

Translate this page into:
Design, characterization and quantum chemical computations of a novel series of pyrazoles derivatives with potential anti-proinflammatory response
⁎Corresponding author. cbustos@uach.cl (Carlos Bustos), ximena.zarate@uautonoma.cl (Ximena Zarate), maschotte@gmail.com (Eduardo Schott)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The synthesis and characterization of the full family of 11 pyrazoles were performed by means of UV–Vis, FTIR, 1H NMR, 13C NMR, two-dimensional NMR experiments and DFT simulations. As pyrazoles are known for showing diverse biological actions, they were also tested in the NCI-60 cancer cell line panel, showing moderate to good activity against different cell lines. Furthermore, the anti-proinflammatory activity test of a set of pyrazoles of the form (E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-R-phenyl-1H-pyrazole was performed, this is based on the study of the blockage of the increase in intracellular [Ca2+] observed in response to platelet-activating factor (PAF) treatment of four pyrazoles (i.e. 6, 8, 9 and 10), which successfully displayed [Ca2+] channel inhibition. Therefore, the obtained intracellular [Ca2+] signal results indicate that the pyrazole family characterized in this study, in particular compounds 6 and 10, are potent blockers of the PAF-initiated Ca2+ signaling that mediates the hyperpermeability typically observed during the development of inflammation.
Keywords
Anti-proinflammatory
Platelet-activating factor
Pyrazoles
NCI-60
DFT

1 Introduction
The biological activity of pyrazole derivatives, see Fig. 1a, has been a focus of interest in recent years, because they have been subject of a variety of medical investigations. Those medical investigations mainly have been oriented to determine the potential pharmacological properties of these molecules. The pyrazole nuclei is present in numerous compounds with biological activity (Elguero et al., 2001), including very successful drugs, v. g. Celebrex (Penning et al., 1997) and Viagra (Terrett et al., 1996). Additionally, pyrazoles are used as drugs for the treatment of anxiety (Wustrow et al., 1998), as antipyretics, analgesics and anti-inflammatories (Menozzi et al., 1997; Surendra Kumar et al., 2016) and have shown antimicrobial activity (Abunada et al., 2009; B’Bhatt and Sharma, 2017; Kumar et al., 2014) and antiparasitic activity (Escario et al., 1988). Compounds of this family have been described as potent inhibitors of PDE4B or PDE4D (Card et al., 2005), GABA antagonist receptors, insecticides (Sammelson et al., 2004), others are potential ligands for the CB1 receptor in PET trials (Kumar et al., 2004) and have also shown growth inhibitory activity (Baraldi et al., 2003).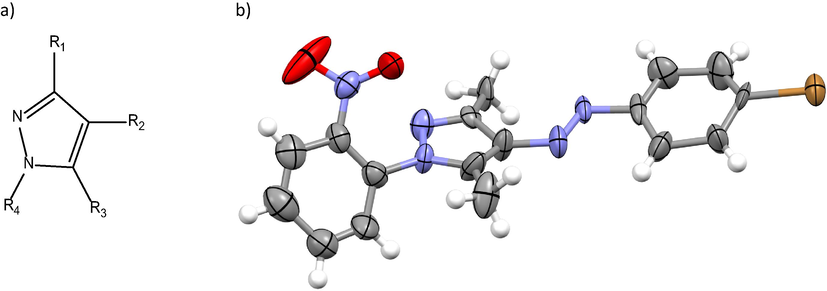
a) Structure of the pyrazole ring and b) Ortep view of compound 9.
Although various methods have been developed to prepare pyrazoles, the regioselective synthesis remains a challenge for chemists (Elguero, 1996, 1984; Makino et al., 1998). The most commonly reported method is the reaction of hydrazines with 1,3-dicarbonyl compounds, where the reaction often results in a mixture of isomers, as the reactivity of the carbonyl groups is not sufficiently different. An alternative is the replacement of 1,3-dicarbonyl compounds with α,β-ethynyl ketones or α-ketoesters, which allows a much more rigorous control of regioselectivity (Bishop et al., 2003; Miller and Reiser, 1993; Norris et al., 2005). Another important method is the 1,3-dipolar cycloaddition of diazoalkanes or nitriloimines with olefins or alkynes. This reaction has limited applications in the synthesis of pyrazoles as the 1,3-dipoles are difficult to prepare and are potentially explosive. However, this method has high regioselectivity (Aggarwal et al., 2003; Foti et al., 1999; Huisgen and Huisgen, 1963; Jung and Trifunovich, 1992; Nakano et al., 1989; Padwa, 2009). The reaction of hydrazones with activated alkenes (Grigg et al., 1987; Le Fevre and Hamelin, 1980; Osella et al., 1995) or alkynes (Bardakos et al., 1975) has also been reported to produce pyrazolidines or pyrazoles, respectively, and there is believed that the mechanism of reaction involves a 1,3-dipole molecule generated in situ (Grigg et al., 1987). In addition, the reaction requires rigorous conditions, since hydrazones have low nucleophilicity and the activation of the alkene or alkyne is often complicated. In contrast, nitroolefins are strong electrophiles widely used in organic synthesis (Barrett, 1991; Barrett and Graboski, 1986; Denmark et al., 2001) and have been found to react with diazo compounds to give pyrazoles (Mancera et al., 1988; Parham et al., 1961; Parham and Bleasdale, 1951). Therefore, it can be concluded that hydrazones and nitroolefins are ideal precursors to synthesize pyrazoles, mainly because they are available at low cost or are confortably prepared (Deng and Mani, 2006).
In literature, the synthesis of an extensive family of 3- (2- (R-phenyl) hydrazono) pentane-2,4-dione (β-diketohydrazones) has been described. These compounds are prepared by a coupling reaction of one diazonium salt with a β-diketone (Yao, 1964). Those compounds retain the dicarbonyl system of β-diketone used as a precursor, contain a resonance assisted intramolecular hydrogen bond (RAHB) that increases the electrophilic character of the carbonyl group involved, thus the compounds do not undergo tautomerization processes.
Previous reports show that in acidic media β-diketohydrazone react with substituted phenylhydrazines to generate pyrazoles of the form (E) −3,5-dimethyl-1- (R 1 -phenyl) −4- (R 2 -phenyldiazenyl) − 1H-pyrazoles (Bustos et al., 2012, 2009).
On the other hand, among the valuable biological activities that pyrazoles have shown, it has been reported that they exhibit anti-inflammatory properties. In this sense, as the development of inflammation depends on an increase in [Ca2+]i, several pyrazole analogues have been found to be potent blockers of store-operated Ca2+ channels and T-type Ca2+ channels (Cav3.1, Cav3.2 and Cav3.3) (Bezençon et al., 2017; Dago et al., 2018). The anti-inflammatory effect of pyrazoles may be related to the modulation of Ca2+ signals, however this relation has not been determined. It is very well known that inflammation is a complex process mediated by the interaction of several signals and, among them, platelet-activating factor (PAF) is an important and potent pro-inflammatory agent. Endothelial cells of post-capillary venules are one of the most relevant targets of PAF during inflammation (Jiang et al., 2008), where it triggers a strong Ca2+-mediated increase in the endothelial cell barrier to macromolecules, leading to edema (Tiruppathi et al., 2002).
In the herein report, our aim is the design and synthesis of a complete family of pyrazoles derived from 3-(2-(4-bromophenyl)hydrazinylidene)pentane-2,4-dione by treatment with substituted arylhydrazines, in acid medium. These pyrazoles have a general name (E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-R-phenyl-1H-pyrazole (with R = 4-OCH3 (1), 4-H (2), 4-F (3), 4-Cl (4), 4-CN (5), 4-NO2 (6), 3-Cl (7), 3-NO2 (8), 2-NO2 (9), 2,4- (NO2)2 (10), C6F5-NHNH2 (11)). The products obtained in these reactions were characterized by analytical techniques (MP, MS-HPLC), spectroscopic (UV–visible, FTIR, 1H NMR, 13C NMR), bidimensional experiments such as DEPT-135, HMBC and HMQC and theoretical calculations in the Density Functional Theory framework. Finally, the previously reported biological activity that pyrazoles have shown, encourage us to carry out antiproliferative tests over a set of 60 cell lines including brain cancer, breast cancer and prostate cancer, among a large list, as well as anti-proinflammatory studies where the inhibitory activity of these compounds on the response initiated by PAF, in primary cultures of mesenteric post-capillary venules endothelial cells was evaluated.
2 Chemistry
2.1 Experimental
All of the synthesis reagents, p-bromoaniline, acetylacetone, sodium nitrite, sodium hydroxide, hydrochloric acid, sodium acetate and substituted arylhydrazines, R-C6H4-NH-NH2 were purchased from Sigma-Aldrich Corporation. Solvents, methanol, ethanol, CHCl3 were obtained from common commercial sources (Merck, Fisher and T.J. Baker) and used without purification. The precursor 3-(2-(4-bromophenyl)hydrazinylidene)pentane-2,4-dione was prepared as described in the literature (Bertolasi et al., 2006; Yao, 1964). Its structure was corroborated by FTIR spectroscopy.
2.2 Physical measurements
The melting points were recorded on an Electrothermal digital recorder, Model IA 9000 SERIES. Elemental analysis was performed on a PerkinElmer 2400. The molecular mass of these compounds was determined by dissolution chromatography on a Micromass equipment, model ZQ. The determination was carried out in 0.1% formic acid solution in methanol. The UV–visible spectra were obtained using concentrated solutions of CHCl3, 5 × 10−4 mol/L and diluting to 5 × 10–5 mol/L. Recording was performed on a Perkin Elmer spectrophotometer, model Lambda 35 in quartz cells of 10 mm path length, in the range of 1100–220 nm. The infrared spectra were obtained in KBr pellets in a Jasco model FTIR-4200, the measurement was performed between 4000 and 550 cm−1. 1H, 13C NMR, DEPT-135, HMQC and HMBC spectra were recorded by conventional procedures in 5 mm diameter glass tubes in CDCl3 solution on a Bruker spectrometer, Model AC 200P 400 MHz spectrophotometer, using as reference signal of the solvent CDCl3.
2.3 Synthesis
3.5 mmol (1.0 g) of 3-(2-(4-bromophenyl)hydrazinylidene)pentane-2,4-dione are added to a distillation flask, 3.5 mmol of an arylhydrazine, R-C6H4-NH-NH2 [98%, 0.63 g, 4-CH3O (1);97%, 0.39 g, 4-H (2);97%, 0.58 g, 4-F (3);98%, 0.64 g, 4-Cl (4);97%, 0.61 g, 4-CN (5);97%, 0.55 g, 4-NO2 (6);≥ 97%, 0.65 g, 3-Cl (7);98%, 0.67 g, 3-NO2 (8); 97%, 0.56 g, 2-NO2 (9);0.70 g, 2,4- (NO2)2 (10);97%, 0.72 g, C6F5-NHNH2 (11)], 2 mL of hydrochloric acid and 30 mL of ethanol. The mixture was refluxed with stirring for 36 h, see Fig. 2. It is then cooled to room temperature and the solid obtained is filtered with vacuum, washed with water (500 mL) and dried in a vacuum oven at 40 °C. All compounds were recrystallized from solvents such as ethanol, acetone, and/or ethanol mixture with tetrahydrofuran.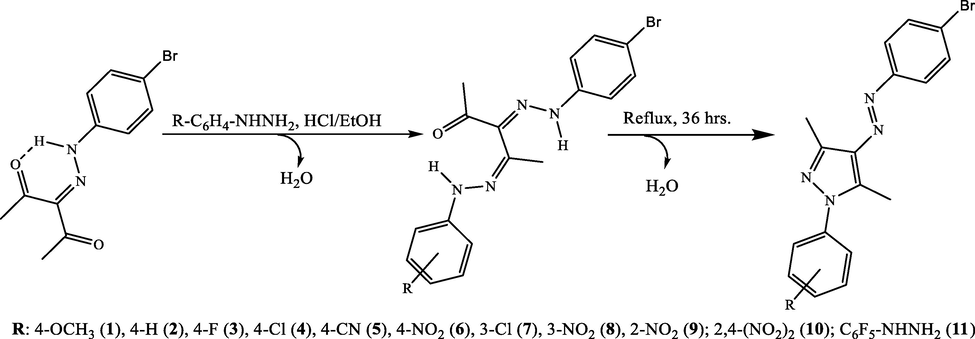
General synthetic procedure and structure of the synthetized pyrazoles.
2.4 Data Collection
Single crystals X-Ray diffraction data set was collected at 294 K for 9. Compound 9 up to a max 2θ of ca. 52° on an Enraf-Nonius CAD diffractometer, using monochromatic MoKα radiation, λ = 0.71073 Å. Data processing was performed using WinGX program in the diffractometer package (Farrugia, 1999). Fig. 1 exhibits an ORTEP view of the compound. Table S1, shows selected bonds length and torsion angles. The data collection and structural refinement are given in Table S1. The structures were solved by direct methods using the SIR-2004 program (Burla et al., 2005). All calculations to solve the structures to refine the model proposed and to obtain results were carried out with the computer programs SHELXL97 (Sheldrick, 1990). Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication, CCDC No. 1,589,910 for 9. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK. Fax: +44 1223 336 033. E-mail: data_request@ccdc.cam.ac.uk. Web page: http://www.ccdc.cam.ac.uk.
2.5 Chemical characterization
(E)-4-((4-bromophenyl)diazenyl)-1-(4-methoxyphenyl)-3,5-dimethyl-1H-pyrazol (1): Yield: 61.3%, crude. Recrystallized from ETOH. Melting point: 148.0–148.2° C. Molecular mass (g/mol), calculated for C18H17BrN4O: 384.26 g/mol (79Br) and 386.26 g/mol (81Br); found 384.00 g/mol (79Br) and 386.00 g/mol (81Br). Elemental Analysis: Calculated: C, 56.12; H, 4.45; Br, 20.74; N, 14.54; O, 4.15; Found: C, 56.10; H, 4.43; Br, 20.54; N, 14.74; O, 4.15. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 439 (3.32); λ2, 347 (4.39); λ3, 240 (4.21). Infrared spectrum in KBr , cm−1 Tablets: (C—H) Arom .: 3078w, 3060w; 3002w; (C—H) Aliph .: 2985w, 2962w, 2939w, 2919w, 2837w; (N⚌N), (C⚌N) and/or (C⚌C): 1590w, 1571w, 1553w, 1519 s; (N—N): 1412 m. 1H NMR (400 MHz, CDCl3) δ 7.69 (w, J = 8.0 Hz, 2H), 7.59 (w, J = 8.2 Hz, 2H), 7.39 (w, J = 8.1 Hz), 7.01 (J = 8.1 Hz, 2H), 3.87 (s, 3H), 2.59 (s, 3H), 2.56 (s, 3H). NMR (101 MHz, CDCl3) δ 159.57, 152.56, 143.87, 139.53, 136.05, 132.23 (+), 132.20, 126.57 (+), 123.55, 123.52 (+), 114.55 (+), 55.74 (+), 14.18 (+), 11.40 (+).
(E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-phenyl-1H-pyrazole (2): Yield: 77.2%, crude. Recrystallized from ETOH. Melting point: 134.6–134.9° C. Molecular mass (g/mol) calculated for C17H15N4Br: 354.23 g/mol (79Br) and 356.23 g/mol (81Br); found 354.00 g/mol (79Br) and 356.00 g/mol (81Br). Elemental Analysis: Calculated: C, 57.48; H, 4.26; Br, 22.49; N, 15.77; Found: C, 57.40; H, 4.16; Br, 22.59; N, 15.85. UV–visible spectrum in CHCl3 5 × 10−5 mol/L mol/L, λ (nm) (logε): λ1, 431 (3.32); λ2, 344 (4.38); λ3, 240 (4.14). Infrared spectrum in KBr pellets, , cm-1: (C—H) Arom .: 3083w, 3068w; 3048w; (C—H) Aliph .: 2987w, 2961w, 2923w; (N⚌N), (C⚌N) and/or (C⚌C): 1597 m, 1584w, 1570 m, 1552 m, 1509 s; (N—N): 1414 s. 1H NMR (400 MHz, CDCl 3) δ 7.70 (w, J = 8.1 Hz, 2H), 7.60 (w, J = 8.3 Hz, 2H), 7.55–7.37 (m, 5H), 2.65 (s, 3H), 2.58 (s, 3H). NMR (101 MHz, CDCl 3) δ 152.53, 144.08, 139.62, 139.18, 136.34, 132.25 (+), 129.42 (+), 128.32 (+), 125.05 (+), 123.67, 123.55 (+), 14.26 (+), 11.52 (+).
(E)-4-((4-bromophenyl)diazenyl)-1-(4-fluorophenyl)-3,5-dimethyl-1H-pyrazole (3): Yield: 64.6%, crude. Recrystallized from EtOH. Melting point: 145.8–146.6° C. Molecular mass (g/mol), calculated for C17H14BrFN4: 372.22 g/mol (79 Br) and 374.22 g/mol (81 Br); found 372.00 g/mol (79Br) and 374.00 g/mol (81Br). Elemental Analysis: Calculated: C, 54.71; H, 3.78; Br, 21.41; F, 5.09; N, 15.01; Found: C, 54.61; H, 3.18; Br, 21.36; F, 5.14; N, 15.01. UV–visible spectrum in 5 × 10−5 mol/L, λ (nm) (logε): λ1, 430 (3.30); λ2, 346 (4.42); λ3, 244 (4.12). Infrared spectrum in KBr Tablets, cm−1: (C—H) Arom .: 3086w, 3071w. 3055w; (C—H) Aliph .: 2992w, 2965w, 2928w; (N⚌N), (C⚌N) and/or (C⚌C): 1582w, 1568w, 1555 m, 1514 s; (N—N): 1407 m. 1H NMR (400 MHz, CDCl3) δ 7.69 (w, J = 8.1 Hz, 2H), 7.59 (w, J = 8.2 Hz, 2H), 7.47 (dw, J = 8.2 Hz, 4H), 2.61 (s, 3H), 2.56 (s, 3H). 1H NMR (CDCl3) δ 162.21 (w, J = 248.6 Hz), 152.41, 144.14, 139.55, 136.23, 135.26 (w, J = 3.1 Hz), 132.23 (+), 126.89 (+) J = 8.7 Hz), 123.74, 123.53 (+), 116.34 (+) (w, J = 23.0 Hz), 14.18 (+), 11.38 (+).
(E)-4-((4-bromophenyl)diazenyl)-1-(4-chlorophenyl)-3,5-dimethyl-1H-pyrazole (4): Yield: 76.8%, crude. Recrystallized from acetone. Melting point: 192.0–192.5° C. Molecular mass (g/mol), calculated for C17H14BrClN4: 388.68 g/mol (79Br) and 390.68 g/mol (81Br); found 388.00 g/mol (79Br) and 390.00 g/mol (81Br). Elemental Analysis: Calculated: C, 52.40; H, 3.62; Br, 20.50; Cl, 9.10; N, 14.38; Found: C, 52.48; H, 3.60; Br, 20.42; Cl, 9.11; N, 14.39. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 429 (3.39); λ2, 347 (4.45); λ3, 244 (4.22). Infrared spectrum in KBr , cm−1 Tablets: (C—H) Arom .: 3097w, 3085w. 3052w; (C—H) Aliph .: 2990w, 2963w, 2928w; (N⚌N), (C⚌N) and/or (C⚌C): 1591w, 1584w, 1567w, 1553 m; 1504 s; (N—N): 1414 s. 1H NMR (400 MHz, CDCl3) δ 7.69 (w, J = 7.6 Hz, 2H), 7.60 (w, J = 8.0 Hz, 2H), 7.47 (dw, 4H), 2.73 (s, 3H), 2.56 (s, 3H). NMR (101 MHz, CDCl3) δ 152.43, 144.38, 139.57, 137.72, 136.47, 134.08, 132.28 (+), 129.60 (+), 126.10 (+), 123.85, 123.58 (+), 14.25 (+), 11.52 (+).
(E)-4-(4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1H-pyrazol-1-yl)benzonitrile (5): Yield: 82.6%, crude. Recrystallized from EtOH: THF (10: 3). Melting point: 213.9–214.4° C. Molecular mass (g/mol), calculated for C18H14BrN 5: 379.24 g/mol (79Br) and 381.24 g/mol (81Br); found 379.00 g/mol (79Br) and 381.00 g/mol (81Br). Elemental Analysis: Calculated: C, 56.86; H, 3.71; Br, 21.01; N, 18.42; Found: C, 56.82; H, 3.715; Br, 21.12; N, 18.30. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 431 (3.30); λ2, 347 (4.50); λ3, 248 (4.16). Infrared spectra of KBr Tablets, cm-1: (C—H) Arom .: 3109w, 3054w. 3047w; (C—H) Aliph .: 2988w, 2963w, 2928w, 2854w; (C≡N): 2226 s; (N⚌N), (C⚌N) and/or (C⚌C): 1603 m, 1583 m, 1567w, 1556 m; 1514 s; (N—N): 1405 s. 1H NMR (400 MHz, CDCl3) δ 7.81 (w, J = 8.0 Hz, 2H), 7.70 (dw, J = 7.7 Hz, 4H), 7.61 (w, J = 8.1 Hz, 3H), 2.74 (s, 3H), 2.57 (s, 3H). NMR (101 MHz, CDCl3) δ 152.30, 145.13, 142.74, 139.93, 137.10, 133.45 (+), 132.34 (+), 124.55 (+), 124.23, 123.66 (+), 118.23, 14.41(+), 11.82 (+).
(E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-(4-nitrophenyl)-1H-pyrazole (6): Yield: 68.6%, crude. Recrystallized from acetone. Melting point: 201.7–202.2° C. Molecular mass (g/mol), calculated for C17H14BrN5O2: 399.23 g/mol (79Br) and 401.23 g/mol (81Br); found 399.00 g/mol (79Br) and 401.00 g/mol (81Br). Elemental Analysis: Calculated: C, 51.02; H, 3.53; Br, 19.96; N, 17.50; O, 7.99; Found: C, 51.22; H, 3.32; Br, 19.92; N, 17.53; O, 8.01. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 442 (3.33); λ2, 354 (4.52); λ3, 244 (4.14). Infrared spectra of KBr Tablets, cm-1: (C—H) Arom .: 3076w; (C—H) Aliph .: 2971w, 2929w, 2852w; (N⚌N), (C⚌N) and/or (C⚌C): 1606w, 1592 s, 1568 m, 1561 m; 1524 s, 1507 m; (N—N): 1407 s. 1H NMR (400 MHz, CDCl3) δ 8.39 (w, J = 8.6 Hz, 2H), 7.76 (w, J = 8.6 Hz, 2H), 7.71 (w, J = 8.2 Hz, 2H) J = 8.3 Hz, 2H), 2.77 (s, 3H), 2.58 (s, 3H). NMR (101 MHz, CDCl3) δ 152.29, 146.51, 145.34, 144.29, 140.13, 137.28, 132.36 (+), 125.03 (+), 124.33, 124.27 (+), 123.69 (+), 14.46 (+), 11.94 (+).
(E)-4-((4-bromophenyl)diazenyl)-1-(3-chlorophenyl)-3,5-dimethyl-1H-pyrazole (7): Yield: 74.1%, crude. Recrystallized from EtOH. Melting point: 145.7–146.2° C. Molecular mass (g/mol), calculated for C17H14BrClN4: 388.68 g/mol (79Br) and 390.68 g/mol (81Br); found 388.00 g/mol (79Br) and 390.00 g/mol (81Br). Elemental Analysis: Calculated: C, 52.40; H, 3.62; Br, 20.50; Cl, 9.10; N, 14.38; Found: C, 51.18; H, 3.36; Br, 19.80; N, 17.59; O, 8.07. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 430 (3.31); λ2, 346 (4.44); λ3, 244 (4.16). Infrared spectra of KBr Tablets, cm−1: (C—H) Arom .: 3081w, 3063w; (C—H) Aliph .: 2986w, 2962w, 2921w, 2853w; (N⚌N), (C⚌N) and/or (C⚌C): 1594 s, 1586w, 1571 s, 1557w, 1541w, 1504 s; (N—N): 1409 s. 1H NMR (400 MHz, CDCl3) δ 7.69 (w, J = 7.7 Hz, 2H), 7.59 (w, J = 7.9 Hz, 2H), 7.55 (s, 1H), 7.47–7.66 (m, 3H), 2.67 (s, 3H), 2.56 (s, 3H). NMR (101 MHz, CDCl3) δ 152.39, 144.41, 140.21, 139.72, 136.52, 135.15, 132.26 (+), 130.33 (+), 128.32 (+), 125.13 (+), 123.88, 123.58 (+), 122.80 (+), 14.28 (+), 11.54 (+).
(E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-(3-nitrophenyl)-1H-pyrazole (8): Yield: 85.0%, crude. Recrystallized from EtOH/THF (5: 3). Melting point: 190.3–191.3° C. Molecular mass (g/mol), calculated for C17H14BrN5O2: 399.23 g/mol (79Br) and 401.23 g/mol (81Br); found 399.00 g/mol (79Br) and 401.00 g/mol (81Br). Elemental Analysis: Calculated: C, 51.02; H, 3.53; Br, 19.96; N, 17.50; O, 7.99; Found: C, 51.18; H, 3.34; Br, 19.94; N, 17.50; O, 8.04.UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 431 (3.30); λ2, 342 (4.46); λ3, 241 (4.31). Infrared spectra of KBr Tablets, cm−1: (C—H) Arom .: 3103w, 3075w; (C—H) Aliph .: 2965w, 2924w; (N⚌N), (C⚌N) and/or (C⚌C): 1583w, 1560w, 1540 s, 1502 s; (N—N): 1415 m. 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.27 (w, J = 8.2 Hz, 1H), 7.91 (w, J = 8.0 Hz, 1H), 7.71 (w, J = 8.0 Hz, 3H), 7.61 (w, J = 8.1 Hz, 2H), 2.74 (s, 3H), 2.58 (s, 3H). NMR (101 MHz, CDCl3) δ 152.30, 148.80, 145.02, 140.27, 139.86, 136.88, 132.33 (+), 130.39 (+), 130.05 (+), 124.19, 123.66 (+), 122.52 (+), 119.40 (+), 14.35 (+), 11.65 (+).
(E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-(2-nitrophenyl)-1H-pyrazole (9): Yield: 83.4%, crude. Recrystallized from EtOH. Melting point: 162.3–162.7° C. Molecular mass (g/mol), calculated for C17H14BrN5O2: 399.23 g/mol (79Br) and 401.23 g/mol (81Br); found 399.00 g/mol (79Br) and 401.00 g/mol (81Br). Elemental Analysis: Calculated: C, 52.40; H, 3.62; Br, 20.50; Cl, 9.10; N, 14.38; Found: C, 52.38; H, 3.14; Br, 20.40; Cl, 9.18; N, 14.30. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 439 (3.25); λ2, 342 (4.40); λ3, 244 (4.20). Infrared spectrum in KBr , cm-1 Tablets: (C—H) Arom .: 3088w; (C—H) Aliph .: 2989w, 2959w, 2922w, 2866w; (N⚌N), (C⚌N) and/or (C⚌C): 1611 m, 1584 m, 1571w, 1557 m; 1528 s, 1509 m; (N—N): 1418 s. 1H NMR Spectrum (400 MHz, CDCl3) δ 8.07 (w, J = 8.1 Hz, 1H), 7.79–7.52 (m, 7H), 2.53 (s, 3H), 2.52 (s, 3H). NMR (101 MHz, CDCl3) δ 152.32, 146.26, 145.47, 140.84, 136.05, 133.68 (+), 132.41, 132.23 (+), 130.21 (+), 129.63 (+), 125.59 (+), 123.93, 123.59 (+), 14.12 (+), 10.74 (+).
(E)-4-((4-bromophenyl)diazenyl)-1-(2,4-dinitrophenyl)-3,5-dimethyl-1H-pyrazole × EtOH (10): Yield: 93.5%, crude. Recrystallized from EtOH. Melting point: 139.5–140.0° C. Molecular mass (g/mol), calculated for C17H13BrN6O4: 444.23 g/mol (79Br) and 446.23 g/mol (81Br); found 444.00 g/mol (79Br) and 446.00 g/mol (81Br). Elemental Analysis: Calculated: C, 45.86; H, 2.94; Br, 17.95; N, 18.88; O, 14.37; Found: C, 45.80; H, 3.00; Br, 17.92; N, 18.90; O, 14.38. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 442 (3.41); λ2, 340 (4.38); λ3, 244 (4.27). Infrared spectrum in KBr Tablets, cm-1: (C—H) Arom .: 3125w, 3101 m, 3008w; (C—H) Aliph .: 2969w, 2928w; (N⚌N), (C⚌N) and/or (C⚌C): 1609 s, 1565 m, 1542 s, 1533 s, 1509 m; (N—N): 1410 s. 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 8.60 (w, J = 8.7 Hz, 1H), 7.79 (w, 2.60 (s, 3H), 2.53 (s, 3H), 1.23 (t, J = 7.0 Hz, 2H), 7.61 (w, 3H). NMR (101 MHz, CDCl3) δ 152.16, 147.09, 147.05, 145.85, 140.52, 137.20, 136.84, 132.37 (+), 129.92 (+), 127.79 (+), 124.58, 123.73 (+), 121.40, 58.57 (-), 18.55 (+), 14.17 (+), 10.94 (+).
(E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-(perfluorophenyl)-1H-pyrazole (11): Yield: 86.6%, crude. Recrystallized from EtOH. Melting point: 153.5–154.4° C. Molecular mass (g/mol), calculated for C17H10BrF5N4: 444.18 g/mol (79Br) and 446.18 g/mol (81Br); found 444.00 g/mol (79Br) and 446.00 g/mol (81Br). Elemental Analysis: Calculated: C, 45.87; H, 2.26; Br, 17.95; F, 21.34; N, 12.59; Found: C, 45.80; H, 2.20; Br, 17.93; F, 21.47; N, 12.61. UV–visible spectrum in CHCl3 5 × 10−5 mol/L, λ (nm) (logε): λ1, 431 (3.21); λ2, 339 (4.39); λ3, 244 (4.06). Infrared spectra of KBr Tablets, cm-1: (C—H) Arom .: 3080w; (C—H) Aliph .: 2990w, 2967w, 2926w; (N⚌N), (C⚌N) and/or (C⚌C): 1583w, 1571 m, 1536 s, 1510 m; (N—N): 1409 s. 1H NMR (400 MHz, CDCl3) δ 7.67 (w, J = 7.8 Hz, 2H), 7.58 (w, J = 8.2 Hz, 2H), 2.54 (s, 3H), 2.49 (s, 3H). NMR (101 MHz, CDCl3) δ 152.20, 146.64, 145.34 (dt, J = 16.2, 4.1 Hz), 143.92–143.11 (m), 142.80 (dt, J = 15.5, 3.7 Hz), 141.98, 141.83–140.84 (m), 139.65–138.96 (m), 137.12–136.59 (m), 135.97, 132.34 (+), 124.38, 123.67 (+), 114.35 (tw, J = 14.9, 4.5 Hz), 14.20 (+), 10.24 (+).
2.6 Computational details
The Gaussian 03 computational package (Frisch et al., n.d.) was used to perform ground-state geometry optimization calculations employing Becke's three-parameter hybrid exchange functional and the Lee Yang Parr nonlocal correlation functional CAMB3LYP (Yanai et al., 2004). The LANL2DZ basis set (Hay and Wadt, 1985) with an effective core potential for I and a 6-31G* basis set was used for C, O, N, Br, Cl, F and H atoms (Rassolov et al., 2001) were employed. The vibrational frequencies were calculated in order to corroborate the arrival to a minimum point in the potential energy surface. Time-dependent density functional theory (TDDFT) calculations were also performed using this methodology, and the first 200 singlet excited states were calculated. Calculations by the first-principles method were used to obtain accurate excitation energies and oscillator strengths. Solvent effect was considered using the polarizable continuum model (PCM) using methanol as solvent (Cossi et al., 2002).
3 Biological assays
3.1 In vitro anti-tumor assays.
The NCI's in vitro anti-tumor screening protocol consists of 60 human tumor cell lines against which compounds 4–11 were tested with 3–4,5-dimethylthiazol-2-yl-2,5-diphenyl-tetrazolium bromide (MTT) assay (Shoemaker, 2006). Cancer cells were treated with 1 μM of test compounds. Dimethyl sulfoxide (DMSO) was used as a vehicle control and six wells were prepared for each compound. After treatment, the supernatant was carefully aspirated and 150 μl of DMSO was added to each well. The absorbance was measured at 590 nm.
3.2 Primary cultures of venular endothelial cells
Microvascular endothelial cells were isolated from the mesenteric arterial beds of male Sprague-Dawley rats (200–230 g). Rats were bred and maintained in the Research Animal Facility of the Pontificia Universidad Católica de Chile and all studies were approved by the Institutional Bioethics Committee. Primary cultures of endothelial cells of post-capillary venules were prepared using a similar procedure to that described previously by Lillo et al. (Lillo et al., 2018). Briefly, rats were anaesthetized with xilazine and ketamine (10 and 90 mg/kg, i.p., respectively) and the mesenteric arterial bed was isolated as described by Gaete et al. (Gaete et al., 2012). Isolated mesenteric beds were perfused with a sterile Tyrode buffer solution containing a mixture of antibiotics and antimycotics (Anti-Anti solution; Thermo Fisher Scientific, Waltham, MA, USA) to remove the blood from the vessels. Several post-capillary venules were isolated from the mesenteric tissue, and thus, incubated in a physiologic saline solution containing 0.2% collagenase type I and 0.1% bovine serum albumin (BSA) for 1 h at 37 °C, diluted (5-fold) with M−199 medium, and centrifuged. Pelleted cells were resuspended in M−199 medium, centrifuged, and resuspended again in M−199 medium containing 20 mg/ml endothelial cell growth supplement from bovine pituitary and 20% fetal bovine serum. Then, cells were seeded onto 35-mm culture dishes or onto sterile glass coverslips. Four hours later, nonadherent cells were removed, and the remaining adherent endothelial cells were kept at 37 °C in a 5% CO2–95% air atmosphere at nearly 100% relative humidity. Experiments were performed with post-capillary venule endothelial cells at 70 to 80% of confluence (∼2 days of culture) in which the culture media was replaced by a MOPS-buffered Tyrode saline solution (pH 7.4).
4 Results and discussion
4.1 Synthesis
The β-diketohydrazone, 3-(2-(4-bromophenyl)hydrazinylidene)pentane-2,4-dione (Bustos et al., 2009, 2007; Yao, 1964), was used as a precursor to obtain a family of pyrazole derivatives. The general name for the obtained family of pyrazoles is (E)-(3,5-dimethyl-1-(4-R-phenyl)-(4-Br-phenyldiazenyl) −1H-pyrazoles pyrazole (with R = 4-OCH3 (1), 4-H (2), 4-F (3), 4-Cl (4), 4-CN (5), 4-NO2 (6), 3-Cl (7), 3-NO2 (8), 2-NO2 (9), 2,4-(NO2)2 (10), C6F5-NHNH2 (11)).
The mechanism of the pyrazoles obtention is shown in the SI as an electronic movement. The reactions were carried out under reflux for 36 h in ethanol solution, in stoichiometric ratio 1:1, using hydrochloric acid as catalyst. Compounds 1–11 were recrystallized from solvents such as ethanol, acetone, and ethanol/THF mixtures.
The characterization was performed using analytical techniques (MP, MM and EA) shown in Table S2 in the SI; where the most relevant point is related with the determination of the calculated molecular mass of pyrazole isotopic molecules, which contain isotopes of 79Br and 81Br. This molecular mass was obtained by mass spectrometry in solution, whose chromatograms are shown in a relation m/z near 1:1, see Fig. S1-S11 in SI, which is consistent with the percentages of abundance of each isotope of bromine in nature. The spectroscopic characterization was performed using UV–visible, IR, 1H NMR, 13C NMR and two-dimensional NMR experiments (HMBC and HMQC).
4.2 X-Ray diffraction
Suitable crystals for X-Ray diffraction were obtained for molecule 9, an ortep view is shown in Fig. S3. In the SI are provided the crystallographic and refinement data. In Table S3 are shown the selected bond distances, angles, torsion angles and DFT results of the geometrical parameters of the whole family of compounds. The discussion of the DFT data is provided in the computational simulations section (vide infra). As shown in Fig. S12, the azo bridge adopts E conformation. In case of 9, as shown in the ortep view, due to the steric hindrance generated by the –NO2 group, the structure does not show planarity. As it has been previously reported for these types of compounds, in the crystallographic structure of the complete supramolecular structure, there are no conventional intermolecular and intramolecular hydrogen bonds. When the substituent is located at a different position than ortho, the whole structure shows higher degree of planarity, vide infra in the theoretical study.
4.3 Spectroscopic study
4.3.1 UV–visible pyrazole spectroscopy
A summary of the absorption bands found in the UV–Vis spectra for 1–11 are shown in Table 1. All superimposed UV–Vis spectra are shown in Fig. 3 and the corresponding spectrum for each compound is shown in the SI (Figs. S13-S23). The theoretical UV–Vis spectra are also shown in the SI (Figs. S24-S34). Each molecule has a C—N⚌N—C fragment, which contains a -N⚌N- double bond. This double bond is conjugated on one end to the phenyl group and, on the other end to the pyrazole ring. The spectra recorded in chloroform show three absorption bands λ1, λ2 and λ3 centered on the ranges 430–442 nm, logε: 3.25–3.39; 339–354 nm, logε: 4.38–4.50 and 240–248 nm, logε: 4.06–4.27, respectively. These bands are attributed to π → π* transitions centered on different regions of the molecules. The first transition is centered on the fragment C—N⚌N—C that connects both pyrazole rings; the second is centered on the pyrazole ring and the third is due to internal transitions of the substituted aromatic rings.
Comp.
λ1(logε)
λ1Th
f
%
λ1(logε)
λ1Th
f
%
λ1(logε)
λ1Th
f
%
1
439 (3.32)
456
0.088
H
→
L
96
347 (4.39)
337
0.266
H-2
→
L
95
240 (4.21)
240
0.01
H-9
→
L
25
H-2
→
L + l
33
H-2
→
L + 2
21
2
431 (3.32)
457
0.042
H
→
L
99
344 (4.38)
316
0.035
H-2
→
L
97
240 (4.14)
248
0.1
H-6
→
L
18
H
→
L + 2
75
3
430 (3.30)
457
0.035
H
→
L
99
346 (4.42)
377
1.335
H-1
→
L
99
244 (4.12)
259
0.24
H
→
L + 2
86
4
429 (3.39)
458
0.045
H
→
L
99
347 (4.45)
378
1.4 16
H-1
→
L
98
244 (4.22)
270
0.22
H
→
L + l
74
H-4
→
L
21
5
431 (3.30)
460
0.047
H
→
L
99
347 (4.50)
381
1.567
H-1
→
L
95
248 (4.16)
268
0.36
H-2
→
L + 1
84
6
442 (3.33)
464
0.056
H
→
L
97
354 (4.52)
360
0.726
H
→
L + l
92
244 (4.14)
246
0.23
H
→
L + 3
75
7
430 (3.31)
458
0.067
H
→
L
99
346 (4.44)
376
1.395
H-1
→
L
98
244 (4.16)
247
0.14
H
→
L + 2
79
H-5
→
L
16
8
431 (3.30)
418
0.030
H
→
L
99
342 (4.46)
374
1.397
H
→
L + l
99
241 (4.31)
246
0.15
H
→
L + 3
80
H-4
→
L + l
13
9
439 (3.25)
443
0.137
H
→
L
98
342 (4.40)
361
0.156
H-2
→
L
77
244 (4.20)
245
0.15
H-4
→
L + l
14
H
→
L + l
19
H
→
L + 4
79
10
442 (3.41)
420
0.133
H
→
L + l
94
340 (4.38)
358
0.699
H-1
→
L + 2
80
244 (4.27)
259
0.11
H-10
→
L
23
H-3
→
L
11
H-8
→
L
19
H-7
→
L + l
28
11
431 (3.21)
456
0.078
H
→
L
99
339 (4.39)
456
1.337
H-1
→
L
98
244 (4.06)
244
0.22
H-6
→
L
19
H-10
→
L + l
39
H
→
L
29
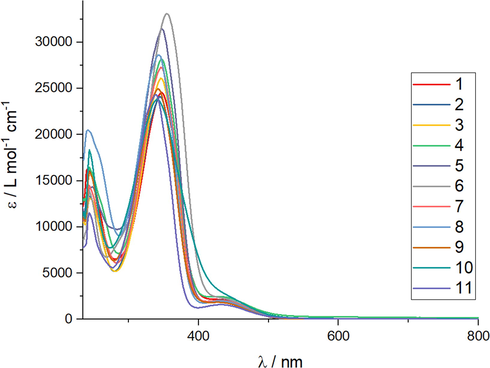
UV–vis in CH3Cl for 1–11.
4.3.2 Infrared Spectroscopy of Pyrazoles
The most important IR bands shown in the solid state KBr pellets are summarized in the SI, see Table S4 and Figs. S46-S56. The full set of spectra can be found in the SI. In general, the weak ν(CH) strechings of the aromatic protons are shown in the spectra over 3000 cm−1 and, immediately below 3000 cm−1, the weak aliphatic stretches ν(C—H) emerge from the system 3,5-dimethylpyrazole, also involving the p-CH3 group for compound 1. Additionally, in the region of 1611–1502 cm−1 are observed several bands attributed to the stretches ν(C⚌N), ν(N⚌N) or ν(C⚌C). The ν(N—N) stretches of the pyrazole ring are located around 1418–1405 cm−1. The influence of the substituent over the IR signals is small or neglectable, thus as can be observed in Table S4, there is no clear tendency of the most important signals as the substituent is changed. Finally, in case of 5 with CN as substituent, the characteristic absorption for the ν(C≡N) is observed at 2226 cm−1.
4.3.3 Nuclear Magnetic Resonance Spectroscopy
All 1H NMR spectra of the pyrazoles (1–11) were recorded in CDCl3. The full set of spectra and a summary of the signals can be found in each 1H NMR spectrum in the SI, see Figs. S46-S56.
According to the obtained data, the most important features of the 1H NMR spectra of the pyrazoles are three. First, variable multiplicity resonances are observed in the range 8.87–7.19 ppm, due to the aromatic protons. Second, a singlet located in the range 2.53–2.58 ppm, due to the protons 1C–1H of the methyl group are observed. Third, a singlet in the range 2.49–2.77 ppm attributed to the 5C–5H protons of the second methyl group are observed. Furthermore, compound 1 shows a resonance at 3.87 ppm due to the methoxy group of the p-OCH3 substituent. Although the substituent over the phenyl pyrazol ring is changed, there are small chemical shifts measured in the 1H NMR spectra, see Table S5. This effect is attributed to the non-planarity observed in the structure; therefore, the delocalization of the ring is not over the whole structure. In this sense, the phenyl ring over the pyrazole acts as one region and the azophenylpyrazole act as a separate region in the molecule.
The signals found in the 13C NMR spectra are summarized in Table S6 in the SI and full set of spectra are also found in the SI, see Figs. S57-S67. Compounds 1–11 show the methyl signals of the pyrazole ring, which follow the same trend as the proton signal, showing almost static positions. Additionally, 1 and 5 show the typical resonances of the R groups located on the aromatic ring. Furthermore, 10 shows the resonances for the ethanol crystallization molecule.
Those compounds that contain fluorine atoms (3 and 11) show typical resonances of the C-F coupling.
In general, the signals of the aromatic carbons attached to the fluorine atom of the ring 4-F-C6H4- in case of 3, appear as a doublet centered at 162.21 ppm (Jgem = 248.6 Hz), 116.34 ppm (Jo = 23.0 Hz)), 126.89 ppm (Jm = 8.7 Hz) and 135.26 ppm (Jp = 3.1 Hz), respectively, see Fig. 4. The intensities of these signals allow to determine the tertiary or quaternary nature of each type of carbon and, together, with the progressive decrease of the values of J (Jgem > Jo > Jm > Jp), which is related to the intensity of the C-F coupling. In this way, the carbons of this ring could be unequivocally assigned. A detail of what is observed in the 13C NMR spectra of (3 and 11) is shown in the SI.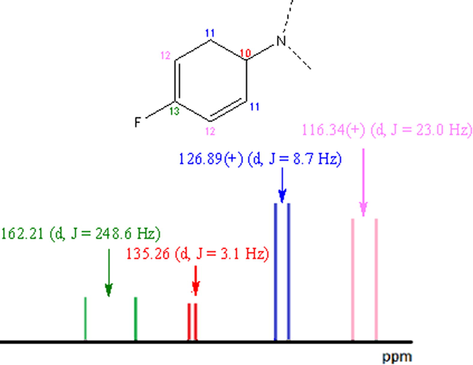
C-F coupling observed in the 13C NMR spectrum of the 4-F-C6H4-N- ring of (IV).
On the other hand, a more complex situation is observed in case of 11. The signals of the quaternary carbons of the C5F5- ring of 11 should appear as a complex multiplet due to geminal C-F coupling and long distance. In Fig. S67 of the supplementary information, an outline of the resonances of the C5F5- ring carbons found in the 13C-spectrum of 11 is shown. As observed in the spectrum, these measured signals are mixed with the resonances of the phenyldiazenyl group carbons. In fact, this spectrum shows three doublet pairs of multiplets, dm, located at 144.10 ppm (Jgem = 256.09 Hz), 142.36 ppm (Jgem = 259.03 Hz) and 138.11 ppm (Jgem = 259.17 Hz). The J values are in agreement with the C-F geminal couplings and similar to the Jgem value found in the signal located at 162.21 (Jgem = 248.6) of 3. Finally, carbon C10, does not show a geminal C-F coupling and appear as triplet of doublets, td, at 114.35 ppm (Jo = 14.9 Hz, Jm = 4.5 Hz), this observed td symmetry is probably due to Jm ∼ Jp.
The DEPT-135 spectrum was measured for all the compounds. In Table S7 in the SI, all the signals of the carbons are assigned as CH3– (+), CH2– (−) and CH– (+) are tabulated. In general, the DEPT-135 spectra of the compounds 1–11 show positive resonances of the carbon atoms. However, in case of 11, two additional signals are observed (18.35(+) ppm and 58.36(−)), which are assigned to crystallization molecules of ethanol. Even though the sample was dried for several days under high vacuum at 90⁰C, those signals remain appearing in the DEPT-135 spectra. The full set of spectra can be found in the SI, Fig. S69-S79.
The HMBC spectra show all the expected interactions, where the most outstanding characteristic is the appearance of an invariable pattern of interactions of the methyl groups with the immediately neighboring carbons, which reveal the presence of the pyrazole ring. This NMR behavior is found in every studied sample, as shown in the SI. A deeper analysis is performed for 1 in Fig. S80, where the mentioned interactions are emphasized, and in addition, the interaction of the substituent 4-CH3O- with the carbon 13C of the aromatic ring is also observed. Furthermore, the two-dimensional HMQC spectra shows a characteristic pattern of interactions, as shown in Fig. S81 for 1. The full set of HMQC and HMBC spectra can be found in the SI, see Figs. S80-S103.
5 In vitro anti-tumor assays
Primary in vitro one dose anticancer assay was carried out over the full NCI 60 cell panel. This panel includes 60 cell lines, which can be classified as leukemia, melanoma, non-small lung cancer, colon cancer, brain cancer, breast cancer, ovarian cancer, kidney cancer and prostate cancer cell lines. The NCI, USA, protocol was used for all the experiments. One-dose data are reported for all studied compounds as a mean graph of the percent growth of treated cells, see Fig. 5 and SI, see Figs. S104-S110. In this sense, the obtained value for the one-dose assay is expressed as growth relative to the control (cells not treated with any drug), and also relative to the number of cells present when the experiment is started. Thus, growth inhibition can be detected and related to values, which are expressed between 0 and 100. On the other hand, lethality is related to negative values. Therefore, as an example, an obtained value of 100 is related to no growth inhibition. On the other hand, a value of 10 means 90% growth inhibition, whereas a value of 0 is related to no net growth during the experiment time. A value of − 50 would mean 50% lethality and finally a value of − 100 means all cells are dead. All the results related to the one-dose measurements can be found in the SI. In Figure 5, are shown the one-dose result value obtained for all compounds. As can be observed, all the studied compounds show certain degree of growth inhibition against different cell lines. Furthermore, all compounds also show certain degree of lethality against different cell lines. The median obtained value, shows that most commonly, the studied compounds show more lethality than growth inhibition. Although the studied compounds show certain degree of similarity, there is no correlation between the activity and the structure. It is important to notice that highly selective growth inhibition activity was observed against UACC-257 (melanoma cell line), NCI-H522 (lung cancer cell line), A549/ATCC (lung cancer cell line) and HT29 (colon cancer cell line). These values can be related with certain degree of tumor-tissue-type (disease-oriented) selectivity. It has been previously reported an over-expression of growth factor kinases, such as EGFR, HER2/Neu, PDGFR, VEGFR, among others (Sikorski and Trzybiński, 2013). Furthermore, lethality is observed against OVCAR-3 and RXF-393. Over those cell lines, every compound shows activity, however, it is only modest.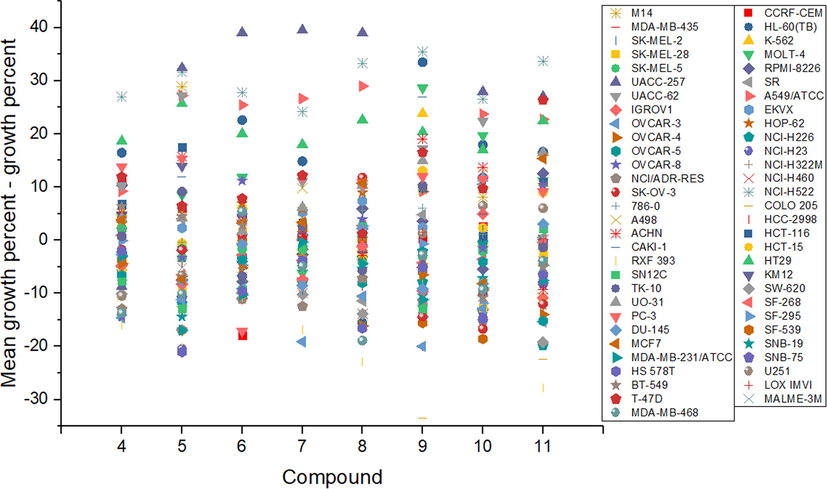
Percentage of cell growth of NCI 60 cancer cell lines displayed by compounds 4–11.
![Concentration-dependent inhibition of PAF-activated Ca2+ signaling by pyrazole analogues. The effect of different concentrations (0.1 nM to 1000 nM) of compounds 6, 8, 9 and 10 on the response to PAF was evaluated in primary cultures of venular endothelial cells. A and B, Time course of the increase in [Ca2+]i induced by PAF in control conditions and after the treatment for 15 min with compounds 6 (A) and 8 (B). Note that compound 6 strongly inhibited the PAF-activated Ca2+ signal and compound 8 evoked a delay in the beginning of the response. Horizontal bars indicate the stimulation period. C to F, Analysis of the maximum increase in [Ca2+]i observed in response to PAF before and after application of a concentration range from 0.1 nM to 1000 nM of compounds 6 (C), 8 (D), 10 (E) or 9 (F) for 15 min.](/content/184/2020/13/8/img/10.1016_j.arabjc.2020.05.042-fig6.png)
Concentration-dependent inhibition of PAF-activated Ca2+ signaling by pyrazole analogues. The effect of different concentrations (0.1 nM to 1000 nM) of compounds 6, 8, 9 and 10 on the response to PAF was evaluated in primary cultures of venular endothelial cells. A and B, Time course of the increase in [Ca2+]i induced by PAF in control conditions and after the treatment for 15 min with compounds 6 (A) and 8 (B). Note that compound 6 strongly inhibited the PAF-activated Ca2+ signal and compound 8 evoked a delay in the beginning of the response. Horizontal bars indicate the stimulation period. C to F, Analysis of the maximum increase in [Ca2+]i observed in response to PAF before and after application of a concentration range from 0.1 nM to 1000 nM of compounds 6 (C), 8 (D), 10 (E) or 9 (F) for 15 min.
6 Anti-proinflammatory activity
We focused the [Ca2+]i study on the analysis of four selected compounds (6, 8, 9 and 10), which hold nitro (–NO2) substituent in their structures. In control conditions, stimulation of endothelial cells with PAF initiated a rapid increase in [Ca2+]i that peaked at 50 to 60 s and gradually declined thereafter (see Fig. 6, a and b). Cells were treated with a concentration range from 0.1 nM to 1 µM of each pyrazole analogues and only one concentration was tested in each experiment. Application of either compound 6, 8, 9 or 10 during 15 min before the stimulation, inhibited the PAF-induced Ca2+ signal in a concentration-dependent manner. Furthermore, compounds 6 and 10 completely abolished the response at 1 µM (Fig. 6, c and e). On the other hand, compounds 8 and 9 only attenuated the increase in [Ca2+]i at this concentration (Fig. 6, d and f). Interestingly, although the inhibition observed in the presence of compound 8 was partial, the treatment with this pyrazole analogue evoked a clear delay in the onset of the PAF-elicited Ca2+ signal (Fig. 6, b and d). These results are consistent with the previously described anti-inflammatory properties of pyrazoles and indicate that the pyrazole family characterized in this study, in particular compounds 6 and 10, are potent blockers of the PAF-initiated Ca2+ signaling that mediates the hyperpermeability typically observed during the development of inflammation (Tiruppathi et al., 2002). Although the inhibition of store-operated Ca2+ channels or T-type Ca2+ channels may contribute to the blockade of the response to PAF, the mechanism involved in this process remain to be determined.
7 Theoretical study
Finally, DFT and TDDFT calculations were performed over the whole family of compounds to model the FTIR and the UV–Vis absorption spectra of each compound, with the aim to get deeper insights of the observed measured values. All the cartesian coordinates of the family of studied compounds are in the SI. The RMSD value for 9 (XRay structure and DFT structure) is of 3.8, showing the good agreement between the experimental and theoretical results. The bromophenyldiazenyl fragment of the molecule is planar, whereas the phenyl fragment shows a torsion angle between 27 and 67°. In the SI can be found all the superimposed FTIR of each compound with the DFT obtained spectrum. A good agreement between the experimental and the theoretical spectra is observed. Therefore, the observed signals in the FTIR spectrum can be assigned to the corresponding vibration, as previously discussed, see Table 1. Furthermore, the UV–Vis transitions were modeled by means of TDDFT calculations. All the FMO involved in the calculated transitions can be found in the SI, see Fig. S111-S115. The first calculated excitation corresponds to a H-L transition and the second calculated transition, which is the most intense transition, involves the electron promotion form HOMO-1 or HOMO-2 to the LUMO, which can be assigned as a n-π* transition. Finally, the third observed band involves low laying MOs and higher unoccupied MOs and corresponds to a π-π* transition. Finally, the reactivity indexes were measured for the whole family of studied compounds, see Table S8. As shown, the most reactive compounds correspond to 6, 8, 9 and 10. Specifically, it is worthy to highlight that 6 and 10 show the lowest value of chemical potential and the largest value of electrophilicity, which correlates with the fact, that these compounds are much more active in the biological trials. All the geometrical coordinates are included in the SI.
8 Conclusions
It was possible to synthesize, with good yields, a family of 11 pyrazoles, of the type of (E)-4-((4-bromophenyl)diazenyl)-3,5-dimethyl-1-R-phenyl-1H-pyrazole, by reacting 3-(2-(4-bromophenyl) hydrazinylidene)pentane-2,4-dione with substituted arylhydrazines. The compounds have geometry E with respect to the double bond -N⚌N- linking the pyrazole ring with the p-bromophenyl ring, as was shown in the X-Ray diffraction and DFT calculations.
In the herein report, we provided novel pyrazol derivatives that successfully displayed a promising anti-proinflammatory activity, i.e. 6, 8, 9 and 10, which was revealed by the blockage of the intracellular [Ca2+] increase after PAF treatment, in terms of the [Ca2+] channel inhibition. The obtained intracellular [Ca2+] signal results are consistent with the previously described anti-inflammatory properties of pyrazoles and indicate that the pyrazole family characterized in this study, in particular compounds 6 and 10, are potent blockers of the PAF-initiated Ca2+ signaling that mediates the hyperpermeability typically observed during the development of inflammation (Tiruppathi et al., 2002). Although the inhibition of store-operated Ca2+ channels or T-type Ca2+ channels may contribute to the blockade of the response to PAF, the mechanism involved in this process remain to be determined.
Furthermore, the pyrazoles were studied in the NCI-60 cancer cell line panel, showing moderate to good activity against different cell lines.
Acknowledgements
FONDECYT 1201880, 1171118, 1180565. Millennium Science Initiative of the Ministry of Economy, Development and Tourism, Chile, grant Nuclei on Catalytic Processes towards Sustainable Chemistry (CSC). ANID/FONDAP/15110019. Universidad del Atlántico (primera convocatoria interna, que otorga apoyo económico para el desarrollo de trabajos de grado en investigación formativa – nivel pregrado y postgrado).
References
- Synthesis and antimicrobial evaluation of some new pyrazole, pyrazoline and chromeno[3,4-c]pyrazole derivatives. J. Braz. Chem. Soc.. 2009;20:975-987.
- [CrossRef] [Google Scholar]
- A novel one-pot method for the preparation of pyrazoles by 1,3-dipolar cycloadditions of diazo compounds generated in situ. J. Org. Chem.. 2003;68:5381-5383.
- [CrossRef] [Google Scholar]
- Synthesis and antimicrobial activity of pyrazole nucleus containing 2-thioxothiazolidin-4-one derivatives. Arab. J. Chem.. 2017;10:S1590-S1596.
- [CrossRef] [Google Scholar]
- Synthesis and growth inhibition activity of alpha-bromoacrylic heterocyclic and benzoheterocyclic derivatives of distamycin A modified on the amidino moiety. Bioorg. Med. Chem.. 2003;11:965-975.
- [Google Scholar]
- Enhydrazine, 10. Einige aliphatische Enhydrazone. Chem. Ber.. 1975;108:2161-2170.
- [CrossRef] [Google Scholar]
- Heterosubstituted nitroalkenes in synthesis. Chem. Soc. Rev.. 1991;20:95.
- [CrossRef] [Google Scholar]
- Conjugated Nitroalkenes: Versatile Intermediates in Organic Synthesis. Chem. Rev.. 1986;86:751-762.
- [CrossRef] [Google Scholar]
- Interplay between steric and electronic factors in determining the strength of intramolecular N-H⋯O resonance-assisted hydrogen bonds in β-enaminones. Acta Crystallogr. Sect. B Struct. Sci.. 2006;62:1112-1120.
- [CrossRef] [Google Scholar]
- Discovery and evaluation of Cav3.2-selective T-type calcium channel blockers. Bioorganic Med. Chem. Lett.. 2017;27:5326-5331.
- [CrossRef] [Google Scholar]
- Regioselective Synthesis of 1,3,5-Substituted Pyrazoles from Acetylenic Ketones and Hydrazines. Synthesis (Stuttg).. 2003;2004:43-52.
- [CrossRef] [Google Scholar]
- SIR2004: an improved tool for crystal structure determination and refinement. J. Appl. Crystallogr.. 2005;38:381-388.
- [CrossRef] [Google Scholar]
- (E)-3,5-Dimethyl-1-p-tolyl-4-(p-tolyldiazenyl)-1H-pyrazole. Acta Crystallogr. Sect. E Struct. Reports Online. 2012;68:o353-o354.
- [CrossRef] [Google Scholar]
- 3,5-Dimethyl-1-(4-nitro-phen-yl)-4-[(E)-(2,3,4,5,6-penta-fluoro-phen-yl) diazen-yl]-1H-pyrazole. Acta Crystallogr. Sect. E Struct. Reports Online. 2007;63:o1138-o1139.
- [CrossRef] [Google Scholar]
- Facile Synthesis Of Isoxazoles And Pyrazoles From Β-Diketohydrazones. J. Chil. Chem. Soc.. 2009;54:267-268.
- [Google Scholar]
- A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nat. Biotechnol.. 2005;23:201-207.
- [CrossRef] [Google Scholar]
- New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys.. 2002;117:43.
- [CrossRef] [Google Scholar]
- Preliminary structure-activity relationship (SAR) of a novel series of pyrazole SKF-96365 analogues as potential store-operated calcium entry (SOCE) inhibitors. Int. J. Mol. Sci.. 2018;19
- [CrossRef] [Google Scholar]
- Reaction of N-monosubstituted hydrazones with nitroolefins: a novel regioselective pyrazole synthesis. Org. Lett.. 2006;8:3505-3508.
- [CrossRef] [Google Scholar]
- Tandem double intramolecular [4 + 2]/[3 + 2] cycloadditions of nitroalkenes. Org. Lett.. 2001;3:2907-2910.
- [CrossRef] [Google Scholar]
- New complexes with pyrazole-containing ligands and different metallic centres. Comparative study of their fluxional behaviour involving M-N bond rupture. New J. Chem.. 2001;25:1050-1060.
- [CrossRef] [Google Scholar]
- Antiparasitic activity of nine pyrazole derivatives against Trichomonas vaginalis, Entamoeba invadens and Plsmodium berghei. Ann. Trop. Med. Parasitol.. 1988;82:257-262.
- [CrossRef] [Google Scholar]
- WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr.. 1999;32:837-838.
- [CrossRef] [Google Scholar]
- First synthesis of a bromonitrilimine. Direct formation of 3- bromopyrazole derivatives. Tetrahedron Lett.. 1999;40:2605-2606.
- [CrossRef] [Google Scholar]
- Ca 2+-activated K + channels of small and intermediate conductance control eNOS activation through NAD(P)H oxidase. Free Radic. Biol. Med.. 2012;52:860-870.
- [CrossRef] [Google Scholar]
- X=Y-ZH Systems as potential 1,3-dipoles : Part 13. Prototropic generation of azomethine imines from hydrazones. Tetrahedron. 1987;43:5873-5886.
- [Google Scholar]
- Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys.. 1985;82:299.
- [CrossRef] [Google Scholar]
- 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chemie Int. Ed. English. 1963;2:565-598.
- [CrossRef] [Google Scholar]
- Three-dimensional localization and quantification of PAF-induced gap formation in intact venular microvessels. Am. J. Physiol. Circ. Physiol.. 2008;295:H898-H906.
- [CrossRef] [Google Scholar]
- Efficient synthesis of 2′,3′-dideoxynucleosides and 2′,3′-dideoxy C-nucleosides from D-glucosamine. Tetrahedron Lett.. 1992;33:2921-2924.
- [CrossRef] [Google Scholar]
- Synthesis of [O-methyl-11C]1-(2-chlorophenyl)-5-(4- methoxyphenyl)-4-methyl-1H-pyrazole-3-carboxylic acid piperidin-1-ylamide: A potential PET ligand for CB1 receptors. Bioorganic Med. Chem. Lett.. 2004;14:2393-2396.
- [CrossRef] [Google Scholar]
- Synthesis and Antimicrobial Studies of Pyrimidine Pyrazole Heterocycles. Adv. Chem.. 2014;2014:1-12.
- [CrossRef] [Google Scholar]
- Addition de phénylhydrazones aux olépines en milieu neutre. Tetrahedron. 1980;36:887-891.
- [CrossRef] [Google Scholar]
- Critical contribution of Na+-Ca2+ exchanger to the Ca2+-mediated vasodilation activated in endothelial cells of resistance arteries. FASEB J.. 2018;32:2137-2147.
- [CrossRef] [Google Scholar]
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, P.M.W. Gill, B.G. Johnson, M.A.R., J.R. Cheeseman, T.A. Keith, G.A. Petersson, J.A. Montgomery, K. Raghavachari, M.A., Al-Laham, V.G. Zakrzewski, J.V. Ortiz, J.B. Foresman, J. Cioslowski, B.B.S., A. Nanayakkara, M. Challacombe, C.Y. Peng, P.Y. Ayala, W. Chen, M.W. Wong, J.L., Andres, E.S. Replogle, R. Gomperts, R.L. Martin, D.J. Fox, J.S. Binkley, D.J.D., J. Baker, J.P. Stewart, M. Head-Gordon, C. Gonzalez, J.A.P., n.d. Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, EUA.
- Stereoselective Syntheses of Nitropyrazolines by 1,3-Dipolar Cycloaddition of Diazoalkanes to Sugar Nitro Olefins. J. Org. Chem.. 1988;53:5648-5651.
- [Google Scholar]
- ω-Dialkylaminoalkyl ethers of phenyl-(5-substituted 1-phenyl-1H-pyrazol-4-yl)methanols with analgesic and anti-inflammatory activity. J. Heterocycl. Chem.. 1997;34:963-968.
- [Google Scholar]
- The synthesis of Electron Donor-Acceptor Substituted Pyrazoles. J. Heterocycl. Chem.. 1993;30:755-763.
- [Google Scholar]
- A synthetic route to bicyclic pyrazolenines via 3-chloropyrazolines and the ring opening of pyrazolenines to diazoalkenes. J. Org. Chem.. 1989;54:5912-5919.
- [Google Scholar]
- New hydroxy-pyrazoline intermediates, subtle regio-selectivity and relative reaction rate variations observed during acid catalyzed and neutral pyrazole cyclization. Org. Biomol. Chem.. 2005;3:1844-1849.
- [CrossRef] [Google Scholar]
- Electronic interactions in organometallic dimers. An electrochemical approach. J. Organomet. Chem.. 1995;488:1-7.
- [Google Scholar]
- Padwa, A., 2009. 1,3-Dipolar cycloaddition chemistry. Volumes 1 and 2. Edited by Albert Padwa. John Wiley and Sons. New York, 1984. Volume 1: XIII + 817 pages. Volume 2: XIII + 704 pages. ISBN 0-471-08364-X (set). $295.00 for the two-volume set. J. Heterocycl. Chem. 23, 1899–1899. https://doi.org/10.1002/jhet.5570230658.
- The Condensation of Diazo Compounds with Nitroölefins. II. 3-Bromo- and 3-Nitropyrazoles. J. Am. Chem. Soc.. 1951;73:4664-4666.
- [CrossRef] [Google Scholar]
- Reaction of Diazo Compounds with Nitroolefins. VI. The Reaction of Diphenyldiazomethane with 1-Nitropropene. J. Org. Chem.. 1961;26:1805-1807.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib) J. Med. Chem.. 1997;40:1347-1365.
- [CrossRef] [Google Scholar]
- 6–31G* basis set for third-row atoms. J. Comput. Chem.. 2001;22:976-984.
- [CrossRef] [Google Scholar]
- GABA receptor antagonists and insecticides: common structural features of 4-alkyl-1-phenylpyrazoles and 4-alkyl-1-phenyltrioxabicyclooctanes. Bioorg. Med. Chem.. 2004;12:3345-3355.
- [CrossRef] [Google Scholar]
- Phase annealing in SHELX-90: direct methods for larger structures. Acta Crystallogr. Sect. A. 1990;46:467-473.
- [CrossRef] [Google Scholar]
- The NCI60 human tumour cell line anticancer drug screen. Nature Reviews Cancer. 2006:813-823.
- [CrossRef] [Google Scholar]
- Networks of intermolecular interactions involving nitro groups in the crystals of three polymorphs of 9-aminoacridinium 2,4-dinitrobenzoate • 2,4-dinitrobenzoic acid. J. Mol. Struct.. 2013;1049:90-98.
- [CrossRef] [Google Scholar]
- Anti-inflammatory and antimicrobial activities of novel pyrazole analogues. Saudi J. Biol. Sci.. 2016;23:614-620.
- [CrossRef] [Google Scholar]
- Sildenafil (Viagra), a potent and selective inhibitor of Type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem.. 1996;6:1819-1824.
- [Google Scholar]
- Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul. Pharmacol.. 2002;39:173-185.
- [CrossRef] [Google Scholar]
- Pyrazolo[1,5-a]pyrimidine CRF-1 receptor antagonists. Bioorg. Med. Chem. Lett.. 1998;8:2067-2070.
- [Google Scholar]
- A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem. Phys. Lett.. 2004;393:51-57.
- [CrossRef] [Google Scholar]
- Azohydrazone Conversion. II. The Coupling of Diazonium Ion with β-Diketones. J. Am. Chem. Soc.. 1964;29:2959-2963.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.05.042.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




