

Translate this page into:
Design, synthesis, cytotoxic evaluation and molecular docking of novel 1, 3, 4-thiadiazole sulfonamides with azene and coumarin moieties as carbonic anhydrase inhibitors
⁎Corresponding author at: Chemistry Department, Faculty of Science, King Khalid University, 9004 Abha, Saudi Arabia. bondock@mans.edu.eg (Samir Bondock) bondock@kku.edu.sa (Samir Bondock)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
New thiadiazole sulfonamide derivatives were designed as human carbonic anhydrase inhibitors (hCAIs) to develop robust and novel anticancer agents. Tail modification approach was considered in designing the target compounds which were synthesized following the two-step procedure starting from 5-acetyl-3-N-(4-sulfamoylphenyl)-2-imino-1,3,4-thiadiazoline. Cytotoxic evaluation revealed the potent diazene derivative 2 with IC50 1.18 μM, 5.28 μM and 7.15 μM against MCF-7, Caco2 and HepG-2, respectively. Moreover, the dihydroxyphenyl triazene derivative 5 demonstrated IC50 3.03 μM, 5.66 μM and 12.50 μM against Caco2, HepG-2 and MCF-7, respectively. Similarly, the carbohydrazide coumarin 18 showed IC50 of 2.00 μM and 12.30 μM against Caco2 and HepG2, respectively. Molecular docking using hCAIX and hCAXII were adopted to explain the achieved cytotoxicity on molecular level with their in silico ADME evaluation.
Keywords
Thiadiazole
Benzenesulfonamide
Carbonic anhydrase inhibitors
Anticancer
Molecular docking

1 Introduction
The development of new anticancer agents has been a global hotspot in drug discovery (M. Wu 2022). This leads to an upward trajectory of assembling diverse scaffolds to be synthesized and evaluated as a new generation of target drugs (M. Szumilak 2021). One of the most attractive targets for new anticancer agent development is the carbonic anhydrases isoenzymes. Human carbonic anhydrases (hCAs) are metalloenzymes that control the balance between carbon dioxide and bicarbonate ions through hydration-dehydration mechanism. Their activity required zinc (II) ion as a crucial cofactor to regulate the cellular pH (Supuran 2008, Y. Le Duc 2017). Among its fifteen human isoforms, both transmembranal hCAIX and hCAXII are strongly correlated with cancer prognosis and thus being an interesting target for novel anticancer drugs especially with their established protein structures (Supuran 2010, A. Nocentini 2018). Both CA isoenzymes were reported to be overexpressed in many solid tumors with strong correlation with the encountered hypoxic conditions associated with tumor growth and metastasis (S. K.Chia 2001, M. Kciuk 2022). Moreover, several studies indicated the privilege of using CA inhibitors as co-treatment with other anticancer agents to enhance the responsiveness (C. Ward 2018). An urge to develop more potent and selective inhibitors of the tumor associated isoforms has been of researchers‘ interest (A. Bonardi 2020).
Carbonic anhydrase inhibitors are classified into two main classes according to the presence of zinc binding group (ZBG); classical and non-classical inhibitors. The thiadiazole sulfonamide derivative acetazolamide AAZ is a clinically proven hCA inhibitor that possesses the common structural features of a classical inhibitor (Fig. 1) (Rankin 2007). The classical inhibitors usually bear ZBG primarily sulfonamide moiety attached to a tail group through linker moiety while the non-classical inhibitors bind directly to the enzyme such as polyamines, phenols and coumarin (A. Maresca 2009, C.L. Lomelino 2016).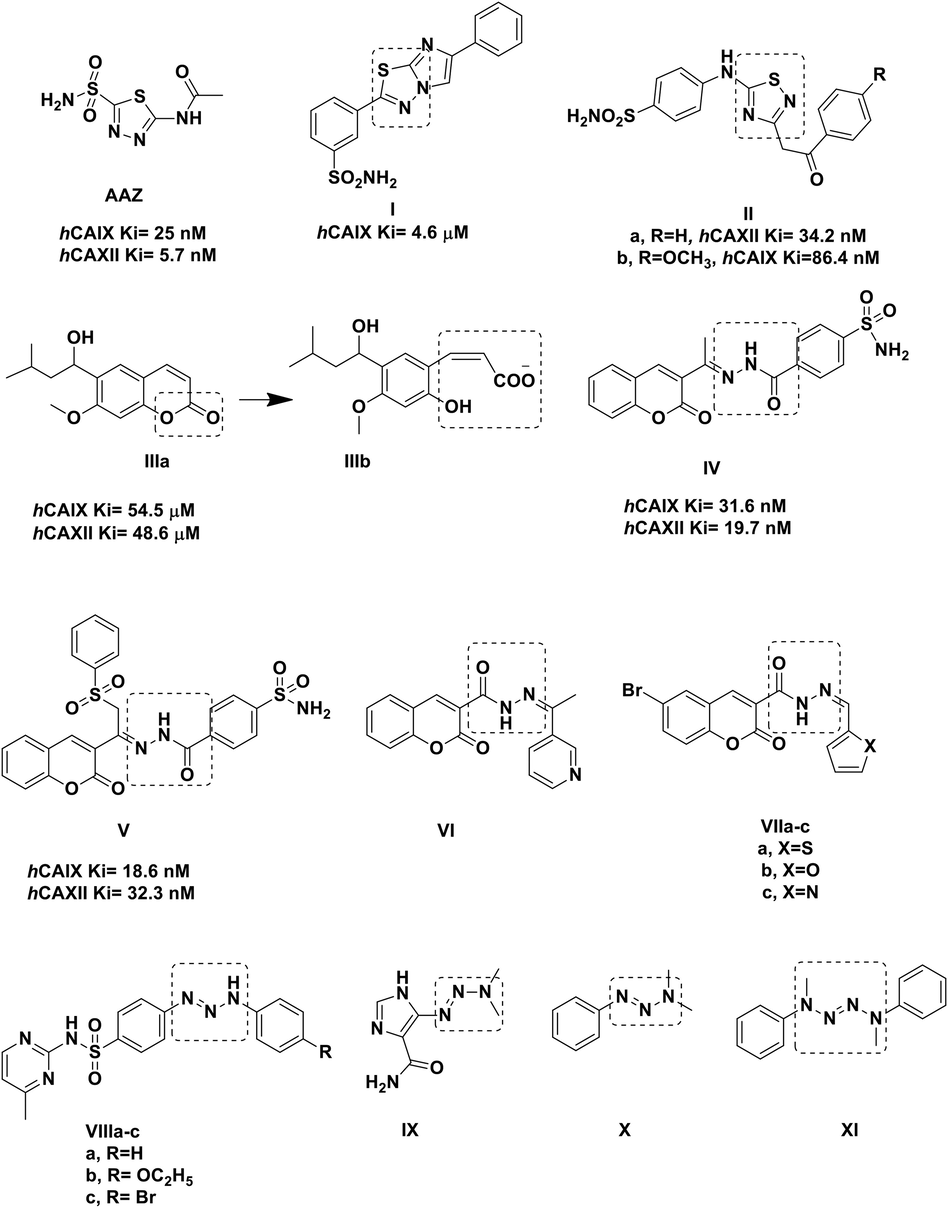
Reported thiadiazole and coumarin derivatives with carbonic anhydrase and anticancer activity.
Hybridizing thiadiazole group with the ZBG benzenesulfonamide (A.M. Soliman 2020, C. Yamali 2021, M. M. Ghorab 2021, M.M. Wassel 2021, S., Bondock. 2021.) was followed in derivatives I-II to design potent classical hCAIs. Compound I exhibited hCAIX Ki of 4.6 μM (A. Swain 2021) while IIa showed hCAXII Ki of 34.2 nM and IIb had hCAIX Ki of 86.4 nM (R. Kumar 2022). On the other hand, the non-classical coumarin-based hCAI IIIa showed selective inhibition towards hCAIX and hCAXII through its hydrolyzed product cis-2-hydroxycinnamic acid derivative IIIb by binding directly to their active sites Asn67 and Phe131 to block their entrance (Fig. 1) (A. Maresca 2009). Therefore, the hydrolyzed product IIIb showed nanomolar inhibitory effect compared to its parent micomolar inhibitory level. Moreover, the 3- substituted coumarin moiety connected to benzenesulfonamide (ZBG) using the hydrazine-carbonyl linkage IV was reported to have appreciated inhibitory activity towards both hCAIX and hCAXII giving Ki 31.6 nM and 19.7 nM, respectively (M. A. Abdelrahman 2021). Similarly, in compound V, adding a phenylsulfonyl group to the linkage moiety of IV enhanced its inhibition towards hCAIX yet, with considerable effect on hCAXII as well (M. A. Abdelrahman 2021) (Fig. 1). In the same context, both thiadiazole core and coumarin derivatives have been acknowledged for their diverse biological activities (A. Lacy 2004, M.A. Musa 2008, A. Thakur 2015, S. Janowska 2020, S. Janowska 2022) specially their anticancer effect. For instance, the 3-substituted coumarin carbohyrdazide derivative VI showed GI50 of 66.8, 10.0 and 36.8 μM for breast MCF-7, lung NCI-H460 and CNS SF-268 cell lines, respectively (R. M. Mohareb 2011). Similarly, the carbohyrdazide coumarin derivatives VIIa and VIIb showed remarkable IC50 against HepG2 of 3.60 and 4.18 μM, respectively while VIIc demonstrated IC50 of 2.02 μM against Pan-1 cell line (T. Nasr 2014).
Additionally, the triazene linkage had been stated in several hCAIs such as VIIIa-c that showed hCAI Ki of 34.3, 10.2 and 101.4 nM, respectively and hCAII Ki 48.1, 40.3 and 18.3 nM, respectively (S. Bilginer 2020). Moreover, several reported anticancer agents included triazene linkage in their structure such as dacarbazine IX and X (A. Gescher 1987). Likewise, tetrazenes motifs had been reported in several anticancer agents such as simtrazene (centrazene) XI (A.Vogel 1963, A.Sloboda 1964).
Based on the aforementioned information we got interested in designing new anticancer agents as classical hCAIs (Fig. 2). The designed compounds were relevant to AAZ as they contain the ZBG, benzenesulfonamide moiety instead of sulfonamide group, directly attached to the thiadiazole linker. The purpose of adding the benzene ring to the benzene sulfonyl moiety is to increase the size of the target compounds to fill the active site of hCAIX and hCAXII to increase their antagonistic activity (A.M. Soliman 2020, C. Yamali 2021, M. M. Ghorab 2021, M.M. Wassel 2021). Moreover, it could form hydrophobic interaction with the active site amino acids. Additionally, we aimed to increase the length of the thiadiazole linker moiety by incorporating hydrazine carbonyl, triazene or tetrazene spacers to increase the size of the designed compounds to fill the active sites of hCAIX and hCAXII consequently increase the inhibitory activity of the target compounds. Specially, triazine moiety was part of a linker in previously reported hCAIs (Fig. 1) (S. Bilginer 2020) and the hydrazine carbonyl moiety was part of a linker in the known hCAIX and hCAXII (Fig. 1) (M. A. Abdelrahman 2021). Additionally, we investigated the effect of replacing the carbonyl moiety of AAZ with its non-classical isostere hydrazine group. Finally, we used different aromatic moieties as a tail part for the purpose of forming pi-pi interactions with the amino acids of the active site of the target enzyme isoforms. Appreciating the conversion of coumarin into its active motif cis-2-hydroxycinnamic acid; different coumarin moieties were included in the tail part to enhance their binding with the crucial residues regardless zinc coordination (A. Maresca 2009).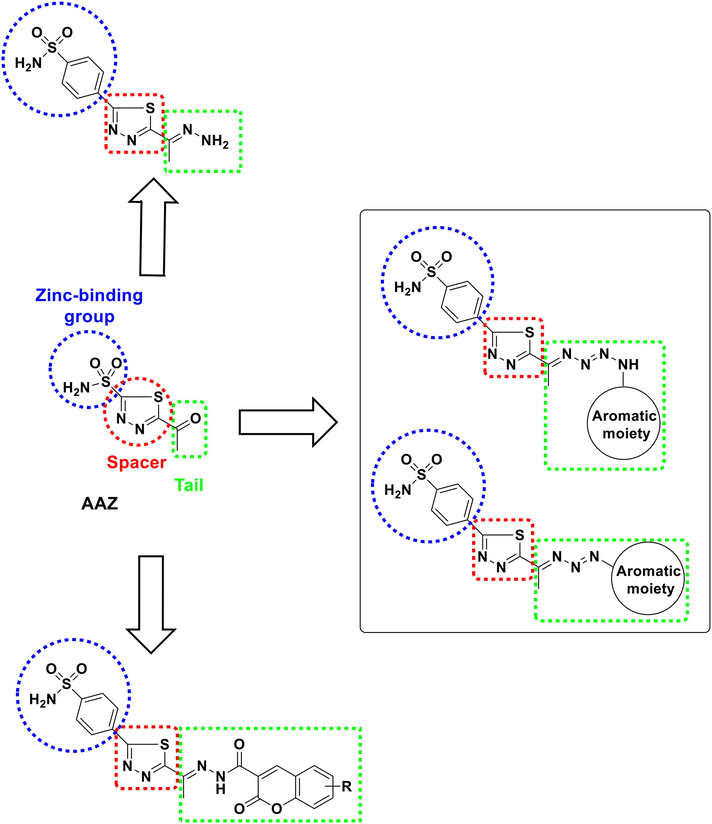
Rationale design of the target compounds.
2 Results and discussion
2.1 Chemistry
The synthetic strategy for constructing the target two series of modified thiadiazole sulfonamides incorporates azene or coumarin fragments (vide Schemes 1, 2).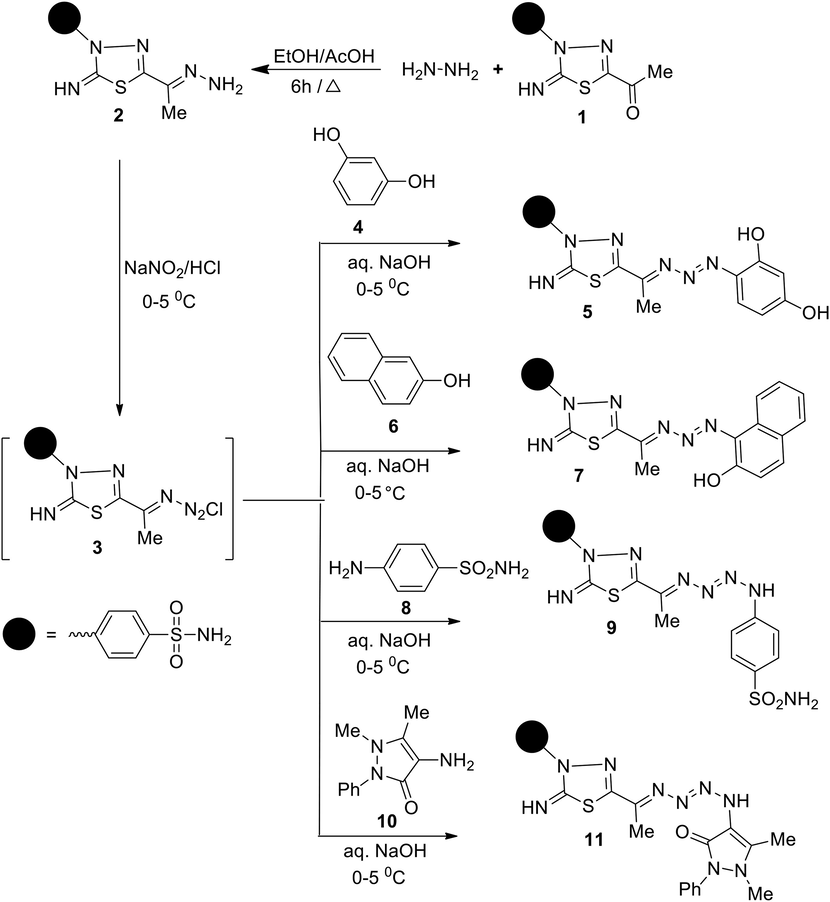
Synthesis of thiadiazole sulfonamides based azene fragments 5, 7, 9 and 11.
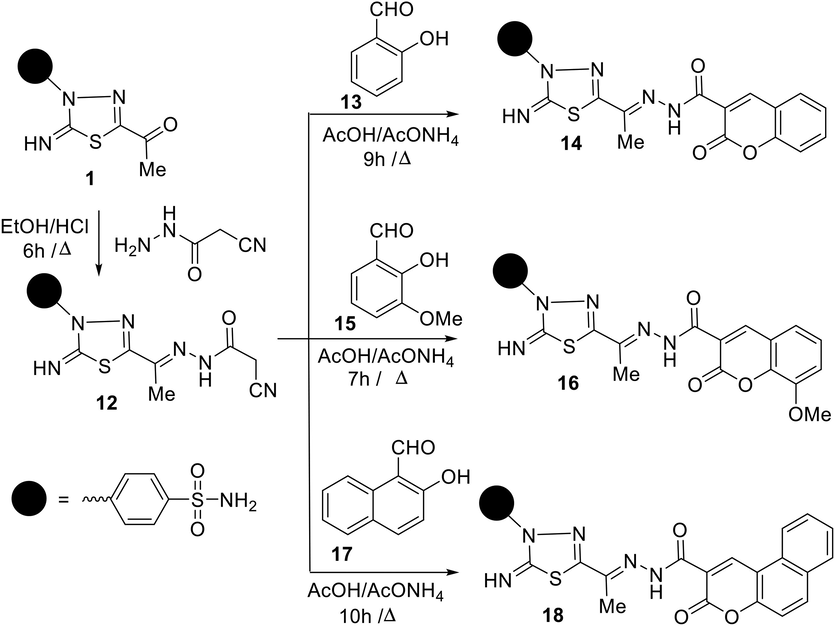
Synthesis of thiadiazole sulfonamides based coumarin fragments (14, 16 and 18).
2.1.1 Synthesis of thiadiazole sulfonamides-based azene fragments (5, 7, 9, and 11)
Initially, the 4-(5-(1-hydrazonoethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide 2 as a commencing material was prepared via the condensation of 1,3,4-thiadiazoline 1 with hydrazine hydrate in boiling ethanol. Subsequently, the hydrazone 2 was subjected to a diazotization reaction with nitrous acid, generated in situ from the treatment of sodium nitrite with HCl, to afford the non-isolable intermediate 4-(5-(1-chlorotriaz-2-enylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide 3. The diversity-oriented synthesis of 1,3,4-thiadiazole incorporating a triazene/tetrazene moiety 5, 7, 9, and 11 were realized using 3 as a precursor. Coupling 3 with 1,3-dihydroxybenzene 4 in a basic medium gave 4-(5-(1-((2,4-dihydroxyphenyl)triaz-2-en-1-ylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfon amide 5. Similarly, the reaction of 3 with 2-hydroxynaphthalene 6 led to 4-(5-(1-((2-hydroxy naphthalene-1-yl)triaz-2-en-1-ylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfon amide 7 (Scheme 1). In this connection, the scope of the reaction of 3 was extended to perform with amines (vide Scheme 1). Asymmetrical tetrazenes appended to 1,3,4-thiadiazole sulfonamides 9 and 11 were efficiently prepared via the treatment of 3 with sulfanilamide 8 and 4-aminoantipyrine 10, respectively, in a basic medium.
The spectral and analytical data provided confirmatory evidence for structures 5 and 7. The IR spectrum of 5 showed absorption bands at 3299–3260, 3107, 1624, 1587, and 1488 cm−1 related to NH2, NH, C = N, conjugated C = C, and N = N functions, respectively. The 1H NMR spectrum of 5 revealed a singlet signal at δ 2.38 ppm due to CH3, two singlet signals at 9.49 and 9.70 ppm due to imine and hydroxy protons, and a multiplet signal at region 7.34–8.50 ppm for the aromatic protons, NH2 and hydroxyl protons. Its 13C NMR spectrum exhibited 14 carbon peaks which agree with its molecular structure. The signals that appeared at δ 13.89, 157.91, and 158.56 ppm were attributed to CH3, C = N, and thiadiazole-C2, respectively. The mass spectra of 5 and 7 showed parent ion peaks at m/z = 433 and 467, respectively, and exhibited several identical peaks at m/z = 324, 296, 255, 197, 172, 156, and 57. For example, the major fragmentation peaks in the MS of compound 15 can be accounted for by the cleavage in the 1,2,3-triazene side chain (Fig. 3).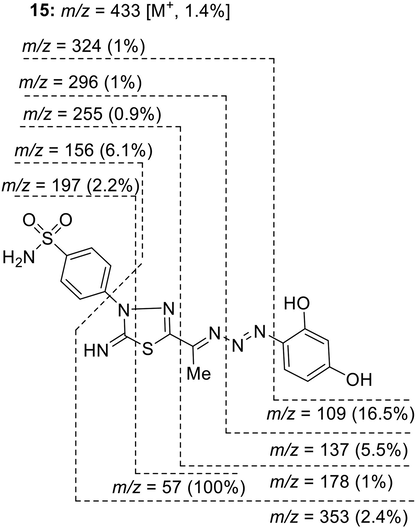
Mass fragmentation pattern of compound 5.
The structures of tetrazene derivatives 9 and 11 were established according to their spectral data. The IR spectrum of 11, as an example, showed characteristic bands at 3303–3249, 3145, 3114, 1686, 1588 and 1488 cm−1 related to NH2, two NH, amidic C = O, C = N, and N = N groups, respectively. The 1H NMR spectrum of compound 11 showed, besides signals for the aromatic proton, six singlet signals at δ 2.45, 2.74, 3.19, 7.41, 9.48, 9.71 ppm to assigned two CH3, CH3N, NH2, imine NH, and NH protons, respectively. Its 13C NMR spectrum displayed the existence of 17 carbon signals. The Sp3-hybridized C-atoms appeared at δ 14.06, 15.56, 38.75 ppm due to three methyl carbons. The characteristic signals of the amidic C = O and the imine carbon (C = NH) resonate at δ 161.87 and 158.56 ppm, respectively. The ESI MS of 11 showed the molecular ion (m/z 526), which is in agreement with a molecular formula (C21H22N10O3S2).
2.1.2 Synthesis of thiadiazole sulfonamides-based coumarin fragments (13, 15, and 17)
The primary step for the preparation of new functionalized 1,3,4-thiadiazole-coumarin conjugates building is the synthesis of 4-(5-(1-(2-(2-cyanoacetyl)hydrazono)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzene sulfonamide 12. Compound 12 was prepared in spectacular yield (88%) from the condensation reaction of 5-acetylthiadiazoline 1 with cyanoacetohydrazide in refluxing ethanol in the presence of hydrochloric acid as a catalyst (Scheme 2). The spectral analyses of compound 12 supported the proposed chemical structure, whereas the IR spectrum exhibited bands at 3303–3251, 3197, 3115, 2270, 1687, 1595 cm−1 attributable to NH2, NH, imine NH, CN, amidic C = O, and C = N functions, respectively. Meanwhile, the 1H NMR spectrum showed, besides the assigned signals of aromatic protons and NH2 protons (δ 7.24–8.25 ppm), four singlet signals at 2.26, 4.14, 9.40, 11.60 ppm assignable to CH3, CH2, imine NH, and amide-NH protons, respectively. After profound explication of the 1H NMR spectrum, the author was rationally able to demonstrate that compound 12 existed as a diastereomeric mixture, due to restricted rotation about azomethine bond (C = N), in the ratio 70:30 (E/Z), which was proved by the existence of extra four singlet signals at δ 2.29, 3.94, 9.42, 11.25 ppm due to CH3, CH2, imine NH, and amide-NH protons for Z-isomer as showed in Fig. 4. Further, the 13C NMR spectrum of 12 displayed the presence of two sets of 11 carbon peaks that support the existence of a mixture of E/Z-diastereoisomers. The signals resonate at about δ 12.18, 24.94, 115.72, 165.88 ppm assigned to CH3, CH2, CN, and amidic C = O carbons, respectively. The preferential formation of the E-isomer may be attributed to its stability due to less steric hindrance and low energy than Z-isomer. Additionally, the assignment of E/Z-isomer is based on comparing the chemical shift values of NH and CH2 protons in amide 12 with their similar compounds reported earlier in the literature (A. Swain 2021).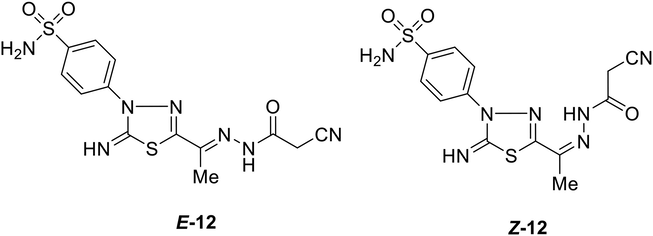
Chemical structures of both E-and Z-isomers of derivative 12.
The conduct of cyanoacetamide moiety of compound 12 towards a variety of substituted 2-hydroxybenzaldehydes was explored as a scaffold for the development of novel coumarins based on 1,3,4-thiadiazole. Thus, the Knoevenagel condensation of amide 12 with each of 2-hydroxybenzaldehyde 13 and 2-hydroxy-3-methyoxybenzaldehyde 15 in boiling acetic acid containing ammonium acetate yielded the respective 4-(2-imino-5-(1-(2-(2-oxo-2H-chromene-3-carbonyl)hydrazono)ethyl)-1,3,4-thiadiazol-3(2H)-yl)benzene sulfonamide 14 and 4-(2-imino-5-(1-(2-(8-methoxy-2-oxo-2H-chromene-3-carbonyl)hydrazono)ethyl)-1,3,4-thiadiazol-3(2H)-yl) benzenesulfonamide 16 (Scheme 2).
The structures of 2-oxo-chromenes 14 and 16 were assured via their correct elemental analyses and spectral data. IR spectra were free of CN group and showed, in each case, characteristic bands at 3339–3239, 3196–3102, 1708–1706, and 1699–1670, 1608–1607 cm−1 assignable to NH2, two NH, coumarin C = O, amidic C = O, and C = N groups, respectively. The 1H NMR spectrum of 14, as a typical example, showed, alongside phenyl proton signals, three singlet signals accompanied proton–deuterium (H/D) exchange at δ 11.96, 9.08, 7.49 ppm due to amidic-NH, imine-NH, and NH2 protons, and two singlets appeared at δ 2.32, 8.70 ppm due to CH3 and chromene-H4, respectively. The highly downfield shift of the amidic-NH proton is attributed to the intramolecular H-bond between the amidic N–H and C = O group of the chromene ring. Its 13C NMR spectrum of 14 revealed the existence of 18C-signals. The Sp3-hybridized carbon atom resonated at δ 12.70 ppm due to the methyl carbon. The signals at δ 153.96, 156.40, 158.81, and 158.92 ppm were assigned to azomethine (C = N), imine carbon (C = NH), amidic C = O, and chromene C = O carbons, respectively. The ESI MS of 14 showed the molecular ion (m/z 484), which agrees with a molecular formula (C20H16N6O5S2). Further, the mass fragmentation pattern of compound 14 is shown in Scheme 3.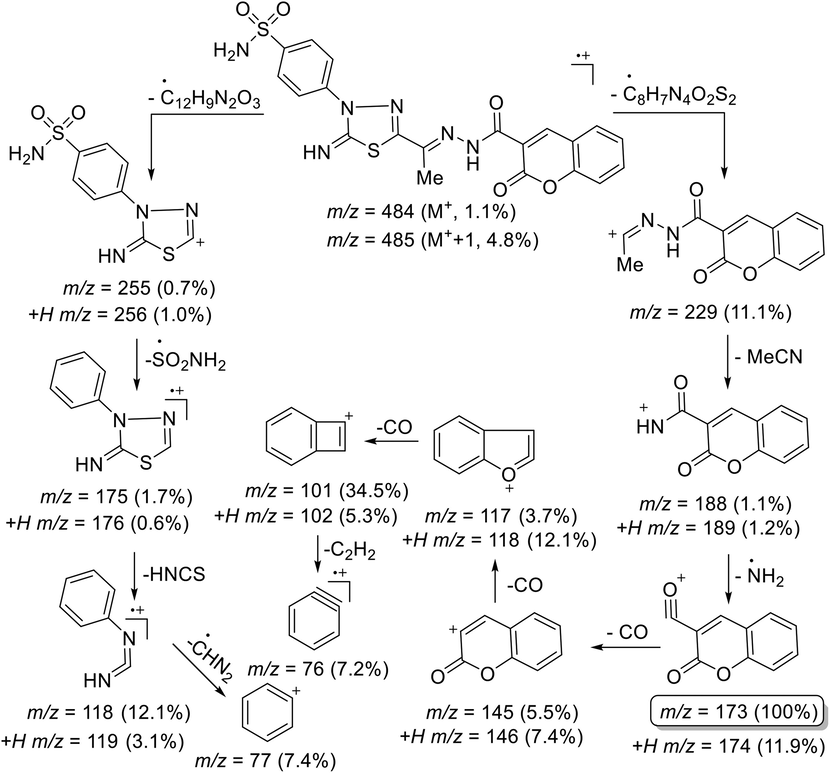
Mass fragmentation pattern of compound 14.
Similarly, treatment of amide 12 with 2-hydroxynapthaldehyde 17 in boiling acetic acid containing ammonium acetate yielded 4-(2-imino-5-(1-(2-(3-oxo-3H-benzo[f]-chromene-2-carbonyl)hydrazono)ethyl)-1,3,4-thiadi-azol-3(2H)-yl)benzene sulfonamide 18 as a sole product (Scheme 2). Elemental analyses and spectrum data were used to confirm the structure of compound 18. The ESI MS of 18 exhibited a parent peak at m/z = 534 (M+) corresponding to its formula (C24H18N6O5S2). The IR spectrum of 18 showed new absorption bands at 3330–3239, 3195–3117, 1701, 1688, and 1584 cm−1 assignable to NH2, two NH, coumarin C = O, amidic C = O, and C = N functions, respectively. Its NMR spectrum exhibited five singlet signals at δ 1.93, 7.66, 8.66, 9.58, 11.95 ppm characteristic for the CH3, NH2, coumarin-H4, imine NH, and amidic NH protons, respectively, alongside aromatic multiplets in the region δ 7.48–8.35 ppm. The 13C NMR spectrum of 18 displayed one methyl carbon peak at δ 12.67 ppm in addition to characteristic amidic (C = O) and coumarin (C = O) peaks at 158.63, 161.30 ppm among a total of 22 carbon signals.
2.2 Bio-evaluation
2.2.1 Cytotoxicity evaluation
Considering the reported overexpression of hCAIX in case of colorectal, gastric, and breast cancers, the cytotoxic potential of the synthesized derivatives 1, 2, 5, 7, 9, 11, 12, 14, 16, and 18 was tested against three different human cancerous cell lines (SK. Chia 2001, J. Chen 2005, AM. Niemela 2007). The tested cell lines were hepatoblastoma cell line HepG-2, colorectal adenocarcinoma cell lines Caco2, breast ductal carcinoma MCF-7 and the diploid lung fibroblast cell lines WI 38 as normal cell line (L. Hayflick 1961) using staurosporine as a reference. Staurosporine is a natural indolocarbazole alkaloid with a proven cytotoxic effect against several cancerous cell lines (J. Boix 1997, A.L. McKeague 2003). The best cytotoxicity was attained by the hydrazonoethyl 2, benzochromene 18 and dihydroxyphenyl 5 derivatives (Table 1 and Fig. 5). Derivative 2 showed better cytotoxicity than staurosporine against HepG-2, Caco2 and MCF-7 with IC50 7.15, 5.28 and 1.18 μM, respectively compared to staurosporine IC50 13.60, 8.18 and 6.19 μM. Similarly, derivative 18, 5 accomplished better HepG-2 and Caco2 growth inhibition than staurosporine in addition to comparable inhibition of WI 38 growth.
Compounds
HepG-2
Caco2
MCF-7
WI 38
IC50 (μM)
±SD
IC50 (μM)
±SD
IC50 (μM)
±SD
IC50 (μM)
±SD
Staurosporine
13.60
0.80
8.18
0.70
6.19
0.30
25.20
1.30
1
59.60
3.40
11.10
0.60
14.90
0.70
91.50
4.70
2
7.15
0.40
5.28
0.30
1.18
0.10
35.00
2.30
5
5.66
0.30
3.03
0.20
12.50
0.60
28.30
1.50
7
14.16
0.92
16.11
1.42
10.17
1.11
65.82
4.35
9
9.12
0.62
12.19
1.32
37.12
1.68
49.90
3.68
11
47.80
2.70
30.40
1.50
29.80
1.30
49.90
2.60
12
51.00
2.90
18.60
0.90
45.60
2.10
72.80
3.80
14
37.20
2.10
25.40
1.30
39.50
1.80
149.00
7.70
16
19.30
1.10
14.80
0.70
10.00
0.40
47.10
2.40
18
12.30
0.70
2.00
0.40
42.60
2.30
25.60
1.30
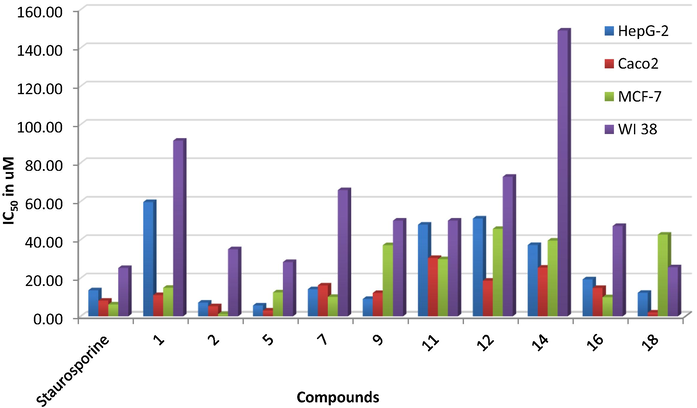
The in vitro cytotoxic evaluation of derivatives 1, 2, 5, 7, 9, 11, 12, 14, 16, and 18 using four human cancerous cell lines presented as IC50 in μM.
Structurally, changing the acetyl terminus 1 with an unsubstituted diazene 5 significantly improved the cytotoxic activity among the three tested cancerous cell lines (Fig. 5). However, adding cyanoacetyl moiety 12 to the free diazene group dropped its cellular growth inhibitory potential over the four cell lines. Moreover, a slight reduction of cytotoxic activity was established upon adding benzochromene moiety to the diazene tail 18, yet; it still showed better cytotoxic activity than staurosporine. Comparably, the 8-methoxy chromene 16 exhibited better cytotoxic activity against the three cancerous cell lines than its parent unsubstituted chromene 14 tail (Fig. 6). In contrast, switching the acetyl terminus of 1 into a triazene linkage connected to a phenolic moiety like dihydroxyphenyl 5 or hydroxynaphthalene 7 improved its cytotoxicity among the three cancerous cell lines. Nonetheless, the dihydroxyphenyl derivative 5 displayed better activity than its hydroxyphenyl analogue 7 almost 2–5 times amongst the tested cell lines. In the same context, the tetrazene containing tails 9 and 11 displayed improved cytotoxicity in the case of HepG-2 than their acetyl parent derivative 1. Furthermore, additional benzenesulfonamide moiety 9 enhanced the cytotoxic effect of the tetrazene analogues in HepG-2 and Caco2 without significant change on WI 38 in contrast to its heterocyclic benzopyrazole analogue 10. Further explanation of those derivatives' binding conformation was discussed in the molecular docking section.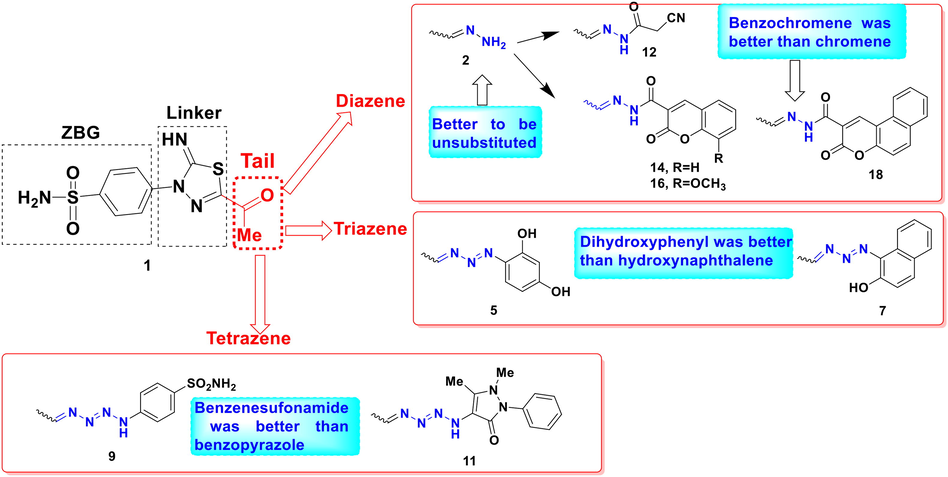
Schematic representation of structure activity relationship of derivatives 1, 2, 5, 7, 9, 11, 12, 14, 16, and 18 based on their in vitro cancerous cell lines cytotoxicity.
2.2.2 In silico molecular docking discussion
To further investigate the achieved in vitro cancerous cell lines inhibition results, molecular docking was used to test derivatives 1, 2, 5, 7, 9, 11, 12, 14, 16, and 18 abilities to inhibit the human tumor-associated carbonic anhydrase (hCA) isoforms IX and XII. The X-ray crystals of the tumor-associated hCAIX and hCAXII were downloaded from the Protein Data Bank https://www.rcsb.org using PDB: 3IAI (V. Alterio 2009) and 1JD0 (D.A. Whittington 2001) for hCAIX and hCAXII, respectively. The molecular docking protocol was validated before commencing the actual docking of 1, 2, 5, 7, 9, 11, 12, 14, 16, and 18 by self-docking the corresponding co-crystallized ligand AAZ to get the lowest RMSD value with the same interaction pattern with the lowest possible binding energy score. The achieved RMSD of hCAIX was 0.37 Å with an energy score of −8.83 Kcal/mol in contrast to 0.96 Å RMSD of hCAXII with anenergy score of −9.01 Kcal/mol (Fig. S1 at supplementary material).
The achieved molecular docking results of the targeted thiadiazole derivatives revealed the capability of the tested derivatives to coordinate with zinc inside both hCA isoforms catalytic sites by their oxygen and/or deprotonated nitrogen of the sulfonamide moiety (Figs. 7-12 and table S1 at supplementary material). The terminal sulfonamide amine was reported to be deprotonated at the physiological pH exposing a negative charge to coordinate with the positive zinc ion and hindering its cofactor role (K. Kanamori 1983, N. Chiaramonte 2018). The produced zinc coordination of the evaluated derivatives had an average distance of 1.90–3.21 Å. Moreover, all derivatives attained binding energy scores surpassed the co-crystallized AAZ with the best values achieved by 9 and 16 for hCAIX and hCAXII, respectively (Table 2), which translated into strong inhibitory effect on HepG2, Caco2 and MCF-7 (Table 1). Furthermore, the aliphatic tailed 11 had the privilege over AAZ to form H-bonds with one of the catalytic histidine triad His94 of hCAIX with a distance 3.48 Å (Table S1) (Supuran 2010). Interestingly, all derivatives formed one or more H-bonds with the active site Thr199 of both isoforms with a maximum distance of 3.10 Å in a similar way to AAZ. Similarly, derivatives 9, 12, 14, 16 and 18 formed H-bonds and hydrophobic interaction with Leu198 with an average bond distance of 3.8 Å in the case of hCAIX. Comparably, all derivatives formed a hydrogen bond with Leu198 in the case of hCAXII with maximum bond distance of 3.59 Å (Table 2 and Table S1 at supplementary material).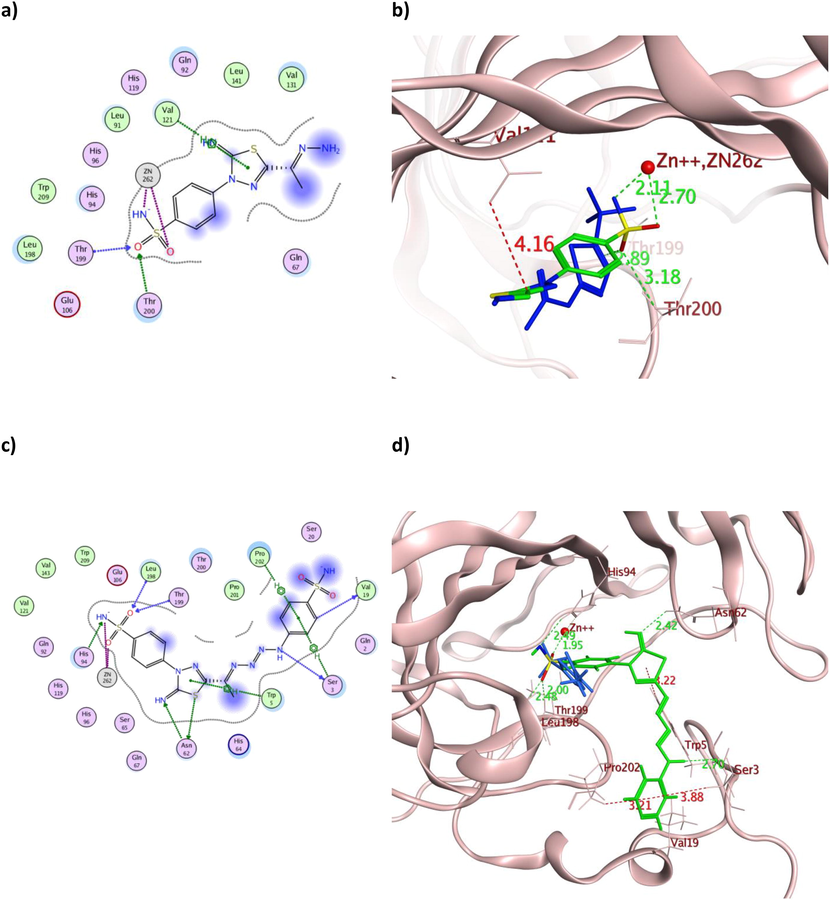
The 2D and 3D presentation of derivatives 2 (a and b) and 9 (c and d) binding conformations to hCAIX using PDB:3IAI. The tested derivatives and the co-crystallized AAZ appeared as a green and blue stick model, respectively, with the H-bonds, illustrated as a dotted green line. In contrast, the hydrophobic interactions appeared as a red dotted line with their corresponding distance in Å.
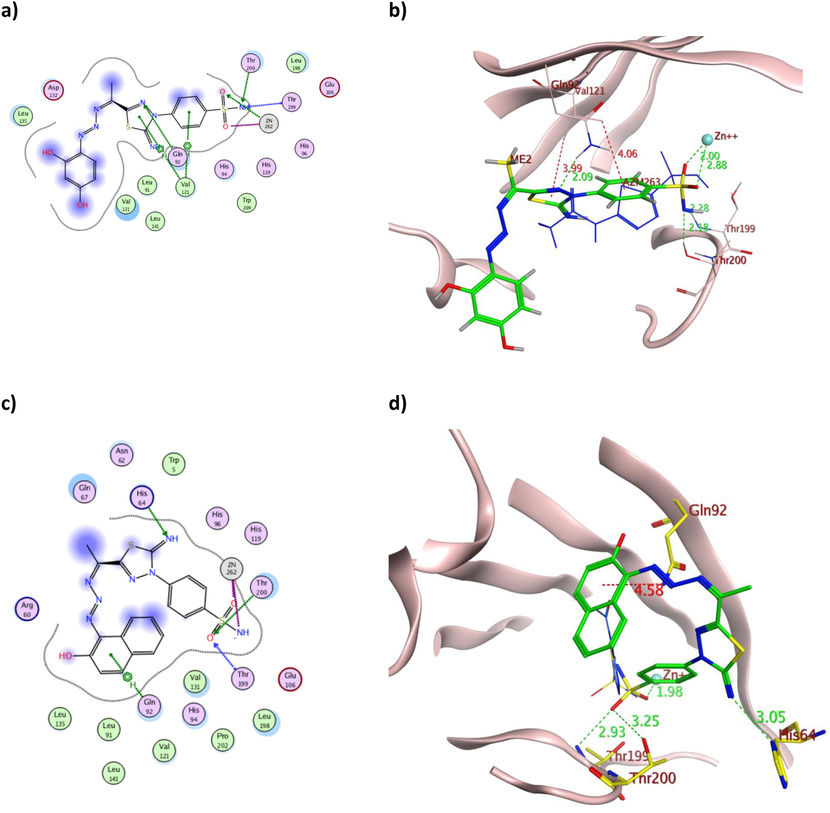
The 2D and 3D presentation of derivatives 5 (a and b) and 7 (c and d) binding conformations to hCAIX using PDB:3IAI. The tested derivatives and the co-crystallized AAZ appeared as a green and blue stick model, respectively, with the H-bonds, illustrated as a dotted green line. In contrast, the hydrophobic interactions appeared as a red dotted line with their corresponding distances in Å.
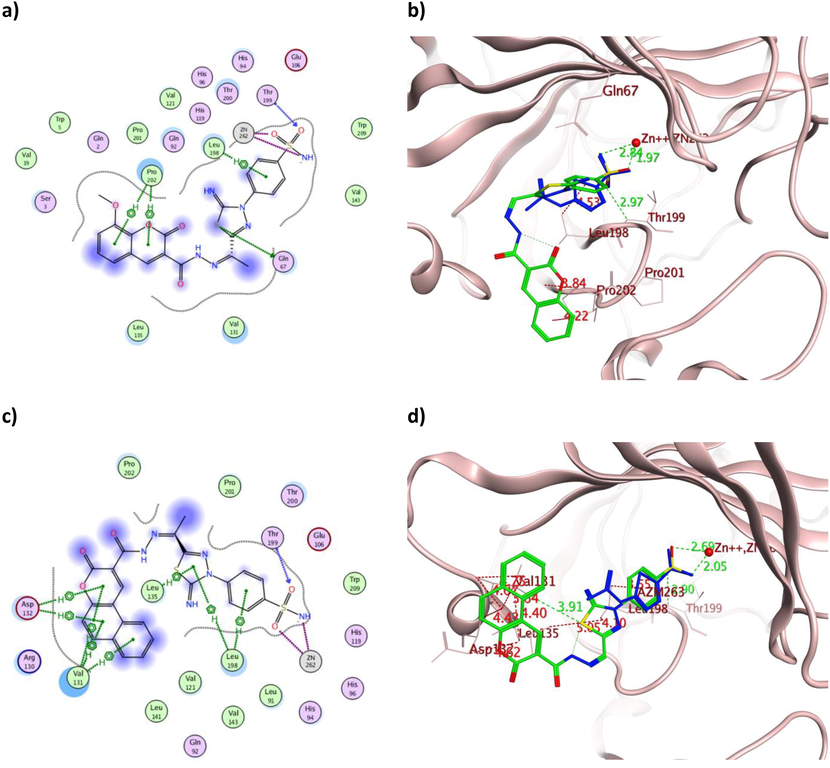
The 2D and 3D presentation of derivatives 16 (a and b) and 18 (c and d) binding conformations to hCAIX using PDB:3IAI. The tested derivatives and the co-crystallized AAZ appeared as a green and blue stick model, respectively, with the H-bonds, illustrated as a dotted green line. In contrast, the hydrophobic interactions appeared a red dotted line with their corresponding distances in Å.
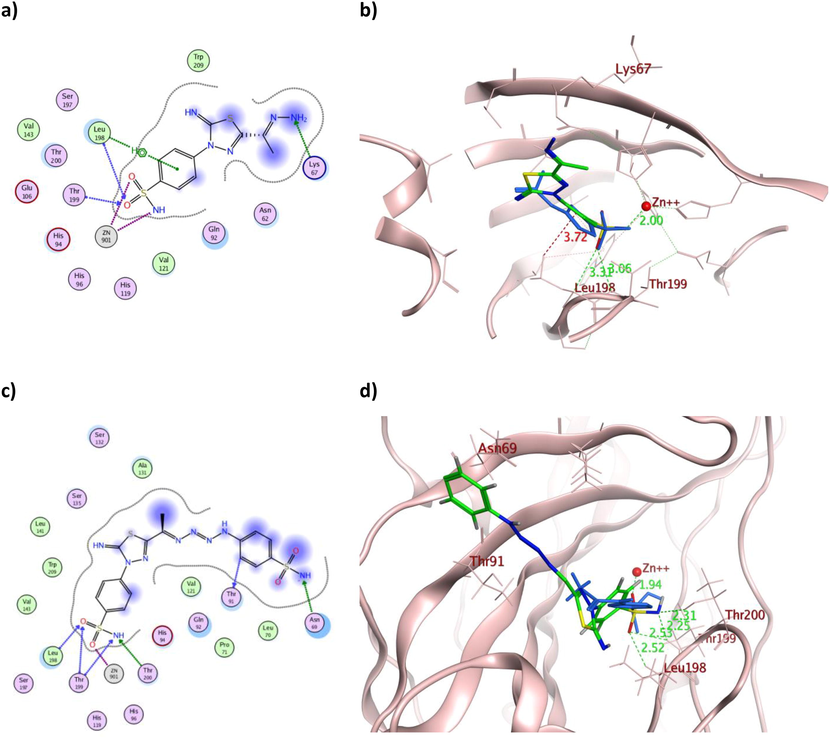
The 2D and 3D presentation of derivatives 2 (a and b) and 9 (c and d) binding conformations to hCAXII using PDB:1JD0. The tested derivatives and the co-crystallized AAZ appeared as a green and blue stick model, respectively, with the H-bonds, illustrated as a dotted green line. In contrast, the hydrophobic interactions appeared as a red dotted line with their corresponding distance in Å.
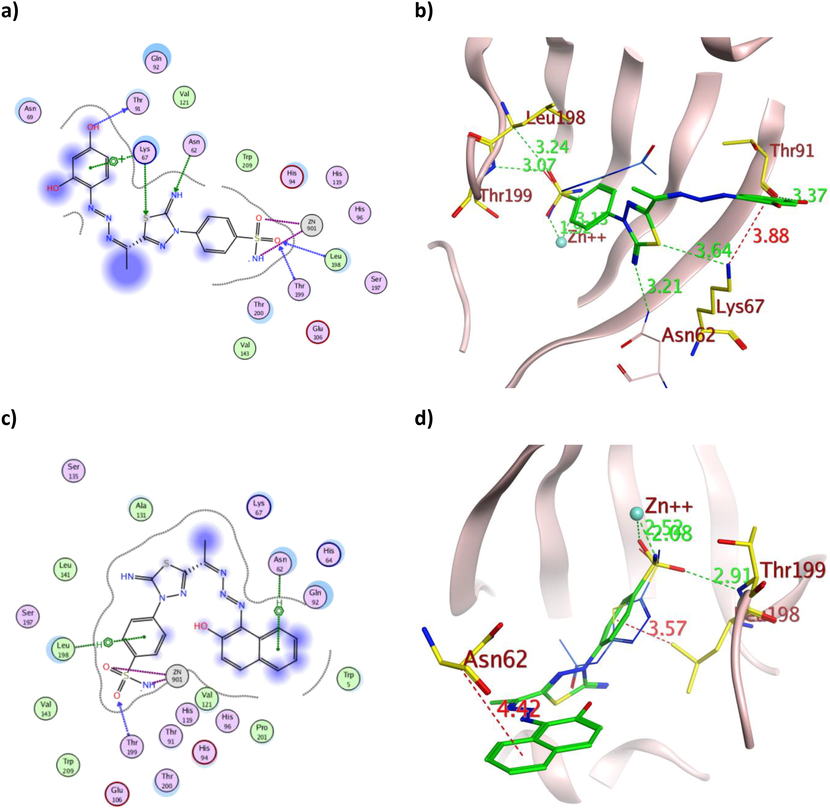
The 2D and 3D presentation of derivatives 5 (a and b) and 7 (c and d) binding conformations to hCAXII using PDB:1JD0. The tested derivatives and the co-crystallized AAZ appeared as a green and blue stick model, respectively, with the H-bonds illustrated as a dotted green line. In contrast, the hydrophobic interactions appeared as a red dotted line with their corresponding distance in Å.
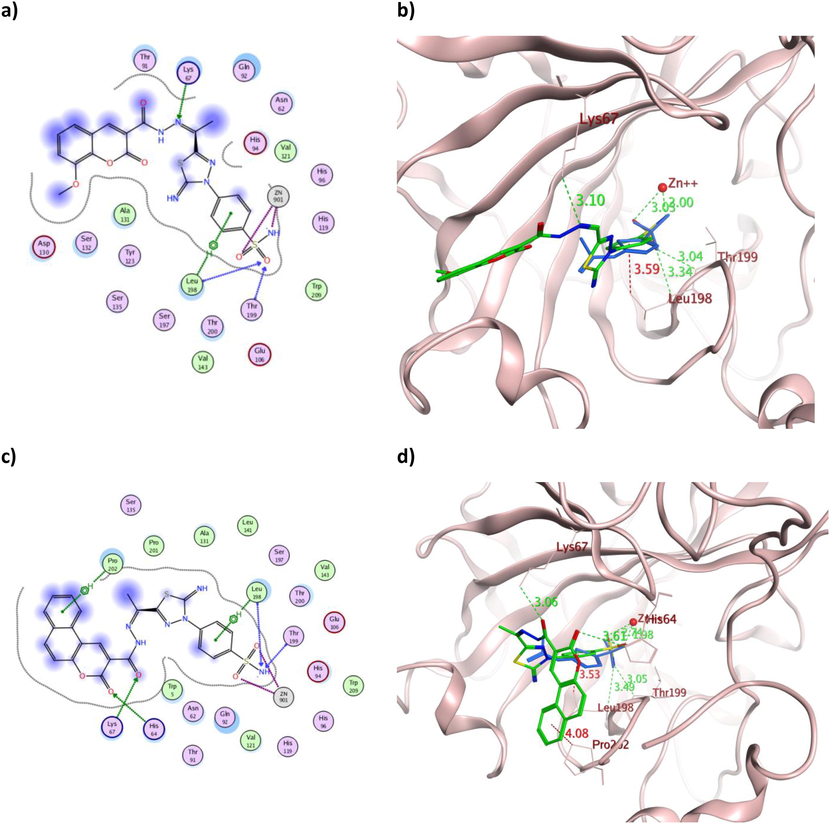
The 2D and 3D presentation of derivatives 16 (a and b) and 18 (c and d) binding conformations to hCAXII using PDB:1JD0. The tested derivatives and the co-crystallized AAZ appeared as a green and blue stick model, respectively, with the H-bonds, illustrated as a dotted green line. In contrast, the hydrophobic interactions appeared as a red dotted line with their corresponding distance in Å.
hCAIX
hCAXII
Cpd
Binding energy (Kcal/mol)
Interacting residues
Interaction type
Distance (Å)
Energy (Kcal/mol)
Binding energy (Kcal/mol)
Interacting residues
Interaction type
Distance (Å)
Energy (Kcal/mol)
AAZ
−8.83
Thr
200
H-acceptor
2.84
−1.60
−9.01
Thr
200
H-acceptor
2.81
−1.10
Thr
199
H-acceptor
2.85
−4.90
Thr
199
H-acceptor
2.96
−4.30
Zn
262
ionic
2.05
−15.30
Zn
901
ionic
1.96
−17.00
Zn
262
ionic
2.88
−5.40
Zn
901
ionic
3.03
−4.30
Leu
198
pi-H
4.21
−0.70
Leu
198
pi-H
3.52
−0.60
Leu
198
pi-H
4.34
−0.60
2
−10.59
Thr
199
H-acceptor
2.89
−1.60
−10.12
Lys
67
H-acceptor
3.19
−3.20
Thr
200
H-acceptor
3.18
−1.30
Leu
198
H-acceptor
3.31
−0.60
Zn
262
ionic
2.70
−6.80
Thr
199
H-acceptor
3.06
−3.80
Zn
262
ionic
2.11
−14.20
Zn
901
ionic
3.09
−3.90
Val
121
pi-H
4.16
−0.80
Zn
901
ionic
2.00
−16.10
Leu
198
pi-H
3.72
−0.40
5
−12.03
Gln
92
H-acceptor
3.04
−1.00
−11.29
Thr
91
H-donor
3.37
−1.00
Zn
262
H-acceptor
2.88
−0.40
Lys
67
H-acceptor
3.64
−0.70
Thr
199
H-acceptor
2.99
−1.50
Asn
62
H-acceptor
3.21
−0.50
Thr
200
H-acceptor
3.10
−3.90
Leu
198
H-acceptor
3.24
−0.50
Zn
262
ionic
2.00
−16.10
Thr
199
H-acceptor
3.07
−3.90
Val
121
pi-H
3.99
−1.00
Zn
901
ionic
1.92
−17.70
Val
121
pi-H
4.06
−0.20
Zn
901
ionic
3.13
−3.70
Lys
67
pi-cation
3.88
−0.50
7
−11.27
His
64
H-acceptor
3.05
−6.40
−11.01
Thr
199
H-acceptor
2.91
−4.90
Thr
199
H-acceptor
2.93
−2.00
Zn
901
ionic
2.52
−8.60
Thr
200
H-acceptor
3.25
−1.20
Zn
901
ionic
2.08
−14.70
Zn
262
ionic
1.98
−16.60
Asn
62
pi-H
4.42
−0.60
Zn
262
ionic
3.21
−3.20
Leu
198
pi-H
3.57
−0.30
Gln
92
pi-H
4.58
−0.60
9
−12.26
Ser
3
H-donor
3.70
−0.40
−11.60
Asn
69
H-acceptor
3.45
−2.00
Val
19
H-donor
3.56
−0.20
Thr
199
H-acceptor
2.53
−1.10
Asn
62
H-donor
3.88
0.00
Thr
199
H-acceptor
2.25
−5.30
Asn
62
H-acceptor
2.42
−1.80
Thr
200
H-acceptor
2.31
−1.90
Leu
198
H-acceptor
2.48
−0.30
Thr
91
H-donor
3.31
−0.30
Thr
199
H-acceptor
2.00
−4.60
Zn
901
ionic
1.94
−17.5
His
94
H-acceptor
2.49
−0.80
Leu
198
H-acceptor
2.52
−0.50
Zn
262
ionic
1.95
−17.20
Zn
262
ionic
2.49
−6.50
Ser
3
pi-H
3.88
−0.20
Trp
5
pi-H
2.70
−0.60
Pro
202
pi-H
3.21
−0.20
16
−12.02
Gln
67
H-donor
4.12
0.00
−12.46
Lys
67
H-acceptor
3.10
−0.60
Thr
199
H-acceptor
2.97
−4.70
Leu
198
H-acceptor
3.34
−0.60
Zn
262
ionic
1.97
−16.80
Thr
199
H-acceptor
3.04
−4.00
Zn
262
ionic
2.84
−5.70
Zn
901
ionic
3.03
−4.30
Leu
198
pi-H
4.53
−0.20
Zn
901
ionic
2.00
−16.20
Pro
202
pi-H
4.22
−0.40
Leu
198
pi-H
3.59
−0.60
Pro
202
pi-H
3.84
−0.50
18
−12.02
Thr
199
H-acceptor
2.90
−5.00
−11.97
Lys
67
H-acceptor
3.06
−8.50
Zn
262
ionic
2.69
−6.90
His
64
H-acceptor
3.61
−0.50
Zn
262
ionic
2.05
−15.20
Leu
198
H-acceptor
3.49
−0.50
Val
131
pi-H
4.07
−0.60
Thr
199
H-acceptor
3.05
−9.70
Val
131
pi-H
3.64
−0.40
Zn
901
ionic
2.74
−6.50
Val
131
pi-H
4.40
−0.30
Zn
901
ionic
1.98
−16.60
Asp
132
pi-H
4.43
−0.80
Leu
198
pi-H
3.53
−0.30
Asp
132
pi-H
4.62
−0.30
Pro
202
pi-H
4.08
−0.20
Leu
135
pi-H
5.05
−0.40
Leu
198
pi-H
4.10
−0.30
Leu
198
pi-H
3.55
−0.40
On the other hand, molecular docking managed to explain the astonishing cytotoxic effect of 2 with the hydrazone terminus on the tested cancerous cell lines, which in addition to its zinc coordination, one of its sulfonamide oxygen atoms accepted H-bonds from the active site Thr199 in both isoforms with additional H-bond formation with Thr200 and Leu198 for hCAIX and hCAXII, respectively (Fig. 7a-b and 10a-b). Those formed H-bonds could fix the molecule orientation in proximity to zinc and stabilize the formed coordination. Meanwhile, additional hydrophobic interaction was formed with Val121 in the case of hCAIX by its thiazole and Leu198 in the case of hCAXII by the phenyl moiety. Those extra hydrophobic interactions might assist in preserving 2 favored structural orientations inside the active site of both isoforms. Likewise, 9 with the tetraaz-2-ene benzenesulfonamide tail showed the usual zinc coordination along with multiple H- bond formation with eight and five residues of the active sites of hCAIX and hCAXII, respectively (Fig. 7c-d and 10c-d). Its calculated binding energy was −12.26 Kcal/mol and −11.60 Kcal/mol for hCAIX and hCAXII, respectively, which exceeded AAZ and could justify its interesting in vitro IC50 values.
The triazene containing derivatives 5 and 7 demonstrated hydrogen bond formation with the crucial Thr199 of both isoforms with a maximum bond distance of 3.07 Å (Table 2), similar to AAZ. Moreover, they formed additional hydrogen bonds with hCAIX Thr200 and Gln92 (Fig. 8a and 7c) with the overall binding energy of −12.03 and −11.27 Kcal/mol for 5 and 7, respectively. The astonishing IC50 values of 5 over 7 on the tested cell lines were rationalized by its multiple interactions and proper positioning within the binding sites of both hCA isoforms (Figs. 8 and 11). Furthermore, the dihydroxyphenyl moiety of 5 formed H-bond and arene-cation interaction with Thr91 and Lys67, respectively (Fig. 11a-b). In contrast, the hydroxynaphthalene moiety of 7 formed only one arene-H bond with Asn62 of slightly weak energy −0.60 Kcal/mol and 4.42 Å distance (Fig. 11c-d). Additionally, the binding energy of 5 was better than 7 in both isoforms, which matched the difference in their in vitro cytotoxicity.
Comparable binding energy to both hCA isoforms was achieved by 16 and 18 with an average of −12.02 Kcal/mol, which translated into analogous in vitro IC50 values. Apart from the established zinc coordination, the aromatic moieties of 16 formed three hydrophobic bonds with the active site Leu198 and Pro202 (Fig. 9a-b) and one with Leu198 (Fig. 12a-b) in the case of hCAIX and hCAXII, respectively. In contrast, 18 showed several hydrophobic interactions with Val131, Asp132, Leu135 and Leu198 in the case of hCAIX (Fig. 9c-d) and with Leu198 and Pro202 in the case of hCAXII (Fig. 12c-d). Other hydrogen bonds were formed between the sulfonamide moiety of 18 and Thr199 in both isoforms, with additional interaction between its carbonyl and His64 and Lys67 in hCAXII. The illustrated multiple interactions of 18 over 16 with comparable binding energy in the case of both hCA isoforms were extrapolated into better IC50 values of HepG-2, Caco2 and WI38.
To evaluate the possibility of the synthesized derivatives to interact with hCAI (Y. Takaoka 2013) and hCAII (A.H. Robbins 2009) as off-targets, molecular docking simulation was done for further investigation. The achieved simulation results demonstrated that despite possessing better binding energy than AAZ, the synthesized derivatives showed unfavorable interaction pattern (Table 3). The typical zinc coordination in both isoforms with sulfonamide terminus was observed among the derivatives with average distance 2.44 Å. Nonetheless, derivatives 2 and 18 with the highest cytotoxic activity formed H-bond with the crucial Thr199 in case of hCAI (Fig. 13a and 13b). On the other hand, both derivatives only demonstrated zinc coordination without further interactions with hCAII binding site residues (Fig. 13c and 13d). Consequently, the most potent cytotoxic derivatives 2 and 18 were expected to exhibit low off-target interactions.
Compound
Binding energy (Kcal/mol)
Ligand
hCAI
Binding energy (Kcal/mol)
ligand
hCAII
Interacting residues
Interaction type
Distance (Å)
Energy (Kcal/mol)
Interacting residues
Interaction type
Distance (Å)
Energy (Kcal/mol)
AAZ
−9.03
O3
Gln
92
H-acceptor
2.63
−3.00
−8.74
O1
Thr
199
H-acceptor
2.95
−4.50
O1
Zn
302
ionic
2.17
−13.30
N1
Zn
301
ionic
2.14
−13.80
O2
Zn
302
ionic
2.63
−7.50
O2
Zn
301
ionic
2.66
−7.20
5-ring
Leu
198
pi-H
3.62
−0.60
5-ring
His
200
pi-pi
3.64
0.00
1
−12.27
N
His
67
H-acceptor
3.26
−2.80
−11.81
O
Thr
199
H-acceptor
3.03
−4.10
O
Thr
199
H-acceptor
2.88
−4.80
N
Zn
301
ionic
3.14
−3.60
O
Zn
302
ionic
1.85
−19.30
O
Zn
301
ionic
1.87
−18.90
2
−10.7
O
Thr
199
H-acceptor
2.93
−4.50
−10.36
O
Zn
301
ionic
2.87
−5.40
O
Zn
302
ionic
1.93
−17.60
O
Zn
301
ionic
1.94
−17.40
N
Zn
302
ionic
2.84
−5.70
5
−11.23
N
Thr
199
H-acceptor
3.08
−8.80
−10.33
O
Thr
199
H-acceptor
2.99
−3.20
O
Zn
302
ionic
1.99
−16.40
N
Zn
301
ionic
2.09
−14.60
O
Zn
302
ionic
2.87
−5.40
7
−11.93
O
Thr
199
H-acceptor
2.90
−4.70
−11.07
O
Thr
199
H-acceptor
2.88
−3.40
O
Zn
302
ionic
2.56
−8.20
O
Zn
301
ionic
2.49
−8.90
N
His
200
ionic
3.73
−1.10
N
Zn
301
ionic
2.28
−11.60
N
Zn
302
ionic
2.12
−14.10
9
−12.32
O
Lys
57
H-acceptor
3.27
−2.30
−12.17
O
Zn
301
ionic
1.94
−17.40
O
Thr
199
H-acceptor
2.89
−4.80
O
Zn
301
ionic
2.95
−4.80
O
Lys
57
ionic
3.27
−2.90
O
Arg
58
ionic
3.82
−0.90
O
Lys
57
ionic
3.87
−0.80
O
Arg
58
ionic
3.21
−3.20
N
Lys
57
ionic
3.12
−3.70
O
Arg
58
ionic
3.73
−1.10
O
Zn
302
ionic
3.35
−2.50
O
Arg
58
ionic
3.49
−1.90
N
Zn
302
ionic
2.05
−15.30
6-ring
His
200
pi-pi
3.91
0.00
11
−12.87
O
Thr
199
H-acceptor
2.89
−4.70
−12.89
O
Zn
301
ionic
2.91
−5.10
O
Zn
302
ionic
2.54
−8.40
O
Zn
301
ionic
1.95
−17.30
N
His
200
ionic
3.55
−1.70
N
Zn
302
ionic
2.22
−12.40
12
−11.78
N
His
67
H-acceptor
2.95
−2.60
−10.85
N
Asn
67
H-acceptor
3.15
−3.70
O
Zn
302
ionic
1.90
−18.20
O
Zn
301
ionic
1.93
−17.70
O
Zn
302
ionic
3.28
−2.90
O
Zn
301
ionic
3.05
−4.10
14
−12.03
N
Thr
199
H-acceptor
3.13
−7.40
−11.69
O
Thr
199
H-acceptor
2.88
−4.90
O
Zn
302
ionic
1.91
−18.00
O
Zn
301
ionic
2.00
−16.20
O
Zn
302
ionic
3.22
−3.20
N
Zn
301
ionic
2.77
−6.20
16
−12.91
O
Thr
199
H-acceptor
2.91
−4.70
−12.25
N
Leu
198
H-acceptor
3.36
−0.70
O
Zn
302
ionic
3.12
−3.70
O
Zn
301
ionic
2.89
−5.30
N
Zn
302
ionic
2.06
−15.10
O
Zn
301
ionic
1.96
−16.90
18
−11.68
N
His
67
H-acceptor
3.11
−1.00
−11.69
O
Zn
301
ionic
2.84
−5.70
O
Thr
199
H-acceptor
2.97
−4.20
O
Zn
301
ionic
1.97
−16.70
O
Zn
302
ionic
1.96
−17.00
N
Zn
302
ionic
2.77
−6.20
5-ring
Gln
92
pi-H
4.14
−2.10
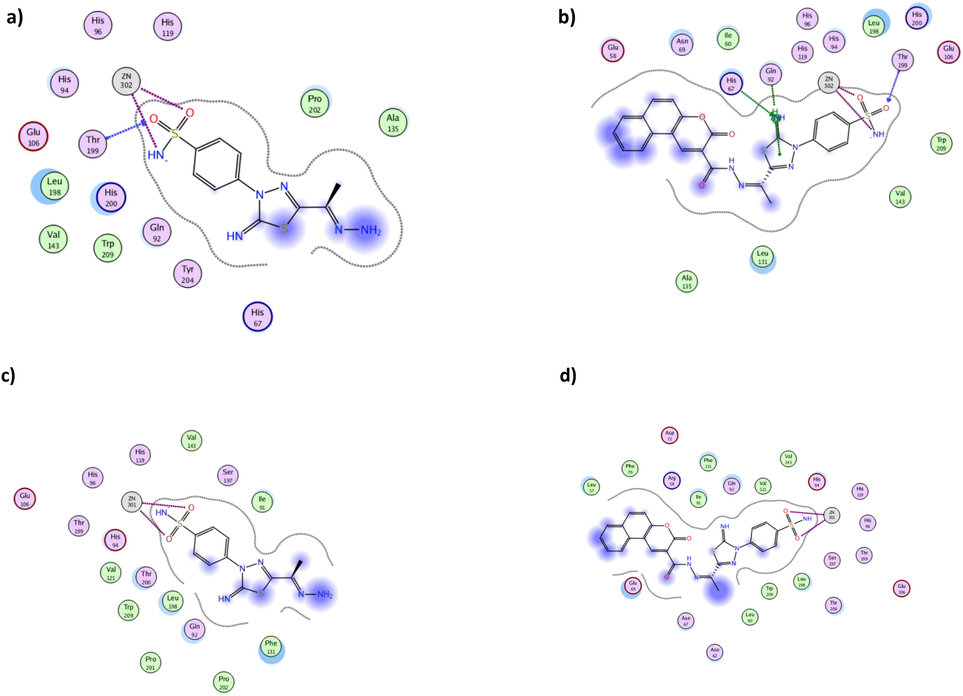
The 2D interactions of derivatives 2 (a, c) and 18 (b, d) using hCAI (PDB 3W6H) and hCAII (PDB 3HS4), respectively.
2.2.3 Physicochemical, pharmacokinetics properties and ADME prediction
The calculated physicochemical descriptors predicted pharmacokinetics properties and drug-likeness of 1, 2, 5, 7, 9, 11, 12, 14, 16, and 18 were determined by the Swiss Institute Bioinformatics free web SwissADME® (Table 4) (Bioinformatics 2017). As stated in Table 4, the calculated physicochemical values of the tested derivatives complied with Lipinski‘s rule of five, and thus these compounds could be administered orally (Lipinski. 2004). Fortunately, all the tested derivatives demonstrated predicted Log P(o/w) values less than 5; however, their topological polar surface area TPSA exceeded 120 Å2 which was reflected in their low gastrointestinal absorption and skin permeation (Kelder 1999, S. J. D. Veber 2002). Luckily, these TPSA values prevented their blood–brain barrier penetration, which could avoid undesirable CNS side effects. Moreover, all derivatives were unaffected by the drug efflux pump P-glycoprotein or the hepatic cytochrome enzymes.
Compounds
1
2
9
5
7
11
12
14
16
18
Physicochemical properties
Molecular weight in g/mole
298.3
312.4
495.6
433.5
467.5
526.6
379.4
484.5
514.5
534.6
Number of heavy atoms
19
20
32
29
32
36
25
33
35
37
Number of rotatable bonds
3
3
7
5
5
7
6
6
7
6
Number of H-bond acceptors
6
6
11
10
9
9
8
9
10
9
Number of H-bond donors
2
3
4
4
3
3
3
3
3
3
Molar refractivity
69.2
75.1
117.1
106.4
121.8
135.9
89.6
121.0
127.5
138.5
TPSA (Å2)
155.5
176.8
256.1
215.9
195.8
214.5
203.7
210.1
219.4
210.1
Log Po/w
1.34
0.82
2.88
2.05
2.05
2.95
0.9
2.4
2.4
3.56
Pharmacokinetics properties
GI absorption
Low
Low
Low
Low
Low
Low
Low
Low
Low
Low
BBB permeant
No
No
No
No
No
No
No
No
No
No
Pgp substrate
No
No
No
No
No
No
No
No
No
No
CYP1A2 inhibitor
No
No
No
No
No
No
No
No
No
No
CYP2C19 inhibitor
No
No
No
No
No
No
No
No
No
No
CYP2C9 inhibitor
No
No
No
No
No
No
No
No
No
No
CYP2D6 inhibitor
No
No
No
No
No
No
No
No
No
No
CYP3A4 inhibitor
No
No
No
No
No
No
No
No
No
No
Skin permeation as log Kp (cm/s)
-7.7
-7.84
-7.7
-7.34
-7.34
-7.84
-8.43
-7.28
-7.49
-6.7
Drug Likeness
Lipinski #violations
0
0
0
0
0
0
0
1
1
1
Ghose #violations
0
0
0
0
0
0
0
1
1
2
Veber #violations
1
1
1
1
1
1
1
1
1
1
Egan #violations
1
1
1
1
1
1
1
1
1
1
Muegge #violations
1
1
1
1
1
1
1
1
1
1
Bioavailability Score
0.55
0.55
0.55
0.55
0.55
0.55
0.55
0.55
0.17
0.17
Lead likeness #violations
0
0
0
1
1
0
1
1
1
2
3 Experimental section
3.1 Synthesis and spectroscopic characterization
3.1.1 Synthesis of (E)-4-(5-(1-hydrazineylideneethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl) benzenesulfonamide (2)
A solution of 1 (2.98 g, 0.01 mol) and N2H4 (1.5 mL, 0.03 mol) in EtOH (40 mL) containing AcOH (3 drops) under reflux for 6 h. The precipitate was filtered and recrystallized from EtOH to get product 2 as a brown powder (78%); M.P 207–210 °C. IR vmax/cm−1 = 3416, 3285, 3233, 3119 (2NH2), 3065 (NH), 1645 (C = N), 1585 (C = C), 1323, 1305 (SO2); 1H NMR (DMSO‑d6): δppm = 2.05 (s,3H, CH3), 7.32 (s, 1H, 2N-Hb), 7.33 (s, 1H, 2N-Ha), 7.36 (s, 2H, NH2), 8.26 (d, J = 8.5 Hz, 2H, Ar-H2,6), 8.34 (d, J = 8.5 Hz, 2H, Ar-H3,5), 9.49 (s, 1H, NH); 13C NMR (DMSO‑d6): δppm = 11.55 (CH3), 120.24 (2CHAr-C2,6), 126.80 (2CHAr-C3,5), 131.35 (Ar-C4), 139.75 (Ar-C1), 145.89 (C = N), 151.28 (thiadiazole-C5), 159.23 (thiadiazole-C2); MS m/z (%): 312 (M+, 10.3), 270 (1.3), 251 (2.5), 227 (2.2), 205 (1.4), 193 (1.5), 176 (9.6), 158 (2.4), 145 (2.2), 129 (7.1), 111 (8.9), 88 (2.2), 65 (11.7), 57 (1 0 0); Anal. Calc. For C10H12N6O2S2 (312.37): C, 38.45; H, 3.87; N, 26.90%; Found: C, 38.46; H, 3.86; N, 26.91%.
3.1.2 General procedure for the synthesis of triaz-2-en-1-ylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzene-sulfonamide derivatives (5, 7)
A solution of compound 3 (1.07 g, 3.4 mmol) in DMF (5 mL) and concentrated HCl (0.75 mL, 7. 5 mmol) cooled to 0–5 °C was diazotized with sodium nitrite (0.26 g, 3.7 mmol) in water (5 mL). After 10 min, a solution of 1,3-dihydroxybenzene 4 or 2-hydroxynaphthalene 6 (3.4 mmol), dissolved in an aqueous solution of 10% sodium hydroxide (10 mL) was added to diazonium salt solution with stirring over 10 min. Then, the pH was adjusted to 7 with the addition of dilute acetic acid. After stirring the mixture for a further 1 h, the formed green precipitate was filtered off, washed with water, dried and recrystallized from ethanol to afford compounds 5 and 7.
3.1.2.1 4-(5-(1-(2,4-Dihydroxyphenyl)triaz-2-en-1-ylidene)-ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzene-sulfonamide (5)
light green powder; yield 61 %; mp 229–230 °C; IR vmax/cm−1 = 3299, 3260 (NH2), 3245, 3199 (2OH), 3107 (NH), 1624 (C = N), 1587 (C = C), 1488 (N = N), 1331, 1305 (SO2); 1H NMR (DMSO‑d6): δppm = 2.38 (s, 3H, CH3), 7.32 (s, 2H, NH2), 7.8–8.11 (m, 4H, Ar-H + OH), 8.22 (d, J = 8.6 Hz, 2H, Ar-H2,6), 8.33 (d, J = 8.6 Hz, 2H, Ar-H3,5), 9.49 (s, 1H, NH), 9.70 (s, 1H, OH); 13C NMR (DMSO‑d6): δppm = 13.89 (CH3), 118.42 (Ar-C1′), 119.55 (Ar-C3′), 121.40 (Ar-C5′), 121.50 (2CHAr-C2,6), 127.37 (2CHAr-C3,5), 127.57 (Ar-C4), 134.56 (1CHAr-C6′), 142.18 (Ar-C1), 143.83 (Ar-C4′), 148.57 (Thiadiazole-C5), 154.30 (Ar-C2′), 157.91 (C = N), 158.56 (Thiadiazole-C2); MS m/z (%): 433 (M+, 1.4), 432 (0.9), 409 (1.4), 393 (1.3), 369 (2.2), 358 (1.9), 342 (1.0), 312 (2.6), 281 (1.2), 257 (1.4), 225 (1.0), 205 (1.0), 179 (1.2), 163 (2.1), 139 (2.7), 121 (6.5), 90 (5.6), 63 (10.1), 57 (1 0 0); Anal. Calc. For C16H15N7O4S2 (433.46): C, 44.34; H, 3.49; N, 22.62%; Found: C, 44.33; H, 3.48; N, 22.63%.
3.1.2.2 4-(5-(1-(2-Hydroxynaphthalen-1-yl)triaz-2-en-1-ylidene)ethyl)-2-imino-1,3,4-thiadi azol-3(2H)-yl)benzenesulfonamide (7)
Green powder; yield 42 %; mp 176–177 °C; IR vmax/cm−1 = 3298, 3253 (NH2), 3235 (OH), 3112 (NH), 1637 (C = N), 1585 (C = C), 1487 (N = N), 1329, 1305 (SO2); 1H NMR (DMSO‑d6): δppm = 2.30 (s, 3H, CH3), 7.20 (s, 2H, NH2), 7.5–8.00 (m, 6H, Ar-H), 8.25 (d, J = 8.6 Hz, 2H, Ar-H2,6), 8.34 (d, J = 8.6 Hz, 2H, Ar-H3,5), 9.48 (s, 1H, NH), 9.73 (s, 1H, OH); 13C NMR (DMSO‑d6): δppm = 13.75 (CH3), 119.05 (2CHAr-C2,6), 119.58 (Ar-C1′), 121.42 (1CH, napthalen-C3), 121.58 (1CH, napthalen-C7), 123.10 (1CH, napthalen-ArC8), 126.43(1CH, napthalen-C6), 126.58 (naphthalen-C5), 126.91 (napthalen-C8a), 127.30 (2CHAr-C3,5), 127.99 (Ar-C4), 135.04 (napthalen-C4a), 141.14 (Ar-C1), 142.18 (1CH, napthalen-C4), 148.63 (Thiadiazole-C5), 155.53 (C = N), 155.71 (Thiadiazole-C2), 158.56 (C-OH); MS m/z (%): 467 (M+, 2.9), 459 (2.2), 407 (2.0), 354 (2.2), 311 (3.3), 283 (2.9), 253 (2.6), 234 (2.0), 206 (1.9), 195 (3.1), 177 (2.6), 151 (4.6), 132 (2.6), 91 (15.0), 63 (25.4), 57 (1 0 0); Anal. Calc. For C20H17N7O3S2 (467.52): C, 51.38; H, 3.67; N, 20.97%; Found: C, 51.37; H, 3.66; N, 20.98%.
3.1.3 General procedure for the synthesis of tetraaz-2-en-1-ylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide derivatives (9, 11)
A solution of compound 2 (1.07 g, 3.4 mmol) in DMF (5 mL) and conc. HCl (0.75 mL, 7. 5 mmol) was cooled to 0–5 °C and diazotized with NaNO2 (0.26 g, 3.7 mmol) in water (5 mL). After 10 min, a solution of sulfanilamide 8 or 4-aminoantipyrine 10 (3.4 mmol) dissolved in 10% aq.NaOH (10 mL) was added to diazonium salt solution with stirring over 10 min. Then the reaction mixture pH was adjusted to 7 by adding dilute AcOH. After stirring for a further 1 h. The formed green precipitate was filtered off, washed with water, dried and recrystallized from EtOH to afford compounds 9 or 11, respectively.
3.1.3.1 4-(4-(1-(2-Imino-4-(4-sulfamoylphenyl)-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethylidene) tetraaz-2-en-1-yl)benzenesulfonamide (9)
Green powder (49%); M.P 263–264 °C; IR vmax/cm−1 = 3330, 3255, 3179, 3133 (2NH2), 3125, 3088 (2NH), 1633 (C = N), 1587 (C = C), 1488 (N = N), 1328, 1305 (SO2); 1H NMR (DMSO‑d6): δppm = 2.30 (s, 3H, CH3), 7.34 (s, 2H, NH2), 7.49 (s, 2H, NH2), 7.91–7.98 (m, 4H, Ar-H), 8.24 (d, J = 8.5 Hz, 2H, Ar-H2,6), 8.35 (d, J = 8.5 Hz, 2HAr-H3,5), 9.48 (s, 1H, NH), 9.71 (s, 1H, NH); 13C NMR (DMSO‑d6): δppm = 13.75 (CH3), 119.58 (2CHAr-C2,6), 121.42 (2CHAr-C2′,6′), 126.88 (2CHAr-C3,5), 126.91 (Ar-C4), 127.30 (2CHAr-C3′,5′), 127.36 (Ar-C4′), 141.13 (Ar-C1), 147.48 (Ar-C1′), 148.63 (C = N), 154.32 (thiadiazole-C5), 158.56 (thiadiazole-C2); MS m/z (%): 495 (M+, 2.5), 492 (1.4), 440 (1.4), 401 (1.4), 384 (1.8), 368 (6.1), 339 (2.6), 313 (9.1), 285 (2.7), 262 (1.4), 237 (2.0), 211 (2.6), 199 (1.8), 183 (1.9), 161 (2.1), 144 (1.4), 121 (8.7), 110 (8.2), 78 (5.6), 57 (1 0 0); Anal. Calc. For C16H17N9O4S3 (495.55): C, 38.78; H, 3.46; N, 25.44%; Found: C, 38.77; H, 3.47; N, 25.45%.
3.1.3.2 4-(5-(1-(4-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)tetraaz-2-en-1-ylidene)ethyl)-2-imino-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (11)
Green powder (45%); M.P 218–219 °C; IR vmax/cm−1 = 3303, 3249 (NH2), 3145, 3114 (2NH), 1686 (amidic C = O), 1588 (C = N), 1544 (C = C), 1488 (N = N), 1327, 1305 (SO2); 1H NMR (DMSO‑d6): δppm = 2.45 (s, 3H, CH3), 2.74 (s, 3H, CH3), 3.19 (s, 3H, CH3-N), 7.41 (s, 2H, NH2), 7.24–7.91–7.95 (m, 5H, Ar-H), 8.25 (d, J = 8.5 Hz, 2H, Ar-H2,6), 8.35 (d, J = 8.5 Hz, 2HAr-H3,5), 9.48 (s, 1H, NH), 9.71 (s, 1H, NH); 13C NMR (DMSO‑d6): δppm = 14.06 (CH3), 15.56 (CH3), 38.75 (CH3), 119.58 (2CHAr-C2,6), 120.49 (pyrazole-C4), 122.35 (Ar-C4′), 125.63 (2CHAr-C2′,6′), 126.88 (2CHAr-C3′,5′), 127.30 (2CHAr-C3,5), 127.36 (Ar-C4), 129.40 (pyrazole-C5), 136.03 (Ar-C1′), 141.13 (Ar-C1), 148.63 (thiadiazole-C5), 155.54 (C = N), 158.56 (thiadiazole-C2), 161.87 (C = O); MS m/z (%): 526 (M+, 0.3), 507 (0.4), 488 (0.2), 465 (0.4), 452 (0.4), 429 (0.3), 407 (0.5), 388 (0.2), 361 (0.2), 339 (1.3), 315 (0.3), 284 (0.9), 257 (4.8), 231 (0.3), 219 (1.1), 198 (0.4), 182 (0.9), 153 (3.3), 113 (7.5), 85 (30.9), 69 (65.8), 57 (1 0 0): Anal. Calc. For C21H22N10O3S2 (526.59): C, 47.90; H, 4.21; N, 26.60%; Found: C, 47.91; H, 4.22; N, 26.59%.
3.1.4 Synthesis of 4-(5-(1-(2-(2-cyano-acetyl)-hydrazineylidene)-ethyl)-2-imino-1,3,4-thia diazol-3(2H)-yl)benzenesulfonamide (12)
A mixture of compound 1 (2.00 g, 0.006 mol) and 2-cyanoacetohydrazide (0.66 g, 0.006 mol) in absolute EtOH (50 mL), containing five drops of conc. HCl was refluxed for 6 h. The resulting precipitate was filtered off, washed with EtOH, dried, and recrystallized from EtOH to produce compound 12 as orange powder (88%); M.P 265–267 °C; IR vmax/cm−1 = 3303, 3251 (NH2), 3197, 3115 (2NH), 2270 (CN), 1687 (C = O), 1595 (C = N), 1582 (C = C), 1328, 1305 (SO2); 1H NMR (DMSO‑d6): δppm = (anti-configuration) 2.26 (s, 3H, CH3), 4.14 (s, 2H, CH2), 7.24 (d, J = 8.5 Hz, 2H, Ar-H2,6), 7.40 (s, 2H, NH2), 8.25 (d, J = 8.5 Hz, 2H, Ar-H3,5), 9.40 (s, 1H, NH), 11.60 (s, 1H, amide-NH); (syn-configuration) 2.29 (s, 3H, CH3), 3.94 (s, 2H, CH2), 7.24 (d, J = 8.5 Hz, 2H, Ar-H2,6), 7.40 (s, 2H, NH2), 8.25 (d, J = 8.5 Hz, 2H, Ar-H3,5), 9.42 (s, 1H, NH), 11.25 (s, 1H, amide-NH); 13C NMR (DMSO‑d6): δppm = 12.18 (CH3), 24.94 (CH2), 115.72 (CN), 120.72 (2CH Ar-C2,6), 126.47 (2CH Ar-C3,5), 126.91 (Ar-C4), 141.92 (Ar-C1), 145.47 (thiadiazole-C5), 148.51 (C = N), 159.90 (thiadiazole-C2), 165.88 (amidic C = O); MS m/z (%): 379 (M+, 28.9), 378 (13.3), 352 (0.3), 323 (0.4), 311 (4.2), 298 (1.7), 283 (0.8), 270 (0.3), 247 (0.2), 213 (0.5), 197 (2.0), 191 (0.4), 175 (0.9), 156 (12.3), 133 (3.8), 108 (14.2), 89 (14.8), 67 (35.1), 57 (1 0 0); Anal. Calc. For C13H13N7O3S2 (379.41): C, 41.15; H, 3.45; N, 25.84%; Found: C, 41.14; H, 3.47; N, 25.83%.
3.1.5 General procedure for the synthesis of coumarin derivatives (14, 16 and 18)
A mixture of compound 12 (0.7 g, 0.0018 mol) and 0.0018 mol of salicylaldehyde derivatives (13, 15 or 17) in AcOH (15 mL), containing (0.13 g, 0.0018 mol) of CH3COONH4, was refluxed for 7–10 h. The formed precipitate was filtered off, washed with EtOH, dried and recrystallized from EtOH to get the desired products 14, 16 and 18, respectively.
3.1.5.1 4-(2-Imino-5-(1-(2-(2-oxo-2H-chromene-3-carbo-nyl)hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (14)
The compound was obtained as a brown powder (80%) from 2-hydroxybenzaldehyde 13 (0.22 mL) with 9 h reflux. M.P 318–320 °C; IR vmax/cm−1 = 3339, 3254 (NH2), 3196, 3102 (2NH), 1708 (C = O), 1670 (C = O, amidic), 1607 (C = N), 1545 (C = C), 1307, 1266 (SO2); 1H NMR (DMSO‑d6): δppm = 2.32 (s, 3H. CH3), 7.33 (s, 2H, NH2), 7.51–7.88 (m, 4H, Ar-H), 8.10 (d, J = 8.5 Hz, 2H, Ar-H2,6), 8.39 (d, J = 8.5 Hz, 2HAr-H3,5), 8.70 (s, 1H, chromene-H4), 9.08 (s, 1H, NH), 11.96 (s, 1H, amidic-NH); 13C NMR (DMSO‑d6): δppm = 12.70 (CH3), 116.75 (chromene-C3), 117.00 (chromene-C8), 118.65 (2CHAr-C2,6), 118.91 (chromene-C4a), 119.93 (chromene-C4), 125.77 (chromene-C6), 127.04 (chromene-C5), 127.35 (chromene-C7), 130.04 (2CH, Ar-C3,5), 130.78 (Ar-C4), 141.29 (Ar-C1), 146.99 (thiadiazole-C5), 153.96 (C = N), 154.45 (chromene-C8a), 156.40 (thiadiazole-C2), 158.81 (amidic, C = O), 158.92 (chromene, C = O); MS m/z (%): 484 (M+, 4.8), 483 (1.0), 456 (1.1), 431 (0.5), 409 (0.4), 382 (0.5), 354 (1.3), 338 (0.5), 324 (1.5), 296 (0.4), 259 (0.5), 237 (0.4), 213 (1.6), 197 (1.3), 176 (0.9), 172 (1 0 0), 155 (3.2), 137 (1.1), 116 (3.8), 95 (5.0), 65 (5.9), 58 (3.6); Anal. Calc. For C20H16N6O5S2 (484.51): C, 49.58; H, 3.33; N, 17.35%; Found: C, 49.57; H, 3.34; N, 17.34%.
3.1.5.2 4-(2-Imino-5-(1-(2-(8-methoxy-2-oxo-2H-chromene-3-carbonyl)hydazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (16)
The compound was obtained from 2-hydroxy-3-methoxy benzaldehyde 15 (0.28 g, 0.001 mol) with 7 h under reflux. Brown powder (77%); M.P 300–302 °C; IR vmax/cm−1 = 3339, 3239 (NH2), 3195, 3120 (2NH), 1706 (C = O), 1699 (C = O, amidic), 1608 (C = N), 1574 (C = C), 1325, 1272 (SO2); 1H NMR (DMSO‑d6): δppm = 1.91 (s, 3H. CH3), 3.95 (s, 3H, OCH3), 7.30 (s, 2H, NH2), 7.43–7.77 (m, 3H, Ar-H), 8.06 (d, J = 8.5 Hz, 2H, Ar-H2,6), 8.22 (d, J = 8.5 Hz, 2HAr-H3,5), 8.35 (s, 1CH, chromen-H4), 9.04 (s, 1H, NH), 11.95 (s, 1H, NH); 13C NMR (DMSO‑d6): δppm = 12.69 (CH3), 56.77 (OCH3), 117.27 (chromene-C7), 118.76 (chromene-C3), 119.47 (2CHAr-C2,6), 121.97 (chomarene-C6), 125.61 (chromene-C5), 125.94 (chromene-C4a), 127.01 (2CHAr-C3,5), 127.34 (Ar-C4), 141.28 (chromene-C8a), 143.73 (Ar-C1), 143.92 (C = N), 147.00 (thiadiazole-C5), 149.78 (chromene-C3), 156.22 (thiadiazole-C2), 158.49 (amidic C = O),161.11 (C = O); MS m/z (%): 514 (M+, 5.1), 513 (1.1), 475 (0.6), 453 (0.9), 426 (0.9), 387 (0.6), 372 (0.7), 353 (6.5), 324 (1.1), 310 (1.1), 297 (0.7), 265 (0.8), 238 (0.7), 225 (0.9), 202 (1 0 0), 180 (0.9), 151 (2.1), 128 (2.6), 82 (7.0), 55 (23.5); Anal. Calc. For C21H18N6O6S2 (514.53): C, 49.02; H, 3.53; N, 16.33%; Found: C, 49.01; H, 3.52; N, 16.32%.
3.1.5.3 4-(2-Imino-5-(1-(2-(3-oxo-3H-benzo[f]chromene-2-carbonyl)-hydrazineylidene)ethyl)-1,3,4-thiadiazol-3(2H)-yl)benzenesulfonamide (18)
The compound was obtained from 2-hydroxynaphthaldehyde 17 (0.29 g, 0.001 mol) with 10 h reflux. Dark Brown powder (79%); M.P 335–337 °C; IR vmax/cm−1 = 3330, 3239 (NH2), 3195, 3117 (2NH), 1701 (C = O), 1688 (C = O, amidic), 1584 (C = N), 1549 (C = C), 1336, 1307 (SO2); 1H NMR (DMSO‑d6): δppm = 1.93 (s, 3H. CH3),7.66 (s, 2H, NH2), 7.48–8.07 (m, 6H, chromene-H), 8.12 (d, J = 8.5 Hz, 2CH, Ar-H2,6), 8.35 (d, J = 8.5 Hz, 2H, Ar-H3.5), 8.66 (s, 1H, chromene-H4), 9.58 (s, 1H, NH), 11.95 (s, 1H, amidic-NH); 13C NMR (DMSO‑d6): δppm = 12.67 (CH3), 113.33 (benzochromene-C3), 116.84 (2CHAr-C2,6), 117.32 (1CH-benzochromene-C10), 117.38 (benzochromene-C4a), 122.91 (1CH, benzochromen-C5), 125.43 (1CH, benzochromen-C7), 127.32 (1CH, benzochromene-C6), 129.35 (1CH, benzochromene-C8), 129.58 (Ar-C9a), 129.89 (2CHAr-C3,5), 130.49 (benzochromene-C9), 137.17 (Ar-C4), 140.08 (Ar-C5a), 141.25 (Ar-C1), 144.51 (1CH-benzochromene-C4), 144.62 (C = N), 146.58 (thiadizole-C5), 151.62 (benzohromene-C10a), 156.17 (thiadiazole-C2), 158.63 (amidic, C = O), 161.30 (C = O); MS m/z (%): 534 (M+, 2.0), 521 (1.9), 495 (1.9), 473 (2.5), 450 (1.7), 378 (3.8), 340 (1.6), 315 (2.3), 285 (3.3), 256 (2.7), 239 (7.7), 220 (1.9), 191 (1.8), 171 (4.9), 143 (3.0), 125 (8.5), 107 (8.3), 98 (37.3), 75 (9.2), 61 (12.5), 57 (1 0 0); Anal. Calc. For C24H18N6O5S2 (534.57): C, 53.92; H, 3.39; N, 15.72%; Found: C, 53.93; H, 3.40; N, 15.71%.
3.2 Biological evaluation
3.2.1 Cytotoxicity evaluation
A volume of 3 mL of the medium containing the tested cells in the log phase was placed in each well of the multi-well plate without phenol red and serum. The final count of the tested cells did not exceed 106 cells/cm2. Then a volume of MTT (M−5655) equivalent to 10% of the culture medium was added and incubated for 2–4 h. The formed formazan crystals were dissolved in the MTT solubilizing solution (M−8910) with occasional shaking equal to the original culture medium. The absorbance of the resulting solution was measured spectrophotometric at wavelength 570 nm. A blank experiment was performed using a medium without cells at every test (S. Bondock, 2023).
3.2.2 Molecular docking analysis
Human carbonic anhydrase IX and XII X-ray crystal structures were downloaded from the Protein Data Bank using PDB: 3IAI and 1JD0 for hCAIX and hCAXII, respectively (A. Sloboda 1964, J. Boix 1997). Their crystal structures were prepared by removing the unnecessary chains, ligands and water molecules, preserving zinc ions. They were corrected, and 3D protonated at cutoff 15 Å using amber10:EHT force field of Molecular Operating Environment (MOE 2014.0901) software. The binding site was selected at the co-crystallized ligand site at a radius of 4.5 Å then molecular docking was performed using Triangle Matcher, London dG, GBVI/WSA as the placement, rescoring function 1 and 2, respectively as the docking algorithm. The tested derivatives were drawn using Chemdraw Ultra 12.0, then transferred as smiles to the MOE builder window, added their hydrogens and energy minimized at the same forcefield.
4 Conclusion
The tail approach was adopted to design and synthesize novel thiadiazole benzenesulfonamide derivatives as hCAIs. The diazene derivative 2, dihydroxyphenyl triazene derivative 5 and carbohydrazide coumarin 18 were the most potent cytotoxic agents among the other derivatives. Derivative 2 achieved IC50 1.18 μM, 5.28 μM, and 7.15 μM against MCF-7, Caco2 and HepG-2, respectively. Moreover, the dihydroxyphenyl triazene derivative 5 demonstrated IC50 3.03 μM, 5.66 μM and 12.50 μM against Caco2, HepG-2 and MCF-7, respectively. Similarly, the carbohyrdazide coumarin 18 showed IC50 of 2.00 μM and 12.30 μM against Caco2 and HepG2, respectively. Molecular docking was used to clarify the possible mechanism of the synthesized derivatives to inhibit both hCAIX and hCAXII that complied with their cytotoxicity. Their sulfonamide terminus coordinated with the zinc ion in the established way to inhibit both isoenzymes with overall favorable binding energy than AAZ. Moreover, the promising derivatives 2, 5 and 18 formed H-bonds with the active site residue Thr199 in both isoforms with an approximate distance of 3.0 Å. Both physicochemical and pharmacokinetic properties of the synthesized compounds were evaluated in silico using ADME demonstrating promising leads to get optimized hCAIs with potent anticancer effects.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Small group Research Project under grant number RGP1/152/44.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- M. A. Abdelrahman, H. S. I., A. Nocentini, 2021. Novel 3-substituted coumarins as selective human carbonic anhydrase IX and XII inhibitors: Synthesis, biological and molecular dynamics analysis. European Journal of Medicinal Chemistry. 209, 112897
- V. Alterio, A. D. F., G. De Simone, 2009. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci U S A 106, 16233-16238
- S. Bilginer, H. I. G., B. Anil, 2020. Synthesis and in silico studies of triazene-substituted sulfamerazine derivatives as acetylcholinesterase and carbonic anhydrases inhibitors. Archiv der Pharmazie. 354
- Bioinformatics, S. I. o., 2017. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. . 7, 42717
- J. Boix, N. L., V.Yuste, 1997. Characterization of the cell death process induced by staurosporine in human neuroblastoma cell lines. Neuropharmacology. 36, 811-821
- A. Bonardi, A. N., S. Bua, 2020. Sulfonamide Inhibitors of Human Carbonic Anhydrases Designed through a Three-Tails Approach: Improving Ligand/Isoform Matching and Selectivity of Action. Journal of medicinal chemistry. 63, 7422-7444
- Advances in the synthesis and chemical transformations of 5-acetyl-1, 3, 4-thiadiazolines. J. Sulphur Chem.. 2021;42(2):202-240.
- [Google Scholar]
- Novel asymmetrical azines appending 1, 3, 4-thiadiazole sulfonamide: synthesis, molecular structure analyses, in silico ADME, and cytotoxic effect. RSC Adv.. 2023;13:10353-10366.
- [Google Scholar]
- J. Chen, C. R., J. Hoffmann, 2005. Expression Of Carbonic Anhydrase 9 At The Invasion Front Of Gastric Cancers. Gut. . 54, 920-927
- S. K.Chia, C. C. W., P.H. Watson, 2001. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol 19, 3660–3668
- SK. Chia, C. W., PH. Watson, 2001. Prognostic Significance Of A Novel Hypoxia-Regulated Marker, Carbonic Anhydrase Ix, In Invasive Breast Carcinoma. J Clin Oncol. 19, 3660–3668
- N. Chiaramonte, M. N. R., E. Teodori, C.T. Supuran, 2018. Amino acids as building blocks for carbonic anhydrase inhibitors. Metabolites. 8, 36-57. https://doi.org/https://doi.org/10.3390/metabo8020036.
- A. Gescher, M. D. T., 1987. The metabolism of triazene antitumor drugs Pharmac. Ther. . 32, 191-205
- M. M. Ghorab, A. M. S., S. Bua, and C. T. Supuran, 2021. Biological evaluation, radiosensitizing activity and structural insights of novel halogenated quinazoline-sulfonamide conjugates as selective human carbonic anhydrases IX/XII inhibitors. Bioorganic Chemistry. 107
- L. Hayflick, P. M., 1961. The Serial Cultivation of Human Diploid Cell Strains. Experimental Cell Research. 25, 585-621
- S. Janowska, A. P., M. Wujec, 2020. Cytotoxic Properties of 1,3,4-Thiadiazole Derivatives—A Review. Molecules. 25
- S. Janowska, D. K., A. Bielawska, 2022. New 1,3,4-Thiadiazole Derivatives with Anticancer Activity. Molecules. 27, 1814
- K. Kanamori, J. D. R., 1983. Nitrogen-15 nuclear magnetic resonance study of benzenesulfonamide and cyanate binding to carbonic anhydrase. Biochem. . 22, 2658-2664
- M. Kciuk, A. G., S. Mujwar, 2022. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J Enzyme Inhib Med Chem. 37, 1278–1298
- Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharma. Res.. 1999;16:1514-1519.
- [Google Scholar]
- R. Kumar, A. K., C. Supuran, 2022. Novel benzenesulfonamide-bearing pyrazoles and 1,2,4-thiadiazoles as selective carbonic anhydrase inhibitors. Arch. Pharm. 355, 2100241
- A. Lacy, R. O. K., 2004. Studies on coumarins and coumarin-related compounds to determine their therapeutic role in the treatment of cancer. . Curr. Pharm. Des. 10, 3797–3811
- Y. Le Duc, E. L., C.T. Supuran, 2017. Carbonic anhydrases activation with 3-amino-1H-1, 2, 4-triazole-1-carboxamides: discovery of subnanomolar isoform II activators. Bioorg. Med. Chem. 25, 1681-1686
- lead- and drug like compounds: the rule of five revolution. Drug Discovery Today: Technol.. 2004;1:337-341.
- [Google Scholar]
- C.L. Lomelino, C. T. S., R. McKenna, 2016. Non-classical inhibition of carbonic anhydrase. Int. J. Mol. Sci. 17, 1150
- A. Maresca, C. T., C.T. Supuran, 2009. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 131, 3057-3062
- A.L. McKeague, D. J. W., J. Nelson 2003. Staurosporine-induced apoptosis and hydrogen peroxide-induced necrosis in two human breast cell lines. British Journal of Cancer. 88, 125-131
- R. M. Mohareb, D. H. F., O. K. Sakka 2011. Novel synthesis of hydrazide-hydrazone derivatives and their utilization in the synthesis of coumarin, pyridine, thiazole and thiophene derivatives with antitumor activity Molecules. 16, 16-27
- M.A. Musa, J. S. C., M.O.F. Khan, 2008. A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr. Med. Chem. 15, 2664–2679
- T. Nasr, S. B., M. Youns, 2014. Anticancer activity of new coumarin substituted hydrazide–hydrazone derivatives. European Journal of Medicinal Chemistry. 76, 539-548
- AM. Niemela, P. H., JP. Mecklin, 2007. Carbonic anhydrase IX is highly expressed in hereditary nonpolyposis colorectal cancer. Cancer Epidemiol Biomarkers Prev. . 16, 1760–1766
- A. Nocentini, C. T. S., 2018. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008e2018). Expert Opin. Ther. Pat. . 28, 729-740
- Rankin, G. O., 2007. Acetazolamide. xPharm: The Comprehensive Pharmacology Reference. 1-5.
- A.H. Robbins, C. G., J. Domsic, 2009. Human carbonic anhydrase II complexed with acetazolamide. RCSB PDB, RCSB: The Protein Data bank
- A.Sloboda, A. W. V., 1964. A laboratory evaluation of 1,4-dimethyl-1,4-diphenyl-2-tetrazene (centrazene), a carcinostatic compound. Cancer research. 24, 461-469
- A.M. Soliman, M. M. G., S. Bua, and C. T. Supuran, 2020. Iodoquinazolinones bearing benzenesulfonamide as human carbonic anhydrase I, II, IX and XII inhibitors: Synthesis, biological evaluation and radiosensitizing activity. European Journal of Medicinal Chemistry. 200,
- Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov.. 2008;7:168-181.
- [Google Scholar]
- A. Swain, K. A., P. Singh, 2021. Design and synthesis of benzenesulfonamide‐linked imidazo[2,1‐b][1,3,4]thiadiazole derivatives as carbonic anhydrase I and II inhibitors. Arch Pharm. e2100028
- M. Szumilak, A. W.-O., A. Stanczak, 2021. Hybrid drugs—a strategy for overcoming anticancer drug resistance? Molecules. 26, 2601
- Y. Takaoka, Y. K., A. Morito, 2013. Crystal structure of 19F probe-labeled hCAI in complex with acetazolamide. RCSB PDB. RCSB: The Protein Data Bank, RCSB: The Protein Data Bank
- A. Thakur, R. S., V. Jaitak, 2015. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 101
- S. J. D. Veber, H. C., 2002. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates Journal of medicinal chemistry. 45, 2615-2623
- A.Vogel, A. E. S., A.S. Tomcufcik 1963. Effect of a New Anticancer Agent, 1,4-Dimethyl-1,4-Diphenyl-2-Tetrazene, on a Transplantable Mammary Adenocarcinoma (72j) and Spontaneous Mouse Breast Tumours. Nature. 197, 85-86
- C. Ward, J. M., M. Gray, 2018. Carbonic Anhydrase IX (CAIX), Cancer, and Radiation Responsiveness. Metabolites. 8
- M.M. Wassel, A. R., G.A.E. Ali, 2021. Novel adamantane-pyrazole and hydrazone hybridized: Design, synthesis, cytotoxic evaluation, SAR study and molecular docking simulation as carbonic anhydrase inhibitors. Journal of molecular structure. 1223, 128966
- D.A. Whittington, A. W., B. Ulmasov, 2001. Crystal Structure Of The Extracellular Domain Of Human Carbonic Anhydrase XII Complexed With Acetazolamide. Proc Natl Acad Sci U S A. 98, 9545-9550
- M. Wu, M. W., H. Jia, 2022. Extracellular vesicles: Emerging anti-cancer drugs and advanced functionalization platforms for cancer therapy. Drug Delivery,. 29, 2513-2538
- C. Yamali, H. S., Y. Uesawa, 2021. Comprehensive Study On Potent And Selective Carbonic Anhydrase Inhibitors: Synthesis, Bioactivities And Molecular Modelling Studies Of 4-(3-(2-Arylidenehydrazine-1-Carbonyl)-5-(Thiophen-2-Yl)-1h-Pyrazole-1-Yl) Benzenesulfonamides, . Eur. J. Med. Chem. 217, 113351
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104956.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




