13C NMR spectrum of 6a revealed a signal at δ 48.82 ppm due to –NH-CH of alanine moiety.
Furthermore, the 4-hydrazineyl derivatives 5a,b served as other intermediate precursors for further condensation reactions. Treatment of 5a,b with different aldehydes namely; 4-methylbenzaldehyde, 2,5-dimethoxybenzaldehyde and thiophene-2-carbaldehyde in ethanol/acetic acid mixture led to the formation of the corresponding hydrazide Schiff’s bases 7a–c. 1H NMR spectra of compounds 7a–c represented singlet signals of the azomethine proton –N = CH at the range δ 8.39–8.77 ppm alongside the aromatic protons. While, the signal of the hydrazide NH2 group was vanished and the signal of the NH group was shifted downfield at the range δ 12.01–12.20 ppm confirming the formation of the Schiff’s bases 7a–c. Moreover, 13C NMR data of 2,5-dimethoxybenzylidene derivatives 7c showed two signals at δ 55.73 and 56.54 ppm corresponding to the (2 OCH3) groups in addition to the expected signals due to the aromatic carbons. The 4-hydrazineyl derivatives 5a,b were further cyclized into the higher polycyclic triazolo derivatives 8a,b by a condensation reaction with formic acid. 1H NMR and 13C NMR spectra of 8a,b were in agreement with the molecular structures of the compounds. Also, 5a,b were condensed with active methylene compounds, namely: ethyl cyanoacetate, ethyl acetoacetate and acetyl acetone to give the corresponding 4-pyrazolone derivatives 9a,b, 10a,b and the 4-pyrazole derivatives 11a,b; respectively (Scheme 2).
The molecular structures of the 4-pyrazolone compounds 9a,b and 10a,b were confirmed by the existence of a singlet signal at the range δ 2.97–3.06 ppm corresponding to the pyrazolone-CH2 in their 1HNMR spectra. On the other hand, an additional band at 1695–1674 cm−1 for the C = O group of the pyrazolone nucleus was found in their IR spectra. Also, 13C NMR spectra of 9a,b and 10a,b represented a signal at the range δ 170.17–172.92 ppm assignable for pyrazolone-C = O group. Furthermore, 1H NMR spectra of the 4-pyrazole derivatives 11a,b showed four singlet signals at the range δ 2.22–2.99 ppm referring to the pyrazole-2CH3 in addition to the parent pyridine-2CH3. Also, the four signals of the 4CH3 of 11a,b were exhibited in their 13C NMR spectra at the range δ 13.72–24.71 ppm alongside the parent signals. Further support for the suggested structures of the new compounds was gained by their mass spectra, which were in accordance with the proposed structures representing their correct molecular ion peaks.
2.2 Biological evaluation
2.2.1 Antimicrobial activity
Since the strategy of our work was based on achieving new pyridotheinopyrimidine compounds possessing dual cytotoxic and antimicrobial activities, all the new compounds were evaluated as antimicrobial agents versus a panel of microbial strains. They were examined against three gram-positive bacteria viz. Staphylococcus aureus 25923, Bacillus subtilis 6633, Bacillus cereus 33018, two gram-negative bacteria viz. Escherichia coli 8739, Salmonella typhimurium 14028, three yeasts viz. Candida albicans 10231, Candida tropicals 750, Saccharomycese cerevisiae and two fungi viz. Aspergillus flavus, Aspergillus niger EM77 (KF774181). The zones of inhibition in (mm) and the MIC values in (μg/mL) were determined for the target compounds and the reference drugs Amoxicillin trihydrate and clotrimazole (Table 1 and 2). N.A. = No Activity (mean diameter of inhibition zones < 11 mm). Minimum Inhibitory Concentrations (MIC) (μg/mL). N.A. = No Activity (mean diameter of inhibition zones < 11 mm). Minimum Inhibitory Concentrations (MIC) (μg/mL).
Gram + ve Bacteria
Gram-ve Bacteria
Compd. No.
S. aureus 25,923
B. subtilis
6633
B. cereus 33,018
E. coli
RCMB 010,052
S. typhimurium
ATCC 14,028
2a
20 (64)
18 (64)
N.A.
N.A.
N.A.
2b
27 (32)
23 (64)
23 (32)
22 (64)
21(64)
3a
14 (128)
14 (128)
NA
NA
NA
3b
12 (128)
13 (128)
NA
NA
NA
4a
22 (64)
17 (64)
21 (64)
28 (8)
27 (16)
4b
24 (64)
21 (64)
22 (32)
26 (32)
24 (32)
5a
19 (64)
18 (64)
14 (128)
12 (128)
11 (128)
5b
20 (64)
25 (32)
24 (32)
19 (64)
18 (128)
6a
35 (4)
32 (8)
29 (16)
27 (16)
31 (16)
6b
19 (64)
24 (32)
20 (64)
22 (64)
24 (64)
6c
25 (32)
28 (16)
25 (32)
27 (8)
26 (16)
7a
13 (128)
25 (32)
19 (64)
23 (32)
21(64)
7b
25 (32)
26 (32)
22 (64)
20 (64)
20 (64)
7c
27 (16)
25 (16)
27 (32)
14 (64)
28 (16)
8a
24 (32)
22 (64)
22 (64)
23 (64)
20 (64)
8b
30 (4)
29 (8)
28 (16)
30 (8)
28 (16)
9a
33 (4)
29 (8)
31 (16)
27 (8)
30 (16)
9b
28 (8)
26 (16)
25 (16)
26 (8)
28 (16)
10a
25 (64)
24 (32)
24 (32)
18 (64)
20 (64)
10b
22 (64)
N.A.
13 (128)
16 (64)
N.A.
11a
24 (32)
25 (32)
22 (64)
24 (64)
27 (32)
11b
26 (32)
24 (32)
21 (32)
25 (32)
25 (32)
Amoxicillin
Trihydrate30(4)
29(8)
28 (16)
27 (8)
28 (16)
Yeasts
Fungi
Compd. No.
C. albicans ATCC 10,231
C. tropicals ATCC 750
S.
cerevisiae
A. flavus
A. niger EM77 (KF774181)
2a
NA
NA
22 (32)
28 (32)
26 (32)
2b
22 (64)
21 (64)
23 (32)
25 (32)
26 (32)
3a
N.A.
N.A.
16 (128)
11 (128)
14 (64)
3b
N.A.
N.A.
15 (128)
N.A.
N.A.
4a
29 (8)
28 (8)
21 (64)
27 (32)
17 (64)
4b
27 (32)
24 (32)
24 (32)
17 (64)
19 (64)
5a
18 (128)
14 (128)
17 (128)
11 (128)
14 (128)
5b
19 (64)
18 (128)
17 (64)
23 (32)
22 (64)
6a
29 (8)
31 (8)
37 (8)
33 (8)
35 (8)
6b
26 (64)
24 (64)
16 (128)
14 (128)
18 (64)
6c
32 (8)
29 (8)
36 (8)
29 (16)
28 (16)
7a
23 (64)
22 (64)
24 (32)
18 (64)
19 (64)
7b
20 (64)
20 (64)
23 (32)
15 (64)
14 (64)
7c
30 (8)
28 (8)
20 (64)
30 (8)
29(8)
8a
24 (32)
26 (32)
24 (32)
18 (64)
18(64)
8b
30 (8)
30 (8)
37 (8)
35 (8)
36 (8)
9a
33 (8)
28 (8)
35 (8)
31 (8)
30 (8)
9b
37 (8)
33 (8)
33 (8)
28(16)
27 (16)
10a
23 (64)
14 (128)
24 (32)
18 (64)
20 (64)
10b
22 (64)
N.A.
18(64)
15 (64)
17 (64)
11a
21 (64)
24 (32)
19 (64)
26 (32)
22 (64)
11b
24 (32)
23 (32)
21 (64)
22 (32)
21 (64)
Clotrimazole
28 (16)
30 (8)
35 (8)
30(8)
31 (8)
On the basis of the MIC values, it is apparent that there was wide variability in the antimicrobial potency of the evaluated derivatives. It has been found that the derivatives; alanine amino acid 6a, the polycyclic triazolo derivatives 8b and the 4-aminopyrazolones 9a, 9b exhibited potent activity against the five tested bacterial strains with MIC values range (4–16) μg/mL, that were equivalent to that obtained by the reference drug amoxicillin trihydrate. While, the 4-chloro- analogue 4a and the glycine derivative 6c showed equipotent antibacterial activity to that of amoxicillin trihydrate against the tested gram-negative strains with MIC values 8 μg/mL and 16 μg/mL for E. coli and S. typhimurium, respectively.
With respect to the tested yeast and fungal strains; 4a, 6a, 6c, 7c, 8b, 9a and 9b revealed more potent activity against Candida albicans than clotrimazole with MIC values 8 μg/mL (MICclotrimazole = 16 μg/mL), and they exhibited equipotent activity to clotrimazole against Candida tropical with MIC values 8 μg/mL. In addition, Saccharomycese cerevisiae showed significant sensitivity towards the target compounds 6a, 6c, 8b, 9a, and 9b similar to its sensitivity towards clotrimazole with MIC values 8 μg/mL. While, compounds 6a, 7c, 8b, and 9a revealed the most potent growth inhibitory activity against Aspergillus flavus and Aspergillus niger, they displayed the same activity against the two fungi with MIC values 8 μg/mL similar to that of clotrimazole. However, compounds 2a, 3a, 3b, and 10b showed weak activity against some of the tested bacterial and fungal strains and were inactive to the others. On the other hand, the rest of the target compounds (2b, 4b, 5a;b, 6b, 7a;b, 8a, 10a;b, and 11a;b) exhibited antimicrobial activity varied in potency from moderate to weak with MIC values range (32–128) μg/mL.
2.2.2 In vitro cytotoxic activity of the target pyridothienopyrimidine derivatives
The antiproliferative potency of the target compounds 2a,b–11a,b was evaluated against HepG-2 and MCF-7 cancer cell lines utilizing MTT method. The commercially available doxorubicin and cisplatin served as reference drugs. The concentrations of the compounds that induced 50% inhibition of cell viability (IC50, μM) were determined and listed in Table 3.
Compound
No.
IC50 (µM)
HepG-2
MCF-7
WISH
2a
11.01 ± 0.75
23.02 ± 1.76
2b
3.11 ± 0.21
12.90 ± 0.70
494.00 ± 17.90
3a
5.71 ± 0.32
60.30 ± 3.85
3b
15.10 ± 1.03
35.60 ± 3.03
4a
3.17 ± 0.22
11.07 ± 0.19
107.01 ± 4.20
4b
48.70 ± 3.34
21.70 ± 0.85
5a
13.01 ± 0.89
28.03 ± 1.75
5b
8.37 ± 0.51
71.61 ± 3.51
6a
1.80 ± 0.12
14.21 ± 0.22
121.02 ± 5.10
6b
4.02 ± 0.37
22.50 ± 1.90
6c
17.80 ± 1.22
29.66 ± 2.65
7a
21.30 ± 1.46
23.80 ± 2.80
7b
2.63 ± 0.18
20.51 ± 2.30
302.00 ± 11.40
7c
2.15 ± 0.15
19.71 ± 1.60
213.01 ± 7.50
8a
3.93 ± 0.27
41.10 ± 3.30
8b
6.10 ± 0.42
22.40 ± 2.70
9a
1.27 ± 0.09
10.80 ± 0.74
278.02 ± 9.70
9b
5.73 ± 0.39
38.11 ± 3.02
10a
17.40 ± 1.20
80.61 ± 3.95
10b
9.02 ± 0.48
64.60 ± 2.90
11a
9.68 ± 0.66
20.80 ± 1.08
11b
16.30 ± 1.12
36.61 ± 3.20
Doxorubicin
2.85 ± 0.21
3.58 ± 0.33
432.10 ± 19.30
Cisplatin
---
20.70 ± 0.83
2.2.2.1 Structure-activity relationship (SAR)
The biological data revealed that the tested derivatives displayed more promising cytotoxic activity against HepG2 cancer cell line of IC50 range; 1.81–48.70 µM more than MCF-7 cells of IC50 range 10.80–80.61 µM, compared to doxorubicin of IC50s; 2.85, 3.58 µM, respectively. With respect to HepG2, it has been noted that the conjugation of the aminopyrazolinone nucleus to the parent 2-(4-methoxyphenyl)-pyridothienopyrimidine scaffold as compound 9a exhibited 2.2 folds more significant activity than doxorubicin of IC50 value; 1.27 µM, IC50doxorubicin 2.85 µM. Furthermore, tagging the latter parent core with glycine amino acid as compound 6a represented 1.5 folds more potent cytotoxic activity than the reference drug of IC50 value; 1.80 µM. Also, the Schiff’s bases 7b,c revealed slightly more potent cytotoxicity than that of doxorubicin with IC50s; 2.63, 2.15 µM, respectively. While, the parent 2b, the 4-chloro derivative 4a, the tetracyclic pyridothienotriazolopyrimidine derivative 8a and the valine amino acid derivative 6b showed a slight decrease in the potency than the reference drug with IC50 values; 3.11–4.02 µM respectively. Additionally, the resultant data also showed that the 4-pyrazolone derivative 9b, the pyridothienopyrimidin-4-one derivative 3a, the 4-hydrazino derivative 5b and the tetracyclic 8b produced cytotoxic activity ranging from half to third that of doxorubicin with IC50 values range; 5.71–8.37 µM. A further apparent decrease in the cytotoxicity was observed by the rest of the compounds (2a, 3b, 4b, 5a, 6c, 7a, 10a,b and 11a,b), they gave a wide variety of IC50 values ranging from 9.02 to 48.70 µM.
On the other hand, MCF-7 cancer cells appeared to be less sensitive to the tested compounds in comparing to doxorubicin. Whereas, all the target compounds exhibited lower cytotoxicity with IC50 values range; 10.80–80.61 µM than that of doxorubicin with IC50 value; 3.58 µM. While taking cisplatin as a reference drug, most of the evaluated derivatives represented promising cytotoxic effects. Fortunately, the 4-pyrazolone derivative 9a, the most potent cytotoxic agent against HepG2 cancer cell line, was the most potent agent against MCF-7 cancer cells with IC50; 10.80 µM compared with IC50cisplatin; 20.70 µM. Moreover, the parent pyridothienopyrimidin-4-one 2b, the 4-chloro analogue 4a, the glycine derivative 6a and the Schiff’s bases 7c exhibited more potent activity than cisplatin with IC50 values range; 11.07–19.70 µM. Approximate equipotent activity to that of the reference drug cisplatin was gained by the compounds 2a, 4b, 6b, 7a, 7b, 8b and the 4-pyrazole derivative 11b of IC50 values; 20.51–23.80 µM. While, the rest derivatives (3a;b, 5a;b, 6c, 8a, 10a;b, 11a) exhibited less potent activity than cisplatin with IC50 values range; 28.03–80.61 µM.
Since the frequency and the severity of the adverse effects to the normal healthy cells at therapeutic levels is considered as one of the most critical factors that characterize different anticancer drugs from each other, the target compounds 2b, 4a, 6a, 7b, 7c and 9a, which revealed the most potent cytotoxic activity against both of HepG2 and MCF-7 cancer cell lines were selected to evaluate their cytotoxic activity against the normal human amnion-derived (WISH) cell line via MTT assay to determine their safety profiles. The tested compounds showed low toxicity against the normal cells with IC50 values ranging from 107.01 to 494.10 µM, which are much higher than their IC50 values range 1.27–20.51 µM against the cancer cell lines (Table 3) confirming their promising safety profile.
2.2.3 In vitro enzyme inhibition assay of EGFRWT, EGFRL858R and EGFRT790M
Several reports indicated that high expression of EGFR is detected in breast cancer and hepatocellular carcinoma (Shetty et al., 2021; Song et al., 2017; Masuda et al., 2012). Thus, EGFRWT kinase assay was performed for the target pyridothienopyridine compounds (2b, 4a, 6a, 7b, 7c and 9a), which investigated the most potent cytotoxic activities against both MCF-7 and HepG-2 cancer cells in order to study their postulated mechanism of action. Erlotinib as one of the most potent EGFRWT inhibitors was utilized as a reference drug (Lyseng-Williamson, 2010) and the results were expressed as IC50 values (µM) (Table 4). Interestingly, all the tested derivatives showed promising inhibitory activity of EGFRWT kinase with IC50 values range; 0.021–0.117 µM. The resultant data demonstrated that the 4-aminopyrazolone derivative 9a as EGFRWT inhibitor was more potent than the standard drug of IC50; 0.021 µM, IC50 erlotinib; 0.027 µM. Also, the Schiff’s base 7c showed significant kinase inhibitory activity with IC50 = 0.032 µM, which decreased slightly than Erlotinib. Whereas, the glycine derivative 6a and the Schiff’s base 7b revealed further lowering in the inhibitory activity with IC50 values; 0.046, 0.051 µM, respectively. However, the precursor 4-chloro analogue 4a appeared to be 3 folds less potent than Erlotinib with IC50; 0.083 µM. Also, a detectable drop was also recognized by the pyrimidin-4-one derivative 2b which revealed IC50; 0.117 µM. It was obvious that the nitrogen of the pyrazolone ring in 9a and the nitrogen in the side chains, as the glycine amino acid of 6a and the hydrazide Schiff’s base of 7b and 7c, which attached at position-4 of the parent pyridothienopyrimidine system, provide an important role in EGFR suppression.
Compound No.
EGFRWT
IC50 (µM)
2b
0.117 ± 0.014
4a
0.083 ± 0.009
6a
0.046 ± 0.003
7b
0.051 ± 0.008
7c
0.032 ± 0.007
9a
0.021 ± 0.004
Erlotinib
0.027 ± 0.005
Furthermore, the compounds representing the most potent inhibitory activity against EGFRWT; 6a, 7b, 7c, 9a were also evaluated as inhibitors against the mutant forms EGFRL858R and EGFRT790M in comparison to erlotinib as a reference drug. The obtained results were summarized in Table 5. It has been observed that the 4-aminopyrazolone derivative 9a displayed more potent inhibitory activity than erlotinib against both mutant forms EGFRL858R and EGFRT790M of IC50s = 0.053, 0.081 µM, respectively, IC50 erlotinib = 0.069, 0.550 µM, respectively. On the other hand, the Schiff’s base 7c employed lower activity than that of erlotinib against EGFRL858R of IC50 = 0.252 µM, but its activity surpassed the reference drug by 2.5 times against the mutant EGFRT790M of IC50 = 0.219 µM. A detectable reduction in the inhibitory activity against both mutant forms by the glycine derivative 6a and the Schiff’s base 7b which appeared less potent than erlotinib representing IC50s = 1.132, 0.801 µM and 0.361, 0.719 µM, respectively.
Compound No.
EGFRL858R
IC50 (µM)EGFRT790M
IC50 (µM)
6a
1.132 ± 0.060
1.801 ± 0.079
7b
0.361 ± 0.020
0.719 ± 0.032
7c
0.252 ± 0.010
0.219 ± 0.010
9a
0.053 ± 0.005
0.081 ± 0.004
Erlotinib
0.069 ± 0.006
0.550 ± 0.090
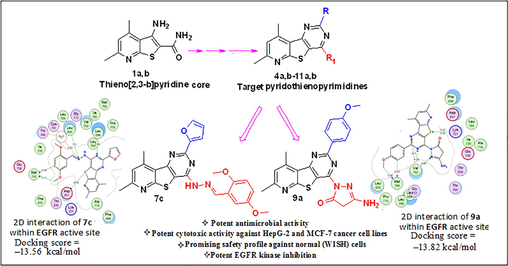
In the light of the gained data, compounds 9a, 7c could be considered as basic scaffolds for the generation of further new candidates used for the treatment of different cancer types harboring EGFRT790M or EGFRL858R mutations, thus might overcome EGFR-TKIs drug resistance.
2.3 Computational studies
2.3.1 Molecular docking study
Molecular modeling studies were performed to study the binding mode of compounds (2b, 4a, 6a, 7b, 7c, and 9a) to the active sits of the target EGFRWT compared with erlotinib as an EGFR inhibitor using (MOE, 2019.0102) software. At the first, from the protein data bank (
https://www.rcsb.org/structure/1M17), the X-ray crystallographic structure of EGFRWT kinase domain complexed with erlotinib (PDB ID: 1 M17) was downloaded. Then the binding interaction of erlotinib to the EGFRWT active site was examined. It showed strong bond interactions with Leu768 & Met769, moreover, it formed hydrogen bonds through a water molecule to Cys751, Thr766 & Thr830 (Fig. 3).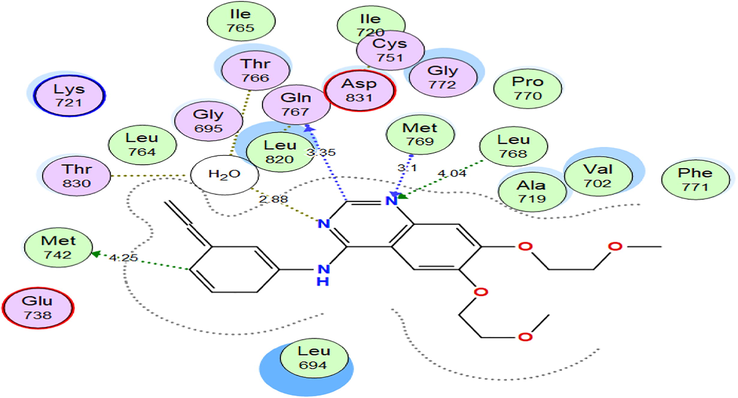
2D interactions of ERL within EGFRWT active site.
The Docking setup was first established by self-docking of the co-crystallized ligand erlotinib in the vicinity of the binding site of the enzyme. The root mean square deviation (RMSD) was 1.03086 Å and erlotinib docking score was –11.7806 kcal/mol (Fig. 4).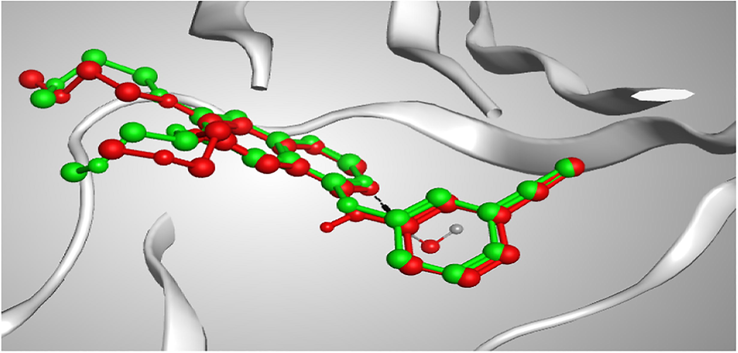
3D representation of the superimposition of the co-crystallized (red) and the docking pose (green) of erlotinib in the active site of EGFRWT enzyme.
While, all the tested compounds showed good binding energy scores indicating their fitness in the binding pocket of the enzyme. Compounds, 9a, 7c and 6a showed the highest binding scores (–13.82, –13.56 & –13.31 kcal/mol, respectively). The 4-pyrazolone derivative 9a showed strong hydrogen bond interactions through both nitrogen atoms of the pyrimidine ring with Val702 amino acid which also showed binding with the pyrazole nitrogen. Furthermore, Lys721 bound to S atom of the thiophene ring and finally Met769 amino acid bound to CH of benzene ring and the methoxy group through non-classical hydrogen bonding. Moreover, the nitrogen atoms of the pyrimidine ring of the Schiff’s base 7c exhibited hydrogen-bond interactions with Leu694 amino acid, the sulpher atom of thiophene ring bound to Leu820 Asp831 amino acids and to Thr830 via a water molecule and also, the imine nitrogen atom bound to Cys751 & Thr766 through water molecules. Through water molecules, the glycine derivative 6a interacted with Cys751, Thr766, Thr830 amino acids, while the pyridine nitrogen bound to Leu768 (Table 6 & Figs. 5-7).
Compound
Docking score (kcal/mol)
Amino acids
Interacting groups
Type of
interactionLength
2b
–9.24
Leu768
Cys751 (water)
Thr766 (water)
Thr830 (water)N (Pyridine)
O (C = O)
O (C = O)
O (C = O)H-bond acceptor
H-bond acceptor
H-bond acceptor
H-bond acceptor3.78
2.86
2.86
2.86
4a
–9.56
Gln767
Met769Cl
SHalogen bond
H-bond acceptor3.74
3.65
6a
–13.31
Leu768
Cys751 (water)
Thr766 (water)
Thr830 (water)N (Pyridine)
O (OH)
O (OH)
O (OH)H-bond acceptor
H-bond acceptor
H-bond acceptor
H-bond acceptor3.78
2.97
2.97
2.97
7b
–12.53
Met742
Thr830
Thr830
Asp831N (Pyridine)
S
S (thiophene)
NHσ-hole bond
σ-hole bond
σ-hole bond
H-bond donor3.99
3.00
4.08
2.90
7c
–13.56
Leu694
Cys751 (water)
Thr766 (water)
Leu820
Thr830 (water)
Asp831N (Pyrimidine)
N (=N)
N (=N)
N (=N)
N (=N)
SH-bond acceptor
H-bond acceptor
H-bond acceptor
H-bond acceptor
H-bond acceptor
σ-hole bond3.84
2.83
2.83
3.50
2.83
4.09
9a
–13.82
Val702
Val702
Val702
Lys721
Met769
Met769N (Pyrazole)
N (Pyrimidine)
N (Pyrimidine)
S
CH3 (OCH3)
CH (AromaticH-bond acceptor
H-bond acceptor
H-bond acceptor
H-bond acceptor
H-bond (non-classical)
H-bond (non-classical)3.66
4.24
4.30
3.51
3.26
3.53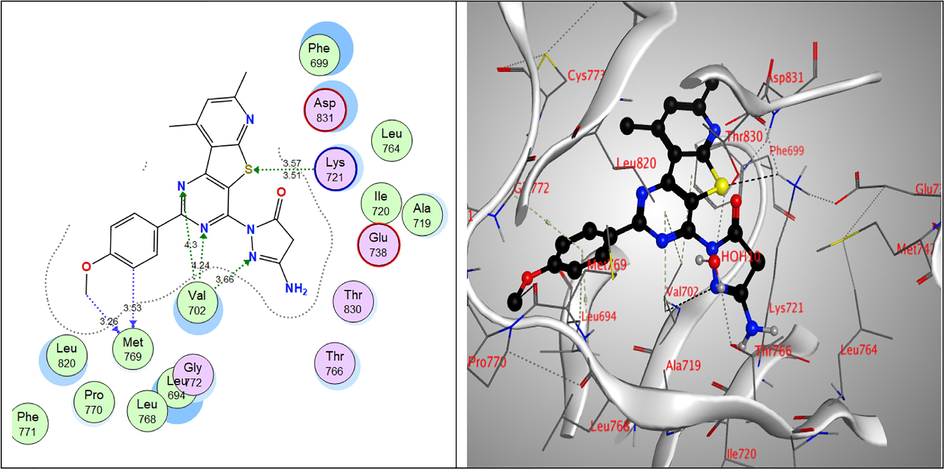
2D & 3D interactions of 9a within EGFRWT active site.
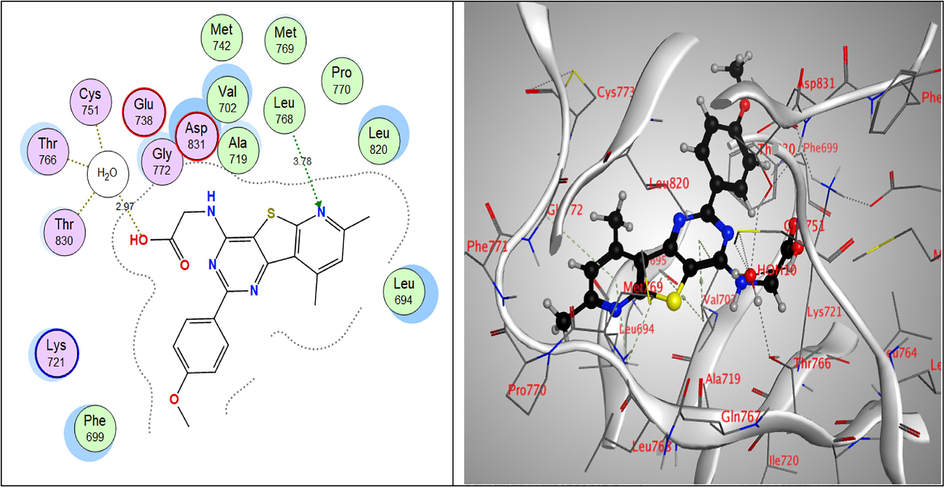
2D & 3D interactions of 6a within EGFRWT active site.
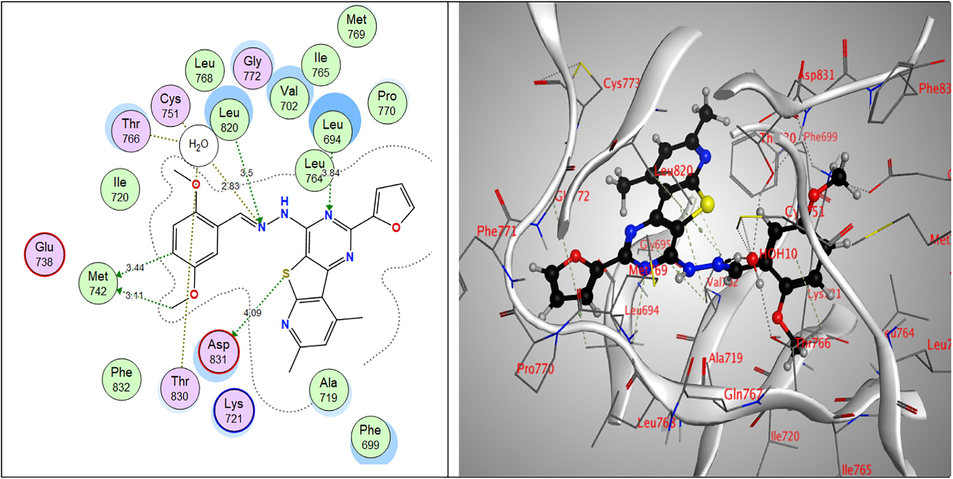
2D & 3D interactions of 7c within EGFRWT active site.
2.3.2 ADME study
Six physicochemical parameters were studied for the tested compounds 2a, 4a, 6a, 7b, 7c, and 9a which are Lipophilicity (LIPO), Size, Polarity (POLAR), Insolubility (INSOL), Unsaturation (UNSAT), and Flexibility (FLEX). The results are presented in the bioavailability radar chart (Fig. 8), in which the pink-colored zone indicates the suitability of physicochemical properties to have a good in vivo bioavailability. All the tested derivatives showed only one violation which indicates a good bioavailability profile except compound 7b which revealed two violations that decreased its probability in a good bioavailability outline.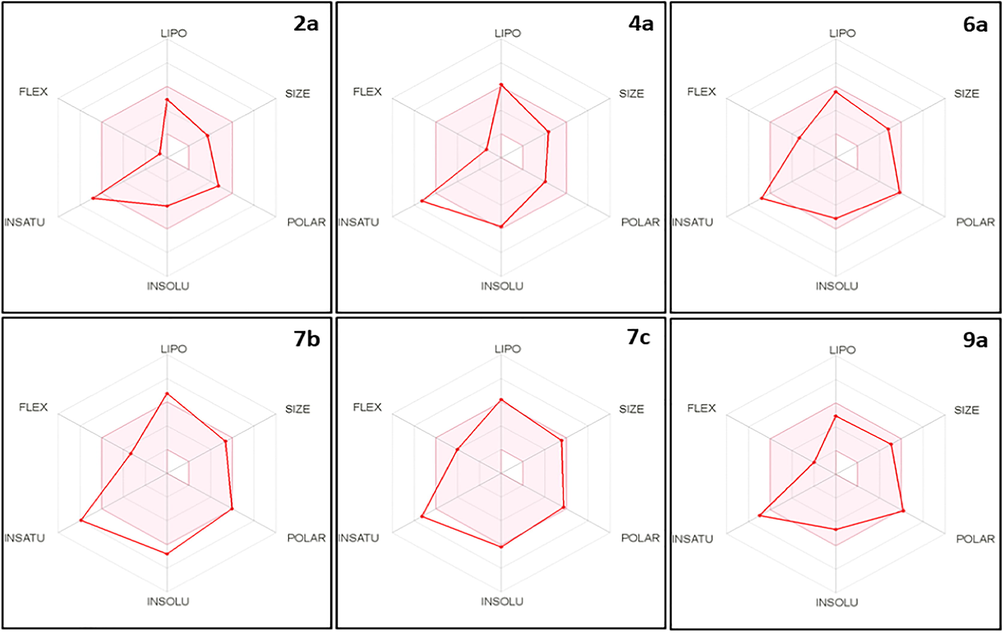
The bioavailability radar chart for the tested compounds (the colored zone is the suitable physicochemical space for oral bioavailability).
The calculated water solubility displayed that all derivatives of moderate solubility except compounds 7b and 7c which were poorly water soluble. The predicted log P values were intermediate (2.51 for 2b, 3.15 for 6a, 3.24 for 9a), slightly high for 4a (4.71), 7c (4.63) and high for 7b compound (5.39).
The Swiss ADME server provides a BOILED EGG chart to indicate the human intestinal absorption (white part), blood brain barrier penetration (the yellow part) and the probability of the tested compound to act as substrate for permeability glycoprotein (PGP) which is an efflux pump for many drugs * (Sharom et al., 1997) (blue color if it’s a possible substrate or red color if it’s not). The results are summarized in (Fig. 9), compounds 7b and 7c were predicted to be not absorbed from the GIT but all others are expected to be of good oral absorption which is indicated by their presence in the white area. All compounds are not present in the yellow area signifying the low probability to penetrate the blood–brain barrier (BBB) and not expected to cause CNS side effects. Finally, compounds 4a, 6a and 9a are colored in red which means that they are anticipated to not be a substrate for PGP and not to be susceptible for efflux from the cells, while other derivatives are colored in blue which indicated their high probability to be substrates for PGP.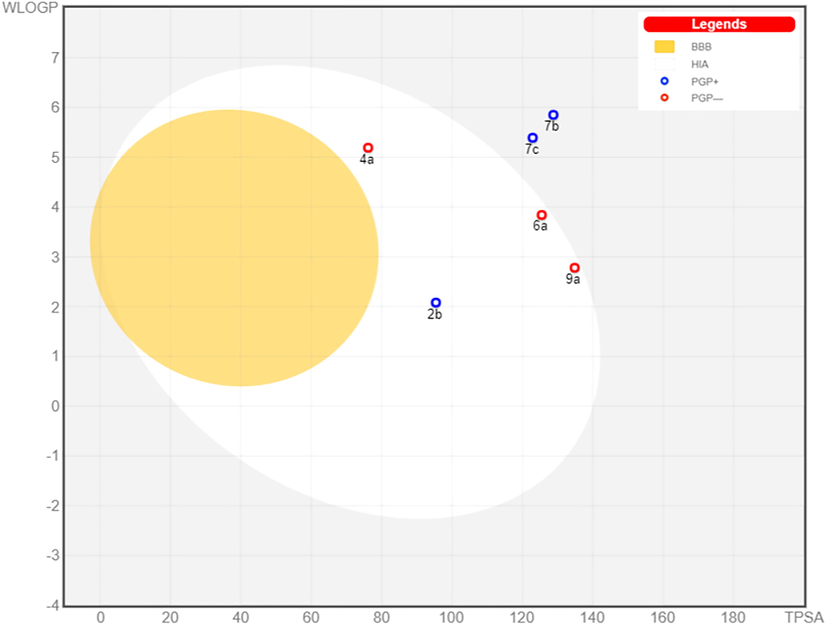
BOILED-EGG chart for the tested compounds.
Eventually, we applied the Lipinski’s rule of five (Giménez, et al., 2010) which predicts the drug-likeness and good oral bioavailability for the tested compounds. All compounds showed no violation in the applied four rules except compound 7b which showed only one violation in having a log P value above 5, the rule states that no violation or even one violation indicates the expected good oral bioavailability. In a conclusion, although all compounds were parallel to Lipinski’s rule of five but the radar chart excluded compound 7b and the BIOLED-Egg chart excluded both 7b and 7c compounds, so, both compounds need future further practical study to evaluate their actual oral bioavailability behavior. Table 7 summarized the obtained data. Log P = 2.51 (<5) Molecular weight = 299.35 g/mol (〈500) No of H-bond donor groups (OHs + NHs) = 2 (≤5) No of H-bond acceptor atoms (Os + Ns) = 5 (≤10) Log P = 3.74 (<5) Molecular weight = 355.84 g/mol (〈500) No of H-bond donor groups (OHs + NHs) = 0 (≤5) No of H-bond acceptor atoms (Os + Ns) = 4 (≤10) Log P = 3.15 (<5) Molecular weight = 394.45 g/mol (〈500) No of H-bond donor groups (OHs + NHs) = 2 (≤5) No of H-bond acceptor atoms (Os + Ns) = 7 (≤10) Log P = 5.39 (>5) Molecular weight = 445.56 g/mol (〈500) No of H-bond donor groups (OHs + NHs) = 1 (≤5) No of H-bond acceptor atoms (Os + Ns) = 6 (≤10) Log P = 4.63 (<5) Molecular weight = 459.52 g/mol (〈500) No of H-bond donor groups (OHs + NHs) = 1 (≤5) No of H-bond acceptor atoms (Os + Ns) = 8 (≤10) Log P = 3.24 (<5) Molecular weight = 418.47 g/mol (〈500) No of H-bond donor groups (OHs + NHs) = 2 (≤5) No of H-bond acceptor atoms (Os + Ns) = 8 (≤10)
Compound NO.
Properties
Comment
2b
No violation
4a
No violation
6a
No violation
7b
One violation
7c
No violation
9a
No violation
3 Conclusion
In summary, a new set of pyridotheinopyrimidine compounds were designed, synthesized and evaluated for their dual antimicrobial and anticancer activities. All the new derivatives were evaluated as antimicrobial agents against a panel of bacterial and fungal strains. The MIC values of the compounds revealed that the 4-chloro analogue 4a, amino acid derivatives 6a,c, the Schiff’s base 7c, the tetracyclic derivative 8b and the 4-pyrazolone derivatives 9a,b exhibited the most potent antimicrobial activity compared with amoxicillin trihydrate and clotrimazole as standard antibacterial and antifungal drugs. Moreover, the in vitro cytotoxicity evaluation of the new derivatives against HepG2 and MCF-7 cell lines revealed the promising activity of the tested compounds against HepG2 cancer cells of IC50 range; 1.80–48.70 µM more than MCF-7 cells of IC50 range; 10.80–80.61 µM. The parent 2b, 4a, 6a, the Schiff’s bases 7b,c and 9a elicited the most potent cytotoxic activity against HepG-2 cell line of IC50 values range; 1.80–5.63 µM compared with doxorubicin as a positive control (IC50; 2.85 µM) and IC50 values ranging from 10.80 to 20.50 µM compared with cisplatin as another positive control of IC50 value 20.70 µM. Further cytotoxic screening was carried out for the most potent compounds 2b, 4a, 6a, 7b, 7c and 9a against the normal cell line (WISH), which revealed their selective cytotoxicity against cancer cells and confirmed their promising safety profile. Additionally, the EGFRWT kinase inhibition assay for the latter derivatives showed the significant inhibitory activity of these compounds with IC50 values range; 0.021–0.117 µM in comparison to erlotinib as standard EGFR inhibitor of IC50; 0.027 µM. Moreover, the 4-pyrazoline derivative 9a exhibited more potent inhibitory activity than that of erlotinib with an IC50 value of 0.021 µM. While, a slight decrease in the activity was detected by 7c and 6a, which gave IC50 values 0.032 and 0.046 µM, respectively.
Furthermore, the most promising EGFRWT inhibitors 6a, 7b, 7c, 9a were also evaluated as inhibitors against the mutant forms EGFRL858R and EGFRT790M in comparison to erlotinib as a reference drug. The compound 9a exhibited more potent suppression effect against both EGFRL858R and EGFRT790M than erlotinib of IC50s = 0.053, 0.081 µM, respectively; IC50 erlotinib; 0.041, 0.550 µM, respectively followed by 7c of IC50s = 0.252, 0.219 µM, respectively. The molecular docking study confirmed that the binding modes of compounds 2b, 4a, 6a, 7b, 7c, 9a were consistent with the EGFRWT inhibitory activities. It was obvious that many of the target compounds have significant antimicrobial and/or anticancer activity and the most potent derivatives of dual activity were the 4-pyrazolone derivative 9a, the glycine derivative 6a, and the Schiff’s base 7c.
Based on the gained results, pyridotheinopyrimidine scaffolds could be considered as a promising template for further developing and optimizing new analogues of more potent dual antimicrobial and anticancer agents via EGFR inhibitory effect.
4 Experimental
4.1 Chemistry
The general information about the instruments utilized in the determination of the melting points, spectral data (IR, 1H NMR,13C NMR, and Mass), and the elemental analysis of the target compounds are mentioned in detail in the Supplementary material. The starting compound 3-amino-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide (1) was prepared as the reported method (Youssefyeh, et al., 1984)
4.1.1 Synthesis of 2,3-dihydropyridothienopyrimidin-4(1H)-ones 2a,b
A mixture solution of compound 1 (0.02 mol) and the appropriate aldehyde namely; 4-methoxybenzaldehyde or furan-2-carbaldehyde (0.02 mol) in glacial acetic acid (30 mL) was refluxed for 8 h. After reaction completion, the solvent was evaporated till dryness under reduced pressure and the obtained precipitate was filtered, washed several times with water, and recrystallized from ethanol to give compounds 2a,b.
4.1.1.1 2-(4-Methoxyphenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (2a)
Yield 82%, brownish powder, m.p. 210 °C; IR (KBr, υmax/cm−1): 3424, 3254 (2NH), 3029 (CH-aromatic), 2958, 2836 (CH-aliphatic), 1656 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.49 (s, 3H, CH3), 2.70 (s, 3H, CH3), 3.71 (s, 3H, OCH3), 5.81 (s,1H, CH-pyrimidinone), 6.91 (d, 2H, J = 8.4 Hz, Ar-H), 7.02 (s, 1H, NH, D2O exchangeable), 7.06 (s, 1H, Ar-H), 7.44 (d, 2H, J = 8.4 Hz, Ar-H), 8.32 (s,1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.73, 24.38 (2CH3), 55.63 (OCH3), 66.13 (CH-pyrimidinone), 114.07, 122.12, 123.01, 128.27, 128.58, 133.84, 137.61, 145.23, 145.33, 159.05, 159.86 (Ar-C), 162.11 (C = O); MS, m/z (%): 339 (M+., 37); Anal. Calc. for C18H17N3O2S (339.41): Calcd. C, 63.70; H, 5.05; N, 12.38; S, 9.45; found C, 63.42, H, 4.88; N, 12.10; S, 9.14.
4.1.1.2 2-(Furan-2-yl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (2b)
Yield 76%, brownish powder, m.p. 245 °C; IR (KBr, υmax/cm−1): 3304, 3225 (2NH), 3029 (CH-aromatic), 2975, 2931 (CH-aliphatic), 1660 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.51 (s, 3H, CH3), 2.68 (s, 3H, CH3), 5.90 (s, 1H, CH-pyrimidinone), 6.31–6.37 (m, 2H, Ar-H), 7.06 (s, 1H, Ar-H), 7.16 (s, 1H, NH, D2O exchangeable), 7.61 (s, 1H, Ar-H), 8.35 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.64, 24.33 (2CH3), 61.41 (CH-pyrimidinone), 107.52, 108.00, 110.81, 122.05, 123.02, 143.35, 144.59, 145.08, 154.25, 159.98, 161.62 (Ar-C), 161.75 (C = O); MS, m/z (%): 299 (M+., 25); Anal. Calc. for for C15H13N3O2S (299.35): Calcd. C, 60.19; H, 4.38; N, 14.04; S, 10.71; found C, 59. 91; H, 4.13; N, 13.87; S, 10.42.
4.1.2 Synthesis of pyridothienopyrimidin-4(3H)-one derivatives 3a,b
A mixture of compounds 2a,b (0.01 mol), and benzoquinone (0.01 mol) in absolute ethanol (50 mL) was refluxed for 9 h. The solid formed was collected by filtration, washed with hot water, and recrystallized from DMF/EtOH to give compounds 3a,b.
4.1.2.1 2-(4-Methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(3H)-one (3a)
Yield 66 %, greyish powder, m.p. 340 °C; IR (KBr, υmax/cm−1): 3435 (NH), 2921 (CH-aliphatic), 1668 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.54 (s, 3H, CH3), 2.91 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 7.07 (d, 2H, J = 8.0 Hz, Ar-H), 7.19 (s, 1H, Ar-H), 8.17 (d, 2H, J = 8.0 Hz, Ar-H), 12.83 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.43, 24.51 (2CH3), 56.09 (OCH3), 114.52, 122.21, 123.33, 127.70, 130.57, 145.41, 153.21, 154.56, 159.30, 159.88, 160.16 (Ar-C), 162.59 (C = O); MS, m/z (%): 337 (M+., 48); Anal. Calc. for for C18H15N3O2S (337.40): Calcd. C, 64.08; H, 4.48; N, 12.45; S, 9.50; found C, 64.23; H, 4.26; N, 12.69; S, 9.31.
4.1.2.2 2-(Furan-2-yl)-7,9- dimethylpyrido[3′,2′:4,5] thieno[3,2-d]pyrimidin-4(3H)-one (3b)
Yield 62%, greyish powder, m.p. 350–351 °C; IR (KBr, υmax/cm−1): 3376 (NH), 3008 (CH-aromatic), 2952 (CH-alicyclic), 1658 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.46 (s, 3H, CH3), 2.82 (s, 3H, CH3), 7.08–8.01 (m, 4H, Ar-H), 13.0 (s,1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.11, 24.51 (2CH3), 113.18, 115.14, 121.29, 124.58, 134.80, 145.59, 147.16, 147.21, 150.03, 152.05, 159.92, 160.13 (Ar-C), 162.09 (C = O); MS, m/z (%): 297 (M+., 26); Anal. Calc. for C15H11N3O2S (297.33): Calcd. C, 60.59; H, 3.73; N, 14.13; S, 10.78; found C, 60.32; H, 3.48; N, 14.01; S, 10.5.
4.1.3 Synthesis of 4-chloropyridothienopyrimidine derivatives 4a,b
A mixture of compounds 3a,b (5 mmol), phosphorus oxychloride (15 mL), and phosphorous pentachloride (5 mmol) was heated on a boiling water bath for 12 h. After the reaction completion, the mixture solution was left to cool then poured gradually with continuous stirring onto crushed ice. The obtained solid was filtered, washed with water, and recrystallized from DMF to give the corresponding chloro derivatives 4a,b.
4.1.3.1 4-chloro-2-(4-methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine (4a)
Yield 87%, brownish powder, m.p. 270 °C; IR (KBr, υmax/cm−1): 3021 (CH-aromatic), 2991 (CH-aliphatic), 1620 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.47 (s, 3H, CH3), 2.77 (s, 3H, CH3), 3.77 (s, 3H, OCH3), 6.92 (d, 2H, J = 10.4 Hz, Ar-H), 7.13 (s, 1H, Ar-H), 8.09 (d, 2H, J = 10.4 Hz, Ar-H); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.71, 24.35 (2CH3), 55.61 (OCH3), 114.73, 121.22, 122.95, 123.81, 127.15, 129.90, 145.57, 154.09, 159.94, 160.89, 161,09, 161.89 (Ar-C);); MS, m/z (%): 355 (M+., 63); Anal. Calc. for C18H14ClN3OS (355. 84): Calcd. C, 60.76; H, 3.97; N, 11.81; S, 9.01; found C, 60.34; H, 3.72; N, 12.11; S, 8.74.
4.1.3.2 4-chloro-2-(furan-2-yl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine (4b)
Yield 80%, greyish powder, m.p. 261 °C; IR (KBr, υmax/cm−1): 3107 (CH-aromatic), 2919 (CH– aliphatic), 1618 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.50 (s, 3H, CH3), 2.77 (s, 3H, CH3),), 6.66 (t, 1H, Ar-H), 7.07 (s, 1H, Ar-H), 7.19 (d, 1H, J = 9.6 Hz, Ar-H), 7.93 (d, 1H, J = 12.4 Hz, Ar-H); 13C NMR (100 MHz, DMSO‑d6, δ ppm):19.09, 24.52 (2CH3), 113.08, 114.63, 122.54, 123.50, 125.59, 145.70, 148.11, 150.88, 153.39, 153.50, 159.87, 162.18, 162.24 (Ar-C); MS, m/z (%): 315 (M+., 25); Anal. Calc. for C15H10ClN3OS (315.78): Calcd. C, 57.05; H, 3.19; N, 13.31; S, 10.15; found C, 56.78; H, 2.89; N, 13.53; S, 9.88.
4.1.4 Synthesis of 4-hydrazineylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine derivatives 5a,b
A mixture of the chloro derivatives 4a,b (5 mmol), and hydrazine hydrate (20 mmol) in ethanol (70 mL) was refluxed for 4 h. After the reaction completion, the excess solvent was evaporated till dryness under reduced pressure. Then the obtained precipitate was washed with water, collected by filtration, and recrystallized from EtOH/H2O to give the corresponding target compounds 5a,b.
4.1.4.1 4-Hydrazineyl-2-(4-methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d] pyrimidine (5a)
Yield 88%, yellowish powder, m.p. 220 °C; IR (KBr, υmax/cm−1): 3380, 3335 (NH, NH2), 3086 (CH-aromatic), 2918, 2832 (CH-aliphatic), 1622 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.51 (s, 3H, CH3), 2.61 (s, 3H, CH3), 3.84 (s, 3H, OCH3), 4.94 (s, 2H, NH2, D2O exchangeable), 7.05 (d, 2H, J = 8.7 Hz, Ar-H), 7.22 (s, 1H, Ar-H), 8.37 (d, 2H, J = 8.6 Hz, Ar-H), 9.02 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.90, 24.55 (2CH3), 55.70 (OCH3), 114.12, 116.29, 122.44, 123.47, 129.54, 131.12, 135.03, 145.21, 146.47, 158.22, 159.60, 159.78, 161.35 (Ar-C); MS, m/z (%): 351 (M+., 58);Anal. Calc. for C18H17N5OS (351.43): Calcd. C, 61.52; H, 4.88; N, 19.93; S, 9.12; found C, 61.24; H, 4.65; N, 19.68; S, 8.83.
4.1.4.2 2-(Furan-2-yl)-4-hydrazineyl-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine (5b)
Yield 89%, yellowish powder, m.p. 218 °C; IR (KBr, υmax/cm−1): 3324, 3288 (NH, NH2), 3109 (CH-aromatic), 2921 (CH-aliphatic); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.58 (s, 3H, CH3), 2.98 (s, 3H, CH3), 4.94 (s, 2H, NH2, D2O exchangeable), 6.65–7.90 (m, 4H, Ar-H), 9.25 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.23, 24.51 (2CH3), 110.40, 114.64, 122.41, 123.45, 125.53, 145.42, 147.20, 150.47, 153.50, 159.82, 160.01, 161.36, 163.08 (Ar-C); MS, m/z (%): 311 (M+.,100); Anal. Calc. for C15H13N5OS (311. 36): Calcd. C, 57.86; H, 4.21; N, 22.49; S, 10.30; found C, 57.59; H, 3.99; N, 22.21; S, 10.03.
4.1.5 Synthesis of (pyridothienopyrimidin-4-yl) amino acid derivatives 6a-c
A mixture of the chloro derivatives 4a,b (1 mmol), and the appropriate amino acids (1 mmol), namely; glycine, valine, and alanine in DMSO (20 mL) containing anhydrous sodium carbonate (0.2 g) was stirred on a water bath at 80 °C for 6 h. Then, the reaction solution was poured onto ice/water and the reaction medium was neutralized with dil. HCl (pH; 7). The formed precipitate was collected by filtration, washed with water, and recrystallized from dioxane to give the corresponding compounds 6a-c.
4.1.5.1 (2-(4-Methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-yl)glycine (6a)
Yield 61%, brownish powder, m.p. 256 °C, IR (KBr, υmax/cm−1): 3395 (broad, OH), 3344 (NH), 3073 (CH-aromatic), 2957, 2920 (CH-aliphatic), 1670 (C = O) ; 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.47 (s, 3H, CH3), 2.87 (s, 3H, CH3), 3.75 (s, 3H, OCH3), 4.11 (s, 2H, –NH-CH2), 6.99 (s, 1H, Ar-H), 7.06 (d, 2H, J = 8.4 Hz, Ar-H), 7.47 (1H, NH, D2O exchangeable), 8.21 (d, 2H, J = 8.5 Hz, Ar-H), 12.23 (s, 1H, OH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.51, 24.45 (2CH3), 44.86 (–NH-CH2), 55.63 (OCH3), 109.81, 114.87, 122.52, 123.50, 129.67, 131.33, 138.42, 145.41, 146.94, 148.22, 157.08, 159.70, 161.56 (Ar-C), 176.14 (C = O); MS, m/z (%): 394 (M+., 52); Anal. Calc. for C20H18N4O3S (394.45): Calcd. C 60.90; H, 4.60; N, 14.20; S, 8.13; found C, 60.63; H, 4.35; N, 13.92; S, 7.84.
4.1.5.2 (2-(4-Methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-yl)valine (6b)
Yield 63%, brownish powder, m.p. 244 °C; IR (KBr, υmax/cm−1): 3400 (broad, OH), 3323 (NH), 3090 (CH-aromatic), 2960, 2850 (CH-aliphatic), 1676 (C = O),; 1H NMR (400 MHz, DMSO‑d6, δ ppm): 0.99 (d, 6H, J = 8.4 Hz, –CH–(CH3)2), 1.90 (m, 1H, –CH–(CH3)2), 2.44 (s, 3H, CH3), 2.70 (s, 3H, CH3), 3.04 (d, 1H, J = 6.8 Hz, –NH-CH), 3.85 (s, 3H, OCH3), 7.16 (d, 2H, J = 12.8 Hz, Ar-H), 7.32 (s, 1H, Ar-H), 7.59 (s, 1H, NH, D2O exchangeable), 8.30 (d, 2H, J = 12.8 Hz, Ar-H), 12.81 (s, 1H, OH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.30, 19.59, 24.49 (4CH3), 45.79 (–CH–(CH3)2), 55.60 (OCH3), 67.97 (–NH-CH2), 105.30, 114.27, 122.45, 123.48, 131.20, 137.61, 140.45, 143.80, 145.47, 159.83, 161.40, 164.67 (Ar-C), 176.73 (C = O); MS, m/z (%): 436 (M+., 39); Anal. Calc. for C23H24N4O3S (436.53): Calcd. C, 63.28; H, 5.54; N, 12.83; S, 7.34; found C, 63.54; H, 5.25; N, 13.06; S, 7.59.
4.1.5.3 (2-(Furan-2-yl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-yl)alanine (6c)
Yield 62%, brownish powder, m.p. 197 °C; IR (KBr, υmax/cm−1): 3393 (broad, OH), 3312 (NH), 3061 (CH-aromatic), 2919, 2852 (CH-aliphatic), 1671 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 1.52 (d, 3H, J = 5.2 Hz, –CH-CH3), 2.83 (s, 3H, CH3), 2.90 (s, 3H, CH3), 3.34 (m, 1H, –NH-CH), 6.69 (s, 1H, NH, D2O exchangeable), 6.76–8.22 (m, 4H, Ar-H), 12.83 (s, 1H, OH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.52, 20.17, 24.45 (3CH3), 48.82 (–HN-CH), 100.21, 114.13, 122.43, 123.55, 131.56, 135.62, 141.33, 143.91, 145.48, 159.80, 162.50, 165.03 (Ar-C), 176.75 (C = O); MS, m/z (%): 368 (M+., 46); Anal. Calc. for C18H16N4O3S (368.41): Calcd. C, 58.68; H, 4.38; N, 15.21; S, 8.70; found C, 58.34; H, 4.74; N, 15.01; S, 8.52.
4.1.6 Synthesis of 4-(2-(Arylidene)hydrazineyl)pyridothienopyrimidine derivatives 7a-c
A mixture of the hydrazide compounds 5a,b (1 mmol) and different aldehydes, namely; 4-methylbenzaldehyde, 2,5-dimethoxybenzaldehyde, and thiophene-2-carbaldehyde (1 mmol) in ethanol/glacial acetic acid solution (1:1, 20 mL) was refluxed for 12 h. The reaction solution was concentrated and poured onto cold water and the formed solid was filtered, washed with water, and crystallized from ethanol to give the corresponding compounds 7a-c.
4.1.6.1 2-(4-Methoxyphenyl)-7,9-dimethyl-4-(2-(4-methylbenzylidene)hydrazineyl)pyrido [3′,2′:4,5]thieno[3,2-d]pyrimidine (7a)
Yield 66%, yellowish powder, m.p. 125 °C; IR (KBr, υmax/cm−1): 3303, 3186 (NH), 3084 (CH-aromatic), 2954, 2919 (CH-aliphatic), 1632 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.38 (s, 3H, CH3), 2.62 (s, 3H, CH3), 3.07 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 7.05–8.23 (m, 9H, Ar-H,), 8.43 (s, 1H, -N = CH), 12.09 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.93, 21.60, 24.46 (3CH3), 55.61 (OCH3), 108.03, 114.45, 114.62, 122.25, 123.72, 126.81, 127.12, 129.50, 129.90, 132.37, 139.45, 145.51, 146.85, 156.30, 159.04, 159.96, 161.30, 167.98 (Ar-C); MS, m/z (%): 453 (M+., 38); Anal. Calc. for C26H23N5OS (453. 56): Calcd. C, 68.85; H, 5.11; N, 15.44; S, 7.07; found C, 68.55; H, 4.86; N, 15.18; S, 7.32.
4.1.6.2 2-(4-Methoxyphenyl)-7,9-dimethyl-4-(2-(thiophen-2-ylmethylene)hydrazineyl)pyrido [3′,2′:4,5]thieno[3,2-d]pyrimidine (7b)
Yield 68%, brownish powder, m.p. 116 °C; IR (KBr, υmax/cm−1): 3341, 3155 (NH), 3099 (CH-aromatic), 2956, 2927 (CH-aliphatic), 1608 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.58 (s, 3H, CH3), 2.99 (s, 3H, CH3), 3.88 (s, 3H, OCH3), 7.03–8.30 (m, 8H, Ar-H), 8.77 (s, 1H, -N = CH), 12.01 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.91, 24.54 (2CH3), 56.07 (OCH3), 108.90, 114.52, 114.94, 122.30, 123.82, 127.64, 128.35, 129.51, 131.20, 132.43, 145.47, 146.82, 157.31, 159.16, 159.93, 160.30, 162.21, 166.93 (Ar-C); MS, m/z (%): 445 (M+., 40); Anal. Calc. for C23H19N5OS2 (445.56): Calcd. C, 62.00; H, 4.30; N, 15.72; S, 14.39; found C, 61.73; H, 4.05; N, 15.44; S, 14.11.
4.1.6.3 4-(2-(2,5-Dimethoxybenzylidene)hydrazineyl)-2-(furan-2-yl)-7,9-dimethylpyrido [3′,2′:4,5]thieno[3,2-d]pyrimidine (7c)
Yield 68%, Yellowish powder, m.p. 126 °C; IR (KBr, υmax/cm−1): 3253, 3143 (NH), 3105 (CH-aromatic), 2993, 2949 (CH-aliphatic), 1613 (CH = N); 1H NMR (DMSO‑d6, δ ppm): 2.50 (s, 3H, CH3), 2.89 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 6.63–7.83 (m, 7H, Ar-H), 8.39 (s, 1H, –CH = N), 12.20 (s, 1H, NH, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.66, 24.50 (2CH3), 55.73, 56.54 (2OCH3), 108.76, 110.54, 112.34, 112.47, 113.37, 116.39, 122.57, 122.91, 123.57, 138.34, 145.44, 146.82, 152.15, 152.86, 153.18, 153.64, 156.44, 157.20, 160.02, 164.50 (Ar-C, –CH = N); MS, m/z (%): 459 (M+., 60); Anal. Calc. for C24H21N5O3S (459.52): Calcd. C, 62.73; H, 4.61; N, 15.24; S, 6.98; found C, 62.51; H, 4.34; N, 15.01; S, 7.21.
4.1.7 Synthesis of pyridothienotriazolopyrimidine derivatives 8a,b
A solution of compound 5a,b (2 mmol) in formic acid (10 mL) was refluxed for 6 h. The excess solvent was evaporated under reduced pressure till dryness. Then the obtained solid was washed with water, collected by filtration, and recrystallized from DMF/H2O to give the corresponding compounds 8a,b.
4.1.7.1 5-(4-Methoxyphenyl)-7,9-dimethylpyrido [3′,2′:4,5] thieno[2,3-e] [1,2,4] triazolo[4,3-c] pyrimidine (8a)
Yield 79%, brown solid, m.p. 240 °C; IR (KBr, υmax/cm−1): 3098 (CH-aromatic), 2943, 2863 (CH-aliphatic), 1619 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.54 (s, 3H, CH3), 2.81 (s, 3H, CH3), 3.88 (s, 3H, OCH3), 7.02 (d, 2H, J = 8.8 Hz, Ar -H), 7. 22 (s, 1H, Ar-H), 8.01 (d, 2H, J = 8.8 Hz, Ar-H), 9.51 (s, 1H, 1,2,4-triazole-H3); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.41, 24.45 (2CH3), 56.16 (OCH3), 114.20, 122.44, 123.61, 129.87, 129.51, 131.27, 136.80, 137.91, 142.73, 145.52, 159.92, 162.43 (Ar-C); MS, m/z (%): 361 (M+., 33); Anal. Calc. for C19H15N5OS (361.42): Calcd. C, 63.14; H, 4.18; N, 19.38; S, 8.87; found C, 62.88; H, 3.89; N, 19.11; S, 8.58.
4.1.7.2 5-(Furan-2-yl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[4,3-c] pyrimidine (8b)
Yield 75%, brown solid, m.p. 275 °C; IR (KBr, υmax/cm−1): 3103 (CH-aromatic), 2919, 2851 (CH-aliphatic), 1620 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.45 (s, 3H, CH3), 2.66 (s, 3H, CH3), 6.86–8.11 (m, 4H, Ar-H), 9.74 (s, 1H, 1,2,4-triazole-H3); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.20, 24.57 (2CH3), 112.20, 115.06, 122.40, 123.84, 129.50, 132.22, 138.50, 142.71, 143,66, 145.50, 154.73, 159.90, 161.23, 165.41 (Ar-C); MS, m/z (%): 321 (M+., 45); Anal. Calc. for C16H11N5OS (321.36): Calcd. C, 59.80; H, 3.45; N, 21.79; S, 9.98; found C, 59.53; H, 3.23; N, 21.51; S, 9.69.
4.1.8 Synthesis of 5-amino-2-(pyridothienopyrimidin-4-yl)-2,4-dihydro-3H-pyrazol-3-one derivatives 9a,b
A mixture of compound 5a,b (1 mmol), and ethyl cyanoacetate (1 mmol) in ethanol/glacial acetic acid solution (3:1; 20 mL) was refluxed for10h. The reaction solution was evaporated under reduced pressure till dryness, then treated with water. The solid obtained was filtered, washed with water, and recrystallized from ethanol to give the corresponding compounds 9a,b.
4.1.8.1 5-Amino-2-(2-(4-methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5] thieno[3,2-d]pyrimidin-4-yl)-2,4-dihydro-3H-pyrazol-3-one (9a)
Yield 85%, Yellowish powder, m.p. 249 °C; IR (KBr, υmax/cm−1): 3212 (NH2), 3092 (CH-aromatic), 2933, 2843 (CH-aliphatic), 1695 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.60 (s, 3H, CH3), 2.97 (s, 3H, CH3), 3.06 (s, 2H, pyrazolone-CH2), 3.85 (s, 3H, OCH3), 7.08–8.45 (m, 5H, Ar-H), 9.58 (s, 2H, NH2, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm):19.61, 24.50, (2CH3), 55.88 (OCH3), 61.57 (pyrazolone-CH2), 110.93, 114.52, 120.15, 122.39, 123.90, 129.88, 145.43, 146.21, 154.98, 157.39, 159.98, 161.58, 162.11 (Ar-C), 170.17 (C = O); MS, m/z (%): 418 (M+., 28); Anal. Calc. for C21H18N6O2S (418.48): Calcd. C, 60.27; H, 4.34; N, 20.08; S, 7.66; found C, 60.01; H, 4.06; N, 19.81; S, 7.38.
4.1.8.2 5-Amino-2-(2-(furan-2-yl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-yl)-2,4-dihydro-3H-pyrazol-3-one (9b)
Yield 78%, brownish powder, m.p. 225 °C, IR (KBr, υmax/cm−1): 3208 (NH2), 3099 (CH-aromatic), 2933, 2843 (CH-aliphatic), 1677 (C = O); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.56 (s, 3H, CH3), 2.89 (s, 3H, CH3), 2.98 (s, 2H, pyrazolone-CH2), 6.64–8.02 (m, 4H, Ar-H), 9.73 (s, 2H, NH2, D2O exchangeable); 13C NMR (100 MHz, DMSO‑d6, δ ppm): 19.87, 24.51 (2CH3), 65.59 (pyrazolone-CH2), 107.93, 110.91, 113.72, 122.19, 123.89, 145.45, 146.29, 151.99, 153.02, 154.67, 154.91, 159.98, 161.58 (Ar-C), 170.90 (C = O); MS, m/z (%): 378 (M+., 60); Anal. Calc. for C18H14N6O2S (378.41): Calcd. C, 57.13; H, 3.73; N, 22.21; S, 8.47; found C, 56.88; H, 3.48; N, 21.97; S, 8.18.
4.1.9 Synthesis of 2-(pyridothienopyrimidin-4-yl)-5-methyl-2,4-dihydro-3H-pyrazol-3-one derivatives 10a,b
A mixture of compound 5a,b (1 mmol), and ethyl acetoacetate (1 mmol) in ethanol/glacial acetic acid solution (1:1, 20 mL) was refluxed for 10 h. Upon reaction completion, the mixture solution was concentrated, poured onto ice/water. The obtained solid was filtered, washed with water, and recrystallized from acetone to give the corresponding derivatives 10a,b.
4.1.9.1 2-(2-(4-Methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-yl)-5-methyl-2,4-dihydro-3H-pyrazol-3-one (10a)
Yield 90%, brownish powder, m.p 241 °C; IR (KBr, υmax/cm−1): 3089 (CH-aromatic), 2959, 2922, (CH-aliphatic), 1676 (C = O), 1590 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 1.96 (s, 3H, CH3), 2.55 (s, 3H, CH3), 2.73 (s, 3H, CH3), 3.01 (s, 2H, pyrazolone-CH2), 3.83 (s, 3H, OCH3), 7.05 (d, 2H, J = 16.7 Hz, Ar-H), 7.21 (s, 1H, Ar-H), 8.36 (d, 2H, J = 16.4 Hz, Ar-H); 13C NMR (100 MHz, DMSO‑d6, δ ppm); 18.93, 19.81, 24.95 (3CH3), 31.59 (pyrazolone-CH2), 55.85 (OCH3), 107.73, 114.12, 114.49, 122.40, 123.87, 129.73, 130.62, 145.47, 147.21, 160.01, 160.59, 161.62, 161.99 (Ar-C), 172.92 (C = O); MS, m/z (%): 417 (M+., 10); Anal. Calc. for C22H19N5O2S (417. 49): Calcd. C, 63.29; H, 4.59; N, 16.78; S, 7.68; found C, 63.02; H, 4.34; N, 16.51; S, 7.39.
4.1.9.2 2-(2-(Furan-2-yl)-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-yl)-5-methyl-2,4-dihydro-3H-pyrazol-3-one (10b)
Yield 87%, brownish powder, m.p. 232 °C; IR (KBr, υmax/cm−1): 3096 (CH-aromatic), 2962, 2921 (CH-aliphatic), 1674 (C = O), 1595 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.26 (s, 3H, CH3), 2.62 (s, 3H, CH3), 2.72 (s, 3H, CH3), 2.97 (s, 2H, pyrazolone-CH2), 6.79–8.02 (m, 4H, Ar-H); 13C NMR (100 MHz, DMSO‑d6, δ ppm); 18.72, 19.86, 24.91 (3CH3), 32.03 (pyrazolone-CH2), 107.72, 108.16, 112.14, 122.17, 123.81, 128.72, 130.15, 145.50, 147.21, 159.79, 161.50, 162.64, 163.93 (Ar-C), 172.57 (C = O); MS, m/z (%): 377 (M+.,74); Anal. Calc. for C19H15N5O2S (377.42): Calcd. C, 60.47; H, 4.01; N, 18.56; S, 8.49; found C, 60.30; H, 3.86; N, 18.38; S, 8.21.
4.1.10 Synthesis of 4-(3,5-dimethyl-1H-pyrazol-1-yl)pyridothienopyrimidines 11a,b
A mixture of compounds 5a,b (1 mmol), and acetylacetone (1 mmol) in glacial acetic acid (10 mL) was refluxed for 8 h. After the reaction completion, the mixture solution was cooled, poured onto ice/water and the solid obtained was collected by filtration and recrystallized from acetone to give the corresponding derivatives 11a,b.
4.1.10.1 4-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-(4-methoxyphenyl)-7,9-dimethylpyrido[3′,2′:4,5] thieno[3,2-d] pyrimidine (11a)
Yield 86%, greenish powder, m.p. 248 °C; IR (KBr, υmax/cm−1): 3087 (CH-aromatic), 2918, 2837 (CH-aliphatic), 1623 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.31 (s, 3H, CH3), 2.59 (s, 3H, CH3), 2.85 (s, 3H, CH3), 2.99 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 6.30 (s, 1H, pyrazole-H4), 7.05 (d, 2H, J = 8.4 Hz, Ar-H), 7.23 (s, 1H, Ar-H), 8.30 (d, 2H, J = 8.4 Hz, Ar-H); 13C NMR (100 MHz, DMSO‑d6, δ ppm); 13.72, 16.21, 19.75, 24.70 (4CH3), 55.85 (OCH3), 108.33, 110.55, 114.63, 116.60, 122.55, 123.52, 131.10, 137.43, 140.45, 143.11, 145.47, 149.80, 155.76, 160.14, 161.62, 166.90 (Ar-C); MS, m/z (%): 415 (M+., 44); Anal. Calc. for C23H21N5OS (415.52): Calcd. C, 66.48; H, 5.09; N, 16.85; S, 7.72; found C, 66.21; H, 4.88; N, 16.58; S, 7.43.
4.1.10.2 4-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-(furan-2-yl)-7,9-dimethylpyrido[3′,2′:4,5]thieno [3,2-d] pyrimidine (11b)
Yield 65%, brownish powder, m.p. 240 °C; IR (KBr, υmax/cm−1): 3097 (CH-aromatic), 2966, 2919 (CH-aliphatic), 1620 (C = N); 1H NMR (400 MHz, DMSO‑d6, δ ppm): 2.22 (s, 3H, CH3), 2.58 (s, 3H, CH3), 2.75 (s, 3H, CH3), 2.86 (s, 3H, CH3), 6.20 (s, 1H, pyrazole-H4), 6.67–7.89 (m, 4H, Ar-H); 13C NMR (100 MHz , DMSO‑d6, δ ppm); 13.94, 15.96, 19.72, 24.71 (4CH3), , 105.61, 110.50, 111.46, 112.83, 122.34, 123.88, 131.15, 137.40, 140.73, 142.58, 145.46, 152.19, 159.88, 160.97, 162.61, 166.60 (Ar-C); MS, m/z (%): 375 (M+., 100); Anal. Calc. for C20H17N5OS (375.45): Calcd. C, 63.98; H, 4.56; N, 18.65; S, 8.54; found C, 63.71; H, 4.31; N, 18.37; S, 8.82.
4.2 Antimicrobial assay
All synthesized compounds were screened for their in vitro antimicrobial activity against five bacterial strains (S. aureus 25923, B. subtilis 6633, B. cereus 33018, E. coli 8739, S. typhimurium 14028), three yeasts (C. albicans 10231, C. tropicals 750, S. cerevisiae) and two fungi (Aspergillus flavus, Aspergillus niger EM77). The diameter of inhibition zone (DIZ) assay was performed by agar disk diffusion method (Penna et al. 1998) and the (MIC) values were determined by using broth dilution method (Wiegand et al. 2008). More details were provided in the supplementary material.
4.3 In vitro anticancer screening
The in vitro cytotoxicity potency of the target compounds 2a,b–11a,b was screened against HepG-2 and MCF-7 cancer cell lines by MTT assay (van Meerloo et al., 2011). The cytotoxicity was estimated as IC50 in μM for the tested compounds and the reference drugs (doxorubicin and cisplatin) listed in Table 3. More details were provided in Supplementary material.
4.4 EGFR kinase inhibitory assay
EGFRWT kinase inhibitory assay was performed for the target compounds 2b, 4a, 6a, 7b, 7c, and 9a with erlotinib as a reference inhibitor, by using the EGFRWT kinase assay kit (Cat. # 40321), while compounds 6a, 7b, 7c and 9a were further tested against EGFRL858R and EGFRL858R using Kinase Assay Kit Catalog # 40,324 and EGFR(T790M) Kinase Assay Kit Catalog # 40,323 in comparison to erlotinib. The assay kit is designed to measure EGFR Kinase activity for screening applications using Kinase-Glo® MAX as a detection reagent using Kinase-Glo® MAX as a detection reagent. The luminescence was measured using the microplate reader (Infinite M200 microplate reader, Tecan, Männedorf, Switzerland) (Aiebchun et al., 2021). All assays were performed in triplicate and the relative inhibition (%) of inhibitors were then calculated compared to the control with no inhibitor. Then the IC50 values (the concentration which provides 50% enzyme inhibition) and their standard deviation (SD) for the tested compounds and the reference drug were determined in (μM) and listed in Table 4, 5. More details were provided in the supplementary material.
4.5 Computational studies
4.5.1 Molecular modeling studies
The molecular modeling studies were carried out using Molecular Operating Environment (MOE, 2019.0102) software. The RMSD gradient of 0.1 kcal∙mol−1Å−1 was reached using the MMFF94x force field and the partial charges were automatically calculated. (Yan et al. 2020; Stamos et al., 2002). More details were provided in the supplementary material.
4.5.2 ADME study
SwissADME is a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. http://www.swissadme.ch/index.php ).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Design, synthesis and docking study of pyridine and thieno[2,3-b] pyridine derivatives anticancerPIM-1 kinase inhibitors. Bioorg. Chem.. 2018;80:674-692.
- [CrossRef] [Google Scholar]
- Identification of vinyl sulfone derivatives as egfr tyrosine kinase inhibitor: in vitro and in silico studies. Molecules. 2021;26(8):2211.
- [CrossRef] [Google Scholar]
- Al-Trawneh, S.A., Tarawneh, A.H., Gadetskaya, A.V., Seo, E., Al-Ta’ani, M.R., Al-Taweel, S.A., El-Abadelah, M.M., 2021. Synthesis and cytotoxicity of thieno[2,3-b]pyridine derivatives toward sensitive and multidrug-resistant leukemia cells. Acta Chim. Solv. 68(2), 458-465. 10.17344/acsi.2020.6609.
- Thieno[2,3-b]pyridine derivatives: a new class of antiviral drugs against Mayaro virus. Arch. Virol.. 2017;162(6):1577-1587.
- [CrossRef] [Google Scholar]
- A synthesis, in silico, in vitro and in vivo study of thieno[2,3-b]pyridine anticancer analogues. Med. Chem. Commun.. 2015;6:1987-1997.
- [CrossRef] [Google Scholar]
- Discovery of novel tricyclic pyrido [3′, 2′: 4, 5] thieno [3, 2-d] pyrimidin-4-amine derivatives as VEGFR-2 inhibitors. Bioorg Chem.. 2015;60:1-12.
- [CrossRef] [Google Scholar]
- Discovery of potent anti-proliferative agents targeting EGFR tyrosine kinase based on pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4-amine scaffold. Chem. Pharm. Bull.. 2015;63:1015.
- [CrossRef] [Google Scholar]
- Synthesis of novel 3-substituted pyridothienopyrimidine derivatives with biological evaluation as antimicrobial agents. Chem. Res. Chin. Univ.. 2016;32:967-972.
- [CrossRef] [Google Scholar]
- Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg. Chem.. 2019;88:102944
- [CrossRef] [Google Scholar]
- Synthesis of new pyridothienopyrimidinone and pyridothienotriazolopyrimidine derivatives as pim-1 inhibitors. J. Enzyme Inhib. Med. Chem.. 2018;33(1):58-66.
- [CrossRef] [Google Scholar]
- Identification of anticancer agents based on the thieno[2,3-b]pyridine and 1H-pyrazole molecular scaffolds. Bioorg. Med. Chem.. 2017;25(2):658-664.
- [CrossRef] [Google Scholar]
- Synthesis and antimicrobial activity of some new substituted pyrido[3′, 2′: 4, 5] thieno[3,2-d]-pyrimidinone derivatives. Russ. J. Bioorg. Chem.. 2014;40:308-313.
- [CrossRef] [Google Scholar]
- Peptides with dual antimicrobial and anticancer activities. Front. Chem.. 2017;5:5.
- [CrossRef] [Google Scholar]
- Synthesis and antibacterial activities of different five-membered heterocyclic rings incorporated with pyridothienopyrimidine. ACS Omega. 2020;5(11):6163-6168.
- [CrossRef] [Google Scholar]
- Evaluation of blockbuster drugs under the rule-of-five. Die Pharmazie-An Int. J. Pharm. Sci.. 2010;65(2):148-152.
- [CrossRef] [Google Scholar]
- Ghattas, A.B.A.G., Khodairy, A., Moustafa, H.M., Hussein, B.R.M., 2015. Synthesis and biological evaluation of some novel thienopyridines. J. Pharm. Appl. Chem. 1(1), 21–26. http://dx.doi.org/10.12785/jpac/010103.
- AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N. Engl. J. Med.. 2015;372(18):1689-1699.
- [CrossRef] [Google Scholar]
- Lainetti, P.d.F., Leis-Filho, A.F., Laufer-Amorim, R., Battazza, A., Fonseca-Alves, C.E., 2020. Mechanisms of resistance to chemotherapy in breast cancer and possible targets in drug.
- The synthesis review of the approved Tyrosine kinase inhibitors for anticancer therapy in 2015–2020. Bioorg. Chem.. 2021;105011
- [CrossRef] [Google Scholar]
- Synthesis, preliminary structure-activity relationships, and in vitro biological evaluation of 6-aryl-3-amino-thieno[2,3-b]pyridine derivatives as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett.. 2013;23(8):2349-2352.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of N-arylbenzo [b] thieno [3, 2-d] pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. Eur. J. Med. Chem.. 2012;58:171-183.
- [CrossRef] [Google Scholar]
- Advances in drug development for hepatocellular carcinoma: clinical trials and potential therapeutic targets. J. Exp. Clin. Cancer Res.. 2021;40:172.
- [CrossRef] [Google Scholar]
- Erlotinib: a pharmacoeconomic review of its use in advanced non-small cell lung cancer. Pharmacoeconomics.. 2010;28(1):75-92.
- [CrossRef] [Google Scholar]
- Madhusudana, K., Shireesha, B., Naidu, V.G., Ramakrishna, S., Narsaiah, B., Rao, A.R., Diwan; P.V., 2012. Anti-inflammatory potential of thienopyridines as possible alternative to NSAIDs. Eur. J. Pharm. 678, 48-54. 10.1016/j.ejphar.2011.12.019.
- Thiophenethieno[2,3-b]pyridine-chitosan nanorods; synthesis, characterization, BSA-Binding and kinetic interactions with BSA, antibacterial and in-vitro release studies. J. Mol. Struct.. 2020;1219:128611
- [CrossRef] [Google Scholar]
- Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat.. 2012;136(2):331-345.
- [CrossRef] [Google Scholar]
- Surmounting the resistance against EGFR inhibitors through the development of thieno[2,3-d]pyrimidine-based dual EGFR/HER2 inhibitors. Eur. J. Med. Chem.. 2018;155:316-336.
- [CrossRef] [Google Scholar]
- Synthesis, docking studies, and in vitro evaluation of some novel thienopyridines and fused thienopyridine–quinolines as antibacterial agents and DNA gyrase inhibitors. Molecules. 2019;24(20):3650.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of new pyridothienopyrimidine derivatives as antibacterial agents and escherichia coli topoisomerase II inhibitors. Antibiotics. 2020;9(10):695.
- [CrossRef] [Google Scholar]
- Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open 11. 2016;1(3):e000060
- [CrossRef] [Google Scholar]
- Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org. Biomol. Chem.. 2020;18(38):7608-7634.
- [CrossRef] [Google Scholar]
- Antimicrobial activity of Eupatorium species growing in Argentina. J. Herbs. Spices Med. Plants.. 1998;5:21-28.
- [Google Scholar]
- EGFR plasma mutation in prediction models for resistance with EGFR TKI and survival of non-small cell lung cancer. Clin. Transl. Med.. 2019;8(1):4.
- [CrossRef] [Google Scholar]
- The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers (Basel). 2019;11(9):1242.
- [CrossRef] [Google Scholar]
- Dual VEGFR-2/PIM-1 kinase inhibition towards surmounting the resistance to antiangiogenic agents via hybrid pyridine and thienopyridine-based scaffolds: Design, synthesis and biological evaluation. Bioorg Chem.. 2019;92:103189
- [CrossRef] [Google Scholar]
- New pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(3H)-one hybrids linked to arene units: synthesis of potential MRSA, VRE, and COX-2 inhibitors. Can. J. Chem.. 2021;99(11):900-909.
- [CrossRef] [Google Scholar]
- 2-Aryl-2-hydroxyethylamine substituted 4-oxo-4, 7-dihydrothieno[2, 3-b]pyridines as broad-spectrum inhibitors of human herpesviruspolymerases. Bioorg. Med. Chem. Lett.. 2007;17:3349-3353.
- [CrossRef] [Google Scholar]
- Anticancer and antimicrobial potential of enterocin 12a from Enterococcus faecium. BMC Microbiol.. 2021;21(1):1-14.
- [CrossRef] [Google Scholar]
- The P-glycoprotein efflux pump: how does it transport drugs? J. Membrane Biol.. 1997;160(3):161-175.
- [Google Scholar]
- Safety and tolerability of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in oncology. Drug Saf.. 2019;42(2):181-198.
- [CrossRef] [Google Scholar]
- Recent advances on epidermal growth factor receptor as a molecular target for breast cancer therapeutics. Anticancer Agents Med. Chem.. 2021;21(14):1783-1792.
- [CrossRef] [Google Scholar]
- Synthesis, antitumor activity, and docking analysis of new pyrido[3′,2′:4,5]furo(thieno)[3,2-d]pyrimidin-8-amines. Molecules. 2019;24:3952.
- [CrossRef] [Google Scholar]
- Hepatocellular carcinoma treated with anti-epidermal growth factor receptor antibody nimotuzumab: A case report. Medicine (Baltimore).. 2017;96(39):e8122
- [CrossRef] [Google Scholar]
- Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem.. 2002;277(48):46265-46272.
- [CrossRef] [Google Scholar]
- Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.. 2021;71(3):209-249.
- [CrossRef] [Google Scholar]
- Rethink of EGFR in cancer with Its Kinase Independent function on board. Front Oncol. 2019;9:800.
- [CrossRef] [Google Scholar]
- Cell sensitivity assays: the MTT assay. Methods Mol Biol.. 2011;731:237-245.
- [CrossRef] [Google Scholar]
- Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol.. 2016;9(1):1-7.
- [CrossRef] [Google Scholar]
- Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc.. 2008;3:163-175.
- [CrossRef] [Google Scholar]
- Design and synthesis of tetrahydropyridothieno[2,3-d]pyrimidine scaffold based epidermal growth factor receptor (EGFR) kinase inhibitors: the role of side chain chirality and Michael acceptor group for maximal potency. J. Med. Chem.. 2010;53:7316-7326.
- [CrossRef] [Google Scholar]
- Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun.. 2018;9:3490.
- [CrossRef] [Google Scholar]
- Structural basis of AZD9291 selectivity for EGFR T790M. J. Med. Chem.. 2020;63(15):8502-8511.
- [CrossRef] [Google Scholar]
- Pyrido [3', 2': 4, 5] thieno [3, 2-d]-N-triazines: A new series of orally active antiallergic agents. J. Med. Chem.. 1984;27(12):1639-1643.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103751.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




