

Translate this page into:
Design, modification, and bio-evaluation of salazinic acid derivatives
⁎Corresponding authors. nn.hong@hutech.edu.vn (Ngoc-Hong Nguyen), huydt@hcmue.edu.vn (Thuc-Huy Duong)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Data on synthesized derivatives of salazinic acid are scarce, with existing reports addressing only derivative hexaacetyl salazinic acid. This study investigated a set of novel potential antidiabetic agents. Analogs of salazinic acid were designed and synthesized using bromination, nucleophilic addition, Friedel-Crafts alkylation, and esterification. Ten synthetic compounds were prepared and structurally elucidated, including eight new compounds (1a-1c, 2a, 3a, 3b, 4a, 4b) and two known analogs. Under bromination, salazinic acid (1) enabled the following reaction chain: oxidation, decarboxylation, and substitution. This yielded products 1a-1c, which were found to have unprecedented scaffolds. Parmosidone F (5) was prepared from 1 with orsellinic acid via Friedel-Crafts alkylation, confirming a previously reported biosynthesis route. These analogs were evaluated for enzyme inhibition of α-glucosidase, and all showed more potent activity than that of acarbose, a positive control (IC50 332 μM), with IC50 values in the range 9.32–39.96 μM. An in silico molecular docking model confirmed that, in terms of enzyme inhibition, the compounds ranked as follows: 3b > 4b > 4a > 1c > 2a > 1b > 1a > 3a. The kinetics of enzyme inhibition showed 4a and 5 to be a non-competitive-type and mixed-type inhibitors, respectively.
Keywords
Lichen
Bromination
Nucleophilic addition
α-glucosidase inhibition
Molecular docking
Kinetic

1 Introduction
Heterocyclic compounds that contain nitrogen, sulfur, and oxygen are potential α-glucosidase inhibitors (Dhameja and Gupta, 2019). In molecular docking models (AutoDock), Schrodinger packages perform well. The relevant approach uses a Lamarckian genetic algorithm with an empirical binding free energy function to predict the bound conformation of the stable ligand to the receptor that is its macromolecular target (Morris et al., 1998). In silico molecular docking has been used in the design of drug-delivery systems (Hassan et al., 2019; Brancolini et al., 2012), in α-glucosidase inhibition (Nguyen et al., 2017; Ghani et al., 2018; Shaikh et al., 2019), and to investigate antitumor agents (Gomha et al., 2016), FabH antibacterial inhibitors (Uwabagira et al., 2019), and Pim-1 Kinase inhibitors with a flexible-receptor docking protocol (Li et al., 2019). It has also been used in covalent docking prediction (London et al., 2015), for algebraic graph-theory-based models in physical modeling and molecular studies (Nguyen et al., 2019), for incorporating a docking scoring function into consensus scoring for use in virtual screening (VS) (Perez – Castillo et al., 2019), and in the critical assessment of docking programs and scoring functions (Warren et al., 2006).
α-Glucosidase plays important roles in the digestion of carbohydrates and the biosynthesis of glycoprotein (Rocha et al., 2020). It is commonly used in screening of natural therapeutic agents for the control of postprandial hyperglycemia (Chandran et al., 2016). Commercial drugs, including voglibose and acarbose, are potent α-glucosidase inhibitors currently used for treatment of diabetes (Hollander, 1992). However, their long-term intake has undesirable side effects. There is therefore an ongoing search for novel α-glucosidase inhibitors with fewer adverse properties. A wide range of α-glucosidase inhibitors have been isolated from medicinal plants, in the form of flavonoids, alkaloids, terpenoids, anthocyanins, glycosides, and phenolic compounds, among others (Chiasson et al., 2002; Kumar et al., 2011).
Depsidone, commonly isolated from lichens, has a unique scaffold (Lang et al., 2007; Shrestha et al., 2014; Moreira et al., 2015, Duong et al., 2020; Devi et al., 2020) and has been shown to have several biological functions, being a blocker of UV rays (Russo et al., 2008), antioxidant (Chomcheon et al., 2009), antifungal (Millot et al., 2017), inhibitor of malignant cancer cells (Khumkomkhet et al., 2009; Boustie et al., 2011), antiviral, antimicrobial, and enzyme inhibitor (Rajachan et al., 2014; Abdou et al., 2010; Boustie et al., 2005; Bucar et al., 2004; Neamati et al., 1997). Salazinic acid, a major β-orcinol-type depsidone isolated from Parmotrema lichens including P. tinctorum, P. delicatulum (Eifler-Lima et al., 2000), and P. dilatatum (Devi et al., 2020), is a member of the rare depsidone class. A hydroxymethylene moiety in its β-ring has yielded compounds with applications as an antibacterial, antifungal (Ankith et al., 2017), antimicrobial (Candan et al., 2007), and antioxidant (Bhattacharyya et al., 2016). It has been reported to exhibit α-glucosidase inhibition, with an IC50 value of 44.3 μM (Verma et al., 2012). Devi and co-workers reported potent α-glucosidase inhibition by four natural salazinic acid derivatives of P. dilatatum. However, only a single hexaacetyl derivative of salazinic acid has been reported (Selvaraj et al., 2015). In this study, we demonstrated the preparation of salazinic acid derivatives via bromination, esterification, and imine formation. We conducted a structural elucidation, evaluated α-glucosidase inhibition and conducted a molecular docking analysis as well as kinetic study of the inhibitory mode.
2 Experimental
2.1 General experimental procedures
NMR spectra were recorded on a Bruker Avance III (500 MHz for 1H NMR and 125 MHz for 13C NMR) using residual solvent signals as internal references: acetone‑d6 at δH 2.05, δC 29.84 and chloroform-d at δH 7.26, δC 77.18. HRESIMS were recorded on Bruker MicrOTOF-QII and Agilent ESIQTOF mass spectrometers. Salazinic acid was isolated from the lichen Parmotrema indicum following the procedure in Do et al. (2021). Solvents were distilled before use. All reagents were purchased from Sigma-Aldrich. All reactions were monitored on silica gel 60 F254 TLC plates (Merck). Thin layer chromatography (TLC) was carried out on precoated 60 F254 silica gel or 60 RP–18 F254S silica gel (Merck). Spots were visualized by spraying with 10% H2SO4 solution followed by heating. Gravity column chromatography was performed using 60 silica gel (0.040–0.063 mm, Himedia). Saccharomyces cerevisiae α-glucosidase (E.C 3.2.1.20), acarbose, and 4-nitrophenyl β-D-glucopyranoside (pNPG) were obtained from Sigma-Aldrich Co (Saint Louis, USA).
2.2 Isolation of salazinic acid from P. indicum
The material (3.8 kg) was cleaned, air-dried, ground, and macerated with ethyl acetate extract at room temperature. The filtrated solution was concentrated under reduced pressure to afford a crude ethyl acetate extract (719.52 g). The filtrated solution was evaporated to dryness and 152.32 g of the precipitate was collected. The crude extract was re-extracted using n-hexane and n-hexane–ethyl acetate extract (1:1, v/v) as solvents, affording three extracts: n-hexane (H, 112 g), n-hexane:ethyl acetate (HEA, 56 g), and a residual (EAR, 70 g).
The EAR was subjected to silica gel CC and eluted with a solvent system of n-hexane-EtOAc-AcOH (1:1:0.02, v/v/v), yielding fractions EAR1-EAR10. EAR10 (7.8 g) was subjected to further silica gel CC using n-hexane-chloroform-EtOAc-acetone-AcOH (1:2:2:2:0.01, v/v/v/v/v) as eluent. This afforded fractions EAR10.1-EAR10.5. Fraction EA10.1 (870 mg) was loaded into a Sephadex LH-20 CC and eluted with methanol to obtain fractions EA10.1.1-EA10.1.3. EA10.1.3 (670 mg) was washed with acetone, yielding compound 1 (480 mg).
2.3 Synthesis of analogues 1a-1c: General procedure
Sodium bromide (21 mg, 0.204 mmol) was added to a solution of salazinic acid (1, 60 mg, 0.155 mmol) in acetic acid (3 mL) at room temperature and dissolved under stirring. Hydrogen peroxide (0.021 mL, 0.182 mmol, 30%) was added each hour for 3.5 h at room temperature with stirring. The reaction was monitored using TLC. Saturated sodium hydrogen carbonate was used to stop the reaction then the neutralized mixture was partitioned between ethyl acetate-water (1:1). The organic layer was pooled, washed with brine, and dried over anhydrous Na2SO4. The residue was absorbed onto silica gel in column chromatography using CHCl3-EtOAc-acetone-AcOH (40:8:5:2, v/v/v/v) as eluent. This yielded compounds 1a (28%), 1b (25%), and 1c (21%).
5,9-Dibromo-1,4,10-trihydroxy-8-methyl-3,7-dioxo-1,3-dihydro-7H-benzo[6,7][1,4]dioxepino[2,3-e]isobenzofuran-11-carbaldehyde (1a). A white amorphous powder with an isolated yield of 28%. Tables 1 and 2 present the 1H and 13C NMR data for 1a: HRESIMS m/z calcd for C17H7Br2O9 [M−H]-: 512.8456, found: 512.8438.
No
1a (Acetone‑d6)
1b (Acetone‑d6)
1c (Acetone‑d6)
2a (Acetone‑d6)
2b (Acetone‑d6)
3a (Acetone‑d6)
3b (Acetone‑d6)
4a (DMSO‑d6)
4b (DMSO‑d6)
1 (DMSO‑d6)
5
6.70 (1H, s)
6.84 (1H, s)
7.00 (1H, s)
6.83 (1H, s)
6.78 (1H, s)
6.78 (1H, s)
6.88 (1H, s)
8
10.57 (1H, s)
10.57 (1H, s)
10.61 (1H, s)
9.03 (1H, s)
9.04 (1H, s)
8.22 (1H, br)
8.24 (1H, br)
10.45 (1H, s)
9
2.59 (3H, s)
2.49 (3H, s)
2.49 (3H, s)
2.40 (3H, s)
2.54 (3H, s)
2.53 (3H, s)
2.46 (3H, s)
2.38 (3H, s)
2.38 (3H, s)
2.45 (3H, s)
8′
4.87 (2H, s)
5.09 (2H, s)
4.88 (2H, m)
5.25 (2H, s)
4.83 (2H, br)
4.85 (2H, s)
4.53 (2H, s)
4.53 (2H, s)
4.64 (2H, s)
9′
7.41 (1H, br s)
7.07 (1H, s)
10.73 (1H, s)
6.43 (1H, s)
7.05 (1H, br s)
8.48 (1H, s)
8.83 (1H, s)
4.77 (1H, s)
4.79 (1H, s)
6.80 (1H, s)
11′
1.99 (3H, s)
2′'-6′'
7.17 (2H, d, 8.0)
7.32 (2H, t, 7.6)
6.92 (2H, d, 8.8)
6.97 (2H, d, 8.7)
3′'-5′'
7.14 (2H, m)
7.01 (2H, d, 8.0)
7.45 (2H, d, 8.8)
7.34 (2H, d, 8.7)
4′'
6.83 (1H, t, 7.2)
6.90 (1H, t, 7.2)
1*
2*-6*
7.71 (2H, d, 7.2)
7.64 (2H, d, 7.6)
3*-5*
7.40 (2H, m)
7.54 (2H, t, 7.6)
4*
7.31 (1H, d, 7.6)
7.46 (1H, m)
4-OH
12.29 (1H, s)
12.26 (1H, s)
12.25 (1H, s)
12.26 (1H, s)
12.06 (1H, s)
2′-OH
12.71 (1H, s)
12.85 (1H, s)
10.35 (1H, br)
10.34 (1H, br)
8′-OH
4.32 (1H, br s)
4.30 (1H, br s)
9′-OH
8.11 (1H, s)
8.30 (1H, s)
NH-1′'
9.91 (1H, br s)
10.40 (1H, br s)
10.87 (1H, s)
10.87 (1H, s)
NH-1*
7.44 (1H, br s)
No
1a (Acetone‑d6)
1b (Acetone‑d6)
1c (Acetone‑d6)
2a (Acetone‑d6)
2b (Acetone‑d6)
3a (Acetone‑d6)
3b (Acetone‑d6)
4a (DMSO‑d6)
4b (DMSO‑d6)
1 (DMSO‑d6)
1
113.6
111.9
113.1
112.9
113.7
114.4
112.9
112.2
112.2
112.2
2
159.4
155.4
156.2
162.7
161.3
161.3
161.6
nd
160.3
160.0
3
112.3
103.8
100.6
111.2
112.0
111.0
109.8
108.3
108.3
nd
4
162.2
157.9
158.6
163.0
162.9
162.6
163.0
161.8
161.9
164.0
5
110.4
115.5
116.5
118.3
118.5
117.8
117.7
116.1
116.2
117.9
6
148.1
142.0
143.1
149.5
149.9
146.6
145.8
144.4
144.4
152.0
7
162.9
160.2
161.1
161.7
161.7
160.8
161.9
161.2
160.3
160.3
8
193.9
195.0
194.9
135.4
134.2
132.1
136.2
193.1
9
22.3
21.3
22.3
21.1
21.9
21.3
21.3
18.4
20.8
21.4
1′
110.0
109.6
104.9
110.4
110.6
109.7
124.3
110.6
112.2
110.6
2′
151.4
153.1
153.4
154.2
154.3
158.1
158.5
158.6
158.1
152.1
3′
118.9
122.3
122.9
123.2
125.4
123.3
130.5
126.8
123.0
123.8
4′
147.9
148.4
142.1
147.1
147.7
148.9
149.4
144.4
144.4
145.1
5′
147.3
148.3
145.1
139.4
139.4
137.4
138.6
138.1
142.8
144.7
6′
138.1
137.4
129.2
137.8
137.6
122.2
119.8
138.1
142.8
137.6
7′
166.9
167.1
165.2
165.2
161.6
160.5
168.4
nd
165.0
8′
54.0
58.4
55.0
56.2
53.0
53.1
52.0
52.0
52.8
9′
105.1
100.1
189.8
104.4
98.5
141.4
134.1
87.4
86.4
95.0
10′
161.9
171.1
11′
80.9
23.3
1′'
146.2
144.4
143.2
143.3
2′'-6′'
114.0
110.3
113.8
113.3
3′'-5′'
128.8
129.6
132.0
129.3
4′'
120.6
121.4
113.7
123.0
1*
139.2
141.7
2*-6*
128.7
122.5
3*-5*
135.7
129.2
4*
126.2
127.0
9,11-Dibromo-1,4,10-trihydroxy-5-(hydroxymethyl)-8-methyl-7H-benzo[6,7][1,4]dioxepino[2,3-e]isobenzofuran-3,7(1H)-dione (1b). A white amorphous powder with an isolated yield of 25%. See Tables 1 and 2 for 1H and 13C NMR data on 1b: HRESIMS m/z calcd for C17H9Br2O9 [M−H]-: 514.8613, found: 512.8612.
2,4,7-Tribromo-3,8-dihydroxy-9-(hydroxymethyl)-1-methyl-11-oxo-11H-dibenzo[b,e][1,4]dioxepine-6-carbaldehyde (1c). A white amorphous powder with an isolated yield of 21%. See Tables 1 and 2 for 1H and 13C NMR data on 1c: HRESIMS m/z calcd for C16H8Br3O7 [M−H]-: 548.7820, found: 548.7836.
2.4 Synthesis of analogues 2a and 2b: General procedure
Salazinic acid (1, 30 mg, 0.077 mmol) and corresponding carboxylic trichloroacetic acid (582 mg, 3.56 mmol) and acetic acid (213.5 mg, 3.56 mmol) were added to 6 mL of DMF dissolved with 3.3 mg of AlCl3. The mixture was heated at 85 °C for 2 h under stirring. The reaction was periodically monitored using TLC. The mixture was partitioned between EtOAc-H2O (1:1, v/v) and was further processed as described in Section 2.3. The residue was subjected to silica gel CC and eluted with CHCl3- EtOAc-acetone-AcOH (40:8:5:2, v/v/v/v) to yield compounds 2a (21%) and 2b (32%).
(11-Formyl-1,4,10-trihydroxy-8-methyl-3,7-dioxo-1,3-dihydro-7H-benzo[6,7][1,4]dioxepino[2,3-e]isobenzofuran-5-yl)methyl 2,2,2-trichloroacetate (2a). A white amorphous powder with an isolated yield of 21%. Tables 1 and 2 present the 1H and 13C NMR data for 2a: HRESIMS m/z calcd for C19H12Cl3O10 [M−H + H2O-CO2]-: 504.9496, found: 504.9497.
Galbinic acid (2b). Another white amorphous powder with an isolated yield of 32%. The NMR data on 2b were consistent with an earlier report (Elix and Engkaninan, 1975; Devi et al., 2020).
2.5 Synthesis of analogues 3a, 3b, 4a, and 4b: General procedure
Salazinic acid (1, 40 mg, 0.103 mmol) was added to a solution of phenylhydrazine (44.6 mg, 0.412 mmol) in 10 mL of ethanol-acetic acid (7:0.08, v/v). The mixture was stirred for 3 h at 60 °C. The reaction was periodically monitored using TLC. The mixture was worked to yield an organic residue, which was subjected to silica gel CC and eluted with n-hexane-EtOAc-acetone (1:6:2, v/v/v) to yield compounds 3a (41%) and 3b (33%). Salazinic acid was reacted with 4-bromophenyl hydrazine (77.0 mg, 0.412 mmol) or 4-chlorophenyl hydrazine (73.7 mg, 0.412 mmol) to afford compounds 4a (64%) or 4b (47%), respectively.
(E)-5,11-Dihydroxy-6-(hydroxymethyl)-9-methyl-3-phenyl-12-((2-phenylhydrazineylidene)methyl)-8H-benzo[6,7][1,4]dioxepino[2,3-f]phthalazine-4,8(3H)-dione (3a). A white amorphous powder with an isolated yield of 41%. Tables 1 and 2 present the 1H and 13C NMR data for 3a: HRESIMS m/z calcd for C30H21N4O7 [M−H]-: 549.1409, found: 549.1410.
3,8-dihydroxy-9-(hydroxymethyl)-1-methyl-11-oxo-4-((E)-(2-phenylhydrazineylidene)methyl)-6-((Z)-(2-phenylhydrazineylidene)methyl)-11H-dibenzo[b,e][1,4]dioxepine-7-carboxylic acid (3b). A white amorphous powder with an isolated yield of 33%. See Tables 1 and 2 for the 1H and 13C NMR data on 3b: HRESIMS m/z calcd for C30H21N4O7 [M−H]-: 549.1409, found: 549.1409.
(E)-11-((2-(4-Bromophenyl)hydrazineylidene)methyl)-1,4,10-trihydroxy-5-(hydroxymethyl)-8-methyl-7H-benzo[6,7][1,4]dioxepino[2,3-e]isobenzofuran-3,7(1H)-dione (4a). A white amorphous powder with an isolated yield of 64%. See Tables 1 and 2 for the 1H and 13C NMR data on 4a: HRESIMS m/z calcd for C24H16BrN2O9 [M−H]-: 557.0019, found: 557.0057.
(E)-11-((2-(4-chlorophenyl)hydrazineylidene)methyl)-1,4,10-trihydroxy-5-(hydroxymethyl)-8-methyl-7H-benzo[6,7][1,4]dioxepino[2,3-e]isobenzofuran-3,7(1H)-dione (4b). A white amorphous powder with an isolated yield of 64%. See Tables 1 and 2 for the 1H and 13C NMR data on 4b: HRESIMS m/z calcd for C24H16ClN2O9 [M−H]-: 511.0544, found: 511.0564.
2.6 Synthesis of compound 5: General procedure
AlCl3 (5.5 mg, 0.041 mmol) was added to a mixture of salazinic acid (1, 40 mg, 0.103 mmol) and orsellinic acid (17.3 mg, 0.103 mmol) in DMF solvent (2 mL). This was stirred at 90 °C for 3 h and the reaction was periodically monitored using TLC. The mixture was partitioned between ethyl acetate and water (50 mL each). Organic layers were pooled, washed with brine, and dried over anhydrous Na2SO4. The residue was purified by normal phase silica gel column chromatography eluted with CHCl3-EtOAc-acetone-AcOH (100:40:24:8, v/v/v/v), yielding compound 5 (31%). The NMR data were consistent with a previous report (Devi et al., 2020).
2.7 In silico molecular docking: General procedure
The target compounds 1, 1a-1c, 2a-2b, 3a-3b, 4a-4b, and 5 were tested in silico for inhibition of α-glucosidase. Acarbose was used as control. AutoDockTools-1.5.6rc3 was used to model the docking of one receptor to each ligand. The crystal structures of the target proteins and receptors were assumed to be 5KEZ (PDB: https://doi.org/10.2210/pdb5KEZ/pdb), based on the protein data bank. The crystal structure of enzyme 5KEZ was detected as a hydrolase inhibitor, converting 4-nitrophenyl-α-D-glucopyranoside (p-NPG) to 4-nitrophenol and α-D-glucopyranose. If the enzyme was inactive or inhibited, the hydrolase conversion of 4-nitrophenyl-α-D-glucopyranoside (p-NPG) was not observed or proceeded slowly. The inhibitory effect of the ligand was evaluated based on this reaction. One strategy for controlling Type-2 diabetes is to slow the release of glucose from food and limit its absorption into the bloodstream. 5KEZ:PDB exhibited HPA (Human pancreatic alpha-amylase) inhibition and constitutes a novel enzyme inhibitor. This model can be used to detect glycosidase inhibitors that are highly selective for active carbohydrate enzymes (Jongkees et al., 2017).
The receptor was first tested for removal of small molecules like water, small ligands, and heteroatoms. The crystal structures were saved as format files (receptor.pdb) using the Discovery Studio 2019 Client (DSC) package. In the case of ligands, the minimum energy of conformation was investigated using the Molecular Mechanics & Force Fields (MMFF94) method, with optimal conformation discovery via the Avogadro package. This optimal conformation was saved. The AutoDock 4.2 package was used for docking of the ligand to the receptor. In the target procedure, a polar hydrogen and Kollman charge were added to the atoms. For ligands, this was added to all polar hydrogens, the Gasteiger charges were computed, non-polar hydrogen was merged, and the files were saved in pdbqt format. The values used were a grid point spacing of 0.375 Å, 120 x120 × 120 user-specified grid points, and X = –8,214, Y = 20.667, Z = -19.179 coordinates of the central grid point of maps. A Lamarckian genetic algorithm docked the conformation ligand to the macromolecular target. The maximum negative free energy of binding was selected to correspond to the most stable conformation after 2,500,000 energy evaluations and 200 runs (Thiratmatrakul et al., 2014). The Discovery Studio and Molegro (MMV) packages were used to visualize the results and present them as tables and figures. The docking procedure is described in Scheme S1.
2.8 α-Glucosidase inhibition assay
The α-glucosidase (0.2 U/mL) and substrate (5.0 mM p-nitrophenyl-α-D-glucopyranoside) were dissolved in 100 mM pH 6.9 sodium phosphate buffer. The inhibitor (50 µL) was preincubated with α-glucosidase, then the substrate (40 µL) was added to the reaction mixture. The enzymatic reaction was carried out at 37 °C for 20 min and stopped by the addition of 0.2 M Na2CO3 (130 μL). Enzymatic activity was quantified by measuring absorbance at 405 nm. All samples were analyzed in triplicate at five different concentrations around the IC50 values, and the mean values were retained. The inhibition percentage (%) was calculated as follows:
Inhibition (%) = [1 – (A sample/A control)] × 100.
2.9 Inhibitory type assay of 4a and 5 on α-glucosidase
The mechanisms by which compounds 4a and 5 inhibits alpha-glucosidase were determined through Lineweaver-Burk plots (Microsoft Excel 2010, Washington, USA), using methods similar to those reported by Tran and co-workers (Tran et al., 2021). Enzyme inhibition at different concentrations of 4a and 5 was evaluated by the effect on the substrate. In Lineweaver–Burk double reciprocal plots of 1/enzyme velocity (1/V) vs. 1/substrate concentration (1/[S]), the inhibition type was determined using pNPG concentrations of 1 mM, 2 mM, and 4 mM in the presence of test compound concentrations of 0, 4.5, 9.0, 18.0, and 36.0 µM for 4a and 0, 3.16, 6.33, and 12.65 µM for 5. All experiments used three replicates. The mixtures were incubated at 37 °C and the 405 nm optical density was measured at 1 min interval for 30 min using a Clariostar Labtech microplate reader (Ortenberg, Germany). Optimal concentrations of the test compounds were chosen based on IC50 values. The inhibition constants were obtained graphically from secondary plots (Microsoft Excel 2010, Washington, USA).
2.10 HPLC experiments proving the purity of compounds 4a and 5
The compounds 4a and 5 were analyzed through a HPLC-DAD using the modified method (Duong et al., 2017). Samples were injected separately. The mobile phase consisted of (ACN + 0.1% HCOOH) as solvent A and (H2O + 0.1% HCOOH) as solvent B with a gradient of 5% − 10% A over 5 min, 10 30% A over 15 min, 30 80% A over 10 min, 80 100% A over 5 min, then 100% A for 5 min. The flow rate was 1 mL/min, and 10 µL of each sample was injected. The HPLC chromatograms were showed in Figs. S33-34.
3 Results and discussion
3.1 Structural elucidation of synthetic compounds
In this study, ten salazinic acid derivatives were successfully synthesized. These included eight new compounds, 1a-1c, 2a, 3a, 3b, 4a, and 4b, and two known analogs, 2b and 5 (Fig. 1). Synthesis proceeded via four different routes, shown in Scheme 1. In the first route, salazinic acid (1) underwent bromination with sodium bromide and hydrogen hydroxide to yield 1a-1c. The chemical structures of 1a-1c (Tables 1 and 2) were identified from 1H NMR, 13C NMR, HMBC, and HRESI mass spectra. The 1H NMR data showed 1a-1c and 1 to be highly similar. 1a and 1 showed two structural differences: the absence of the aromatic methine proton (δH 6.88, δC 117.9, H-5) and the hydroxymethylene group (δH 4.64, δC 52.8, H2-8′), suggesting that both H-5 and 8′-CH2OH were replaced by bromine in 1a. This was confirmed by the HRESI mass data. In 1b, proton H-5 and the formyl group 3-CHO (δH 10.45, δC 193.1) were replaced by bromine, suggesting that the reaction occurred at C-3 and C-5 of 1. Scheme 2 proposes a possible mechanism for the formation of 1a-1c. First, bromination takes place at C-5, forming the intermediate 1a-1 (Abe et al., 2002; D’Aleo et al., 2013). This intermediate becomes oxidized, then decarboxylated, then brominated at C-3, forming 1b. A similar procedure occurs at C-8′ of the intermediate 1a-1, yielding 1a (Lei and Wang. 2008; Zhao et al., 2018; Perry et al., 2017; Quibell et al., 2018; Kong et al., 2019) (Scheme 2). 1b and 1c differ structurally in the addition of an extra aldehyde group (δC 189.8, δH 10.73) and the disappearance of the hemiacetal proton H-9′ from 1c. This suggests that the 5-member lactone of 1 becomes hydrolyzed, enabling decarboxylative bromination at C-1′ (Munoz and Murelli, 2012) and the formation of 1c (Scheme 2). In combination, these reactions give 1a-1c their unique scaffolds.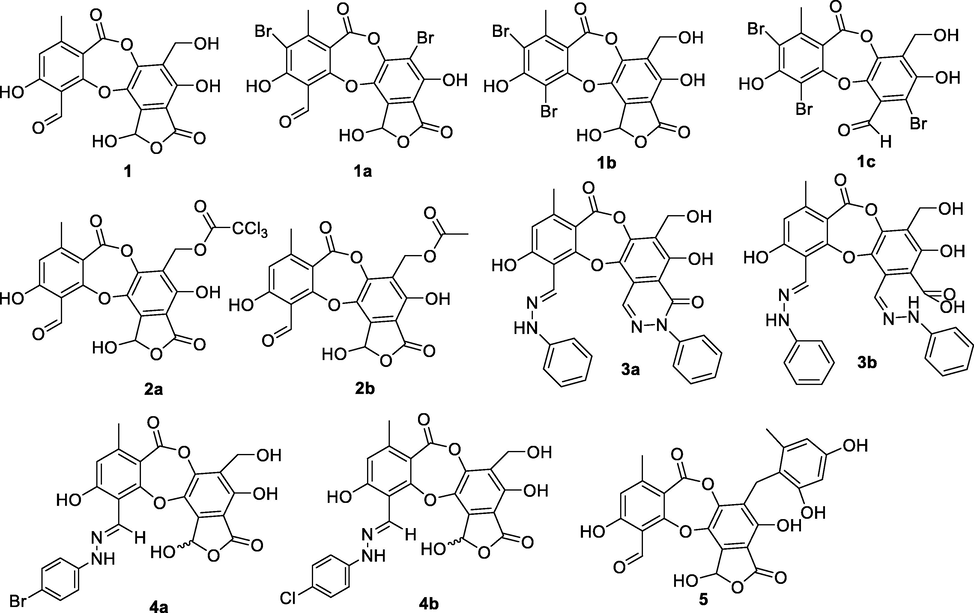
Chemical structure of compounds 1a-1c, 2a-2b, 3a-3b, 4a-4b, and 5.
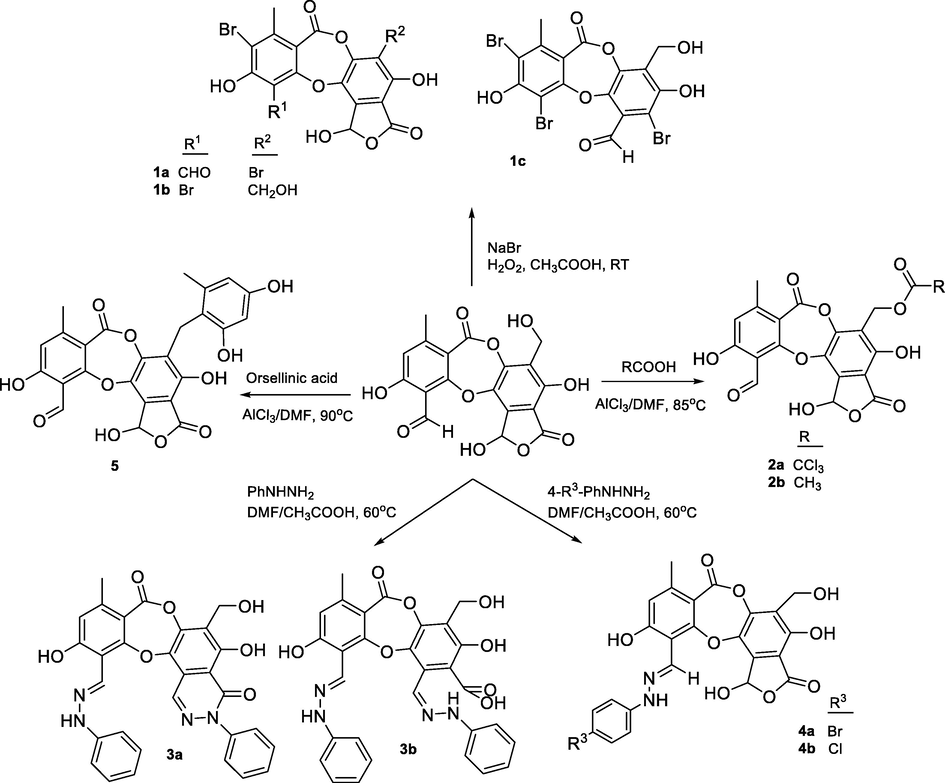
General synthetic routes to form 1a-1c, 2a-2b, 3a-3b, 4a-4b, and 5.
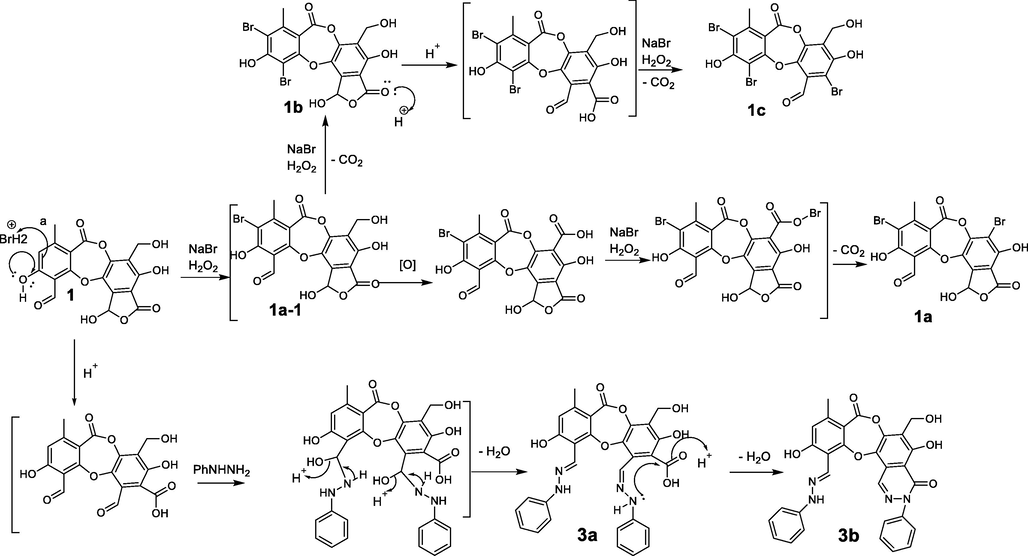
The proposed mechanism of the bromination and nucleophilic addition to form 1a-1c and 3a-3b.
Salazinic acid (1) esterification yielded 2a when using trichloroacetic acid and 2b when using acetic acid. NMR data showed 2a and 1 to be similar, with the only structural difference being observed in the B-ring. The H2-8′ was shifted downfield (δH 4.88 in 2a compared with δH 4.64 in 1), suggesting that esterification of 1 appeared at C-8′.
Hydrazone formation was applied to compound 1, and yielded 3a-3b and 4a-4b. Their chemical structures were elucidated using 1H NMR, 13C NMR, HMBC, and HRESI mass spectra. The active site 3-CHO of 1 was shown to react with 4-chlorophenylhydrazine or 4-bromophenylhydrazine to form 4a and 4b. Interestingly, compounds 3a and 3b were both obtained when phenylhydrazine was used as reagent under the same conditions. This indicated that phenylhydrazine is more reactive than either 4-chlorophenylhydrazine or 4-bromophenylhydrazine. Proton H-8 was shifted upfield in 3a-3b and 4a-4b (δH 9.03–9.33) relative to 1 (δH 10.45), indicating that the reaction occurred at 3-CHO. This was supported by the substitution of the imine carbon (δC 135.4) in 3a-3b and 4a-4b for the 3-CHO group (δC 193.1) in 1 and was backed up by the HMBC correlations of H-8 to C-2, C-3, and C-4. Spectroscopic analysis of 3a indicated that a further reaction occurred at C-9′. This was supported by a downfield shift of H-9′ (δH 8.48, δC 141.4 in 3a compared with δH 6.80, δC 95.0 in 1), together with the HMBC correlations of NH-1* (δH 7.44) to C-9′ (δC 141.4), C-2* (δC 128.7), and C-6* (δC 128.7), and of H-9′ (δH 8.48) to C-1′ (δC 109.7) and C-6′ (δC 122.2). NMR data showed strong structural similarities between 3b and 3a. The absence of the NH-1* proton in 3b indicated that a 6-member lactam was produced by the nucleophilic attack of an active amino group on carbonyl carbon C-7′ (Saundane and Kalpana. 2015; Rafeeq et al., 2015). Scheme 2 presents a possible mechanism for the formation of 3a and 3b.
A reaction between compound 1 and orsellinic acid yielded compound 5. NMR data showed 5 to be identical to parmosidone F (Devi et al., 2020). This supported the biosynthesis reaction proposed by Devi and co-workers. First, orsellinic acid becomes decarboxylated to orcinol and this intermediate undergoes Friedel-Crafts alkylation of the activated 3′ –CH2OH in 1 to form compound 5.
3.2 In vitro α-glucosidase inhibition of compounds 1–5
Salazinic acid (1) and its derivatives 1a-1c, 2a-2b, 3a-3b, 4a-4b, and 5 were tested for inhibition of α-glucosidase (Table 3). Most of the synthetic products showed inhibition twice as potent as that of the mother compound (IC50 34.80 µM). In the Br-substituted products 1a-1c, the presence of bromine at C-3 enhanced inhibition. However, as the IC50 values of these compounds were similar, the structural changes in the B-ring may have little effect on potency. The significant increase in potency of compound 2a may be attributed to the presence of chlorine. Compounds 3a, 3b, 4a, and 4b exhibited excellent potency, suggesting that imine moieties play an important role in α-glucosidase inhibition. Compound 5 was the most potent (IC50 9.72 µM), which was consistent with the findings of Devi et al., (2020), who isolated natural Friedel-Crafts alkylated depsidones similar to 5. They attributed the increase in potency to the transformation of the CH2OH group in 1 to CH2-Ph in 5, enhancing the activity of salazinic acid derivatives. In our study, the replacement of the 3-CHO of 1 by substituents in 1a-1c, 3a-3b, and 4a-4b significantly enhanced α-glucosidase inhibition.
Compounds
IC50 (µM)
1a
14.34 ± 0.64
1b
15.57 ± 1.23
1c
16.35 ± 1.56
2a
34.96 ± 1.12
2b
14.68 ± 1.80
2c
39.14 ± 0.49
3a
11.87 ± 0.36
3b
14.67 ± 0.16
4a
11.36 ± 0.26
4b
19.44 ± 1.12
5
9.72 ± 1.80
1
34.80 ± 0.85
Acarbose (positive control)
332 ± 3.9
3.3 In silico molecular docking
To elucidate the mechanisms of α-glucosidase inhibition, the binding modes in the active site were investigated. Figs. 2-21 and S1-S5, and Tables 4-5, show the interactions of the α-glucosidase enzyme with all ligands and with 1, 1a-1c, 2a-2b, 3a-3b, 4a-4b, 5, and the acarbose control. The α-glucosidase enzyme corresponded to the crystal structure of enzyme 5KEZ: PDB. Figs. 2-21 and Tables 4-5 showed docking of the most active ligands to 5KEZ: PDB. Compounds 3b, 2a, 3a, 1a, 1c, 1b, and 5 were shown In vitro to exhibit strong inhibition of this α-glucosidase enzyme. Further details are presented in Figs. S1−S16 in the supporting information. As can be seen from Table 4, the docking of conformation ligands with the target enzyme were ranked as follows: 5 > 3b > 4b > 4a > 1c > 1b > 1a > 1 > 2a > 3a > 2b. This suggested that a lower value of free energy of binding was associated with stronger and more stable docking with the receptor, and therefore a lower IC50 value. Fig. 2 shows conformation ligands 1, 1a-c, 2a-b, 3a-b, 4, 4a-b, 5 and acarbose docked to the same pocket enzyme in the crystal structure of α-glucosidase enzyme 5KEZ: PDB. Fig. 3 shows the docking poses of ligands including pose 90 (the most stable conformation ligand of entry 1), pose 7 (1a), pose 56 (1b), pose 88 (1c), pose 150 (2a), pose 196 (2b), pose 104 (3a), pose 65 (3b), pose 164 (4a), pose 15 (4b), pose 13 (5), and pose 164 (acarbose). The RMSD values shown in Table 5 confirmed the validity of the model. The poses of the most stable conformation ligands were ranked as follows: 3b > 1c > 2a > 1b > 1a > 3a. [a] The crystal structure of 5KEZ: PDB code was downloaded from the protein data bank. [b] The free energy of binding, receptor-ligand presented in the unit of kcal.mol−1 and calculated by Auto Dock Tools-package. [c] The inhibition constant calculated by Auto Dock Tools- and reported in the unit of µM. [d] The number of hydrogen bonds was indicated by Discovery Studio 2019 Client package. [e] They have visualized in Discovery Studio 2019 Client package and the unit of Angstrom, respectively. [a]. Unit of Å, performing by PyMOL software, and demonstration in Fig. 2. [b]. Red color in Fig. 2. [c]. Blue color. [d]. Yellow color. [e]. Green color. [f]. Magenta. [g]. Cyan color. [h]. Orange color. [i]. Chocolate color. [j]. Marine color. [k]. Deep purple color. [l]. Line green color. [m]. Hot pink color.
The docking poses of ligands, 1a-1c, 2a-2b, 3a-3b, 4a-4b, 5 and Acarbose docked to the same cavity in a pocket enzyme of receptor, 5KEZ. Pose 65 or ligand 3b was reference ligand.
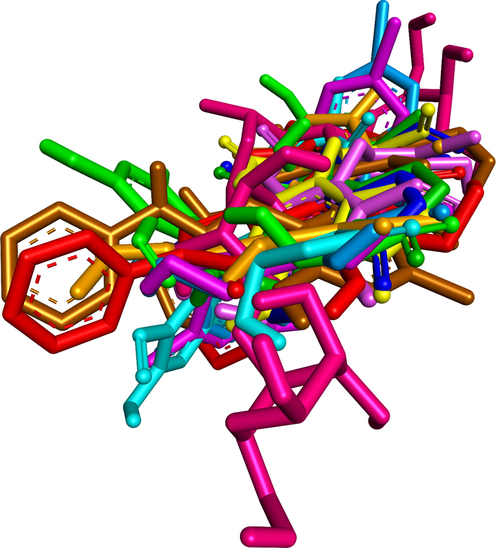
Ranked docking poses of ligands 1, 1a-1c, 2a-2b, 3a-3b, 4a-4b, 5 and acarbose aligned and calculated the values of RMSD by PyMOL software.
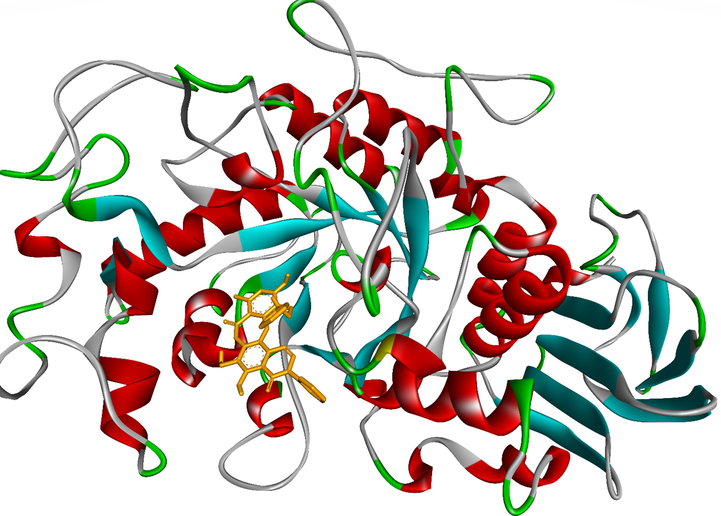
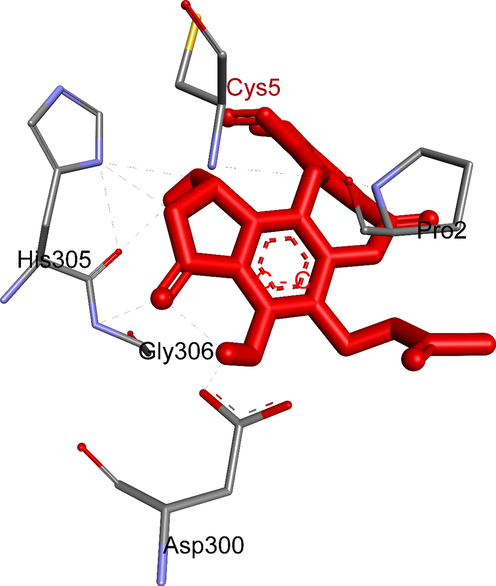
The most stable conformation ligand, 3b (ranked pose 65) was immersed in receptor, one enzyme 5KEZ: PDB.
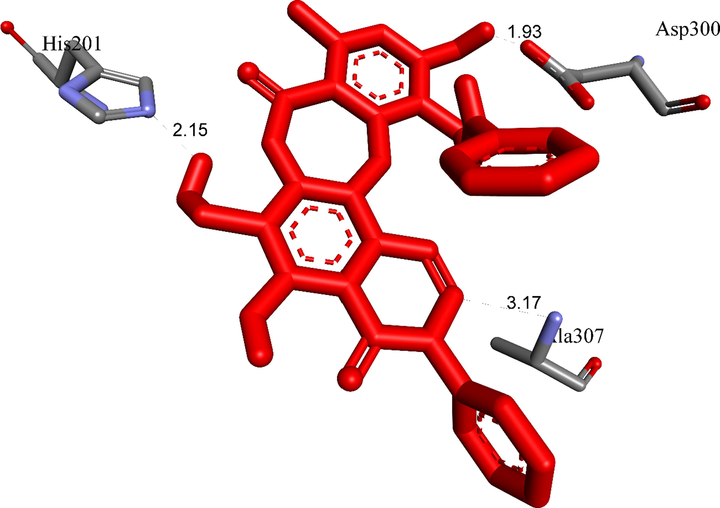
The most stable conformation of ligand 3b formed two hydrogen bonds with active sites of receptor, 5KEZ.
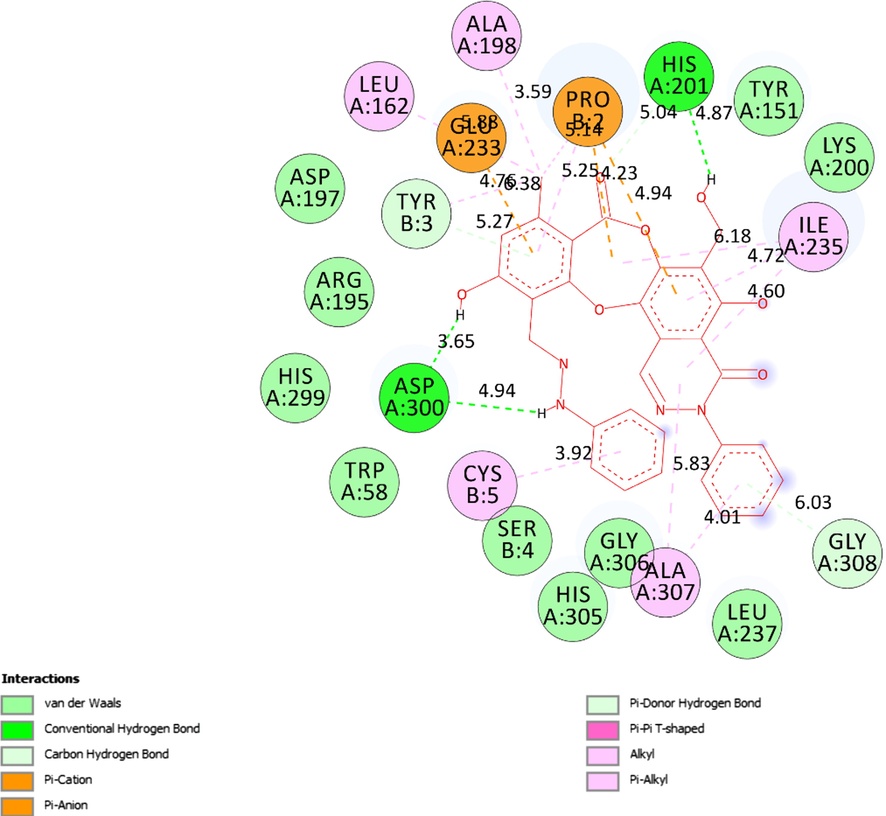
The interactions between active sites of receptor (5KEZ) and the active site atoms on ligand (3b) were indicated like hydrogen bonds (light green color), Val der Waals, pi-cation, pi-anion, pi-donor hydrogen bond, pi-pi T-shaped, alkyl and pi-alkyl on a 2D diagram.
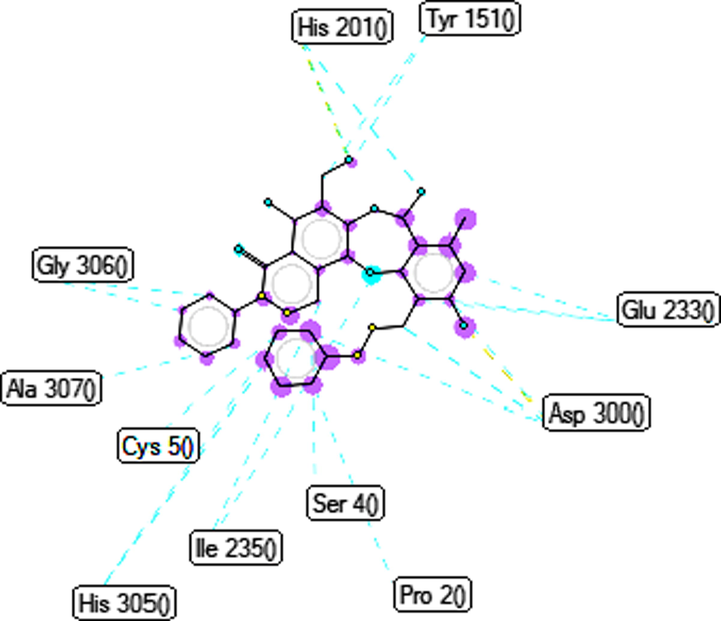
The ligand map showed secondary interactions between the most stable conformation 3b and receptor, 5KEZ.
The most stable conformation, ranked pose 150/200, 2a anchored in receptor (5KEZ: PDB), one crystal structure of α-glucosidase enzyme.
Five hydrogen bonds formed from residual amino acids of receptor, 5KEZ to active sites in ligand, 2a.
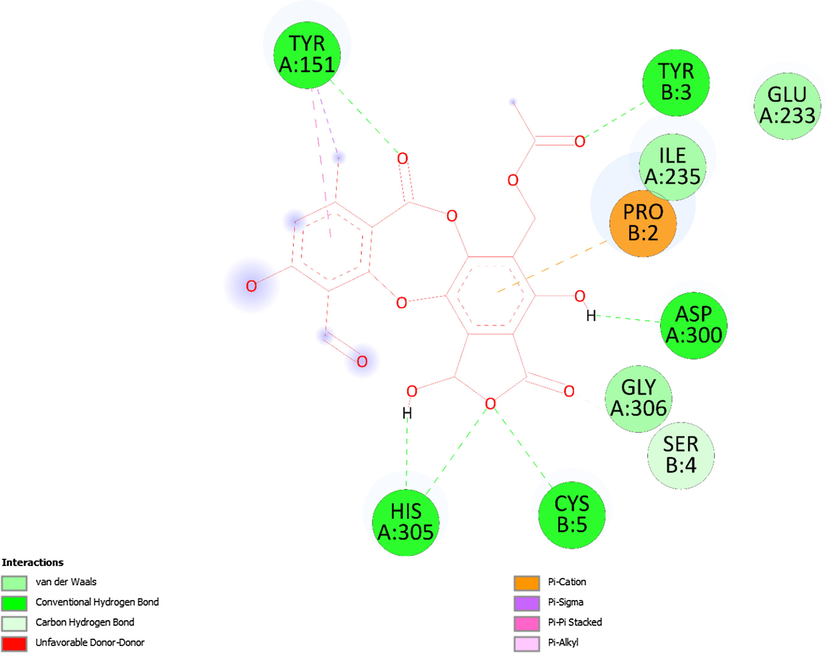
The interactions between the best conformation of 2a, pose 150 and receptor, 5KEZ indicated on a 2D diagram, which included hydrogen bonds, Van der Waals, unfavorable acceptor–acceptor, pi-donor hydrogen bond, alkyl, and pi-alkyl.
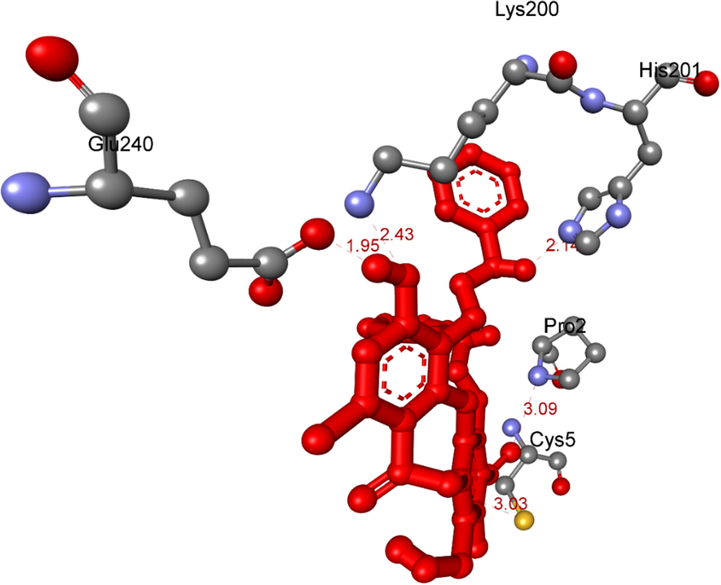
The hydrogen bonds linked from residual amino acids of target protein of receptor, 5KEZ to active atoms in a docking pose 104, one conformation of the ligand 3a.
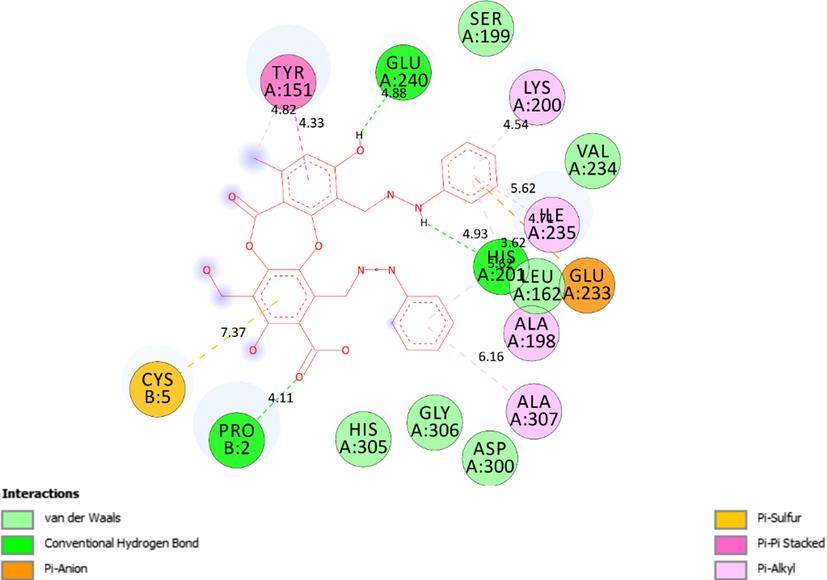
The interactions between the best conformation, pose 104 of ligand 3a and receptor 5KEZ indicated on a 2D diagram.
The hydrogen bond bound from residual amino acids of protein of enzyme, 5KEZ to pose 7/200, ranked pose of ligand 1a.
The significant interactions established on 2D diagram between residual amino acids of receptor, 5KEZ and active sites of pose 7, a ranked pose 7/200 of ligand 1a.
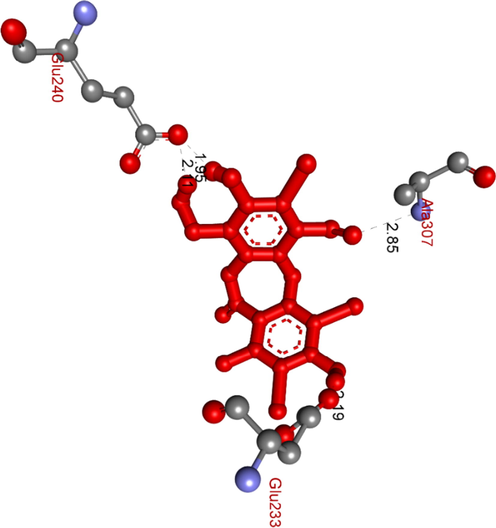
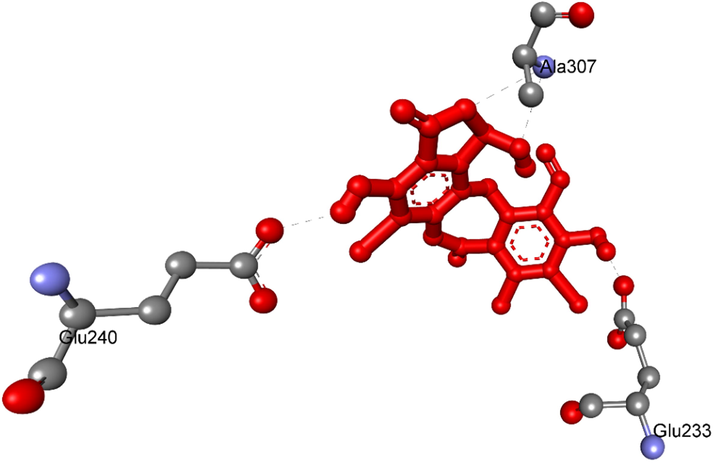
The hydrogen bonds formed from residual amino acids of receptor 5KEZ to active site binding atoms on the most stable conformation ligand 1c, pose 88.
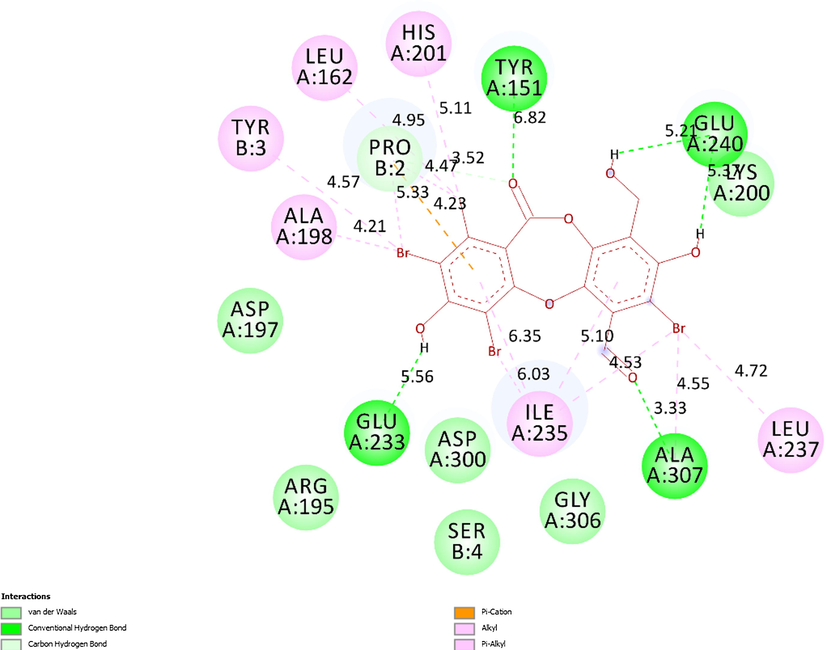
The 2D diagram presented the remarkable interactions between pose 88, ligand 1c and receptor 5KEZ.
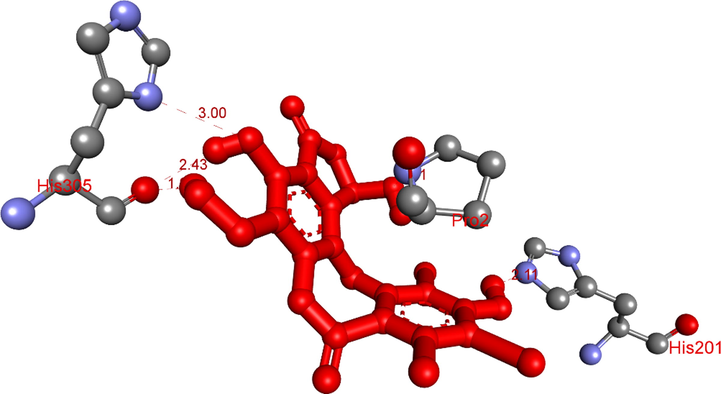
The hydrogen conducted from the conformation ligand 1b, pose 56 to amino acids of protein of enzyme, 5KEZ.
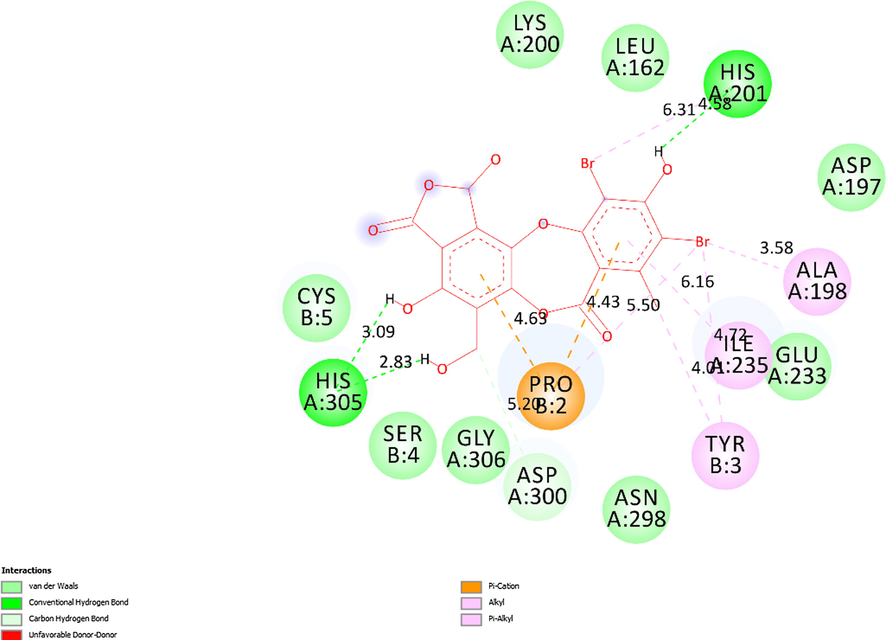
The 2D diagram indicated the essential interactions from residual amino acids of receptor, 5KEZ to active sites on the docking pose 88, ligand 1b.
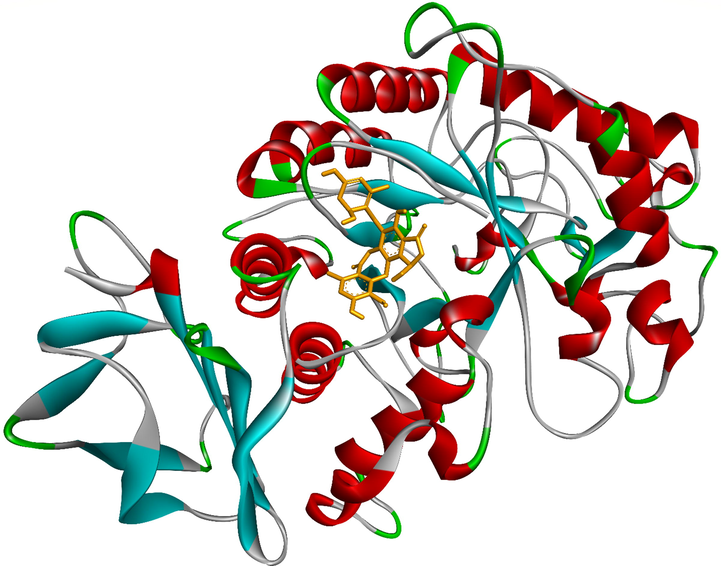
Ranked pose 13, the best stable conformation of ligand 5 docked to crystal structure of alpha-glucosidase, 5KEZ.
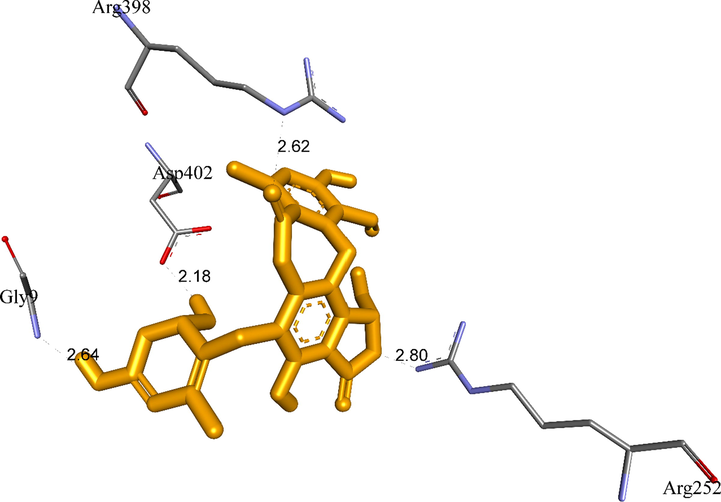
The active amino acids of enzyme 5KEZ bound to active atoms in pose 13 of ligand 5.
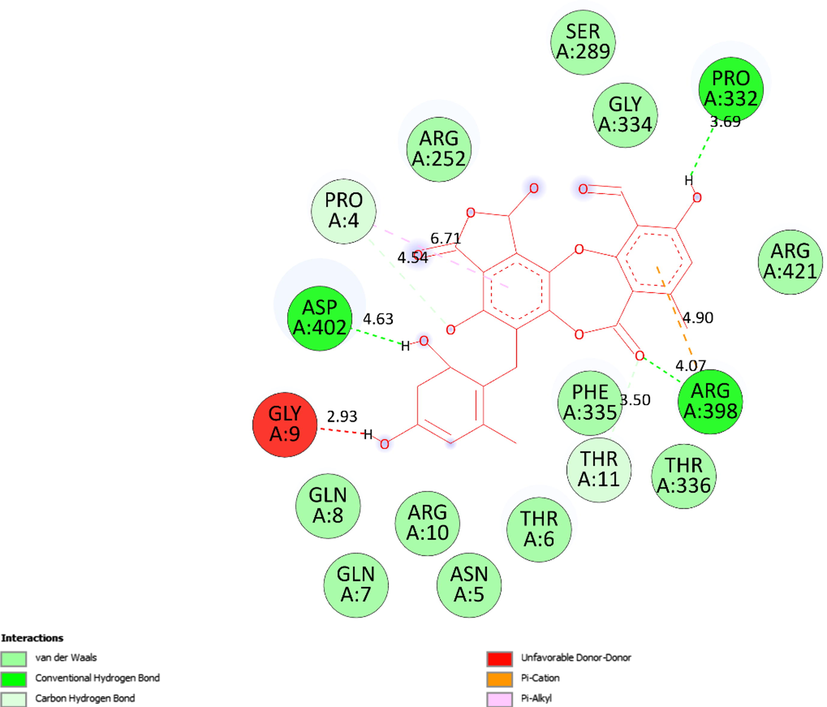
The 2D diagram showed the essential interactions between pose 13 of ligand 5 and receptor 5KEZ.
Entry
Free Energy of Binding[b]
IC50 in vitro
Ki[c]
The number of hydrogen bonds[d]
The property and bond length[e]
1
−7.08
Pose 90/20034.80 ± 0.85
6.42
4
A:Ala307:N–1: O (2.86 Å); Ala307:N–1:O (2.68 Å); 1:H– A:Glu233:O (1.89 Å); 1:H–A:His201:N (2.09 Å).
1a
−7.30
Pose 714.34 ± 0.64
4.48
2
A:Ala307:N − 1a:O (3.10 Å); :Ala307:N − 1a :O (3.09 Å); 1a :H − A:Glu233:O (2.15 Å); 1a :H − A:Glu240:O (2.14 Å).
1b
−7.81
Pose 5615.57 ± 1.23
1.90
5
A:His305:N–1b:O (3.0 Å); :Pro2:N–1b :O (3.11 Å); 1b :H–A:His201:N (2.11 Å); 1b :H–A:His305:O (1.98 Å); 1b :H– A:His305:O (2.43 Å).
1c
−8.03
Pose 8816.35 ± 1.56
1.30
5
A:Lys200:N–1c:O (3.05 Å); Ala307:N–1c:O (2.85 Å); 1c:H–A:Glu233:O (2.19 Å); 1c:H– A:Glu240:O (2.11 Å); 1c:H–A: Glu240:O (1.95 Å
2a
−6.90
Pose 15014.68 ± 1.80
8.75
8
A:His305:N–2a:O (2.75 Å); A:His305:N–2a :O (2.07 Å); A:Gly306:N–2a :O (3.07 Å); B:Pro2:N–2a :O (3.03 Å); B:Pro2:N–2a :O (2.83 Å); B:Cys5:N–2a:O (2.97 Å); 2a :H–A:His305:O (2.20 Å); 2a :H–A:Asp300:O (1.68 Å).
2b
−7.45
Pose 19639.14 ± 0.49
3.43
1
2b:H − A:Glu233:OE1 (2.48 Å).
3a
− 6.45
Pose 10413.87 ± 0.36
18.80
5
A:Lys200:N − 3a:O (2.43 Å); B:Pro2:N − 3a:O (3.09 Å); B:Cys5:S − 3a:O (3.03 Å); 3a:H − A:His201:N (2.14 Å); 3a:H − A:Glu240:O (1.94 Å).
3b
−9.79
pose 6514.67 ± 0.16
0.07
3
3b:H − A:Asp300:O (1.93 Å); 3b:H − A:His201:N (2.12 Å); A:Ala307:N – 3b:N (3.17 Å)
4a
−8.56
Pose 16413.36 ± 0.26
0.53
3
A:Ala307:N–4a :O (2.96 Å); 4a :H–A:Glu233:O (2.13 Å); 4a :H–A:His201:N (2.16 Å).
4b
−8.95
Pose 1519.44 ± 1.12
0.28
5
A:Ala198:N − 4b:O (3.18 Å); A:Ala307:N − 4b:O (2.73 Å); 4b:H − A:His305:O (1.87 Å); 4b:H − A:Glu233:O (1.93 Å); 4b:H − A:His201:N (2.02 Å).
5
−9.24
Pose 139.72 ± 1.80
0.17
5
A:Gly9:N − 5:O (2.64 Å); A:Arg252:N − 5:O (2.80 Å); A:Arg398:N − 5:O (2.62 Å); 5:H − A:Pro332:O (2.29); 5:H - A:Asp402:O (2.18 Å).
Acarbose
−3.25
Pose 164332 ± 3.9
>100
11
A:His305:N − Acarbose :O (2.74 Å); A:Ala307:N − Acarbose:O (3.10 Å); B:Pro2:N − Acarbose :O (2.63 Å); B:Tyr3:N − Acarbose:O (2.91 Å); B:Ser4:N − Acarbose:O (2.92 Å); Acarbose:H − A:GLU240:OE1 (2.11 Å); Acarbose:H − A:ASP300:O (2.12 Å); Acarbose:H − A:HIS305:O (2.02 Å); Acarbose:H − A:HIS305:O (2.02 Å); Acarbose :H − A:GLU240:O (1.69 Å); Acarbose:H − A:GLY238:O (1.99 Å).
RMSD[a]
Pose 90, liand 1[c]
Pose 7, ligand 1a[d]
Pose 56, ligand 1b[e]
Pose 88, ligand 1e[f]
Pose 150, ligand 2a[g]
Pose 196, ligand 2b[h]
Pose 104, ligand 3a[i]
Pose 164, ligand 4a[j]
Pose 15, ligand 4b[k]
Pose 13, ligand 5[l]
Pose 164, Acarbose[m]
Pose 65, ligand 3b[b]
3.618
4.059
4.453
2.840
3.589
3.854
2.290
4.295
4.337
4.312
4.424
Ranked pose 65, ligand 3b: Figs. 4-7 and Table 4 show the docking profile of 3b. Pose 65 gave the most stable docking with receptor 5KEZ, with free energy of binding of −9,79 kcal.mol−1 and inhibitor constant of 0.067 µM. It was also strongly hydrophilic (shown by the blue range). Pose 65 therefore showed strong thermodynamic bonding to 5KEZ. As seen in Fig. 5 and Table 4, three hydrogen bonds formed from residual amino acids of the A chain of enzyme 5KEZ: Asp300, His201, and Ala 307. These bound to N and O atoms. As seen in Fig. 6, significant ligand interactions between pose 65 and 5KEZ included hydrogen bonds, pi-cation, alkyl, pi-anion, pi-pi T shaped, pi-donor hydrogen, carbon-hydrogen bonds, and Van der Waals forces. The structure had three parts: a cap group (protein identification), a linker (aromatic or aliphatic chains), and functional groups that interacted with the structure enzyme. The cap group (protein identifications) formed interactions including pi-pi T-shaped ligands from the Ile 235: A chain to the aromatic ring, alkyl or pi alkyl ligands from the Ala307: A chain and Cys5: B chain, pi-cation or pi-anion ligands from the Glu233: A chain and Pro2: B chain to aromatics, from the Tyr3: B chain to the aromatic chain via carbon-hydrogen bonding, and from the Gly308: A chain to the aromatic chain via pi-donor hydrogen bonding. The link from Leu162, Ala198 to the methyl group was via pi-pi T shaped ligands, from the Pro2: B chain to the methyl group via pi-cation or pi-anion interactions, from the Pro2: B chain to the oxygen heterocyclic (pi-cation or pi-anion interactions), from the Ile235: A chain with oxygen heterocyclic via pi-pi-T-shaped ligands, from the Ile235: A chain to the nitrogen heterocyclic (pi-pi-T shaped), and from the Ala307: A chain to the nitrogen heterocyclic via alkyl or pi-alkyl interactions. The functional group at the identification part of the pose confirmed the hydrogen bonds, which formed from amino acids Asp300 and His201: A chain to the hydrogen atoms of phenolic hydroxyl group, hydrogen of an amine group, and hydroxy benzyl of the pose. Ligand 3b or pose 65 is the best candidate among ligands to delivery. The ligand map in Fig. 7, indicated the secondary interactions like hydrogen bonds (brown color), steric (light blue), and overlap (violet circles on atoms of ligand 3b) interactions between pose 65 and residual amino acids (distance of less than 4 Å) of the receptor, 5KEZ. Those green lines presented the steric effects to the strength of interactions of the stable pose 65 to the receptor, 5KEZ. The size of violet circles on atoms controlled the strength of overlap interactions and contributed to steric hindrance.
Ranked pose 150, ligand 2a: the interaction profile of pose 150 and target enzyme showed in Figs. 8-10 and Table 4. As seen in Fig. 8, pose 150 anchored to a pocket enzyme with the values of ΔGo and Ki of −6.90 Kcal.mol−1 and 8.75 µM, respectively. The values of IC50 in vitro and Ki in silico are equivalent. The hydrophilic ability of this pose is also indicated by eight hydrogen bonds, which are linked from active amino acids of A or B chain as seen in Table 4 and Fig. 9. Among hydrogen bonds, 2a:H–A:ASP300:O was the strongest due to its shortest bond length of 1.68 Å. The significant interactions between this pose and 5KEZ indicated in the 2D diagram in Fig. 10. The cap group of pose recognized via Pro 2: B chain with an aromatic ring- pi cation, an electrostatic interaction, Tyr151: A chain with an aromatic ring via pi-pi- stacked. The linker interaction linked from Tyr151: A chain to the methyl group of pose via a pi-sigma ligand interaction. The functional group of pose formed from His305: A chain, Cys5: B chain, Asp300: A chain, Tyr3: B chain, and Tyr151: A chain to hydro atom of alcohol group, oxygen atom of ether group, hydrogen atom of phenolic hydroxy, oxygen of ketone group, and oxygen atom of lactone ring. It indicated that pose 150, a conformation ligand 2a interacted well with target enzyme.
Ranked pose 104, ligand 3a: the ranked pose 104/200, the most stable conformation ligand docked to target enzyme with values of the free Gibbs energy and inhibition constant of − 6.45 Kcal.mol−1 and 18.80 µM. As shown in Table 4 and Fig. 11, pose 104 formed five hydrogen bonds from active amino acids such as Lys200, His201, Glu240: A chain, and Pro2, and Cys5: B chain to oxygen and hydrogen atoms in pose 104. Among hydrogen bonds, one hydrogen bond, 3a:H − A:GLU240:O is the strongest due to its shortest bond length of 1.94 Å. Those hydrogen bonds determined the hydrophilic ability of pose because they were classified by electrostatic interactions. The important interactions between this pose and the crystal structure of the enzyme exposed in the 2D diagram as seen in Fig. 12. Pose 104 considered strong interaction with target enzyme because three parts of pose 104 identified full ligand interactions. The cap group detected by Ala307, Ala198, Ile 235, Lys200 of A chain via pi-alkyl ligand interactions, Tyr151: A chain via pi-pi stacked, Glu233: A chain via pi-cation, an electrostatic, and Cys5: B chain via pi sulfur interaction. The linker was identified by Tyr151 via pi-alkyl interaction. The functional groups in this pose were recognized by hydrogen bonds which bound from Pro2: B chain, His 201: A chain, and Glu240: A chain to the oxygen atom of carboxylic group, the hydrogen atom of an amine, and the hydrogen atoms of phenolic hydroxy group in pose 104. Other residual interactions were weak Van der Waals interactions, which linked from this pose to wall of the cell via residual amino acid around poses such as His305, Gly306, Asp300, Leu162, Val234, and Ser199 of A chain.
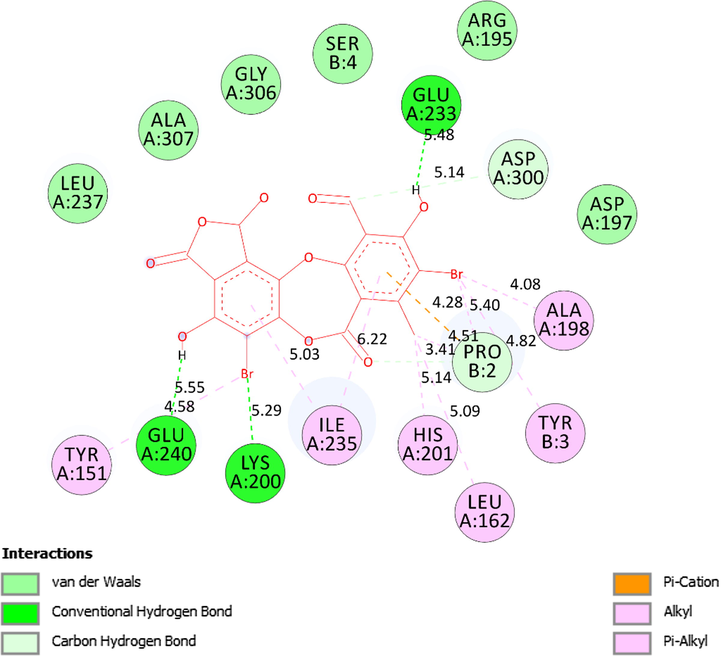
Ranked pose 7, ligand 1a: the interaction profile between ranked pose 7, the most stable conformation ligand was selected and docked to receptor after performing docking as shown in Figs. 13-14 and Table 4. The values of ΔGo and Ki of binding between the best docking pose 7 and receptor 5KEZ conducted −7.30 Kcal.mol−1 and 4.48 µM, respectively as shown in Table 4. As seen in Fig. 13 and Table 4, pose 7 and receptor, 5KEZ showed four hydrogen bonds from active amino acids such as Ala307, Glu233, Glu240: A chain of the pocket enzyme to active atoms in the pose. They are hydrophilic interactions because the hydrogen bonds were electrostatic interactions. The essential interactions between pose 7 and enzyme were indicated in the 2D diagram in Fig. 15. Pose 7 interacted well with enzyme due to three parts of pose 7, which had strong ligand interactions with enzyme, 5KEZ. The cap group of pose 7 recognized by Ile235: A chain and via pi-alkyl or alkyl, and Pro2: B chain via pi-cation (electrostatic interaction), the aromatic identifications of pose 7. The linker part of pose discovered by Tyr151: A chain, His201: A chain, Leu162: A chain, Pro2: B chain, Tyr3; B chain, Ala198: A chain via alkyl or pi-alkyl, and Asp300: B chain via carbon-hydrogen bond. The functional groups of this pose were identified via hydrogen bonds, which linked from Glu240, Lys200, and Glu233: A chain to hydrogen atom of a phenolic hydroxy group, bromine atom of the phenyl ring, and hydrogen atom of a phenolic hydroxy group, respectively. In other sites, the bromine atoms which linked to phenyl rings indicated more ligand interactions with receptors.
Ranked pose 88, ligand 1c: ranked pose 88/200, the best stable conformation ligand of ligand 1c among 200 conformations, which were docked to the enzyme, 5KEZ had the values of free Gibb’s energy and inhibition constant of −8.03 Kcal.mol−1 and 1.03 µ, respectively. As seen in Fig. 15 and Table 4, active atoms in pose 88 and pocket enzyme performed 5 hydrogen bonds, electrostatic interactions, and hydrophilic interactions. The essential ligand interactions between this pose and the crystal structure of an enzyme were indicated in the 2D diagram in Fig. 16. Generally, this pose interacted well with enzyme because it has full ligand interactions at three parts of this pose. The protein identification of ligand or cap group detected aromatic rings of ligand via category interactions such as alkyl or pi-alkyl from Ile235: A chain to aromatic rings in the structure of this pose, pi-cation, an electrostatic interaction, or hydrophilic interaction (yellow lines) from Pro2: B chain to the aromatic ring. The linker part in this pose has been identified via alkyl or pi-alkyl (pink color lines) from Ile235, Ile237, Ala307, His201, Leu162, Ala198 of A chain, and Tyr3, Pro2 of B chain to bromine atoms and methyl group in the pose. The functional group of this pose was classified by hydrogen bonds linked from Ala307, Tyr151, Glu233, and Glu240 to aldehyde, carbonyl, phenolic hydroxyl group, and alcohol benzyl group in pose (green lines). Another special site, this pose has fully characteristic identifications via cap group: aromatic ring, oxygen heterocyclic of 7 members, linker part: (bromine atoms or methyl), and functional groups. Bromine atoms in this pose made more ligand interactions.
Ranked pose 56 or ligand 1b: Ranked pose 56 docked to the pocket enzyme with the calculated values of ΔGo and Ki of −7.81 Kcal.mol−1 and 1.9 µM, respectively as shown in Table 4. As seen in Table 4 and Fig. 17, pose 56 formed 5 hydrogen bonds with target enzyme, 5KEZ. They are electrostatic interactions, which proved more hydrophilic ligand. The hydrogen bond, 1b:H–A: His305: O was the strongest among them owing to its shortest bond length of 1.98 Å. As shown in Fig. 18, in the 2D diagram, pose 56 of ligand 1b did not interacted ligand as well as pose 65 (ligand 3b) because it did not identify fully the characteristic conformation ligand for instance without ligand interactions with oxygen heterocyclic of seven members, lactone ring. The cap group of this pose identified the aromatic ring via ligand interaction styles such as pi-cation (electrostatic interaction) via Pro2: B chain, alkyl, or pi-alkyl via Tyr3: B chain, Ile235: A chain. The linker part of this pose detected bromo atoms by pi-cation style via Pro 2B with bromo atom, pi-alkyl, or alkyl interactions via Tyr3: B chain, Ala198: A chain, and His201: A chain. The functional groups have been detected by hydrogen bonds via His201, His 305: A chain to hydrogen atoms of phenyl rings. Pose 56 of ligand 1b was the potential drug delivery due to full identification ligand.
Ranked pose 13, ligand 5: The interaction profile of pose 13, the best stable conformation of ligand 5 was shown in Figs. 19-20. In Fig. 19, the best docking pose ligand 5 or pose 13 docked to the same cavity of enzyme 5KEZ as well as other poses with the values of ΔGo and Ki of −9.24 Kcal.mol−1 and 0.17 µM, respectively. It made clear that this pose interacted with the receptor, 5KEZ in the thermodynamic site. As shown in Fig. 20 and Table 4, five hydrogen bonds linked from active amino acids of A chain of protein of enzyme 5KEZ to active atoms in this pose. Among them, the hydrogen bond, 5:H - A: Asp402: O was the strongest due to its shortest bond length of 2.18 Å. As seen in Fig. 20, the significant interactions between this pose and 5KEZ indicated in the 2D diagram such as hydrogen bond (hydrophobic interaction) pi-cation (electrostatic interaction), pi-alkyl, carbon-hydrogen bond, Van der Waals, and unfavorable donor–donor. In this case, unfavorable interaction (red color), which formed from Gly9: A chain to hydrogen atom of alcohol group of 1,3-dienyl cyclohexyl ring of pose 13, made the system of ligand interactions weaker. The system energy of pose 13 and 5KEZ increased and the complex, which formed from pose 13 to receptor, 5KEZ was unstable. Therefore, pose 13 or ligand 5 did not interact well with enzyme.
Compared docking model and ligand interactions
Ligand piHA-Dm was a type of structure of peptide, composing five amino acids linked to a head-to-side-chain thioether macrocycle. piHA-Dm exposed selected inhibitor of novel typical peptide and occupied in Human pancreatic alpha-amylase (HPA). The HPA/piHA-Dm complex have deposited under the code PDB:5KEZ (target enzyme in silico docking.) It inhibited enzyme 5KEZ with the value of inhibition constant of 7.0 ± 3.5 nM (Jongkees et al., 2017). Ligand 3b or pose 65 was the best ligand, which inhibited enzyme 5KEZ with the value of inhibition constant of 0.067 μM or 67 nM, which indicated ligand 3b inhibited around ten-fold lower than ligand piHA-Dm. Insightful analysis of ligand interactions between ligand piHA-Dm or ligand 3b (pose 65) and crystal structure of enzyme 5KEZ illustrated that ligand piHA-Dm interacted well with five residual amino acids such as Asp197 and 300, His101 and 201, and Glu233 in enzyme 5KEZ. While as shown in the 2D diagram of Fig. 6, ligand 3b (pose 65) interacted well with 5KEZ via Asp300 of A chain: two hydrogen bonds and His 201 of A chain: one hydrogen bond, Glu233: A chain: pi-charge interaction. Other ligand interactions that make ligand 3b to enzyme 5KEZ different from ligand piHA-Dm to the same enzyme included alkyl or pi-alkyl interactions as Leu162, Ala198, Ile235, Ala307: A chain, and Cys 5: B chain, and pi-charge infractions from Pro 2: B chain to ligand 3b as show in Fig. 6. As indicated in Fig. 7, one ligand map presented hydrogen bonds from His 201 and Asp300: A chain (brown color lines) to active atoms in ligand 3b (pose 65). Many steric interactions (green lines) bound from His201, Asp300, Glu233, Tyr151, Gly306, Ala307, His305, Ile235, Cys50, Ser4, and Pro2 of enzyme to ligand 3b, which indeed proved that ligand 3b and target enzyme, 5KEZ interacted strongly through docking processing. At the interaction site, ligand 3b interacted with the same enzyme more strongly than that of ligand piHA-Dm.
The validation of molecular docking model
Ranked docking poses of ligands and Acarbose docked the same cavity of enzyme pocket, 5KEZ (Fig. 2). Pose 65 or ligand 3b, the best docking ligand was selected as reference ligand. Other ligands and ligand Acarbose aligned to pose 65 and calculated the values of RMSD by PyMOL software as illustrated in Fig. 3. The calculation results of redocking can be found in Fig. 3 and Table 5. As shown in Fig. 3, docking poses bound to the same cavity of enzyme without being superimposed because they are not analogue structures. As seen in Table 5, the values of RMSD of pair poses changed from 2.290 Å (ligand 3a, ligand 3b) to 4.453 Å (ligand 1b, ligand 3b). Additionally, the value of RMSD between 3a and 3b was lowest due to their analogue structures. The values of RMSD were considered more suitable and they proved that the docking parameters, which were valuation in terms of conformation, interactions, and reproducing orientation, were predicted well in molecular docking model of interesting compounds (Hidalgo-Figueroa el al., 2021).
From these in vitro data, compounds 4a and 5 were identified as the most active. To elucidate the inhibition mechanism, we investigated activity levels at the concentrations of 0, 4.5, 9.0, 18.0, and 36.0 µM for 4a and of 0, 3.16, 6.33, and 12.65 µM for 5. Lineweaver–Burk plots of 4a showed linearity at each concentration, intersecting the y-axis in the first quadrant (Fig. 22). The kinetics of enzyme inhibition confirmed that 4a acted as a non-competitive inhibitor. The kinetic analysis showed that an increase in the concentration of 4a had no effect on the Km (1.20), but decreased the vmax from 0.07 to 0.03 µM.min−1. The inhibition constant (Ki) was 28.44 ± 2.50 µM. For compound 5, Lineweaver–Burk plots gave a group of lines with different slopes and intercepts, intersecting the y-axis in the second quadrant (Fig. 23A). The kinetics of enzyme inhibition indicated that 5 acted as a mixed-type inhibitor. The inhibition constants of 5 binding with the free enzyme (Ki) and with the enzyme-substrate complex (K’i) were determined to be 8.22 ± 0.32 μM and 20.32 ± 0.51 μM, respectively (Fig. 23B and Fig. 23C). The value of Ki was lower than that of K’i, indicating that the binding affinity of α-glucosidase-5 exceeded that of α-glucosidase-PNPG-5 complex.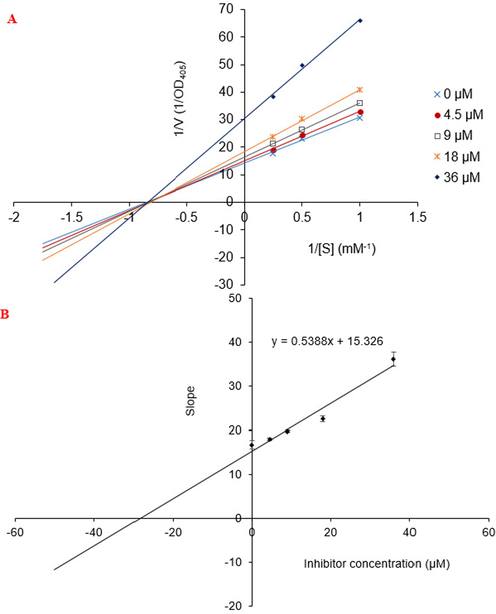
Lineweaver-Burk plot for alpha-glucosidase inhibition by compound 4a (A) and the secondary plot of slope vs the inhibitor concentration (B).
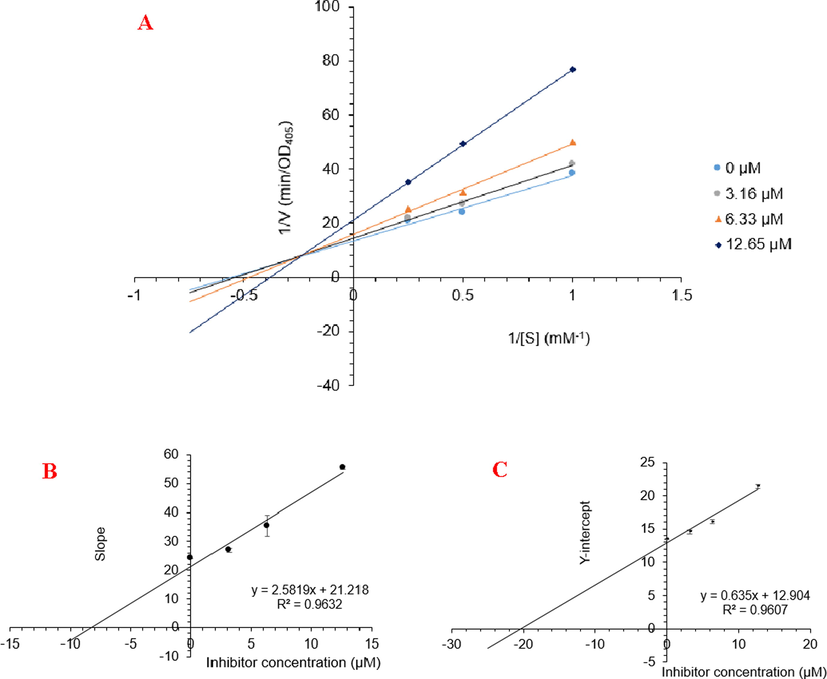
Lineweaver-Burk plot (A) for α-glucosidase inhibition by 5 and the secondary plots of slope and Y-intercept vs inhibitors concentration (B and C)
4 Conclusions
Ten compounds were synthesized using bromination, esterification, Friedel-Crafts alkylation, and imine nucleophilic addition of salazinic acid (1). The derivatives were evaluated for α-glucosidase inhibition. The majority of the compounds proved to be potential α-glucosidase inhibitors, with IC50 values in the range 9.32–39.96 μM. All were significantly lower than those of the mother compound 1. The results demonstrated that salazinic acid modification increased the potency of α-glucosidase inhibition. Compounds 3b, 4a, and 5 were the strongest inhibitors. An in silico docking model indicated that ligands 3b and 4a were the best candidates for drug delivery systems. A kinetic study of 4a and 5 indicated that 4a was a non-competitive-type inhibitor and 5 was a mixed-type inhibitor.
Acknowledgements
The study was funded by Saigon University (TĐ2021-15).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Botryorhodines A-D, antifungal and cytotoxic depsidones from Botryosphaeria rhodina, an endophyte of the medicinal plant Bidens pilosa. Phytochemistry. 2010;71(1):110-116.
- [Google Scholar]
- Abe, M., You, Y., Detty, M.R., 2002. 21-Telluraporphyrins. 2. Catalysts for bromination reactions with hydrogen peroxide and sodium bromide. Organometallics 21(21), 4546–4551.
- Antibacterial and antifungal activity of three Ramalina species. J. Drug Delivery Ther.. 2017;7(5):27-32.
- [Google Scholar]
- Lichen secondary metabolites and its biological activity. Am. J. Pharm. Tech. Res.. 2016;6(6):29-44.
- [Google Scholar]
- Lichens—a promising source of bioactive secondary metabolites. Plant Genetic Resources. 2005;3:273-287.
- [Google Scholar]
- Bioactive lichen metabolites: Alpine habitats as an untapped source. Phytochem. Rev.. 2011;10:287-307.
- [Google Scholar]
- Anti-proliferative lichen compounds with inhibitory activity on 12(S)-HETE production in human platelets. Phytomedicine. 2004;11(7):602-606.
- [Google Scholar]
- Antimicrobial activity of extracts of the lichen Parmelia sulcata and its salazinic acid constituent. Zeitschrift für Naturforschung C. 2007;62(7):619-621.
- [Google Scholar]
- Antidiabetic activity of Syzygium calophyllifolium in Streptozotocin-nicotinamide induced type-2 diabetic rats. Biomed. Pharmacother.. 2016;82:547-554.
- [Google Scholar]
- Acarbose for prevention of type 2 diabetes mellitus: The STOPNIDDM randomised trial. Lancet. 2002;359:2072-2077.
- [Google Scholar]
- Aromatase inhibitory, radical scavenging, and antioxidant activities of depsidones and diaryl ethers from the endophytic fungus Corynespora cassiicola L36. Phytochemistry. 2009;70(3):407-413.
- [Google Scholar]
- Green halogenation of aromatic heterocycles using ammonium halide and hydrogen peroxide in acetic acid solvent. Can. J. Chem.. 2013;91(8):79-683.
- [Google Scholar]
- Salazinic acid-derived depsidones and diphenylethers with α-glucosidase inhibitory activity from the lichen Parmotrema dilatatum. Planta Med.. 2020;86(16):1216-1224.
- [Google Scholar]
- Do, T.H., Nguyen, T.T., Dao, T.B.N., Vo, H.C., Huynh, B.L.C., Nguyen, T.A.T., Mai, D.T., Vo, T.P.G., Sichaem, J., Nguyen, N.H., Duong, T.H., 2021. A new diphenyl ether from Parmotrema indicum Hale growing in Vietnam. Natural Product Research 1–7. Article in Press. https://doi.org/10.1080/14786419.2021.1907748
- Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem.. 2019;176:343-377.
- [Google Scholar]
- α-Glucosidase inhibitory depsidones from the lichen Parmotrema tsavoense. Planta Med.. 2020;86(11):776-781.
- [Google Scholar]
- New erythritol derivatives from the fertile form of Roccella montagnei. Phytochemistry. 2017;137:156-164.
- [Google Scholar]
- NMR spectral data of salazinic acid isolated from some species of Parmotrema. Magn. Reson. Chem.. 2000;38:472-474.
- [Google Scholar]
- The structure of galbinic acid. A depsidone from the lichen Usnea undulata. Aust. J. Chem.. 1975;28(8):1793-1797.
- [Google Scholar]
- Carbazoles and hydrazone-bridged thiazole-pyrrole derivatives as new inhibitors of α-glucosidase. Chemitry Select. 2018;3(27):7921-7925.
- [Google Scholar]
- Synthesis, characterization and molecular docking of novel bioactive thiazolyl-thiazole derivatives as promising cytotoxic antitumor drug. Molecules. 2016;21(1):1-17.
- [Google Scholar]
- Molecular docking-guided ungual drug-delivery design for amelioration of onychomycosis. ACS Omega. 2019;4(5):9583-9592.
- [Google Scholar]
- Synthesis, molecular docking, dynamic simulation and pharmacological characterization of potent multifunctional agent (dual GPR40-PPARγ agonist) for the treatment of experimental type 2 diabetes. Eur. J. Pharmacol.. 2021;907:174244
- [Google Scholar]
- Rapid Discovery of potent and selective glycosidase-inhibiting de novo peptides. Cell Chem. Biol.. 2017;24(3):381-390.
- [Google Scholar]
- Antimalarial and cytotoxic depsidones from the fungus Chaetomium brasiliense. J. Nat. Prod.. 2009;72(8):1487-1491.
- [Google Scholar]
- Electrochemical synthesis of enaminones via a decarboxylative coupling reaction. Green Chem.. 2019;21(14):3796-3801.
- [Google Scholar]
- α–Glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn. Rev.. 2011;5:19-29.
- [Google Scholar]
- Excelsione, a depsidone from an endophytic fungus isolated from the New Zealand endemic Tree Knightia excels. J. Nat. Prod.. 2007;70(2):310-311.
- [Google Scholar]
- Oxidation of alcohols using H2O2 as oxidant catalyzed by AlCl3. Catal. Commun.. 2008;9(5):740-742.
- [Google Scholar]
- Discovery of novel Pim-1 kinase inhibitors with a flexible-receptor docking protocol. J. Chem. Inf. Model. 2019;59(10):4116-4119.
- [Google Scholar]
- Covalent docking predicts substrates for haloalkanoate dehalogenase superfamily phosphatases. Biochemistry. 2015;54(2):528-537.
- [Google Scholar]
- Antifungal and anti-biofilm activities of ccetone lichen extracts against Candida albicans. Molecules. 2017;22:651-662.
- [Google Scholar]
- Chemistry and biological activity of Ramalina lichenized fungi. Molecules. 2015;20(5):8952-8987.
- [Google Scholar]
- Chemical modulation of agonistic display in Betta splendens. J. Comput. Chem.. 1998;90(2):198-202.
- [Google Scholar]
- Acid-mediated coupling of γ-hydroxybutenolides and aldehydes: synthesis of a new class of spirocyclic ketal-lactones. Tetrahedron Lett.. 2012;53(50):6779-6781.
- [Google Scholar]
- Depsides and depsidones as inhibitors of HIV-1 integrase: Discovery of novel inhibitors through 3D database searching. J. Med. Chem.. 1997;40(6):942-951.
- [Google Scholar]
- Quinoliniumolate and 2H–1,2,3-triazole derivatives from the stems of Paramignya trimera and their α-glucosidase inhibitory activities: In vitro and in silico studies. J. Nat. Prod.. 2017;80(7):2151-2155.
- [Google Scholar]
- AGL-Score: Algebraic graph learning score for protein-ligand binding scoring, ranking, docking, and screening. J. Chem. Inf. Model. 2019;59(7):3291-3304.
- [Google Scholar]
- Transition-metal-free decarboxylative iodination: New routes for decarboxylative oxidative cross-couplings. J. Am. Chem. Soc.. 2017;139(33):11527-11536.
- [Google Scholar]
- CompScore: Boosting structure-based virtual screening performance by incorporating docking scoring function components into consensus scoring. J. Chem. Inf. Model. 2019;59(9):3655-3666.
- [Google Scholar]
- Transition-metal-free decarboxylative bromination of aromatic carboxylic acids. Chem. Sci.. 2018;9(15):3860-3865.
- [Google Scholar]
- Green and efficient synthesis of 2-(4-oxo-3,4-dihydroquinazolin-2-yl)-2,3-dihydropthalazine-1,4-dione. Indian J. Chem.. 2015;54B:412-417.
- [Google Scholar]
- Bioactive depsidones from the Fungus Pilobolus heterosporus. Planta Med.. 2014;80(17):1635-1640.
- [Google Scholar]
- A systematic review on anti–diabetic properties of chalcones. Curr. Med. Chem.. 2020;27:2257-2321.
- [Google Scholar]
- Russo, A., Piovano, M., Lombardo, L., Garbarino, J., Cardile, V., Lichen metabolites prevent UV light and nitric oxide-mediated plasmid DNA damage and induce apoptosis in human melanoma cells. Life Sci. 83, 468-474.
- Synthesis and biological evaluation of novel indolo[2,3-c]isoquinoline derivatives. Med. Chem. Res.. 2015;24(4):1681-1695.
- [Google Scholar]
- In vitro antioxidant activities of salazinic acid and its derivative hexaacetyl salazinic acid. Int. J. Res. Eng. Technol.. 2015;4(2):345-355.
- [Google Scholar]
- Shrestha, G., Raphael, J., Leavitt, S.D., Clairm L.L.m 2014. In vitro evaluation of the antibacterial activity of extracts from 34 species of North American lichens. Pharm. Biol. 52(10), 1262–1266.
- Carbazole-linked 1,2,3-triazoles: In vitro β-glucuronidase inhibitory potential, kinetics, and molecular docking studies. Chem. Select.. 2019;4(20):6181-6189.
- [Google Scholar]
- Synthesis, biological evaluation and molecular modeling study of novel tacrine-carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem.. 2014;75(21):21-30.
- [Google Scholar]
- Alpha-glucosidase inhibitory diterpenes from Euphorbia antiquorum growing in Vietnam. Molecules. 2021;26:2257-2264.
- [Google Scholar]
- Glucosidase inhibitory and radical scavenging properties of lichen metabolites salazinic acid, sekikaic acid and usnic acid. Hacettepe J. Biol. Chem.. 2012;40:7-21.
- [Google Scholar]
- Potent pharmacophoric aminothiazole derivatives as FabH inhibitors for antibacterial activity: In vitro and in silico approach. SN Appl. Sci.. 2019;1(11):1375-1396.
- [Google Scholar]
- A critical assessment of docking programs and scoring functions. J. Med. Chem.. 2006;49(20):5912-5931.
- [Google Scholar]
- Synthesis of benzaldehyde and benzoic acid by selective oxidation of benzyl alcohol with Iron(III) tosylate and hydrogen peroxide: A solvent-controlled reaction. Catal. Lett.. 2018;148(10):3082-3092.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103535.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




