

Translate this page into:
Determination of the absolute configurations and anti-angiogenic activities of new diarylheptanoid glucosides from Curcuma phaeocaulis
⁎Corresponding authors at: State Key Laboratory of Southwestern Chinese Medicine Resources, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China. feifeifly555@126.com (Fei Liu), xiling0505@126.com (Liang Xiong)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Diarylheptanoids, potential therapeutic agents and dietary supplements, are the main active compounds in the genus Curcuma, however, determination of the absolute configurations of the flexible polyhydric main chains in linear diarylheptanoids is still a challenge. In this study, an exploration of the phytochemical constituents of Curcuma phaeocaulis led to the isolation of eight novel linear diarylheptanoids (1–8). Enzymatic hydrolysis, preparation of acetonide derivatives, preparation of MTPA esters, and electronic circular dichroism calculations were comprehensively performed in order to determine their absolute configurations. In in vitro assays, compounds 2, 3, and 6 exhibited anti-angiogenic activities, and compounds 2, 3, and their aglycones inhibited the proliferation of HepG2 cells. These findings suggest that diarylheptanoid glucosides of C. phaeocaulis may be useful for suppression of hepatoma growth and metastasis.
Keywords
Curcuma phaeocaulis
Diarylheptanoids
Absolute configurations
Anti-angiogenic activities

1 Introduction
Diarylheptanoids, also called curcumins, are an important type of natural compounds firmly bearing a 1,7-diphenylheptane skeletons. They are characteristic components that exist in the genera Curcuma, Zingiber, Alpinia, Alnus, Betula, and Myrica. Potential therapeutic agents and dietary supplements, they have been applied extensively in traditional Chinese, Ayurveda, and Siddha medicines for centuries (Li et al., 2019). Additionally, they are also a very popular spices used to make curries, notably in India and other Asian countries due to their flavor and golden color (Kocaadam & Şanlier, 2017; Núñez et al., 2020). The diverse therapeutic attributes of these substances have been extensively investigated, encompassing their potential as antineoplastic, anti-inflammatory, antioxidative, anti-estrogenic, hepatoprotective, anti-leishmanial, and neuroprotective agents. (Lv & She, 2010; Sun et al., 2020; Alberti et al., 2018). More significantly, diarylheptanoids demonstrate remarkable non-toxicity even when administered at elevated dosages, thus further underscoring their immense potential as therapeutic agents. Clinical trials of diarylheptanoids have found that they are safe for human consumption at a doses up to 12 g/day (Li et al., 2011; Mathew & Hsu, 2018; Patel et al., 2020). Over the last two decades, more than 200 linear diarylheptanoids have been isolated from plants, but determination of the absolute configurations of their flexible polyhydric chains is still a challenge due to the geometric uncertainty of the flexible moieties and the trace amounts of natural products (Sun et al., 2020; Zhu et al., 2021; Devidas et al., 2022; Diao et al., 2019; Yao et al., 2018).
Curcuma phaeocaulis Valeton is primarily distributed in the southwest of China. Due to its ability to eliminate blood stasis and reduce pain, it has been utilized as an herbal medication for thousands of years in China. Recently, C. phaeocaulis has drawn considerable attention because of its pharmacological activities, such as anti-inflammatory (Oh et al., 2014), anti-tumor (Chen et al., 2011), anti-angiogenic (Liao et al., 2021), antioxidant, and anti-bacterial effects (Shi et al., 2021). Diarylheptanoids are one of the major types of the bioactive constituents of C. phaeocaulis, but the anti-angiogenic activity of C. phaeocaulis diarylheptanoids has been rarely studied. A large amount of evidence has suggested that anti-angiogenesis is closely associated with tumor growth and metastasis. Thus, the diarylheptanoids of C. phaeocaulis and their anti-angiogenic activity deserve attention. In this study, eight new linear diarylheptanoids (1–8) were isolated (Fig. 1) and their absolute configurations were determined utilizing a series of chemical and spectroscopic methods. In addition, their anti-angiogenic activities were also evaluated.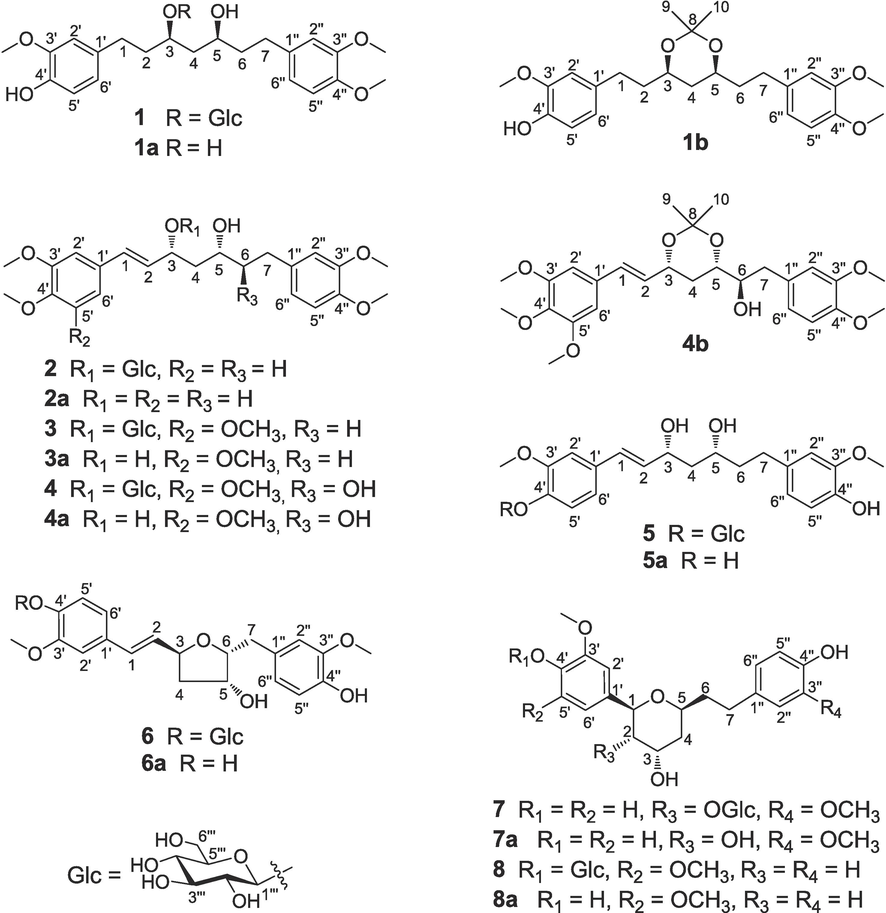
Structures of compounds 1–8 and their derivatives (1a–8a, 1b, and 4b).
2 Experimental
2.1 General experimental procedures
NMR data were acquired by Bruker Avance Neo 600 MHz NMR spectrometer. Optical rotation were measured by Rudolph Autopol-I automated polarimeter. An Agilent Cary 600 FT-IR microscope was used to capture IR spectra. A Bruker TIMS-TOF-MS instrument afforded HRESIMS data. An Applied Chirascan-plus circular dichroism spectrometer was used to record CD data. Sephadex LH-20 (40–70 μm), D-101 macroporous adsorbent resin, and silica gel (200–300 mesh) were used for column chromatography. Snailase was used for glycoside hydrolysis. Reversed-phase semi-preparative HPLC analysis was performed with an Agilent 1100 instrument with a Welch Ultimate XB-C18 column (10 × 250 mm2, 5 μm).
2.2 Plant material
C. phaeocaulis was obtained from Sanjiang Town, Chongzhou, Chengdu, Sichuan, China in March 2018. Dr. Jihai Gao identified the sample, which was stored in the School of Pharmacy in Chengdu University of Traditional Chinese Medicine (voucher specimen: CP-20180303).
2.3 Extraction and isolation
Fifty kilograms of dried C. phaeocaulis rhizomes was extracted three times (3 h each time) with 95% EtOH under reflux. After evaporating the solvent under reduced pressure, the extract was reconstituted in water and subjected to sequential partitioning with petroleum ether, followed by EtOAc, and finally n-BuOH. Subsequently, the n-BuOH soluble fraction (500 g) was subjected to chromatographic separation using a macroporous adsorbent resin (D-101) column. Elution was performed in a stepwise manner using H2O, followed by 20%, 50%, 70%, and 95% ethanol in H2O, resulting in the generation of five distinct fractions (A–E). With the use of reversed-phase MPLC, the fraction C was separated into 17 subfractions by eluting it with MeOH (30–80%) in H2O (F1–F17).
The subfraction F2 was subjected to additional separation using silica gel chromatography with CH2Cl2/MeOH (200:1–0:1) to produce 10 subfractions (F2-1–F2-10). Six subfractions (F2-4-1–F2-4-6) were obtained from F2-4 by elution on silica gel using gradient CH2Cl2/MeOH (200:1–0:1). Compound 6 (4.7 mg, tR = 72 min) was obtained by purifying F2-4-3 successively through Sephadex LH-20 column chromatography (CH2Cl2/MeOH, 1:1) and RP semipreparative HPLC (37% MeOH in H2O). Four subfractions (F2-7-1–F2-7-4) were obtained by chromatography of on a Sephadex LH-20 column (CH2Cl2/MeOH, 5:1). Compound 5 (1.3 mg) was prepared from F2-7-3 by preparative TLC (CH2Cl2/MeOH, 5:1) followed by RP semipreparative HPLC (37% MeOH in H2O).
Subfraction F3 was subjected to silica gel chromatography using a gradient of CH2Cl2/MeOH (200:1–0:1) to produce 12 subfractions (F3-1–F3-12). F3-12 was further separated on a Toyopearl HW-40F column with MeOH/H2O (1:1) as the eluent, affording five subfractions (F3-12-1–F3-12-5). From F3-12-1, compounds 4 (1.5 mg), 7 (1.3 mg), and 8 (1.3 mg) were yielded progressively through preparative TLC (CH2Cl2/MeOH, 6:1) followed by RP semipreparative HPLC (45% MeOH in H2O).
Twelve subfractions (F6-1–F6-12) were produced from F6 by silica gel column chromatography with CH2Cl2/MeOH (200:1–0:1). Successive separation of F6-9 by silica gel chromatography with CH2Cl2/MeOH (200:1–0:1) and preparative TLC (CH2Cl2/MeOH, 8:1) yielded four subfractions (F6-9-1–F6-9-4). Using RP semi-preparative HPLC (48% MeOH in H2O), compounds 2 (3.0 mg, tR = 89 min) and 3 (3.0 mg, tR = 102 min) were isolated from F6-9-3, while compound 1 (3.0 mg, tR = 88 min) was isolated from F6-9-4.
2.4 Physicochemical properties and spectroscopic data of compounds 1–8
Curcuminoside A (1): yellowish amorphous powder; [α]20D− 27.5 (c 0.09, MeOH); IR (ATR) νmax 3363, 2925, 2854, 1514, 1463, 1421, 1259, 1234, 1154, 1075, 1025, 801, 695 cm−1; UV (MeCN) λmax (logε) 200 (3.78), 227 (3.07), 281 (2.64) nm; (+)-HRESIMS m/z 575.2460 [M + Na]+ (calcd for C28H40O11Na, 575.2468); Tables 1 and 2 show 1H and 13C NMR data.
Position
1
2
3
4
5
6
7
8
1
2.65 m
6.59 d (15.9)
6.59 d (15.9)
6.58 d (15.9)
6.53 d (15.8)
6.51 d (15.6)
4.51 d (9.8)
4.72 dd (11.8, 2.0)
2
1.86 m
5.96 dd (15.9, 8.4)
6.04 dd (15.9, 8.4)
6.01 dd (15.9, 8.4)
6.11 dd (15.8, 7.2)
6.16 dd (15.6, 7.2)
3.80 dd (9.8, 3.0)
1.73 m,
1.88 m
3
3.88 m
4.65 m
4.65 m
4.66 m
4.42 m
4.83 m
4.27 q (3.0)
4.23 m
4
1.70 m,
1.86 m
1.78 m,
1.93 m
1.77 m,
1.94 m
1.83 m,
2.03 m
1.70 m,
1.80 m
1.97 m,
2.18 m
1.76 m,
1.89 dd (14.6, 3.0)1.55 m,
1.80 m
5
3.73 m
3.71 m
3.72 m
3.64 m
3.73 m
4.21 t (3.6)
3.86 m
3.90 m
6
1.75 m
1.78 m
1.78 m
3.72 m
1.75 m,
1.78 m
4.10 td (6.9, 3.6)
1.69 m,
1.76 m
1.55 m,
1.69 m
7
2.62 m,
2.69 m
2.61 m,
2.69 m
2.62 m,
2.70 m
2.67 dd (13.7, 7.9),
2.84 dd (13.7, 5.4)2.58 m,
2.68 m
2.80 dd (13.7, 6.9),
2.93 dd (13.7, 6.9)2.60 m
2.64 m
2′
6.78 d (2.0)
7.05 d (2.0)
6.74 s
6.71 s
7.05 d (2.0)
7.06 d (2.0)
7.01 d (2.0)
6.72 s
5′
6.68 d (8.0)
6.91 d (8.4)
–
7.12 d (8.4)
7.10 d (8.4)
6.78 d (8.0)
6′
6.62 dd (8.0, 2.0)
6.95 dd (8.4, 2.0)
6.74 s
6.71 s
6.93 dd (8.4, 2.0)
6.93 dd (8.4, 2.0)
6.91 dd (8.0, 2.0)
6.72 s
2′′
6.81 d (2.0)
6.80 d (2.0)
6.80 d (2.0)
6.85 brs
6.77 d (2.0)
6.92 d (1.9)
6.72 d (2.0)
6.99 d (8.5)
3′′
–
–
–
–
–
–
–
6.66 d (8.5)
5′′
6.84 d (8.2)
6.77 d (8.2)
6.78 d (8.2)
6.78 brs
6.67 d (8.0)
6.71 d (8.0)
6.67 d (8.0)
6.66 d (8.5)
6′′
6.73 dd (8.2, 2.0)
6.73 dd (8.2, 2.0)
6.74 dd (8.2, 2.0)
6.78 brs
6.63 dd (8.0, 2.0)
6.74 dd (8.0, 1.9)
6.59 dd (8.0, 2.0)
6.99 d (8.5)
1′′′
4.33 d (7.8)
4.38 d (7.8)
4.38 d (7.8)
4.36 d (7.8)
4.91 d (7.4)
4.88 d (7.4)
3.67 d (7.8)
4.86 d (7.8)
2′′′
3.19 m
3.22 m
3.22 m
3.20 m
3.50 m
3.48 m
3.04 m
3.48 m
3′′′
3.35 m
3.31 m
3.31 m
3.32 m
3.43 m
3.40 m
2.96 m
3.19 m
4′′′
3.27 m
3.27 m
3.27 m
3.34 m
3.41 m
3.40 m
3.18 m
3.41 m
5′′′
3.21 m
3.22 m
3.23 m
3.22 m
3.48 m
3.46 m
3.05 m
3.41 m
6′′′
3.65 m,
3.83 m
3.67 m,
3.89 m
3.67 m,
3.89 m
3.66 m,
3.88 m
3.70 m,
3.90 m
3.69 m,
3.88 m
3.61 m,
3.76 m
3.66 m,
3.77 m
OMe-3′
3.80 s
3.85 s
3.85 s
3.84 s
3.88 s
3.86 s
3.88 s
3.86 s
OMe-4′
–
3.84 s
3.77 s
3.76 s
–
–
–
–
OMe-5′
–
–
3.85 s
3.84 s
–
–
–
3.86 s
OMe-3′′
3.79 s
3.77 s
3.77 s
3.76 s
3.79 s
3.84 s
3.78 s
–
OMe-4′′
3.81 s
3.75 s
3.76 s
3.75 s
–
–
–
–
Position
1
2
3
4
5
6
7
8
1
31.6
134.8
134.8
134.8
131.1
131.4
78.0
75.0
2
37.8
128.0
129.6
129.6
132.2
130.6
80.7
41.5
3
78.5
77.7
77.5
77.5
72.3
79.8
68.9
65.5
4
43.5
44.1
44.0
40.1
45.4
43.3
39.1
39.5
5
70.0
69.1
69.1
71.2
69.9
73.4
71.9
72.6
6
40.6
40.6
40.6
76.1
41.0
85.6
38.7
39.2
7
32.4
32.4
32.4
40.0
32.4
36.1
32.4
31.8
1′
135.1
131.3
134.1
134.0
133.6
133.4
133.0
141.5
2′
113.2
110.8
105.0
105.0
111.4
111.4
112.8
105.0
3′
148.9
150.5
154.6
154.6
150.9
150.8
148.8
154.1
4′
145.7
150.5
139.0
139.0
147.7
147.7
147.5
135.3
5′
116.2
112.8
154.6
154.6
117.9
117.8
115.9
154.1
6′
121.8
121.2
105.0
105.0
120.8
120.9
121.9
105.0
1′′
136.8
136.6
136.6
133.5
135.1
131.8
135.0
134.6
2′′
113.3
113.6
113.6
113.0
113.2
114.1
113.3
130.4
3′′
150.3
150.3
150.3
150.1
148.8
148.8
148.4
116.1
4′′
148.5
148.5
148.5
148.8
145.5
145.8
145.4
156.4
5′′
113.6
113.1
113.1
114.7
116.1
116.0
116.0
116.1
6′′
121.7
121.8
121.8
122.9
121.8
122.7
121.8
130.4
1′′′
103.9
100.6
100.7
100.7
102.7
102.7
103.6
105.4
2′′′
75.4
75.2
75.2
75.2
74.9
74.9
75.1
75.7
3′′′
78.1
78.2
78.2
78.1
78.2
78.2
77.9
78.3
4′′′
71.8
71.9
71.9
71.9
71.4
71.3
71.3
71.3
5′′′
77.9
78.0
78.0
78.1
77.9
77.8
77.4
77.8
6′′′
63.0
63.0
63.0
63.0
62.5
62.5
62.4
62.4
OMe-3′
56.4
56.4
56.6
56.6
56.7
56.7
56.4
57.0
OMe-4′
–
56.5
56.5
56.5
–
–
–
–
OMe-5′
–
–
56.6
56.6
–
–
–
57.0
OMe-3′′
56.4
56.4
61.2
61.2
56.3
56.3
56.3
–
OMe-4′′
56.5
56.5
56.4
56.4
–
–
–
–
Curcuminoside B (2): yellowish amorphous powder; [α]20D − 45.0 (c 0.05, MeOH); IR (ATR) νmax 3364, 2933, 1590, 1514, 1456, 1419, 1262, 1234, 1138, 1075, 1023, 804, 764, 680 cm−1; UV (MeCN) λmax (logε) 201 (3.81), 268 (3.16) nm; (+)-HRESIMS m/z 587.2459 [M + Na]+ (calcd for C29H40O11Na, 587.2468); Tables 1 and 2 show 1H and 13C NMR data.
Curcuminoside C (3): yellowish amorphous powder; [α]20D − 32.5 (c 0.05, MeOH); IR (ATR) νmax 3363, 2939, 1583, 1463, 1421, 1396, 1318, 1269, 1121, 1042, 859, 696 cm−1. UV (MeCN) λmax (logε) 200 (3.82), 223 (3.55), 274 (3.16) nm; (+)-HRESIMS m/z 617.2565 [M + Na]+ (calcd for C30H42O12Na, 617.2574); Tables 1 and 2 show 1H and 13C NMR data.
Curcuminoside D (4): yellowish amorphous powder; [α]20D − 14.2 (c 0.02, MeOH); IR (KBr) νmax 3421, 2923, 2847, 1624, 1506, 1454, 1378, 1264, 1123, 1026 cm−1; UV (MeCN) λmax (logε) 200 (3.89), 221 (3.60), 274 (3.15) nm; (+)-HRESIMS m/z 633.2516 [M + Na]+ (calcd for C30H42O13Na, 633.2523); Tables 1 and 2 show 1H and 13C NMR data.
Curcuminoside E (5): yellowish amorphous powder; [α]20D − 25.0 (c 0.08, MeOH); IR (ATR) νmax 3178, 3106, 1644, 1588, 1427, 1283, 1223, 1104, 933, 872, 773, 647 cm−1; UV (MeCN) λmax (logε) 196 (3.33), 264 (1.84) nm; (+)-HRESIMS m/z 559.2134 [M + Na]+ (calcd for C27H36O11Na, 559.2155); Tables 1 and 2 show 1H and 13C NMR data.
Curcuminoside F (6): yellowish amorphous powder; [α]20D − 35.3 (c 0.02, MeOH); IR (ATR) νmax 3323, 2889, 2791, 1513, 1453, 1377, 1265, 1090, 922, 804 cm−1; UV (MeCN) λmax(logε) 199 (3.87), 264 (3.36) nm; (+)-HRESIMS m/z 557.1995 [M + Na]+ (calcd for C27H34O11Na, 557.1999); Tables 1 and 2 show 1H and 13C NMR data.
Curcuminoside G (7): yellowish amorphous powder; [α]20D − 27.8 (c 0.02, MeOH), IR (KBr) νmax 3418, 2925, 2849, 1637, 1517, 1457, 1386, 1272, 1035, 802 cm−1; UV (MeCN) λmax (logε) 201 (2.81), 279 (1.75) nm; (+)-HRESIMS m/z 575.2101 [M + Na]+ (calcd for C27H36O12Na, 575.2104). Tables 1 and 2 show 1H and 13C NMR data.
Curcuminoside H (8): yellowish amorphous powder; [α]20D− 40.0 (c 0.04, MeOH), IR (KBr) νmax 3332, 2928, 2855, 1671, 1598, 1457, 1416, 1379, 1236, 1118, 1066, 826 cm−1; UV (MeCN) λmax (logε) 196 (3.52), 221 (2.81), 281 (2.31) nm; (+)-HRESIMS m/z 559.2135 [M + Na]+ (calcd for C27H36O11Na, 559.2155). Tables 1 and 2 show 1H and 13C NMR data.
2.5 Enzymatic hydrolysis of compounds 1–8
Based on a method described in the literature (Tian et al., 2021), compounds 1 (1.5 mg), 2 (1.5 mg), 3 (1.5 mg), 4 (1.2 mg), 5 (0.8 mg), 6 (2.4 mg), 7 (0.7 mg), and 8 (0.9 mg) were suspended in H2O (4.0 mL) and hydrolyzed for 48 h at 37°C with snailase (10.0 mg). The reaction mixture was subjected to three successive extractions with EtOAc (3 × 8 mL). Every EtOAc extract was undergone RP semipreparative HPLC eluted with 60% MeOH to afford the aglycones 1a (0.9 mg), 2a (0.7 mg), 3a (0.8 mg), 4a (0.8 mg), 5a (0.4 mg), 6a (1.0 mg), 7a (0.4 mg), and 8a (0.4 mg). The structures of these aglycones were confirmed through the HRESIMS as well as 1H and 13C NMR data (Tables 3 and 4). 1a: [α]20D − 12.5 (c 0.02, MeOH); (+)-HRESIMS m/z 413.1932 [M + Na]+ (calcd for C22H30O6Na, 413.1940). 2a: [α]20D + 15.0 (c 0.04, MeOH); (+)-HRESIMS m/z 425.1937 [M + Na]+ (calcd for C23H30O6Na, 425.1940). 3a: [α]20D + 8.3 (c 0.03, MeOH); (+)-HRESIMS m/z 455.2044 [M + Na]+ (calcd for C24H32O7Na, 455.2046). 4a: [α]20D − 10.0 (c 0.04, MeOH); (+)-HRESIMS m/z 471.1990 [M + Na]+ (calcd for C24H32O8Na, 471.1995). 5a: [α]20D + 6.3 (c 0.02, MeOH); (+)-HRESIMS m/z 397.1624 [M + Na]+ (calcd for C21H26O6Na, 397.1627). 6a: [α]20D + 9.1 (c 0.03, MeOH); (+)-HRESIMS m/z 395.1467 [M + Na]+ (calcd for C21H24O6Na, 395.1471). 7a: [α]20D − 16.0 (c 0.03, MeOH); (+)-HRESIMS m/z 413.1571 [M + Na]+ (calcd for C21H26O7Na, 413.1576). 8a: [α]20D + 20.0 (c 0.02, MeOH); (+)-HRESIMS m/z 397.1618 [M + Na]+ (calcd for C21H26O6Na, 397.1627). The sugars isolated from every water layer were determined to be D-glucose due to their positive [α]20D values.
Position
1a
2a
3a
4a
5a
6a
7a
8a
1
2.57 m
6.50 d (15.8)
6.50 d (15.9)
6.51 d (15.9)
6.48 d (15.8)
6.36 d (15.6)
4.41 d (9.8)
4.71 d (11.8, 2.0)
2
1.69 m
6.07 dd (15.8, 7.2)
6.14 dd (15.9, 7.2)
6.13 dd (15.9, 7.2)
6.01 dd (15.8, 7.2)
6.04 dd (15.6, 7.5)
3.54 dd (9.8, 3.0)
1.73 m,
1.86 m
3
3.75 m
4.41 m
4.42 m
4.44 m
4.42 m
4.65 m
4.12 q (3.0)
4.24 m
4
1.62 m
1.71 m,
1.78 m
1.71 m,
1.79 m
1.77 m,
1.92 m
1.70 m,
1.78 m
1.80 m,
2.01 m
1.73 m,
1.91 m
1.57 m,
1.78 m
5
3.75 m
3.71 m
3.71 m
3.64 m
3.72 m
4.08 m
3.62 m
3.92 m
6
1.74 m
1.78 m
1.79 m
3.67 m
1.78 m
3.95 m
1.61 m,
1.73 m
1.64 m,
1.73 m
7
2.67 m
2.61 m,
2.70 m
2.61 m,
2.70 m
2.68 m,
2.85 m
2.58 m,
2.67 m
2.62 dd (13.7, 7.0),
2.78 dd (13.7, 7.0)2.61 m
2.65 m
2′
6.78 d (2.0)
7.01 d (1.9)
6.70 s
6.69 s
6.98 d (2.0)
6.97 d (2.0)
7.02 d (2.0)
6.67 s
5′
6.68 d (8.0)
6.90 d (8.3)
6.73 d (8.1)
6.67 d (8.1)
6.79 d (8.0)
6′
6.61 dd (8.0, 2.0)
6.93 dd (8.3, 1.9)
6.70 s
6.69 s
6.83 dd (8.1, 2.0)
6.75 dd (8.1, 2.0)
6.90 d (8.0, 2.0)
6.67 s
2′′
6.80 d (2.0)
6.80 d (2.0)
6.80 d (2.0)
6.87 d (2.0)
6.76 d (2.0)
6.82 d (2.0)
6.74 d (2.0)
7.01 d (8.5)
3′′
6.68 d (8.5)
5′′
6.84 d (8.2)
6.79 d (8.2)
6.78 d (8.2)
6.81 d (8.2)
6.66 d (8.0)
6.64 d (8.0)
6.68 d (8.0)
6.68 d (8.5)
6′′
6.73 dd (8.2, 2.0)
6.72 dd (8.2, 2.0)
6.72 dd (8.2, 2.0)
6.79 dd (8.2, 2.0)
6.62 dd (8.0, 2.0)
6.63 d (8.0, 2.0)
6.61 d (8.0, 2.0)
7.01 d (8.5)
OMe-3′
3.82 s
3.84 s
3.84 s
3.85 s
3.86 s
3.75 s
3.88 s
3.86 s
OMe-4′
3.83 s
3.77 s
3.78 s
OMe-5′
3.84 s
3.85 s
3.86 s
OMe-3′′
3.79 s
3.77 s
3.76 s
3.76 s
3.78 s
3.71 s
3.80 s
OMe-4′′
3.80 s
3.76 s
3.75 s
3.76 s
OH-5
4.91 d (3.9)
Position
1a
2a
3a
4a
5a
6a
8a
1
32.3
131.3
134.5
134.5
131.8
129.7
75.4
2
40.6
131.4
131.5
131.6
130.4
128.6
41.2
3
70.7
72.4
72.2
72.1
72.5
77.8
65.6
4
43.5
45.4
45.4
40.0
45.5
42.3
39.5
5
70.7
69.8
69.8
71.9
70.0
71.3
72.7
6
40.8
40.8
40.8
76.5
41.1
83.6
39.2
7
32.3
32.4
32.4
41.5
32.4
35.0
31.8
1′
135.1
131.7
132.9
132.8
130.5
128.1
135.2
2′
113.1
110.7
104.8
104.8
110.5
109.6
104.6
3′
148.8
150.5
154.6
154.6
149.1
147.7
149.1
4′
145.5
150.3
138.8
138.8
147.5
146.4
135.8
5′
116.2
112.9
154.6
154.6
116.2
115.4
149.1
6′
121.7
120.9
104.8
105.0
121.0
119.8
104.6
1′′
136.7
136.6
136.6
133.5
135.2
130.3
134.4
2′′
113.2
113.6
113.6
113.0
113.2
113.4
130.4
3′′
150.4
150.3
150.3
150.2
148.8
147.2
116.1
4′′
148.6
148.6
148.6
148.8
145.4
144.6
156.3
5′′
113.5
113.1
113.2
114.6
116.1
115.2
116.1
6′′
121.8
121.7
121.7
122.8
121.8
121.3
130.4
OMe-3′
56.6
56.4
56.7
56.6
56.4
55.5
56.8
OMe-4′
56.5
56.5
56.5
OMe-5′
56.7
56.6
56.8
OMe-3′′
56.3
56.4
61.2
61.1
56.3
55.5
OMe-4′′
56.4
56.5
56.4
56.4
2.6 Preparation of acetonide derivatives 1a and 4a
Compound 1a (0.6 mg) was dissolved in DMF (2.5 mL), and (1S)-(+)-camphorsulforic acid (CSA) (10 mg) and 2,2-dimethoxypropane (1 mL) were added respectively based on a method described in the literature (Xiong et al., 2011). Following a 5-h stirring period at room temperature, the reaction mixture was quenched by the addition of triethylamine, and then the mixture was subjected to N2 blowing to produce a crude product that was then purified using RP HPLC (60% MeOH in H2O) to produce acetonide derivative 1b (0.4 mg). Similarly, 4a (0.7 mg) yielded acetonide derivative 4b (0.6 mg).
Compound 1b: (+)-HRESIMS m/z 453.2251 [M + Na]+ (calcd for C25H34O7Na, 453.2253), 1H NMR (CD3OD, 600 MHz): δH 6.82 (1H, d, J = 8.0 Hz, H-5′′), 6.77 (1H, d, J = 2.0 Hz, H-2′′), 6.72 (1H, d, J = 2.0 Hz, H-2′), 6.70 (1H, dd, J = 8.0, 2.0 Hz, H-6′′), 6.66 (1H, d, J = 8.0 Hz, H-5′), 6.58 (1H, dd, J = 8.0, 2.0 Hz, H-6′), 3.80 (3H, s, OMe-3′), 3.78 (3H, s, OMe-4′′), 3.77 (3H, s, OMe-3′′), 1.38 and 1.37 (each 3H, s, acetonide-CH3); 13C NMR (CD3OD, 150 MHz): δC 99.8 (acetonide-C), 30.4 and 20.4 (acetonide-CH3).
Compound 4b: (+)-HRESIMS m/z 511.2304 [M + Na]+ (calcd for C27H36O8Na, 511.2308). 1H NMR (DMSO‑d6, 600 MHz): δH 6.84 (1H, d, J = 8.2 Hz, H-5′′), 6.81 (1H, d, J = 2.0 Hz, H-2′′), 6.74 (1H, s, H-2′), 6.74 (1H, s, H-6′), 6.72 (1H, dd, J = 8.2, 2.0 Hz, H-6′′), 6.47 (1H, d, J = 15.9 Hz, H-1), 6.25 (1H, dd, J = 15.9, 7.2 Hz, H-2), 4.51 (1H, s, OH-6), 3.78 (3H, s, OMe-3′), 3.78 (3H, s, OMe-5′), 3.77 (3H, s, OMe-4′), 3.71 (3H, s, OMe-3′′), 3.64 (3H, s, OMe-4′′), 1.42 and 1.38 (each 3H, s, acetonide-CH3); 13C NMR (DMSO‑d6, 150 MHz): δC 98.2 (acetonide-C), 30.1 and 19.8 (acetonide-CH3).
2.7 Preparation of (S)-MTPA (4c) and (R)-MTPA (4d) esters
A sample of 4b (0.3 mg) was dissolved in 100 μL of pyridine‑d5 in NMR sample tube in order to acquire (S)-MTPA ester 4c. (R)-MTPA-Cl (5 μL) was added, and the mixture was allowed to sit at room temperature overnight. The mixtures were utilized for 1H NMR analysis without undergoing any purification steps. (R)-MTPA ester 4d was yielded by the reaction of 4b (0.3 mg) with (S)-MTPA-Cl (5 μL) in a similar manner. The Δ(S-R) values between the (S)- and (R)-MTPA esters were determined via 1H NMR.
Compound 4c: 1H NMR (pyridine‑d6, 600 MHz): δH 6.86 (1H, d, J = 15.9 Hz, H-1), 6.48 (1H, dd, J = 15.9, 7.2 Hz, H-2), 5.88 (1H, m, H-6), 4.75 (1H, m, H-3), 4.36 (1H, m, H-5), 3.44 (1H, m, H-7a), 3.34 (1H, m, H-7b), 1.76 (1H, m, H-4a), 1.64 (1H, m, H-4b), 1.60 and 1.55 (each 3H, s, acetonide-CH3).
Compound 4d: 1H NMR (pyridine‑d6, 600 MHz): δH 6.87 (1H, d, J = 15.9 Hz, H-1), 6.54 (1H, dd, J = 15.9, 7.2 Hz, H-2), 5.72 (1H, m, H-6), 4.78 (1H, m, H-3), 4.52 (1H, m, H-5), 3.24 (1H, m, H-7a), 3.14 (1H, m, H-7b), 1.79 (1H, m, H-4a), 1.73 (1H, m, H-4b), 1.66 and 1.63 (each 3H, s, acetonide-CH3).
2.8 Cell culture
HepG2 and HUVECs were purchased from Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China) and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cells were grown at 37℃ in a humidified atmosphere with 5% CO2.
2.9 Cell viability test
The viability of HUVEC and HepG2 cells was evaluated using the MTT method based on a method described in the literature (Zhao et al., 2021). In brief, The incubation of HUVEC and HepG2 cells were preformed by seeding into 96-well plates at 5 × 103 cells/mL for 24 h. After that, the cells were subjected to treatment with the test compounds at varying concentrations of 3.125, 6.25, 12.5, 25, and 50 μM. Following 48 h of treatment, the cells were incubated in 20 μL of 5 mg/mL MTT for a duration of 4 h. After collecting the supernatant, 150 µL of DMSO was added to dissolve the formazan crystals. A Varioskan Flash Multiskan Mk3 microplate reader was used to measure absorbance at 570 nm.
2.10 Wound healing assay
HUVECs were plated on the bottoms of the 6-well plates and cultured for 24 h to allow for the formation of a fully confluent monolayer based on a method described in the literature (Zhao et al., 2021; Zhou et al., 2020). Inverted fluorescence microscopy was used to examine the wound after cells were vertically scraped using a 200 μL pipette tip. After that, cells were exposed to different concentrations (25 or 50 μM) of the test compounds for an another 24 h. The healing area of every wound were calculated by ImageJ software version 1.8.0.
2.11 Tube formation assay
The formation of tubular structures by HUVECs was analyzed using a method in the literature (Zhou et al., 2020). Matrigel solution (Corning® Matrigel® Matrix; Cat. No: 354234) was defrosted at 4°C overnight. Every well of 96-well plates was prechilled and coated with 50 μL/well of Matrigel. After 1 h of incubation at 37°C, HUVECs (8 × 105 cells/mL) were suspended in a 1% serum medium with the tested compounds (25 or 50 μM). After that, the cells were seeded on the Matrigel-coated medium and incubated for 6 h at 37°C. A Leica DMI3000B microscope was used to photograph the tube-like structures, and the ImageJ software version 1.8.0 software was used to quantify tube formation by measuring the lengths of the tubular structures.
2.12 Statistical analysis
Data are displayed as the mean ± standard deviation (SD). Every experiment was carried out a minimum of three times. The data from various groups were compared by one-way analysis of variance ANOVA. The figures were created using the GraphPad Prism version 5.0 program. P < 0.05 was used to determine if a comparison was statistically significant.
3 Results and discussion
3.1 Structure elucidation
Compound 1 was was purified and dried as a yellowish, amorphous powder. The molecular formula (C28H40O11) was determined by a quasi-molecular ion at m/z 575.2460 [M + Na]+ (calculated for C28H40O11Na, 575.2468) in its HRESIMS data. Distinct absorption peaks for hydroxy groups (3363 cm−1) and aromatic rings (1514 and 1463 cm−1) were detected in the IR spectrum of compound 1. The 1H NMR data (Table 1) of 1 indicated the presence of two 1,3,4-trisubstituted aromatic rings [δH 6.78 (1H, d, J = 2.0 Hz, H-2′), 6.68 (1H, d, J = 8.0 Hz, H-5′), 6.62 (1H, dd, J = 8.0, 2.0 Hz, H-6′), 6.81 (1H, d, J = 2.0 Hz, H-2′′), 6.84 (1H, d, J = 8.2 Hz, H-5′′), and 6.73 (1H, dd, J = 8.2, 2.0 Hz, H-6′′)], three aromatic methoxy groups [δH 3.79 (3H, s), 3.80 (3H, s), and 3.81 (3H, s)], and a glucosyl unit [δH 4.33 (1H, d, J = 7.8 Hz, H-1′′′), 3.19 (1H, m, H-2′′′), 3.35 (1H, m, H-3′′′), 3.27 (1H, m, H-4′′′), 3.21 (1H, m, H-5′′′), 3.65 (1H, m, H-6′′′a), and 3.83 (1H, m, H-6′′′b)]. The glucosyl unit was identified as the β form based on the coupling constant of the anomeric proton (J = 7.8 Hz). In addition to the units mentioned above, the 1H NMR data showed resonances corresponding to two oxygenated methines [δH 3.88 (1H, m, H-3) and 3.73 (1H, m, H-5)] and five methylenes [δH 1.70–2.70 (10H, m)]. There were 28 carbon signals in the 13C NMR and DEPT spectra for compound 1 (Table 2), which are attributed to the above-mentioned protonated units and six aromatic quaternary carbons. Based on these NMR data, compound 1 was a diarylheptanoid glucoside that similar to (3R,5R)-3,5-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-heptane 3-O-β-D-glucopyranoside (Yokosuka et al., 2002). The 1H–1H COSY correlations of H-1/H-2/H-3/H-4/H-5/H-6/H-7, together with the HMBC correlations of H-1 with C-2, C-1′, C-2′, and C-6′; H-7 with C-5, C-6, C-1′′, C-2′′, and C-6′′; OMe-3′ with C-3′; OMe-3′′ with C-3′′; OMe-4′′ with C-4′′, and H-1′′′ with C-3 (Fig. 2) further determined the planar structure of 1 as 3,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(3,4-dimethoxyphenyl)heptane 3-O-β-glucopyranoside.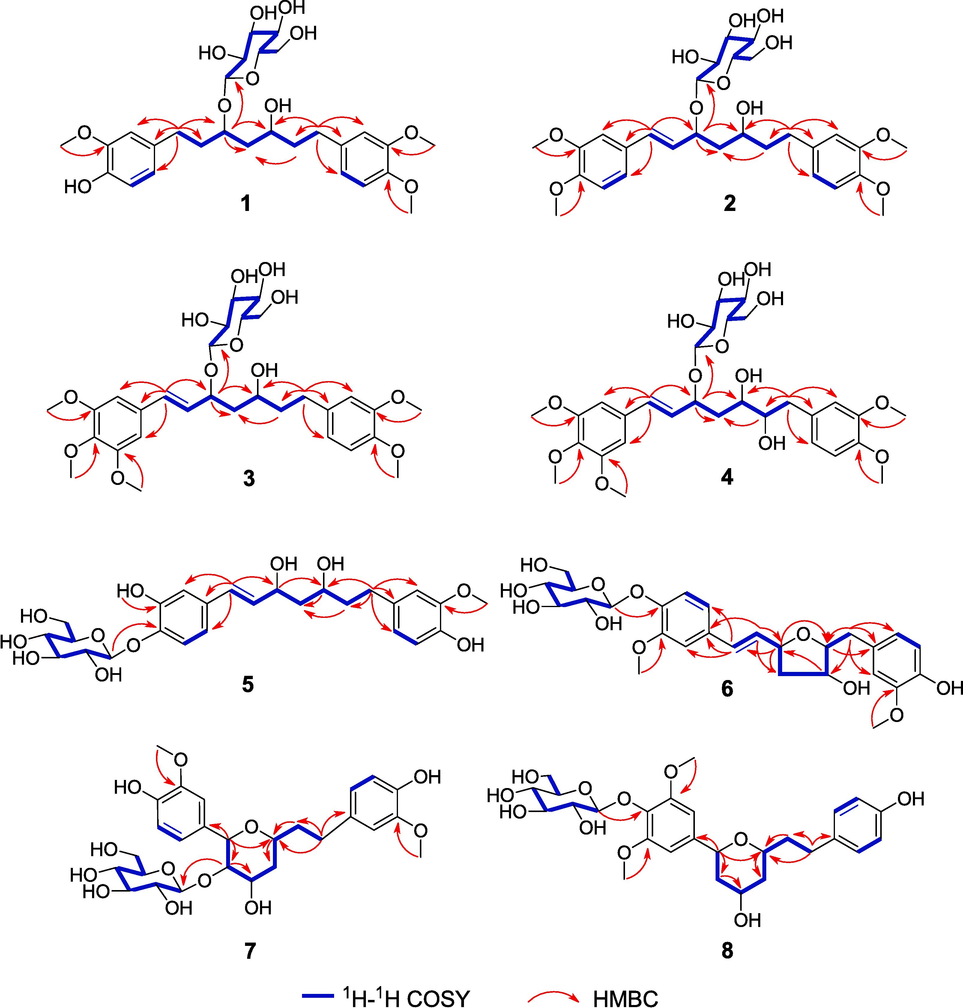
Key HMBC and 1H–1H COSY correlations of compounds 1–8.
Compound 1 was hydrolyzed by snailase to afford 1a (Fig. 1) and a sugar, which were analyzed for the purpose of establishing the absolute configuration. The sugar was identified as D-glucose by TLC comparison with an authentic sugar sample and the positive specific optical rotation {[α]20D + 42.5 (c 0.04, H2O)} (Xiong et al., 2013). The 1H (Table 3) and 13C NMR (Table 4) and HRESIMS data of 1a led to its characterization of 1a as 3,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(3,4-dimethoxyphenyl)-heptane. Next, to determine the relative configurations of OH-3 and OH-5 in 1a, its acetonide derivative (1b) was further synthesized. The 13C chemical shifts of the two methyl groups in the acetonide derivative of a 1,3-diol could determine the relative configuration of the 1,3-diol (Rychnovsky et al., 1997; Boger et al., 1997; Sugawara et al., 1996; Kim et al., 2012). When the 1,3-diol unit was syn, the two methyl carbons showed two distinctive chemical shifts around δC 19 and 30, while when the 1,3-diol unit is anti, the two methyl carbons displayed nearly identical signals near δC 25. In compound 1b, the 13C NMR data of the two methyl groups (δC 30.4, δH 1.37; δC 20.4, δH 1.38) indicated the presence of a syn 1,3-diol unit in compound 1a. Electronic circular dichroism (ECD) calculations were further carried out to determine the absolute configuration of 1a. The results indicated that an agreement between the calculated ECD spectrum of (3R,5S)-1a and the experimental ECD spectrum of 1a (Fig. 3). Therefore, compound 1 was determined to be (3R,5S)-3,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(3,4-dimethoxyphenyl)heptane 3-O-β-D-glucopyranoside and was named curcuminoside A.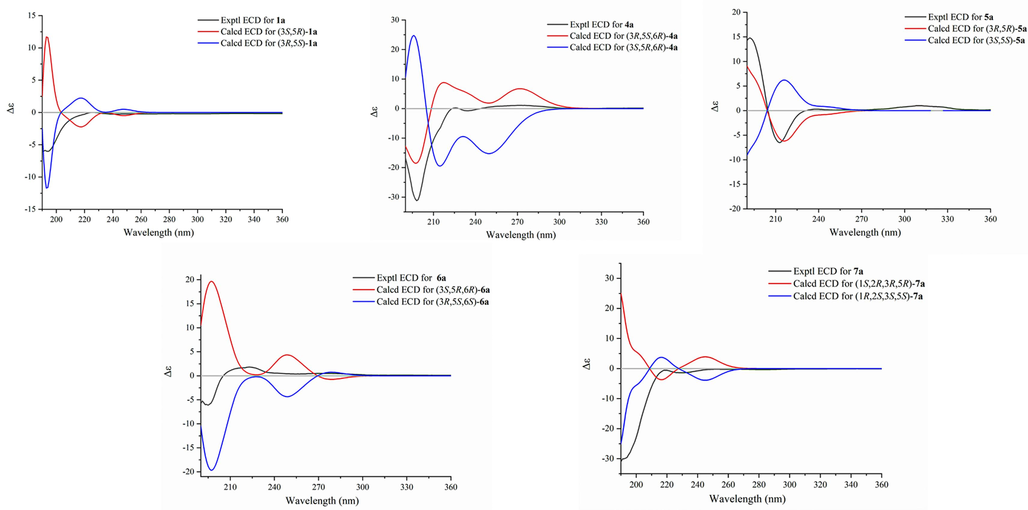
Experimental and calculated ECD spectra of compounds 1a and 4a-7a.
Compound 2 showed a positive quasi-molecular ion peak at m/z 587.2459 [M + Na]+ (calculated for 587.2468, C29H40O11Na), indicating that its molecular formula was C29H40O11. The 1H and 13C NMR spectra of 2 exhibited similarities to those of 1 except that the signals for an aromatic methoxy group [δH 3.84 (3H, s, OMe-4′); δC 56.5 (OMe-4′)] and a trans-double bond in 2 [δH 5.96 (1H, dd, J = 15.9, 8.4 Hz, H-2), 6.59 (1H, d, J = 15.9 Hz, H-1); δC 128.0 (C-2), 134.8 (C-1)] replace those for an aromatic hydroxy group and two methylene groups in 1. The HMBC correlations from an olefinic proton (δH 6.59) to C-3, C-1′, C-2′, and C-6′ and from another olefinic proton (δH 5.96) to C-1′ suggested that the trans-double bond was located at C-1 and C-2. The HMBC correlations of four methoxy groups (δH 3.85, 3.84, 3.77, and 3.75) with C-3′, C-4′, C-3′′, and C-4′′, respectively, indicate that the four methoxy groups were substituted at C-3′, C-4′, C-3′′, and C-4′′ (Fig. 2). Additionally, the glucosyl unit was linked to C-3 based on an HMBC signal of the anomeric proton (δH 4.38) with C-3. Compound 2 was hydrolyzed to produce the aglycone 2a and D-glucose using the same procedure as described for compound 1. Compound 2a displayed nearly identical 1H NMR, 13C NMR, and HRESIMS data with those of a known diarylheptanoid, (3S,5S)-1,7-bis(3,4-dimethoxyphenyl)-1-hepten-3,5-diol (Jang et al., 2019). These spectroscopic data reveal that compound 2a and (3S,5S)-1,7-bis(3,4-dimethoxyphenyl)-1-hepten-3,5-diol had the same planar structure and relative configuration. However, 2a had a positive optical rotation value ([α]20D + 15.0), which was opposite to that of (3S,5S)-1,7-bis(3,4-dimethoxypheny-l)-1-hepten-3,5-diol ([α]20D –7.0). In addition, the trend of the experimental ECD spectrum of 2a is opposite to that of (3S,5S)-1,7-bis(3,4-dimethoxypheny-l)-1-hepten-3,5-diol reported in the literature. Thus, compound 2a and (3S,5S)-1,7-bis(3,4-dimethoxyphenyl)-1-hepten-3,5-diol were a pair of enantiomers, and compound 2 was determined to be (3R,5R)-3,5-dihydroxy-1,7-bis(3,4-dimethoxyphenyl)-1-hepten 3-O-β-D-glucopyranoside (curcuminoside B).
Compound 3 had a molecular formula of C30H42O12 as indicated by its HRESIMS data. The spectroscopic data of 3 were similar to those of 2 except that 3 had resonances of an additional aromatic methoxy group at C-5′ (δH 3.85; δC 56.6). The structure of 3 was further verified by HSQC, 1H–1H COSY, and HMBC data analysis (Fig. 2). Particularly, based on the HMBC cross-peaks, the locations of the five methoxy groups were confirmed to be at C-3′, C-4′, C-5′, C-3′′, and C-4′′, respectively. The hydrolysis of 3 yielded 3a that had the identical 1H and 13C NMR data (Tables 3 and 4) as those of (3S,5S)-1-(3,4,5-trimethoxyphenyl)-7-(3,4-dimethoxyphenyl)-1-hepten-3,5-diol, but an opposite optical rotation (Jang et al., 2019). In addition, the trend of the experimental ECD spectrum of 3a is opposite to that of (3S,5S)-1-(3,4,5-trimethoxyphenyl)-7-(3,4-dimethoxyphenyl)-1-hepten-3,5-diol reported in the literature. Thus, compound 3 was determined to be (3R,5R)-3,5-dihydroxy-1-(3,4,5-trimethoxyphenyl)-7-(3,4-dimethoxyphenyl)-1-hepten 3-O-β-D-glucopyranoside and was named curcuminoside C.
Compound 4 was also a yellowish amorphous powder and had a molecular formula of C30H42O13 based on the HRESIMS data (m/z 633.2516 [M + Na]+; calculated for C30H42O13Na, 633.2523). The 1H and 13C NMR spectra of 4 suggested that it was similar to compound 3 except for an additional hydroxy group at C-6 (δH 3.72, δC 76.1) in 4 (Tables 1 and 2). This conjecture was confirmed by the 1H–1H COSY correlations of H-1/H-2/H-3/H2-4/H-5/H-6/H2-7 and the HMBC correlations of H-6 with C-4, C-5, C-7, and C-1′′. To determine the absolute configurations, compound 4 was hydrolyzed by snailase to yield 4a. The 1H NMR (Table 3), 13C NMR (Table 4), and HRESIMS data of 4a led to the characterization of 4a as 3,5,6-trihydroxy-1-(3,4,5-trimethoxyphenyl)-7-(3,4-dimethoxyphenyl)-1-hepten. The corresponding acetonide derivative 4b was prepared in the same manner of 1b to establish the relative configurations of OH-3 and OH-5 in 4a. In the 13C NMR spectrum of compound 4b, the 13C data of the two acetonide methyl groups (δC 30.4; δC 20.3) indicated the presence of a syn 1,3-diol in compound 4a. The absolute configuration of C-6 was directly determined by the modified Mosher’s method (Zhai et al., 2022). Treatment of 4b with (R)- and (S)-MTPA chlorides afforded the (S)- and (R)-MTPA esters (4c and 4d). The pattern of the ΔδS−R values (especially ΔδH-5: −0.15, ΔδH-7: +0.23) indicated the 6R-configuration in 4b (Fig. 4). Lastly, the absolute configurations of C-3 and C-5 were elucidated by comparing the experimental ECD spectrum of 4a with the computed ECD spectra of two possible stereoisomers [(3R,5S,6R)-4a and (3S,5R,6R)-4a] (Fig. 3). The result indicated that compound 4a had the 3R,5S,6R configuration. Thus, the structure of compound 4 was identified as (3R,5S,6R)-3,5,6-trihydroxy-1-(3,4,5-trimethoxyphenyl)-7-(3,4-dimethoxyphenyl)-1-hepten 3-O-β-D-glucopyranoside (curcuminoside D).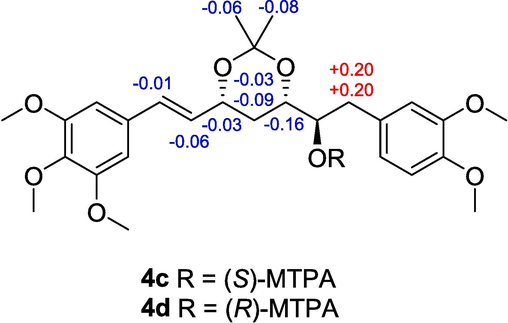
ΔδH values (δS − δR) for 4c and 4d.
The spectroscopic data of compound 5 revealed that it was another diarylheptanoid glucoside with a molecular formula of C27H36O11. Its 1H and 13C NMR data (Tables 1 and 2) indicated the presence of two 1,3,4-trisubstituted aromatic rings, a 1,7-disubstituted 3,5-dihydroxy-1-hepten unit, a β-glucosyl, two aromatic methoxy groups, and an aromatic hydroxy group. The structure of 5 was further determined by the HMBC correlations and the 1H–1H COSY correlations as shown in Fig. 2. The HMBC correlation from H-1′′′ to C-4′ confirmed the position of the β-glucosyl moiety at C-4′. Compound 5 was further hydrolyzed by snailase to produce its aglycone (5a), which had the same planar structure as neohexahydrocurcumin (Maehara et al., 2011). However, the 1H and 13C NMR spectra of 5a were different from those of neohexahydrocurcumin, especially H-3 and H-5, thus suggesting that 5a was an epimer of neohexahydrocurcumin. This deduction indicated a syn orientation for OH-3 and OH-5 in 5a, which was the same for compounds 2a and 3a. In addition, since 2a, 3a, and 5a all had a positive optical rotation values ([α]20D + 15.0, +8.3, and + 6.3, respectively), and they had very similar structures, the absolute configuration of 5a was presumed to be 3R,5R. This deduction was confirmed by comparing the calculated and experimental ECD data (Fig. 3). Thus, compound 5 was determined to be (3R,5R)-3,5-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-1-hepten 4′-O-β-D-glucopyranoside (curcuminoside E).
Compound 6 had a molecular formula of C27H34O11 with one more degree of unsaturation than compounds 1–5, implying that an oxygen ring was present in the 1,7-disubstituted 1-hepten chain in 6. The 1H and 13C NMR data of 6 (Tables 1 and 2) were similar to those of compound 5 except that an oxymethine (δH 4.10, δC 85.6) in 6 replaced the methylene at C-6 (δH 1.75 and 1.78, δC 41.0) in 5. These NMR data, combined with the degrees of unsaturation of 6, indicated that C-3 and C-6 were linked by an ether bond to form a tetrahydrofuran ring in 6. This deduction was confirmed by the 1H–1H COSY correlations of H-1/H-2/H-3/H2-4/H-5/H-6/H2-7, together with the HMBC correlation from H-3 to C-6 (Fig. 2). In addition, the HMBC correlations of H-5 with C-3, C-4, C-6 and C-7 revealed a hydroxy group located at C-5. To further elucidate the structure of 6, it was hydrolyzed to afford the aglycone (6a). In the NOESY spectrum of 6a, correlations of H-5/H-2 and H-6/H-2 indicated the relative configurations of H-3, H-5, and H-6 in the tetrahydrofuran ring (Fig. 5). By comparing the calculated and experimental ECD data, the absolute configuration of 6a was determined to be 3R,5S,6S (Fig. 3). Accordingly, curcuminoside F (6) was determined to be (3R,5S,6S)-3,6-epoxy-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-1-hepten 4′-O-β-D-glucopyranoside.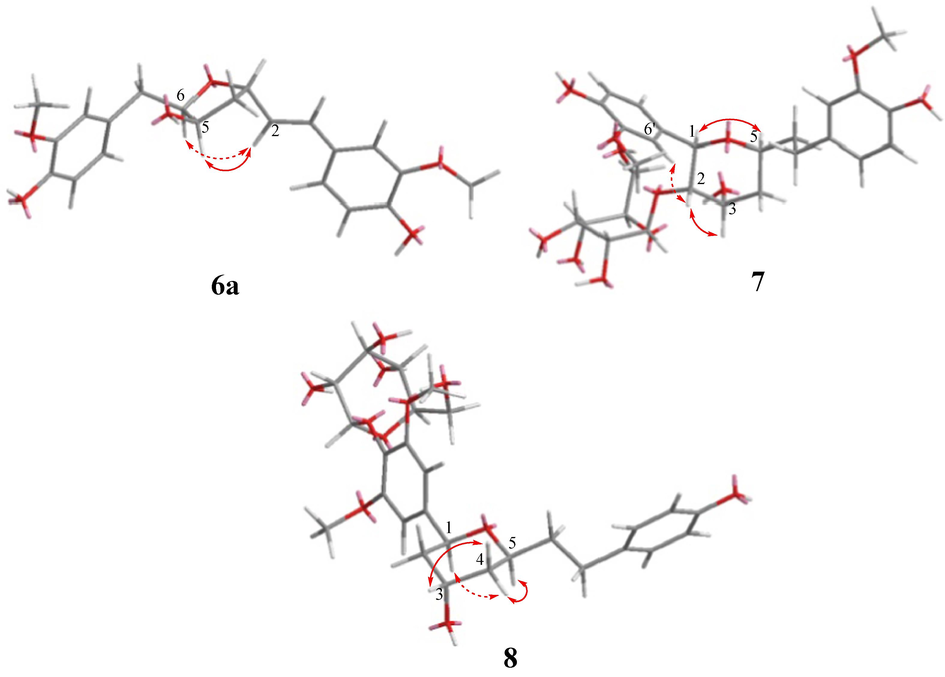
Key NOESY correlations of compounds 6a, 7, and 8.
Compound 7 was also a diarylheptanoid glucoside with an oxygen ring in the chain between two aromatic rings. The 1H and 13C NMR data (Tables 1 and 2) of 7 indicated the presence of two 4-hydroxy-3-methoxyphenyl groups and a β-glucosyl unit. In addition, the NMR data showed resonances corresponding to four oxygenated methines [δH 4.51 (1H, d, J = 9.8 Hz, H-1), 3.80 (1H, dd, J = 9.8, 3.0 Hz, H-2), 4.27 (1H, q, J = 3.0 Hz, H-3) and 3.86 (1H, m, H-5)] and three methylenes [δH 1.69–2.60 (6H, m)] in the aliphatic chain. Further analysis of the 2D NMR data showed that the structure of compound 7 was 1,5-epoxy-2,3-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)heptane 2-O-β-D-glucopyranoside. In particular, the HMBC correlation of H-1 with C-5 together with 1H–1H COSY correlations of H-1/H-2/H-3/H2-4/H-5/H2-6/H2-7 confirmed that a tetrahydropyrane ring was linked to the benzene ring. The HMBC correlation of H-1′′′/C-2 confirmed the position of the β-glucosyl. The relative configurations of 7 were determined by analysis of NOESY data and 1H−1H coupling constants (Fig. 5). The NOESY correlation of H-1 with H-5 revealed the same orientation of H-1 and H-5. Further, the trans-orientation of H-1 and H-2 was established by the large coupling constant between them (J1,2 = 9.8 Hz), which was determined by the NOESY correlation of H-2 with H-6′. Finally, the NOESY correlation of H-2 with H-3, together with the small coupling constant of J2,3 (3.0 Hz), indicated that H-2 and H-3 were located in the same face of the tetrahydropyrane ring (Suciati et al., 2013). The absolute configuration of 7 was established by enzymatic hydrolysis and ECD calculations. Specifically, the experimental ECD spectrum of the aglycone (7a) agreed well with the calculated ECD spectrum of (1R,2S,3S,5S)-7a (Fig. 3). Thus, compound 7 was determined to be (1R,2S,3S,5S)-1,5-epoxy-2,3-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)heptane 2-O-β-D-glucopyranoside and was named curcuminoside G.
The spectroscopic data of 8 suggested that it was an analogue of 7. Comparison of their 1H and 13C NMR data (Tables 1 and 2) revealed that two 1,3,4-trisubstituted phenyl moieties in 7 were replaced by a symmetric 1,3,4,5-tetrasubstituted phenyl ring and a para-substituted phenyl ring in 8. Additionally, an oxymethine (C-2) in 7 was replaced by a methylene in 8. As shown in Fig. 2. The HMBC correlations of MeO-3′/C-3′ and MeO-5′/C-5′ indicated that two methoxy groups were located at C-3′ and C-5′, and the HMBC correlation of H-1′′′/C-4′ confirmed the position of the β-glucosyl at C-4′. The key NOESY correlations confirmed that 7 and 8 had the same relative configurations (Fig. 5). Using the same procedure as described for compound 7, compound 8 was hydrolyzed to produce the aglycone 8a and D-glucose. Compound 8a displayed nearly identical 1H NMR, 13C NMR, HRESIMS, and optical rotation data with those of a known diarylheptanoid, (1S,3S,5S)-1,5-epoxy-3-hydroxy-1-(4-hydroxy-3,5-dimethoxyphenyl)-7-(4-hydroxyphenyl)heptane (Chen et al., 2015). Thus, curcuminoside H (8) was determined to be (1S,3S,5S)-1,5-epoxy-3-hydroxy-1-(4-hydroxy-3,5-dimethoxyphenyl)-7-(4-hydroxyphenyl)heptane 4′-O-β-D-glucopyranoside.
3.2 Diarylheptanoid glucosides inhibited proliferation of HUVECs
Compounds 1–4 and 6 were measured for their inhibitory effects on proliferation of HUVECs by the MTT assay. As shown in Fig. 6, compounds 2, 3, and 6 showed moderate cytotoxicity against HUVECs with viability rates of 56.22 ± 7.58 %, 36.66 ± 9.17 %, and 59.03 ± 2.14 % at a concentration of 50 μM (P < 0.01).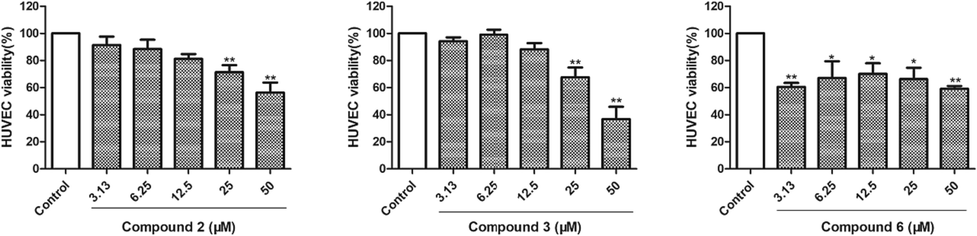
HUVECs proliferation was inhibited by compounds 2, 3, and 6. The data of three independent experiments are presented as the mean ± SD. **P < 0.01 vs. untreated control, *P < 0.05 vs. untreated control.
3.3 Compounds 2 and 3 inhibited migration of HUVECs
The inhibitory effects on HUVEC migration of the diarylheptanoid glucosides were evaluated by wound healing assay (Fig. 7). The migration rate of the control group was 44.90 ± 3.45%. whereas compounds 2 and 3 significantly inhibited HUVEC migration dose-dependently (P < 0.01). After treatment of 2 for 24 h, the migration rates at 25 and 50 μM were decreased to 32.0 ± 2.92% and 15.5 ± 2.4%, respectively. Compound 3 at 25 and 50 μM decreased the migration rates to 23.37 ± 2.44% and 10.33 ± 0.23% (P < 0.01), respectively.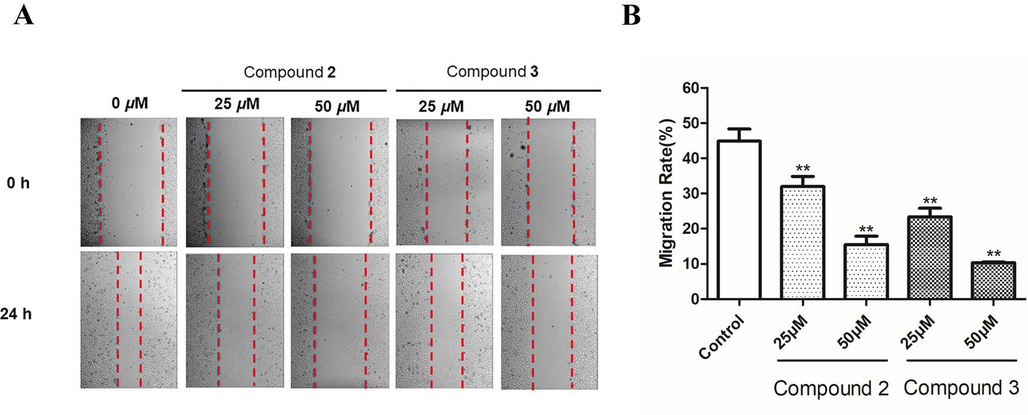
The migration of HUVECs was inhibited by compounds 2 and 3. (A) Wound healing was observed using microscopy at 0 and 24 h. (B) The healing area in the wound healing assay was computed by ImageJ software version 1.8.0. The data of three independent experiments are shown as the mean ± SD. **P < 0.01 vs. untreated control.
3.4 Compounds 2 and 3 inhibited tube formation of HUVECs
The development of a sophisticated network of vasculatures from endothelial cell tubes is one of the key stages in angiogenesis. Therefore, the inhibitory effects on the early tube formation in HUVECs of diarylheptanoid glucosides were assayed (Fig. 8). Compared with the control group, compounds 2 and 3 reduced the tube formation of HUVECs (P < 0.01). Notably, the tube length decreased from 100% to 62.17 ± 4.35% (P < 0.01) in response to the highest concentration of 3 (50 μM). Taken together, the above assays demonstrated the anti-angiogenic effects of compounds 2 and 3, and the anti-angiogenic activity of 3 is better than that of 2. Their difference in activity suggests that the amount of methoxy groups at aromatic ring may plays an important role in anti-angiogenic activity. Interestingly, a diarylheptanoid isolated from Alpinia officinarum showed more potent anti-angiogenic activity than compounds in our research (Hu et al., 2019), probably due to the presence of a ketone group at C-3, making its structure more similar to that of curcumin.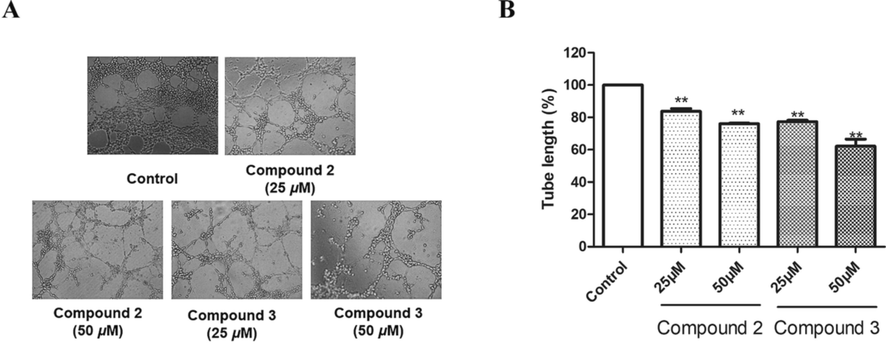
The tube formation of HUVECs inhibited by compounds 2 and 3. (A) The Growth of HUVECs on Matrigel was used to evaluate the effects of compounds 2 and 3 on tube formation in the HUVECs. The images of the tubular structures were captured under 100 × magnification. (B) Tube lengths in the control group, compound 2 group, and compound 3 group. The data of three independent experiments are presented as the mean ± SD. **P < 0.01 vs. untreated control, *P < 0.05 vs. untreated control.
3.5 Diarylheptanoid glucosides inhibited the proliferation of HepG2 cells
As shown in Fig. 9, compounds 2, 2a, 3, and 3a showed moderate cytotoxicity against HepG2 cells with viability rates of 34.75 ± 2.60%, 71.40 ± 0.21%, 52.94 ± 2.82%, and 66.31 ± 3.31% at a concentrations of 50 μM (P < 0.01), respectively. Interestingly, compounds 2 and 3 showed stronger cytotoxicity than their aglycones (2a and 3a, respectively).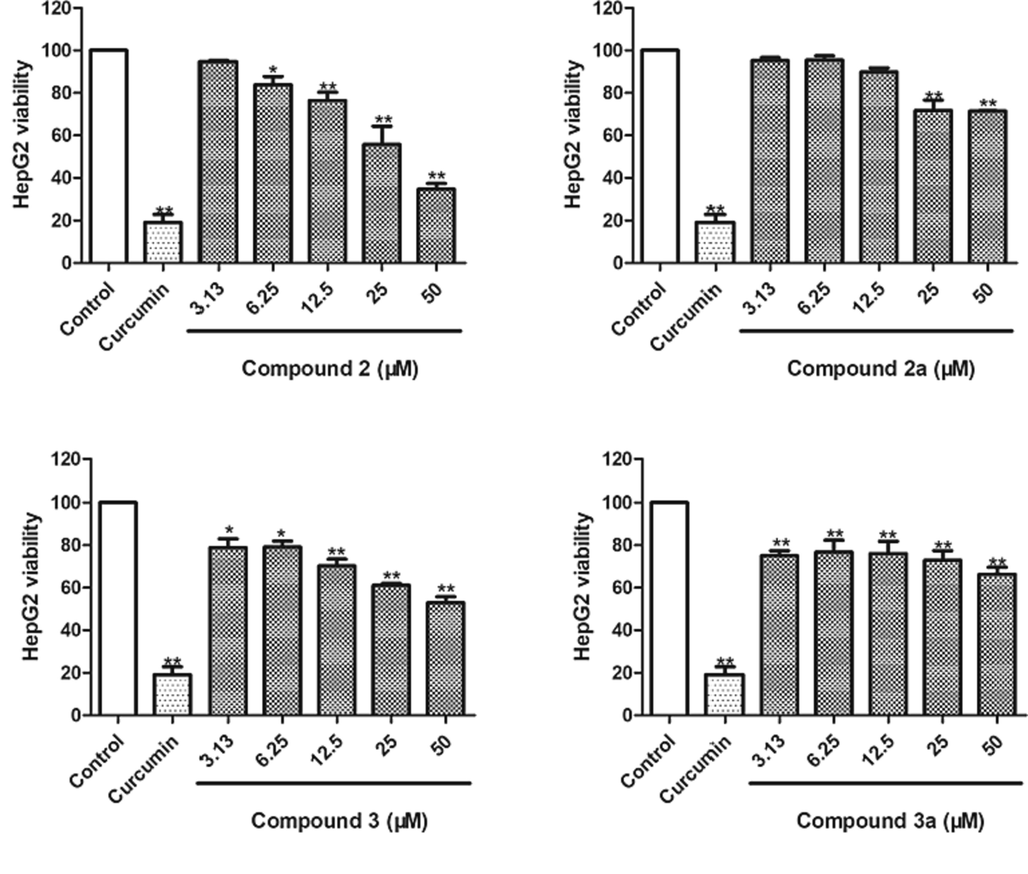
Cytotoxicity of compounds 2, 2a, 3, and 3a against HepG2 cells. Curcumin was used as a positive control (50 μM). The data of three independent experiments are presented as the mean ± SD. **P < 0.01 vs. untreated control, *P < 0.05 vs. untreated control.
4 Conclusions
Due to their extraordinary biological activity and hypotoxicity at large dosages, curcumin-containing herbal medications are not only formally recognized in pharmacopoeias but also offered as dietary supplements. However, determining the absolute configurations of most linear diarylheptanoids that have flexible polyhydroxy moieties is difficult. In this study, eight new diarylheptanoids (1–8) were isolated from C. phaeocaulis. Notably, their absolute configurations were successfully determined by comprehensive application of NOESY experiments, enzymatic hydrolysis, preparation of acetonide derivatives, the modified Mosher’s method, and CD calculations. Compounds 2, 3, and 6 showed inhibitory effects on the proliferation of HUVECs, and compounds 2, 2a, 3, and 3a showed moderate inhibition on the proliferation of HepG2 cells. Moreover, compounds 2 and 3 inhibited migration and tube formation of HUVECs.
Funding
This work was supported by the the National Natural Science Foundation of China (NNSFC; Grant Nos. 82022072, 81903777, and 82104371), the Innovation Team Program of Jinan City (Grant No. 202228038), the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (Grant No. ZYYCXTD-D-202209), the Natural Science Foundation of Sichuan Province (Grant Nos. 2023NSFSC1773 and 2022NSFSC1332), the “Xinglin Scholar” Plan of Chengdu University of Traditional Chinese Medicine (Grant No. QJRC2022020), and the Scientific and Technological Industry Innovation Team of Traditional Chinese Medicine of Sichuan Province (Grant No. 2022C001).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Characterization of diarylheptanoids: An emerging class of bioactive natural products. J. Pharm. Biomed. Anal.. 2018;147:13-34.
- [CrossRef] [Google Scholar]
- Determination of the relative and absolute stereochemistry of fostriecin (CI-920) J. Org. Chem.. 1997;62:1748-1753.
- [CrossRef] [Google Scholar]
- Five new diarylheptanoids from the rhizomes of Curcuma kwangsiensis and their antiproliferative activity. Fitoterapia.. 2015;102:67-73.
- [CrossRef] [Google Scholar]
- Anti-tumor potential of ethanol extract of Curcuma phaeocaulis Valeton against breast cancer cells. Phytomedicine.. 2011;18:1238-1243.
- [CrossRef] [Google Scholar]
- Two undescribed diarylheptanoids from green husk of Carya illinoinensis as acetylcholinesterase inhibitors. Nat. Prod. Res.. 2022;36:1161-1169.
- [CrossRef] [Google Scholar]
- A new diarylheptanoid and a new diarylheptanoid glycoside isolated from the roots of Juglans mandshurica and their anti-inflammatory activities. Nat. Prod. Res.. 2019;33:701-707.
- [CrossRef] [Google Scholar]
- PHMH, a diarylheptanoid from Alpinia officinarum attenuates VEGF-induced angiogenesis via inhibition of the VEGFR-2 signaling pathway. Food Funct. 2019;10:2605-2617.
- [CrossRef] [Google Scholar]
- Diarylheptanoids from Curcuma phaeocaulis Suppress IL-6-Induced STAT3 Activation. Planta Med. 2019;85:94-102.
- [CrossRef] [Google Scholar]
- Bahamaolides A and B, antifungal polyene polyol macrolides from the marine actinomycete Streptomyces sp. J. Nat. Prod.. 2012;75:959-967.
- [CrossRef] [Google Scholar]
- Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit. Rev. Food Sci. Nutr.. 2017;57:2889-2895.
- [CrossRef] [Google Scholar]
- Qualitative and quantitative analysis of curcuminoids in herbal medicines derived from Curcuma species. Food Chem.. 2011;126:1890-1895.
- [CrossRef] [Google Scholar]
- Analysis of bisabolocurcumin ether (a terpene-conjugated curcuminoid) and three curcuminoids in Curcuma species from different regions by UPLC-ESI MS/MS and their in vitro anti-inflammatory activities. J. Funct. Foods.. 2019;52:186-195.
- [CrossRef] [Google Scholar]
- Vinegar-processed Curcuma phaeocaulis promotes anti-angiogenic activity and reduces toxicity in zebrafish and rat models. Pharm. Biol.. 2021;59:408-415.
- [CrossRef] [Google Scholar]
- Naturally occurring diarylheptanoids. Nat. Prod. Commun.. 2010;5:1687-1708.
- [CrossRef] [Google Scholar]
- Microbial conversion of curcumin into colorless hydroderivatives by the endophytic fungus Diaporthe sp. associated with Curcuma longa. Chem. Pharm.. 2011;59:1042-1044.
- [CrossRef] [Google Scholar]
- Characterization, Classification and Authentication of Turmeric and Curry Samples by Targeted LC-HRMS Polyphenolic and Curcuminoid Profiling and Chemometrics. Molecules. 2020;25:2942.
- [CrossRef] [Google Scholar]
- Suppression of inflammatory cytokine production by ar-turmerone isolated from Curcuma phaeocaulis. Chem. Biodivers.. 2014;11:1034-1041.
- [CrossRef] [Google Scholar]
- Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Crit. Rev. Food Sci. Nutr.. 2020;60:887-939.
- [CrossRef] [Google Scholar]
- Two-dimensional NMR analysis of acetonide derivatives in the stereochemical assignment of polyol chains: the absolute configurations of dermostatins A and B. J. Org. Chem.. 1997;62:2925-2934.
- [CrossRef] [Google Scholar]
- Ethanol extracts from twelve Curcuma species rhizomes in China: Antimicrobial, antioxidative and anti-inflammatory activities. S. Afr. J. Bot.. 2021;140:167-172.
- [CrossRef] [Google Scholar]
- Secondary metabolites of the sponge-derived fungus Acremonium persicinum. J. Nat. Prod.. 2013;76:1432-1440.
- [CrossRef] [Google Scholar]
- YM-47522, a novel antifungal antibiotic produced by Bacillus sp. II. Structure and Relative Stereochemistry. J. Antibiot.. 1996;49:345-348.
- [CrossRef] [Google Scholar]
- Diarylheptanoid: A privileged structure in drug discovery. Fitoterapia.. 2020;142:104490
- [CrossRef] [Google Scholar]
- New lignans from the fruits of Leonurus japonicus and their hepatoprotective activities. Bioorg Chem.. 2021;115:105252
- [CrossRef] [Google Scholar]
- Lignans and neolignans from Sinocalamus affinis and their absolute configurations. J. Nat. Prod.. 2011;74:1188-1200.
- [CrossRef] [Google Scholar]
- Phenolic glucosides from Dendrobium aurantiacum var. denneanum and their bioactivities. Molecules.. 2013;18:6153-6160.
- [CrossRef] [Google Scholar]
- Anti-inflammatory diarylheptanoids and phenolics from the rhizomes of kencur (Kaempferia galanga L.) Ind. Crop Prod.. 2018;125:454-461.
- [CrossRef] [Google Scholar]
- New diarylheptanoids and diarylheptanoid glucosides from the rhizomes of Tacca chantrieri and their cytotoxic activity. J. Nat. Prod.. 2002;65:283-289.
- [CrossRef] [Google Scholar]
- Structures and absolute configurations of butenolide derivatives from the isopod-associated fungus Pidoplitchkoviella terricola. Phytochemistry. 2022;193:112981
- [CrossRef] [Google Scholar]
- Anisotanols A—D, Four Norsesquiterpenoids with an Unprecedented Sesquiterpenoid Skeleton from Anisodus tanguticus. Chin. J. Chem.. 2021;39:3375-3380.
- [CrossRef] [Google Scholar]
- Stachydrine promotes angiogenesis by regulating the VEGFR2/MEK/ERK and mitochondrial-mediated apoptosis signaling pathways in human umbilical vein endothelial cells. Biomed. Pharmacother.. 2020;131:110724
- [CrossRef] [Google Scholar]
- Antioxidant and Cytoprotective Effects of New Diarylheptanoids from Rhynchanthus beesianus. J. Agric. Food Chem.. 2021;69:6229-6239.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105572.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




